
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnvironmentally-friendly processing of thermosets by two-stage sequential aza-Michael addition and free-radical polymerization of amine–acrylate mixtures
Gustavo
González
a,
Xavier
Fernández-Francos
a,
Àngels
Serra
b,
Marco
Sangermano
c and
Xavier
Ramis
 *a
*a
aThermodynamics Laboratory, ETSEIB Universitat Politècnica de Catalunya, Av. Diagonal 647, 08028, Barcelona, Spain. E-mail: ramis@mmt.upc.edu; Fax: +34 934017389; Tel: +34 934016592
bDepartment of Analytical and Organic Chemistry, Universitat Rovira i Virgili, C/Marcel·lí Domingo s/n, 43007, Tarragona, Spain
cDept. of Material Science and Chemical Engineering, Politecnico di Torino, C.so Duca degli Abruzzi 24, 10129 Torino, Italy
First published on 17th August 2015
Abstract
A new dual-curing, solvent-free process is described for the preparation of tailor-made materials from off-stoichiometric amine–acrylate formulations. The first stage reaction is a self-limiting click aza-Michael addition between multifunctional amine and acrylate monomers with an excess of acrylate groups. The second stage reaction is a photoinduced radical polymerization of the unreacted acrylate groups. By selecting the structure of the monomers and the stoichiometry of the formulations, mechanical and thermal characteristics of the intermediate and final materials can be tuned. The materials obtained after the first curing stage can be gelled or ungelled and loosely or tightly crosslinked at the end of the second curing stage. The methodology used allows to obtain storable and processable intermediate polymers and final networks with optimum properties for different applications. The presence of amines in the reaction medium overcomes the intrinsic oxygen inhibition of acrylate free-radical polymerizations, resulting in a quasi complete cure.
1. Introduction
UV-curing is a widely accepted technology, because of its distinct advantages: solvent-free formulations, rapid cure rates, and low-energy consumption.1 Photopolymerization of monomer acrylates to form thermosetting materials is the most commonly used UV-system for diverse applications such as dental materials, protective coatings, printing inks, adhesive, varnishes and composites.1–3 However, acrylate polymerization shows two significant drawbacks: the inhibition of curing by oxygen4 and the insufficient cure in the areas which cannot be reached by light.5 To avoid these limitations, a dual curing process, combining photochemical and thermal curing, can be performed. Acrylate systems processed this way can achieve full cure, but with a high-energy cost and thermal stress generation during cooling down. Thermal stresses can be very significant in coating applications, due to the mismatch between the thermal expansion coefficient of the coating and the substrate and can only be completely avoided on curing at room temperature. There are many conventional strategies to reduce oxygen inhibition in photoinduced polymerization: working in an inert environment, increasing the photoinitiator content, increasing the duration and the intensity of light exposure, use of multiple photoinitiators with different rates of initiation or addition of oxygen scavengers.4 This last strategy may be interesting when these oxygen scavengers are part of the reagents or are formed in situ during the reaction.Dual curing processing, combining different and compatible polymerization mechanisms, can be used in many applications to obtain a stable material after the first stage that maintains the ability, upon application of a second stimulus, to further react and achieve the final properties desired. Nair et al.6 developed new two-stage reactive systems based on the combination of base-catalyzed thiol-acrylate Michael addition and UV-induced free radical polymerization of excess acrylates. Peng et al.7 prepared polymeric substrates with the ability to record holographic data via base-catalyzed thiol–acrylate Michael addition followed by thiol–allyl radical photopolymerization. Ruiter et al.8 proposed an elegant procedure to produce dual-curable coatings at room temperature based on the sequential and selective activation of free radical and cationic polymerization by UV irradiation at different wavelengths.
Michael addition reactions are a prized tool in materials science due to their click nature, which meet some requirements: they do not produce by-products, they are orthogonal, quick, and highly efficient and are performed under mild conditions. The Michael reaction typically refers to the base catalyzed addition of a nucleophile (Michael donor) to an activated electrophile (Michael acceptor) such as α,β-unsaturated carbonylic compounds. Although thiols are generally more nucleophilic than amines, a base is often added to deprotonate them. On the contrary, since amines can act as both nucleophiles and bases, an additional base may not be necessary in aza-Michael additions.9,10
Taking all of these into account, in the present publication we report the first preparation and characterization of thermosets based on aza-Michael reaction and radical photopolymerization dual curing of off-stoichiometric amine–acrylate formulations, with custom-tailored properties after the first curing stage and of the final material. During the first stage amino and acrylate groups react through an aza-Michael mechanism to form a partially reacted material. This reactive process proceeds at room temperature and may be catalyzed by the tertiary amines contained in the reagents or formed by the reaction of a primary amine or a secondary amine with two or one equivalents of the acceptor, respectively. In the second stage, the photoinitiated free radical polymerization of the excess of acrylic groups, embedded within the material formed in the first stage, results in a crosslinked network (Scheme 1). The tertiary amines present in the reaction medium may also act as oxygen scavengers or as co-initiators, when a type II photoinitiator is used, during the second stage of curing.4
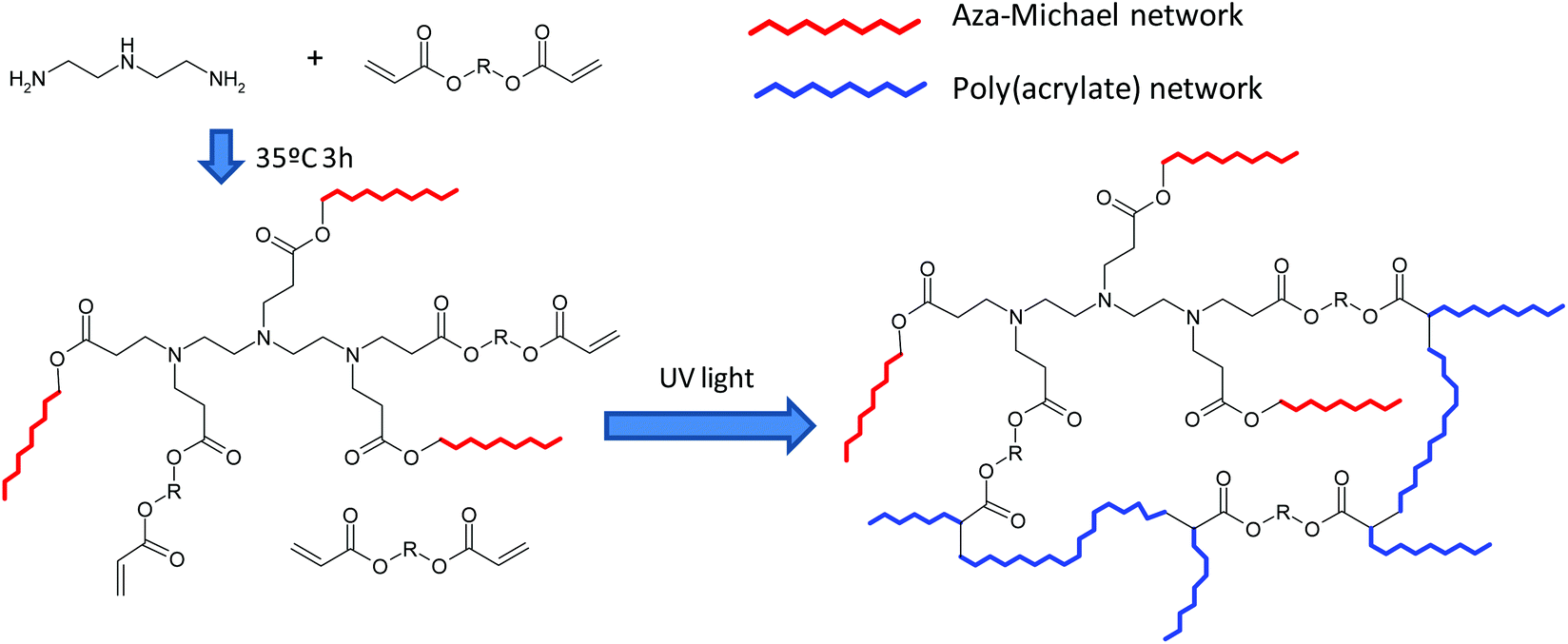 | ||
| Scheme 1 Methodology for dual-network formation from amine–acrylate mixtures. | ||
Mixtures of acrylates of different functionalities and structures, a simple or hyperbranched amine and a type I photoinitiator have been used as reference materials. For comparison purposes methacrylates and a type II photoinitiator have also been tested, to realize the broad application of the proposed procedure. The kinetics of aza-Michael addition and the conversion reached at the end of the two stages have been studied by FTIR spectroscopy and the gelation by thermomechanical analysis. The materials obtained have been characterized by calorimetry and dynamomechanical analysis.
