
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHypercrosslinked porous polymer nanosheets: 2D RAFT agent directed emulsion polymerization for multifunctional applications†
Wuxue
Zhao‡
a,
Zongsheng
Hou‡
a,
Zhaoquan
Yao
a,
Xiaodong
Zhuang
*a,
Fan
Zhang
*a and
Xinliang
Feng
ab
aShanghai Key Lab of Electrical Insulation and Thermal Ageing & Shanghai Electrochemical Energy Devices Research Center, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: zhuang@sjtu.edu.cn; fan-zhang@sjtu.edu.cn
bCenter for Advancing Electronics Dresden & Department of Chemistry and Food Chemistry, Technische Universitaet Dresden, 01062 Dresden, Germany
First published on 26th August 2015
Abstract
Two-dimensional hyper-cross-linked microporous polymers (2D HCPs) have been readily synthesized by emulsion polymerization using trithiocarbonate functionalized graphene oxide as a 2D reversible addition–fragmentation chain transfer (RAFT) agent. After a gel-type and permanently porous poly(vinylbenzyl chloride-co-divinylbenzene) precursor resin was grafted to the graphene surface, an FeCl3-promoted Friedel–Crafts reaction was carried out for benzyl chloride groups to construct a porous structure confined within 2D nanosheets. The resulting HCPs exhibited superhydrophobic behavior, and had a predominance of micropores with a specific surface area of up to 1224 m2 g−1. Moreover, they showed improved thermal stability in comparison with unadorned HCPs obtained without using the graphene template. Their H2 and CO2 capacities at 77 and 273 K reached 1.27 and 9.74 wt%, respectively, at a relative pressure of 0.99. In addition, the as-synthesized 2D HCPs were used as carbon precursors to generate 2D porous carbons with a high specific surface area (871 m2 g−1) and high-performance electrochemical energy storage (144 F g−1 at 0.5 A g−1).
Introduction
Porous polymers1–6 with organic porous structures have attracted tremendous attention owing to their controllable porous features associated with prominent physical properties and potential applications, such as in light harvesting,7 sensing,8 gas separation9 and storage,10 catalysis,11–13 and energy storage and conversion.14 Among them, hyper-cross-linked porous polymers (HCPs) represent one of the fastest developing types of porous polymers—not only because of their high specific surface areas and gas storage properties but also due to the easy availability of many controllable polymerization methods and the wide variety of functional monomers.15 Most of the reported HCPs have been amorphous, and were synthesized by traditional radical10,16–18 and oxidation19 polymerization methods, followed by an additional step for further cross-linking. Most of the previous studies on HCPs have focused on pore construction, tuning the gas19–21 or small-molecule22,23 separation/storage properties through optimizing the reaction conditions such as the monomer ratio, catalyst percentages, the temperature, and the reaction time.10,24 However, effort dedicated to morphology control has rarely been reported for HCPs. Only quasi-zero-dimensional microsphere17,25 and three-dimensional monolith22,23 HCP types have been successfully synthesized. Two-dimensional (2D) HCPs are still under exploration owing to the lack of synthetic methods or 2D templates and the thermodynamic instability of 2D porous polymers in comparison with their zero- and three-dimensional porous counterparts.Graphene, which consists of a honeycomb-like hexagonal array of sp2-bonded carbon atoms, exhibits many remarkable properties such as superior electrical conductivity, a large surface area, excellent mechanical flexibility, and high thermal/chemical stability.26,27 Graphene oxide (GO), one of the most widely synthesized derivatives of graphene and made from graphite by a modified Hummers’ method, is an ideal 2D template for the preparation of 2D porous polymers because GO can be easily functionalized with many functional groups and solution processing.28–33 It is well known that the properties of graphene-based nanocomposites34 are highly dependent on the good dispersibility and interfacial interactions of graphene. In order to produce a homogeneous graphene dispersion during the preparation procedure, many attempts have been made to attach small molecules and linear polymers to the graphene surface by means of covalent bonds and weak interactions.35 Several grafting routes have been explored using either GO or functionalized GO with good dispersibility in various organic solvents as templates for the preparation of graphene-based 2D brushes and “sheet–coil” polymers.36 Ruoff and co-workers have prepared GO-based macroinitiators by the functionalization of GO with α-bromoisobutyryl bromide for controlled styrene grafting via atom transfer radical polymerization (ATRP).37,38 Almost all the reported studies have focused on linear polymers, such as polystyrene,39,40 poly(N-vinylcarbazole),41 polymethacrylamide,42,43 and poly(N-isopropylacrylamide),44 grafting from or grafting to graphene surfaces. To the best of the authors’ knowledge, the preparation of 2D porous polymers using the 2D chain-transfer agent (CTA) promoted reversible addition–fragmentation chain transfer (RAFT) polymerization method has not yet been reported.
