
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New biomaterials from renewable resources – amphiphilic block copolymers from δ-decalactone†
Kuldeep K.
Bansal
a,
Deepak
Kakde
a,
Laura
Purdie
a,
Derek J.
Irvine
b,
Steven M.
Howdle
c,
Giuseppe
Mantovani
a and
Cameron
Alexander
 *a
*a
aSchool of Pharmacy, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: Cameron.alexander@nottingham.ac.uk
bDepartment of Chemical and Environmental Engineering, University of Nottingham, NG7 2RD, UK
cSchool of Chemistry, University of Nottingham, NG7 2RD, UK
First published on 28th August 2015
Abstract
The synthesis of polymers for biomedical applications via environmentally benign routes and with sustainable feedstocks is an area of intense interest. Here we describe the synthesis, characterisation and drug carrier potential of novel polymeric materials obtained from a non-toxic, low cost and easily accessible renewable monomer, δ-decalactone. A range of different polymers and copolymers of δ-decalactone was synthesised under mild reaction conditions using organic and enzymatic catalysts. Amphiphilic block copolymers of δ-decalactone with poly(ethyleneglycol) (PEG) and terpolymers with poly(pentadecalactone) were shown to self-assemble into micelles and a hydrophobic dye (Nile Red) was incorporated inside the micellar cores via a nanoprecipitation method. The encapsulation properties of the polymeric micelles were explored using Amphotericin B (AmpB) as a model drug. A comparative loading study of AmpB in PEG-b-poly(δ-decalactone) and in PEG-b-poly(ε-caprolactone) micelles demonstrated a higher loading of AmpB in the δ-decalactone co-polymer. In vitro release studies of AmpB from the polymer micelles demonstrated sustained release of AmpB for up to 8 days. A preliminary hydrolytic degradation and cytotoxicity study indicated that the block co-polymer micelles are biodegradable and exhibit low toxicity. These data suggest that the δ-decalactone copolymers are of promise for further development towards biomedical applications.
Introduction
Polymers for drug delivery applications must meet a number of stringent requirements.1–3 These include very low toxicity, predictable degradability, compatibility with both active drugs and excipients, and ‘formulation flexibility’, such that they can be used in a variety of dosing formats. Increasingly, there are demands that these materials should be accessible by rapid and facile synthesis from renewable resources.4–7 This is partially due to future supply constraints for non-biogenic monomers but also a greater desire to include sustainability throughout the biomedical manufacturing process. New chemistries are being devised to generate polymers from non-foodstock sources, or by novel fermentation and bioengineering techniques.8–14 Polymers obtained from renewable resources are already under investigation for drug delivery, and there have been many examples using natural or modified polysaccharides and polypeptides. The degradation profiles of these classes of natural polymers can vary widely dependent on substituents, and so the polyester backbone has been a major focus for drug delivery use as the hydrolysis rates are relatively easy to control. For example, polyesters derived from small hydrophilic units such as glycerol and citric acid have been used for gentamicin delivery,15 while the use of sebacoic acid co-polymerised with glycerol has yielded elastomers suitable for retinal implants.16Other polyesters such as poly(lactic acid), (PLA), poly(lactic-co-glycolic acid) (PLGA), poly(caprolactone) (PCL), are widely used in biomedical settings but despite their successes, these polymers still exhibit some undesirable features. PCL is currently obtained from a non-renewable monomer source,17 while for PLA or PLGA materials, the loading of hydrophobic drugs has been reported to be low compared to long chain polyesters.18 Considering these limitations, as well as the increasing lipophilicity of many candidate drugs emerging from pharmaceutical screens, there is a real need to develop more hydrophobic polyester components for drug delivery, which can be produced from cheap renewable monomers via easy synthetic methodologies.19,20
δ-Decalactone, an FDA approved flavouring agent (FDA 21 CFR -172.515) is a candidate monomer for biomedical polymer applications. This readily available compound is obtained from the plant Cryptocarya massoia and the ring-opening of this monomer to synthesise high molar mass poly(δ-decalactone) (PDL) was recently reported.21 The presence of the alkyl side chain pendant to the polyester backbone in the polymerised δ-decalactone structure is also of potential value in drug delivery applications. This is because it should generate a highly hydrophobic core when formulated as a micellar or nanoparticle delivery system, which may be useful to achieve better drug loading than is possible with other currently-used polymers. In addition, the side-chain is expected to disrupt backbone packing in condensed structures, which might also favour increased drug incorporation.22,23
In this study, we report the synthesis of novel amphiphilic block copolymers of δ-decalactone with poly(ethyleneglycol) (PEG). The reactions were performed under mild conditions in the absence of solvents as a step towards a metal free “green” approach to biomedical materials. Homopolymers of δ-decalactone were also synthesised using propargyl alcohol and cis-1,3-O-benzylideneglycerol (BZD) to introduce model functional end groups for post polymerisation modification. Further co-polymers were prepared using a macrolactone (ω-pentadecalactone- an FDA approved flavouring agent/food additive, FDA 21 CFR -172.515) found naturally in angelica root oil, as used here as a second renewable monomer.24
The micelle-forming properties of the amphiphilic block copolymers were investigated using Nile red (NR) as a hydrophobic dye probe, while the drug loading and release profiles of the PDL block copolymers were evaluated with Amphotericin B (AmpB), a broad spectrum antifungal drug of poor aqueous solubility.