The methodology used in this work is simple, versatile, environmentally-friendly and makes it possible to fine-tune the properties of both the intermediate and final materials. Just by controlling the stoichiometric amine/acrylate ratio in the formulation and the amine and acrylate functionality, a broad range of glass transition temperatures can be achieved, from −38 °C to 40 °C in the intermediate stage and from −5 °C to 150 °C at the end of the second curing stage. Likewise, ungelled or gelled materials can be obtained after the first curing stage. All materials are stable after aza-Michael addition and can be safely stored before further application in a second step.
2. Experimental section
2.1. Materials
All chemicals and reagents were purchased from Sigma-Aldrich and used as received unless otherwise noted. As diacrylate reactive monomers bisphenol A glycerolate (1 glycerol/phenol) diacrylate (BGDA, Mw = 484.5 g mol−1), 1,6-hexanediol diacrylate (HDDA, Mw = 226.3 g mol−1) and trimethylolpropane triacrylate (TMPTA, Mw = 296.3 g mol−1) were used. As a dimethacrylate reactive monomer triethylene glycol dimethacrylate (TEGDMA, Mw = 286.3 g mol−1) was used. A hyperbranched poly(ethyleneimine) (LP, Mw = 800 g mol−1) (Lupasol™ FG supplied by Basf) and diethylenetriamine (DETA, Mw = 103 g mol−1) were used as aza-Michael reagents. The ratio of primary/secondary/tertiary amine groups in LP and DETA is 1.0/0.82/0.53 and 1.0/0.5/0.0 respectively and their degree of branching is 0.56 and 0, respectively.11 Different catalytic systems were used: dimethoxy-2-phenylacetophenone (DMPA) as a type I photoinitiator and benzophenone (BP) as a type II photoinitiator. Scheme 2 shows the chemical structure of the compounds used.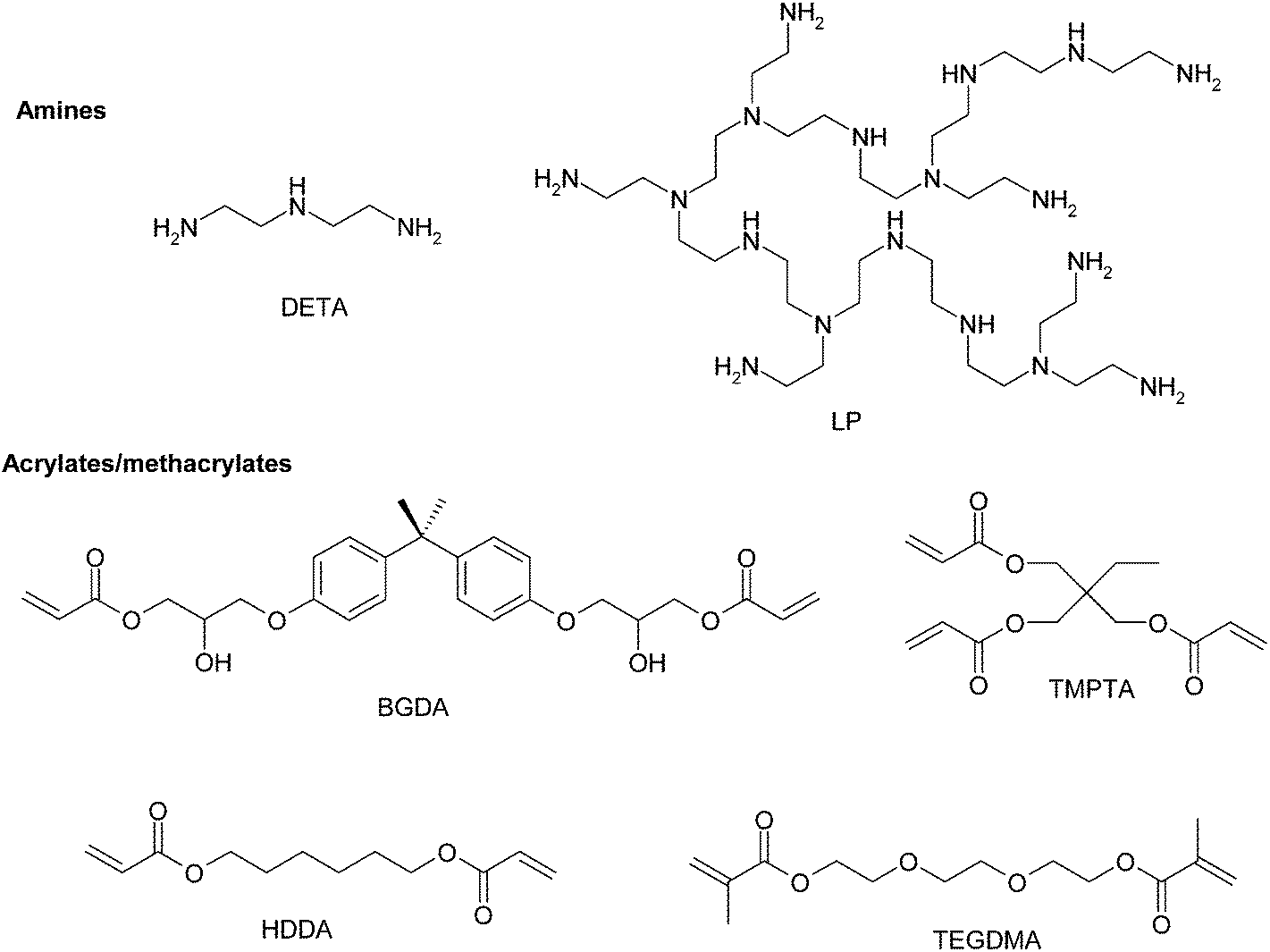 | ||
| Scheme 2 Chemical structures of the amines and acrylates used. | ||
2.2. Sample preparation
The photoinitiator was added to the diacrylates mixture and stirred mechanically until the mixture became clear at room temperature. Then the amine was added to the aforementioned mixture, stirred mechanically and degassed under vacuum at 35 °C for 15 minutes.For the aza-Michael reaction, one can define r as the stoichiometric ratio between acrylate groups and reactive amine hydrogens. If α is defined as the acrylate group conversion, then the conversion of amine groups is calculated as r·α. In the case of r > 1, the expected maximum conversion of acrylate groups in the aza-Michael will be equal to 1/r.
Formulations containing different ratios between reactive amine hydrogens and double bonds are then coded as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :r. The notation used in this paper also reflects the composition in wt% of the acrylates. As an example, LP/BGDA25/HDDA75 1:8 formulation, contains 25 g of BGDA per 75 g of HDDA and 1 N–H bond coming from the LP per 8 acrylate groups.
:r. The notation used in this paper also reflects the composition in wt% of the acrylates. As an example, LP/BGDA25/HDDA75 1:8 formulation, contains 25 g of BGDA per 75 g of HDDA and 1 N–H bond coming from the LP per 8 acrylate groups.
Formulations 1:8, 1:4, 1:2 and 1:1 (corresponding to a maximum of 12.5, 25, 50 and 100% conversion of double bonds in the aza-Michael reaction with respect to the total curing process) and a 3 wt% of a photoinitiator with respect to the diacrylate mixture were prepared. Neat acrylate formulations without amine were also prepared for comparison purposes. Mixtures of acrylates containing a 3 wt% of DMPA as a photoinitiator were used as reference materials, but for comparison purposes methacrylates and a type II photoinitiator were also studied. In these cases, the formulations were prepared in a similar manner to those previously reported for acrylate mixtures and the notation was also similar.