In this work, a highly soluble trithiocarbonate-based CTA was used to functionalize GO to act as a 2D CTA as well as a 2D template. Then, poly(vinylbenzyl chloride-co-divinylbenzene) (PVD) grafted GO was prepared by RAFT emulsion polymerization based on the as-prepared 2D CTA. After further hyper-cross-linking by an FeCl3-promoted Friedel–Crafts reaction of the benzyl chloride groups, graphene-based sandwich-type HCPs with a 2D morphology (GHCPs) were successfully prepared on a large scale (∼100 g per synthesis). The resulting GHCPs exhibited superhydrophobic behavior (contact angle >150°) and had high specific surface areas of up to 1224 m2 g−1. They also exhibited an enhanced H2 capacity at 77 K (1.27 versus 0.85 wt%) and CO2 capacity at 273 K (9.74 versus 8.80 wt%) at a relative pressure of 0.99 in comparison with unadorned HCP obtained without using the graphene template. The high specific surface areas and the enhanced H2 and CO2 capacities can be the contributions of the 2D graphene template. Owing to the carbon backbone of graphene, GHCPs were used to prepare porous carbon nanosheets without removing the template. The as-prepared porous carbon nanosheets exhibited the typical 2D morphology and a high surface area (871 m2 g−1), and were successfully used in electrochemical capacitors (144 F g−1 at 0.5 A g−1). These results indicate that this method is a good approach for the preparation of 2D HCPs and 2D porous carbons with multifunctional applications.
Experimental section
Reagents and chemicals
Natural flake graphite, 1-dodecanethiol, carbon disulfide, aliquot 336, 1,3-diaminopropane, N-hydroxy-succinimide (NHS), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl), divinylbenzene (DVB), 2,2′-azobis(2-methylpropio-nitrile) (AIBN), and poly(vinyl alcohol) (PVA) were purchased from Aladdin (Shanghai). Vinylbenzyl chloride (VBC) was purchased from TCI. 1,2-Dichloroethane (DCE), tetrahydrofuran (THF), FeCl3, and other chemicals were purchased from Sinopharm Chemical Reagent Co. (Shanghai). THF, DCE and triethylamine were dried by standard methods before use.Functionalization of graphene oxide with amine groups45
GO was prepared from natural graphite flakes by a modified Hummers’ method.46 GO sheets (1.0 g) were added to dry DMF (250 mL). After ultrasonication for 24 h, NHS (3.42 g, 30 mmol) and EDC·HCl (5.75 g, 3.2 mmol) were added to GO dispersion at 0 °C. After stirring for 2 h, 1,3-diaminopropane (5.0 mL) was added, and the solution was stirred overnight at room temperature. After filtration and washing with water and ethanol, the resulting powder was freeze-dried and denoted as GO-NH2.Preparation of 2D CTA
2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDAT), which is a widely used and highly soluble chain transfer agent (CTA) for RAFT polymerization, was synthesized according to reported methods.47,48 GO-NH2 (1.0 g) was added to dry DMF (250 mL) and sonicated for a few hours. Then, triethylamine (5.0 mL) was added to the dispersion. Acylchloride-DDAT was prepared as follows: DDAT (5.0 g) and excess thionyl chloride (20 mL) were added to a 50 mL three-neck round-bottom flask and refluxed overnight under a nitrogen atmosphere. Then, unreacted thionyl chloride was completely removed under reduced pressure and washed with anhydrous THF (8 mL × 3). Lastly, the acylchloride-DDAT in THF (10 mL) was transferred to the above GO-NH2 dispersion very slowly under vigorous stirring in an ice bath. The mixtures were stirred at room temperature for 24 h. The resulting DDAT functionalized GO (GO-DDAT) was washed with CH2Cl2 and ethanol, and dried overnight under vacuum at 50 °C.RAFT emulsion polymerization
VBC/DVB modified GO precursor resin was prepared by conventional RAFT emulsion polymerization.49,50 The aqueous phase consisted of distilled water (50 mL), PVA (0.38 g) and NaCl (0.17 g). The organic phase comprised GO-DDAT (100 mg), VBC (6.37 g, 41.7 mmol), DVB (0.16 g, 1.2 mmol), and AIBN (33 mg). The organic phase was suspended in the aqueous phase at 80 °C by a stirring speed of 425 rpm under a N2 atmosphere. After 8 h, the beads were filtered and washed three times with water, methanol and diethyl ether. After drying under vacuum overnight at 60 °C, the copolymer grafted GO (GO-PVD) was produced. Bare PVD without using a graphene template was also synthesized using the same method.Synthesis of 2D hypercrosslinked microporous polymer
