
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determining the effect of side reactions on product distributions in RAFT polymerization by MALDI-TOF MS†
Antonio
Veloso
,
Wendy
García
,
Amaia
Agirre
,
Nicholas
Ballard
,
Fernando
Ruipérez
,
José C.
de la Cal
and
José M.
Asua
*
POLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta R&D Ctr, Avda. Tolosa-72, Donostia-San Sebastián 20018, Spain. E-mail: jm.asua@ehu.es
First published on 25th June 2015
Abstract
Reversible Addition–Fragmentation chain Transfer (RAFT) polymerization has emerged as one of the most versatile reversible deactivation radical polymerization techniques and is capable of polymerizing a wide range of monomers under various conditions. One of the most important factors governing the success of a RAFT polymerization is the fraction of living chains at the end of the reaction, which can be maximized by using a low amount of initiator. From the point of view of the process, it is tempting to perform the polymerization in solution, which allows a better mixing. However, in this work it is shown that this choice may be negative for the quality of the polymer. Detailed analysis using Matrix Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF MS) of poly(n-butyl acrylate) (pBA) obtained at high conversion in the RAFT solution polymerization revealed that in addition to the polymer chains, formed by the RAFT mechanism, there were two distinct populations resulting from chain transfer to solvent and transfer to polymer followed by β-scission. Complementary results from Size Exclusion Chromatography coupled with Multi Angle Light Scattering detector (SEC/MALS), quantum chemical calculations, and a mathematical model that predicts product distributions, were also used to further confirm the assigned structures. The results highlight the scope and limitation on the living fraction of chains due to chain transfer events using RAFT polymerization and reversible deactivation radical polymerizations in general, and furthermore, yielded information about the fate of midchain radicals formed by intramolecular transfer to polymer.
Introduction
The large commercial demand for acrylic polymers has prompted research toward improving processes for the polymerization of acrylates. Most poly(acrylates) are produced by free radical polymerization that, in comparison to anionic and cationic polymerizations, provides poor control of the polymerization.1 Reversible deactivation radical polymerization techniques2–8 allow a much better control of this microstructure. Most of these methods utilize a dynamic equilibrium between the growing radicals and dormant species. Among these techniques, reversible-addition–fragmentation chain transfer (RAFT) polymerization is attractive because it has the advantage of being compatible with a wide range of monomers,9 notably including acidic monomers, and it can be performed in an extensive range of solvents and under a broad range of conditions. In addition, recent work has highlighted the potential of RAFT polymerization for the synthesis of multiblock copolymers due to the high living fraction of polymer chains that can be obtained by exploiting the degenerative transfer mechanism of the polymerization.10,11 In RAFT polymerization, thiocarbonylthio compounds with generic formula R–S–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–Z (referred to as RAFT agents) are employed as highly active chain transfer agents which allow for growth of all chains throughout the polymerization.
S)–Z (referred to as RAFT agents) are employed as highly active chain transfer agents which allow for growth of all chains throughout the polymerization.
Standard free radical initiators are used to generate radicals which react with the thiocarbonylthio compound to generate an intermediate radical, as can be seen in Scheme 1. This intermediate radical then fragments via a β-scission process which results in reformation of the thiocarbonylthio group at the dormant chain end and an active, propagating radical. Through this reversible addition–fragmentation process an equilibrium between dormant and active chains is established and control over molecular weight and end group functionality can be achieved.
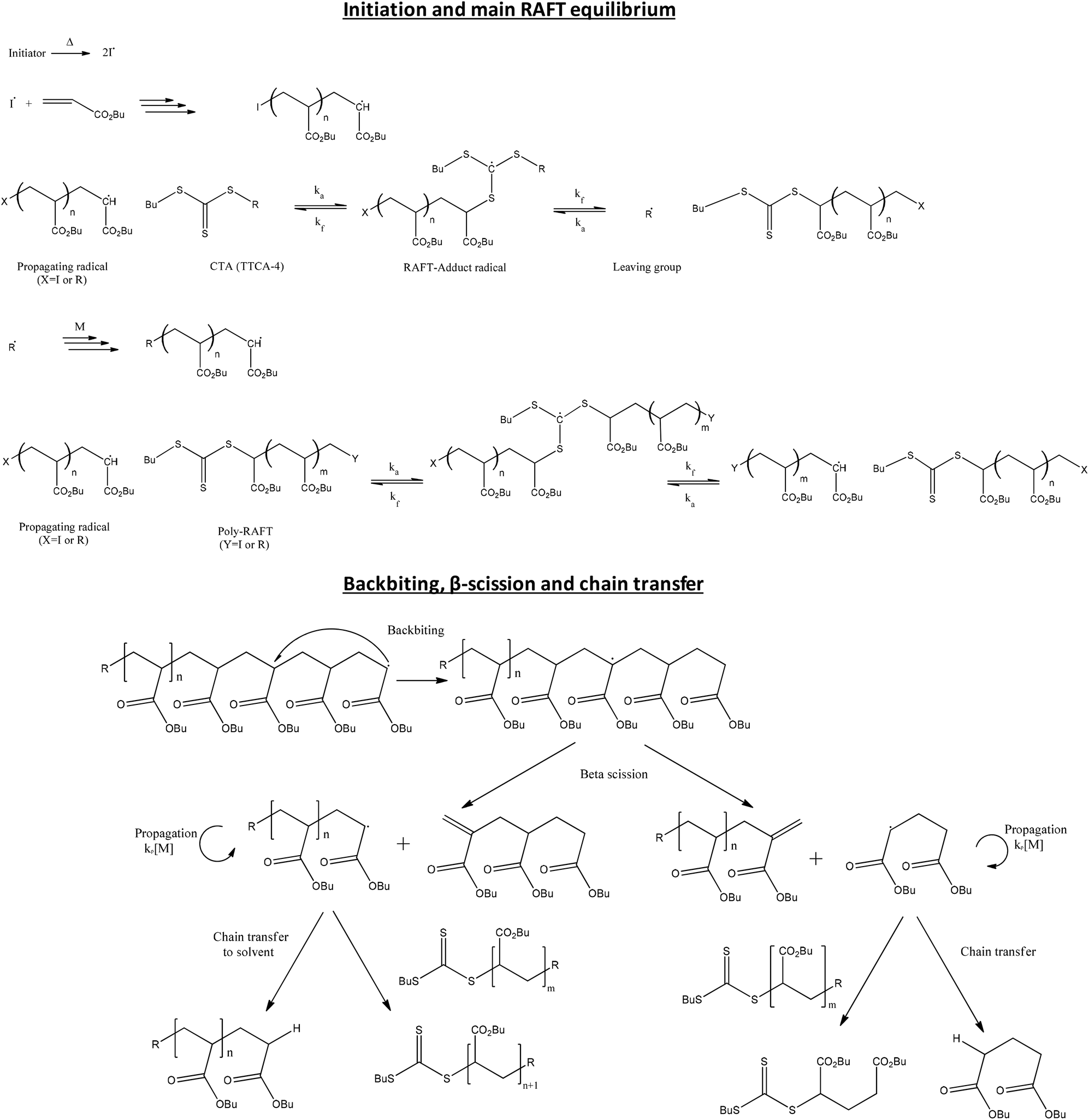 | ||
| Scheme 1 Mechanism of the RAFT polymerization of pBA using trithiocarbonate as CTA (e.g. TTCA-4) and products formed from of backbiting and β-scission. | ||
The factors governing the success of a RAFT polymerization are the fraction of living chains at the end of the reaction and the achievement of a narrow molecular weight distribution (MWD). Under conditions where termination reactions lead to a significant number of dead chains, the MWD can be narrowed and the fraction of living chains increased by simply lowering the [initiator]/[RAFT agent] ratio.
The choice of RAFT agent and initiator can also play a key role in the control of the polymers structure and can avoid undesired products which can result from reactions of the RAFT intermediate radicals.8,9,12–15
From the point of view of the process, it is beneficial to perform the reaction in solution, particularly for higher Tg polymers, which allows for a better mixing during the reaction. However, solution polymerization may present two major drawbacks in comparison to bulk polymerization. Firstly, it increases the possibility of chain transfer to solvent, which increases the fraction of dead chains. Secondly, in solution polymerization of acrylic monomers, the lower monomer concentration promotes intramolecular transfer to polymer (backbiting)16,17 and this results in a large fraction of tertiary radicals which can subsequently react to form branch points, or undergo β-scission to yield macromonomers, thus impacting on the kinetics, molecular weight distribution and end group functionality of the polymerization (see Scheme 1).18–20
In order to ascertain the extent to which the product distribution is affected by the side reactions influenced by the process conditions, a comprehensive analysis of the product mixture is needed. Matrix assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS)21–24 is the most promising technique for such analysis.
Despite the widespread use of acrylic polymers in RAFT polymerization and the importance associated with maintaining the living nature of the polymeric RAFT agents, relatively little has been published regarding the detailed characterization of pBA-RAFT polymers by MALDI-TOF MS25,26 especially at higher conversion, which is the region of interest for most applications. In addition, most work to date has focused on the reactivity of the RAFT intermediate radical and potential side products arising from various modes of termination. In the RAFT polymerization of BA using cumyl dithiobenzoate Venkatesh et al.26 used MALDI-TOF as a tool in order to explore the effects of termination of the RAFT intermediate during monomer-free, model experiments and observed the formation of 3- and 4-arm star structures. Similarly, Ah Toy et al.14 studied the RAFT polymerization of methyl acrylate and observed significant number of species arising from oxidation of the RAFT agent as well as two armed stars which were thought to arise by disproportionation of the RAFT intermediate radical. Chirowodza et al.27 synthesized polymer/clay hybrid materials by grafting polymer chains from the surface of LAPONITE® clay via surface-initiated RAFT polymerization of BA, and the grafted polymer chains were analyzed using MALDI-TOF MS. The end-group structures of the free and grafted polymers were studied. Their work expands the application range for MALDI-TOF MS, as it shows that the analysis of polymers attached to a solid surface via electrostatic interactions is possible without detaching them.
In order to gain a greater insight into the effects of chain transfer reactions and intermediate radicals on the product distribution in RAFT polymerization of acrylates, in this work we study in detail poly(n-butyl acrylate) (pBA) produced using a trithiocarbonate RAFT agent in solution and in bulk by means of MALDI-TOF MS combined with other techniques including Nuclear Magnetic Resonance (NMR) and Size Exclusion Chromatography coupled with a Multi Angle Light Scattering detector (SEC/MALS). The results are analyzed by means of a detailed mathematical model, highlighting the importance of the process conditions on the fraction of living chains as well as on the individual molecular weight distributions of the distinct species formed in the polymerization.