Fully cured samples for dynamic mechanical analysis and thermal analysis assays were prepared in a polypropylene mould by isothermal dual curing in an oven at 35 °C for 3 hours (aza-Michael addition) followed by a UV-induced curing process at ambient temperature (photopolymerization of the unreacted acrylate/methacrylate groups). Each side of the sample was irradiated during UV-curing for 15 min with a monochromatic UV lamp of 365 nm wavelength and 4 mW cm−2 of intensity. Prismatic rectangular samples (1 × 13 × 20 mm3) were prepared using this procedure. Longer curing times were tested in both stages, but the curing process did not advance substantially. The samples prepared were characterized at the end of both curing stages.
2.3. Real-time conversion
A Brucker Vertex 70 FTIR spectrometer equipped with an attenuated total refection (ATR) accessory (Golden gate™, Specac Ltd) which is temperature controlled (heated single-reflection diamond ATR crystal) was used to monitor the evolution of acrylate/methacrylate groups during isothermal dual curing at 35 °C of the formulations. Real-time spectra were recorded at 35 °C in absorbance mode with a resolution of 4 cm−1 and a wavelength ranging from 400 to 4000 cm−1, averaging 20 scans for each spectrum. A Hamamatsu Lightning Cure LC5 (Hg-Xe lamp) with one beam conveniently adapted to an ATR accessory was used to irradiate the samples during the second curing stage. A wire-wound rod was used to set a sample thickness of 25 μm. The dual curing process was performed in a similar way to the samples described above, three hours without irradiation (first stage) followed by 1 minute of UV-irradiation (second stage). It was observed that final acrylate/methacrylate conversion was barely modified using higher irradiation times. Spectra were recorded during the first stage and at the end of the second stage. Neat formulations without amine were cured under an inert atmosphere.The spectra were corrected taking into account the dependence of the penetration on the wavelength and normalized using the area of the carbonyl ester band at 1720 cm−1.12 The band at 1407 cm−1 (band of the CH2 scissor deformation mode)13 was used for monitoring of acrylate groups and the conversion of these groups was determined as:
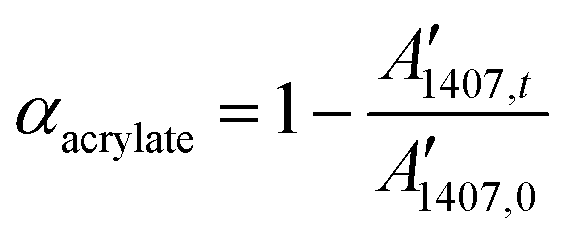 | (1) |
Formulations containing methacrylates and acrylates were evaluated in a similar way. The acrylate conversion was determined from the normalized change of absorbance at 1407 cm−1 (eqn (1)) and the methacrylate conversion was calculated by taking into account the acrylate/methacrylate composition in the formulation, and the overall methacrylate/acrylate conversion was obtained by the change of absorbance at 809 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C deformation) and the acrylate conversion.14
C deformation) and the acrylate conversion.14
2.4. Gelation
A Mettler thermo-mechanical analyzer SDTA840 for thermomechanical analysis was used to determine the gel point during aza-Michael addition (first stage).A silanized glass fiber disc about 5 mm in diameter was impregnated with the liquid formulation and sandwiched between two aluminium discs. The sample was placed at 35 °C for 3 h and subjected to an oscillatory force from 0.005 to 0.01 N with an oscillation frequency of 0.083 Hz. The gel time was taken as the onset in the decrease of the oscillation amplitude measured by the probe. The conversion of acrylate groups at the gel point, αgel, was determined as the conversion reached in FTIR at the gel time.
Assuming that no substitution effects are present (i.e. no different primary and secondary amine reactivity or size/structure dependent reactivity), conversion of acrylate groups at gelation during the aza-Michael reaction of an amine crosslinking agent and a bifunctional acrylate can be estimated from the well-known Flory–Stockmayer relationship for ideal polycondensation systems:
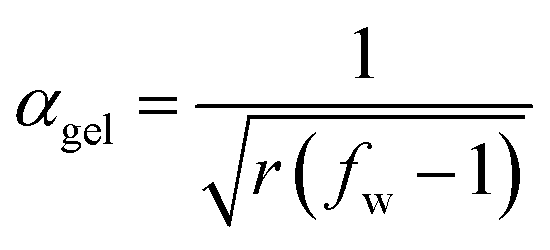 | (2) |
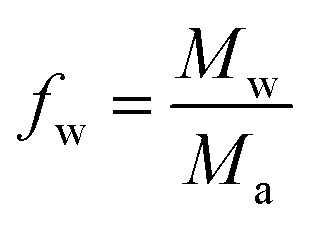 | (3) |
In the above expression, Mw is the mass-average molecular weight of the hyperbranched polymer (equal to 800 g mol−1 for Lupasol FG) and Ma is the mass of the ethyleneimine repeating unit (equal to 43 g mol−1 in the case of a hyperbranched poly(ethyleneimine). For the derivation of this parameter one has to consider that, overall, each polymerized ethyleneimine monomer contributes to one reactive group in the final HBP structure.16Eqn (2) is analogous to the well-known expression of Miller and Macosko17,18 for the crosslinking of bifunctional monomers with a mixture of crosslinking agents with different functionalities.
Eqn (2) has been used to determine, for bifunctional acrylate formulations, the theoretical conversion of acrylate groups at the gel point, αgel, using a fw of 18.6 (Mw/Ma = 800/43) for Lupasol FG and a f of 5 for diethylenetriamine (each DETA molecule has 5 N–H bonds that can react via an aza-Michael reaction with an acrylate group).
It should be noted that deviations from the ideal behaviour predicted by these expressions may be present as a consequence of nonidealities such as intra-molecular reactions among others, especially in the case of hyperbranched crosslinking agents.15,16 However, such considerations fall out of the scope of this work and the predicted values will be taken as reasonable estimates of the real effect.
2.5. Differential scanning calorimetry (DSC)
Calorimetric analyses were carried out on a Mettler DSC-822e thermal analyser. The calorimeter was calibrated using an indium standard (heat flow calibration) and an indium–zinc standard (temperature calibration).Samples of approximately 10 mg were placed in aluminium pans with pierced lids and isothermally cured in an oven for three hours at 35 °C (aza-Michael addition, first stage of dual curing). The glass transition temperatures (Tgs) of the obtained materials after the first curing step and of the amines and acrylate mixtures before curing were determined, by means of a scan at 10 °C min−1 under a nitrogen atmosphere, as the temperature of the half-way point of the jump in the heat capacity when the material changed from a glassy to the rubbery state under a N2 atmosphere and the error is estimated to be approximately ± 1 °C.
The glass transition temperatures of the formulations before curing (Tg0s) were determined using the Tgs of the different reagents (amines and acrylate mixtures), the weight composition and the Fox equation. An accurate experimental Tg0 cannot be obtained, since the aza-Michael addition starts at the first moment when acrylates and amines are mixed. The experimental Tg values of −60 °C and −95 °C for LP and DETA respectively were used to determine the Tg0s of the initial mixtures.
2.6. Dynamic mechanical analysis (DMA)
DMA was carried out with a TA Instruments DMA Q800. Prismatic rectangular samples (ca. 1 × 13 × 20 mm3) obtained after the dual curing process were analyzed using a low-friction three point bending clamp, at a frequency of 1 Hz and 0.05% strain, at 3 °C min−1 from −150 to 250 °C. The glass transition temperatures were assigned to the peak temperature of the tanδ curve. The storage moduli in the rubbery state, E′r, were determined at tanδ peak + 50 °C. This parameter is roughly proportional to the crosslinking density, as deviations from the ideal rubber elasticity model are frequent in densely crosslinked thermosetting systems.18
2.7. Thermogravimetric analysis (TGA)
Thermogravimetric analysis was carried out with a Mettler TGA/SDTA 851e/LF/1100 thermobalance. Samples, obtained by dual curing, with an approximate mass of 10 mg were degraded between 30 and 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere (50 cm3 min−1 measured under normal conditions).3. Results and discussion
3.1. Effect of the amine
The amine–acrylate polymer consisting of a diacrylate mixture (25 wt% BGDA and 75 wt% HDDA) and a multifunctional amine (LP or DETA) were chosen to demonstrate the potential value of the dual cure aza-Michael reaction/free radical polymerization to prepare tailor-made materials, storable and processable after the first curing step, with a broad range of properties after completion of the second curing process. LP and DETA were selected to highlight the effect of the degree of branching of the amine and the ratio of primary/secondary/tertiary amine groups on the dual curing and properties of the prepared materials.Table 1 shows the conversions after both curing stages and relevant thermal-mechanical properties before and after each step of curing for formulations containing LP and DETA and a 3 wt% of DMPA as a photoinitiator. Fig. 1 shows, for the same formulations, the evolution of acrylate conversion corresponding to the aza-Michael reaction during the first curing stage, determined by in situ FTIR/ATR monitoring. Some characteristic trends can be observed: (1) most of the reaction process takes place during the first 30 minutes, (2) after 180 minutes the reaction reaches a plateau, (3) formulations containing DETA react faster and reach a higher degree of conversion than those containing LP and (4) formulations with a lower amine content react to a relatively higher extent so that the aza-Michael reaction is closer to completion (see Table 1).
