
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThienopentathiepine: a sulfur containing fused heterocycle for conjugated systems and their electrochemical polymerization†
Sashi
Debnath
,
Anjan
Bedi
and
Sanjio S.
Zade
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, 741246, India. E-mail: sanjiozade@iiserkol.ac.in
First published on 25th September 2015
Abstract
A series of conjugated building blocks based on the thieno[3,4-f][1,2,3,4,5]pentathiepine (C4S6) core has been synthesized by a new synthetic approach. The structural and optoelectronic properties of C4S6-derivatives are tuned by a judicious choice of end-capping. Chalcogenophene-capped derivatives (2d, 2e, 2f) were successfully electrochemically polymerized. The optical band gaps (Eoptg) of P1, P2 and P3 were found to be in the range of 1.7–1.8 eV. A spectroelectrochemical study of the polymers showed reversibility in the formation of singly (polaron) and doubly charged (bipolaron) species.
Introduction
Thiophene-based conjugated systems represent one of the most widely investigated groups of π-conjugated materials1,2 because of the potential industrial applications of these materials as active materials in organic electronic devices such as organic light-emitting diodes (OLEDs),3 field-effect transistors (OFETs),4,5 and photovoltaic cells (OPVs).6,7 Structural modifications of the conjugated backbone are keystones to control the energy levels of the frontier orbitals and the optoelectronic properties of the resulting materials.8 Developing a synthetic approach for new π-conjugated building blocks with the desired optoelectronic properties is imperative as conjugated materials with suitable consequences are limited.9 Among the numerous chalcogenophene-based polymers, polythiophene (PT) derivatives have received growing attention because of their fundamental advantages such as (i) stronger donor characteristics, (ii) rigidity/crystallinity, (iii) good chemical stability, and (iv) high conductivity in the doped state.Several strategies have been adopted to introduce new building blocks for conjugated materials with the desired properties. One of the important strategies includes synthesis of chalcogen containing heterocycles having multiple heteroatoms in the fused ring systems. An amplified spatial electronic arrangement because of the presence of multiple sulfur/selenium atoms in the molecular building blocks delocalizes the charge efficiently. In this regard, we have reported diselenolodiselenole (C4Se4) as a new building block for organic electronics, containing four selenium atoms in the central core.10
Chenard et al. reported the first synthesis of thieno[3,4-f][1,2,3,4,5]pentathiepine (C4H2S6) in 2% yield where 3,4-dibromothiophene was used as a precursor.11 In spite of the accessibility of the thienopentathiepine (C4S6) derivatives, synthetic reports are scant.12–14 Although its nearest analogue benzopentathiepine has been explored to some extent its electrochemical properties were not investigated.15–17 Recently, Swager and coworkers reported multiple sulfur containing fused dithiolodithiole (C4S4) derivatives focusing on their interesting structural and optoelectronic properties, where the formation of thienopentathiepines was observed as side products.18 Here we report a new synthetic approach for the synthesis of thieno[3,4-f][1,2,3,4,5]pentathiepine derivatives as novel conjugated building blocks containing a fused pentathiepine ring in bicyclic heterocycles. Optoelectronic and structural properties were tuned by a judicial choice of end capping of the central C4S6 core. Chalcogenophene capped derivatives (2d, 2e, 2f) were electrochemically polymerized and the resulting polymers were successfully studied by spectroelectrochemistry.
Results and discussion
Synthesis of thieno[3,4-f][1,2,3,4,5]pentathiepine (C4S6) derivatives (2a–2f) involves heating of 1,4-disubstituted 1,3-butadiyne (1a–1f) with elemental sulfur near about the melting point of sulfur (125 °C) for 0.5 hour in a closed-flask (Scheme 1). Without using any initiator/catalyst, the yields were obtained in the range of 12–31%. This new synthetic approach is quick and simple compared to that of Chenard et al. and also benign in terms of precursors. The product yields did not improve by increasing the reaction time (up to 12 hours). In the presence of a solvent (1,2-dichloroethane) very low yields (2–3%) were obtained. The reaction at higher temperature (>150 °C) afforded C4S4 derivatives as previously reported by Swager et al.18 The precursor diynes 1a–1f were prepared either by a previously reported procedure10,19,20 or by new synthetic strategies (see the ESI†). The conversion of diynes to C4S6 derivatives might proceed through the radical mechanism (Fig. S1†).21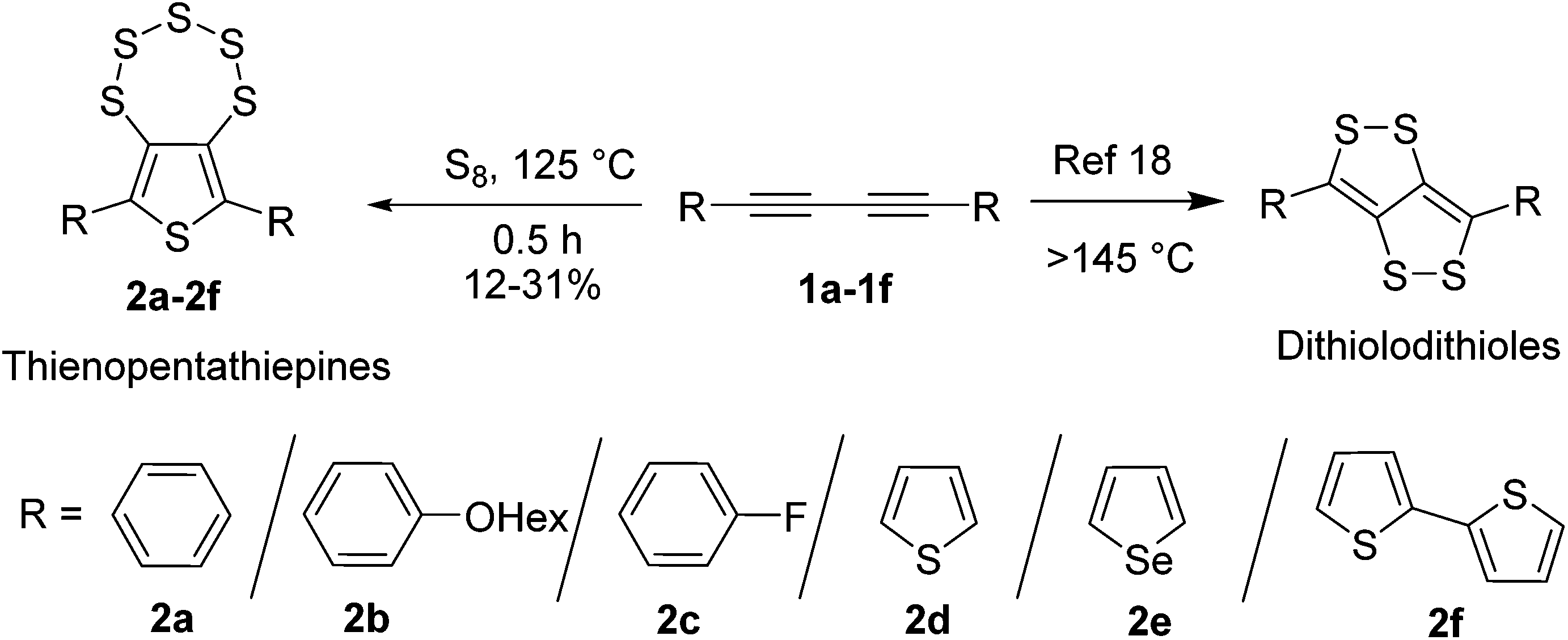 | ||
| Scheme 1 Synthesis of C4S6 derivatives 2a–2f. | ||
Single crystal X-ray structures of 2a and 2c–2e
Crystals of 2a, and 2c–2e suitable for single crystal X-ray crystallography were obtained from slow evaporation of their dichloromethane solutions. In the molecular structure of 2a, terminal phenyl rings are twisted from the central thiophene core by dihedral angles of ∼37° and ∼45° (Fig. S2a†), whereas the packing diagram shows formation of 1D chains via intermolecular interactions (S3–C8 = 3.40 Å and S4–S6 = 3.47 Å) (Fig. S2c†). Similarly in the molecular structure of 2c (Fig. S2b†), both p-fluoro substituted phenyl rings are twisted by a dihedral angle of ∼43° from the central thiophene of the C4S6-core. The structure of 2c exhibits resolute S⋯S intermolecular interaction (S4–S3 = 3.39 Å and S4–S5 = 3.39 Å). In the structures 2a and 2c, the S⋯S interaction leads to the arrangement of a virtual –(S–S⋯S–S)n– polymeric chain along the b- and a-axes, respectively (Fig. S2c and S2d†).In the molecular structure of 2d and 2e, the terminal thiophene and selenophene rings are anti to the central thiophenes (Fig. 1).22 In the thiophene-capped C4S6-derivative 2d, the torsional angles of the terminal thiophenes with the central thiophene of the C4S6-core are ∼9° and ∼23°, respectively; whereas in the selenophene-capped C4S6-derivative 2e, the corresponding torsional angles are ∼3° and ∼7°. It is congruent with the earlier reports23,24 which showed that selenophene based conjugated systems are more planar and rigid because of the low aromaticity of selenophene and the higher polarizability of Se. In the crystal packing of 2d, three molecules are connected by four S⋯S interactions (S1–S8 = 3.56 Å (two), S6–S6 = 3.48 Å, and S3–S7 = 3.47 Å) (Fig. 1b) to form a 2D zip-lock structure.25,26 In compound 2e, Se⋯C (Se2–C10 = 3.55 Å (forming π-stacking dimer) and Se2–C11 = 3.59 Å) and S⋯S (S5–S6 = 3.43 Å) interactions form a typically 2D closed packed arrangement (Fig. 1d). In 2e, the S⋯S interaction constructs a –(S–S⋯S–S)n– polymeric chain.
 | ||
| Fig. 1 (a) and (c) ORTEP diagram of 2d and 2e, (b) and (d) packing of 2d and 2e. The ellipsoids are drawn at the 50% probability level in (a) and (c). | ||
Optical and electrochemical properties of 2a–2f
Compounds 2a–2f exhibited two distinct absorption bands in their absorption spectra with absorption maxima (λmax) ranging from 258 to 277 nm (high energy bands) and 316 to 429 nm (relatively broad and low energy bands) (Fig. 2a). TD-DFT calculations on 2d and 2e indicate that the lower energy absorption bands include (i) the π → π* (HOMO → LUMO) transition of the terthiophene/selenophene–thiophene–selenophene backbone and (ii) the transition from the π-orbital of terthiophene/selenophene–thiophene–selenophene to the π* orbital of the central thiophene ring coupled with the antibonding orbitals of S–S bonds of the pentathiepine heterocycle (Tables S2 and S3†). The higher energy band mainly originates from (i) the transition from the π orbital (HOMO) to the antibonding orbitals of the pentathiepine heterocycle and (ii) the non-bonding orbital on the S atoms of pentathiepine to the π* orbital (LUMO). Compounds with electron donating groups exhibited a bathochromic shift in the absorption spectra. The absorption spectrum of selenophene-capped 2e showed a red shift (of 27 nm) compared to that of thiophene-capped 2d because of the better conjugation in 2e. The bithiophene-capped C4S6-derivative 2f showed lower-energy absorption bands due to the extended conjugation. | ||
| Fig. 2 (a) UV-vis spectra in DCM and (b) electrochemical properties of compounds 2a–2f in 0.1 M TBAPF6 in dry DCM as the solvent using a Pt-disk working electrode, a Pt-wire counter electrode and a Ag/AgCl reference electrode. | ||
Cyclic voltammograms (CV) of 2a–2f showed irreversible oxidation peaks ranging from 0.80 V to 1.60 V vs. Ag/AgCl (Fig. 2b).27 Although selenophene is electron rich compared to thiophene, 2e exhibited higher oxidation potential than 2d. The planar conjugated backbone of 2e could result in superior delocalization of the electrons throughout the molecular plane, which might decrease the electron density on the C4S6 unit. The UV-vis and CV data were used to measure the band gap (Eg) and HOMO/LUMO energy levels (Table 1).
| Compounds | % Yield | λ max(nm) | E oxonset(V) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (eV) a (eV) |
E
HOMOb(eV) |
E
LUMOc(eV) |
|---|---|---|---|---|---|---|
| a E optg = 1240/λonset. b E HOMO = −4.44 + Eoxonset. c E LUMO = EHOMO + Eoptg. | ||||||
| 2a | 27 | 258, 316 | 1.53 | 3.40 | −5.97 | −2.57 |
| 2b | 31 | 266, 330 | 1.14 | 3.22 | −5.58 | −2.36 |
| 2c | 22 | 258 | 1.49 | 3.41 | −5.93 | −2.52 |
| 2d | 17 | 270, 366 | 0.90 | 2.87 | −5.34 | −2.47 |
| 2e | 12 | 277, 393 | 1.01 | 2.73 | −5.44 | −2.71 |
| 2f | 21 | 261, 429 | 0.76 | 2.45 | −5.20 | −2.75 |
Electrochemical properties of the polymers
Under repeated CV cycles, compounds 2d, 2e and 2f polymerized smoothly via progressive growth of a visible polymer film a platinum-disk working electrode (Scheme 2, Fig. 3, and S3†). The polymer films were investigated for their scan rate dependence at 50–250 mV s−1 for P1 and 50–300 mV s−1 for P2 and P3. All the polymers showed linear scaling of the anodic currents with the increasing value of scan rate (Fig. 3c, d and S3†), as expected from an electrode-supported electroactive film. Cyclic voltammograms of P1, P2, and P3 polymer films on a Pt working electrode showed quasi-reversible p-doping/dedoping processes over a positive potential range in the DCM/0.1 M TBAPF6 solvent/electrolyte system at 50 mV s−1 scan rate (Fig. 4b). The onsets of the oxidation potentials (Eoxonset) of P1, P2 and P3 were found to be 0.67, 0.49 and 0.43 V, respectively, corresponding to the HOMO levels at −5.11, −4.93 and−4.87 eV, respectively. The LUMO levels, estimated from the HOMO and the onsets of the absorption peaks, were found to be −3.35, −3.27, and−3.07 eV for P1, P2, and P3, respectively (Fig. 4, Table 2). P2 exhibited a lower oxidation potential than P1, which can be attributed to the presence of more polarizable Se.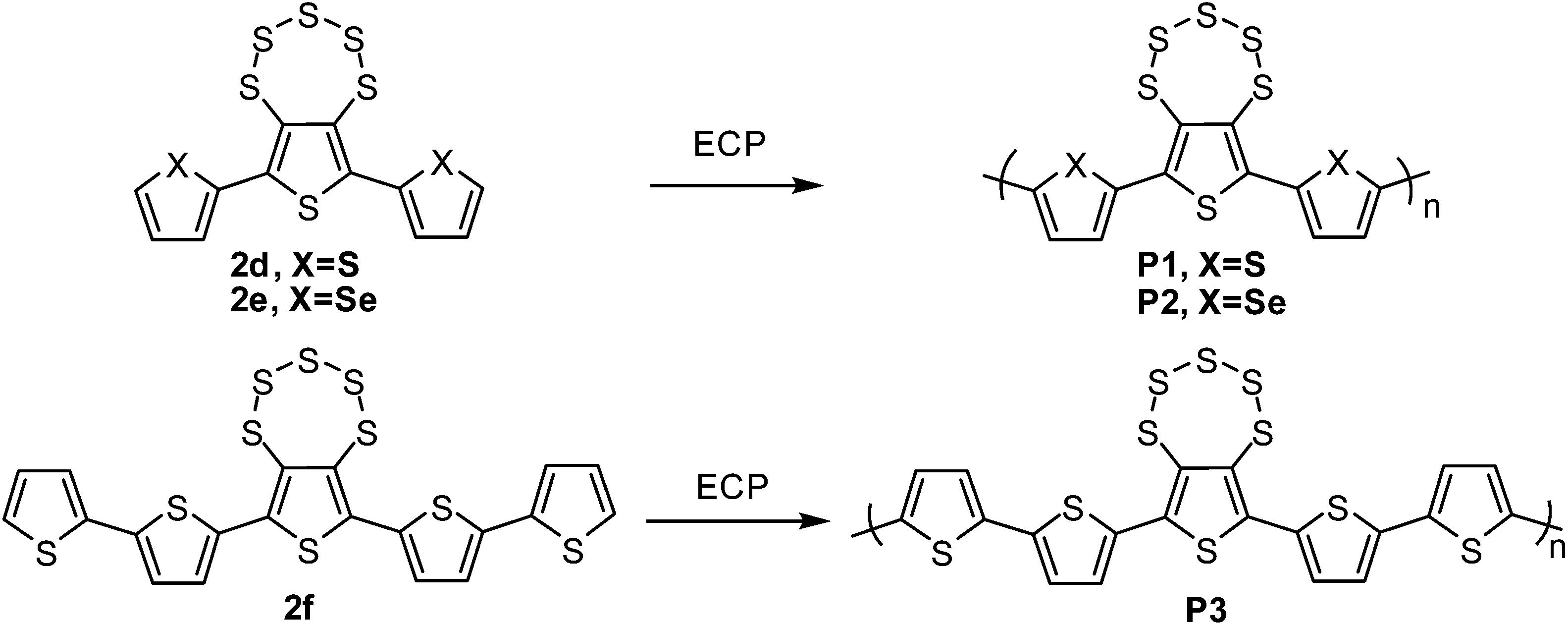 | ||
| Scheme 2 Synthesis of polymers P1, P2 and P3. | ||
 | ||
| Fig. 3 (a) and (b) Multisweep electropolymerization of 2d and 2e on a Pt electrode in DCM and 0.1 M TBAPF6 at 50 mV s−1vs. Ag/AgCl wire. (c) and (d) CV of P1 and P2 in monomer free DCM and 0.1 M TBAPF6 as a function of scan rate. | ||
 | ||
| Fig. 4 (a) Absorption spectra of P1–P3 in thin film. (b) CV of P1, P2 and P3, using the DCM/TBAPF6 solvent/electrolyte system. | ||
| Polymer | λ max(nm) | E optg (eV) | E oxonset(V) | E HOMO (eV) | E LUMO (eV) |
|---|---|---|---|---|---|
| P1 | 439 | 1.76 | 0.67 | −5.11 | −3.35 |
| P2 | 550 | 1.69 | 0.49 | −4.93 | −3.27 |
| P3 | 518 | 1.80 | 0.43 | −4.87 | −3.07 |
Spectroelectrochemistry
Polymers were chronoamperometrically deposited on an ITO-coated glass electrode from 1 × 10−2 M monomer solutions in DCM and the spectroelectrochemical properties were investigated in situ in the monomer-free electrolyte solution under a variety of voltage pulses. The absorption spectra in the UV–vis–NIR region of P1, P2 and P3 films (Fig. 5, S4†) revealed good electrochromic nature at distinct applied potentials in the DCM/TBAPF6 solvent/electrolyte couple. The onset values of the lowest energy bands correspond to the optical band gaps (Eoptg) of 1.76, 1.69 and 1.80 eV for P1, P2 and P3, respectively (Table 2). P2 exhibited a smaller band gap than P1 because of the presence of selenophene in P2 instead of thiophene in P1. Notably, these polymers having the fused pentathiepine ring as a substituent on thiophene possess a smaller band gap compared to polythiophene (2.0 eV) and polyselenophene (1.9 eV).2,28,29 The band gaps of all three polymers remained in between the band gaps of poly(3-hexylthiophene) (1.9 eV) and poly(3-hexylselenophene) (1.6 eV).30,31 The lowering in the band gap of these polymers can be attributed to the delocalization of the π electron density over the pentathiepine rings and interchain interactions due to the presence of multiple sulphur atoms. | ||
| Fig. 5 Spectroelectrochemistry of (a) P1 and (b) P2 thin films prepared on ITO-coated glass as a function of applied potential between −0.3 and +1.3 V for P1, and −0.2 and +1.3 V for P2 in DCM. | ||
In the spectroelectrochemical experiments, switching of the polymers from neutral to oxidized states was associated with the color change from deep brown to transparent-peach for P1, purple to transparent-gray for P2, and violet to transparent-gray for P3. With the increasing applied potential, the intensity of the absorption band in the visible region (arises due to the π–π* transition) was gradually decreased and two new bands gradually appeared in the higher wavelength region. New peaks at 820, 905, and 750 nm for P1, P2 and P3, respectively, in their absorption spectra (Fig. 5 and S4†) indicate the formation of singly charged species (polaron), while the lower energy bands at 1180, 1480, and 1160 nm correspond to the formation of doubly charged species (bipolaron), respectively.32,33 The deviation in spectroelectrochemical experiments precisely comes at 850 nm (Fig. 5a and b) which is due to the lamp change (deuterium to tungsten lamp) during the NIR region to VIS region transformation.
Conclusion
In conclusion, a series of conjugated compounds, thieno[3,4-f][1,2,3,4,5]pentathiepine derivatives, was synthesized by a simple synthetic method of heating diaryl diynes with elemental sulfur. The structural and optoelectronic properties of C4S6-derivatives were tuned successfully by the choice of end capping. Multiple close S⋯S contacts due to the presence of a fused pentathiepine ring in C4S6-derivatives and their polymers may be beneficial for the extended conjugation and charge transport. The C4S6-derivatives with thiophene- and selenophene-capping were successfully electrochemically polymerized. The low band gap polymers P1–P3 exhibited well-defined spectroelectrochemistry and sufficient stability in the oxidized state, which can be exploited for organic electronic devices.Experimental section
Most of the reagents and solvents of reagent grade were obtained from commercial sources and used without any further purification. NMR spectra were recorded on a Jeol-ECS 400 MHz spectrometer using CDCl3/C6D6 as the solvent and chemical shifts are reported in parts per million (δ scale) relative to tetramethylsilane (TMS) as the internal standard. Columns were equipped with silica gel (100–200 mesh). A nonaqueous Ag/AgCl electrode was made by dipping silver wire into a FeCl3/HCl solution. Electrochemical analysis was carried out with a Princeton Applied Research 263A potentiostat using a platinum (Pt) disk electrode (dia. 1.6 mm) as the working electrode, a platinum wire as the counter electrode, and an AgCl coated Ag wire as the reference electrode. Pt disk electrodes were gleamed with alumina, water, and acetone and were dried with nitrogen to remove any incipient oxygen. The electrolyte used was 0.1 M TBAPF6 in DCM. Films were electrodeposited in 0.1 M TBAPF6 in DCM by cyclic voltammetry (CV) between various applied potentials at 50 mV s−1 for 20 cycles. 2d, 2e and 2f were electrochemically polymerized on indium tin oxide (ITO) coated glass as a working electrode. Before examining the optical properties of polymer films, the films were rinsed with DCM. UV-vis-NIR spectra were recorded on a HITACHI U-4100 UV-vis-NIR spectrophotometer.Fine crystals of 2a and 2c–2e were collected on a SuperNova, Dual, Cu/Mo at zero, Eos diffractometer. Using Olex2,34 the structure was solved with the Superflip35 structure solution programme applying Charge Flipping and refined with the ShelXL36 refinement package applying least squares minimization.
Synthesis of 1e
A tetrabutylammonium fluoride solution (3 mL, 1 M in THF) was added dropwise into a stirred solution of trimethyl(selenophen-2-ylethynyl)silane (500 mg, 2.19 mmol) in 10 mL THF and stirred for 10 min at rt. This solution was added into a three necked round-bottom flask containing Cu(OAc)2 (553 mg, 3.06 mmol) and 10 mL of pyridine/methanol mixture (1:1 v/v) and refluxed for 2 h. The reaction mixture was allowed to cool at rt and added to crushed-ice and 3.6 mL of 9 M H2SO4. The resulting solution was extracted with diethyl ether (3 × 25 mL). The ether layers were mixed, dried over anhydrous Na2SO4 and concentrated to produce a deep brown crude product. Column chromatography of the crude on silica gel using hexane as the eluent gave 1e as a pale yellow solid (170 mg, 50%). 1H NMR (400 MHz, δ, ppm, CDCl3): 8.07 (d, 2H, J = 4.6 Hz), 7.54 (d, 2H, J = 3.44 Hz), 7.24–7.21 (m, 2H,). 13C NMR (100 MHz, CDCl3): δ 136.6, 134.9, 129.6, 126.0, 79.6, 79.5.
Synthesis of 1f
Compound 1f was synthesized by using a similar synthetic procedure to 1e using (2,2′-bithiophen-5-ylethynyl)trimethylsilane (563 mg, 2.15 mmol), which afforded 1f as a pale brown solid (300 mg, 73%). 1H NMR (400 MHz, δ, ppm, C6D6): 6.83 (d, 2H, J = 3.84 Hz), 6.80 (d, 2H, J = 3.84 Hz), 6.65 (d, 2H, J = 5.36 Hz), 6.57–6.52 (m, 4H). 13C NMR (100 MHz, C6D6): δ 140.9, 136.5, 135.8, 128.1, 125.5, 125.0, 123.8, 120.6, 79.8, 78.1.General procedure for the synthesis of 2a–2f
Butadienes (1 mmol) and sulfur powder (8 mmol) were charged into a Schlenk flask and it was stoppered. The reaction mixture was heated at 125 °C for 0.5 h. The reaction was cooled and the resulting residue was purified by column chromatography on 100–200 mesh silica gel (2% CH2Cl2 in hexane) to obtain the products.2a: Yellowish brown solid. Yield: 106 mg, 27%. M.p.: 154 °C. UV-vis in DCM: λmax (nm) (104 × ε (M−1 cm−1)) = 258 (4.5), 316 (3.4). 1H NMR (400 MHz, CDCl3): δ 7.55–7.51 (m, 2H), 7.49–7.43 (m, 3H). 13C NMR (100 MHz, CDCl3): δ 148.3, 137.9, 132.5, 130.0, 129.1, 128.5. HRMS (ESI+): calculated for C16H10S6, 393.9107; found 393.9029.
2b: Yellow solid. Yield: 184 mg, 31%. M.p.: 122 °C. UV-vis in DCM: λmax (nm) (104 × ε (M−1 cm−1)) = 266 (5.9), 330 (4.7). 1H NMR (400 MHz, CDCl3): δ 7.46 (d, 2H, J = 8.4 Hz), 6.97 (d, 2H, J = 8.4 Hz), 4.00 (t, 2H), 1.80 (m, 2H), 1.51–1.43 (m, 2H), 1.38–1.32 (m, 4H), 0.91 (t, 3H). 13C NMR (100 MHz, CDCl3): δ 159.9, 147.9, 136.8, 131.2, 124.7, 114.4, 68.1, 31.5, 29.1, 25.7, 22.5, 14.0. HRMS (ESI+): calculated for C28H34O2S6, 594.0883; found 594.0229.
2c: Yield: 94 mg, 22%. M.p.: 143 °C. UV-vis in DCM: λmax (nm) (10−4 × ε (M−1 cm−1)) = 258 (7.8). 1H NMR (400 MHz, CDCl3): δ 7.53–7.48 (m, 2H), 7.19–7.13 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 164.2 (d, J = 199.3 Hz), 138.1, 134.5 (d, J = 6.5 Hz), 131.9 (d, J = 6.5 Hz), 128.5, 115.8 (d, J = 18.05 Hz). HRMS (ESI+): calculated for C16H8F2S6, 429.8918; found 429.8120.
2d: Yellowish brown solid. Yield: 68 mg, 17%. M.p.: 167 °C. UV-vis in DCM: λmax (nm) (10−4 × ε (M−1 cm−1)) = 270 (9.5), 366 (4.4). 1H NMR (400 MHz, CDCl3): δ 7.45 (d, 2H, J = 3.80 Hz), 7.40 (d, 2H, J = 3.84 Hz), 7.18–7.07 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 140.5, 136.7, 133.8, 128.5, 128.4, 127.3. HRMS (ESI+): calculated for C12H6S8, 405.8235; found 404.8191.
2e: Yellowish brown solid. Yield: 60 mg, 12%. M.p.: 179 °C. UV-vis in DCM: λmax (nm) (10−4 × ε (M−1 cm−1)) = 277 (10.5), 393 (3.2). 1H NMR (400 MHz, CDCl3): δ 8.16 (d, 2H, J = 5.32 Hz), 7.61 (d, 2H, J = 3.8 Hz), 7.34–7.30 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 142.4, 138.0, 136.3, 134.9, 130.2, 129.4. HRMS (ESI+): calculated for C12H6S6Se2, 501.7124; found 501.7051.
2f: Deep orange solid. Yield: 118 mg, 21%. M.p.: 194 °C. UV-vis in DCM: λmax (nm) (10−4 × ε (M−1 cm−1)) = 261 (4.0), 429 (4.5). 1H NMR (400 MHz, C6D6): δ 6.96–6.92 (m, 4H), 6.81 (d, 2H, J = 3.8 Hz), 6.70 (d, 2H, J = 3.84 Hz), 6.61 (m, 2H). 13C NMR (100 MHz, C6D6): δ 150.4, 148.4, 147.2, 143.4, 139.3, 139.2, 127.3, 124.8, 124.3, 120.0. HRMS (ESI+): calculated for C20H10S10, 569.7990; found 569.8073.
Theoretical methods
Single point TD-DFT calculations were performed on the structure obtained from the single crystal X-ray diffraction of 2d and 2e by using the Gaussian 09 programme37 at the B3LYP/6-31G(d) level.Acknowledgements
We thank DST, India for funding. SD thanks UGC and AB thanks CSIR for a research fellowship.References
- W. Ni, X. Wan, M. Li, Y. Wang and Y. Chen, Chem. Commun., 2015, 51, 4936 RSC.
- Handbook of Thiophene-Based Materials, ed. I. F. Perepichka and D. F. Perepichka, Wiley-VCH, Chichester, UK, 2009 Search PubMed.
- P. Furuta, J. Brooks, M. E. Thompson and J. M. J. Fréchet, J. Am. Chem. Soc., 2003, 125, 13165 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- Y. S. Yang, T. Yasuda, H. Kakizoe, H. Mieno, H. Kino, Y. Tateyama and C. Adachi, Chem. Commun., 2013, 49, 6483 RSC.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef CAS PubMed.
- K. Feng, X. Xu, Z. Li, Y. Li, K. Li, T. Yu and Q. Peng, Chem. Commun., 2015, 51, 6290 RSC.
- Y. Liang, D. Feng, Y. Wu, S. T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792 CrossRef CAS PubMed.
- X. He, J. Borau-Garcia, A. Y. Y. Woo, S. Trudel and T. Baumgartner, J. Am. Chem. Soc., 2013, 135, 1137 CrossRef CAS PubMed.
- A. Bedi, S. Debnath and S. S. Zade, Chem. Commun., 2014, 50, 13454 RSC.
- B. L. Chenard, R. L. Harlow, A. L. Johnson and S. A. Vladuchick, J. Am. Chem. Soc., 1985, 107, 3871 CrossRef CAS.
- L. S. Konstantinova, O. A. Rakitin, C. W. Rees, L. I. Souvorova, D. G. Golovanov and K. A. Lyssenko, Org. Lett., 2003, 5, 1939 CrossRef CAS PubMed.
- S. A. Amelichev, L. S. Konstantinova, O. A. Rakitin and C. W. Rees, Mendeleev Commun., 2006, 16, 289 CrossRef PubMed.
- M. J. Earle, A. G. Massey, A.-R. AL-Soudani and T. Zaidi, Polyhedron, 1989, 8, 2817 CrossRef CAS.
- B. L. Chenard and T. J. Miller, J. Org. Chem., 1984, 49, 1221 CrossRef CAS.
- D. Aebisher, E. M. Brzostowska, N. Sawwan, R. Ovalle and A. Greer, J. Nat. Prod., 2007, 70, 1492 CrossRef CAS PubMed.
- E. M. Brzostowska and A. Greer, J. Org. Chem., 2004, 69, 5483 CrossRef CAS PubMed.
- D. J. Schipper, L. C. H. Moh, P. Müller and T. M. Swager, Angew. Chem., Int. Ed., 2014, 53, 5847 CrossRef CAS PubMed.
- I. D. Campbell and G. Eglinton, Org. Synth., 1965, 45, 39 CrossRef CAS.
- Y. Arakawa, S. Nakajima, R. Ishige, M. Uchimura, S. Kang, G.-i. Konishi and J. Watanabe, J. Mater. Chem., 2012, 22, 8394 RSC.
- G. Zhang, H. Yi, H. Chen, C. Bian, C. Liu and A. Lei, Org. Lett., 2014, 16, 6156 CrossRef CAS PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318 CrossRef CAS PubMed.
- Y. H. Wijsboom, A. Patra, S. S. Zade, Y. Sheynin, M. Li, L. J. W. Shimon and M. Bendikov, Angew. Chem., Int. Ed., 2009, 48, 5443 CrossRef CAS PubMed.
- M. Turbiez, P. Freère, M. Allain, N. Gallego-Planas and J. Roncali, Macromolecules, 2005, 38, 6806 CrossRef CAS.
- Y. Liu, C. Di, C. Du, Y. Liu, K. Lu, W. Qiu and G. Yu, Chem. – Eur. J., 2010, 16, 2231 CrossRef CAS PubMed.
- X. C. Li, H. Sirringhaus, F. Garnier, A. B. Holmes, S. C. Moratti, N. Feeder, W. Clegg, S. J. Teat and R. H. Friend, J. Am. Chem. Soc., 1998, 120, 2206 CrossRef CAS.
- S. Ahn, K. Yabumoto, Y. Jeong and K. Akagi, Polym. Chem., 2014, 5, 6977 RSC.
- T. T. Ong, S. C. Ng and H. S. O. Chan, Polymer, 2003, 44, 5597 CrossRef CAS.
- B. Dong, Y. Xing, J. Xu, L. Zheng, J. Hou and F. Zhao, Electrochim. Acta, 2008, 53, 5745 CrossRef CAS PubMed.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowea and S. Tierneya, Chem. Commun., 2007, 5061 RSC.
- A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422 RSC.
- W. T. Neo, L. M. Loo, J. Song, X. Wang, C. M. Cho, H. S. O. Chan, Y. Zonga and J. Xu, Polym. Chem., 2013, 4, 4663 RSC.
- S. Kaur, N. J. Findlay, A. L. Kanibolotsky, S. E. T. Elmasly, P. J. Skabara, R. Berridge, C. Wilsonc and S. J. Coles, Polym. Chem., 2012, 3, 2277 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2:a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- SHELXL, G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds, crystallographic data and refinement parameters of single crystal X-ray structures, the ORTEP and packing diagram of 2a and 2c, spectroelectrochemistry of P3, TDDFT calculated molecular orbitals and excitations of 2d and 2e. CCDC 1049965, 1050201, 1050202 and 1050203. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5py01133g |
| This journal is © The Royal Society of Chemistry 2015 |