
Syntheses, structures and gas sorption properties of two coordination polymers with a unique type of supramolecular isomerism†
Xiao-Lin
Qi
,
Jia-Wen
Ye
,
Rui-Biao
Lin
,
Pei-Qin
Liao
,
Si-Yang
Liu
,
Chun-Ting
He
,
Jie-Peng
Zhang
* and
Xiao-Ming
Chen
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry & Chemical Engineering, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhangjp7@mail.sysu.edu.cn; Fax: +86-20-84112245; Tel: +86-20-84115358
First published on 5th December 2014
Abstract
The solvothermal reaction of a short bridging ligand 1H-pyrazole-4-carboxylic acid (H2pc) and Zn(NO3)2 in a mixed solvent containing N,N-dimethylacetamide (DMA) at 110 °C produced two genuine supramolecular isomers [Zn2(pc)2(DMA)] (1) and [Zn2(pc)2]·DMA (2). Single-crystal X-ray diffraction studies showed that 1 is a densely packed layered structure with DMA molecules coordinated on the layered surface, and 2 is a porous three-dimensional framework structure with DMA molecules filled inside the pore without coordination to any metal ion, which represents a rare case of coordination-sphere isomerism in coordination polymers. Removal of the coordinated DMA molecules in 1 occurs at high temperatures, accompanying the irreversible formation of an unidentified nonporous phase. On the other hand, the guest DMA molecules in 2 can be readily removed under mild conditions to give a new porous phase [Zn2(pc)2] (2′). Gas-sorption measurements of 2′ revealed that the framework has significant flexibility and selective CO2 adsorption at room temperature.
Introduction
Porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) have attracted much attention partly because of their rich structural diversity and potential applications.1 Supramolecular isomerism in coordination polymers involves the existence of more than one type of network superstructure from the same building blocks, which is caused from the structural uncertainty in the self-assembly processes, but this can also be regarded as an invaluable opportunity for the study of the self-assembly and crystal growth processes, as well as the structure–property relationship of these interesting materials.2According to the type of structural difference, supramolecular isomerism in PCPs can be categorized into many classes, such as structural isomerism, topological isomerism, interpenetration isomerism, etc.3 For example, in the isomers [Cu(SCN)(dpt)] (dpt = 2,4-bis(4-pyridyl)-1,3,5-triazine) reported by Champness et al., Cu(I) cations coordinated to SCN anions and dpt ligands to construct N3S and N2S2 coordination spheres, respectively, forming a 3D porous framework structure with 65·8 topology and a stair-like chain.3b With the same coordination geometries, the building units can be linked to form extended networks with different topologies, which can be defined as topological isomers. For example, Maverick et al. found that the Cu(II) ion tends to coordinate to two β-ketonate and two pyridyl groups from four 3-(4-pyridyl)pentane-2,4-dionate ligands to form a square-planar 4-connected node, which can be linked to form a two-dimensional (2D) sql network or 3D nbo network depending on the solvent used in the syntheses.3c With identical coordination environments and topologies, porous networks may interpenetrate differently to form interpenetration isomers.3j,k Comparison experiments indicate that a larger interpenetration number usually leads to an increase in the low-pressure adsorption performance, although the porosity and saturated adsorption capacity are decreased,4 also the interpenetration number is an important influencing factor for framework flexibility.5 In PCPs, supramolecular isomerism usually involves the coordination frameworks rather than the whole crystal. Strictly, most supramolecular isomers in PCPs are not genuine supramolecular isomers because there are different guest molecules in the pore systems.3
Herein, we report two new coordination polymers, namely [Zn2(pc)2(DMA)] (1) and [Zn2(pc)2]·DMA (2) (H2pc = 4-carboxylpyrazole, DMA = N,N-dimethylacetamide), showing a new type of supramolecular isomerism, in which the crystals have the same chemical composition but the coordination frameworks consist of different components. Because the DMA molecules serve as terminal coordination molecules and guests, respectively, the two isomers display totally different porosity and sorption behavior.