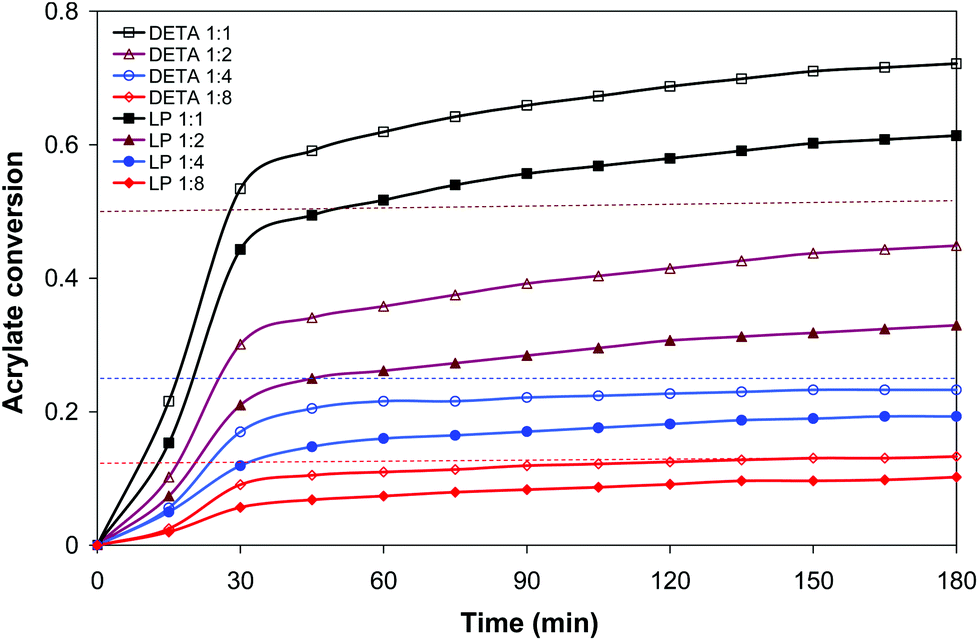 | ||
| Fig. 1 Acrylate conversion (from FTIR/ATR experiments) during aza-Michael addition (first curing stage) against time obtained by the isothermal reaction at 35 °C of BGDA25/HDDA75 formulations with a 3 wt% of DMPA as a photoinitiator and different molar ratios of LP and DETA, indicated in the legend. Horizontal dashed lines indicate the maximum acrylate conversion if aza-Michael addition would be completed for 1:8, 1:4 and 1:2 formulations. | ||
| Formulation | N–H/acrylate ratio | w amine/wacrylate (%) | T go (°C) | Stage 1 | Stage 2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Acrylate conversion (%) | Aza-Michael reaction (%) | T g (°C) | Acrylate conversion (%) | T g∞ (°C) | E ′r (MPa) | ||||
| BGDA25/HDDA75 (Neat) | −94.0 | 0 | 0 | −94.0 | 90 | 102 | 164 | ||
| LP/BGDA25/HDDA75 | 1:8 |
3.2 | −93.1 | 10 | 80 | −92.4 | 91 | 72 | 100 |
| 1:4 |
6.5 | −92.2 | 19 | 76 | −60.5 | 97 | 64 | 87 | |
| 1:2 |
12.9 | −90.7 | 33 | 66 | −59.6 | 90 | 53 | 65 | |
| 1:1 |
25.8 | −87.9 | 62 | 62 | −32.6 | 99 | 35 | 44 | |
| DETA/BGDA25/HDDA75 | 1:8 |
2.0 | −94.0 | 12 | 100 | −92.6 | 99 | 70 | 92 |
| 1:4 |
4.0 | −94.0 | 23 | 92 | −85.6 | 99 | 56 | 73 | |
| 1:2 |
8.0 | −94.1 | 45 | 90 | −66.5 | 93 | 39 | 44 | |
| 1:1 |
16.0 | −94.1 | 73 | 73 | −44.7 | 99 | 16 | 33 | |
The evolution of conversion during aza-Michael addition suggests that this reaction is chemically controlled in the early stages, up to 30 minutes, but diffusion is controlled at higher reaction times. The inspection of the reaction medium showed a significant increase in viscosity above 30 minutes of curing, which slow downs the reaction. Vitrification of the material must be disregarded since the temperature of the reaction is above the Tg of the growing chains. After three hours of reaction, the samples were stored at room temperature for six months, and it was observed that the Tg of the materials barely changed during storage. This result suggests that the aza-Michael reaction may not proceed further (if incomplete), the materials are stable over time and that they can be stored safely before the activation of the second curing stage by UV-irradiation.
Although the aza-Michael reaction generally has a high efficiency, in some amine-rich formulations, especially when LP is used, the conversion is not complete (see Table 1). Many authors have attributed a similar behaviour, to the different reactivities of primary and secondary amines, to the steric hindrance of the polymer backbone that reduces the reactivity of the secondary amines formed and to the diffusivity of the growing chain in the reaction medium.10,19,20 Moreover, it can be taken into account that the Michael reaction takes place through two equilibrium steps thermodynamically controlled by the strengths of the base and the type of nucleophile (in our case the amine can act as both nucleophile and base).9 Wu et al.10 investigated the effect of chemistry of trifunctional amines on the mechanisms of aza-Michael addition polymerizations with diacrylates. They established that the reactivity sequence of the three types of amines was secondary amines (original) > primary amines > secondary amines (formed), but the reactivity sequence changed to primary amines > secondary amines (original) > secondary amines (formed) when the steric hindrance of secondary amines (original) was increased. These authors also observed that secondary amines, formed during aza-Michael addition, were unable to participate in the polymerization process, and remained unreacted.
According to these results, it can be rationalized that the differences observed in the reactivity of LP and DETA formulations, can be related to the different primary and secondary amine contents and to the accessibility of N–H bonds. Of all the potentially reactive amine hydrogens, LP contains 29% of original secondary amines, 35.5% that may react as primary amines and 35.5% of formed secondary amines. DETA contains only a 20% of original secondary amines, 40% that may react as primary amines and 40% of formed secondary amines. In the case of DETA, the contribution of original secondary amines and primary amines (reacting as such) is 60%, and this ratio coincides well with the change of slope in the conversion curves, taking into account the reagents ratio. Although this contribution is somewhat higher for LP, the topological restrictions imposed by the densely branched structure of LP, with a significant amount of tertiary amine groups, reduce the reactivity of the original secondary amines and possibly further more in the case of the formed amines, resulting in a higher number of unreacted secondary amines, original or formed. Amine-poor formulations reach a quasi complete aza-Michael reaction for both types of amines, given the lower topological restrictions and the overall higher mobility of the reactive species. It has to be considered that only a small part of the reagents are involved in the aza-Michael reaction and the molecular weight of the growing polymer is significantly lower in amine-poor formulations.
During the second curing stage, the acrylate conversion increases significantly due to the free-radical homopolymerization of the excess of acrylate monomers (see Table 1). All formulations reach acrylate conversion between 90 and 100%, similar or even higher than that reached by the neat formulation without amine. As seen in Table 1, there is a significant increase in Tg after the first curing stage, especially in the amine-rich formulations, but the results suggest that the intermediate network structure (if present) does not interfere with or even favours crosslinking during the second curing stage.
It can be concluded that the proposed dual curing methodology in general leads to nearly completely cured materials. However, the fact that unreacted amine groups are present in the material after both reaction processes, especially in amine-rich formulations, makes these materials sensitive to moisture absorption and oxidation, and this may restrict their application. Nevertheless, a proper selection of amine reagents and a careful choice of amine–acrylate ratio would help to overcome this drawback.