The VBC/DVB modified GO precursor resin beads (3.25 g) were swollen in DCE (40 mL) for 3 h. The mixture was cooled in an ice bath before adding FeCl3 (2.0 g). After achieving a uniform dispersion of FeCl3 throughout the precursor beads in 2 h, the mixture was heated to 80 °C for 12 h. After the resulting hypercrosslinked beads were filtered, the catalyst was washed out using methanol, a mixture of acetone and HCl (0.5 M), and deionized water. After drying under vacuum at 60 °C, the resulting 2D hypercrosslinked microporous polymer (2D-HCP) was produced. In the control experiment, GHCP-1, GHCP-2 and GHCP-3, denoting 100 mg, 200 mg and 800 mg of GO-DDAT, were used in above procedure respectively. According to the same procedure, bare PVD was also used to prepare HCP in order to compare with GHCPs.Characterization
FT-IR spectra, using KBr tablets containing the sample, were recorded on a Spectrum 100 (PerkinElmer, Inc., USA) spectrometer with a scan range of 4000–400 cm−1. The thermal degradation was analyzed by means of thermogravimetric analysis (TGA), which was measured by using a Q5000IR (TA Instruments, USA) thermogravimetric analyzer with a heating rate of 20 °C min−1 under nitrogen flow. Scanning electron microscopy (SEM) measurements were performed on a FEI Sirion-200 (FEI Co., USA) field emission scanning electron microscope. Transmission electron microscopy (TEM) images were taken with an EOL-2100 (JEOL Ltd, Japan) electron microscope at an operating voltage of 200 kV. The suspensions of the samples were dropped onto a copper grid covered with lacey support films. The nitrogen content was determined with a Vario ELIII/Isoprime (Elementar Co., Germany) isotope ratio mass spectrometer. The Brunauer–Emmett–Teller (BET) specific areas were measured on an ASAP 2010 M+C (Micromeritics Inc., USA) surface area and porosimetry analyzer based on N2 adsorption. The hydrogen and carbon dioxide sorptions were measured with Autosorb-iQA3200-4 adsorption equipment (Quantatech Co., USA).Electrochemical capacity characterization was conducted on an EG & potentiostat/galvanostat Model 2273 advanced electrochemical system. A conventional cell with a three-electrode configuration was employed throughout this study. The working electrode was prepared by mixing GHCP-1-900 with carbon black (Mitsubishi Chemicals, Inc.) and a poly-tetrafluoroethylene (PTFE) binder. The weight ratio of GHCP-1-900, carbon black and PTFE was 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. A platinum foil was applied as a counter electrode with a Hg/HgO electrode as a reference electrode. The experiments were carried out in 6 M KOH solution. The potential range was between −1 and 0 V (Ag/AgCl) at different scan rates at ambient temperature.
:10:10. A platinum foil was applied as a counter electrode with a Hg/HgO electrode as a reference electrode. The experiments were carried out in 6 M KOH solution. The potential range was between −1 and 0 V (Ag/AgCl) at different scan rates at ambient temperature.
Results and discussion
The strategy for the synthesis of GHCPs is presented in Scheme 1. First, a GO sheet was functionalized by 1,3-diaminopropane in the presence of NHS and EDC·HCl and then by acylchloride-DDAT in the presence of Et3N. The sulfur content of the produced DDAT-modified GO (DDAT) reached 2.3 wt%. Using GO-DDAT as a 2D CTA, RAFT emulsion polymerization was carried out on graphene surfaces using 1,4-divinylbenzene and 4-vinylbenzyl chloride as monomers in water along with AIBN, PVA, and NaCl. Afterwards, the copolymer-modified GO (GO-PVD) that was produced was hyper-cross-linked by using an FeCl3-catalyzed Friedel–Crafts reaction. Finally, 2D sandwich-type GHCPs were obtained by vacuum drying. In this procedure, GO-DDAT not only showed very good dispersibility in most organic solvents but acted as a 2D template for polymerization on graphene surfaces. In a control experiment, GHCP-1, GHCP-2, and GHCP-3 (denoting 1, 2, and 3 equivalents of GO-DDAT (see the Experimental section)) were obtained using the same procedure. The color of HCP, GHCP-1, GHCP-2, and GHCP-3 darkens as the GO-DDAT content increases (Fig. 1a). Moreover, the uniform color of the GHCPs indicates that the porous polymer has grafted uniformly to the GO flakes. The unadorned hyper-cross-linked porous polymer was also prepared without using the graphene template for comparison in this work (denoted HCP).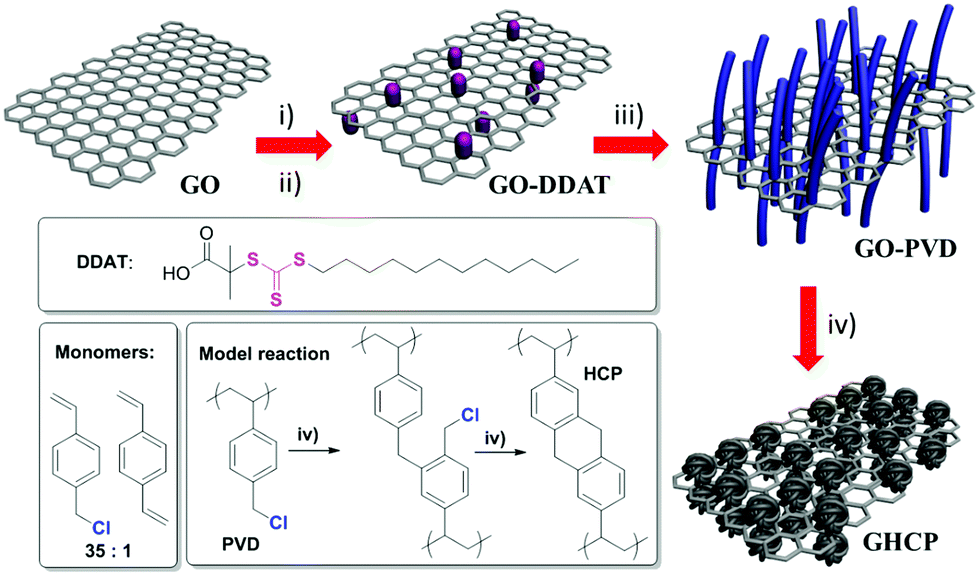 | ||