Experimental section
Materials and methods
The ligand H2pc was synthesized according to the literature method.6 Other reagents and solvents were commercially available and used without further purification. Elemental analyses (C, H, N) were performed with a Vario EL elemental analyzer. FT-IR spectra were obtained using KBr pellets on a Bruker EQUINOX 55 FT-IR spectrometer in the 4000–400 cm−1 region. Powder X-ray diffraction (PXRD) patterns were recorded on a Bruker D8 ADVANCE or a D8 DAVANCI X-ray powder diffractometer (Cu Kα). Thermogravimetry (TG) analyses were performed using a TA Q50 instrument. Each sample was heated from room temperature to 700 °C at a heating rate of 5.0 °C min−1 under a nitrogen atmosphere. Gas sorption isotherms were measured on Belsorp MAX or Micromeritics ASAP 2020M volumetric adsorption apparatus. The sample was placed in the sample tube and dried under high vacuum at 170 °C for 4 h to remove the remnant solvent molecules prior to measurements.Synthesis
Crystal structure determination
Diffraction data were collected on a Bruker Apex CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Absorption corrections were applied by using the multi-scan program SADABS.7 The structures were solved by direct methods and refined with the full-matrix least-squares technique with the SHELXTL program package.8 Anisotropic thermal parameters were applied to all non-hydrogen atoms. All hydrogen atoms were generated geometrically. The disordered bridging ligands and terminal coordinated DMA molecules were subjected to geometric restrains during the refinement.Results and discussion
Syntheses and structures
Both 1 and 2 were synthesized under similar, mild solvothermal conditions, except that H2O and methanol were used as the main solvents, respectively. A small amount of DMA was added to serve as not only a necessary component of 1 and 2, but also a base to deprotonate the H2pc ligands. Without the addition of DMA, no precipitate formed after the solvothermal reactions. Infrared spectra of 1 and 2 show different adsorption peaks around 1640–1450 cm−1, suggesting that the DMA molecules reside in different environments or the carboxylate groups adopt different coordination modes (Fig. S1†).1 crystallizes in the C2/m space group, containing two Zn atoms (Zn1 at a C2v axis, 1/2 occupancy, Zn2 at a mirror plane, 1/2 occupancy), two fully deprotonated pc2− ligands both located at mirror planes with 1/2 occupancies (the molecular axis of one is parallel to its mirror plane, while that of the other is perpendicular to its mirror plane, so the pyrazolate and carboxylate ends of the latter are statistically disordered), and one DMA molecule (lying on the same mirror plane with Zn2, 1/2 occupancy) in the asymmetric unit. As shown in Fig. 1 and S2,† Zn1 is tetrahedrally coordinated by two pyrazolate nitrogen atoms (Zn1–N1 1.994(4), Zn1–N2 2.005(4) Å) from two ordered pc2− ligands and two carboxylate/pyrazolate donors (Zn1–O3 1.995(7) and Zn1–N4 1.909(6) Å) from two symmetrically disordered pc2− ligands. Zn2 coordinates to a chelating carboxylate from the ordered pc2− (Zn2–O1 2.416(5) and Zn2–O2 1.996(3) Å) and a carbonyl oxygen (Zn2–O5 2.000(3) Å) of DMA, as well as two carboxylate oxygen/pyrazolate nitrogen atoms [Zn2–N3 1.910(6) and Zn2–O4 2.048(8) Å] from the symmetrically disordered pc2− to form a seriously distorted triangular-bipyramidal coordination environment. All the Zn–N and Zn–O bond lengths are consistent with the expected values.9 The ordered and symmetrically disordered pc2− ligands are both four coordinated, but they serve as three- and four-connected linkers, respectively, which bridge the Zn atoms along two mutually perpendicular directions to form a thick, porous 2D layer extending across the bc-plane with DMA molecules pointing outwards (Fig. 1a). These layers interdigitate to form the final 3D structure with DMA molecules inserted into pores of adjacent layers, so that no solvent accessible free volume is found in 1 (Fig. 1b).