In order to meet primary requirements for biomedical use, the degradation profiles of the polymers were tested in a preliminary in vitro hydrolysis study, and in vitro cytocompatibility studies were carried out on human cell lines as a first step to determine suitability for in vivo use. The Alamar Blue assay was accordingly used to assess the effects of the co-polymers on the metabolic activity of HCT-116 cells, a cancer cell line chosen to represent a likely target for injected co-polymer formulations.
The monomers and co-polymers evaluated in this work are shown in Scheme 1.
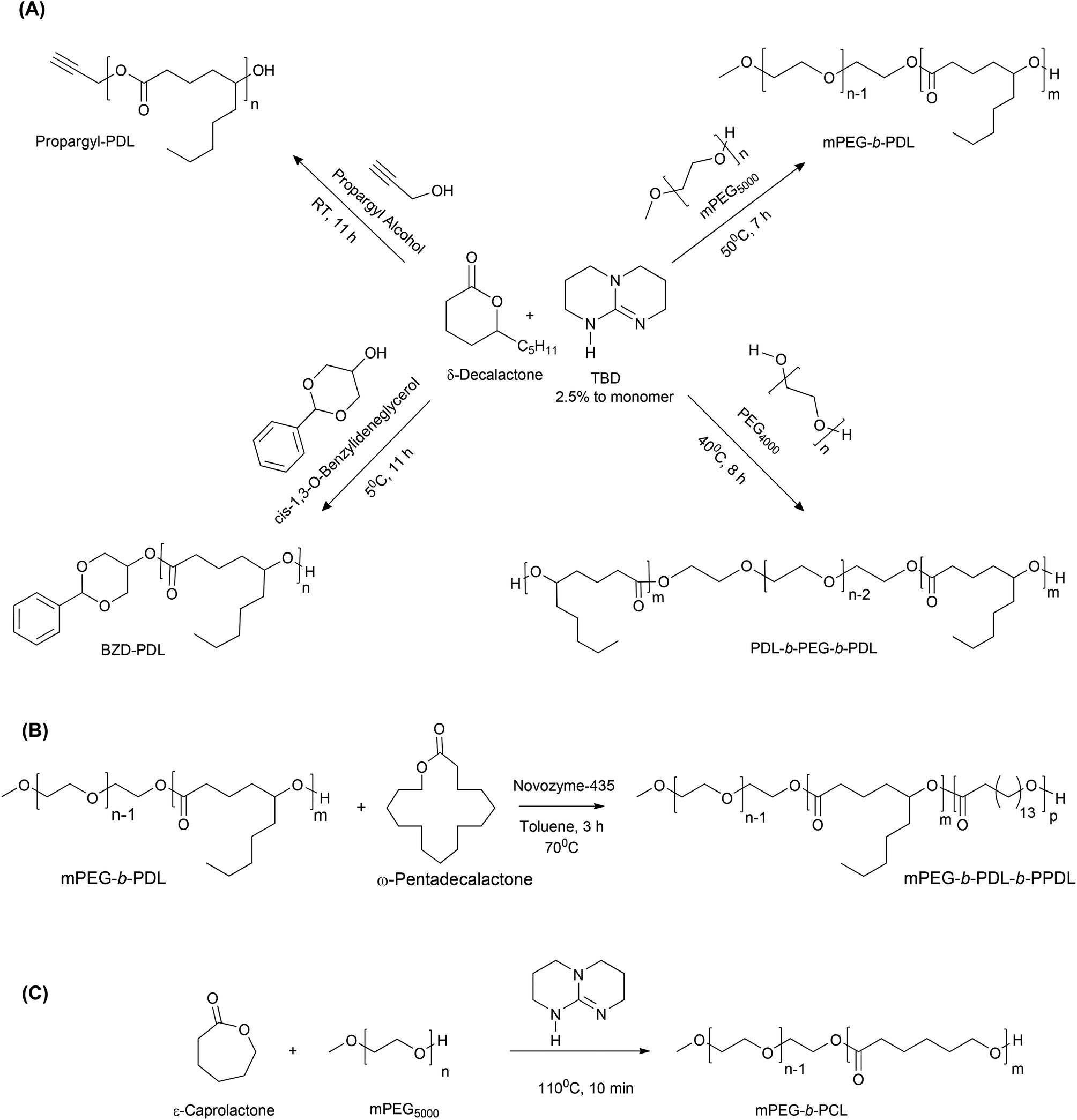 | ||
| Scheme 1 Schematic representation of the ring opening polymerization of (A) δ-decalactone, (B) ω-pentadecalactone and (C) ε-caprolactone using different initiators to generate ranges of homopolymers and copolymers (RT-room temperature, TBD-1,5,7-triazabicyclo[4.4.0]dec-5-ene). | ||
Experimental section
Materials
δ-Decalactone (≥98%), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) (98%), poly(ethyleneglycol) (Mn 4.0 kDa), monomethoxyPEG (Mn 5.0 kDa) (mPEG), propargyl alcohol (99%), cis-1,3-O-benzylideneglycerol (BZD, 97%), benzoic acid (≥99.5%), ω-pentadecalactone (≥98%), novozymes-435 (≥5000 U g−1, recombinant, expressed in Aspergillus niger), pyrene (≥99%), ε-caprolactone (97%), NR (technical grade), AmpB (∼80%), Tween 80 and RPMI-1640 medium were purchased from Sigma Aldrich. All solvents were purchased from Fischer Scientific UK except deuterated solvents and anhydrous toluene, which were purchased from Sigma Aldrich. Opti-MEM® and Alamar Blue® were purchased from Life Technologies, UK. PEG and mPEG were dehydrated by azeotropic distillation using anhydrous toluene. δ-Decalactone was passed through basic alumina before use. Novozymes-435 was dried under vacuum at 50 °C for 24 hours before use. All other chemicals were used as received.Instruments
Particle size and zeta potential measurements
Particle sizes were determined using dynamic light scattering (DLS) and ζ-potential measurements were performed at 25 °C using a Malvern Zetasizer (Nano-ZS) with samples in HPLC grade water and HEPES 10 mM buffer (pH-7.4) respectively. Data analysis was carried out using the Malvern Zetasizer software version 7.03 and mean values obtained from three independent measurements were reported.Synthesis of polymers
| Run | Initiator | M/I ratio | Temperature (°C) | Time (h) | Catalyst (mol%) | Conversion NMR (%) | M n,NMR (kDa) | M n,SEC (kDa) | Đ (Mw/Mn) | T g (°C) | T m (°C) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | cis-1,3-O-Benzylidene glycerol | 100 | 5 | 11 | 2.5 | 89 | 15.3 | 8.8 | 1.21 | NM | NM |
| 2 | Propargyl alcohol | 100 | RT | 11 | 2.5 | 87 | 14.8 | 7.5 | 1.18 | −54.2 | ND |
| 3 | Poly(ethylene glycol) | 150 | 40 | 8 | 2.5 | 89 | 10.3 | 16.2 | 1.15 | −53.3 | 47.0 |
| 4 | Poly(ethylene glycol) methyl ether | 150 | 50 | 7 | 2.5 | 91 | 11.3 | 19.5 | 1.17 | −54.6 | 54.6 |
| 5* | Poly(ethylene glycol) methyl ether | 52 | 110 | 0.17 | 2.0 | 99 | 10.7 | 19.3 | 1.31 | NM | NM |
| 6$ | mPEG-b-PDL | 12 | 70 | 3 | 10.0 ( wt%) | 98 | 12.9 | 21.8 | 1.25 | −52.7 | 54.7, 88.0 |
BZD-PDL. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.53–7.42 (aromatic–CH, m, 2H), 7.38–7.30 (aromatic–CH, m, 3H), 5.52 (acetal CH, s, 1H), 4.94–4.78 (CH–O–C(O), m, 89H), 4.68 (CH2–C
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –O–CO, dd, 1H), 4.31–4.09 (O–C2–CH, m, 4H), 3.64–3.48 (CH2–C–O, m, 4H), 2.37–2.18 (O–CO–CH2, m, 178H), 1.76–1.38 (C2–C2–CH–C2, m, 535H), 1.27 (C2–C2–C2–CH3, m, 546H), 0.96–0.76 (C3, t, 282H).
–O–CO, dd, 1H), 4.31–4.09 (O–C2–CH, m, 4H), 3.64–3.48 (CH2–C–O, m, 4H), 2.37–2.18 (O–CO–CH2, m, 178H), 1.76–1.38 (C2–C2–CH–C2, m, 535H), 1.27 (C2–C2–C2–CH3, m, 546H), 0.96–0.76 (C3, t, 282H).
13C NMR (101 MHz, CDCl3) δ (ppm) 172.98 (CH–O–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O, CH2–CH–O–O), 137.25 (aromatic––CH–O) 128.98 (aromatic–CH), 128.19 (aromatic–CH), 125.97 (aromatic–CH), 101.15 (aromatic–C–H–O), 73.62 (CH2–H–O–CO, CH2–H–OH), 71.21 (O–H2–CH), 69.04 (CH2–H–O–CO), 34.14 (CH–H2–CH2), 33.91 (H2–CH–O–CO, H2–CH–OH), 33.43 (O–CO–H2), 31.60 (H2–CH2–CH3), 24.89 (CH–CH2–H2), 22.48 (O–CO–CH2–H2), 20.77 (H2–CH3), 13.94 (CH2–H3).
O, CH2–CH–O–O), 137.25 (aromatic––CH–O) 128.98 (aromatic–CH), 128.19 (aromatic–CH), 125.97 (aromatic–CH), 101.15 (aromatic–C–H–O), 73.62 (CH2–H–O–CO, CH2–H–OH), 71.21 (O–H2–CH), 69.04 (CH2–H–O–CO), 34.14 (CH–H2–CH2), 33.91 (H2–CH–O–CO, H2–CH–OH), 33.43 (O–CO–H2), 31.60 (H2–CH2–CH3), 24.89 (CH–CH2–H2), 22.48 (O–CO–CH2–H2), 20.77 (H2–CH3), 13.94 (CH2–H3).
Propargyl-PDL. 1H NMR (400 MHz, CDCl3) δ (ppm) 4.94–4.78 (CH–O–C(O), m, 87H), 4.67 (CH2–O, s, 2H), 3.64–3.48 (CH2–C
–O, m, 4H), 2.48 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, s, 1H), 2.37–2.18 (O–CO–CH2, m, 174H), 1.76–1.38 (C2–C2–CH–C2, m, 522H), 1.38–1.14 (C2–C2–C2–CH3, m, 526H), 0.96–0.76 (CH2–C3, t, 270H).
CH, s, 1H), 2.37–2.18 (O–CO–CH2, m, 174H), 1.76–1.38 (C2–C2–CH–C2, m, 522H), 1.38–1.14 (C2–C2–C2–CH3, m, 526H), 0.96–0.76 (CH2–C3, t, 270H).
13C NMR (101 MHz, CDCl3) δ (ppm) 173.04 (CH–O–O, CH2–O–O), 77.23 (H–C–CH2), 75.86 (CH––CH2), 73.69 (CH2–H–O–CO, CH2–H–OH), 34.19 (CH–H2–CH2), 33.94 (H2–CH–O–CO, H2–CH–OH), 33.46 (O–CO–H2), 31.64 (H2–CH2–CH3), 24.93 (CH–CH2–H2), 22.52 (O–CO–CH2–H2), 20.79 (H2–CH3), 13.98 (CH2–H3).
mPEG-b-PDL. 1H NMR (400 MHz, CDCl3) δ (ppm) 4.95–4.84 (CH–O–CO, m, 37H), 4.27–4.17 (CH2–O–CO, t, 2H), 3.65 (O–CH2–CH2–O, s, 497H), 3.38 (O–CH3, s, 3H), 2.32 (O–CO–CH2, m, 75H), 1.75–1.40 (C
2–C2–CH–C2, m, 222H), 1.39–1.18 (C2–C2–C2–CH3, m, 227H), 0.95–0.77 (CH2–C3, t, 138H).
13C NMR (101 MHz, CDCl3) δ (ppm) 173.04 (CH–O–O, CH2–O–O), 73.67 (CH2–H–O–CO, CH2–H–OH), 70.57 (CH2–CH2–O), 65.96 (H2–O–CO) 57.86 (O–CH3), 34.19 (CH–H2–CH2), 33.94 (H2–CH–O–CO, H2–CH–OH), 33.47 (O–CO–H2), 31.64 (H2–CH2–CH3), 24.93 (CH–CH2–H2), 22.52 (O–CO–CH2–H2), 20.80 (H2–CH3), 13.99 (CH2–H3).
FTIR ν (cm−1): 2858 (C–H, stretching), 1729 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, stretching), 1341 (C–H, bending), 1106 (C–O, Stretching).
O, stretching), 1341 (C–H, bending), 1106 (C–O, Stretching).
PDL-b-PEG-b-PDL. 1H NMR (400 MHz, CDCl3) δ (ppm) 4.97–4.80 (CH–O–CO, m, 37H), 4.24 (CH2–O–CO, t, 4H), 3.66 (O–CH2–CH2–O, s, 409H), 2.41–2.18 (O–CO–CH2, m, 78H), 1.75–1.39 (C
2–C2–CH–C2, m, 218H), 1.37–1.12 (C2–C2–C2–CH3, m, 226H), 0.97–0.77 (CH2–C3, t, 140H).
13C NMR (101 MHz, CDCl3) δ (ppm) 173.05 (CH–O–O, CH2–O–O), 73.70 (CH2–H–O–CO, CH2–H–OH), 70.56 (CH2–CH2–O), 64.11 (H2–O–CO), 34.20 (CH–H2–CH2), 33.94 (H2–CH–O–CO, H2–CH–OH), 33.47 (O–CO–H2), 31.65 (H2–CH2–CH3), 24.95 (CH–CH2–H2), 22.52 (O–CO–CH2–H2), 20.79 (H2–CH3), 13.99 (CH2–H3).
1H NMR (400 MHz, CDCl3) δ (ppm) 4.24 (CH2–O–CO, t, 2H), 4.07 (CH2–O–CO, t, 100H), 3.66 (O–CH2–CH2–O, s, 522H), 3.40 (O–CH3, s, 3H), 2.33 (O–CO–C2, t, 100H), 1.74–1.58 (C2–CH2–C2, m, 200H), 1.46–1.32 (CH2–C2–CH2, m, 100H).
13C NMR (101 MHz, CDCl3) δ (ppm) 173.51 (CH2–O–O), 70.57 (CH2–CH2–O), 64.13 (H2–O–CO), 57.69 (O–CH3), 34.11 (O–CO–H2), 28.35 (CH2–CH2–H2), 25.52 (CH2–H2–CH2), 24.57 (H2–CH2–CH2).
1H NMR (400 MHz, CDCl3) δ (ppm) 4.88 (CH–O–CO, m, 38H), 4.28–4.17 (CH2–O–CO, t, 2H), 4.08 (CH2–O–CO, m, 14H), 3.66 (O–CH2–CH2–O, s, 505H), 3.39 (O–CH3, s, 3H), 2.32 (O–CO–CH2, m, 92H), 1.77–1.42 (C2–C2–CH–C2, O–CO–CH2–C2, m, 243H), 1.28 (C2–C2–C2–CH3, CH2–CH2, m, 374H), 0.88 (CH2–C3, t, 121H).
13C NMR (101 MHz, CDCl3) δ (ppm) 173.98 (CH–O–O–CH2), 173.03 (CH–O–O), 73.68 (CH2–H–O–CO), 70.56 (CH2–CH2–O), 64.38 (H2–O–CO), 58.58 (O–CH3), 34.40 (O–CO–H2), 34.19 (CH–H2–CH2), 33.94 (H2–CH–O–CO), 33.46 (O–CO–H2), 31.64 (H2–CH2–CH3), 29.77–29.00 (pentadecalactone–CH2), 28.65 (pentadecalactone–CH2), 25.93 (pentadecalactone–CH2), 24.98 (CH–CH2–H2, pentadecalactone–CH2), 22.51 (O–CO–CH2–H2), 20.81 (H2–CH3), 13.98 (CH2–H3).
Determination of CMC of PDL and PCL block copolymer micelles
The CMC of block copolymers of PDL (i.e. mPEG-b-PDL, PDL-b-PEG-b-PDL and mPEG-b-PDL-b-PPDL) and PCL (i.e. mPEG-b-PCL) were determined via a reported method with slight modification.26 A 6 × 10−7 M stock solution of pyrene in acetone was prepared and a pre-calculated quantity of pyrene was transferred into vials followed by evaporation of acetone under a slow stream of nitrogen. Different concentrations (from 0.001 to 50 μg mL−1) of polymer solutions in water were then added to each vial and left overnight (in the dark) with agitation to equilibrate. Fluorescence spectra of solutions were recorded in the range of 350 to 420 nm at an excitation wavelength of 335 nm. The intensities of emitted light at 373 nm (I1) and 384 nm (I3) were used to calculate the pyrene 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio and plotted against the concentration of polymer used (log scale). All measurements were carried out in triplicate at 25.0 ± 1 °C and mean values were reported. The data were analysed using GraphPad Prism software (version 6.4).
:3 ratio and plotted against the concentration of polymer used (log scale). All measurements were carried out in triplicate at 25.0 ± 1 °C and mean values were reported. The data were analysed using GraphPad Prism software (version 6.4).
Empty and dye loaded micelles preparation from PDL and PCL block copolymers
Micelles of synthesised block copolymers were prepared by a single-step nano-precipitation method with minor modification.27,28 A PDL or PCL block copolymer (50 mg) was dissolved in acetone (5 mL) and this solution was added drop-wise into 10 mL of HPLC grade water under stirring (1000 rpm). The solution was then stirred for 3 hours at room temperature and left overnight (open vial, without stirring) to ensure the complete removal of acetone. Dye-loaded PDL block copolymer micelles were prepared by a similar method in which NR (1.0 mg) was dissolved along with the polymer (50 mg) in acetone (5 mL). The micellar solution was then filtered through a membrane syringe filter (pore size: 0.22 μm) (Millex-LG, Millipore Co., USA) and used for further characterisation. A portion of the dye loaded micelles was freeze dried for the determination of dye content and encapsulation efficiency (EE). Dye content was determined by dissolving a known amount of freeze dried micelles (5.0 mg) in acetone followed by quantification of NR content by UV-Vis spectrophotometry at λmax = 541 nm. The concentration of NR in samples was calculated using a NR standard calibration curve.Preparation of nano-emulsion from PDL homopolymer
An end-functional homopolymer of PDL i.e. propargyl-PDL was used to make an oil-in-water nanoemulsion for comparative studies with blank micelles. The nano-emulsion formulation was prepared by following the same procedure used to prepare blank micelles. This method resulted in the rapid formation of dispersed droplets of propargyl-PDL in water.Preparation and characterisation of blank and AmpB loaded micelles
Empty and AmpB loaded micelles of mPEG-b-PDL and mPEG-b-PCL (50.0 mg each) were fabricated by nanoprecipitation as described above, but using methanol (5 mL) as an organic solvent to solubilise polymer and drug instead of acetone. The initial amount of AmpB used for the encapsulation study was 2.0 mg. During the drug content determination procedure, redissolution of lyophilised mPEG-b-PCL micelles in methanol resulted in the formation of a small amount of insoluble residue. Samples were therefore sonicated for 2 minutes and then centrifuged (2 minutes) at 5000 rpm to remove any residual precipitate. An identical method was followed for lyophilised mPEG-b-PDL micelles. Supernatant (methanol) was collected from each sample and analysed at λmax = 405 nm by UV-Vis spectrophotometry. The amount of AmpB was then calculated using a standard calibration curve for AmpB in methanol.In vitro release study of AmpB from block copolymer micelles
An in vitro release study was performed in modified release media i.e. water containing Tween 80 (1% v/v) to facilitate the solubilisation of AmpB.29 A calculated amount of AmpB-loaded freeze-dried micelles, equivalent to 200 μg of AmpB was redispersed in HPLC grade water (2 mL). The solution was then placed in dialysis tubing (Slide-A-Lyzer, 3.5 kDa molar mass cut-off, Thermo Scientific) and dialysed against 10 mL of release media at 37 °C. The whole release media were replaced with fresh media at predetermined time intervals to maintain sink conditions. Collected release media (samples) were then freeze dried. The dried samples were dissolved in methanol (1 mL) and analysed via UV-Vis spectrophotometry at λ = 405 nm. The amount of AmpB in samples was then calculated using a standard calibration curve of AmpB in methanol (containing 10% v/v of Tween 80). A control sample was prepared by dissolving 200 μg of AmpB in 2 mL of water (containing 2% v/v of Tween 80) (control “A”). An additional control experiment was also carried out by adding 50 μl of mPEG-b-PDL copolymer solution in acetone (100 mg of mPEG-b-PDL in 1 mL of acetone) to the control “A” solution. The solution was then bubbled with nitrogen to remove acetone and volume was made up to 2 mL using water, if required. This sample was named control “B”. The AmpB release profile for control samples was analysed by following the same method used for mPEG-b-PDL micelles.In vitro degradation study of mPEG-b-PDL micelles
The degradation profile of empty mPEG-b-PDL micelles was assessed at two different pH values, pH 7.4 (to model normal body fluid conditions) and pH 4.0 (to simulate acidic build-up during bulk degradation) at 37 °C. Freeze dried empty micelles (8 mg) samples were redispersed in 1 mL of acetate buffer (pH 4.0, 10 mM) and phosphate buffer (pH 7.4, 10 mM) separately. The samples were then incubated at 37 °C under agitation in a water bath. At predetermined time intervals, one sample vial from each pH was collected and freeze-dried. The freeze-dried samples were then dissolved in chloroform, filtered and analysed by SEC to determine the change in polymer average molar mass (Mn).In vitro cytocompatibility study of mPEG-b-PDL and mPEG-b-PCL micelles
HCT116 cells were obtained from ATCC. Cells were routinely maintained in RPMI-1640 medium supplemented with 10% FBS and 2 mM L-glutamine at 37 °C 5% CO2. The cells were seeded at 10000 cells per well in 96 well plates and after 48 hours, the complete medium was replaced with 200 μl Opti-MEM® (Life Technologies). Stock solutions of micelles (5 mg mL−1) were diluted to 3.33, 1.67 and 0.83 mg mL−1 in sterile distilled water (dH2O). To each well, 15 μl of the appropriate micelle dilution or dH2O was added to give final concentrations of 349, 232.5, 116, 58 and 0 μg mL−1. After 24 hours incubation at 37 °C, an Alamar Blue assay (Life Technologies) was conducted according to the manufacturer's instructions. After 1 hour incubation, the fluorescence from each well was measured using a Molecular Devices Flexstation 3 plate reader (λex = 585 nm, λem = 610 nm). The metabolic activities of the cells were reported relative to untreated cells (100% viability). A one-way ANOVA with Tukey's multiple comparison test was conducted for the 349 μg mL−1 sample using GraphPad Prism 6 software.
Results
Synthesis and characterisation of δ-decalactone homopolymers
The synthetic route for synthesis of PDL homopolymers is shown in Scheme 1A. The ROP of δ-decalactone was performed at either 5 °C or room temperature to generate homopolymers with terminal functionality. Conversion was monitored by 1H NMR, by comparing the integral of the CH ester peak at 4.3 ppm of the cyclic monomer (Fig. S1,† peak e), and the corresponding CH signal of the PDL polymer, at 4.9 ppm (Fig. S2,† peak d). Polymerisations were stopped by blocking the organo-catalyst with benzoic acid, followed by precipitation in cold methanol, which removed the unconverted monomer and inactivated catalyst (Fig. S3†). Purified polymer was isolated by centrifugation, and traces of solvent were removed under vacuum. The obtained δ-decalactone homopolymers were amorphous, and therefore no Tm could be observed in DSC analysis, while the Tg of the polymer was measured for propargyl-PDL and found to be ∼−54 °C (Table 1, Fig. S4†). 1H NMR spectra of the synthesised polymers with assigned peaks are shown in Fig. S2.† Proton integration of peaks at 4.9 ppm and 4.67 ppm (for propargyl-PDL), or 5.52 and 4.68 ppm (for BZD-PDL) were used to calculate the average molar mass of polymer (Mn,NMR). 13C NMR spectra were also acquired to characterise the PDL polymers (Fig. S5†). The NMR spectra for the PDL homopolymers were in line with literature data,21 although it was also found that for as-prepared polymers, the integral of the CH–OH end-group proton resonance at 3.5 ppm in 1H NMR was higher than the expected value.SEC using chloroform as the mobile phase gave a unimodal size distribution with relatively narrow polydispersity for both homopolymers. However, the Mn observed by SEC was lower than the molar mass calculated by 1H NMR (Table 1, Fig. S6†).
Synthesis and characterisation of δ-decalactone block copolymers
Block copolymers of δ-decalactone were synthesised at a temperature above the melting point of PEG to avoid the use of added solvents in the reaction (Scheme 1A, Table 1). The target molar mass of PDL chain for both copolymers was 5 kDa. It was observed that increases in the catalyst loading accelerated the conversion of monomer to polymer. Reaction rates were monitored by 1H NMR and the acquired data suggested that the reaction followed first order kinetics (Fig. S7†).As noted above, homopolymer formation was observed from the beginning of the reaction (Fig. S8, Table S1†), suggesting a competing initiator for ROP, either residual moisture or traces of ring-opened decalactone from monomer hydrolysis. The undesired PDL homopolymer was separated from block copolymers by washing the reaction mixture with excess petroleum ether (2–3 times). FTIR, 1H NMR and 13C NMR spectra of the synthesised block copolymers with assigned peaks are shown in Fig. S9, S10 and S11† respectively. Molar masses were calculated from 1H NMR spectra by comparing the number of protons adjacent to the PDL ester linkage at 4.9 ppm with respect to protons of initiator (PEG) at 3.6 (3.3 ppm in the case of mPEG) and the protons adjacent to the ester bond created after ring opening of δ-decalactone by PEG-OH at 4.2 ppm.
Both copolymers showed essentially unimodal distributions in SEC traces however, some tailing was detected for mPEG-b-PDL (Fig. S12†), which may have been due to the presence of residual free mPEG. Synthesised copolymers were also characterised by DSC to determine the change in thermal properties. The block copolymers showed the expected Tm corresponding to PEG, as well as a low Tg attributed to amorphous PDL (Fig. S13†). The Mn detected by SEC, molar mass by NMR, Tg, Tm and other experimental details are summarised in Table 1.
Synthesis and characterisation of mPEG-b-PDL-b-PPDL
A schematic representation of the synthesis of block copolymers of ω-pentadecalactone, using mPEG-b-PDL as initiator, is shown in Scheme 1B. The conversion of monomer to copolymer (mPEG-b-PDL-b-PPDL) was monitored by 1H NMR in which the appearance of a resonance at 4.08 ppm suggested the successful ROP of ω-pentadecalactone. No changes in peak positions of mPEG-b-PDL in 1H NMR spectrum were observed after growth of the PPDL block. The physical state of the polymer was changed from a waxy material (mPEG-b-PDL) to a sticky solid (mPEG-b-PDL-b-PPDL). The 1HNMR spectrum of the purified copolymer was obtained in chloroform-d and the peak positions of the PPDL block matched those reported previously25 (Fig. S14†). Integrals of methylene protons in 1H NMR at 4.0, 4.8 and 3.3 ppm were used to calculate the molar mass, which was found to be 12.9 kDa, compared to Mn 21.8 kDa with Mw/Mn (Đ) of 1.25 obtained by SEC (Table 1). This block copolymer was further characterized by 13C NMR and DSC to confirm the structure and to determine the effect on thermal properties due to the presence of the PPDL block. Peak positions of the PPDL block in 13C NMR spectra also matched the previously reported values25 (Fig. S14†). The Tg of mPEG-b-PDL-b-PPDL copolymer did not change when compared to mPEG-b-PDL however the graph showed two distinct melting peaks which corresponded to the individual PEG and PPDL blocks (Fig. S15†).The melting temperature observed for the PPDL block was ∼88 °C after polymerisation with mPEG-b-PDL while the Tg of this block (PPDL) was not detectable by DSC.25 Characterisation data obtained for this copolymer confirmed the successful synthesis and purification of the desired triblock copolymer (ABC type).
Synthesis and characterisation of mPEG-b-PCL
The synthetic route to produce mPEG-b-PCL is shown in Scheme 1C and the experimental details with characterisation results are reported in Table 1. The target molar mass for mPEG-b-PCL was aimed to be similar to that of mPEG-b-PDL (i.e. 10 kDa), in order to carry out the appropriate comparison of this well-established copolymer with novel block copolymers of δ-decalactone. Copolymer mPEG-b-PCL was characterised by NMR and by SEC (Fig. S16 and S17†). The peak positions in NMR matched prior data30 and integrals suggested an approximate molar mass of 10 kDa.Determination of CMC of PDL and PCL block copolymer micelles
The CMC values of PDL-b-PEG-b-PDL, mPEG-b-PDL, mPEG-b-PCL and mPEG-b-PDL-b-PPDL were determined by changes in the I1 and I3 peak intensity ratio of pyrene with polymer concentration. The obtained curve was fitted using nonlinear regression (sigmoidal, 4PL, X axis log scale) to determine the CMC value (Fig. S18†). The inflection point (IC50) of the sigmoidal curve was considered as the CMC value of the polymer.The CMC values observed for block copolymers are presented in Table 2. Further, 95%-confidence intervals of the CMCs were plotted to visualise any statistical difference in obtained CMC values (Fig. 1). The CMC ranges for the PDL block copolymers were overlapped indicating no statistically significant differences across these materials. The CMC value observed for mPEG-b-PCL copolymer was very close to the previously reported value,31 and was approximately 2.5 times higher compared to the PDL block copolymer micelles (Fig. 1, Table 2).