The gelation during the aza-Michael reaction was studied by isothermal FTIR/TMA combined experiments at 35 °C and the results are summarized in Table 2. The conversion at the gelation point, αgel, is strongly affected by a stoichiometric ratio and by the kind of amine, as predicted in eqn (2). LP formulations reach gelation at lower conversion than DETA formulations due to the higher functionality of LP (fw = 18.6) (f = 5). Table 2 shows that the theoretical conversion at gelation, αtheoreticalgel, and the experimental conversion, αexperimentalgel, follow the same trend: increasing with the amine content. However, the experimental values are always slightly higher. As predicted in eqn (2), DETA/BGDA25/HDDA75 1:8 formulation did not gel. In Table 2 it can also be observed that tgel increases significantly on decreasing the amine content, due to the decrease in the reaction rate caused by the lower concentration of amine groups.
| Formulation | N–H/acrylate ratio | w amine/wacrylate (%) | α experimentalgel | α theoreticalgel | t gel (min) |
|---|---|---|---|---|---|
| LP/BGDA25/HDDA75 | 1:8 |
3.2 | 0.12 | 0.08 | 445.2 |
| 1:4 |
6.5 | 0.16 | 0.12 | 48.5 | |
| 1:2 |
12.9 | 0.20 | 0.17 | 28.2 | |
| 1:1 |
25.8 | 0.26 | 0.23 | 17.5 | |
| DETA/BGDA25/HDDA75 | 1:8 |
2.0 | Not gelled | ||
| 1:4 |
4.0 | 0.25 | 0.25 | 127.4 | |
| 1:2 |
8.0 | 0.40 | 0.35 | 95.8 | |
| 1:1 |
16.0 | 0.62 | 0.50 | 44.6 | |
Following the above line of reasoning for the analysis of the conversion curves, the differences between experimental and theoretical values can be explained by (1) the significantly lower reactivity of formed secondary amine groups and (2) the densely branched structure of LP leading to topological restrictions that also reduce the reactivity of original secondary amines. Both factors tend to increase the conversion at gelation that can be expected for an ideal system.18 One has to take into account also the experimental error arising from the combination of data obtained by different techniques and from the accurate evaluation of acrylate conversion from FTIR data. Nevertheless, the similarity between theoretical and experimental data makes it possible to anticipate which formulations gel during aza-Michael addition and estimate the conversion achieved at the gel point with a fair degree of accuracy.
Table 1 shows some relevant thermo-mechanical properties of the networked poly(amino ester)–poly(acrylate)s obtained at the end of the dual curing process. LP and DETA formulations present a significant increase on Tg during the second stage of curing, depending on the extent of acrylate conversion during this second stage. Even in 1:1 formulations, the presence of unreacted acrylate groups after the aza-Michael reaction leads to a significant increase in crosslinking. In Table 1 it can also be seen that rubbery moduli increase proportionally to the homopolymerization of acrylate groups in the second curing stage. This can be expected from the densely crosslinked structure of amine-free BGDA25/HDDA75 formulation. It should be taken into account that diacrylates have a functionality of 2 in aza-Michael addition, but their functionality is 4 in diacrylate homopolymerization. In consequence, formulations with a higher proportion of acrylates lead to thermosets with higher crosslinking density, while formulations with a higher amine content should lead to more loosely crosslinked materials with lower Tg. The comparison between both sets of systems shows that LP formulations have a higher Tg and crosslinking density than DETA formulations, due to the densely branched structure of LP, leading to the activation of internal branching points and further network mobility restrictions.11,21
Fig. 2 and 3 plot the evolution of tanδ and storage modulus of LP and DETA materials, respectively. In agreement with the data shown in Table 1, in formulations with a higher amine content and therefore a higher contribution of the aza-Michael reaction the network relaxation curves are shifted towards lower temperatures, indicating a decrease in Tg. This effect is more remarkable in DETA due to its lower functionality and the absence of internal branching points, in comparison with LP. It can also be observed that the α-relaxations (related to the Tg) of all materials are unimodal, indicating that all thermosets have homogeneous network structures, but acrylate-rich formulations show a more disperse network, as seen from the broader tanδ peak and modulus drop step. It can be observed that the glass transition temperature of some of the materials, determined as the peak in tanδ curves, is higher than the photocuring temperature. This can be caused by the high exothermicity of the free-radical acrylate polymerization leading to an increase in the temperature within the sample and making it possible to achieve a higher conversion and Tg during the photocuring process. In addition, the breadth of the relaxation curves indicates there is a certain network mobility at low temperatures, that may contribute to promote further reaction at low or moderate temperatures.
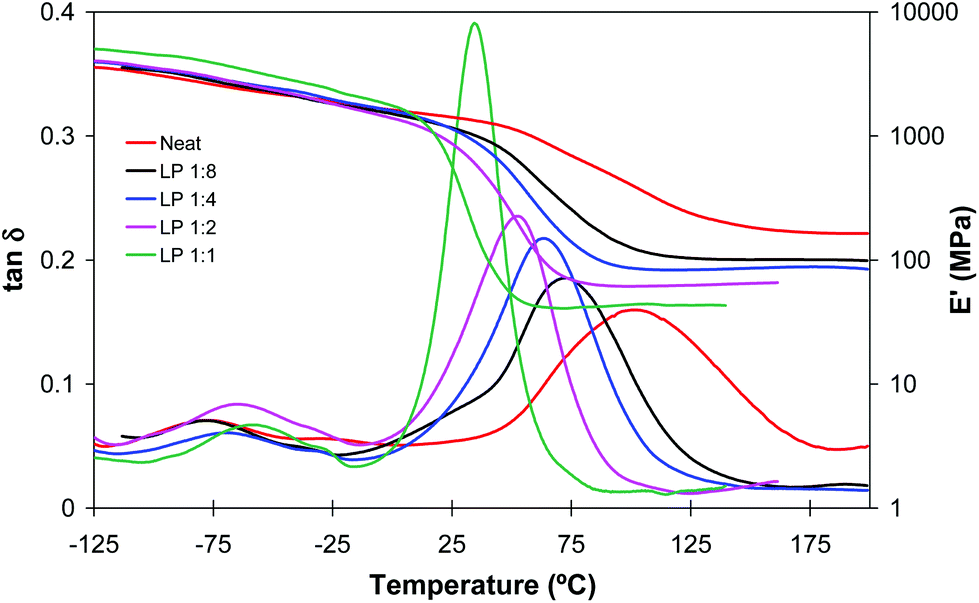 | ||
| Fig. 2 Storage moduli and tanδ curves against temperature of dual cured BGDA25/HDDA75 formulations with a 3 wt% of DMPA as a photoinitiator and different molar ratios of LP/acrylate, indicated in the legend. | ||
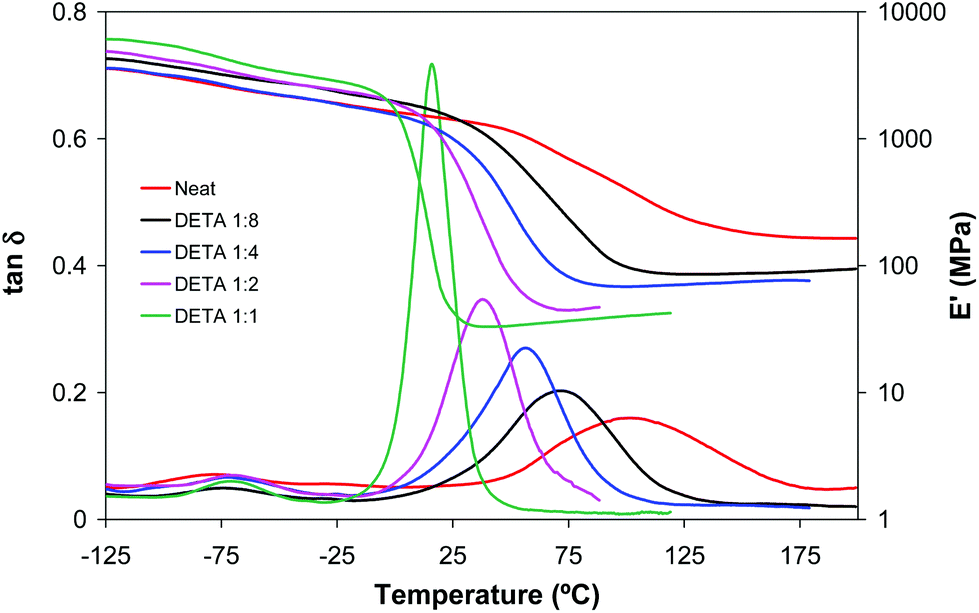 | ||
| Fig. 3 Storage moduli and tanδ curves against temperature of dual cured BGDA25/HDDA75 formulations with a 3 wt% of DMPA as a photoinitiator and different molar ratios of DETA/acrylate, indicated in the legend. | ||
Fig. 2 and 3 also show that the glassy storage modulus is higher in amine rich formulations in spite of their lower degree of crosslinking. This result can be justified in terms of cohesive energy density (CED), higher for DETA and LP than for diacrylate mixture. The CED values calculated using the group contributions according to the method outlined by Van Krevelen22 were 863 MPa for DETA and LP and 703 MPa for BGDA25/HDDA75.