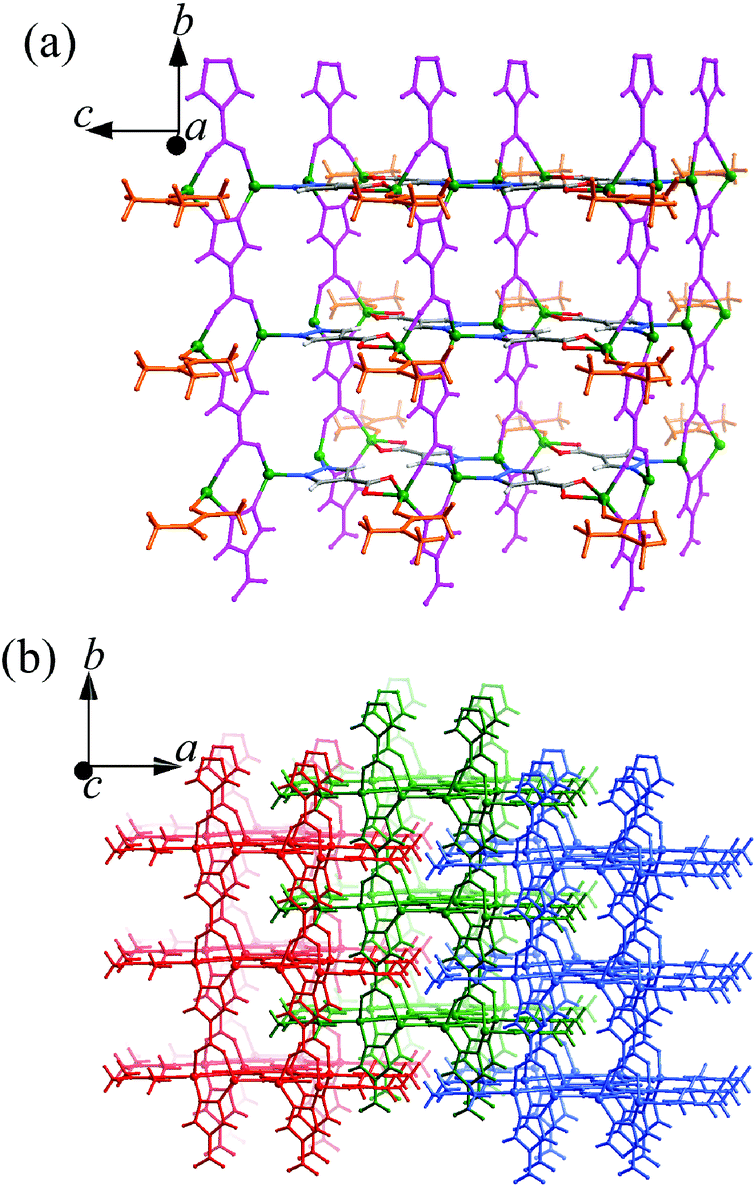 | ||
| Fig. 1 Perspective views of the (a) 2D layer (the symmetrically disordered pc2− ligands are highlighted in magenta, and only one possible orientation is shown; the coordinated DMA molecules are highlighted in orange) and (b) 3D packing (adjacent layers are shown in different colours to highlight the interdigitation) of 1. | ||
2 crystallizes in the Pmma space group containing a Zn atom (Zn1 at a mirror plane, 1/2 occupancy), two fully deprotonated pc2− ligands (both at the C2h axis with 1/4 occupancy so that all the pyrazolate and carboxylate ends are symmetrically disordered) and a non-coordinated DMA molecule (at a mirror plane, 1/2 occupancy) in the asymmetric unit. As shown in Fig. S3,† Zn1 is tetrahedrally coordinated by four disordered carboxylate/pyrazolate donors (Zn1–N/O 1.9300(4)–1.9785(3) Å) from four pc2− ligands. Each pc2− ligand also adopts the same coordination environment linking to four Zn1 atoms with exo-tetradentate coordination to construct a 3D porous framework (void 27%). There are small narrow 3D channels consisting of small cavities of 4.0 × 5.1 × 3.8 Å3 (filled with DMA molecules) and even smaller apertures of 2.5 × 5.0 Å2, 1.6 × 2.4 Å2, and 1.5 × 4.0 Å2 running along the a-, b-, and c-axes, respectively, taking into account the van der Waals radii (Fig. 2).
