
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering thermoresponsive polyether-based nanogels for temperature dependent skin penetration†
M.
Asadian-Birjand
a,
J.
Bergueiro
a,
F.
Rancan
b,
J. C.
Cuggino
a,
R.-C.
Mutihac
a,
K.
Achazi
a,
J.
Dernedde
c,
U.
Blume-Peytayi
b,
A.
Vogt
b and
M.
Calderón
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustrasse 3, 14195 Berlin, Germany. E-mail: marcelo.calderon@fu-berlin.de; Fax: +49-30-838-459368
bClinical Research Center of Hair and Skin Science, Department of Dermatology and Allergy, Charité-Universitätsmedizin Berlin, Charitéplatz 1, 10117 Berlin, Germany
cCharité – Universitätsmedizin Berlin, Institut für Laboratoriumsmedizin, Klinische Chemie und Pathobiochemie, CVK, Augustenburger Platz 1, 13353 Berlin, Germany
First published on 16th June 2015
Abstract
Highly biocompatible thermoresponsive nanogels (tNGs) based on oligo ethylene glycol (OEG) as thermoresponsive unit and dendritic polyglycerol (dPG) as cross-linker, were precisely engineered in terms of size and volume phase transition temperature (VPTT). Preliminary uptake studies into human skin were realized to show the temperature-dependent internalization behavior of these systems.
Transdermal delivery has many advantages in comparison to intravenous or oral administration like patient amenability, reduced side effects, and enhanced activity by avoiding hydrolytic enzymes or hard environmental/metabolic conditions.1–5 Nonetheless efficient transdermal administration of certain hydrophilic molecules of biomedical interest (peptides, proteins, hydrophilic drugs, etc.) is still a challenge due to the protecting barrier function performed by the highly organized stratum corneum (SC) in the skin.6–9 Several techniques based on the disruption of the SC's ordered structure have been developed, of which microneedles,10 laser ablation11 or ultrasound12 are the most commonly used. But all of these techniques present limitations and disadvantages highlighting the need for a more sophisticated way of action in transdermal delivery.
Numerous studies suggest that selective skin penetration and specific targeting effects can be achieved by nanoparticles with controlled size, lipophilicity, surface charge, and stability.13 Recently new soft synthetic, polymer based nanometric architectures have emerged that can potentially solve the transdermal delivery challenge.14,15 New developments have highlighted polymeric nanoparticles as possible drug delivery devices for transdermal applications by accumulation, e.g., by improving drug penetration, taking advantage of the reservoir function in hair follicle (HF) openings, and by performing controlled release.16,17 Of all the polymeric nanoparticles, nanogels (NGs) have gained the most attention. NGs are aqueous dispersions of hydrogel particles in the nanometer range and are formed with physically or chemically crosslinked polymer network chains.18,19 NGs have already shown many interesting intrinsic properties, such as high water content, soft nature, flexibility, cell and tissue compatibility, and excellent water dispersion/solubility.
When responsive polymers are used as building blocks for NGs, these architectures can largely respond to external factors like temperature,20 pH,21 light,22 electrical fields,9etc., and change their properties as a result.23 Particularly, thermoresponsive polymers react to enviromental changes, i.e., temperature can change NG properties like hydrophobic/hydrophilic balance, softness, size, and aggregation behavior, which are all important for biomedical applications.24–26 Because of these changes upon enviromental triggers, the drug encapsulation and release profiles of these systems have been well studied as drug delivery devices.18,27,28 The same concept has already been explored for transdermal drug delivery systems,21,29–32 but, to the best of our knowledge, no study of a temperature dependent dermal uptake of thermoresponsive NGs has been reported yet. Nevertheless, optimization of synthetic methodologies that allow a reproducible synthetic control over size, shape, and physicochemical properties is still a milestone in the synthesis of NGs.22,33,34
More precisely, the development of NGs for transdermal applications calls for an optimization of synthetic routes that enable control over the nanoparticle properties like size, flexibility, deformability, and drug loading capacity. Among the typical methodologies for preparing NGs, free radical dispersion/precipitation polymerization is particularly attractive because it yields particles with sizes between 0.1 to 15 μm in a single reaction step.25,35,36 Therefore this methodology was utilized in this work to synthesize tNGs through a free radical polymerization of acrylated dendritic polyglycerol (dPG-Ac) and ethylene glycol methacrylates, as shown in Fig. 1a and Table S1 (ESI†). dPG-Ac as a water soluble crosslinker and di(ethylene glycol) methyl ether methacrylate (DEGMA) and oligo ethylene glycol methacrylate (OEGMA475) as the thermoresponsive monomers, were chosen, respectively, as building blocks to combine their inherent multifunctionality, responsiveness, and biocompatibility.37–41
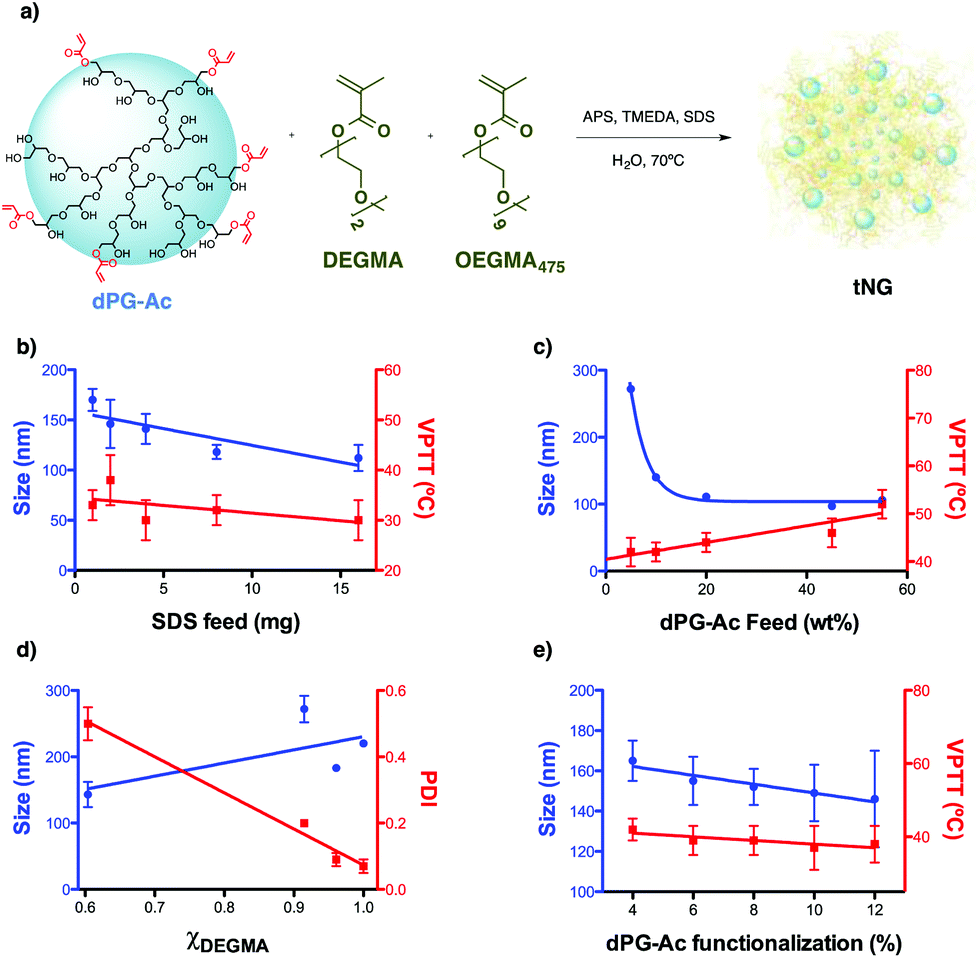 | ||
| Fig. 1 (a) General scheme of the nanogelification reaction and parameters screened for size and VPTT tuning: (b) SDS feed, (c) dPG-Ac feed, (d) monomer ratio, and (e) dPG-Ac functionalization degree. All data are shown in detail in Table S1 (ESI†). | ||
It is difficult to predict the size at which a particle will pass the skin barrier. It is known from the literature that the biggest particle size is 10 nm for not easily deformable materials and higher (above 50 nm) for soft and flexible particles.42 Therefore we aimed for tNGs in the size range of 50–200 nm and with a VPTT of 36–40 °C, slightly higher than the temperature of healthy skin.43 It is expected that such tNGs would not easily penetrate the lipophilic SC barrier below their transition temperature due to their high hydrophilicity. But once the tNGs surpasse their VPTT, for instance in inflamed skin areas, their lipophilicity will change from a hydrophilic to a hydrophobic state. This change is expected to improve the interaction with the hydrophobic structures of the SC that would result in a better penetration in the skin. To achieve tNGs with these requirements, we fine tuned their size and VPTT by varying the synthetic parameters like sodium dodecyl sulphate (SDS) feed, dPG-Ac feed, monomer ratio, and dPG-Ac functionalization degree (Fig. 1b–e, Table S1, ESI†).
The size of the tNGs was tuned first by modifying the SDS feed prior to polymerization. As expected,44 increasing the SDS feed from 1 to 16 mg decreased the tNG's size from 170 to 112 nm, respectively. VPTT showed a slightly decreasing tendency with the SDS feed increasing. With a size close to 150 nm and a transition temperature above 30 °C, a value of 2 mg SDS feed was therefore set as the first fixed parameter (Fig. 1b).
It was found that increasing the dPG-Ac feed ratio effectively decreased the tNG diameter within the range of 272–97 nm (Fig. 1c). This was attributed to the higher crosslinker density within the network, which consequentially decreased the primary chain length. Moreover, the greater degree of crosslinking may have provoked an increase in the polymer network's elastic tension, which conspicuously reduced the swelling of the NGs and, as a result, their hydrodynamic diameter. The dPG-Ac feed ratio within the tNGs was confirmed by 1H-NMR spectroscopy, which showed an increase in the ethylene glycol chain proton signals for the tNGs with a lower dPG-Ac feed ratio (Fig. S1, ESI†). This result indicated a positive correlation of the crosslinker composition inside the tNG with the dPG-Ac feed. Moreover, VPTT was also affected by the dPG-Ac feed. Since VPTT depends on the balance between hydrophobicity and hydrophilicity within the tNG, increasing the dPG-Ac composition also increased the network's hydrophilicity. This caused the phase transition temperature to rise until a dPG-Ac feed ratio of 55 wt%, at which point the VPTT was no longer measurable (Fig. 1c). A compromise between a size below 200 nm and a low transition temperature around 40 °C was found at a dPG-Ac feed ratio of 10 wt%.
Varying the monomer mole fraction (χDEGMA) had a big influence on the polydispersity of the yielded tNGs. Copolymerization with dPG-Ac (7% acrylation degree) only showed an acceptable polydispersity value when monomer mol fractions near 1 were used (χDEGMA = 1, PDI = 0.07) (Fig. 1d). As a result, the synthetic control over tNG's VPTT could be investigated by tailoring the hydrophobic/hydrophilic balance on the macromolecular crosslinker with its acrylation degree.
As expected,35 when the functionalization degree of the dPG-Ac (4–12%) increased, the size of the particles tended to decrease from 165 to 146 nm. Moreover, when the crosslinker's degree of functionalization rose, the VPTT moderately descended from 42 to 37 °C. A higher dPG-Ac functionalization degree increased the network's hydrophobicity and caused the VPTT to slightly drop (Fig. 1e). In summary, a dPG acrylation degree of around 10% was found to be ideal for temperature-dependant skin penetration purposes and yielded tNGs with sizes below 200 nm and convenient transition temperatures between 37–42 °C.
The four screened parameters were used to tune the size and transition temperatures of the synthesized thermoresponsive system in order to allow the design of tNGs that would facilitate the shuttling of active compounds across the skin barrier. Temperature differences between healthy and diseased skin or external heating were expected to improve skin penetration of tNGs due to the change in physicochemical properties at different temperatures.
Consequently, rhodamine B (Rhd) labelled tNGs were synthesized to be trackable in ex vivo human skin experiments. The above-mentioned parameters were applied for the Rhd labelled tNGs synthesis: 2 mg as SDS feed, χDEGMA = 1, 10 wt% dPG-Ac feed, and 9% dPG-Ac acrylation degree. A post-synthetic labelling of tNG with Rhd through ester formation coupling reaction only yielded low conjugation efficiencies and NGs with dye ratios in the sub μmolRhd/mgNG range (data not shown). Therefore, a 2/8 mixture of dye labelled dPG-Ac (Rhd-dPG-Ac) and unlabelled dPG-Ac was employed to introduce the dye with a total crosslinker feed value of 10 wt%. Adding an extra crosslinking unit like Rhd-dPG-Ac, the system's hydrophilicity was increased. Consequently, the size for Rhd-tNG (NG20) changed from the expected 150 to 80 nm. The VPTT, however, did not significantly differ (36 °C) and remained in the desired range when only 2 wt% feed of the hydrophilic crosslinker Rhd-dPG-Ac was used (ESI†). Moreover reversible thermoresponsive behavior was demonstrated when NG20 was analysed via turbidimetry (Fig. S2†). As a negative control for skin internalization experiments, a non-thermoresponsive NG was synthesized by mainly modifying the cross-linking content (ESI†). By employing only Rhd-dPG-Ac as the crosslinker and a monomer ratio of χDEGMA = 0.91, NG21 was obtained with a size of 78 nm, which was comparable to NG20 (Table S1, ESI†), but did not show a phase transition below 75 °C (Fig. 2c). Both NGs were intensively characterized (Fig. 2 and ESI†). The characteristic monomer and crosslinker peaks as well as the lack of vinyl signals in the 1H-NMR spectra revealed a complete polymerization. The sizes were investigated with dynamic light scattering (DLS), transmission electron microscopy (TEM), and liquid/fluid atomic force microscopy (AFM) techniques, which gave correlative sizes as well as a low degree of dispersion (Fig. 2d–g, Fig. S3, ESI†). Dye concentration was determined by UV-Vis spectroscopy which yielded values of 8.26 and 68.3 mmolRhd/mgNG for NG20 and NG21, respectively. The bigger dye amount per NG in the non-thermoresponsive NG was obtained due to the use of only Rhd-dPG-Ac as crosslinker.
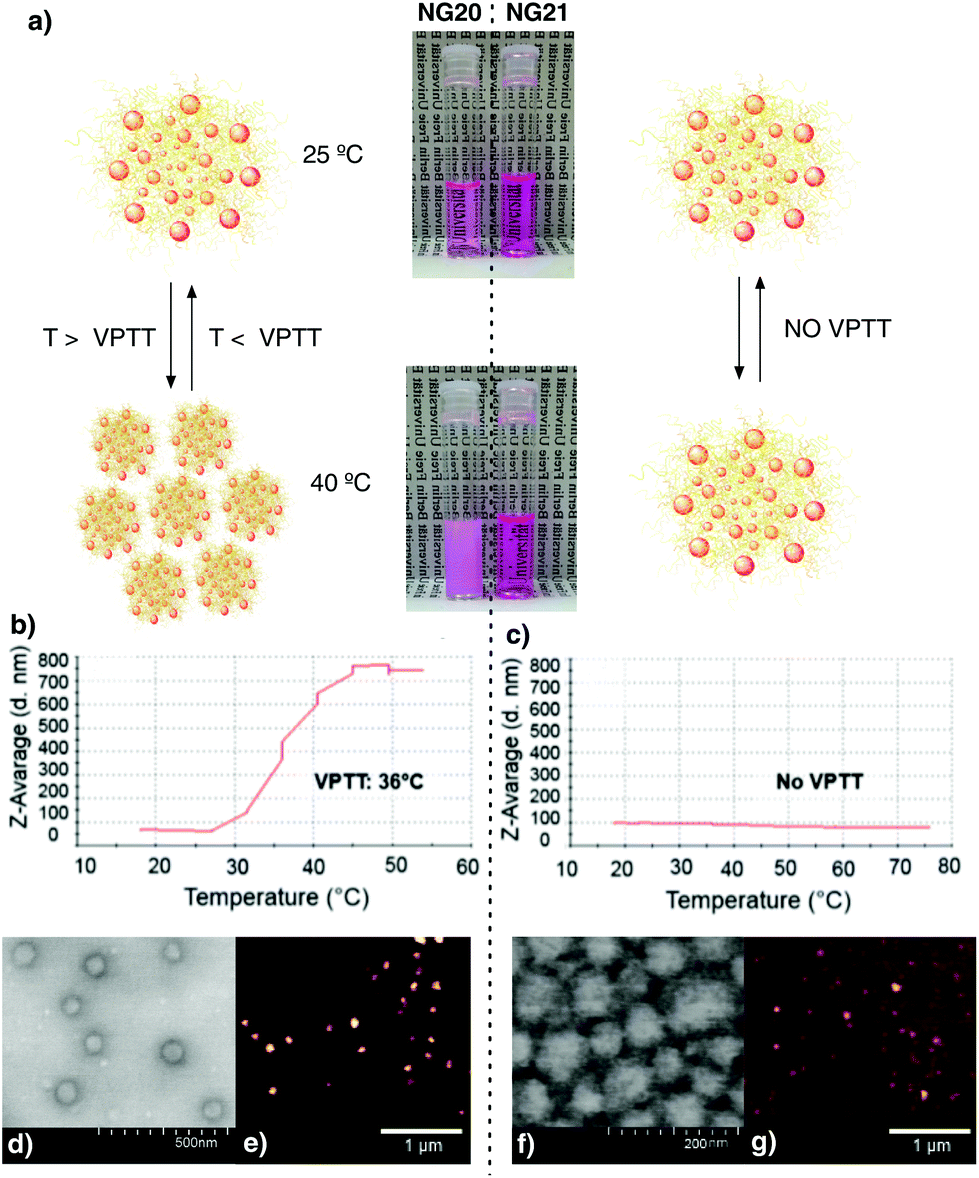 | ||
| Fig. 2 (a) Images of thermoresponsive (NG20, left) and non thermoresponsive (NG21, right) NGs at two temperatures, below (25 °C) and above (40 °C) the VPTT of NG20. DLS temperature-trend measurements of NG20 (b) and NG21 (c). TEM (d and f) and AFM (e and g) images of NG20 and NG21. | ||
To test the tNGs’ biocompatibility, the cytotoxicity in a human keratinocyte cell line (HaCaT) and fibroblast cell line (NIH3T3) was analyzed. Metabolic activity (MTS assay) and real time cell analysis (RTCA) showed IC50 values between 10 and 33 mg mL−1, respectively (Fig. S4, ESI†). These high tolerable doses demonstrate tNGs’ great cytocompatibility and potential for topical applications. Cell uptake experiments in A549 cell line revealed an endosomal uptake pathway on cancer cells (Fig. S5, ESI†).
Rhd-tNG (NG20) was employed to study temperature-dependent transdermal penetration into full-thickness excised human skin after topical application. Non-thermoresponsive Rhd-NG (NG21) was used as the negative control. Both NG samples were applied on the surface of the excised human skin at the same dye concentration of 41.3 mM. The skin penetration profile was analyzed on skin sections after 4 hours incubation at two different temperatures, below (4 °C) and above (37 °C) the VPTT of NG20. Dye fluorescence intensity was observed on the skin surface and in SC of all analyzed cryosections, regardless of the type of NGs and the incubation temperature (Fig. 3). Similar skin penetration profiles were also found in our previous studies with more rigid nanoparticles like polystyrene and silica oxide.45,46 However, fluorescence intensity was also detected in the epidermis, that indicated the presence of tNGs in viable skin (Fig. 3b). Mean fluorescence intensity (MFI) analysis confirmed that there were no significant differences observed in the analyzed SC sections between the thermoresponsive and non-thermoresponsive NGs, regardless of the incubation temperature. On the contrary, higher fluorescence intensities were measured in viable skin for the NG20 samples incubated at 37 °C in comparison to the same NGs at 4 °C incubation temperature and NG21 samples at 37 °C (Fig. 3). Even though diffusion processes might be slowed down at 4 °C, there were no remarkable differences observed between MFI values of NG20 and NG21 in the viable skin regions. These observations reflect the temperature-dependent conformational changes in the tNGs and the resultant alteration of their physicochemical properties like increased hydrophobicity, which allowed them to penetrate the SC and translocate to the epidermis. Accumulation of NGs in the HF canal was also investigated. Higher fluorescence intensity was observed in the HF canal for NG20 incubated at 37 °C than for the same sample incubated at 4 °C (Fig. S6, ESI†). No statistical analysis could be performed because of the low amount of HF in the analyzed skin sample.
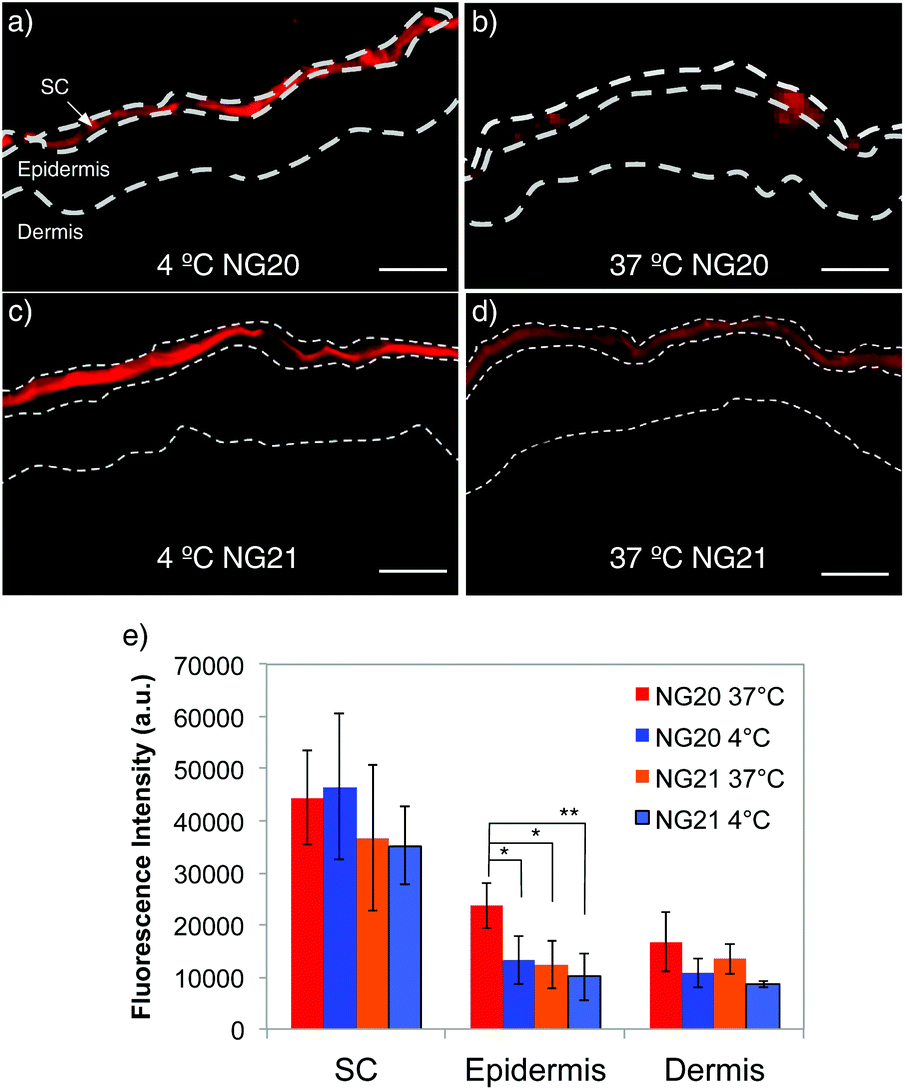 | ||
| Fig. 3 Fluorescence images of skin internalization at 4 and 37 °C of NG20 (a and b) and NG21 (c and d) scale bars = 50 μm. At least 20 sections per sample were analyzed. In approximately 25% of sections fluorescence intensity was detected also in the epidermis. Control images with non treated skin and free Rhd are shown in Fig. S11 (ESI†). (e) Average MFI value of skin sections where fluorescence intensity was detected in both SC and epidermis (ESI†) (*p < 0.05, **p < 0.01). | ||
Conclusions
The dispersion/precipitation polymerization methodology proved to be robust for the synthesis of tNGs. Sizes and VPTTs were modified by changing the surfactant concentration (SDS feed), crosslinker (dPG-Ac) acrylation degree and feed, and OEGMA feed ratio. By varying the studied synthetic parameters, Rhd labeled NGs were synthesized with required sizes and VPTT. Application of tNGs on human skin explants showed, for the first time, a temperature-dependent interaction of soft nanoparticles with the skin barrier and HF canals. Dye labeled NGs easily penetrated into SC and occasionally in the epidermis. The thermoresponsive nanogel NG20 had a better penetration in the epidermis than the non-thermoresponsive one when incubation temperatures were above the VPTT. These first experiments strengthen the rationale for further development of tNGs for applications in dermatotherapy and transdermal drug delivery. While in this study we investigated the influence of temperature on the skin penetration profile of tNGs, in ongoing studies, we are investigating tNGs loaded with non-covalently bound dyes and drugs in order to evaluate the influence of thermal triggers on the tNGs release properties.Acknowledgements
We gratefully acknowledge financial support from the Bundesministerium für Bildung und Forschung (BMBF) through the NanoMatFutur award (13N12561). Moreover, we acknowledge financial support from the Sonderforschungsbereich 1112 (http://www.sfb1112.de/), Projects A04 and C04. Dr. Julian Bergueiro acknowledges Dahlem Research Center for a Dahlem International Network PostDocs fellowship. Furthermore, we thank Prof. Dr. Rainer Haag for fruitful discussion, Pamela Winchester for proof reading the manuscript, and Dr. Maria Molina, Katrin Michel, and Dr. Christoph Böttcher for performing AFM and TEM measurements.Notes and references