Fig. 4 shows the thermogravimetric curves for neat and DETA formulations. It can be observed that the degradation takes place in two steps. The first one at a low temperature range can be related to the degradation of the poly(amino ester) structure due to the high content of labile C–N bonds. The degradation step at a higher temperature, in comparison with the degradation of neat material, can be clearly assigned to the degradation of the poly(acrylate) structure. The mass loss associated with each step agrees approximately with the amino and acrylate content of the formulation. This result suggests that the amines and the diacrylates degrade quasi separately during the first and second steps respectively, but this in turns decreases the thermal stability of the remaining poly(acrylate) network, seen from the broadening towards lower temperatures of the second step. The thermal behaviour observed is related fundamentally to the different stabilities of C–N and C–C bonds, but a contribution of the lower crosslinking density of the materials should also be considered. The degradation profiles of LP formulations were similar (not shown). The materials prepared are more easily degradable than conventional acrylates and can be used in applications where the reworkability of the thermoset is necessary in order to remove the coating and recover valuable substrates.23
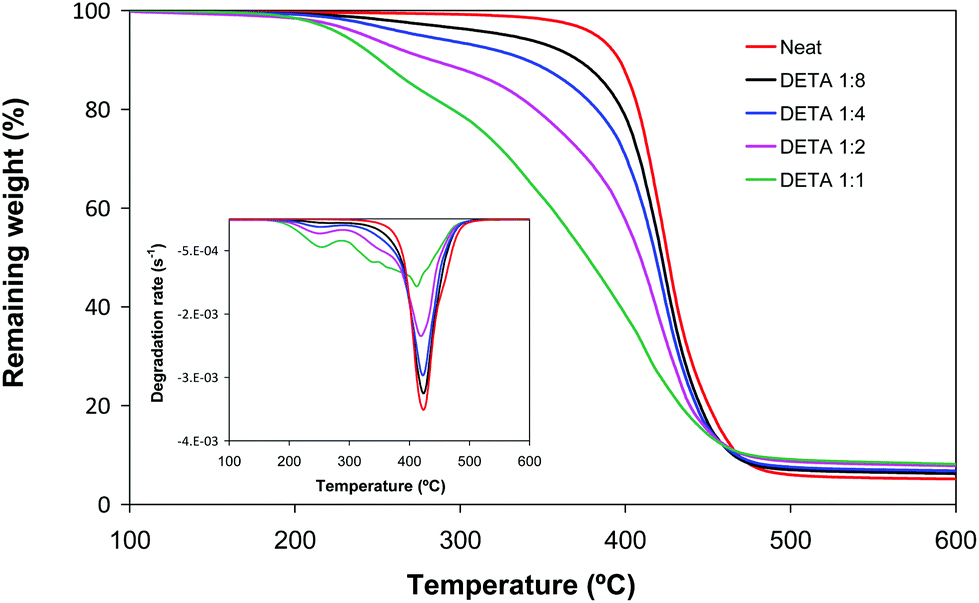 | ||
| Fig. 4 Thermogravimetric curves at 10 °C min−1 under a nitrogen atmosphere of dual cured BGDA25/HDDA75 formulations with a 3 wt% of DMPA as a photoinitiator and different molar ratios of DETA/acrylate, indicated in the legend. | ||
The results obtained demonstrate that both stage 1 and stage 2 polymer properties can be tuned, for target applications, by simply changing the type of monomer, functionality and stoichiometric ratio.
In the subsequent sections we present some results on the influence of other parameters in the proposed dual curing methodology.
3.2. Effect of the acrylates
The objective of this part of the work is to determine the influence of acrylate structure on the structure–property relationships of poly(amino ester)–poly(acrylate) thermosets obtained via aza-Michael addition/acrylate photopolymerization dual curing. The networks are prepared with a constant ratio 1:4 of N–H/acrylate using DETA as a crosslinking agent for the aza-Michael reaction and a 3 wt% of DMPA as a photoinitiator, and systematically varying the rigidity of the acrylate structure and the crosslinking points coming from acrylates with varying weight fractions of the components in the acrylate mixture, the type of acrylate and its functionality. Fundamental trends are established between the chemical structure of the network and the crosslinking density with the glass transition temperature and rubbery modulus.
Table 3 summarizes some relevant results illustrating the effect of changing the relative amounts of BGDA and HDDA. On increasing the BGDA content, the crosslinking density strongly decreases for both DETA/acrylate and neat formulations, in agreement with the lower acrylate content of BGDA (4.1 mol of CC groups per kg) in comparison with HDDA (8.8 mol of CC groups per kg). In general the Tg after stages 1 and 2, increases progressively on increasing the proportion of BGDA, but not in the same way for both steps of curing. In the aza-Michael reaction BGDA and HDDA have a functionality of two, acting therefore as chain extenders, and in consequence the Tg after this reaction is controlled by the rigidity of structure and by the degree of crosslinking coming from amine. Taking into account that all the formulations compared have the same amount and type of amine, the Tg value depends primarily on the rigidity of the chain extender, hence the higher Tg of BGDA-rich formulation after the aza-Michael reaction. During the second curing stage acrylate groups have a functionality of four, being involved in the crosslinking process, so the Tg after this stage is controlled simultaneously by the rigidity and degree of crosslinking. In this case, both factors change Tg in the opposite direction on increasing the BGDA content. Thus, increasing BGDA increases the network rigidity but decreases the crosslinking density. A further factor to be taken into consideration is the final lower degree of acrylate conversion that can be achieved in BGDA-rich formulations, as seen in Table 3, resulting in 25% of unreacted acrylate groups in the BGDA75/HDDA25 formulations. This can be caused by the low mobility of bulky BGDA and steric hindrance of CC bonds in BGDA. It is known that high molecular meth(acrylate) monomers, such as BGDA and others, need the addition of a low molecular meth(acrylate) diluent to reach the adequate viscosity and near complete curing.24
:4 formulations containing 3 wt% of DMPA as a photoinitiator
| Formulation | N–H/acrylate ratio | w amine/wacrylate (%) | T go (°C) | Stage 1 | Stage 2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Acrylate conversion (%) | Aza-Michael reaction (%) | T g (°C) | Acrylate conversion (%) | T g∞ (°C) | E ′r (MPa) | ||||
| BGDA25/HDDA75 (Neat) | −94.0 | 0 | 0 | −94.0 | 90 | 102 | 164 | ||
| BGDA50/HDDA50 (Neat) | −74.3 | 0 | 0 | −74.3 | 89 | 86 | 92 | ||
| BGDA75/HDDA25 (Neat) | −47.8 | 0 | 0 | −47.8 | 83 | 93 | 69 | ||
| DETA/BGDA25/HDDA75 | 1:4 |
4.0 | −94.0 | 23 | 92 | −85.6 | 99 | 56 | 73 |
| DETA/BGDA50/HDDA50 | 1:4 |
3.4 | −75.0 | 25 | 100 | −68.1 | 88 | 64 | 58 |
| DETA/BGDA75/HDDA25 | 1:4 |
2.8 | −49.4 | 25 | 100 | −32.1 | 75 | 67 | 28 |
In a second set of experiments, HDDA was replaced totally and partially by TMPTA, keeping the content of BGDA constant (25 wt%) in the mixture of acrylates. Table 4 shows some of the results obtained. The Tg after the first stage and the Tg and crosslinking density after the second step increase proportionally to the TMPTA content. TMPTA can increase significantly the crosslinking density in both stages of curing for a variety of reasons: (1) it has a higher acrylate content than HDDA (10.1 mol of CC groups per kg vs. 8.8 mol of CC groups per kg) (2) it has a functionality 3 and 6 in aza-Michael and acrylate homopolymerization, respectively (HDDA only 2 and 4) and (3) TMPTA can contribute with an internal branching point that is activated provided that the three acrylate groups in the TMPTA molecule react. In spite of the lower global acrylate conversion after the aza-Michael reaction and at the end of the dual-curing process, the higher crosslinking density induced by the presence of TMPTA dominates.