To begin, we present results for the RAFT polymerization of butyl acrylate in ethanol, a solvent that is particularly useful in synthesis of multiblock polymers made out of monomers of widely different polarity such as BA and acrylic acid (AA)28–30 whose effectiveness is hindered by chain transfer to solvent. After showing the huge impact that transfer reactions can have on the living fraction of chains we subsequently present results under conditions where transfer reactions are considered to have less impact using the same RAFT agent, 2-[(butylsulfanyl) carbonothioyl sulfanyl] propanoic acid (TCCA-4). Solution polymerization are conducted at 20% monomer content in benzene, where solvent transfer is low but transfer to polymer and β-scission reactions remain significant, at 70% in dioxane, as a typical reaction medium, and in bulk where both transfer to polymer and transfer to solvent are low.
Materials and methods
Chemicals
Butyl acrylate (BA, Quimidroga), 4,4′-azobis(4-cyanopentanoic acid) (V-501, Fluka), and 2,2′-azobis(2-methylbutyronitrile) (AMBN, Sigma-Aldrich) were used as received. Ethanol, dioxane, benzene (Scharlab) and HPLC grade tetrahydrofuran (THF, Scharlab) were used as solvents. 2-[(butylsulfanyl) carbonothioyl sulfanyl] propanoic acid (TTCA-4) was prepared according to Ferguson et al.31 MALDI matrices: α-cyano-4-hydroxycinnamic acid (CHCA, Aldrich) and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB, Aldrich). Cationization agents: sodium iodide (NaI, Aldrich), sodium trifluoroacetate (NaTFA, Aldrich) and potassium trifluoroacetate (KTFA, Aldrich).Synthesis of pBA by RAFT polymerization
Solution and bulk RAFT polymerizations were carried out using TTCA-4 as RAFT agent. Scheme 1 shows the reaction of the synthesis. The solution polymerization was carried out by mixing BA, TTCA-4 and V-501 in different solvents at molar ratios 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.1 and total solids content of 20%. Ethanol, dioxane and benzene were used as solvents. The solution was placed in a 0.10 L glass bottle, sealed and purged for 10 min under N2 atmosphere. The reaction was carried out by placing the bottle in a rotating device and immersed in a thermostatic bath at 60 °C by 24 h. Bulk polymerization was performed at 60 °C in a round bottomed flask on a scale of 2 g to alleviate the possibility of any exotherm. The molar ratio of reactants and the reaction temperature was the same as the solution polymerizations but the initiator was replaced with AMBN due to low solubility of V-501 in butyl acrylate. The resulting homopolymer (pBA) was dried in a vacuum oven for 40 °C in order to remove all the residual monomer and the solvent. Then, it was analyzed by MALDI-TOF MS, NMR and SEC/MALS. The theoretical molar mass was calculated according to:
:1:0.1 and total solids content of 20%. Ethanol, dioxane and benzene were used as solvents. The solution was placed in a 0.10 L glass bottle, sealed and purged for 10 min under N2 atmosphere. The reaction was carried out by placing the bottle in a rotating device and immersed in a thermostatic bath at 60 °C by 24 h. Bulk polymerization was performed at 60 °C in a round bottomed flask on a scale of 2 g to alleviate the possibility of any exotherm. The molar ratio of reactants and the reaction temperature was the same as the solution polymerizations but the initiator was replaced with AMBN due to low solubility of V-501 in butyl acrylate. The resulting homopolymer (pBA) was dried in a vacuum oven for 40 °C in order to remove all the residual monomer and the solvent. Then, it was analyzed by MALDI-TOF MS, NMR and SEC/MALS. The theoretical molar mass was calculated according to:where [BA] and [RAFT] are the initial monomer and RAFT agent concentrations, MwBA and MwRAFT are the molar masses of butyl acrylate and RAFT agent, and X is the monomer conversion determined gravimetrically.

Size exclusion chromatography (SEC) coupled with a multi angle light scattering (MALS) and refractive index (RI) detectors
The average molar mass of the pBA was analyzed by SEC/MALS. The equipment was composed by a LC20 pump (Shimadzu) coupled to a miniDAWN Treos multiangle (3 angles) light scattering laser photometer equipped with an He–Ne laser (λ = 658 nm), an Optilab Rex differential refractometer (λ = 658 nm) (all from Wyatt Technology Corp., USA). Separation was carried out using three columns in series (Styragel HR6, Styragel HR4 and Styragel HR2, with pore sizes of 106, 104 and 102 Å, respectively). Filtered toluene (HPLC-grade from Sigma-Aldrich) was used for the calibration of the 90° angle scattering intensity. The detectors at angles other than 90° in the MALS instrument were normalized to the 90° detector using a standard (PS 28770 g mol−1, Polymer Labs), which is small enough to produce isotropic scattering, at a flow rate of THF through the detectors of 1 mL min−1. In addition, the same standard and conditions were used to perform the alignment (interdetector delay volume) between concentration and light scattering detectors and the band broadening correction for the sample dilution between detectors.
The analysis was performed at 35 °C and THF was used as mobile phase at a flow rate of 1 mL min−1. The dried polymer was diluted in HPLC grade THF at concentrations of about 4 mg mL−1 and then was injected into the equipment. The dn dc−1 used for the molar mass calculation was the one corresponding to pBA (dn dc−1 = 0.064 mL g−1).32 Due to the low molecular weight of the polymers synthesized herein, this dn dc−1 value will be an approximation of the true value and may result in some error in the calculation of molecular weights. The SEC/MALS data was analyzed by using the ASTRA software version 6.0.6. (Wyatt Technologies, USA). The absolute molar mass was calculated from the MALS/RI data using the Debye plot (with 1st order Zimm formalism).
Mass spectrometry
MALDI-TOF MS measurements were performed on a Bruker Autoflex Speed system (Bruker, Germany) instrument equipped with a 355 nm Nd:YAG laser. All spectra were acquired in the positive-ion reflectron mode (accelerating voltage 20 kV, pressure 5 × 10−6 mbar). Samples were prepared with CHCA and DCTB. CHCA matrix was dissolved at concentration of 10 g L−1 and DCTB at 40 g L−1 in THF.26 NaI, NaTFA and/or KTFA were added as cationic ionization agents (approximately 10 g L−1 dissolved in THF). The matrix, salt and polymer solutions were premixed in the ratio 10:1:10 (matrix:salt:sample).
Density functional theory calculations
All the density functional theory calculations (DFT) have been carried out with the Gaussian 0933 suite of programs using the M06-2X34 functional. Structure optimizations were performed in gas phase by using the 6-31+G(d) basis set35 and harmonic vibrational frequencies were obtained, at the same level of theory, by analytical differentiation of the gradients in order to determine whether the structures were minima or transition states. These frequencies were then used to evaluate the zero-point vibrational energy (ZPVE) and the thermal corrections, at T = 298 K, to the enthalpy and Gibbs free energy in the harmonic oscillator approximation. The electronic energy was refined by single-point calculations using the 6-311++G(d,p) basis set.36Nuclear magnetic resonance
The degree of polymerization (DPBA) was determined by 1H-NMR spectra recorded on a Bruker nuclear magnetic resonance instrument (400 MHz Spectrometer, Germany) using CDCl3 as the solvent and tetramethylsilane (TMS) as internal standard. Molar mass was calculated according to:![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n,NMR = DPBAMwBA + MwRAFT n,NMR = DPBAMwBA + MwRAFT |
Results and discussion
In order to demonstrate the effect of transfer to solvent on the livingness and molecular weight distribution of polymers produced by RAFT we first synthesized a low molecular weight pBA polymer in ethanol, a known transferring solvent which has been used in the synthesis of multiblock BA/AA copolymers.28–30 The synthesis of pBA by RAFT polymerization in ethanol solution proceeded to 98% conversion. Table 1 presents the number and weight-average molar masses (n, w), the dispersity (Đ) and the polymerization degree (DPn) obtained by the different techniques (MALDI-TOF MS, SEC/MALS and 1H-NMR) together with the theoretical values. It should be noted that, as expected from RAFT polymerization, a low dispersity was achieved and the degree of polymerization is also in good agreement with the theoretical value. For MALDI-TOF MS, the species with the highest ionization efficiency observed in the spectra were considered for molar mass calculations. The slightly higher molecular weight values obtained by SEC/MALS are related to the low-light scattering signal at high elution times (small molecular weights). It is worth noting that in SEC/MALS chromatography, in order to accurately calculate the molar mass, the signal of the detectors should be sufficient in the whole elution range, and at small elution volumes SEC/MALS signal is more sensitive than refractive index (RI), and the opposite happens at large elution times. A consequence is that dispersity is likely underestimated.
n, w), dispersity (Đ) and polymerization degree (DPn) obtained by different techniques (MALDI-TOF MS, SEC/MALS and 1H-NMR) together with the theoretical values for the pBA synthesized in ethanol solution (98% conversion)
| Label |
n (g mol−1) |
w (g mol−1) |
Đ | DPn |
|---|---|---|---|---|
| Theoretical | 2102 | — | — | 15 |
| SEC/MALS | 2900 | 3000 | 1.025 | 21 |
| MALDI | 2393 | 2509 | 1.048 | 17 |
| 1H-NMR | 2289 | — | — | 16 |
Taking into account the different possible reactions of the radicals showed in Table 1S (ESI†), 18 different structures were considered for the assignment of the peaks in the spectra (Fig. 1). Moreover, for each structure, three possible adducts (proton, [M + H]+; sodium, [M + Na]+, [M + 2Na–H]+;37 and potassium, [M + K]+) were considered, therefore, more than 100 possibilities were evaluated in this work.