 | ||
| Fig. 1 Comparison of 95% confidence intervals of the CMC values of synthesised copolymers. Values were obtained using non-linear curve fitting (sigmoidal, 4PL). | ||
| Sample | CMC (μg mL−1) | Z-Average size (d/nm) (±SD) (empty) | Poly(dispersity) index (empty) | Zeta potential (mv) (±SD) (empty) | Z-Average size (d/nm) (±SD) (NR loaded) | Poly(dispersity) index (NR loaded) | NR content in micelles (wt%) (±SD) |
|---|---|---|---|---|---|---|---|
| a Nano-emulsion preparation of homopolymer. | |||||||
| Propargyl PDLa | NA | 149 ± 4 | 0.04 ± 0.02 | −70.5 ± 3.0 | NA | NA | NA |
| PDL-b-PEG-b-PDL | 1.50 | 163 ± 7 | 0.26 ± 0.03 | −6.8 ± 2.6 | 58 ± 5 | 0.32 ± 0.02 | 0.81 ± 0.01 |
| mPEG-b-PDL | 1.33 | 34 ± 4 | 0.12 ± 0.02 | −3.1 ± 0.8 | 38 ± 3 | 0.17 ± 0.02 | 0.69 ± 0.01 |
| mPEG-b-PDL-b-PPDL | 1.19 | 85 ± 5 | 0.28 ± 0.01 | −2.5 ± 0.8 | 83 ± 4 | 0.38 ± 0.02 | 0.71 ± 0.01 |
| mPEG-b-PCL | 3.34 | 36 ± 3 | 0.14 ± 0.02 | −1.2 ± 1.2 | NA | NA | NA |
Preparation and characterisation of empty micelles, nanoemulsions and NR loaded micelles
The nano-precipitation method has previously been employed successfully for the incorporation of hydrophobic molecules inside micellar cores32 and therefore was chosen for preparation of empty and dye loaded micelles. The recoveries (after filtration) of micellar suspensions fabricated from mPEG-b-PDL, mPEG-b-PCL and PDL-b-PEG-b-PDL copolymer ranged from 90 to 95%. However, the recovery of the micellar suspensions of mPEG-b-PDL-b-PPDL copolymer was approximately 60%. The sizes (Z-average diameter) recorded for samples in HPLC grade water are reported in Table 2. The average sizes of mPEG-b-PCL copolymer micelles corresponded well with the sizes of similar copolymers reported in the literature.33 Size and polydispersity indices obtained for empty mPEG-b-PCL micelles were comparable to those of mPEG-b-PDL micelles. Size distribution curves observed for mPEG-b-PDL and mPEG-b-PCL micelles were unimodal whereas micelles prepared from PDL-b-PEG-b-PDL gave bimodal distribution curves (Fig. 2). Micelles prepared from mPEG-b-PDL-b-PPDL copolymer gave the broadest size distribution when compared to the other PDL block copolymers (Fig. 2).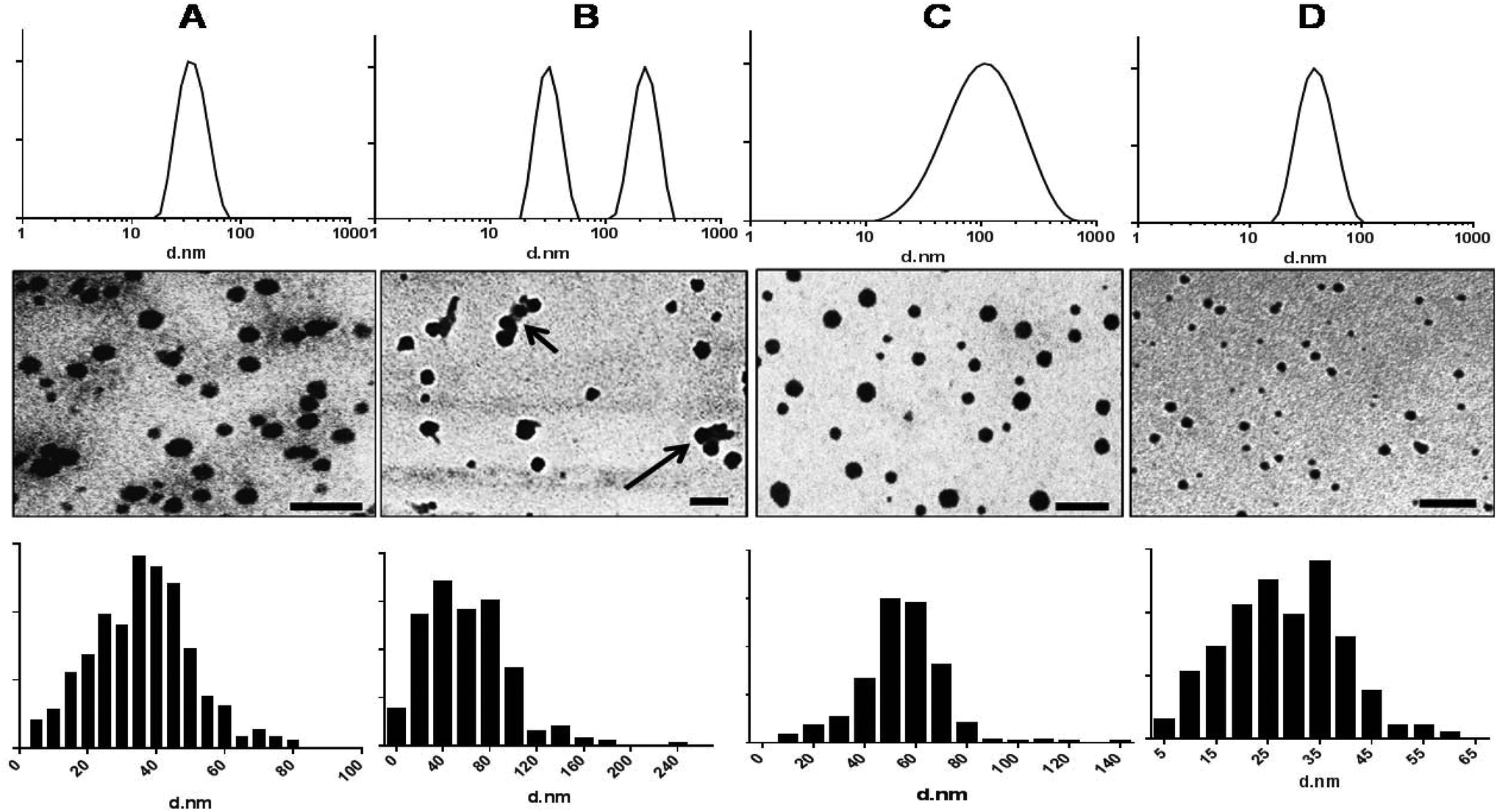 | ||
| Fig. 2 Size distribution curve by intensity determined by DLS, TEM images and size distribution histogram (analysed using ImageJ software) of empty (A) mPEG-b-PDL, (B) PDL-b-PEG-b-PDL, (C) mPEG-b-PDL-b-PPDL and (D) mPEG-b-PCL micelles. Arrows represents the presence of clusters in PDL-b-PEG-b-PDL micelles sample. TEM images were acquired without staining the samples. Scale bar – 200 nm. | ||
The nano-precipitated polymers were also imaged by TEM and it was found that the micelles obtained from amphiphilic block copolymers were roughly spherical in shape. TEM images of mPEG-b-PDL-b-PPDL indicated a broad size range in the sample whereas mPEG-b-PDL and mPEG-b-PCL possessed narrower and more uniform size distributions (Fig. 2). TEM images of the sample PDL-b-PEG-b-PDL indicated the presence of some aggregates (clusters) of the micelles. Thus, it is likely that the bimodal distribution observed in the DLS analysis for this sample was due, at least in part, to some clustered micelles. Analysis (Image J) of TEM images suggested that none of the micelles were larger than 60 nm for mPEG-b-PDL and 80 nm for mPEG-b-PCL samples respectively. However, as expected PDL-b-PEG-b-PDL and mPEG-b-PDL-b-PPDL samples were shown to be larger by TEM image analysis in accord with the DLS data. The nano-emulsion prepared from propargyl-PDL homopolymer were also larger than the micelles prepared from the amphiphilic diblock co-polymers (Fig. S19†).
The zeta potentials for the nano-emulsion (globules) prepared from PDL homopolymer in HEPES buffer (10 mM, pH 7.4) were (−) 70.5 ± 3.0 mv, which suggested charge stabilisation due most likely to carboxyl termini from lower molar mass polymers at the surface (Table 2, Fig. S19†). The zeta potential values for amphiphilic block copolymers were close to neutral (Table 2, Fig. S20†), as expected owing to the neutral PEG corona for these types of micelles.34
The self-assembly behaviour of the PDL block copolymer micelles and their ability to act as carrier vehicles was evaluated by using NR as an encapsulant. The absorption maxima of NR are strongly dependent on the polarity of the surrounding environment. Generally the λmax of NR shifts from a high value to low value with a decrease in the polarity of surrounding media.35 A clear hypsochromic shift in λmax (i.e. 531 nm) of NR encapsulated in micelles was observed when compared with the maximum absorption of dye in acetone solution (λmax – 541 nm) (Fig. S21†). This result suggested the encapsulation of NR inside the hydrophobic PDL core of the micelles.18
The amount of NR encapsulated in each micelle formulation was calculated using UV-Vis spectroscopy. A control sample (water only) showed no solubilisation of NR as anticipated from the highly hydrophobic character of this dye (Fig. S22†). The loading percentages were compared using a one-way ANOVA with Tukey's correction for multiple comparisons. Loading of NR was however, low, with less than 1% dye content in all the formulations (Table 2). The sizes of the NR loaded micelles were not significantly different when compared to the blank micelles except with PDL-b-PEG-b-PDL micelles (Table 2, Fig. S22†).
Preparation and characterisation of AmpB loaded micelles
The aliphatic nature of the PDL backbone suggested that drugs with more flexible carbon chains might be better encapsulated in the micellar cores than the highly aromatic dye NR. Therefore, the anti-fungal compound AmpB was used as a probe drug for PDL co-polymer formulations. AmpB-loaded micelles were prepared by nanoprecipitation from methanol.It was observed that the mPEG-b-PCL copolymer, due to its poor solubility in methanol, produced large particles on nanoprecipitation (Fig. S23-A†), although particles above 220 nm sizes could be removed by filtration (Fig. S23-B†).
The recovery of micellar suspensions after filtration was approximately 30–40% for mPEG-b-PCL copolymer whereas 90–95% of mPEG-b-PDL micellar suspensions were recovered. No significant differences in the sizes of empty micelles were observed when compared with micelles prepared using acetone as solvent. Furthermore, AmpB loading did not significantly change the size of the micelles when compared with blank micelles (Table 3, Fig. 3 and S24†). However, a high polydispersity index was observed for mPEG-b-PCL micelles, possibly due to the presence of aggregates (Fig. S24†). Interestingly, after loading of AmpB in mPEG-b-PCL micelles, a slight reduction in mean size and polydispersity index was observed.
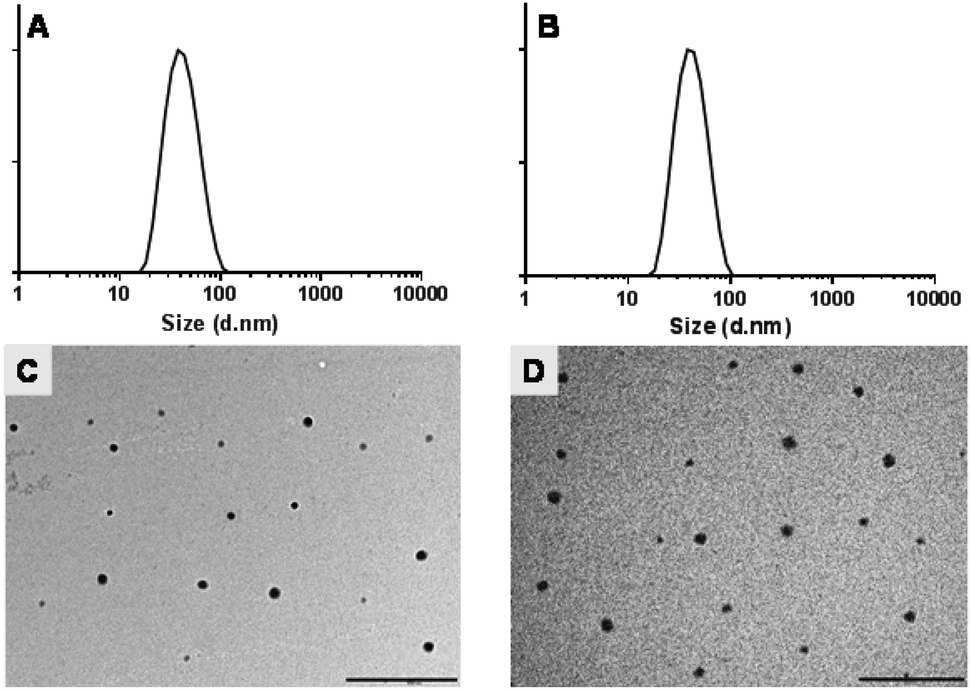 | ||
| Fig. 3 Size distributions curve by intensity of (A) blank mPEG-b-PDL, (B) AmpB loaded mPEG-b-PDL, and TEM image of (C) blank mPEG-b-PDL and (D) AmpB loaded mPEG-b-PDL micelles. The images were taken without staining. Scale bar – 500 nm. | ||
| Sample | Z-Average size (d/nm) (±SD) | Polydispersity index | Zeta potential (mv) (±SD) |
|---|---|---|---|
| mPEG-b-PDL (blank) | 41 ± 2 | 0.15 ± 0.01 | −2.4 ± 1.3 |
| mPEG-b-PDL (loaded) | 44 ± 3 | 0.16 ± 0.02 | −2.8 ± 1.1 |
| mPEG-b-PCL (blank) | 36 ± 2 | 0.30 ± 0.03 | −0.3 ± 1.7 |
| mPEG-b-PCL (loaded) | 32 ± 3 | 0.26 ± 0.02 | −1.2 ± 1.0 |
Zeta potentials observed for all micelle formulations in 10 mM HEPES buffer were almost neutral and no significant change in zeta potential was observed after AmpB loading (Table 3). TEM images of blank and AmpB loaded micelles confirmed the size ranges and suggested that the prepared micelles were roughly spherical in shape (Fig. 3 and S24†). The AmpB content found in mPEG-b-PDL micelles (3.5 ± 0.2 wt%) was seven-fold higher than mPEG-b-PCL micelles (0.5 ± 0.1 wt%) (Fig. 4).
 | ||
| Fig. 4 Graph represents AmpB content (weight% to polymer) observed in micelles, which was determined by UV-Vis spectrophotometry (λmax – 405 nm). Dots represent separate individual value and bar represents the mean value (n = 3). | ||
In vitro release study of AmpB from block copolymer micelles
AmpB is poorly soluble in water and hence Tween 80 (1% v/v) was added to enhance its solubility in the release media. The concentration of Tween 80 used was well above the CMC and hence it was expected that micelles of this surfactant would not diffuse through the dialysis bag (MWCO 3.5–5 kDa). Due to the poor loading of AmpB in mPEG-b-PCL micelles, this formulation was excluded from the release study. The release pattern of mPEG-b-PDL micelles was compared with the Tween 80 micelles formulation.An initial burst release of AmpB (30% approx. within 6 h) was observed with mPEG-b-PDL micelles followed by a slow-release phase, which continued for 8 days. In contrast, control “A” (Tween 80 micelles only) released 100% of AmpB in 3 days, of which 53% of the drug was released in the first 6 h. The control “B” formulation contained AmpB in Tween 80, added to unloaded mPEG-b-PDL micelles showed a more sustained release of AmpB compared to control “A” but faster release compared to AmpB pre-loaded in mPEG-b-PDL micelles (Fig. 5). In the first 6 h, no significant difference in the percentage drug released was observed with control “B” when compared to control “A”. However, due to the likely equilibration of AmpB in to empty mPEG-b-PDL micelles with time, a sustained release was observed with control “B”, which lasted for 5 days.
 | ||
| Fig. 5 Cumulative release (%) of AmpB from different test formulations in water containing Tween 80 (1% v/v) at 37 °C. The release study was performed by a dialysis method and AmpB concentration was estimated using UV-Vis spectrophotometer. Curve fits were based on a two-phase decay model to accommodate the initial burst period and subsequent sustained release. | ||
In vitro degradation study of mPEG-b-PDL micelles
The degradation time of a polyester is an important parameter regarding its fate inside the body and/or on long term storage in solution. A known degradation profile of a polymer is also valuable in designing a sustained drug release system. Therefore, a preliminary study of the hydrolytic degradation of mPEG-b-PDL micelles was performed over a 4 month period.Samples were analysed by SEC to determine the change in molar mass (Mn). An SEC trace of mPEG-b-PDL micelles after 120 days (at pH 7.4) and the loss of Mnversus time are shown in Fig. S25† and Fig. 6 respectively. The change in Mn of mPEG-b-PDL (peak 1 in Fig. S25†) was used to plot the degradation of polymer with time. Complete cleavage of ester fragments from the non-degradable mPEG block (Mn detected for mPEG by SEC was 10.8 kDa) was observed in 53 days at pH 4.0. However, at physiological pH (i.e. 7.4) only a 16% drop in Mn was observed after 4 months.
 | ||
| Fig. 6 Loss in molar mass (Mn) of mPEG-b-PDL micelles with time, incubated at 37 °C at two different pH. The SEC instrument was calibrated using polystyrene standards and chloroform was used as mobile phase. | ||
Effect of PDL-based co-polymers on metabolic activity of human cells in vitro
The potential for cytotoxicity of empty co-polymer micelles was tested on HCT116 cells. Cells were incubated with polymers for 24 h before their metabolic activity (as a proxy for viability) was assessed using an Alamar Blue assay (Fig. 7). | ||
| Fig. 7 Effects of empty micelle formulations on metabolic activity of HCT116 measured using an Alamar Blue assay after 24 hours. N = 6; mean ± SEM. | ||
At the highest polymer concentration no differences in viability between micelles was observed. However, a small but statistically significant reduction in viability was observed for both co-polymers compared to untreated controls (one-way ANOVA P ≥ 0.0001). As the maximum observed reduction in viability was not greater than 20% the micelles were considered to display an acceptably low level of toxicity.
Discussion
In this manuscript, the synthesis of homopolymers and copolymers of δ-decalactone under mild conditions has been demonstrated, with a view to developing a ‘green’ route to biomedical materials. We modified a previously reported procedure and observed that reactions at low temperatures gave higher conversion, but also that no further reaction occurred after ∼91% conversion: these results were analogous to those reported by Martello et al.21PDL homopolymers were prepared to evaluate the difference in physicochemical properties compared to copolymers of PDL. Although not of major significance for our particular study, we noted that during the synthesis of homo- and block copolymers of δ-decalactone, the formation of PDL homopolymer was also always observed. This suggested the presence of an initiator, additional to the added PEG or alcohol in the reaction mixture. NMR spectra of δ-decalactone showed resonances at 3.6 and 4.9 ppm, even from pristine samples, indicating the existence of the open form of the lactone ring and short oligomers with hydroxyl end groups (Fig. S1† inset). We were not able to remove all traces of these species during the purification procedure for the monomer, and thus it is possible that the free hydroxyl groups on the oligomers could have acted as ROP initiators. Additionally in the mechanism of TBD catalysis for ROP proposed by Pratt et al.36 there is a reversible step in which an alcohol is generated, and this hydroxyl group can potentially act as initiator for other molecules activated by TBD. Jaffredo et al. used TBD as catalyst as well as initiator for the ROP of β-butyrolactone37 and suggested that TBD has the potential to act as an initiator.
Fortunately for our study, we found that the difference in solubility of copolymers and homo-polymers in ether offered a route to separate the desired copolymer from the homopolymer impurity. However, we were not able to separate the end-functional BZD-PDL and propargyl-PDL away from the hydroxyl-tipped variants due to lack in solubility differences. Therefore, the Mn values for these polymers reported from NMR included a systematic error owing to the presence of both detectable (BZD, propargyl) and non-detectable (hydroxyl) initiating groups in each polymer sample. In contrast, the Mn values derived from SEC for the PEG-based block copolymers were much larger than the molar masses calculated by 1H NMR. This we attribute to the very different solution properties of the blocks compared to the polystyrene standards, and interaction of PEG with the SEC columns (PLgel Mixed-D). It should be noted that the Mn of mPEG under the SEC conditions used for the copolymers was almost twice the molar mass reported by the supplier (PEG4000Mn ∼ 7.7 kDa, mPEG5000Mn ∼ 10.8 kDa, confirmed by 1H NMR analysis of both PEG starting materials), indicative of the obvious limitations of polystyrene as an SEC standard for block co-polymers.
DSC data for the copolymers suggested semicrystalline polymers had been produced since both Tg and Tm were observed for the PDL copolymers. Subsequently, the syntheses of ABC block copolymers of ω-pentadecalactone were performed with the aim to generate materials with better solubility and intermediate crystallinity. ROP of ω-pentadecalactone using mPEG-b-PDL as an initiator and TBD (1 mol% to monomer) as catalyst was first attempted at 110 °C in bulk. During this reaction, it was found that at higher temperature, the mPEG-b-PDL chain was cleaved and the decalactone ring was regenerated. The depolymerisation (reversible) process observed with PDL was likely associated with the thermodynamic equilibrium of δ-decalactone with TBD at high temperature.21 Therefore, in order to provide comparator block copolymers of PEG-PDL with PPDL, a lipase was used as catalyst to synthesise the blocks with minimal side reactions. The molar mass of the PPDL block was varied in attempts to modulate the solubility of the final copolymer in acetone. It was found that increases in the PPDL block molar mass above 2 kDa for the mPEG-b-PDL-b-PPDL copolymer reduced its apparent solubility in acetone. Therefore, a copolymer containing less than 2 kDa of PPDL block was synthesised.
The organic catalyst TBD has been reported to be very efficient for the ROP of ε-caprolactone30 and therefore was used for the synthesis of a ‘control’ copolymer of PEG and ε-caprolactone. Compared to the ROP of δ-decalactone, no polymer synthesis from additional initiator was observed during ROP of ε-caprolactone with TBD as catalyst, most likely due to the greater purity of the monomer. Other characterisation data for these polymers were in line with prior literature.
Further evaluation of the PDL-based block co-polymers by CMC measurements showed some differences in solution behaviour compared to previously reported block copolymers of PCL. It is well established that the CMCs of amphiphilic block copolymers decrease with increase in the length of core forming block or by a decrease in the length of the shell forming section.38 From the molar masses calculated by 1H NMR, the relative hydrophobicities of the block copolymers based on core- and shell-forming block lengths were predicted to be mPEG-b-PDL < PDL-b-PEG-b-PDL < mPEG-b-PDL-b-PPDL. However, no significant differences in the CMCs were observed across these polymer formulations, suggesting that the differences in the lengths of the hydrophobic chains in each case were not sufficient to alter the self-assembly energetics to a measurable extent. In addition, the CMC for the PDL-b-PEG-b-PDL triblock copolymer was higher than that for mPEG-b-PDL copolymer (Table 2). This unexpected result may have been due to the altered packing in the triblock copolymer, with the reduced interfacial curvature possible compared to a di-block, and thus the reduced tendency to assemble into a well-defined micellar architecture.
CMC values for the block copolymers of PDL were lower compared to those determined for the PEG-b-PCL block copolymer. These data were also unanticipated given previous reports which showed that PEG-b-ε-PDL copolymers exhibited higher CMC values compared with PEG-b-PCL block co-polymers, as a result of the semicrystalline hydrophobic cores in PCL blocks compared to the amorphous ε-PDL segments.22 However, the lower CMC values for the δ-PDL copolymer micelles in our study compared to the similar molar mass PCL block copolymer might partly be attributed to the different alkyl side chain within the δ-PDL structure and a resulting change in packing compared to the ε-PDL reported previously. A similar phenomenon has been noted in which copolymers of poly(lactide) and alkyl chain-substituted lactide monomer displayed lower CMC values compared to their unsubstituted analogues18 In addition, the CMC value of the mPEG-b-PCL co-polymer in this study was ∼3 fold lower than that reported by Glavas et al.22 for a PEG-b-PCL di-block. This suggested that crystallinity of the hydrophobic block was less important than block chain length in this instance, as the molar mass of the hydrophobic block in our PEG-b-PCL was almost 2.5 times higher in that in the previous work.22 It should also be noted that the PEG chains in the work by Glavas et al. were shorter (2 kDa) than those in this study (5 kDa), which would also have changed the micelle structure and stability.
Low CMC values are a prerequisite for avoiding premature release of loaded therapeutic agents from carrier systems before reaching target sites,39 thus the PDL block copolymers from this study were considered suitable for drug incorporation and release experiments. Micelle-like nanoparticles of mPEG-b-PDL and mPEG-b-PCL micelles formed by nanoprecipitation routes were of similar sizes (∼40 nm), whereas the sizes of ABA (PDL-b-PEG-b-PDL) and ABC (mPEG-b-PDL-b-PPDL) tri-block copolymers were considerably larger (85–165 nm). The bimodal size distributions observed with the empty ABA type copolymer (PDL-b-PEG-b-PDL) micelles were probably due to additional hydrophobic–hydrophobic interactions between micelle cores, which led to aggregation and clustering40 This mechanism is in accord with proposals that ABA type block copolymers can assemble in flower-shaped micelles41 leading to a decrease in PEG chain length at the periphery separating the core from the surrounding aqueous environment. In addition, the PDL-b-PEG-b-PDL copolymer was synthesised using a shorter chain PEG (Mn = 4.0 kDa) compared to the di-block (mPEG-b-PCL and mPEG-b-PDL) co-polymers and hence was expected to have a less dense hydrophilic corona. Since PEG imparts steric stability to micelles by minimizing the interfacial free energy of the micellar core and by inhibiting hydrophobic inter-micellar attractions,42 it is likely that hydrophobic block self-association may have occurred with the ABA triblock copolymer micelles. In turn, partial self-association may have generated bimodal size distributions, which is apparent in DLS and TEM images. A similar phenomenon was reported earlier with PCL-b-PEG-b-PCL micelles in which the presence of aggregates (clusters) was proposed.43 Broad size distributions were also observed for the mPEG-b-PDL-b-PPDL tri-blocks after nanoprecipitation (Fig. 2D). Since different sized particles in pharmaceutical formulations for injection would give unpredictable in vivo bio-distribution and aggregates would lead to severe problems in elimination,44 the tri-block co-polymers were not selected for drug incorporation experiments in this particular study. However, since ABC tri-block polymers have the possibility to be differentially functionalised at each end, one might envisage the use of tri-blocks, similar to those we prepared, for targeted polymersome preparation. In such cases, the different hydrophilic blocks might be derivatised with ligands such that combined receptor targeting could be achieved. Targeted triblock polymersomes have been shown to deliver siRNA45 but as the focus for our study was more towards hydrophobic core polymers and drugs, we did not pursue these tri-blocks further.
It was anticipated that the presence of alkyl side-chains pendant to the backbone in the mPEG-b-PDL copolymer might enhance association with aliphatic-rich drug compounds such as AmpB. The mPEG-b-PDL copolymer formed micelles from methanol nanoprecipitations which were of comparable size (∼40 nm) to micelles prepared using acetone as solvent. However, methanol was found not to be suitable for nanoprecipitation of comparator mPEG-b-PCL di-blocks, due to the poor solubility of this copolymer. Large and polydisperse mPEG-b-PCL micelles were formed from this solvent, but a reduction in size and polydispersity was observed for the same di-blocks nano-precipitated in the presence of AmpB. This may have been due to hydrophobic interactions between the PCL core and AmpB, which facilitated the assembly of denser micelles. Similar results have been reported with mPEG-b-PCL micelles when the highly hydrophobic drug fenofibrate was loaded.46
The encapsulation data suggested that the mPEG-b-PDL copolymer incorporated more of the AmpB drug compared to mPEG-b-PCL copolymer. However, the drug content observed in mPEG-b-PDL micelles was low compared to previously reported AmpB formulations.47 This may have been associated with the relative hydrophobicities in the polymer cores used but may simply have been a consequence of unoptimised experimental conditions during the nanoprecipitation reactions. For example, it has been reported that the loading of AmpB in a polymeric drug delivery system can be improved by either increase in the hydrophobic block length or by initial quantity of drug used for loading or both.48 In addition, computational simulation of nanoprecipitation processes49 using models of poly(glyceryl adipate) esters and amphiphilic drugs has shown a marked effect of polymer collapse rate on drug incorporation, and this is highly solvent dependent. It is thus reasonable to assume that the difference in drug loading we observed in mPEG-b-PDL polymers compared to previous materials could be due to both solvent quality and polymer composition.
An in vitro release experiment of AmpB loaded micelles indicated that reversible binding of drug towards carrier (micelles) was important in defining the release profile, based on the partition coefficient of drug between carrier and dispersed phase.50 Nevertheless, the release pattern observed with novel mPEG-b-PDL micelles was more sustained compared to the earlier reported release pattern for AmpB in PLA nanoparticles using the same release media.29
Preliminary in vitro degradation studies suggested that the mPEG-b-PDL micelles degraded more slowly at physiological pH (9% after 30 days) compared to two different reports on PEG-b-PLA block copolymers (27.6%,51 16.3%52 after 30 days). This was likely due to the higher hydrophobicity of mPEG-b-PDL copolymer. However, it was found that the degradation rate was faster than with mPEG-b-PCL copolymers (9.0% after 126 days,53 4.1% after 56 days54). It has been reported that amorphous polymers degrade faster than semicrystalline polymers55 and thus the faster degradation of the PDL block compared to the PCL blocks in our study may have been due to differences in crystallinity.
The in vitro cell activity study results suggested that the mPEG-b-PDL micelles were similar to mPEG-b-PCL comparators in regards to their effects on metabolic activity. The similar cyto-compatibility profile was not unexpected, as in both systems the cells encountered primarily the PEG corona and degradation of the PDL and PCL cores was not significant over the assay period.
Conclusions
In this study, the synthesis and characterisation of homopolymers and novel amphiphilic block copolymers based on renewable monomers has been reported. The self-assembly of amphiphilic block copolymers was demonstrated via NR loading. Further, increased loading of AmpB was demonstrated with mPEG-b-PDL micelles compared to their counterpart mPEG-b-PCL micelles. In vitro release studies showed a prolonged release of AmpB from mPEG-b-PDL micelles when compared with Tween 80 micelles. Preliminary in vitro degradation studies of mPEG-b-PDL micelles indicated the slow degradation of copolymer at physiological pH. In vitro cell metabolic activity assays revealed that the novel mPEG-b-PDL micelles exhibited similar tolerability profiles compared to mPEG-b-PCL micelles. The results suggest that mPEG-b-PDL copolymers might find future application as drug delivery systems in which slow release and long degradation time is required, for example in sub-cutaneously injected depot formulations.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the Indian Government (PhD Scholarship to KB). We thank EPSRC for additional financial support (EPSRC Leadership Fellowship – EP/H005625/1 to CA) and Christy Grainger-Boultby and Paul Cooling for technical assistance.Notes and references
- N. D. Stebbins, J. J. Faig, W. Yu, R. Guliyev and K. E. Uhrich, Biomater. Sci., 2015, 3, 1171–1187 RSC.
- V. Delplace, P. Couvreur and J. Nicolas, Polym. Chem., 2014, 5, 1529–1544 RSC.
- C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 1501–1502 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- C. S. Hege and S. M. Schiller, Green Chem., 2014, 16, 1410–1416 RSC.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- S. A. Miller, Polym. Chem., 2014, 5, 3117–3118 RSC.
- J. J. Garcia and S. A. Miller, Polym. Chem., 2014, 5, 955–961 RSC.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- M. Meier, Green Chem., 2014, 16, 1672–1672 RSC.
- K. Schroder, K. Matyjaszewski, K. J. T. Noonan and R. T. Mathers, Green Chem., 2014, 16, 1673–1686 RSC.
- F. Stempfle, B. S. Ritter, R. Mulhaupt and S. Mecking, Green Chem., 2014, 16, 2008–2014 RSC.
- D. Tang, C. W. Macosko and M. A. Hillmyer, Polym. Chem., 2014, 5, 3231–3237 RSC.
- J. M. Halpern, R. Urbanski, A. K. Weinstock, D. F. Iwig, R. T. Mathers and H. A. von Recum, J. Biomed. Mater. Res., Part A, 2014, 102, 1467–1477 CrossRef PubMed.
- C. D. Pritchard, K. M. Arnér, R. S. Langer and F. K. Ghosh, Biomaterials, 2010, 31, 7978–7984 CrossRef CAS PubMed.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS PubMed.
- T. Trimaille, K. Mondon, R. Gurny and M. Moeller, Int. J. Pharm., 2006, 319, 147–154 CrossRef CAS PubMed.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef CAS.
- M. T. Martello, A. Burns and M. Hillmyer, ACS Macro Lett., 2012, 1, 131–135 CrossRef CAS.
- L. Glavas, P. Olsen, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2013, 14, 4150–4156 CrossRef CAS PubMed.
- P. Olsen, T. Borke, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2013, 14, 2883–2890 CrossRef CAS PubMed.
- D. McGinty, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2011, 49, S193–S201 CrossRef CAS PubMed.
- Z. Jiang, H. Azim, R. A. Gross, M. L. Focarete and M. Scandola, Biomacromolecules, 2007, 8, 2262–2269 CrossRef CAS PubMed.
- J. Aguiar, P. Carpena, J. A. Molina-Bolivar and C. C. Ruiz, J. Colloid Interface Sci., 2003, 258, 116–122 CrossRef CAS.
- M. Gou, K. Men, H. Shi, M. Xiang, J. Zhang, J. Song, J. Long, Y. Wan, F. Luo, X. Zhao and Z. Qian, Nanoscale, 2011, 3, 1558–1567 RSC.
- S. Galindo-Rodriguez, E. Allemann, H. Fessi and E. Doelker, Pharm. Res., 2004, 21, 1428–1439 CrossRef CAS.
- W. Zhou, Y. Wang, J. Jian and S. Song, Int. J. Nanomed., 2013, 8, 3715–3728 Search PubMed.
- M. Bouyahyi, M. P. F. Pepels, A. Heise and R. Duchateau, Macromolecules, 2012, 45, 3356–3366 CrossRef CAS.
- X. Gao, B. Wang, X. Wei, W. Rao, F. Ai, F. Zhao, K. Men, B. Yang, X. Liu, M. Huang, M. Gou, Z. Qian, N. Huang and Y. Wei, Int. J. Nanomed., 2013, 8, 971–982 CrossRef PubMed.
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86–97 CrossRef CAS PubMed.
- W. Xie, W. Zhu and Z. Shen, Polymer, 2007, 48, 6791–6798 CrossRef CAS PubMed.
- T. Merdan, K. Kunath, H. Petersen, U. Bakowsky, K. H. Voigt, J. Kopecek and T. Kissel, Bioconjugate Chem., 2005, 16, 785–792 CrossRef CAS PubMed.
- M. Wyszogrodzka and R. Haag, Chem. – Eur. J., 2008, 14, 9202–9214 CrossRef CAS PubMed.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Macromol. Rapid Commun., 2012, 33, 1938–1944 CrossRef CAS PubMed.
- G. Gaucher, M. H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J. C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- Q. Dai, Y. Yan, C.-S. Ang, K. Kempe, M. M. J. Kamphuis, S. J. Dodds and F. Caruso, ACS Nano, 2015, 9, 2876–2885 CrossRef CAS PubMed.
- S. Cammas, K. Suzuki, C. Sone, Y. Sakurai, K. Kataoka and T. Okano, J. Controlled Release, 1997, 48, 157–164 CrossRef CAS.
- S. Maiti and P. R. Chatterji, Langmuir, 1997, 13, 5011–5015 CrossRef CAS.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2012, 64, 246–255 CrossRef PubMed.
- H. X. Ge, Y. Hu, X. Q. Jiang, D. M. Cheng, Y. Y. Yuan, H. Bi and C. Z. Yang, J. Pharm. Sci., 2002, 91, 1463–1473 CrossRef CAS PubMed.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- E. Gallon, T. Matini, L. Sasso, G. Mantovani, A. Armiñan de Benito, J. Sanchis, P. Caliceti, C. Alexander, M. J. Vicent and S. Salmaso, Biomacromolecules, 2015, 16, 1924–1937 CrossRef CAS PubMed.
- K. K. Jette, D. Law, E. A. Schmitt and G. S. Kwon, Pharm. Res., 2004, 21, 1184–1191 CrossRef CAS.
- H. Van de Ven, C. Paulussen, P. B. Feijens, A. Matheeussen, P. Rombaut, P. Kayaert, G. Van den Mooter, W. Weyenberg, P. Cos, L. Maes and A. Ludwig, J. Controlled Release, 2012, 161, 795–803 CrossRef CAS PubMed.
- K.-C. Choi, J.-Y. Bang, P.-I. Kim, C. Kim and C.-E. Song, Int. J. Pharm., 2008, 355, 224–230 CrossRef CAS PubMed.
- R. Mackenzie, J. Booth, C. Alexander, M. C. Garnett and C. A. Laughton, J. Chem. Theory Comput., 2015, 11, 2705–2713 CrossRef CAS.
- C. Washington, Int. J. Pharm., 1989, 56, 71–74 CrossRef CAS.
- Y. Li, X. R. Qi, Y. Maitani and T. Nagai, Nanotechnology, 2009, 20 Search PubMed.
- M. Stefani, J. Coudane and M. Vert, Polym. Degrad. Stab., 2006, 91, 2554–2559 CrossRef CAS PubMed.
- M. H. Huang, S. M. Li, D. W. Hutmacher, J. T. Schantz, C. A. Vacanti, C. Braud and M. Vert, J. Biomed. Mater. Res., Part A, 2004, 69A, 417–427 CrossRef CAS PubMed.
- S. Cometa, I. Bartolozzi, A. Corti, F. Chiellini, E. De Giglio and E. Chiellini, Polym. Degrad. Stab., 2010, 95, 2013–2021 CrossRef CAS PubMed.
- T. Trimaille, R. Gurny and M. Moeller, J. Biomed. Mater. Res., Part A, 2007, 80A, 55–65 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01203a |
| This journal is © The Royal Society of Chemistry 2015 |