- Y. Tahara, S. Honda, N. Kamiya, H. Piao, A. Hirata, E. Hayakawa, T. Fujii and M. Goto, J. Controlled Release, 2008, 131, 14–18 CrossRef CAS PubMed.
- B. J. Thomas and B. C. Finnin, Drug Discovery Today, 2004, 9, 697–703 CrossRef CAS PubMed.
- L. B. Lopes, C. M. Brophy, E. Furnish, C. R. Flynn, O. Sparks, P. Komalavilas, L. Joshi, A. Panitch and M. V. L. B. Bentley, Pharm. Res., 2005, 22, 750–757 CrossRef CAS PubMed.
- M. R. Prausnitz, S. Mitragotri and R. Langer, Nat. Rev. Drug Discovery, 2004, 3, 115–124 CrossRef CAS PubMed.
- K. Fukushima, A. Ise, H. Morita, R. Hasegawa, Y. Ito, N. Sugioka and K. Takada, Pharm. Res., 2010, 28, 7–21 CrossRef PubMed.
- K. C. Madison, J. Invest. Dermatol., 2003, 121, 231–241 CrossRef CAS PubMed.
- B. W. Barry, Nat. Biotechnol., 2004, 22, 165–167 CrossRef CAS PubMed.
- D. Lee, J. N. Ashcraft, E. Verploegen, E. Pashkovski and D. A. Weitz, Langmuir, 2009, 25, 5762–5766 CrossRef CAS PubMed.
- G. Cevc and U. Vierl, J. Controlled Release, 2010, 141, 277–299 CrossRef CAS PubMed.
- M. R. Prausnitz, Adv. Drug Delivery Rev., 2004, 56, 581–587 CrossRef CAS PubMed.
- W.-R. Lee, S.-C. Shen, K.-H. Wang, C.-H. Hu and J.-Y. Fang, J. Pharm. Sci., 2002, 91, 1613–1626 CrossRef CAS PubMed.
- A. Azagury, L. Khoury, G. Enden and J. Kost, Adv. Drug Delivery Rev., 2014, 72, 127–143 CrossRef CAS PubMed.
- F. Rancan, A. Todorova, S. Hadam, D. Papakostas, E. Luciani, C. Graf, U. Gernert, E. Rühl, B. Verrier, W. Sterry, U. Blume-Peytavi and A. Vogt, Eur. J. Pharm. Biopharm., 2012, 80, 76–84 CrossRef CAS PubMed.
- J. Khandare, M. Calderón, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- J. Lademann, F. Knorr, H. Richter and S. Jung, J. Innov. Opt. Health Sci., 2014, 8, 1530004–1530012 CrossRef.
- F. Rancan, D. Papakostas, S. Hadam, S. Hackbarth, T. Delair, C. Primard, B. Verrier, W. Sterry, U. Blume-Peytavi and A. Vogt, Pharm. Res., 2009, 26, 2027–2036 CrossRef CAS PubMed.
- M. Asadian-Birjand, A. Sousa-Herves, D. Steinhilber, J. C. Cuggino and M. Calderón, Curr. Med. Chem., 2012, 19, 5029–5043 CrossRef CAS PubMed.
- Q. Wang, Y. Zhao, Y. Yang, H. Xu and X. Yang, Colloid Polym. Sci., 2007, 285, 515–521 CAS.
- J. Ramos, A. Imaz, J. Callejas-Fernández, L. Barbosa-Barros, J. Estelrich, M. Quesada-Pérez and J. Forcada, Soft Matter, 2011, 7, 5067–5082 RSC.
- S. Mangalathillam, N. S. Rejinold, A. Nair, V.-K. Lakshmanan, S. V. Nair and R. Jayakumar, Nanoscale, 2012, 4, 239–250 RSC.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- G. Chen and A. S. Hoffman, Nature, 1995, 373, 49–52 CrossRef CAS PubMed.
- M. Giulbudagian, M. Asadian-Birjand, D. Steinhilber, K. Achazi, M. Molina and M. Calderón, Polym. Chem., 2014, 5, 6909–6913 RSC.
- M. Molina, M. Giulbudagian and M. Calderón, Macromol. Chem. Phys., 2014, 215, 2414–2419 CrossRef CAS.
- M. Molina, M. Asadian-Birjand, J. Balach, J. Bergueiro, E. Miceli and M. Calderón, Chem. Soc. Rev., 2015 10.1039/C5CS00199D.
- L. Zha, B. Banik and F. Alexis, Soft Matter, 2011, 7, 5908–5916 RSC.
- J. Bergueiro and M. Calderón, Macromol. Biosci., 2015, 15, 183–199 CrossRef CAS PubMed.
- M. Kim, B. Jung and J.-H. Park, Biomaterials, 2011, 33, 668–678 CrossRef PubMed.
- P. P. Shah, P. R. Desai, A. R. Patel and M. S. Singh, Biomaterials, 2012, 33, 1607–1617 CrossRef CAS PubMed.
- W. Il Choi, J. H. Lee, J.-Y. Kim, J.-C. Kim, Y. H. Kim and G. Tae, J. Controlled Release, 2012, 157, 272–278 CrossRef PubMed.
- N. H. A. Samah and C. M. Heard, Int. J. Pharm., 2013, 453, 630–640 CrossRef CAS PubMed.
- Y. Sasaki and K. Akiyoshi, Chem. Rec., 2010, 10, 366–376 CAS.
- A. L. Sisson and R. Haag, Soft Matter, 2010, 6, 4968–4975 RSC.
- J. C. Cuggino, C. I. Alvarez I, M. C. Strumia, P. Welker, K. Licha, D. Steinhilber, R.-C. Mutihac and M. Calderón, Soft Matter, 2011, 7, 11259–11266 RSC.
- C. K. Ober and K. P. Lok, Macromolecules, 1987, 20, 268–273 CrossRef CAS.
- R. K. Kainthan, S. R. Hester, E. Levin, D. V. Devine and D. E. Brooks, Biomaterials, 2007, 28, 4581–4590 CrossRef CAS PubMed.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- J.-F. Lutz, A. Özgür Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- H. Zhang and M. W. Grinstaff, Macromol. Rapid Commun., 2014, 35, 1906–1924 CrossRef CAS PubMed.
- A. Thomas, S. S. Mueller and H. Frey, Biomacromolecules, 2014, 15, 1935–1954 CrossRef CAS PubMed.
- M. Schneider, F. Stracke, S. Hansen and U. F. Schaefer, Derm.-Endocrinol., 2009, 1, 197–206 CrossRef CAS.
- E. F. J. Ring and K. Ammer, Physiol. Meas., 2012, 33, R33–R46 CrossRef CAS PubMed.
- N. Sanson and J. Rieger, Polym. Chem., 2010, 1, 965–977 RSC.
- A. Vogt, B. Combadiere, S. Hadam, K. M. Stieler, J. Lademann, H. Schaefer, B. Autran, W. Sterry and U. Blume-Peytavi, J. Invest. Dermatol., 2006, 126, 1316–1322 CrossRef CAS PubMed.
- F. Rancan, Q. Gao, C. Graf, S. Troppens, S. Hadam, S. Hackbarth, C. Kembuan, U. Blume-Peytavi, E. Rühl, J. Lademann and A. Vogt, ACS Nano, 2012, 6, 6829–6842 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00924c |
| This journal is © The Royal Society of Chemistry 2015 |