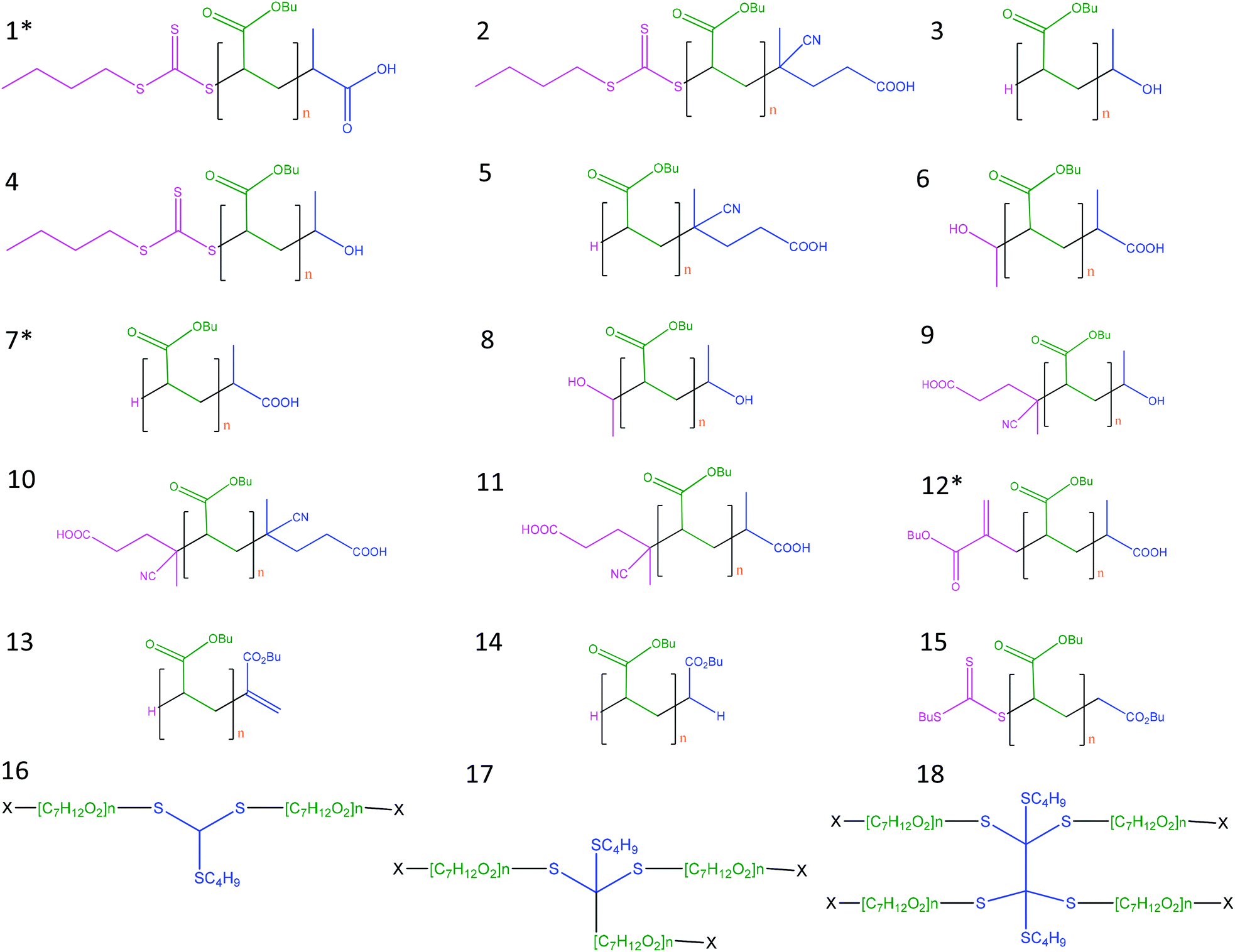 | ||
| Fig. 1 Possible structures derived from the reaction of n-pBA using TTCA-4 as RAFT agent, and initiated with V-501 in ethanol. Structures from 1 to 15 are formed due to the RAFT equilibrium, bimolecular termination by combination or chain transfer to solvent, and 2-, 3- and 4-arm star structures (from 16 to 18) (being X the initiator radical and/or R group) that result from termination of the intermediate RAFT radicals with various radical species. These 2-, 3- and 4-arm star structures have been reported in the literature26 using dithiobenzoate as RAFT agent. * Detected structures 1, 7 and 12. | ||
The MALDI-TOF mass spectra of pBA produced by solution polymerization in ethanol using DCTB as MALDI matrix and NaTFA salt as cationization agent26 are shown in Fig. 2. A CHCA matrix and NaI salt was also used, but better resolution of the spectral peaks was obtained with the aforementioned system. The mass spectrum of this sample consists of numerous groups of peaks (or series of peaks). Fig. 2 presents these groups of peaks in enlargements made at different mass ranges (A) 1500–1580, (B) 1880–1960, and (C) 2520–2600 Da. It can be seen that although the relative intensity of the peaks varies with the mass range, the main peaks appeared in all ranges. It is observed that the molar mass difference between two consecutive peaks of the same kind, e.g., between two consecutive S1 peaks, is 128.08 Da, which corresponds to the mass of one BA monomer unit. Peaks S1 and S3 were assigned to structure 1 in Fig. 1, which is formed in the main equilibrium of the RAFT agent and contains an R group and a RAFT group at either end of the chain. The difference is that S1 corresponds to [M + Na]+ adduct (M being the polymer) and S3 to the [M + 2Na–H]+ adduct. The formation of these adducts has been also reported by Arakawa et al.37 for acrylic acid polymers. A good agreement between the theoretical and experimental isotopic distribution was achieved (Fig. 3).
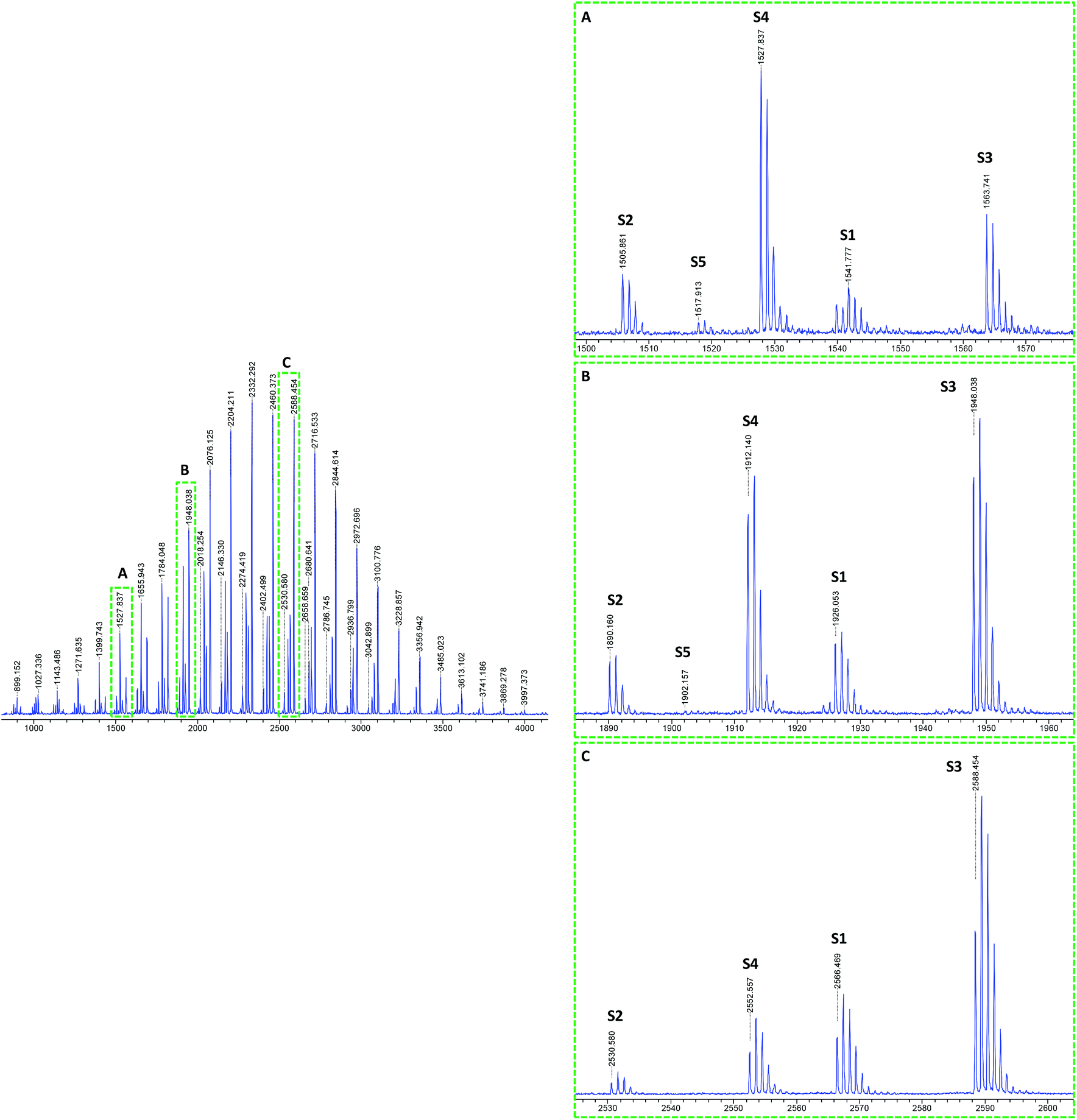 | ||
| Fig. 2 MALDI-TOF mass spectra of RAFT polymerization of BA in ethanol solution (98% conversion) using DCTB matrix and NaTFA cationization agent. Complete spectra in 700–4000 Da mass range (left). Enlargements (right) in different mass ranges (A) 1500–1580, (B) 1880–1960, and (C) 2520–2600 Da show the different detected series from S1 to S5. See Table 2S (ESI†) for details. | ||
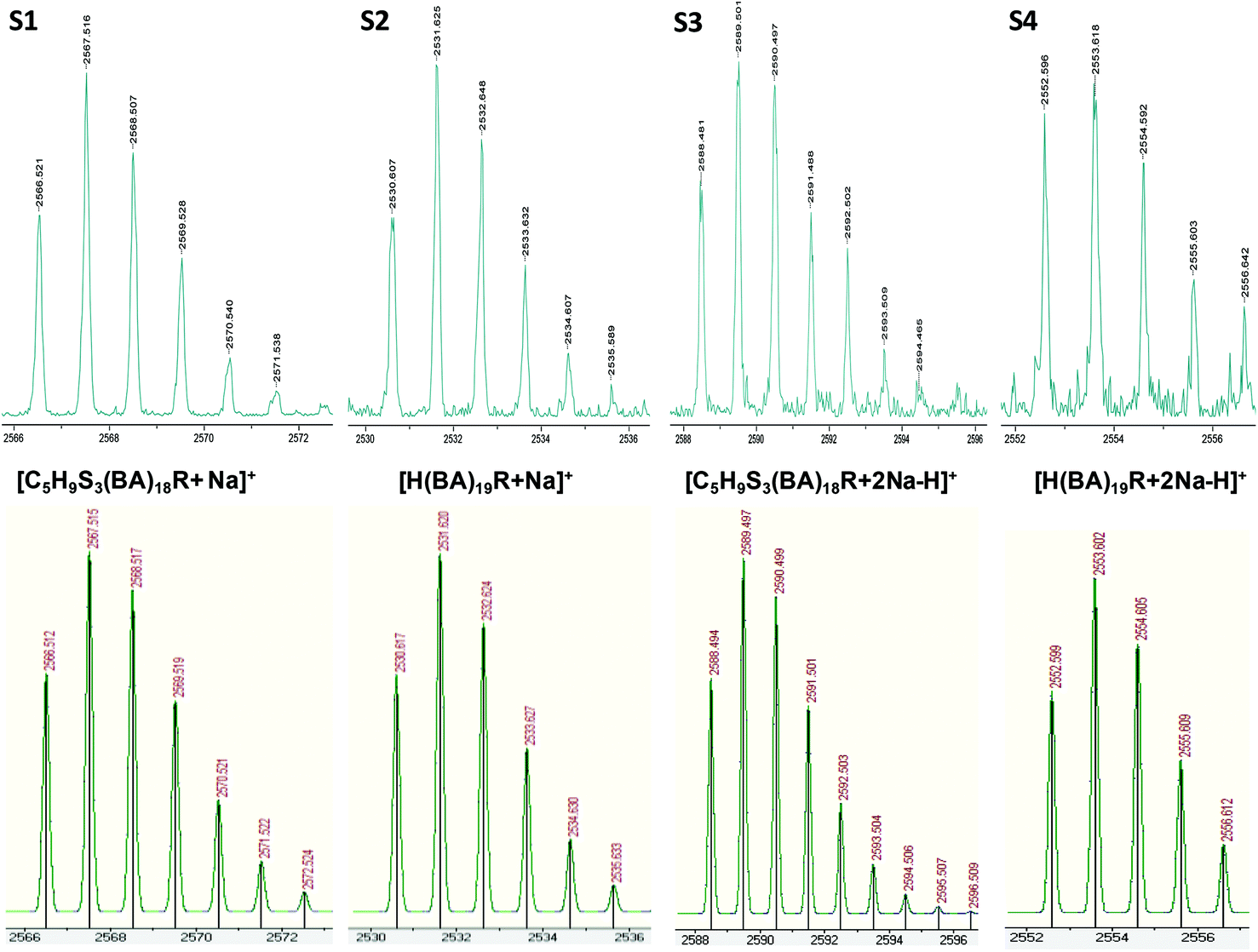 | ||
| Fig. 3 MALDI-TOF mass spectra in reflectron mode of pBA on the basis of the isotopic mass distribution: observed (above) and theoretical (below). The series S1 and S3 were assigned to structure 1 from Fig. 1, and for S2 and S4 were assigned to species 7 from Fig. 1 due to chain transfer to solvent. S1 and S2 correspond to sodiated species [C5H9S3–(C7H12O2)18–C3H5O2 + Na]+ (S1) and [H–(C7H12O2)19–C3H5O2 + Na]+ (S2), while S3 and S4 form different adduct [C5H9S3–(C7H12O2)18–C3H5O2 + 2Na–H]+ (S3) and [H–(C7H12O2)19–C3H5O2 + 2Na–H]+ (S4).37 | ||
The S2 and S4 series were attributed to a hydrogen terminated product containing an R terminal group, labeled species 7 in Fig. 1, with [M + Na]+ and [M + 2Na–H]+ adducts, respectively. The structure can be further confirmed by the good agreement between the theoretical isotopic distribution and the experimental one (Fig. 3). In order to explain species 7, two mechanisms may be proposed: (a) fragmentation of the polymer during the ionization process inside the spectrometer, which could allow the generated radical 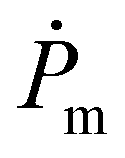 to take a proton from the acidic medium of the matrix preparation; and (b) proton transfer from the solvent to the radical species
to take a proton from the acidic medium of the matrix preparation; and (b) proton transfer from the solvent to the radical species 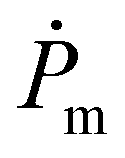 .
.
Characterizing by MALDI and electrospray ionization (ESI) a poly(methyl methacrylate) synthesized by RAFT polymerization, Jiang et al.38 demonstrated that labile end-groups, such as the dithioester groups, can be lost in MALDI-TOF MS experiments during the ionization process. In order to investigate if this may occur in the present case, a study of the energetics of the process using density functional theory (DFT) calculations was performed. This consists of breaking different bonds and reacting with the protons of the medium. The calculations showed that the C–S bond cleavage during MALDI ionization with the laser energy is feasible since the BuS(CS) 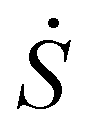 group may be detached from the polymer during the desorption process, forming a new carbanion (ΔH = 51.94 kcal mol−1) which is able to remove a proton from the matrix to form the hydrogenated species (ΔH = −188.19 kcal mol−1). The whole process is thermodynamically favorable (ΔH = −129.32 kcal mol−1). This point should not be underestimated since in many types of reversible deactivation radical polymerization, the control agent is relatively labile and during MALDI ionization may cause the presence of hydrogen terminated products.39 However, if fragmentation of the polymer during the ionization process were the cause of the presence of species 7, the ratio (S1 + S3)/(S2 + S4) should be constant along the MWD because fragmentation during MALDI ionization is proportional to the number of C–S bonds, normally one per chain. Therefore, the fragmentation should be the same for all lengths. However, Fig. 2 shows that this is not the case and the ratio (S1 + S3)/(S2 + S4) increased with the molecular weight. This suggests that chain transfer to solvent is responsible for the hydrogen terminated species (S2 + S4).
group may be detached from the polymer during the desorption process, forming a new carbanion (ΔH = 51.94 kcal mol−1) which is able to remove a proton from the matrix to form the hydrogenated species (ΔH = −188.19 kcal mol−1). The whole process is thermodynamically favorable (ΔH = −129.32 kcal mol−1). This point should not be underestimated since in many types of reversible deactivation radical polymerization, the control agent is relatively labile and during MALDI ionization may cause the presence of hydrogen terminated products.39 However, if fragmentation of the polymer during the ionization process were the cause of the presence of species 7, the ratio (S1 + S3)/(S2 + S4) should be constant along the MWD because fragmentation during MALDI ionization is proportional to the number of C–S bonds, normally one per chain. Therefore, the fragmentation should be the same for all lengths. However, Fig. 2 shows that this is not the case and the ratio (S1 + S3)/(S2 + S4) increased with the molecular weight. This suggests that chain transfer to solvent is responsible for the hydrogen terminated species (S2 + S4).
The explained results about the end-groups S1, S2, S3 and S4 were confirmed by the use of other MALDI matrices and cationization agents: CHCA/NaI, DCTB/NaTFA and DCTB/KTFA (Fig. 1S, ESI†). Venkatesh et al26 selected the DCTB/NaTFA system to characterize pBA polymers synthesized using cumyl dithiobenzoate as RAFT agent and, by generating polymeric radicals using ATRP type chemistry in the presence of RAFT agent, were able to detect 2-, 3- and 4-arm star structures due to the relatively high stability of the intermediate radical. In our case, after comparing the mass spectral peaks obtained using the same matrix system (Fig. 2) and using CHCA and NaI (not shown), it could be confirmed that none of these arm star structures was present, so the trithiocarbonate type RAFT agent used here seems to be more selective and appropriate to avoid undesired products resulting from termination of the RAFT intermediate radical and additionally to avoid oxidation of the RAFT agent.
Fig. 2 shows the presence of small amounts of species S5 that can only be observed at moderate molecular weights. This species was attributed to the product of intramolecular transfer to polymer, followed by β-scission (Scheme 1) leading to a terminally unsaturated species, labeled structure 12 in Fig. 1. In order to verify the backbiting reaction, the low masses were analyzed and the small fragments arising from these reactions were detected as proton and potassium adducts as KTFA salt was used (Fig. 4). Both the low molecular weight radical formed by β-scission in one direction (observed as the corresponding radical having terminated by disproportionation) and the low molecular weight macromonomer formed by scission in the other direction were observed. Interestingly, no higher molecular weight analogues of this compound were observed.
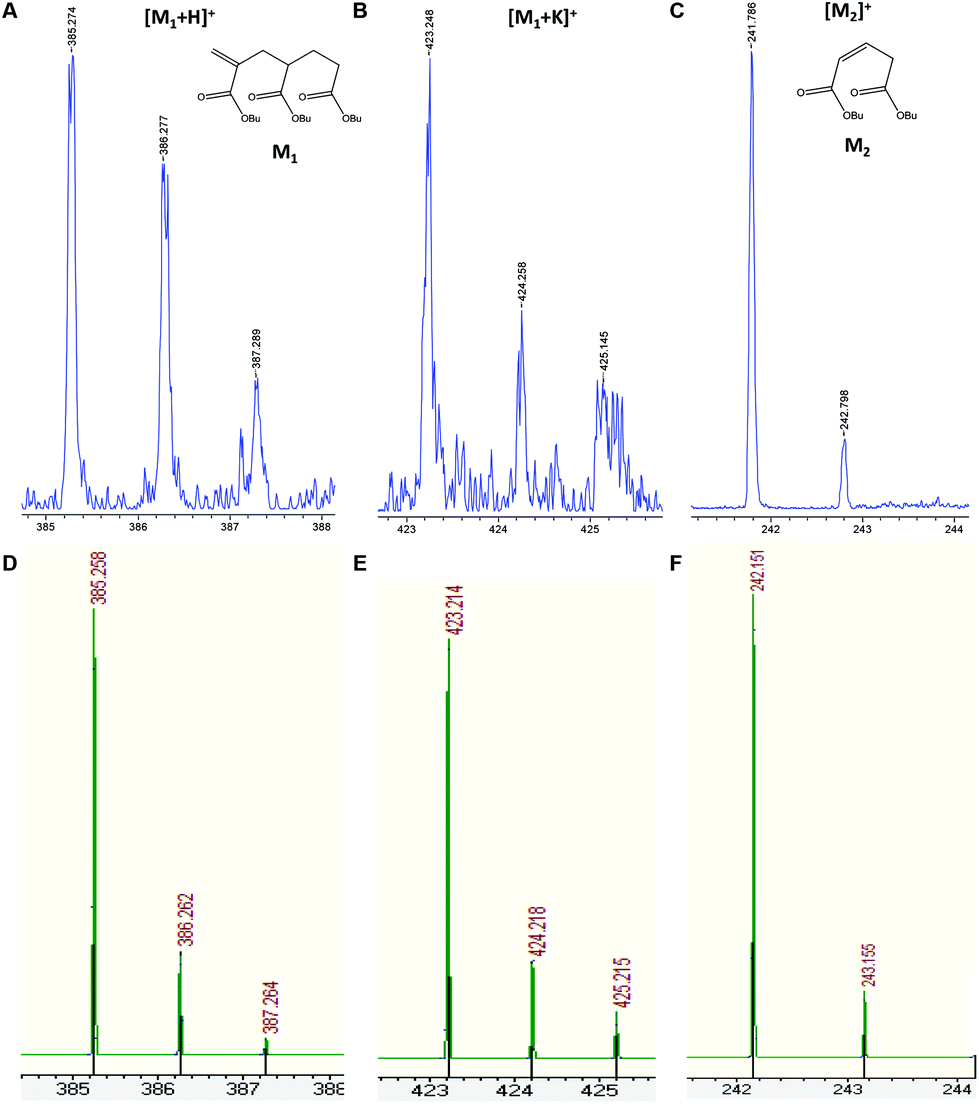 | ||
| Fig. 4 Enlargements of MALDI-TOF mass spectra in reflectron mode of two generated molecules after backbiting and β-scission reactions (Scheme 1), and the corresponding adducts (blue, upper trace). (A) and (B) represent proton and potassium adducts of C21H36O6 (M1, m/z = 384.25 Da) and (C) represents the C13H22O4 structure without forming any adduct (M2, m/z = 242.15 Da). Comparison of each adduct with theoretical isotopic mass distribution is also shown (green, below) (D, E and F). | ||
It has previously been shown that migration of the midchain radical can result in a size selective molecular weight distribution39,40 of this macromonomer in monomer free experiments designed for macromonomer synthesis. The fact that this cannot be seen in this case would suggest that the migration process is negligible in the presence of competing propagation reactions. This result will impact on expected distributions of short and long chain branches in synthesis of acrylic polymers and would suggest that only short arm branches (of two monomer units) would be expected under standard reaction conditions.
In order to determine whether the proposed structures were compatible with the accepted mechanisms for their formation we constructed a mathematical model that takes into account propagation, activation and deactivation reactions, chain transfer to solvent, backbiting and β-scission, and is capable of yielding the complete molecular weight distribution of each individual species (see ESI† for details). Due to the known experimental observations of a reduction in products arising from intramolecular transfer to polymer both in the presence of hydrogen bonding solvents41,42 and under conditions of reversible deactivation,43,44 the rate constant for backbiting obtained from radical polymerization in hydrocarbon solvents was reduced accordingly (see ESI†). The cause of the reduction in the rate coefficient under these conditions is still uncertain and therefore the amount by which it is reduced was estimated from previous literature data. Fig. 5 shows a comparison between the spectra generated from the mathematical model (above) and the most intense series of the MALDI spectra (below) representing the peaks related to series S1 (+S3), S2 (+S4) and S5. Both spectra show similar MWD for each series in the same mass range. The profile of the series S2 is also similar to the experimental one, and displays the experimental trend of a shift to lower masses compared to S1. The shift to lower masses is to be expected if the peak is the result of chain transfer to solvent because this event results in a dead chain whose mass can no longer increase while the main chain distribution will continue to extend throughout the polymerization due to the living nature of the reaction. This is in contrast to what one would expect if the peak was simply due to loss of the labile RAFT group upon MALDI ionization. The comparison of these spectra reinforce the idea that a significant amount of chain transfer to solvent takes during RAFT polymerization of pBA under these conditions. This also highlights that hydrogen terminated species which result from ionization, can be distinguished from products resulting from chain transfer simply by the looking at the relative mass distributions.45,46
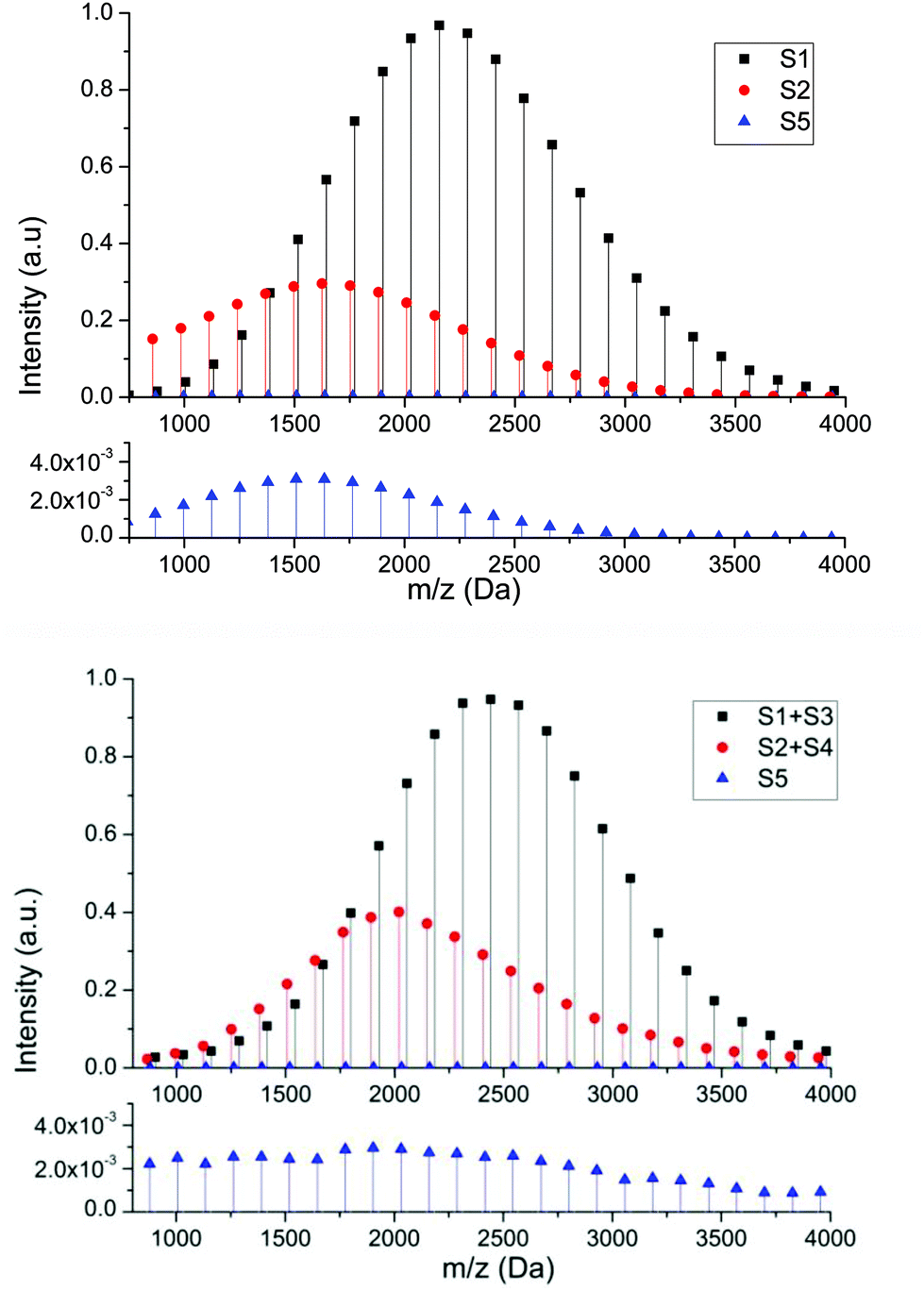 | ||
| Fig. 5 Comparison between the spectra generated from mathematical model (above) and the most intense series of the MALDI spectra (below) for RAFT polymerization of BA in solution of ethanol with TCCA-4 and V-501, representing the three different detected species. In MALDI spectrum the experimental peaks represented as black squares and red circles are the sum of the two adducts for each one. Below each spectrum an enlargement to see S5 behaviour. | ||
Similarly, good agreement between the model and experimental data for the molecular weight distribution for the product of β-scission, S5, can be observed with a greater intensity at lower molecular weights. This higher intensity at lower molecular weights is due to the decrease in molecular weight with respect to the main chain caused by the β-scission reaction, which fragments the polymer, creating a lower molecular weight macromonomer. Although this macromonomer is technically a reactive species,47 due to the lower rate constant for propagation and (mainly) the low concentration the probability of this molecule reacting further is low. Thus, while the main chain distribution is constantly growing, any macromonomer formed tends to remain at lower molecular weight. This tendency becomes more obvious as target chain length increases since the chains grow to a greater extent.
One important conclusion from this analysis is that even at moderate temperatures a significant number of side products can arise that can affect not only the molecular weight distribution but also the livingness of the polymerization. This is particularly important for obtaining high molecular weight polymers and for recently developed processes for synthesis of multiblock copolymers in one-pot systems where conversion is taken to high values between each sequential monomer addition stage.48–50 Formation of multiblocks is only possible if after each extension step (which typically proceeds to high conversion) a high living fraction of chains remains. In RAFT it has been assumed that a high living fraction can be achieved simply by using a low [Initiator]/[RAFT] ratio51,52 but the present results highlight that a substantial fraction of dead chains can arise from alternative reactions.
In order to quantify this effect, the fraction of living chains with RAFT groups attached can be calculated by summing the concentration chains formed from the RAFT agent and of new chains created by transfer to solvent [Cs], β-scission [Cβ] and the concentration of terminated chains according to
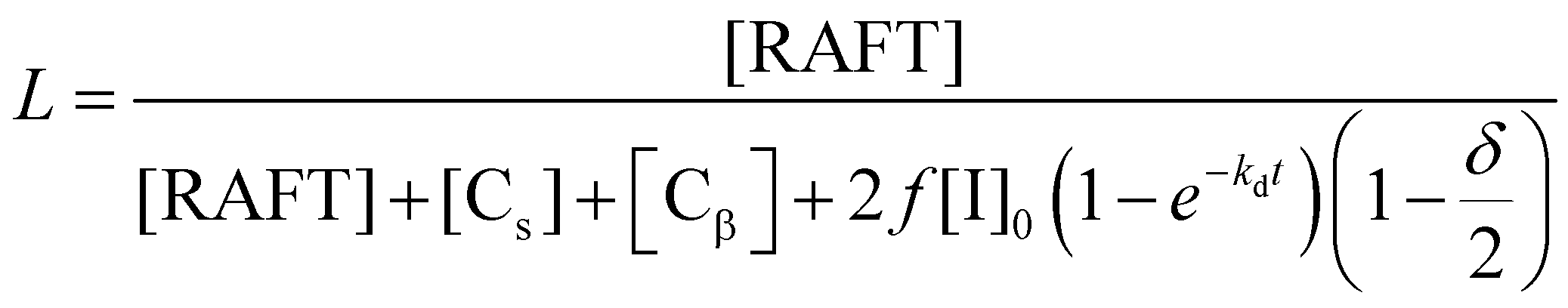
| [Cs] = −Cct[S]ln(1 − X) |
where Cct = kct/kp, [S] is the solvent concentration, Kbb = kbb/kp, Kp3 = kp3/kβ, and kct, kp, kbb, kp3, kβ are the rate coefficients of chain transfer to solvent, propagation, backbiting, propagation of the tertiary radical and β-scission respectively. The derivation of these equations along with that to calculate chain transfer to monomer is given in the ESI.† From this equation it can be seen that if any chain transfer process generates a significant number of chains with respect to the initial RAFT agent concentration then the living fraction of chains will be affected and it follows that the number average degree of polymerization will decrease compared to the ideal case. This is therefore particularly important at high target degrees of polymerization (low [RAFT]0) where small numbers of chains generated will affect the living fraction to a greater extent. This effect applies equally to RAFT as to other reversible deactivation radical polymerization techniques such as NMP and ATRP.
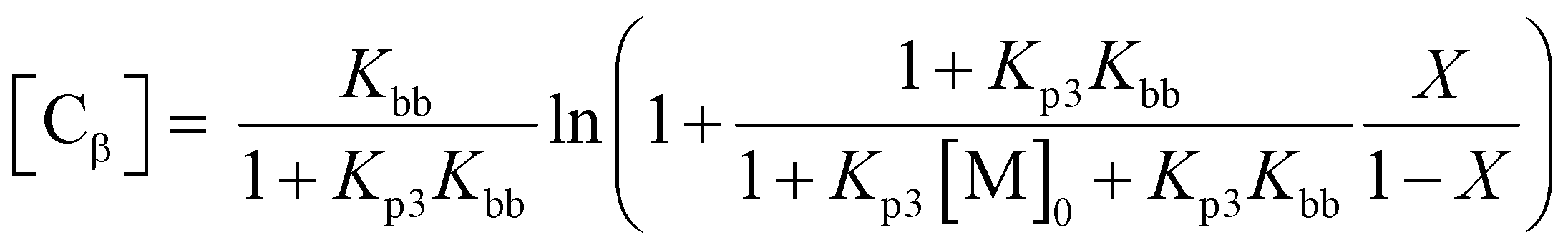
Fig. 6 shows the fraction of dead chains accumulated as a result of chain transfer to solvent for the solution polymerization of butyl acrylate with varying solvents and from the fraction of dead chains arising from β scission reactions of midchain radicals at various temperatures calculated using the parameters given in Table 3S of the ESI.† It can be seen that the process of chain transfer to solvent is less sensitive to changes in conversion than β-scission as may be predicted from the strong effect of monomer concentration on formation of tertiary radicals, and the subsequent scission reaction. With regards to chain transfer to solvent, it can be seen that the solvent choice impacts hugely on the livingness of the polymerization and that even for the short chain lengths obtained with the [M]0/[RAFT]0 ratio used in the simulations, a solvent with very low chain transfer rate coefficient must be used in order to obtain a polymer with high living fraction. For chains arising from β-scission of midchain radicals a large effect of temperature on the number of new chains created can be observed. This is expected due to the high activation energies of backbiting and particularly β-scission and highlights the need to polymerize at low temperatures to avoiding the degradative effects of β-scission.
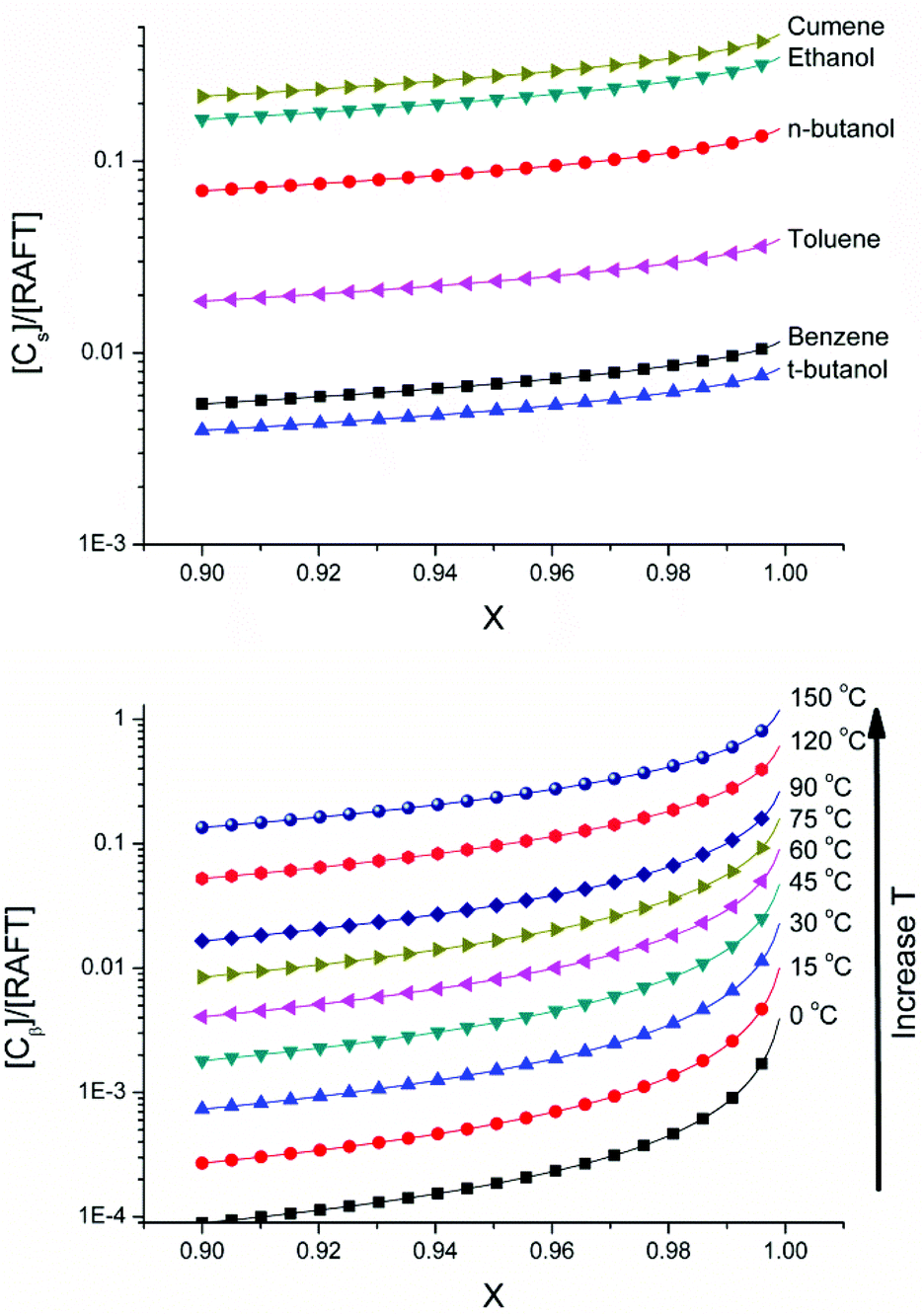 | ||
| Fig. 6 (Above) Modelled effect of solvent and conversion on fraction of dead chains formed by chain transfer to solvent compared to living chains [Cs]/[RAFT]0 for polymerization of butyl acrylate at 60 °C ([M]0 = 1.2 M, ([RAFT]0 = 0.08 M). Values for chain transfer constants were obtained from literature (Cct = kct/kp = 0.17 × 10−4 (t-butanol), 0.22 × 10−4 (benzene), 0.9 × 10−4 (toluene), 2.91 × 10−4 (n-butanol), 4.38 × 10−4 (ethanol), 13.8 × 10−4 (cumene), 3.9 × 10−4 (dioxane)).53,54 (Below) Modelled effect of temperature and conversion on fraction of dead chains formed by β-scission compared to living chains [Cs]/[RAFT]0 for polymerization of butyl acrylate ([M]0 = 1.2 M, ([RAFT]0 = 0.08 M). No adjustment was made for any potential reduction of the rate of backbiting under conditions of reversible deactivation or with hydrogen bonding solvents. | ||
These results emphasize the importance of solvent choice and temperature on the potential for multiblock polymers. To date, such syntheses have only been shown to be successful in solvents with low transfer to solvent rate coefficients such as dimethyl sulfoxide (DMSO) and water. Unfortunately, we could not find any rate coefficients to demonstrate the effect of DMSO in Fig. 6. Furthermore, synthesis of such multiblock copolymers consisting of acrylic monomers, which are highly influenced by chain transfer to polymer events, have been shown to be successful only at low temperatures where backbiting and subsequent scission of the midchain radical can be sufficiently neglected. In this respect the success of the synthesis of a given multiblock copolymer is highly subject to the polymerization conditions.
The results above show that significant deviation from the synthetic conditions that have been reported in the literature (low temperature, low Cct solvents) and the use of monomers which have a greater tendency to undergo chain transfer will result in high fraction of dead chains and limit the potential for multiple chain extension steps. It is also important to note that these chain transfer events will lead to a lower living fraction of polymer chains, and that, for multiblock copolymers, the cumulative effect of chain transfer for each additional block formed will limit the synthetic scope of the procedure if conditions are not selected with great care.
In order to further highlight the importance of avoiding chain transfer and its effects on product distributions, a series of RAFT polymerizations were carried out under conditions where transfer reactions are less favoured. A reaction was conducted at 20 wt% in benzene using identical conditions to those employed for the solution polymerization in ethanol above and led to slightly lower conversion (93%). Fig. 2S† shows the MALDI spectrum for this reaction in which it can be seen that due to the lower conversion, macromonomer formation (species S5) is less prevalent. The use of benzene prevented the formation of any dead chains due to transfer to solvent due to the extremely low transfer constant, so no species S2 was detected. Subsequently, a polymerization was carried out under more “standard” RAFT conditions in dioxane at 70 wt% (MALDI also included in Fig. 2S, ESI†). In this case, chains arising from both transfer to solvent (species S2) and β-scission (species S5) could be observed although at lower concentration than those of the reaction conducted in ethanol due to the higher monomer concentration and lower tendency for dioxane to undergo transfer. Although, in both cases the dead chains were more prevalent at low molecular weight in agreement with the other experiments conducted herein, in dioxane 70 wt% there is higher conversion value and therefore, more β-scission species (S5) are detected. It should also be noted that in both cases the major chain stopping event is not termination. This highlights the importance of both transfer to solvent and transfer to polymer followed by β-scission, which must be considered when accounting for the living fraction of polymers produced by reversible deactivation polymerizations. The MWD values obtained using MALDI and SEC/MALS and the chromatograms are shown in Table 4S and Fig. 3S of the ESI.†
Finally, a reaction was carried out under bulk conditions where both transfer to monomer and intramolecular transfer to polymer are expected to be low. The molar ratio of the reactants was the same as for solution polymerization, but AMBN was used due to the low solubility of V-501 in butyl acrylate. Fig. 7 shows a comparison between the spectra from pBA created in ethanol solution and in bulk. In the complete spectra (left) we can observe fewer peaks in the bulk reaction. In the enlargements of the spectra (right), it is clear that the most intense peaks are due to the RAFT main mechanism, corresponding to species S1 and S3. The species S2 and S4 disappear totally and the intensity of the structure S5 diminishes drastically as expected since transfer to polymer is largely averted and essentially no hydrogen terminated product, which could be formed due to either termination by disproportionation or unavoidable effects of transfer to monomer could be observed.
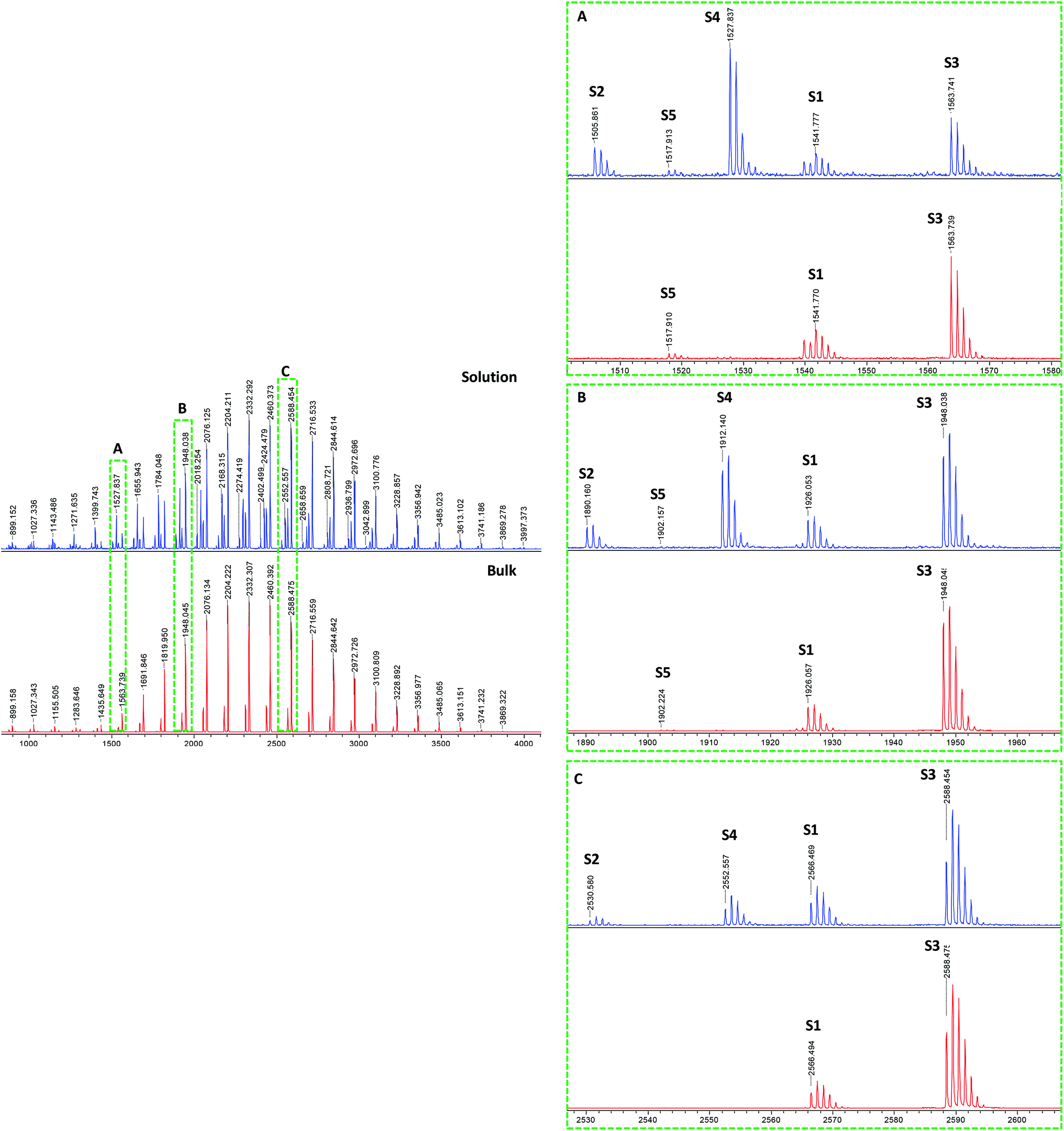 | ||
| Fig. 7 Comparison between MALDI-TOF spectra of the same RAFT reaction in ethanol solvent (98% conversion) and in bulk (99% conversion). Complete spectra in 800–4000 Da mass range (left). Enlargements (right) in different mass ranges (A) 1500–1580, (B) 1880–1960, and (C) 2520–2600 Da show the different detected series from S1 to S5. | ||
In order to confirm that the decrease in hydrogen terminated products arising from chain transfer to solvent (monomer in this case) and terminal double bonds resulting from β-scission of midchain radicals is directly related to the polymerization conditions the theoretical mass distribution was calculated for this reaction using the mathematical model (see equations from ESI†). Fig. 8 shows a comparison between the spectra generated from the mathematical model (above) and the most intense series of the MALDI spectra (below) representing the peaks related to series S1 and S5. In this case, comparison between Fig. 5 and 8 clearly shows that in bulk polymerization, the amount of species due to chain transfer disappear totally. This confirms that transfer to monomer does not have any substantial effect on the molecular weight distribution and confirms that the choice of solvent is critical if high living fraction of chains is necessary. This also further demonstrates that S2 and S4 species were created due to transfer to solvent and not due to the rupture of C–S bond during MALDI ionization process. One point of interest is that the experimental concentration of the S5 species is higher in the case of bulk polymerization than in the solution polymerization. It would be expected that due to the solution polymerization being conducted at lower monomer concentration the reverse would be the case. However, there are several additional factors that this supposition does not take into account. Firstly, from an experimental point of view, although the reaction was performed under conditions designed to minimize any effect arising from the exothermic nature of the reaction in bulk, local temperature rises are inevitable and can lead to increased rate of both backbiting and β scission relative to propagation. Secondly, as the solution polymerization is conducted in ethanol, hydrogen bonding may result in a decrease in the amount of backbiting relative to the bulk polymerization.41,42 Finally, the potential of the midchain radical to undergo chain transfer to solvent for the reaction conducted in ethanol, which has not been considered herein, may also lead to a reduction in the products arising from the midchain radical as has been demonstrated to be important for reactions carried out in the presence high concentrations of chain transfer agent.55,56
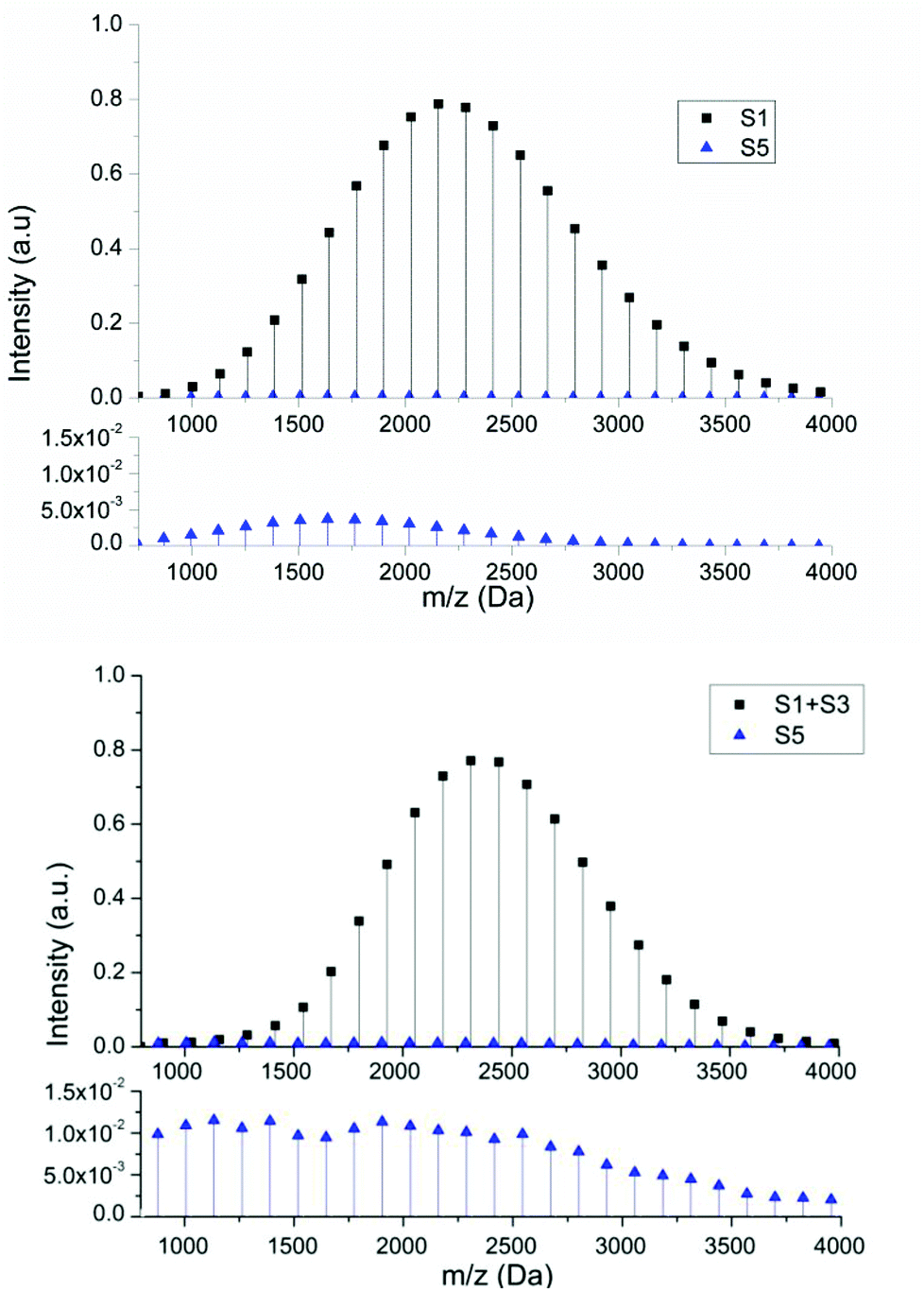 | ||
| Fig. 8 Comparison between the spectra generated from mathematical model (above) and the most intense series of the MALDI spectra (below) for bulk RAFT polymerization butyl acrylate using TCCA-4, representing the three different detected species. In MALDI spectrum the experimental peaks represented as black squares are the sum of the two adducts. In order to see behaviour of S5 species, the enlargements are shown below. | ||
Conclusions
In conclusion, through the synthesis of low molar mass poly(butyl acrylate) by RAFT polymerization under varying conditions and detailed analysis of the resulting polymers by MALDI-TOF we have shown the critical effect of chain transfer events on the living fraction of chains at high conversion in reversible deactivation radical polymerizations. Under conditions where chain transfer is heightened (dilute solution polymerization in ethanol) three products with distinct molecular weight distributions were present in the final reaction mixture. The main population corresponded to polymer chains formed according to the RAFT polymerization mechanism described in Scheme 1. The second population corresponds to dead chains arising from chain transfer to solvent and the third species results from intramolecular transfer to polymer followed by fragmentation of the polymer chain by β-scission. No evidence of detrimental effects of the intermediate RAFT radical leading to 2-, 3-, or 4-arm star products which have been detected by other groups was found. In contrast, polymerization in conditions at which transfer events do not significantly affect the reaction (bulk polymerization at 60 °C) resulted in almost exclusively a single product formed according to the main RAFT equilibrium. Chain transfer to solvent was shown to result in a significant number of dead chains across a wide range of conversion thus limiting the solvent choice for synthesis of multi-block copolymer and high molecular weight polymers where high chain end fidelity is required. Furthermore, from analysis of the products of the β-scission reaction it can be concluded that intramolecular transfer to polymer occurs almost exclusively via a six membered ring transition state and that migration of the midchain radical along the polymer backbone did not occur under the present reaction conditions.Abbreviations
| MALDI | Matrix assisted laser desorption ionization |
| TOF | Time of flight |
| MS | Mass spectrometry |
| MWD | Molar mass distribution |
| SEC/MALS | Size exclusion chromatography/multi angle light scattering |
| BA | Butyl acrylate |
| CRP | Controlled/living polymerization |
| RAFT | Reversible addition–fragmentation chain transfer |
| ATRP | Atom transfer radical polymerization |
| NMP | Nitroxide-mediated controlled free-radical polymerization |
| ITP | Iodine transfer polymerization |
| RITP | Reverse iodine transfer polymerization |
| V-501 | 4,4′-Azobis(4-cyanopentanoic acid) |
| THF | Tetrahydrofuran |
| CHCA | α-Cyano-4-hydroxycinnamic acid |
| TTCA-4 | 2-[(Butylsulfanyl) carbonothioyl sulfanyl] propanoic acid |
| DMSO | Dimethyl sulfoxide |
| AMBN | 2,2′-Azobis(2-methylbutyronitrile |
| NaI | Sodium iodide |
| KTFA | Potassium trifluoroacetate |
| SA | Sinapinic acid |
| MBT | 2-Mercaptobenzothiazole |
| DHB | 2,5-Dihydroxy benzoic acid |
| DCTB | trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile. |
Conflict interest
The authors declare no competing financial interest.Acknowledgements
Diputación Foral de Gipuzkoa, University of Basque Country (UFI 11/56), Basque Government (GVIT373-10), and Ministerio de Economía y Competitividad (CTQ2011-25572) and POLYMAT Industrial Liaison Program (Akzo Nobel, Allnex, Arkema, BASF, Foresa, Nuplex Resins, Solvay, Stahl, Synthomer, Vinavil, and Wacker) are gratefully acknowledged for their financial support. The SGI/IZO-SGIker UPV/EHU is gratefully acknowledged for generous allocation of computational resources.Notes and references
- K. Matyjaszewski, Y. Nakagawa and C. Jasieczek, Macromolecules, 1998, 31, 1535–1541 CrossRef CAS.
- C. J. Hawker, Acc. Chem. Res., 1997, 30, 373–382 CrossRef CAS.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. Le, R. Mayadunne, G. Meijs, C. Moad, G. Moad, E. Rizzardo and S. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, J. Chiefari, Y. Chong, J. Krstina, R. Mayadunne, A. Postma, E. Rizzardo and S. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- M. Dietrich, M. Glassner, T. Gruendling, C. Schmid, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 634–644 RSC.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- E. Rizzardo and D. H. Solomon, Aust. J. Chem., 2012, 65, 945–969 CrossRef CAS.
- D. Keddie, G. Moad, E. Rizzardo and S. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- C. Barner-Kollowik, Handbook of RAFT Polymerization, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, pp. 5–49 Search PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505–2514 Search PubMed.
- P. B. Zetterlund, G. Gody and S. Perrier, Macromol. Theory Simul., 2014, 23, 331–339 CrossRef CAS PubMed.
- K. Matyjazewski, Controlled and Living Radical Polymerizations: Progress in ATRP, NMP and RAFT, American Chemical Society, 1st edn, 2000 Search PubMed.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Macromolecules, 2002, 35, 8300–8306 CrossRef CAS.
- A. Ah Toy, P. Vana, T. P. Davis and C. Barner-Kowollik, Macromolecules, 2004, 37, 744–751 CrossRef.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS PubMed.
- C. Plessis, G. Arzamendi, J. R. Leiza, H. A. S. Schoonbrood, D. Charmot and J. M. Asua, Macromolecules, 2000, 33, 5041–5047 CrossRef CAS.
- A. N. Nikitin, R. A. Hutchinson, G. A. Kalfas, J. R. Richards and C. Bruni, Macromol. Theory Simul., 2009, 18, 247–258 CrossRef CAS PubMed.
- C. Plessis, G. Arzamendi, J. R. Leiza, H. A. S. Schoonbrood, D. Charmot and J. M. Asua, Macromolecules, 2000, 33, 5041–5047 CrossRef CAS.
- R. X. E. Willemse, A. M. van Herk, E. Panchenko, T. Junkers and M. Buback, Macromolecules, 2005, 38, 5098–5103 CrossRef CAS.
- T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7585–7605 CrossRef CAS PubMed.
- C. Schilli, M. G. Lanzendörfer and A. H. E. Müller, Macromolecules, 2002, 35, 6819–6827 CrossRef CAS.
- F. D'Agosto, R. Hughes, M. Charreyre, C. Pichot and R. Gilbert, Macromolecules, 2003, 36, 621–629 CrossRef.
- J. Loiseau, N. Doerr, J. Suau, J. Egraz, M. Llauro and C. Ladaviere, Macromolecules, 2003, 36, 3066–3077 CrossRef CAS.
- A. Favier, C. Ladaviere, M. Charreyre and C. Pichot, Macromolecules, 2004, 37, 2026–2034 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- R. Venkatesh, B. Staal, B. Klumperman and M. Monteiro, Macromolecules, 2004, 37, 7906–7917 CrossRef CAS.
- H. Chirowodza, P. Hartmann and H. Pasch, Macromol. Chem. Phys., 2012, 213, 847–857 CrossRef CAS PubMed.
- M. Jacquin, P. Muller, G. Lizarraga, C. Bauer, H. Cottet and O. Théodoly, Macromolecules, 2007, 40, 2672–2682 CrossRef CAS.
- A. Krieg, C. Pietsch, A. Baungaertel, M. D. Hager, C. R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1669–1676 RSC.
- X. Zhang, F. Boisson, O. Colombani, C. Chassenieux and B. Charleux, Macromolecules, 2014, 47, 51–60 CrossRef CAS.
- C. Ferguson, R. Hughes, D. Nguyen, B. Pham, R. Gilbert, A. Serelis, C. Such and B. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- S. Podzimeck, Light Scattering, Size Exclusion Chromatography and Asymmetric Flow Field Fractionation: Powerful Tools for the Characterization of Polymers, Proteins and Nanoparticles, Wiley, Canada, 2011 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- W. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS PubMed.
- R. Arakawa, S. Egami and S. Okuno, J. Mass Spectrom., 2006, 41, 549–550 CrossRef CAS PubMed.
- X. Jiang, P. Schoenmakers, J. van Dongen, X. Lou, V. Lima and J. Brokken-Zijp, Anal. Chem., 2003, 75, 5517–5524 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Macromolecules, 2012, 45, 6850–6856 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Macromolecules, 2013, 46, 3324–3331 CrossRef CAS.
- K. Liang, R. A. Hutchinson, J. Barth, S. Samrock and M. Buback, Macromolecules, 2011, 44, 5843–5845 CrossRef CAS.
- K. Liang and R. A. Hutchinson, Macromol. Rapid Commun., 2011, 32, 1090–1095 CrossRef CAS PubMed.
- W. Wang, A. N. Nikitin and R. A. Hutchinson, Macromol. Rapid Commun., 2009, 30, 2022–2027 CrossRef CAS PubMed.
- N. Ballard, S. Rusconi, E. Akhmatskaya, D. Sokolovski, J. C. de la Cal and J. M. Asua, Macromolecules, 2014, 47, 6580–6590 CrossRef CAS.
- M. Bednarek, T. Biedron and P. Kubisa, Macromol. Chem. Phys., 2000, 201, 58–66 CrossRef CAS.
- L. Charles, Mass Spectrom. Rev., 2014, 33, 523–543 CrossRef CAS PubMed.
- A. Postma, T. P. Davis, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5307–5318 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Macromolecules, 2014, 47, 5051–5059 CrossRef CAS.
- J. Brandrup and E. H. Immergut, Polymer Handbook, Wiley-Interscience, New York, 4th edn, 1999 Search PubMed.
- J. Loiseau, N. Doërr, J. M. Suau, J. B. Egraz, M. F. Llauro and C. Ladavière, Macromolecules, 2003, 36, 3066–3077 CrossRef CAS.
- M. Gaborieau, S. P. S. Koo, P. Castignolles, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 5492–5495 CrossRef CAS.
- N. Ballard, J. C. de la Cal and J. M. Asua, Macromolecules, 2015, 48, 987–993 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00838g |
| This journal is © The Royal Society of Chemistry 2015 |