:4 formulations containing a 3 wt% of DMPA as photoinitiator
| Formulation | N–H/acrylate ratio | w amine/wacrylate (%) | T go (°C) | Stage 1 | Stage 2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Acrylate conversion (%) | Aza-Michael reaction (%) | T g (°C) | Acrylate conversion (%) | T g∞ (°C) | E ′r (MPa) | ||||
| BGDA25/HDDA75 (Neat) | −94.0 | 0 | 0 | −94.0 | 90 | 102 | 164 | ||
| BGDA25/HDDA 37.5/TMPTA37.5 (Neat) | −78.2 | 0 | 0 | −78.2 | 83 | 115 | 246 | ||
| BGDA25/TMPTA75 (Neat) | −58.2 | 0 | 0 | −58.2 | 77 | 127 | 431 | ||
| DETA/BGDA25/HDDA75 | 1:4 |
4.0 | −94.0 | 23 | 92 | −85.6 | 99 | 56 | 73 |
| DETA/BGDA25/HDDA 37.5/TMPTA37.5 | 1:4 |
4.2 | −78.9 | 21 | 88 | −62.5 | 90 | 76 | 157 |
| DETA/BGDA25/TMPTA75 | 1:4 |
4.5 | −59.8 | 20 | 80 | −41.6 | 88 | 97 | 246 |
Fig. 5 shows the DMA traces of the DETA 1:4 formulations containing BGDA (25 wt%) and varying amounts of HDDA and TMPTA. In addition to the effects exerted by TMPTA on the density of crosslinking and Tg previously discussed (Table 4), it can be observed that the glassy modulus and the β-relaxation increase and decrease respectively on increasing the TMPTA content. This can be rationalized by the higher rigidity and the CED value of TMPTA than HDDA (CEDTMPTA = 798 MPa and CEDHDDA = 562 MPa).22 In Fig. 5, it is observed that the broadness of the tanδ peak increases with the presence of TMPTA, indicating a more heterogeneous network structure, as commonly reported for highly crosslinked thermosets.18
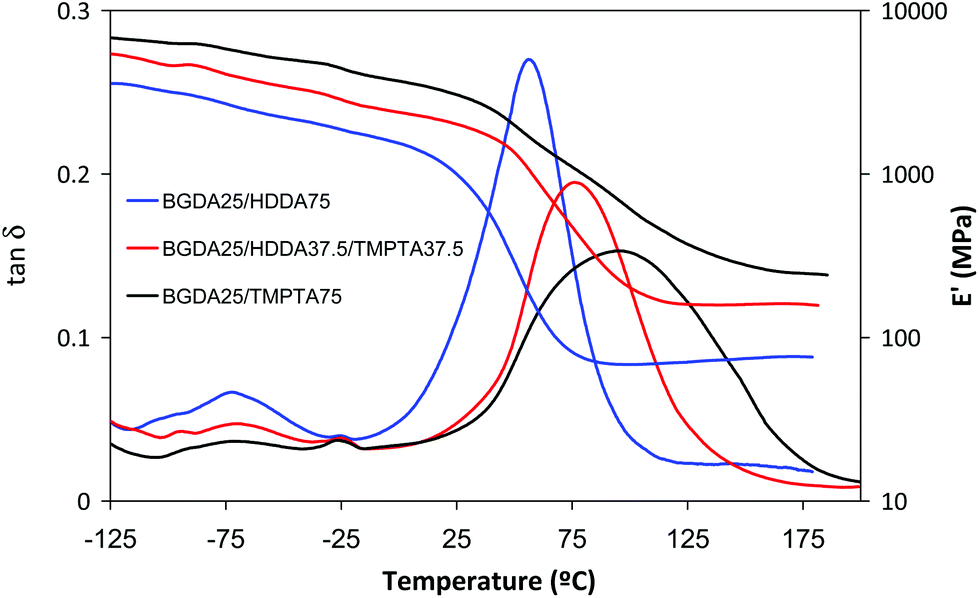 | ||
| Fig. 5 Storage moduli and tanδ curves against temperature of dual cured DETA 1:4 formulations with a 3 wt% of DMPA as a photoinitiator and different amounts of BGDA, HDDA and TMPTA, indicated in the legend. | ||
3.3. Effect of methacrylate
In order to test if methacrylates can be used in the preparation of the thermosets via aza-Michael addition/meth(acrylate) photopolymerization dual curing, we studied the complete replacement of HDDA by TEGDMA in LP/BGDA25/HDDA75 1:4 formulations. A 3 wt% of DMPA as a photoinitiator was again used. The overall acrylate/methacrylate conversion after the aza-Michael reaction was 22%, a value close to the expected one (25%), but it was found that the methacrylate conversion was only 9%, while the acrylate conversion was 87%. This result agrees with the fact that alkyl methacrylates are relatively poor Michael acceptors because of the steric hindrance caused by the methyl at the α position and the reduction of the electrophilic character.9 The Tg achieved after the aza-Michael reaction was 26 °C. At the end of the curing process, the overall acrylate/methacrylate conversion was 99%, showing only a small amount of unreacted acrylate of, about 5%. The final Tg∞ was 150 °C, with a relaxed modulus E′r of 83 MPa. Comparing these results with those obtained previously with LP/BGDA25/HDDA75 1:4 formulation (see Table 1), some differences can be observed when TEGDMA is used:
(1) During stage 1, CC bonds react and aza-Michael takes places in a higher extension, but most of the double bonds reacted are acrylates coming from BGDA. As a consequence, the glass transition temperature is raised further, since the rigid BGDA is mainly incorporated into the structure instead of the more flexible TEGDMA backbone. It should be noticed that they act as chain extenders.
(2) During stage 2, acrylate and methacrylate conversions are practically complete, retaining a small amount of unreacted acrylates in both cases.
(3) The mobility of homopolymerized poly(methacrylate) chains is much lower than that of homopolymerized poly(acrylate) chains, because of the further chain rotational barrier caused by the presence of the side methyl moieties in the repeating units along the chain. Hence the much higher Tg obtained with TEGDMA after the second curing process.
From these results it can be inferred that methacrylate/acrylate mixtures may be suitable for dual curing consisting of a self-limiting click aza-Michael addition followed by acrylate/methacrylate homopolymerization. However, it is difficult to anticipate the intermediate and final properties of the materials because of (1) the different reactivities of both unsaturated monomers, especially during aza-Michael addition, and (2) their different contribution, in terms of network structure and mobility, after the aza-Michael reaction and the subsequent acrylate/methacrylate homopolymerization.
Given the above results, one can hypothesize that the network characteristics after the aza-Michael reaction will be determined by the structure of acrylate used, but the final network will depend fundamentally on the methacrylate structure and on the relative amount of acrylate/methacrylate monomers.
3.4. Effect of type II photoinitiator
Type II photoinitiators are photoreactive substances that require a co-initiator or synergist to produce initiating radicals. This kind of photoinitiator forms an excited state upon irradiation and then abstracts an atom or electron from a donor molecule (co-initiator). The donor molecule then acts as the initiating species of polymerization. A widely used type II photoinitiator consists of benzophenone in combination with tertiary amines. Tertiary amines are typically used as co-initiators, because they react efficiently with benzophenone and also act as oxygen scavengers.4We firstly tested 3% of benzophenone as a photoinitiator of neat 25BGDA/75HDDA acrylate formulation. As expected, the curing process did not take place and almost all CC bonds remained unreacted even after 20 minutes of irradiation. When a 3% of benzophenone was used in DETA/25BGDA/75HDDA 1:2 formulations the results were completely different, and equivalent to those obtained for this same formulation but using a 3% DMPA (Table 1). In general, properties and conversion after the two stages of curing were practically the same for both photoinitiators and only a slight increase in aza-Michael addition was observed using benzophenone. In particular, after the aza-Michael addition an acrylate conversion of 45% could be reached, in comparison with 33% with DMPA (see Table 1). Although the effect of benzophenone during the first curing stage is not clear, the results suggest that the aza-Michael reaction is favoured by its presence. Fig. 6 shows the dynamic mechanical relaxation spectra for DETA/25BGDA/75HDDA 1:2 formulations obtained using DMPA or benzophenone as a photoinitiator. As we can see, both materials have similar properties after dual curing.
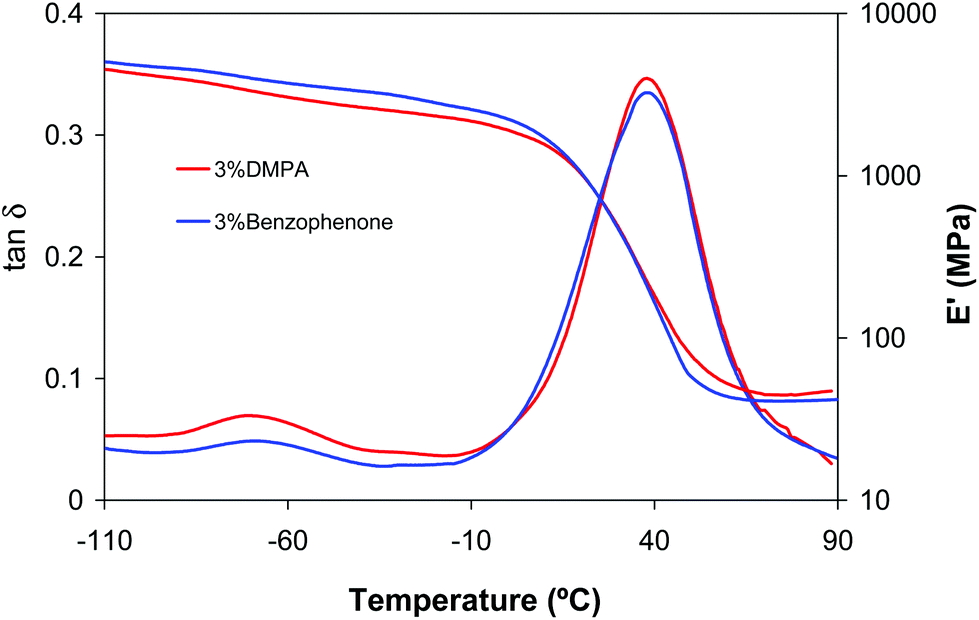 | ||
| Fig. 6 Storage moduli and tanδ curves against temperature of dual cured DETA/BGDA25/HDDA75 1:2 formulations with 3% of DMPA and 3% of benzophenone as a photoinitiator. | ||
It must also be emphasized that benzophenone is active as a photoinitiator in the presence of DETA, allowing acrylate groups to achieve near complete photocuring during the second stage. Since DETA/25BGDA/75HDDA 1:2 formulation does not contain tertiary amines or other hydrogen donors that can act as co-initiators, the tertiary amines formed during aza-Michael addition should be responsible for the observed behaviour.4
4. Conclusions
A new family of poly(amino ester)–poly(acrylate) thermosets based on off-stoichiometric amine–acrylate formulations has been prepared via a novel dual-curing solvent-free process at ambient temperature. The first stage of curing is a self-limiting aza-Michael addition between amine and acrylate groups at ambient temperature and the second stage is an acrylate radical photopolymerization of the acrylates in excess.The novel dual methodology is very versatile and allows to tune, with an adequate selection of the monomers and the stoichiometry of the formulations, the mechanical and thermal properties of the prepared materials after both stages of curing. The obtained materials can be gelled or ungelled and safely stored after the first curing stage, and loosely or tightly crosslinked at the end of the second curing stage. The versatility of the methodology proposed is based on the orthogonality of both stages of curing due to the fact that the excess of acrylate groups are not able to homopolymerize during aza-Michael addition at ambient temperature.
The dual curing methodology can be applied not only to acrylate monomers, but also to acrylate/methacrylate formulations. However, methacrylates are less reactive in aza-Michael reactions and participate preferably in the homopolymerization process.
The presence of tertiary amines formed during aza-Michael addition overcomes the intrinsic oxygen inhibition of acrylate free-radical polymerizations and allows the curing to be performed in non-inert environments. Type II photoinitiators can be used to promote the radical homopolymerization of acrylates because the tertiary amines act as co-initiators.
Acknowledgements
The authors would like to thank MINECO (Ministerio de Economía y Competitividad) and FEDER (Fondo Europeo de Desarrollo Regional) (MAT2014-53706-C03-01 and MAT2014-53706-C03-02) and the Comissionat per a Universitats i Recerca del DIUE de la Generalitat de Catalunya (2014-SGR-67).References
- U. Yaci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef.
- C. G. Roffey, Photogeneration of Reactive Species for UV Curing, John Wiley and Sons, New York, 1977 Search PubMed.
- J.-P. Fouassier, Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications, Hanser Publisher, Munich, 1995 Search PubMed.
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed.
- K. Studer, C. Decker, E. Beck, R. Schwalm and N. Gruber, Prog. Org. Coat., 2005, 53, 126–133 CrossRef CAS PubMed.
- D. P. Nair, N. B. Cramer, J. C. Gaipa, M. K. McBride, E. M. Matherly, R. R. McLeod, R. Shandas and C. N. Bowman, Adv. Funct. Mater., 2012, 22, 1502–1510 CrossRef CAS PubMed.
- H. Peng, D. P. Nair, B. A. Kowalski, W. Xi, T. Gong, C. Wang, M. Cole, N. B. Cramer, X. Xie, R. R. McLeos and C. N. Bowman, Macromolecules, 2014, 47, 2306–2315 CrossRef CAS.
- B. de Ruiter, A. El-ghayoury, H. Hofmeier, U. S. Schubert and M. Manea, Prog. Org. Coat., 2006, 55, 154–159 CrossRef CAS PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS PubMed.
- D. Wu, Y. Liu, C. He, T. Chung and S. Goh, Macromolecules, 2004, 37, 6763–6770 CrossRef CAS.
- X. Fernández-Francos, D. Santiago, F. Ferrando, X. Ramis, J. M. Salla, A. Serra and M. Sangermano, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1489–1503 CrossRef PubMed.
- X. Fernández-Francos, S. G. Kazarian, X. Ramis and A. Serra, Appl. Spectrosc., 2013, 67, 1427–1436 CrossRef PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to infrared and Raman spectroscopy, Academic Press, San Diego, 3rd edn, 1990 Search PubMed.
- V. Kumar, Y. K. Bhardwaj and S. Sabharwal, Prog. Org. Coat., 2006, 55, 316–323 CrossRef CAS PubMed.
- K. Dušek and M. Dušková-Smrčková, Macromolecules, 2003, 36, 2915–2925 CrossRef.
- X. Fernández-Francos and X. Ramis, Eur. Polym. J., 2015, 70, 286–305 CrossRef PubMed.
- D. R. Miller, E. M. Valles and C. W. Macosko, Polym. Eng. Sci., 1979, 19, 272–283 CAS.
- J. P. Pascault, H. Sautereau, J. Verdu and R. J. J. Williams, Thermosetting polymers, Marcel Dekker, New York, 2002 Search PubMed.
- P. Kalita, C. D. Pegu, P. Dutta and P. K. Baruah, J. Mol. Catal. A: Chem., 2014, 394, 145–150 CrossRef CAS PubMed.
- G. Bosica, J. Spireti and C. Borg, Tetrahedron, 2014, 70, 2449–2454 CrossRef CAS PubMed.
- D. Santiago, X. Fernández-Francos, F. Ferrando and S. De la Flor, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 924–933 CrossRef CAS PubMed.
- D. W. Van Krevelen, Properties of polymers, Elsevier, Amsterdam, 1972 Search PubMed.
- J. S. Chem, C. K. Ober and M. N. Poliks, Polymer, 2002, 43, 131–139 CrossRef.
- N. Moszner and U. Salz, Prog. Polym. Sci., 2001, 26, 535–576 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |