
Redox and acidic properties of chalcogenido-substituted mixed-metal polyoxoanions: a DFT study of α-[PW11O39ME]4− (M = Nb, Ta; E = O, S, Se)
Cai Xia
Wu
,
Li Kai
Yan
*,
Ting
Zhang
and
Zhong Min
Su
*
Institute of Functional Material Chemistry, Key Laboratory of Polyoxometalate Science of Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: yanlk924@nenu.edu.cn; zmsu@nenu.edu.cn; Fax: +86-0431-85684009
First published on 22nd December 2014
Abstract
The redox and acidic properties of α-Keggin anions [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) have been investigated by using the density functional theory (DFT) method. The calculated results for the studied clusters are consistent with the hypothesis that the substitution of one O atom by S or Se atoms in polyoxometalates (POMs) modifies the relative energy of the lowest unoccupied molecular orbital, inducing slight changes in the redox properties of POMs. The electronic structures and reduction energies of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) confirm that the substitution of one O atom by S and Se atoms in POMs enhances the redox properties, which is in good agreement with the cyclic voltammetry characterization of α-[PW11NbO40]4− and α-[PW11NbSO39]4−. The bonding energies for adding the first proton to the anions and for ammonia coordination to the protonated α-[PW11O39NbE]4− (E = O, S, Se) were simulated to study the basicity of external oxygen sites and the acidity. The results show that the protonation energy becomes more positive after substitution by S and Se, indicating that the substitution enhances the acidity of POMs. The adsorption energy of ammonia in the case of α-[PW11O39NbE]4− (E = S, Se) is more negative than that of α-[PW11O40NbO]4−, which also provides the same result as mentioned above.
Introduction
Polyoxometalates (POMs) have attracted great attention for many years due to the controllability of their molecular properties (composition, size, shape, acidity and redox potential), which imparts diverse applications in catalysis,1–3 electrochemistry,4 medicine,5,6 magnetism,7,8 and especially in the area of oxidation and acid catalysis. Of the vast range of physicochemical properties that POMs possess, the redox properties have great relevance. Enormous amounts of information on the electronic structures and reduction potentials of POMs have been collected, thus making POMs technically applicable and largely appealing in catalysis.9 Oxidation catalysis by POMs has received much attention because the chemical properties of POMs can be finely tuned, and POMs are thermally stable in comparison with organometallic complexes, organocatalysts, and enzymes. To date, various kinds of POMs have been developed for H2O2- and O2-based green oxidations.10,11 Furthermore, POMs are a potential replacement for corrosive and environmentally toxic liquid acid catalysts, which are currently used in both solution and solid forms12 due to their high catalytic activity in acid-catalyzed reactions,13,15 such as isobutane alkylation by n-butenes.13,14 The first step in many catalytic processes involves the adsorption or release of protons. Therefore, the catalytic activity of POMs is usually connected to their acid strength. The protonation and acid strength of phosphotungstic acid, which are potential indicators of solid acid activity, are still widely debated in the literature.16,17There are many aspects that can affect the redox and acidic properties of POMs. For example, the redox properties of POMs can be altered by the substitution of addenda metals or heteroatoms within the POM framework.18,19 Another emerging strategy to modify the redox and acidic properties of POMs consists of covalent grafting of addenda metallic cations on the surface of metal-saturated POM anions.20 Moreover, redox behavior can be finely adjusted by changing the size and charge of the cluster.21 Alternately, the properties of POMs can be modified through the substitution of oxo ligands by other ligands, such as sulfur and selenium. The corresponding chemistry of the higher congeners of oxygen, namely the chalcogen sulfur, has also received much attention in the development of POMs due to its possible implications in the reduction of protons into hydrogen22–24 and in hydrodesulfurization processes. Indeed, sulfur and selenide compounds have been widely reviewed.25 The inclusion of donor sulfur or selenide is expected to generate new properties for POMs. The chalcogenido-substituted compounds appear less stable in solution than those of the oxo analogues, confirming their higher instability and versatility.26
To date, two strategies have been developed to synthesize POMs containing sulfur and selenium. One of the routes uses a preformed sulfur-bridged [M2S2O2]2+ (M = W or Mo) or [Mo3S4]4+ cationic precursor which reacts with anionic POMs.27,28 Another methodology to prepare POMs containing sulfur and selenium is through the direct exchange of sulfur or selenium for oxygen in POMs. The latter is difficult because the substitution is often accompanied by reduction of the metal center and/or metal–oxygen framework degradation.29 In order to overcome these difficulties, Klemperer and co-workers have chosen to substitute oxygen by sulfur in poorly reducible Ta![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NbO groups previously introduced in a Lindqvist framework. They firstly succeeded in synthesizing the isolable early-transition-metal d0 polyoxothioanions, [TaW5O18S]3− and [NbW5O18S]3−.30 Only one terminal oxygen atom could be replaced by S or Se in the Nb-substituted or Ta-substituted POM. Such an approach has been successfully applied to Keggin-type derivatives. In 1995, Cadot and co-workers synthesized the first Keggin-type anion α-[PW11NbSO39]4− with sulfur replacing the oxygen.31 Later, Radkov and co-workers synthesized a series of group 5 mixed-metal Keggin-type polytungstophosphates [PW11O39ME]4− (M = Nb, E = S, Se; M = Ta, E = S).32
O and NbO groups previously introduced in a Lindqvist framework. They firstly succeeded in synthesizing the isolable early-transition-metal d0 polyoxothioanions, [TaW5O18S]3− and [NbW5O18S]3−.30 Only one terminal oxygen atom could be replaced by S or Se in the Nb-substituted or Ta-substituted POM. Such an approach has been successfully applied to Keggin-type derivatives. In 1995, Cadot and co-workers synthesized the first Keggin-type anion α-[PW11NbSO39]4− with sulfur replacing the oxygen.31 Later, Radkov and co-workers synthesized a series of group 5 mixed-metal Keggin-type polytungstophosphates [PW11O39ME]4− (M = Nb, E = S, Se; M = Ta, E = S).32
Cadot and co-workers have investigated the redox behavior of α-[PW11NbO40]4− and α-[PW11NbSO39]4− by cyclic voltammetry, concluding that the tungsten atoms were reduced at lower potentials due to O/S substitution.31 However, theoretical studies on POMs containing sulfur and selenium are still rare. Density functional theory (DFT) has proved successful in predicting the properties of molecules and materials over the past decade. In our previous work, we have theoretically investigated the properties of [X2Mo5O23]6− (X = PV, SVI, AsV, SeVI), [PTi2W10O40]7−, and [Mo6O17R2]2− (R = organoimido) clusters, such as bonding character, redox properties, protonation, stability, and second-order nonlinear optical (NLO) properties.33 The results showed that the quantum chemistry calculations based on the DFT formalism may be very useful for understanding the properties of POMs, including redox properties and acidity. Poblet and co-workers have focused much attention on the fundamental aspects of POMs and their derivatives, and they have particularly made an effort to gain a deep understanding of their redox properties.34,35 In this work, we present a detailed DFT study on a series of chalcogenido-substituted anions [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) to analyze the electronic structure and the basicity of external oxygen sites aiming at describing the redox properties and acidity of these anions.