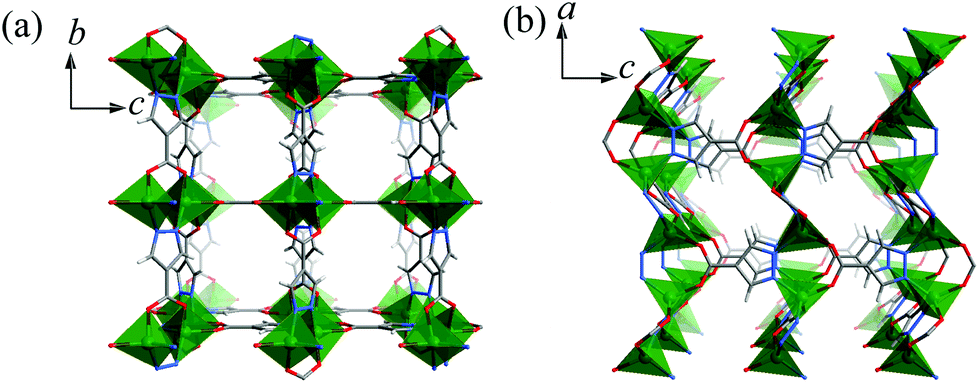 | ||
| Fig. 2 Perspective views of the 3D coordination framework structure of 2 along the (a) a- and (b) b-axes. Note that all pc2− ligands have statistically disordered pyrazolate/carboxylate ends, and only one possible ligand orientation is shown. | ||
In 1 and 2, many of the pyrazolate and carboxylate groups are statistically disordered, because the pyrazolate usually adopts a similar bidentate bridging coordination mode as the carboxylate. Several PCPs based on bifunctional pyrazolate–carboxylate ligands have been reported, some of which also show statistically disordered pyrazolate and carboxylate groups.9,10 The key structural difference between 1 and 2 is the role of the DMA molecules, which reside at different coordination spheres. Interestingly, the H2O and MeOH molecules do not participate in the crystal composition although they are the main solvents and should serve as important additives in the syntheses of the two isomers.2b In 1, the DMA molecules coordinate to the surface of the grid layer, preventing it from extending along the a-axis into a 3D structure. Also because of the coordination of DMA molecules, the pc2− ligands in 1 connect with less Zn atoms as compared with those in 2. On the other hand, the DMA molecules fill inside the grid layers in 2, so that the coordination network is extended into a microporous 3D framework. In most cases, DMA serves merely as a solvent or a basic agent for deprotonating the ligand, rather than a ligand. It should be noted that the crystal compositions of 1 and 2 are the same, meaning that they are genuine supramolecular isomers. Furthermore, supramolecular isomerism based on this type of structural difference has not been reported for coordination polymers in the literature so far.
Thermal stability
Thermal stabilities of 1 and 2 were studied by TG and PXRD. As shown in Fig. S5a,† the PXRD pattern of as-synthesized 1 is consistent with the single crystal X-ray diffraction data. A small weight loss of ca. 2.4% was observed from 180 °C to 220 °C (Fig. S4†), which might be attributed to the DMA molecules coordinated on the external surface of the crystals (the calculated weight loss for all the coordinated DMA molecules is 20%). At 200 °C, displacement and splitting of diffraction peaks in the PXRD patterns between 11° and 13° suggest obvious distortion of the framework (Fig. S5a†). Another weight loss of 17.2% appears in the range of 310–400 °C, which corresponds to all the other coordinated DMA molecules inside the crystals. The widening and displacement of the diffraction peaks above 200 °C indicate that the framework kept distorting during the loss of DMA molecules before decomposition. As proved by PXRD, the original framework cannot be recovered by dipping the desolvated samples (after heating at 350 °C) in DMA for 3 days (Fig. S4a†), which suggests that the framework was irreversibly damaged upon the loss of the coordinated DMA molecules and no porosity can be found in the desolvated structure. For 2, all DMA guest molecules escape at 170–220 °C (Fig. S4†), and the distorted guest-free framework 2′ decomposes over 400 °C (Fig. S3 and S4b†). Obvious framework distortion can be found after the loss of guest molecules and the original framework structure 2 can be recovered by exposing 2′ to MeOH vapor at room temperature, which suggests that 2 is a flexible framework and MeOH molecules can act as guest molecules to fill the pore system and drive the structural transformation of the host framework. On the other hand, structural transformation of 2′ to 2 can be realized by heating 2′ in DMA at 140 °C, instead of dipping 2′ in DMA at room temperature, which can be attributed to the bigger volume of DMA molecules, preventing them from getting across the small apertures at ambient temperature (Fig. S5b†).Although 1 and 2 cannot transform into each other at high temperatures (Fig. S5†), we tried to explore whether the structural transformation of the two isomers could occur by either refluxing or solvothermal reaction in the solvent used for the syntheses of their counterparts. Nevertheless, no structural transformation between 1 and 2 can be observed (Fig. S6†). By comparing the asymmetric units and the packing structure of the two isomers, we can find that the transformation from 1 to 2 requires the departure of the coordinated DMA molecules, the change of the connection number of the pc2− ligands from three to four, and translation of the disordered pc2− ligands, which can only occur after network dissociation (Fig. S7†).
Gas sorption
To examine the pore characteristics and gas adsorption properties of guest-free 2′, CO2, N2, and H2 isotherms were measured (Fig. 3). A type-I-like N2 sorption isotherm at 77 K with an apparent Langmuir surface area of 781 m2 g−1 was obtained. In contrast to the typical type I adsorption isotherm, the sorption amount keeps increasing at high pressures. The adsorption amount at P/P0 = 0.01 and 0.95 is about 125 and 183 cm3(STP) g−1, respectively. For comparison, the value calculated from the single-crystal structure is 122 cm3(STP) g−1 (Fig. 3a). These results further confirm the significant flexibility of 2′ and it can be speculated that 2′ may expand to a state more porous than the as-synthesized one.11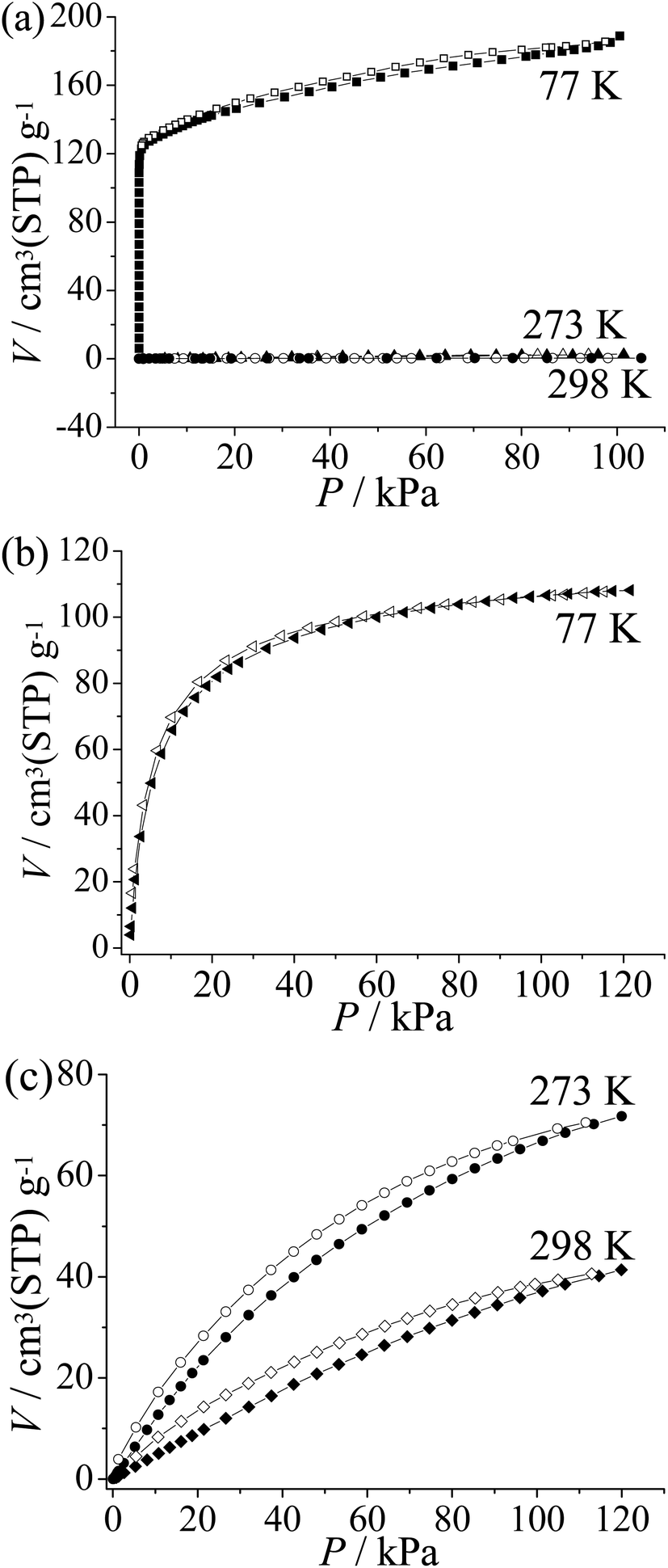 | ||
| Fig. 3 (a) N2, (b) H2 and (c) CO2 adsorption (solid) and desorption (open) isotherms of 2′. | ||
The H2 sorption isotherm of 2′ measured at 77 K shows type-I characteristics. The sorption amount is near to saturation at low pressure (86 cm3(STP) g−1 at 0.26 bar) and the uptake increases gradually to 107 cm3(STP) g−1 or 0.7 wt% at 1 atm (Fig. 3b). The relatively low saturated sorption amount is mainly caused from the small pore volume. The H2 density can be calculated to be 0.064 g cm−3 in the pore system of 2′, which is very close to the density of liquid H2 (0.07 g cm−3).12
CO2 adsorption isotherms were obtained for 2′ at 273 K and 298 K, which show moderate sorption amounts of 67 and 37 cm3(STP) g−1 at 1 atm, respectively (Fig. 3c). Using the Virial fitting method and the Clausius–Clapeyron equation, the CO2 adsorption enthalpy on 2 is calculated to be around 25 kJ mol−1 at zero coverage (Fig. S8†), which is relatively low among PCPs.13 Furthermore, 2 shows only surface adsorption for N2 at 273 K and 298 K (2.0 cm3(STP) g−1 and 0.45 cm3(STP) g−1 at P = 101 kPa respectively). The CO2/N2 selectivity is estimated to be ca. 47 at 273 K and 17 at 298 K by comparing the CO2 and N2 uptake at the relevant partial pressures for flue gas (CO2 0.15 atm; N2 0.75 atm),13b which is similar to that of zeolite 13X (ca. 20) used for CO2 capture.14
Conclusions
In short, two genuine supramolecular isomers, with a layered structure and a flexible microporous 3D framework, have been synthesized under similar solvothermal conditions using water and methanol as the main solvents, although water or methanol does not participate in the crystal composition. More interestingly, DMA molecules, as the minor component of the mixed solvents used for the syntheses, are included in the isomers as either terminal ligands on the layered structure or lattice guest molecules in the pores of the 3D framework, representing a new type of supramolecular isomerism in coordination polymers. Interestingly, the 3D framework isomer can be activated to form an ultramicroporous material with high framework flexibility and adsorption affinity for H2. These results highlight that even subtle modification of the synthetic condition in a very simple metal–ligand system can produce very different outcomes in the context of material structures and properties.Acknowledgements
This work was supported by the “973 Project” (2012CB821706 and 2014CB845602), NSFC (21290173, 21225105 and 21371181) and NSF of Guangdong (S2012030006240).Notes and references
- (a) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed; (b) M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310 CrossRef CAS.
- (a) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS PubMed; (b) J.-P. Zhang, X.-C. Huang and X.-M. Chen, Chem. Soc. Rev., 2009, 38, 2385 RSC; (c) T. A. Makal, A. A. Yakovenko and H.-C. Zhou, J. Phys. Chem. Lett., 2011, 2, 1682 CrossRef CAS.
- (a) P. Jensen, S. R. Batten, B. Moubaraki and K. S. Murray, J. Solid State Chem., 2001, 159, 352 CrossRef CAS; (b) S. A. Barnett, A. J. Blake, N. R. Champness and C. Wilson, Chem. Commun., 2002, 1640 RSC; (c) B. Chen, F. R. Fronczek and A. W. Maverick, Chem. Commun., 2003, 2166 RSC; (d) F.-C. Liu, Y.-F. Zeng, J.-P. Zhao, B.-W. Hu, E. C. Sañudo, J. Ribas and X.-H. Bu, Inorg. Chem., 2007, 46, 7698 CrossRef CAS PubMed; (e) R. Peng, S.-R. Deng, M. Li, D. Li and Z.-Y. Li, CrystEngComm, 2008, 10, 590 RSC; (f) S. Wang, H. Zang, C. Sun, G. Xu, X. Wang, K. Shao, Y. Lan and Z. Su, CrystEngComm, 2010, 12, 3458 RSC; (g) J. J. Jiang, M. Pan, J. M. Liu, W. Wang and C. Y. Su, Inorg. Chem., 2010, 49, 10166 CrossRef CAS PubMed; (h) X. Zhao, H. He, F. Dai, D. Sun and Y. Ke, Inorg. Chem., 2010, 49, 8650 CrossRef CAS PubMed; (i) D. Sun, S. Ma, J. M. Simmons, J. R. Li, D. Yuan and H. C. Zhou, Chem. Commun., 2010, 46, 1329 RSC; (j) S. K. Elsaidi, M. H. Mohamed, L. Wojtas, A. Chanthapally, T. Pham, B. Space, J. J. Vittal and M. J. Zaworotko, J. Am. Chem. Soc., 2014, 136, 5072 CrossRef CAS PubMed; (k) C.-T. He, P.-Q. Liao, D.-D. Zhou, B.-Y. Wang, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2014, 5, 4755 RSC.
- (a) S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H.-C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858 CrossRef CAS PubMed; (b) P. Ryan, L. J. Broadbelt and R. Q. Snurr, Chem. Commun., 2008, 4132 RSC.
- H.-L. Jiang, T. A. Makal and H.-C. Zhou, Coord. Chem. Rev., 2013, 257, 2232 CrossRef CAS PubMed.
- C. Foces-Foces, A. Echevarría, N. Jagerovic, I. Alkorta, J. Elguero, U. Langer, O. Klein, M. Minguet-Bonvehí and H.-H. Limbach, J. Am. Chem. Soc., 2001, 123, 7898 CrossRef CAS PubMed.
- SADABS, SMART, and SAINT, Bruker AXS Inc., Madison, WI, 2002 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- (a) C. Montoro, F. Linares, E. Q. Procopio, I. Senkovska, S. Kaskel, S. Galli, N. Masciocchi, E. Barea and J. A. Navarro, J. Am. Chem. Soc., 2011, 133, 11888 CrossRef CAS PubMed; (b) D. J. Tranchemontagne, K. S. Park, H. Furukawa, J. Eckert, C. B. Knobler and O. M. Yaghi, J. Phys. Chem. C, 2012, 116, 13143 CrossRef CAS.
- (a) E. Quartapelle Procopio, F. Linares, C. Montoro, V. Colombo, A. Maspero, E. Barea and J. A. Navarro, Angew. Chem., Int. Ed., 2010, 49, 7308 CrossRef CAS PubMed; (b) E. Quartapelle Procopio, T. Fukushima, E. Barea, J. A. Navarro, S. Horike and S. Kitagawa, Chem. – Eur. J., 2012, 18, 13117 CrossRef CAS PubMed; (c) N. M. Padial, E. Quartapelle Procopio, C. Montoro, E. Lopez, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290 CrossRef CAS PubMed; (d) X.-L. Qi, C. Zhang, B.-Y. Wang, W. Xue, C.-T. He, S.-Y. Liu, W.-X. Zhang and X.-M. Chen, CrystEngComm, 2013, 15, 9530 RSC; (e) C. Heering, I. Boldog, V. Vasylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757 RSC; (f) W. Y. Gao, R. Cai, L. Meng, L. Wojtas, W. Zhou, T. Yildirim, X. Shi and S. Ma, Chem. Commun., 2013, 49, 10516 RSC.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC.
- (a) L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; (b) M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed.
- (a) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791 CrossRef CAS PubMed; (b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- Q. Yang, S. Vaesen, F. Ragon, A. D. Wiersum, D. Wu, A. Lago, T. Devic, C. Martineau, F. Taulelle, P. L. Llewellyn, H. Jobic, C. Zhong, C. Serre, G. De Weireld and G. Maurin, Angew. Chem., Int. Ed., 2013, 52, 10316 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of the crystal data, additional structural plots, adsorption enthalpies of CO2, and X-ray crystallographic files in the CIF format. CCDC 1031539–1031540. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00190g |
| This journal is © the Partner Organisations 2015 |