| Scheme 1 Preparation of GHCPs: (i) N-hydroxy-succinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), 1,3-diaminopropane, water, 0 °C, 12 h; (ii) N2, acyl chloride-S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (DDAT), dry dimethylformamide, Et3N, 0 °C, 24 h; (iii) N2, water, polyvinyl alcohol (PVA), NaCl, AIBN, 1,4-divinylbenzene, 4-vinylbenzyl chloride, 80 °C, 8 h; (iv) 1,2-dichloroethane, FeCl3, 80 °C, 12 h. | ||
 | ||
| Fig. 1 (a) HCP and the GHCPs. (b, c) SEM and (d, e) TEM images of GHCP-3. | ||
The morphology and microstructure of the as-prepared GHCPs were investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). All of the GHCPs showed similar sheet morphologies (Fig. 1 and S1†). Thus, the results of GHCP-3 are discussed here as a typical example. As shown in Fig. 1b, many free-standing sheets with a morphology similar to that of graphene and with sizes ranging from 500 nm to several micrometers were observed. In addition, these porous polymer sheets exhibited wrinkles and flexible features that stand in contrast to the rigid inorganic porous silica sheets reported previously.51 No free porous polymer particles or naked graphene sheets were observed in either the TEM or SEM investigations. This suggests that, as expected, most of the monomers have been polymerized on the surface of graphene. The control sample (HCP) without the GO-DDAT template exhibited the amorphous nanoparticle structure reported previously (Fig. S2†).52 These results strongly suggest the crucial role of graphene as a substrate in the grafting of HCPs in a 2D manner.53
To analyze the molecular structure of the HCPs, Fourier transform infrared (FT-IR) spectroscopy was employed. Typical FT-IR spectra for GO, GO-DDAT, unadorned HCP, and GHCP-3 are shown in Fig. 2. The broad band at 3430 cm−1 is attributed to O–H stretching vibrations,54 which might originate from carboxylic acid and hydroxyl groups on the GO sheets or from adsorbed water. GO shows two adjacent peaks at 1720 and 1645 cm−1, which correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC stretching vibrations.55 The peak at 1065 cm−1 represents C–O stretching vibrations,56 which indicate the presence of the epoxide group in GO.57 In Fig. 2, unadorned HCP and GHCP-3 show a significant peak at 1610 cm−1 (skeletal vibrations from graphitic domains), attributable to aromatic CC.54 Additionally, the peaks at 2925 and 2855 cm−1 assigned to the methylene stretch indicate the existence of CH2 or CH groups.54 In particular, the peak at 1065 cm−1 for GO shifted to 1108 cm−1 for GO-DDAT and GHCP-3, which might be attributable to hydrogen bond interactions.58 Compared with GO and GO-DDAT, unadorned HCP and GHCP-3 have a distinctive peak at 820 cm−1, corresponding to C–Cl stretching,59 which suggests the existence of a few unreacted Ar–CH2–Cl groups (see the model reaction in Scheme 1).
O and CC stretching vibrations.55 The peak at 1065 cm−1 represents C–O stretching vibrations,56 which indicate the presence of the epoxide group in GO.57 In Fig. 2, unadorned HCP and GHCP-3 show a significant peak at 1610 cm−1 (skeletal vibrations from graphitic domains), attributable to aromatic CC.54 Additionally, the peaks at 2925 and 2855 cm−1 assigned to the methylene stretch indicate the existence of CH2 or CH groups.54 In particular, the peak at 1065 cm−1 for GO shifted to 1108 cm−1 for GO-DDAT and GHCP-3, which might be attributable to hydrogen bond interactions.58 Compared with GO and GO-DDAT, unadorned HCP and GHCP-3 have a distinctive peak at 820 cm−1, corresponding to C–Cl stretching,59 which suggests the existence of a few unreacted Ar–CH2–Cl groups (see the model reaction in Scheme 1).