Computational methods
In this work, all DFT calculations were carried out with the Amsterdam Density Functional program (ADF2012.01).36 Electron correlation was treated within the local density approximation (LDA) in the Vosko–Wilk–Nusair (VWN) parametrization.37 The nonlocal corrections of Becke38 and Perdew39,40 were added to the exchange and correlation energies, respectively. The basis functions used for describing the electrons are Slater-type orbitals with (triple-) TZP quality. The frozen core approximation for metal atoms has been used to reduce the large computational effort. These cores include all orbitals up to 3d for niobium atoms, 4d for tantalum and tungsten atoms. These basis sets have been used in conjunction with the zero-order regular approximation41 (ZORA) to the relativistic effects. A conductor solvent model (COSMO) was introduced to consider the solvent effects42 with acetonitrile as the solvent. The van der Waals radii, i.e. COSMO hole sizes, were set as 1.08 Å, 1.41 Å, 1.40 Å, 1.92 Å, 1.82 Å, 1.93 Å, 2.10 Å, 2.07 Å and 2.17 Å for H, N, O, P, S, Se, W, Nb and Ta, respectively. The geometry optimization of [PW11O39{MVE}]4−(M = Nb, Ta; E = O, S, Se) under Cs symmetry constraints was carried out and the optimized structures are shown in Fig. 1. We divide the models into two groups, the first one with M = Nb and the other one with M = Ta.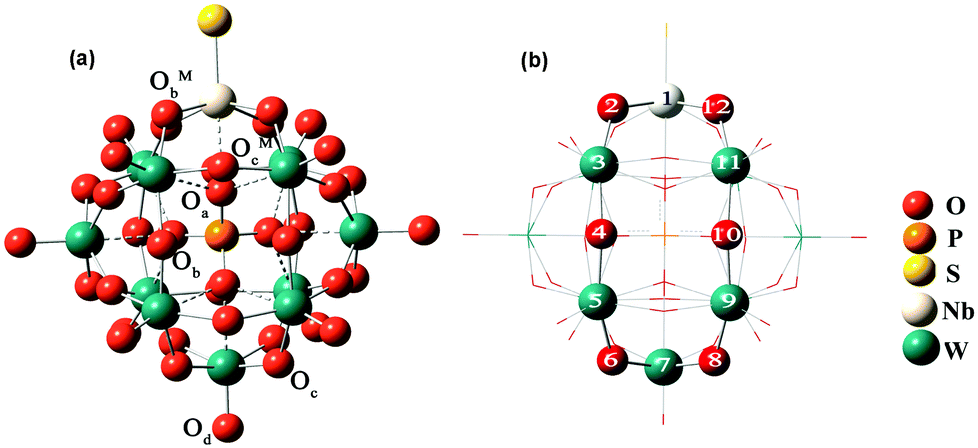 | ||
| Fig. 1 Distinct types of oxygen atoms (a) and model of the NbW5 ring (b) in [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se). | ||
Results and discussion
Substitution effects on the geometrical structures of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se)
There are three types of oxygen atoms in traditional Keggin-type phosphotungstates: center tetrahedral oxygen (Oa), terminal oxygen (Od) double-bonded to a single tungsten atom, and bridging oxygens (Ob and Oc). The bridging oxygens can be divided into oxygen atoms that bridge two tungsten atoms not sharing a central oxygen atom (corner-sharing, Ob), and oxygen atoms that bridge two tungsten atoms sharing the same central oxygen atom (edge-sharing, Oc). In [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se), there are two additional types of bridging oxygens linked with mixed-metals Nb/Ta (corner-sharing, ObM and edge-sharing, OcM), as shown in Fig. 1a. The selected optimized bond distances in [PW11O39ME]4− (M = Nb, Ta, E = O, S, Se) are summarized in Table 1.| ME | NbO | NbS | NbSe | TaO | TaS | TaSe |
|---|---|---|---|---|---|---|
| M–E | 1.757 | 2.219 | 2.350 | 1.767 | 2.228 | 2.362 |
| M–ObM | 2.027 | 2.014 | 2.012 | 2.007 | 1.992 | 1.989 |
| M–OcM | 2.031 | 2.021 | 2.020 | 2.015 | 2.002 | 2.001 |
| W–Od | 1.729 | 1.729 | 1.729 | 1.729 | 1.728 | 1.728 |
| W–Ob | 1.933 | 1.933 | 1.933 | 1.933 | 1.933 | 1.933 |
| W–Oc | 1.944 | 1.943 | 1.943 | 1.943 | 1.943 | 1.943 |
| W–Oa | 2.469 | 2.467 | 2.467 | 2.470 | 2.468 | 2.468 |
| P–Oa | 1.556 | 1.556 | 1.556 | 1.556 | 1.555 | 1.555 |
| M–Oa | 2.557 | 2.570 | 2.568 | 2.526 | 2.543 | 2.541 |
| Δd | 0.020 | 0.018 | 0.018 | 0.019 | 0.016 | 0.016 |
In [PW11O39NbO]4−, the computed bond length of Nb–Od is 1.757 Å, and the bond distances between Nb and the bridging oxygen atoms are calculated to be 2.027 and 2.031 Å, which are larger than the corresponding bonds between W and the bridging oxygen atoms. The bridging oxygen atoms in [PW11O39NbO]4− form two nonequivalent metal–oxygen bonds with one bond being shorter than the other, which leads to the alternating short and long bond length distortions43 within the ring structures NbW5O6 and W6O6 of the POMs (NbW5O6 ring shown in Fig. 1b).
Compared to [PW11O39NbO]4−, the bond lengths between the O atoms and metal atoms in [PW11O39NbS]4− and [PW11O39NbSe]4− show similar trends. The M–E distance increases as E = O (1.757 Å) < S (2.219 Å) < Se (2.350 Å), which is related to the size of the E atom. The bond lengths between niobium and the bridging oxygens decrease with the increasing size of E (for instance, O (2.027 Å) > S (2.014 Å) > Se (2.012 Å) for M–ObM). However, the substitution results in a longer attachment of the ObM and OcM atoms to the nearest W. In addition, the bond lengths between Nb and the center tetrahedral oxygen atoms are lengthened due to the substitution of S or Se. The substitution doesn't influence the bonds (W–Od, W–Ob, W–Oc, W–Oa, P–Oa) far away from Nb and Ta. The substitution effects of S and Se on the structural parameters of [PW11O39TaE]4− are consistent with those of [PW11O39NbE]4−. The distortion magnitude parameter Δd between the average long d(W–O) values and short d(W–O) values in [PW11O39ME]4− are given in Table 1. The calculated Δd values for [PW11O39NbE]4− and [PW11O39TaE]4− decrease in the following order: [PW11O39MO]4− > [PW11O39MS]4− > [PW11O39MSe]4−, suggesting that the strength of the bridging bonds varies in a narrower range in the case of S and Se.
Substitution effects on the electronic properties of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se)
To investigate the chalcogenido-substitution effects on the electronic properties of polyanions, the frontier molecular orbitals (FMOs) of [PW11O39ME]4− (M = Nb; E = O, S, Se) are compared and presented in Fig. 2. Both the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of [PW11O39TaE]4− behave very similarly to those of [PW11O39NbE]4−. In [PW11O39MO]4− (M = Nb and Ta), the HOMOs are formally delocalized over the bridging oxygen ligands, while the LUMOs are mainly delocalized over the tungsten atoms with some antibonding participation of oxygen. Compared with [PW11O39MO]4−, the HOMOs of chalcogenido-substituted [PW11O39MS]4− and [PW11O39MSe]4− are significantly changed. The sulfur or selenium atom has the main contribution to the HOMOs of [PW11O39MS]4− and [PW11O39MSe]4−. The oxygen atoms have almost no contribution to the HOMOs. Namely, the chalcogenido atoms (S and Se) mainly modify the occupied molecular orbitals and the electron-donating ability of sulfur or selenium atoms are stronger than that of the oxygen atom. The LUMOs of [PW11O39MS]4− and [PW11O39MSe]4− are still mainly delocalized over the tungsten atoms. Nb or Ta give little contribution to the LUMO since the electronegativities of Nb5+ and Ta5+ are smaller than that of W6+.35 The HOMO and LUMO energies of [PW11O39ME]4− (M = Nb, Ta; E = O, S and Se) are illustrated in Fig. 3. It can be seen that the HOMO energy of [PW11O39ME]4− increases as the size of E does, which is consistent with the modification by the S or Se atom on the HOMO, while the LUMO energy slightly decreases. Consequently, the energy gap between the HOMO and LUMO decreases from O to Se. In addition, the LUMO energies of [PW11O39NbE]4− are almost equal to those of the corresponding [PW11O39TaE]4−.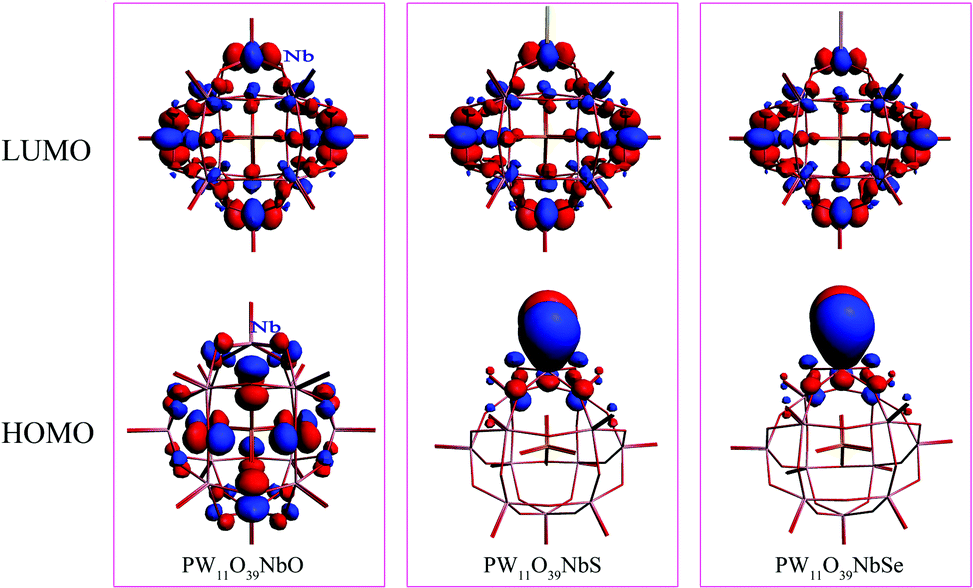 | ||
| Fig. 2 Frontier molecular orbitals for [PW11O39ME]4− (M = Nb; E = O, S, Se). | ||
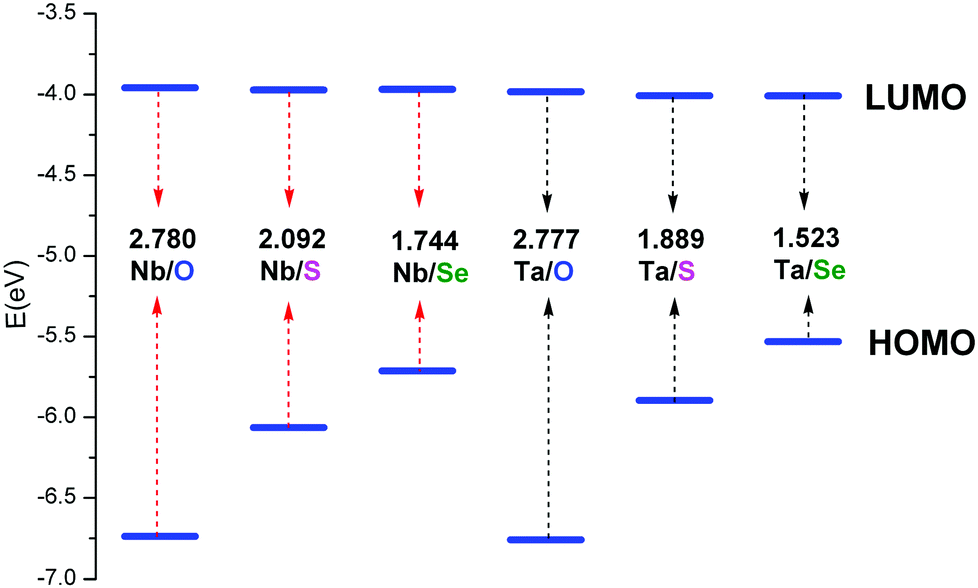 | ||
| Fig. 3 Energies of the frontier molecular orbitals calculated for [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se). | ||
In fully oxidized Keggin-type anions, the LUMO is a mixture of the 2p(O) and 4d of metal atoms with antibonding π character. Given that the MO6 octahedra are distorted from the ideal Oh symmetry in Keggin-type anions, the dxy, dxz and dyz orbitals are destabilized differently depending on the antibonding interactions with the neighbouring p orbitals of oxygen atoms. These antibonding interactions are more obvious between the dxz and dyz orbitals of metals with 2p(Od) because the M–Od bond lengths are shorter and the orientations are more favorable. This leads to the energies of the dxz and dyz orbitals increasing. Therefore, the LUMOs in POMs are always symmetry-adapted combinations of dxy-like orbitals.44 Sulfur or selenium substitution at the terminal site should not alter the LUMO obviously since they weakly interact with dxy-like orbitals, which are the lowest ones. On the other hand, the relative energy and composition of the LUMO correlates well with the electron affinity of each isolated Mn+ ion.35 The ionic radii of the two metals are similar, 0.69 Å for Nb5+ and 0.68 Å for Ta5+. The electronegativities of niobium(V) and tantalum(V) are qualitatively similar, which results in the similar LUMO energy levels in [PW11O39NbE]4− and [PW11O39TaE]4−.
According to cyclic voltammetry results, the reduction peaks of [PW11O39NbS]4− have a weak but significant shift toward negative potentials compared to those of [PW11O39NbO]4−. The weak difference observed between the redox behavior of [PW11O39NbO]4− and [PW11O39NbS]4− was expected because of the minor structural changes introduced by the substitution.31 In order to probe the substitution effect of sulfur and selenium on the redox properties of POMs, the first one-electron reduction energy for [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) and their reduced centers were investigated. Theoretical estimation of the redox energy of POMs requires the determination of the free energy change during the process.
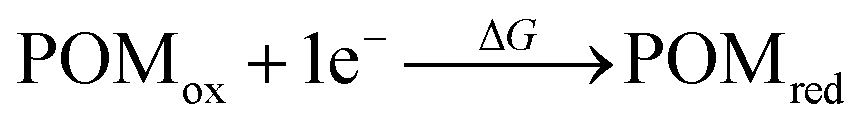 | (1) |
Given that the reduction process of POMs involves the addition of one electron to an almost nonbonding orbital, the entropic and vibrational contributions to ΔG in eqn (1) are negligible, the electronic part is the decision parameter of the reduction processes. The first one-electron reduction energies (REs) are −3.84, −3.86 and −3.87 eV for [PW11O39NbO]4−, [PW11O39NbS]4− and [PW11O39NbSe]4−, respectively, which are in good agreement with the results of cyclic voltammetry. For [PW11O39TaE]4−, REs show a similar trend. There is a linear relationship between the reduction energy and LUMO energy. As a consequence of the slight difference on the LUMO, the one-electron reduction energies also slightly decrease from [PW11O39NbO]4−, [PW11O39NbS]4− to [PW11O39NbSe]4−. As reported by Aparicio,45 the metal replacement in the external framework produces a less delocalized W-like molecular orbital, suggesting that the additional electrons in the monoreduced complexes delocalize in a smaller region. Does the chalcogenido-substitution influence the reduced center? To investigate the reduced center, Mulliken population analysis was performed on the one-electron reduced species. The spin densities of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) are illustrated in Table 2. It can be seen that the extra electrons in monoreduced [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) delocalize over the four tungsten atoms (W3, W5, W9 and W11) in the equatorial plane (Fig. 1b). These four tungsten atoms all belong to one M6O6 ring containing Nb or Ta. Obviously, the chalcogenide-substitution does not change the reduced center.
| ME | NbO | NbS | NbSe | TaO | TaS | TaSe |
|---|---|---|---|---|---|---|
| W3 | 0.2274 | 0.1801 | 0.1809 | 0.2171 | 0.1775 | 0.1741 |
| W5 | 0.2239 | 0.1824 | 0.1836 | 0.2179 | 0.1782 | 0.1750 |
| W9 | 0.1777 | 0.2261 | 0.2259 | 0.1756 | 0.2173 | 0.2120 |
| W11 | 0.1820 | 0.2235 | 0.2232 | 0.1757 | 0.2166 | 0.2113 |
| M | −0.0010 | −0.0015 | −0.0014 | −0.0010 | −0.0017 | −0.0016 |
Substitution effects on the acidic properties of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se)
Firstly, we perform a simple analysis of the Mulliken charges for [PW11O39ME]4−. The main Mulliken charges calculated in gas and solution are summarized in Table 3. The Mulliken charges slightly change for the different types of bridging oxygens in the gas phase. We mainly discuss the charge calculated in solution. For [PW11O39NbO]4−, the basicity of oxygens increases as Od < E < Ob < ObNb < Oc < OcNb < Oa. The results predict that Oa is the most basic site in the whole cluster and Od is the least basic site. The edge-sharing oxygens are more nucleophilic than the corner-sharing ones. In addition, the charges of the bridging oxygens ObNb and OcNb are more negative than those of the corresponding Ob and Oc. In sulfur and selenium-substituted [PW11O39NbS]4− and [PW11O39NbSe]4−, Oa and Od are the most and least basic sites, respectively. The edge-sharing oxygens are still more basic than the corner-sharing ones. In contrast to [PW11O39NbO]4−, the basicity of the bridging oxygens linked to Nb and W are weaker than those of the bridging oxygens linked to two W atoms. The charge of S and Se is more positive than that of the least basic Od, indicating that the electron donating ability is enhanced from O to S and Se. The average negative charges on oxygen atoms in [PW11O39NbS]4− and [PW11O39NbSe]4− are slightly higher compared to those of [PW11O39NbO]4−. The basicity of oxygen in Ta-containing POMs is close to that in Nb-containing POMs.
| ME | NbO | NbS | NbSe | TaO | TaS | TaSe |
|---|---|---|---|---|---|---|
| Gas phase | ||||||
| ObM | −0.902 | −0.889 | −0.888 | −0.908 | −0.891 | −0.891 |
| OcM | −0.898 | −0.881 | −0.880 | −0.908 | −0.887 | −0.886 |
| Ob | −0.901 | −0.901 | −0.901 | −0.901 | −0.901 | −0.901 |
| Oc | −0.901 | −0.900 | −0.900 | −0.901 | −0.900 | −0.900 |
| E | 0.758 | −0.665 | −0.889 | −0.793 | −0.704 | −0.892 |
| Od | −0.726 | −0.722 | −0.721 | −0.725 | −0.721 | −0.720 |
| Oa | −0.981 | −0.980 | −0.980 | −0.984 | −0.980 | −0.980 |
| Solution | ||||||
| ObM | −0.918 | −0.903 | −0.903 | −0.925 | −0.905 | −0.906 |
| OcM | −0.926 | −0.912 | −0.912 | −0.938 | −0.918 | −0.920 |
| Ob | −0.914 | −0.914 | −0.914 | −0.914 | −0.913 | −0.913 |
| Oc | −0.924 | −0.923 | −0.924 | −0.923 | −0.922 | −0.922 |
| E | −0.814 | −0.536 | −0.647 | −0.876 | −0.612 | −0.707 |
| Od | −0.737 | −0.736 | −0.735 | −0.735 | −0.733 | −0.733 |
| Oa | −0.982 | −0.981 | −0.981 | −0.982 | −0.981 | −0.981 |
In order to investigate the location of the first proton, DFT calculations were performed to obtain the protonation energy (PE) needed for the addition of the first proton to the oxygen atoms in the gas phase and in solution. PE is defined as the energy difference between the products (HPOM3− + H2O) and reactants (POM4− + H3O+) involved in the following acid–base reaction.
| POM4− + H3O+ → HPOM3− + H2O | (2a) |
| PE = E(HPOM3−) + E(H2O) − E(POM4−) − E(H3O+) | (2b) |
The protonation of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) could yield several isomers depending on the protonation site. Table 4 lists the values of the relative PEs for the five isomers H–ObM, H–OcM, H–Ob, H–Oc and H–E of [HPW11O39ME]3−. The proton affinity is referenced to the isomer H–Ob. The optimized structures and H⋯O distances for the five isomers of protonated [PW11O39NbO]4− are shown in Fig. 4. As shown in Table 4, the stability of the protonated isomers for [PW11O39NbO]4− in the gas phase is as follows: ObM > Ob > OcM > Oc > E. The most stable protonated isomer features a protonated ObM site. It is followed by the protonation of the bridging Ob, 1.34 kcal mol−1 higher in energy. Thus, protonation is preferred at the corner-sharing oxygen. The same conclusion can be obtained for [PW11O39NbS]4− and [PW11O39NbSe]4−. For the same protonated site, the PE decreases as [PW11O39NbO]4− > [PW11O39NbS]4− > [PW11O39NbSe]4−. Namely, the basicity of the oxygen sites reduces when E changes from O to S and Se. The calculated results for [PW11O39TaE]4− are in agreement with those of [PW11O39NbE]4− except for the case in which H bonds to the terminal E site.
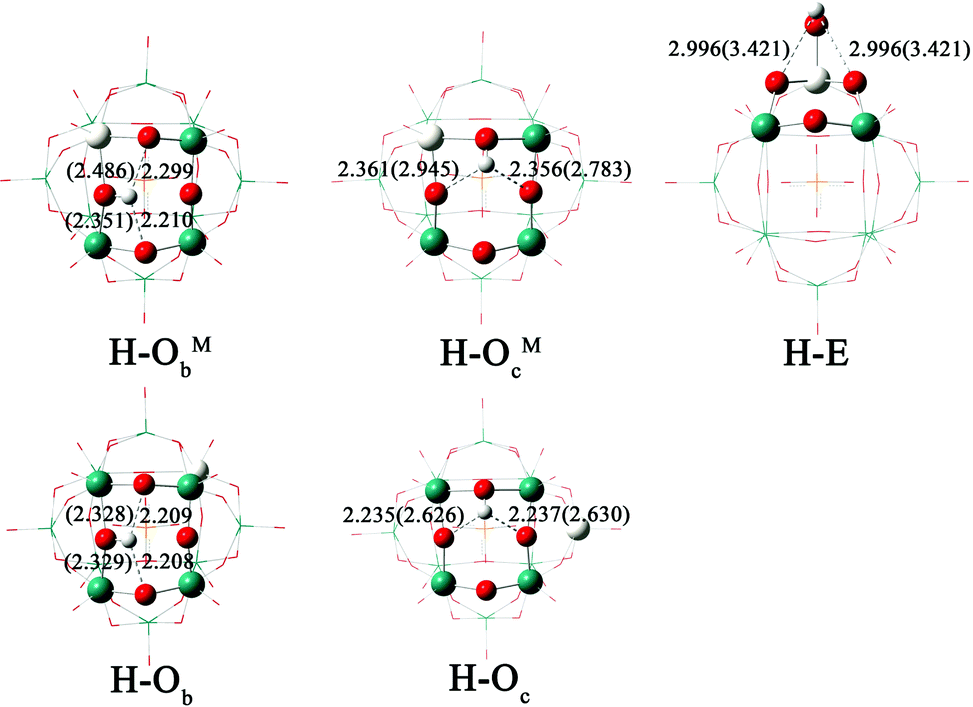 | ||
| Fig. 4 Optimized structures and H⋯O distances (in Å) for the five isomers of [HPW11O39NbO]3−. The values in parentheses represent those in the gas phase. | ||
| ME | NbO | NbS | NbSe | TaO | TaS | TaSe |
|---|---|---|---|---|---|---|
| Gas phase | ||||||
| Oc | 1.69 | 1.64 | 1.63 | 4.05 | 1.71 | 1.70 |
| ObM | −1.34 | −1.52 | −1.50 | −0.72 | −0.83 | −0.81 |
| OcM | 0.75 | 0.33 | 0.29 | 2.36 | 1.83 | 1.68 |
| Ob | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| E | 3.49 | 4.27 | 7.08 | −1.95 | 0.58 | 2.95 |
| Solution | ||||||
| Oc | −0.50 | −0.36 | −0.53 | −0.50 | −0.35 | −0.29 |
| ObM | −1.17 | −0.96 | −0.53 | −0.28 | 0.14 | 0.45 |
| OcM | −1.64 | −1.59 | −1.39 | −1.36 | −0.80 | −0.67 |
| Ob | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| E | −2.96 | 1.60 | 4.36 | −7.28 | −1.98 | 0.56 |
In acetonitrile, the computed PEs for [PW11O39NbO]4− decrease as E > OcM > ObM > Oc > Ob, which are different from those in the gas phase. E becomes the most stable protonated site, which is consistent with the assumption of Poblet44 and is closer to the results of the Mulliken charge analysis. Namely, the edge-sharing oxygens are more nucleophilic than the corner-sharing ones, and the PEs of the bridging oxygens linked to Nb and W (NbOW) are larger than those of the bridging oxygens linked to two W (WOW) atoms. In addition, REDOR experiments demonstrated that the acidic protons are localized on both the bridging (Oc) and terminal (Od) oxygen atoms of H3PW12O40 in the anhydrous state,16 which is in agreement with our results obtained in acetonitrile. This indicates that the solvent is needed to find the local protonated sites in POMs. The energy difference between HPOM3− and POM4− becomes smaller in acetonitrile compared to that in the gas phase. It can be concluded that the solvent introduces a large stabilization effect, especially for nonprotonated species. In the case of [PW11O39TaE]4−, the same conclusion can be drawn. In other words, PE decreases in the following order: [PW11O39TaO]4− > [PW11O39TaS]4− > [PW11O39TaSe]4−. Furthermore, the PEs of [PW11O39ME]4− (M = Nb; E = O, S, Se) are more negative than those of [PW11O39ME]4− (M = Ta; E = O, S, Se), suggesting that the basicity of oxygen atoms in [PW11O39NbE]4− is slightly stronger than that of the corresponding [PW11O39TaE]4−.
As mentioned above, the charge of the oxygen atom, H⋯O interactions and solvent could be important parameters for the PE in this study. To analyze these effects on PE, we take [PW11O39NbO]4− as an example. Fig. 4 illustrates that H⋯O interactions in solution are strengthened in the following order: E < OcNb < ObNb < Oc < Ob, which is closer to the order of PE in the gas phase. In the gas phase, the effect of H⋯O interactions on protonation is larger than that of the Mulliken charge because the Mulliken charge is almost constant between the different types of bridging oxygens. The order of PE in acetonitrile, however, is closer to the results of the Mulliken charge. Thus it can be seen that the effect of the Mulliken charge is critical to PE in solution. As shown in Fig. 4, H⋯O interaction between H and POM is weakened in solution because the proton forms hydrogen bonds with acetonitrile. The solvent influences H⋯O interactions and introduces a large stabilization effect. In brief, the H⋯O interactions determine the order of PE in the gas phase, which is more likely to change the energetic order in the gas phase than that in solution. The solvent and Mulliken charge mainly influence the order of PE in solution.
The majority of the electron density needed to form the O–H bond is derived from the terminal oxygen atoms throughout the molecule.47 The charges of the terminal oxygen atoms decrease as one Od is substituted by S and Se, and the computed PE also decreases in the chalcogenido-substituted POMs. In conclusion, the most basic site in [PW11O39MO]4− is the terminal oxygen bonded to M. However, in POMs containing S or Se, the most basic site is ObM. The basicity of external oxygen sites reduces when E changes from O, S to Se.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logKa. The pKa corresponds to the release of a proton from the external O site in HPOM3−. From eqn (2b), we can qualitatively conclude that the acidity of HPOM3− becomes weaker with the increasing negative values of PE. PE becomes more positive after substitution by S and Se, indicating that the acidity of the POM is enhanced owing to the substitution of S or Se.
logKa. The pKa corresponds to the release of a proton from the external O site in HPOM3−. From eqn (2b), we can qualitatively conclude that the acidity of HPOM3− becomes weaker with the increasing negative values of PE. PE becomes more positive after substitution by S and Se, indicating that the acidity of the POM is enhanced owing to the substitution of S or Se.
In order to further confirm the conjecture mentioned above, DFT calculations were used to compare the acid strengths of the protonated [PW11O39ME]4− (M = Nb; E = O, S, Se) by the adsorption energy of ammonia coordination to the protonated [PW11O39ME]4−. For consistency in comparing the different POMs, ammonia adsorbed at the ObM and Oc sites was considered. The representative optimized structures of ammonia adsorbed at the protonated [PW11O39ME]4− at the ObM and Oc sites are depicted in Fig. 5 and their adsorption energies are shown in Table 5. The predicted adsorption energies for the ObM sites are −115.65 kJ mol−1, −117.91 kJ mol−1 and −117.40 kJ mol−1 for [PW11O39NbO]4−, [PW11O39NbS]4− and [PW11O39NbSe]4−, respectively. The adsorption energies at the Oc sites are −125.08 kJ mol−1, −126.44 kJ mol−1 and −129.83 kJ mol−1 for [PW11O39NbO]4−, [PW11O39NbS]4− and [PW11O39NbSe]4−, respectively, which are slightly stronger than that of the ObM site. This is consistent with the results of the PE calculations. The calculated adsorption energies for [PW11O39ME]4− are close to its analogous α-Keggin [PW12O40]3− computed by Bardin and co-workers.48 The adsorption of ammonia on [HPW11O39NbS]3− and [HPW11O39NbSe]3− is stronger than that of [HPW11O39NbO]3−, so it is reasonable to assume that S or Se substitution in POMs enhances the acidity. This result is in good agreement with the PE calculations, which further confirms our conjecture.
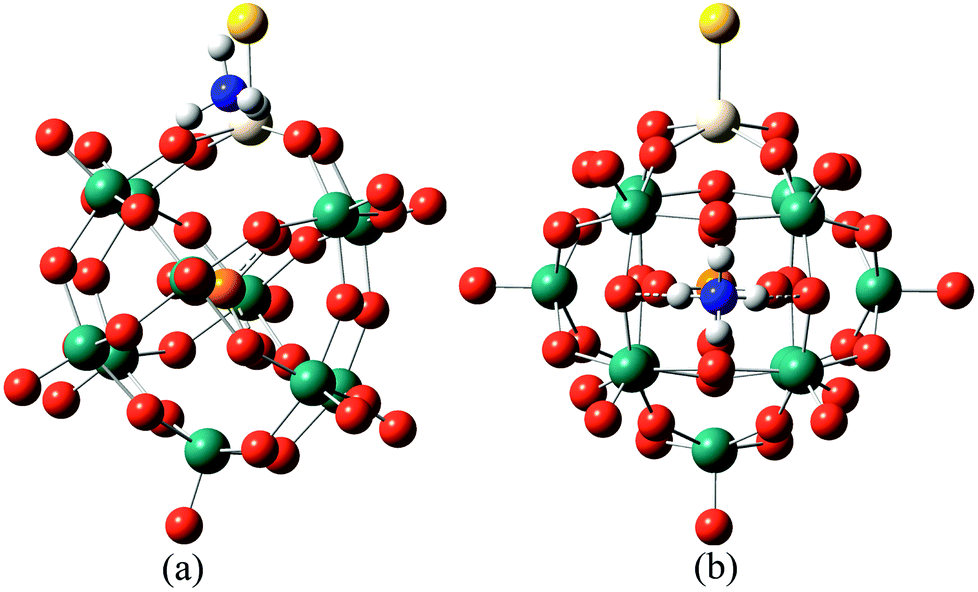 | ||
| Fig. 5 The typically optimized structures of ammonia adsorbed on the protonated [PW11O39NbO]4− at different locations: (a) ammonia adsorbed to an ObM atom. (b) Ammonia adsorbed to an Oc atom. | ||
| ME | NbO | NbS | NbSe |
|---|---|---|---|
| ObM | −115.65 | −117.91 | −117.40 |
| Oc | −125.08 | −126.44 | −129.83 |
Conclusions
DFT calculations were carried out on a series of chalcogenido-substituted mixed-metal Keggin-type anions α-[PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) and the electronic properties and basicity of the oxygen sites were analyzed. The calculated results for the electronic structure and reduction energies of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) both confirm that substitution of one O atom by S and Se in POMs can improve the oxidation ability of POMs, but the effect is not obvious. For example, when one oxygen atom is substituted by a sulfur or selenium atom, the energy of the LUMO and the one-electron reduction energy both fall slightly as E goes down the column in the chalcogen group. The additional electron delocalizes over the four tungsten atoms in the equatorial plane: W3, W5, W9 and W11. These four tungsten atoms all belong to one M6O6 ring containing Nb or Ta. Therefore, the chalcogenido-substitution does not change the reduced center. In addition, the reduction potential of [PW11O39ME]4− (M = Ta; E = O, S, Se) is close to the corresponding [PW11O39ME]4− (M = Nb; E = O, S, Se) since the ionization potentials of niobium(V) and tantalum(V) are expected to be qualitatively similar. As to acidity, the calculations carried out on the five protonated isomers of [PW11O39ME]4− (M = Nb, Ta; E = O, S, Se) suggest that the most basic site in [PW11O39MO]4− is the terminal oxygen bonded to M. However, in POMs containing S or Se, the most basic site is OcM. Moreover, the calculated PEs increase as E goes down the column in the chalcogen group, indicating that the basicity of the oxygen sites reduces when E changes from O, S to Se and the acidities of the substituted POMs are stronger than those of [PW11O39MO]4−. For [PW11O39ME]4− (M = Ta; E = O, S, Se), the orders of basicity of the oxygen sites are different from those in [PW11O39ME]4− (M = Nb; E = O, S, Se). Furthermore, the protonation energies of POMs containing niobium are more negative than those of the tantalum-mixed POMs, indicating that the basicity of the oxygen atoms in [PW11O39ME]4− (M = Nb; E = O, S, Se) are stronger than those of the corresponding [PW11O39ME]4− (M = Ta; E = O, S, Se). Further investigation on the adsorption energy of ammonia in [PW11O39ME]4− (M = Nb; E = O, S, Se) also provides the same results as mentioned above. In conclusion, the electronic properties and redox properties are slightly affected by substitution with S or Se. The acidic properties of POMs are enhanced owing to the introduction of S and Se. POMs consisting of Nb and Ta are stable only in highly alkaline solutions, limiting their aqueous redox chemistry and other applications. The introduction of S or Se atoms to POMs containing Nb and Ta would allow access to tunable pH stability and expand their applications. We expect that it will facilitate and inspire new science in both the transition-metal POMs and the new frontiers of the f-element POMs.Acknowledgements
The authors gratefully acknowledge financial support by the NSFC (21073030 and 21131001), the Program for New Century Excellent Talents in University (NCET-10-318), the Doctoral Fund of the Ministry of Education of China (20100043120007), and the Science and Technology Development Planning of Jilin Province (20100104 and 20100320).Notes and references
- D. Sattari and C. L. Hill, J. Am. Chem. Soc., 1993, 115, 4649–4651 CrossRef CAS.
- K. Kamata, Y. Nakagawa, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 15304–15310 CrossRef CAS PubMed.
- N. S. Antonova, J. J. Carbó, U. Kortz, O. A. Kholdeeva and J. M. Poblet, J. Am. Chem. Soc., 2010, 132, 7488–7497 CrossRef CAS PubMed.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219–237 CrossRef CAS PubMed.
- B. Hasenknopf, Front. Biosci., 2005, 10, 275–287 CrossRef CAS PubMed.
- T. Yamase, J. Mater. Chem., 2005, 15, 4773–4782 RSC.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC.
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed.
- A. M. Khenkin, G. Leitus and R. Neumann, J. Am. Chem. Soc., 2010, 132, 11446–11448 CrossRef CAS PubMed.
- K. Kamata, K. Yonehara, Y. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964–966 CrossRef CAS PubMed.
- K. Kamata, K. Yonehara, Y. Nakagawa, K. Uehara and N. Mizuno, Nat. Chem., 2010, 2, 478–483 CrossRef CAS PubMed.
- T. Blasco, A. Corma, A. Martínez and P. Martínez-Escolano, J. Catal., 1998, 177, 306–313 CrossRef CAS.
- P. Y. Gayraud, I. H. Stewart, S. B. D.-A. Hamid, N. Essayem, E. G. Derouane and J. C. Vedrine, Catal. Today, 2000, 63, 223–228 CrossRef CAS.
- Y. Izumi, K. Hosano and T. Hida, Appl. Catal., A, 1999, 181, 277–282 CrossRef CAS.
- T. Kwon and T. J. Pinnavaia, J. Mol. Catal., 1992, 74, 23–33 CrossRef CAS.
- J. Yang, M. J. Janik, D. Ma, A. Zheng, M. J. Zhang, M. Neurock, R. J. Davis, C. H. Ye and F. Deng, J. Am. Chem. Soc., 2005, 127, 18274–18280 CrossRef CAS PubMed.
- J. Macht, M. J. Janik, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2008, 130, 10369–10379 CrossRef CAS PubMed.
- E. Radkov and R. H. Beer, Polyhedron, 1995, 14, 2139–2143 CrossRef CAS.
- L. Vilà-Nadal, K. Peuntinger, C. Busche, J. Yan, D. Lüders, D. L. Long, J. M. Poblet, D. M. Guldi and L. Cronin, Angew. Chem., Int. Ed., 2013, 125, 9877–9881 CrossRef.
- H. El Moll, B. Nohra, P. Mialane, J. Marrot, N. Dupré, B. Riflade, M. Malacria, S. Thorimbert, B. Hasenknopf, E. Lacôte, P. A. Aparicio, X. López, J. M. Poblet and A. Dolbecq, Chem. – Eur. J., 2011, 17, 14129–14138 CrossRef PubMed.
- X. López, J. A. Fernández and J. M. Poblet, Dalton Trans., 2006, 1162–1167 RSC.
- A. Hijazi, J. C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, C. R. Mayer, V. Artero and E. Cadot, Inorg. Chem., 2011, 50, 9031–9038 CrossRef CAS PubMed.
- S. Duval, S. Floquet, C. Simonnet-Jégat, J. Marrot, R. N. Biboum, B. Keita, L. Nadjo, M. Haouas, F. Taulelle and E. Cadot, J. Am. Chem. Soc., 2010, 132, 2069–2077 CrossRef CAS PubMed.
- B. Keita, S. Floquet, J.-F. Lemonnier, E. Cadot, A. Kachmar, M. Bénard, M.-M. Rohmer and L. Nadjo, J. Phys. Chem. C, 2008, 112, 1109–1114 CAS.
- M. A. Ansari and J. A. Ibers, Coord. Chem. Rev., 1990, 100, 223–266 CrossRef CAS.
- H. E. Moll, J. C. Kemmegne-Mbouguen, M. Haouas, F. Taulelle, J. Marrot, E. Cadot, P. Mialane, S. Floquet and A. Dolbecq, Dalton Trans., 2012, 41, 9955–9963 RSC.
- V. S. Korenev, S. Floquet, J. Marrot, M. Haouas, I.-M. Mbomekallé, F. Taulelle, M. N. Sokolov, V. P. Fedin and E. Cadot, Inorg. Chem., 2012, 51, 2349–2358 CrossRef CAS PubMed.
- A. Muller, V. P. Fedin, C. Kuhlmann, H. D. Fenske, G. Baum, H. Bogge and B. Hauptfleish, Chem. Commun., 1999, 1189–1190 RSC.
- Y. Do, E. D. Simhon and R. H. Holm, Inorg. Chem., 1985, 24, 1831–1838 CrossRef CAS.
- W. G. Klemperer and C. Schwartz, Inorg. Chem., 1985, 24, 4459–4461 CrossRef CAS.
- E. Cadot, V. Béreau and F. Sécheresse, Inorg. Chim. Acta, 1995, 239, 39–42 CrossRef CAS.
- E. Radkov, Y. J. Lu and R. H. Beer, Inorg. Chem., 1996, 35, 551–552 CrossRef CAS.
- L. K. Yan, Z. M. Su, W. Guan, M. Zhang, G. H. Chen, L. Xu and E. B. Wang, J. Phys. Chem. B, 2004, 108, 17337–17343 CrossRef CAS; W. Guan, L. K. Yan, Z. M. Su, S. X. Liu, M. Zhang and X. H. Wang, Inorg. Chem., 2005, 44, 100–107 CrossRef PubMed; L. K. Yan, G. C. Yang, W. Guan, Z. M. Su and R. S. Wang, J. Phys. Chem. B, 2005, 109, 22332–22336 CrossRef PubMed; C. G. Liu, Z. M. Su, W. Guan and L. K. Yan, Inorg. Chem., 2009, 48, 541–548 CrossRef PubMed; Z. L. Lang, G. C. Yang, N. N. Ma, S. Z. Wen, L. K. Yan, W. Guan and Z. M. Su, Dalton Trans., 2013, 42, 10617–10625 RSC.
- I. M. Mbomekallé, X. López, J. M. Poblet, F. Sécheresse, B. Keita and L. Nadjo, Inorg. Chem., 2010, 49, 7001–7006 CrossRef PubMed.
- X. López, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2002, 124, 12574–12582 CrossRef PubMed.
- ADF2012.01, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 34, 7406–7406 CrossRef.
- E. van Lenthe, A. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC; A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS; A. Klamt and V. Jones, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef PubMed; C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 CrossRef.
- L. K. Yan, X. López, J. J. Carbó, R. Sniatynsky, D. C. Duncan and J. M. Poblet, J. Am. Chem. Soc., 2008, 130, 8223–8233 CrossRef CAS PubMed.
- J. M. Poblet, X. López and C. Bo, Chem. Soc. Rev., 2003, 32, 297–308 RSC.
- P. A. Aparicio, J. M. Poblet and X. López, Eur. J. Inorg. Chem., 2013, 1910–1916 CrossRef CAS.
- S. Ganapathy, M. Fournier, J. F. Paul, L. Delevoye, M. Guelton and J. P. Amoureux, J. Am. Chem. Soc., 2002, 124, 7821–7828 CrossRef CAS PubMed.
- I. Efremenko and R. Neumann, J. Phys. Chem. A, 2011, 115, 4811–4826 CrossRef CAS PubMed.
- B. B. Bardin, R. J. Davis and M. Neurock, J. Phys. Chem. B, 2000, 104, 3556–3562 CrossRef CAS.
| This journal is © the Partner Organisations 2015 |