 | ||
| Fig. 2 FT-IR spectra of (a) GO, (b) GO-DDAT, (c) unadorned HCP and (d) GHCP-3. | ||
In order to reveal the porous structures of the 2D HCPs, the N2 adsorption/desorption properties of GHCPs and unadorned HCP were studied (Fig. 3 and Table 1). All the samples showed a distinct step in the N2 adsorption isotherm at a low relative pressure (P/P0 < 0.1), which corresponds to gas sorption in micropores.60 According to the IUPAC classification,61 these samples give rise to type I nitrogen gas sorption isotherms with H3 hysteresis loops, indicating that the materials contain micro- and mesopores. The specific surface areas of 1224 and 1693 m2 g−1 (the former calculated from the Brunauer–Emmett–Teller (BET) theory, the latter from Langmuir's theory) for GHCP-1 are larger than those of HCP (600 and 832 m2 g−1), respectively. This result could be attributable to the graphene template and the resulting 2D morphology. This analysis was further confirmed by a pore size study. Pore size distributions calculated by the nonlocal density functional theory (NL-DFT) method for HCP and the GHCPs are illustrated in Fig. 3b. All of the samples exhibited pore diameters centering around 1.6 nm, implying that they contained mainly micropores (<2.0 nm). The GHCPs also showed several broad peaks between 2.3 and 6.0 nm, implying the existence of a few mesopores (2.0–50.0 nm). These results—when considered together—indicate that the GHCPs prepared in this study are new hierarchical porous organic materials with a 2D morphology.
 | ||
| Fig. 3 (a) Nitrogen adsorption and desorption isotherms of unadorned HCP and the GHCPs at 77 K. (b) The pore size distribution calculated by the NL-DFT method. (c, d) Illustrative images of a water droplet on the superhydrophobic surfaces. | ||
|
S
BETa (m2 g–1) |
S
Langa (m2 g–1) |
V
totb (cm3 g–1) |
H2 uptakec | CO2 uptakec | |||
|---|---|---|---|---|---|---|---|
| 77 K | 87 K | 273 K | 298 K | ||||
| a Surface area calculated from the N2 adsorption isotherm using the BET method and the Langmuir method, respectively. b Total pore volume at P/P0 = 0.99. c Gravimetric gas uptake (wt%) for H2 and CO2 at P/P0 = 0.99. | |||||||
| HCP | 600 | 832 | 0.69 | 0.85 | 0.63 | 8.80 | 5.86 |
| GHCP-1 | 1224 | 1693 | 1.45 | 1.27 | 0.82 | 9.74 | 6.19 |
| GHCP-2 | 971 | 1344 | 0.71 | 1.16 | 0.77 | 9.84 | 6.22 |
| GHCP-3 | 780 | 1083 | 0.78 | 1.09 | 0.67 | 9.06 | 5.82 |
Additionally, the hydrophobic property of the GHCPs was studied to better understand their unique hierarchical porous structures. First, ethanol dispersions of the GHCPs and HCP were sprayed onto glass surfaces. After drying in air at room temperature overnight, water droplets were placed on these surfaces (Fig. 3c and d). All the GHCPs showed a contact angle of 155 ± 4°, which is typically superhydrophobic, and marginally larger than that of HCP (151 ± 1°). The slightly improved contact angle may result from the 2D morphology, and could prove useful for the construction of better superhydrophobic materials.
Hydrogen is an ideal and attractive candidate for industrial applications, such as ammonia synthesis, automobile fuels, and metallurgy, because of its abundance, renewability, and environmentally “clean” aspect.62 Hydrogen isotherms for HCP and the GHCPs up to a maximum relative pressure (P/P0) of 0.99 at 77 K were examined, as shown in Fig. 4a. GHCP-1, GHCP-2, and GHCP-3 exhibited hydrogen capacities of 142, 130, and 122 cm3 g−1 (1.27, 1.16, and 1.09 wt%), respectively, which are all higher than that of HCP (95 cm3 g−1, 0.85 wt%). Obviously, the hydrogen capacities decrease along with the content of the graphene template. These results reveal that graphene contributes to the total hydrogen capacity, and underline the importance of a high graphene surface area for maximal hydrogen uptake capacity (SBET: GHCP-1 > GHCP-2 > GHCP-3). Based on a variant of the Clausius–Clapeyron equation, the hydrogen isosteric heat of adsorption can be calculated (Fig. 4b).63 At low coverage, GHCP-1, GHCP-2, and GHCP-3 exhibited the highest adsorption enthalpies at 6.3, 6.7, and 6.9 kJ mol−1, respectively (Fig. 4b), which are slightly larger than that of unadorned HCP (5.9 kJ mol−1). A more rapid decrease to 4.7 kJ mol−1 for unadorned HCP was shown in the high hydrogen uptake region. The high heat of adsorption may stem from the narrower pores, which allow stronger overall interactions of the guest gas molecules because of additional interactions with the opposite walls.64 The adsorption enthalpies of H2 for the GHCPs increased with the increase in graphene content, indicating that the interaction between H2 and the sample was increasingly dominating that between H2 molecules because of the higher content of the graphene template.
 | ||
| Fig. 4 Volumetric (a) H2 and (c) CO2 isotherms of unadorned HCP and the GHCPs at 77 K and 273 K, respectively. Isosteric heat of adsorption of (b) H2 and (d) CO2 for HCP and the GHCPs at different gas loadings. | ||
In order to study the different gas capacities of the GHCPs, their CO2 storage capacities were investigated. The CO2 adsorption isotherms of HCP and the GHCPs at 273 K are presented in Fig. 4c. GHCP-1, GHCP-2, GHCP-3, and unadorned HCP exhibited CO2 uptake of 9.74, 9.84, 9.06, and 8.80 wt%, respectively (see Table 1). It was found that adsorbed CO2 was not significantly affected in comparison with H2 when employing the graphene template, implying that the interactions between CO2 molecules and samples were unaffected by the graphene template. Unadorned HCP showed CO2 adsorptions of 44.8 cm3 g−1 (273 K) and 29.8 cm3 g−1 (298 K) at a relative pressure (P/P0) of 0.99. Under the same pressure conditions, the CO2 adsorptions of GHCP-1 were 49.6 and 31.5 cm3 g−1 at 273 and 298 K, respectively. The isosteric enthalpies (Qst) for CO2 were also calculated, and are shown in Fig. 4d. The CO2Qst value at lower coverage mainly reflects the interaction strength between CO2 and the sorbent.65 As shown in Fig. 4d, the CO2Qst values of HCP and the GHCPs were found to be almost the same in the range 0–8.0 cm3 g−1, indicating that (1) the interactions between CO2 and the porous samples are weak and (2) morphology and the graphene template have a limited impact on CO2 uptake.66
The thermal behaviors of unadorned HCP and the GHCPs were recorded by thermal gravimetric analysis (TGA) at a heating rate of 20 °C min−1 under a nitrogen atmosphere (Fig. 5a). Unadorned HCP is thermally unstable below 400 °C. Remarkably, the weight loss for the GHCPs started as high as 300 °C, and became rapid only in the range between 400 and 600 °C, which might be attributed to the alkane structure and oxygen-containing groups in the materials. The improved thermal stability of the GHCPs may be attributable to the high thermal stability of the graphene sheets.40 After 600 °C, the residual weight for all samples was almost constant, which suggests that the samples had been converted to stable carbon structures. At 800 °C, the residual weights of HCP, GHCP-1, GHCP-2, and GHCP-3 were 28.9, 33.1, 36.8 and 45.4 wt%, respectively, which also indicates that the graphene templates in the GHCPs enhance the thermal stability. The porous nature of GHCP-1-900 was further studied using nitrogen physisorption measurements. It was found that the isotherms for GHCP-1-900 were type I (Fig. 5b). The BET and Langmuir surface areas of GHCP-1-900 were calculated as 871 and 1212 m2 g−1, respectively. The pore size distribution of GHCP-1-900 is presented as an inset in Fig. 5b. The two peaks at 1.47 and 4.00 nm indicate that the hierarchical porous carbon nanosheets can be easily produced via the direct pyrolysis of 2D HCPs. These results also indicate that the high surface areas of 2D HCP-derived porous carbon nanosheets can be achieved by taking advantage of the 2D structure of graphene sheets. TEM images of GHCP-1-900 are shown in Fig. 5c and d: free-standing sheets with a morphology similar to that of graphene and several micrometers in size can be observed. It is clear that the 2D morphology with visible wrinkles can be well preserved after high temperature pyrolysis of GHCP-1. Interestingly, alternating light and dark regions can be easily seen in the enlarged TEM image, indicating the possible creation of a porous carbon during pyrolysis.
 | ||
| Fig. 5 (a) TGA curves of HCP and the GHCPs at a heating rate of 20 °C min−1 under nitrogen flow. (b) Nitrogen adsorption and desorption isotherms at 77 K (inset: the pore size distribution calculated by the NL-DFT method). (c, d) TEM images of GHCP-1-900. | ||
The high specific surface area and unique 2D nanosheet morphology as well as the porous structure of these newly prepared porous carbon nanosheets hold promise for their application in electrochemical capacitors. Therefore, the electrochemical capacitance of GHCP-1-900 was examined under alkaline conditions (6 M KOH) as a proof of concept. Typically, as shown in Fig. 6a, symmetric and horizontal cyclic voltammetry (CV) curves at a scan rate of 5–100 mV s−1 were observed for GHCP-1-900, indicating ideal capacitive behavior. The capacitive performance was further investigated in galvanostatic charge/discharge cycling experiments (Fig. 6b). On the basis of the discharge curve, the specific capacitances of GHCP-1-900 were calculated to be 144, 116, 112, 127, and 122 F g−1 at 0.5, 1, 2, 5, and 10 A g−1, respectively. For supercapacitor applications, the graphene layer in sandwich-type GHCP-1-900 can act as both a mini-current collector and a long-distance in-plane charge transporter during the charge and discharge processes, by taking advantage of the high electrical conductivity and the 2D electron transport properties of graphene. Additionally, inter-/intra-layer electrolyte transport also contributes to the superior capacitance, given that the active surface of nanosheets can be efficiently exposed to the electrolyte.31
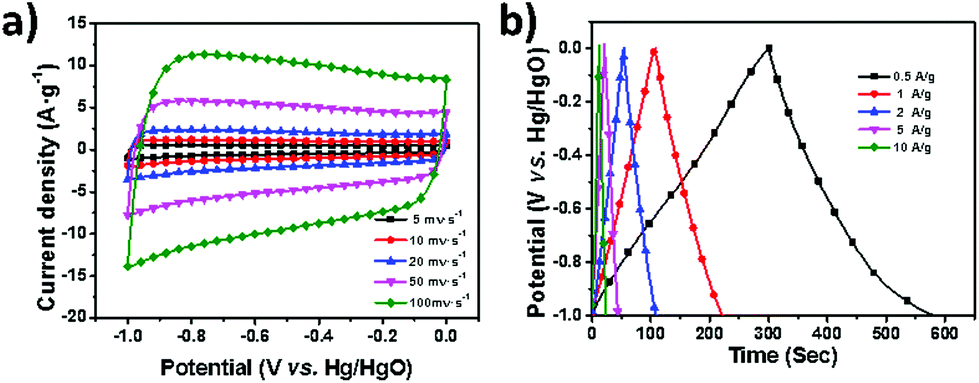 | ||
| Fig. 6 (a) CV curves of GHCP-1-900 at 5–100 mV s−1 in 6 M KOH aqueous solution. (b) Galvanostatic charge/discharge curves for GHCP-1-900 at a current density of 0.5–10 A g−1. | ||
Conclusions
In summary, GHCPs based on vinylbenzyl chloride and divinylbenzene were successfully prepared by a RAFT emulsion polymerization approach and a FeCl3-promoted Friedel–Crafts reaction. A highly soluble trithiocarbonate functionalized GO, which is the key 2D template in this approach, was used as a 2D micro-CTA for RAFT polymerization. The resulting GHCPs exhibited superhydrophobic behavior (contact angle >150°) and a high specific surface area of up to 1224 m2 g−1. They also exhibited an enhanced H2 capacity (1.27 wt%) at 77 K and CO2 capacity (9.9 wt%) at 273 K at a relative pressure of 0.99 in comparison with unadorned HCP without using the graphene template. The high specific surface areas and enhanced H2 and CO2 capacities might come from the contribution of the 2D graphene template and the 2D morphology. The GHCPs were also used to prepare porous carbon nanosheets without a template-removing operation. The as-prepared porous carbon nanosheets were successfully utilized in electrochemical capacitors, which makes this method a good approach for the preparation of 2D HCPs and 2D porous carbons with wide applications.Acknowledgements
This work was financially supported by the National Basic Research Program of China (973 Program: 2013CBA01602 and 2012CB933404), the Natural Science Foundation of China (51403126), and Shanghai Jiao Tong University (211 and 985 Project). We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for providing some measurements.Notes and references
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959 CrossRef CAS PubMed.
- M. S. Silverstein, N. R. Cameron and M. A. Hillmyer, Porous Polymers, John Wiley & Sons, 2011 Search PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530 CrossRef CAS PubMed.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328 CrossRef CAS PubMed.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453 CrossRef CAS PubMed.
- X. Feng, X. S. Ding and D. L. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC.
- L. Chen, Y. Honsho, S. Seki and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 6742 CrossRef CAS PubMed.
- X. M. Liu, Y. H. Xu and D. L. Jiang, J. Am. Chem. Soc., 2012, 134, 8738 CrossRef CAS PubMed.
- H. A. Patel, S. Hyun Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909 CrossRef CAS PubMed.
- M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 7216 CrossRef CAS PubMed.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753 CrossRef CAS PubMed.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471 CrossRef CAS PubMed.
- M. Seo, S. Kim, J. Oh, S.-J. Kim and M. A. Hillmyer, J. Am. Chem. Soc., 2014, 137, 600 CrossRef PubMed.
- F. S. Macintyre, D. C. Sherrington and L. Tetley, Macromolecules, 2006, 39, 5381 CrossRef CAS.
- L. Pan, Q. Chen, J.-H. Zhu, J. Yu, Y. He and B.-H. Han, Polym. Chem., 2015, 6, 2478 RSC.
- J. Germain, J. M. J. Frechet and F. Svec, Chem. Commun., 2009, 1526 RSC.
- J. Germain, J. Hradil, J. M. J. Frechet and F. Svec, Chem. Mater., 2006, 18, 4430 CrossRef CAS.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2011, 23, 1818 CrossRef CAS.
- X.-J. Chen, D. Ngoc Phuoc, J. Zhao, Y.-T. Wang, S.-P. Li and F. Svec, J. Sep. Sci., 2012, 35, 1502 CrossRef CAS PubMed.
- Y. Lv, Z. Lin and F. Svec, Anal. Chem., 2012, 84, 8457 CrossRef CAS PubMed.
- C. D. Wood, B. Tan, A. Trewin, F. Su, M. J. Rosseinsky, D. Bradshaw, Y. Sun, L. Zhou and A. I. Cooper, Adv. Mater., 2008, 20, 1916 CrossRef CAS PubMed.
- Y. Ouyang, H. Shi, R. Fu and D. Wu, Sci. Rep., 2013, 3, 1430 Search PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- X. Zhuang, F. Zhang, D. Wu, N. Forler, H. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 9668 CrossRef CAS PubMed.
- X. Zhuang, Y. Chen, L. Wang, K.-G. Neoh, E.-T. Kang and C. Wang, Polym. Chem., 2014, 5, 2010 RSC.
- X. Zhuang, Y. Chen, G. Liu, P. P. Li, C. X. Zhu, E. T. Kang, K. G. Neoh, B. Zhang, J. H. Zhu and Y. X. Li, Adv. Mater., 2010, 22, 1731 CrossRef CAS PubMed.
- X. Zhuang, F. Zhang, D. Wu and X. Feng, Adv. Mater., 2014, 26, 3081 CrossRef CAS PubMed.
- C. A. Cao, X. Zhuang, Y. Su, Y. Zhang, F. Zhang, D. Wu and X. Feng, Polym. Chem., 2014, 5, 2057 RSC.
- Y. Zhang, X. Zhuang, Y. Su, F. Zhang and X. Feng, J. Mater. Chem. A, 2014, 2, 7742 CAS.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720 CrossRef CAS PubMed.
- H. J. Salavagione, G. Martinez and G. Ellis, Macromol. Rapid Commun., 2011, 32, 1771 CrossRef CAS PubMed.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098 RSC.
- S. H. Lee, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, S. O. Kim, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281 CrossRef CAS PubMed.
- H. M. Etmimi, M. P. Tonge and R. D. Sanderson, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1621 CrossRef CAS PubMed.
- F. Beckert, C. Friedrich, R. Thomann and R. Mülhaupt, Macromolecules, 2012, 45, 7083 CrossRef CAS.
- B. Zhang, Y. Chen, L. Xu, L. Zeng, Y. He, E. T. Kang and J. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2043 CrossRef CAS PubMed.
- Y. Li, X. Li, C. Dong, Y. Li, P. Jin and J. Qi, Biosens. Bioelectron., 2009, 25, 306 CrossRef CAS PubMed.
- Y. Li, X. Li, C. Dong, J. Qi and X. Han, Carbon, 2010, 48, 3427 CrossRef CAS PubMed.
- Y. Yang, X. Song, L. Yuan, M. Li, J. Liu, R. Ji and H. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 329 CrossRef CAS PubMed.
- Y. Yang, J. Wang, J. Zhang, J. Liu, X. Yang and H. Zhao, Langmuir, 2009, 25, 11808 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754 CrossRef CAS.
- B. Zhang, J. Wang, Y. Chen, D. Fruchtl, B. Yu, X. D. Zhuang, N. He and W. J. Blau, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3161 CrossRef CAS PubMed.
- J. H. Ahn, J. E. Jang, C. G. Oh, S. K. Ihm, J. Cortez and D. C. Sherrington, Macromolecules, 2006, 39, 627 CrossRef CAS.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk and E. Stöckel, Chem. Mater., 2007, 19, 2034 CrossRef CAS.
- S. Yang, X. Feng, L. Wang, K. Tang, J. Maier and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 4795 CrossRef CAS PubMed.
- B. Li, F. Su, H.-K. Luo, L. Liang and B. Tan, Microporous Mesoporous Mater., 2011, 138, 207 CrossRef CAS PubMed.
- X. Zhuang, Y. Mai, D. Wu, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 403 CrossRef CAS PubMed.
- P.-G. Ren, D.-X. Yan, X. Ji, T. Chen and Z.-M. Li, Nanotechnology, 2011, 22, 055705 CrossRef PubMed.
- N. N. Chai, J. Zeng, K. G. Zhou, Y. L. Xie, H. X. Wang, H. L. Zhang, C. Xu, J. X. Zhu and Q. Y. Yan, Chem. – Eur. J., 2013, 19, 5948 CrossRef CAS PubMed.
- M. Xue, F. Li, J. Zhu, H. Song, M. Zhang and T. Cao, Adv. Funct. Mater., 2012, 22, 1284 CrossRef CAS PubMed.
- K. K. Sadasivuni, A. Saiter, N. Gautier, S. Thomas and Y. Grohens, Colloid Polym. Sci., 2013, 291, 1729 CAS.
- K. Haubner, J. Murawski, P. Olk, L. M. Eng, C. Ziegler, B. Adolphi and E. Jaehne, ChemPhysChem, 2010, 11, 2131 CrossRef CAS PubMed.
- B.-S. Ko, J.-Y. Sohn and J. Shin, Polymer, 2012, 53, 4652 CrossRef CAS PubMed.
- J. Rouquerol, F. Rouquerol and K. S. Sing, Absorption by Powders and Porous Solids, Academic Press, 1999 Search PubMed.
- R. Pierotti and J. Rouquerol, Pure Appl. Chem., 1985, 57, 603 Search PubMed.
- Y. Luo, S. Zhang, Y. Ma, W. Wang and B. Tan, Polym. Chem., 2013, 4, 1126 RSC.
- Z. Wang, S. W. Yuan, A. Mason, B. Reprogle, D. J. Liu and L. P. Yu, Macromolecules, 2012, 45, 7413 CrossRef CAS.
- W. G. Lu, D. Q. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Brase, J. Guenther, J. Blumel, R. Krishna, Z. Li and H. C. Zhou, Chem. Mater., 2010, 22, 5964 CrossRef CAS.
- Y. Zhao, X. Liu, K. X. Yao, L. Zhao and Y. Han, Chem. Mater., 2012, 24, 4725 CrossRef CAS.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01194a |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |