DOI:
10.1039/C5PY00365B
(Paper)
Polym. Chem., 2015,
6, 3979-3987
Electrochemical synthesis of polymer microgels†
Received
13th March 2015
, Accepted 13th April 2015
First published on 14th April 2015
Abstract
We describe an electrochemical approach for the synthesis of polymer microgels through polymerization of the monomer in the presence of the crosslinker. This electrochemical approach means initiation by the electron transfer processes which occur at the electrodes, in that by controlling the applied potential it is possible to control the generation of free radicals and/or other reactive species. Upon applying a suitable potential above the electrochemical oxidation waves of N-isopropylacrylamide (as a model of the monomer) and N,N′-methylenebisacrylamide (as a model of the crosslinker), the polymerization and crosslinking are able to proceed to obtain nearly monodisperse polymer microgels with high yield. The apparent rate constant was determined to be 1.69 × 10−2 min−1 based on the evolution of light scattering intensity, or 1.43 × 10−2 min−1 based on the average hydrodynamic diameter. The underlying formation mechanism to reach polymer microgels instead of macrogels, even at high monomer concentrations, is possibly due to the limitation of the primary chain length such that bridging between growing microgel regions can be eliminated. The microgel size can be tuned by varying the applied potential. The reaction medium can be recycled, and reused directly without a notable impact on the next cycle of synthesis. This electrochemical approach can be extended to synthesize microgels of poly(acrylamide) or poly(acrylic acid) (as the additional models).
Introduction
Polymer microgels, colloidal particles having a gel structure internally, have been receiving a great deal of attention due to their unique properties that combine colloids with gels.1–5 Current strategies for the synthesis of polymer microgels can be divided into: (i) polymerization of monomers in a homogeneous phase or in a micro- or nanoscale heterogeneous environment; (ii) cross-linking of preformed polymers; (iii) physical self-assembly of interactive polymers; and (iv) template-assisted micro- or nanofabrication.6–11 In particular, the chemical synthesis through polymerization of monomers can provide general opportunities to vary the size and structure of the polymer microgels. The versatility of this strategy has led to the development of a range of polymer microgels with diverse physical, chemical, and biological properties (e.g., functionality and responsiveness) for prospective applications in many fields, such as sensing, catalysis, and biomedicine.12–14
Previous work on the chemical synthesis of polymer microgels through polymerization of monomers has largely focused on radical polymerization, including conventional free radical and controlled/“living” radical polymerizations.6–10 Despite the remarkable breakthroughs, in those polymerization methods the radicals are usually provided by chemical additives as initiators (the feeding molar ratio of initiators/monomers is typically 0.01–0.39) that cannot be recycled; if metal compounds are used as initiators (or activators), the reactions would lead to metal waste products, and likely purification issues as well (because polymer microgels are known for metal extraction).15–17 These limitations highlight the need for development of alternative robust and benign approaches for initiating polymerization of monomers to generate polymer microgels. Radiation provides such a possibility.11 Since the first radiochemical synthesis of polymer microgels by Schnabel and coworkers in 1969,18 various polymer microgels have been synthesized by irradiation in dilute solutions.11,19,20 One of the advantages, among others, of radiochemical synthesis of polymer microgels is no necessity to add any chemical additives as initiators. Unfortunately, the high-energy radiation could also split covalent bonds, which might lead to degradation of the formed polymers into low molecular weight fragments, resulting in a relatively large polydispersity of the soft products. Moreover, in comparison with other laboratory approaches in polymer chemistry, radiochemical synthesis seems to be expensive, because of the requirements in the facility like 60Co γ-plants, electron accelerators or some of the radiation sources. Thus, development of robust/benign approaches for chemical synthesis of polymer microgels through polymerization of monomers still remains a considerable challenge.
In this work, we report an electrochemical approach for the synthesis of polymer microgels. Electrochemistry is long-known to provide a “green” approach for exploring synthetically useful reactions, because it can enable the selective manipulation of molecular oxidation states and the generation of highly reactive species.21,22 In appropriate systems, such highly reactive species can attack monomers and transform them into so-called “active centers”, which will then initiate chain growth processes, so that the production of polymers can be accomplished. Moreover, this approach has the special appeal of a nearly perfect controllability through varying the applied adjustable parameters (e.g., potential and current), making it an interesting approach for polymer synthesis.23–27 Recently, the electrochemical-initiated polymerization was exploited for the production of thin hydrogel layers on conductive surfaces.28–34 While electrochemical-initiated polymerization was recognized in the 1940s and has undergone a fulminating development since then, surprisingly rare attention has been paid to the electrochemical synthesis of polymer microgels. Some electrochemical syntheses of polymers might also be referred to the formation of colloidal particles,35–37 which, however, do not have a gel structure internally and thus should not belong to polymer microgels.
Here, we commence our study using electrochemical synthesis of poly(N-isopropylacrylamide) (PNIPAM) microgels, one of the golden standards of temperature-responsive polymer microgels,1,2 as a first example. The synthesis can be performed in a three-neck round-bottom flask equipped with a commonly-used three-electrode electrochemical system (Fig. 1). We show that nearly monodisperse microgels can be produced with a high yield, and the size of the microgels can be tuned facilely via varying the applied potential. After the synthesis, the reaction medium can be recycled and reused directly without a notable impact on the next cycle of synthesis. To our delight, this electrochemical approach can be extended to the synthesis of other polymer microgels, such as poly(acrylamide) (PAAm) microgels and poly(acrylic acid) (PAA) microgels that are difficult to be reduced from micron in size to sub-micron and even nanoscale directly through polymerization of the corresponding monomers using other approaches.38–40
 |
| | Fig. 1 A typical setup for the electrochemical synthesis of polymer microgels. | |
Experimental
Materials
All chemicals were purchased from Aldrich. N-Isopropylacrylamide (NIPAM) was recrystallized from a hexane–acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) mixture and dried under vacuum. Acrylic acid (AA) was purified by distillation under reduced pressure. Acrylamide (AAm), N,N′-methylenebisacrylamide (MBAAm), acetonitrile (CH3CN), and sodium perchlorate (NaClO4) were used as received without further purification. The water used in all experiments was of Millipore Milli-Q grade.
:1 by volume) mixture and dried under vacuum. Acrylic acid (AA) was purified by distillation under reduced pressure. Acrylamide (AAm), N,N′-methylenebisacrylamide (MBAAm), acetonitrile (CH3CN), and sodium perchlorate (NaClO4) were used as received without further purification. The water used in all experiments was of Millipore Milli-Q grade.
Microgel synthesis
The monomer (i.e., NIPAM, AAm, or AA; 1.0 × 10−3 mol) and the crosslinker MBAAm (5.0 × 10−5 mol) were dissolved in the reaction medium (composed of 0.1 M NaClO4 in CH3CN; 20.0 mL) in a 50 mL three-neck round-bottom flask equipped with a stirrer, a N2 gas inlet, and a three-electrode system. The working electrode was a Pt plate, and the counter electrode consisted of a Pt wire. After purging with N2 for 3 h, the electrochemical polymerization was performed at room temperature (∼22 °C) and at a preconcerted potential versus Ag/AgCl using a potentiostat of a CHI660E electrochemical workstation. The potential of the working electrode was measured by reference to Ag/Ag+. The polymerization was allowed to proceed for 3 h. Then the dispersion was centrifuged twice (20000 rpm, 20 min, 25 °C) with the supernatant discarded (or reused directly for the next cycle of synthesis) and the precipitate redispersed in water. The product was further purified by 3 days of dialysis against water at room temperature. The product was finally freeze-dried, and the yield was calculated by the weight of the product against the ideal weight assuming 100% conversion (i.e., 120.9, 78.8, and 79.8 mg, respectively, for PNIPAM, PAAm, and PAA microgels).
Laser light scattering (LLS) studies
A standard laser light scattering spectrometer (BI-200SM) equipped with a BI-9000 AT digital time correlator (Brookhaven Instruments, Inc.) and a Mini-L30 diode laser (30 mW, 637 nm) as the light source was used. The very dilute microgel dispersions (10.0 μg mL−1; pH = 6.8) were passed through Millipore Millex-HV filters with a pore size of 0.80 μm to remove dust before measurements. In Dynamic LLS (DLS), the Laplace inversion of each measured intensity–intensity time correlation function in a dilute dispersion can lead to a line-width distribution G(Γ). For a purely diffusive relaxation, Γ is related to the translational diffusion coefficient D by (Γ/q2)C→0,q→0 = D, so that G(Γ) can be converted to a translational diffusion coefficient distribution and a <Dh> distribution using the Stokes–Einstein equation, <Dh> = (kBT/3πη)/D, where kB, T, and η are the Boltzmann constant, the absolute temperature, and the solvent viscosity, respectively.41
Other characterization
pH values were measured on a Mettler Toledo SevenEasy pH meter. IR spectra were recorded on a Thermo Electron Corporation Nicolet 380 Fourier transform infrared spectrometer. TEM images were taken on a JEOL JEM-2100 transmission electron microscope at an accelerating voltage of 200 kV.
Results and discussion
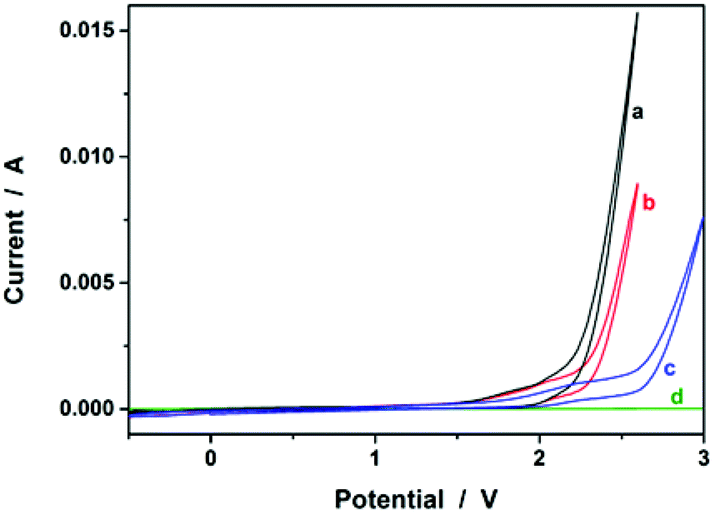
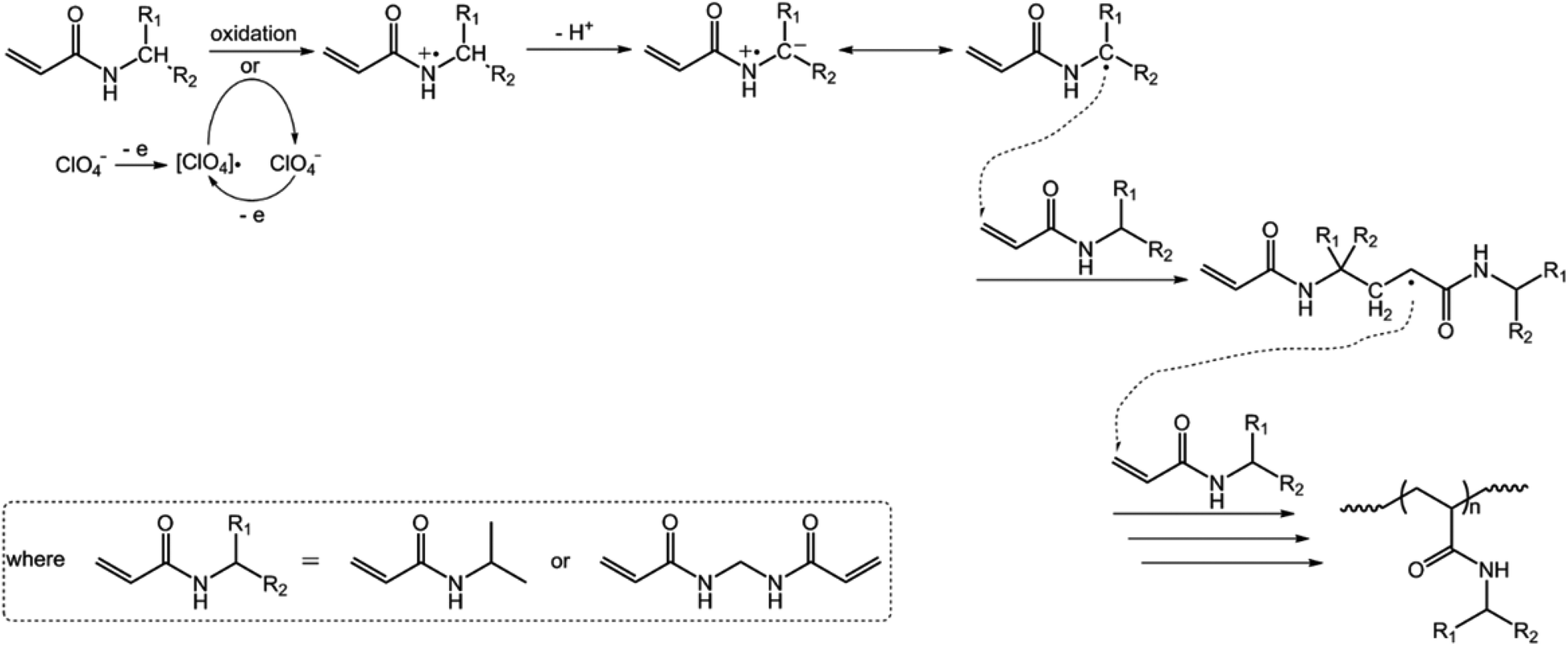
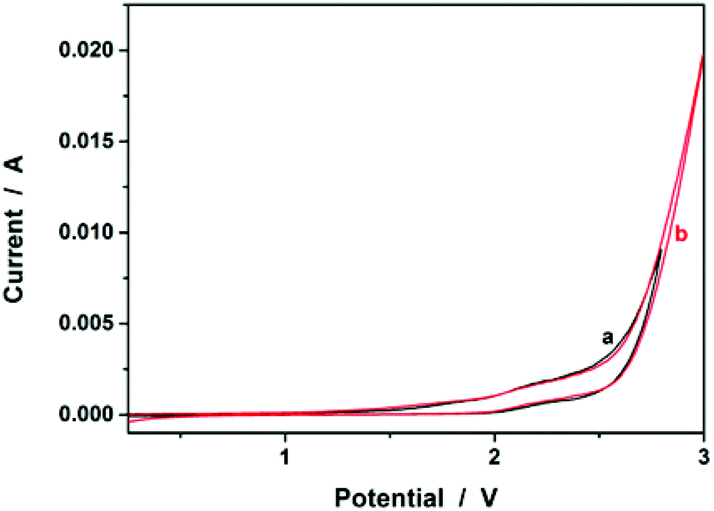
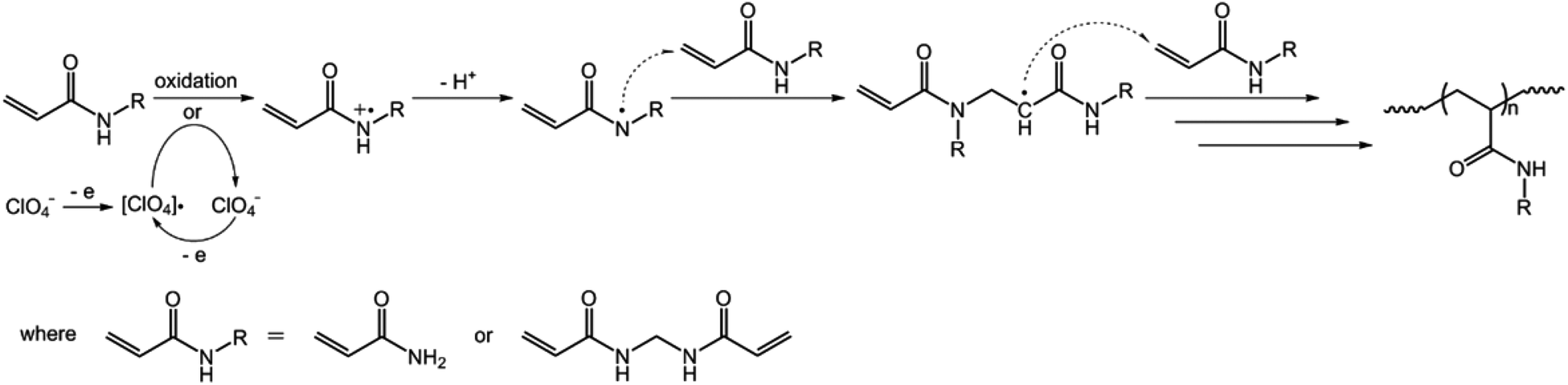
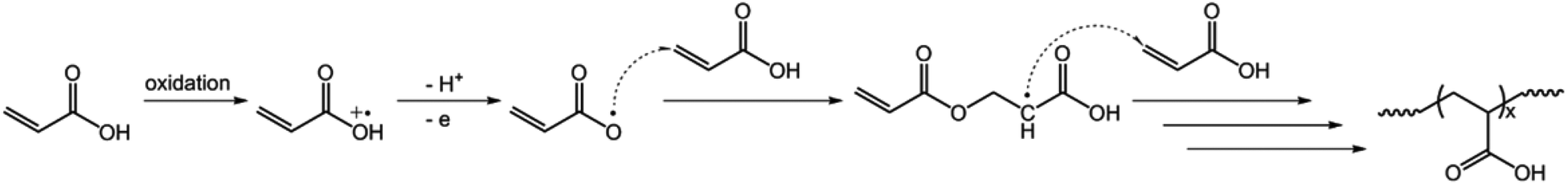
We started our study by electrochemical synthesis of PNIPAM microgels as a first example. Before the synthesis, we took a glance at some key electrochemical properties of the monomer and the crosslinker by scanning the cyclic voltammogram, a specific type of voltammetry that is used for exploring the redox properties of chemicals.42 To obtain the cyclic voltammogram, the voltage is varied in a solution, and the change in current is measured with respect to the change in voltage. Fig. 2 shows the typical cyclic voltammograms of NIPAM (1.0 × 10−3 mol) and MBAAm (5.0 × 10−5 mol) in the reaction medium (a solution of 0.1 M NaClO4 in CH3CN) at room temperature. On the anodic scan, an oxidation reaction starts to develop on NIPAM and MBAAm at about +1.5 V and +1.6 V versus Ag/AgCl, respectively. The suggested mechanism for the anodic oxidation involves the initial formation of the N-localized radicals directly.43–47 Moreover, the oxidation reaction of NaClO4, which was initialized at about +1.7 V versus Ag/AgCl, can be associated with the formation of [ClO4]˙ radicals, which could also attack the monomer/crosslinker to form the N-localized radicals.23 Those N-localized radicals can trigger a cascade of reactions that typically involve the loss of a proton and the formation of C-localized radicals, which would then initiate polymerization on attacking the vinyl groups of the monomer/crosslinker, making them enter into the polymerization process immediately.23,24 Under these circumstances, the polymerization should be able to proceed if applying a suitable potential above the electrochemical oxidation waves. The electrochemical-initiated polymerization mechanism is depicted in Scheme 1.
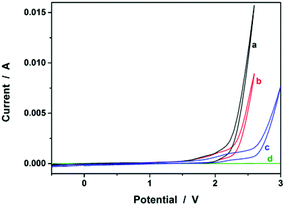 |
| | Fig. 2 Typical cyclic voltammograms of (a) NIPAM (1.0 × 10−3 mol) and (b) MBAAm (5.0 × 10−5 mol) in the reaction medium (a solution of 0.1 M NaClO4 in CH3CN; 20.0 mL). The results of (c) the reaction medium and (d) pure CH3CN are also given for comparison. | |
 |
| | Scheme 1 A proposed electrochemical-initiated polymerization mechanism for the electrochemical synthesis of PNIPAM microgels. | |
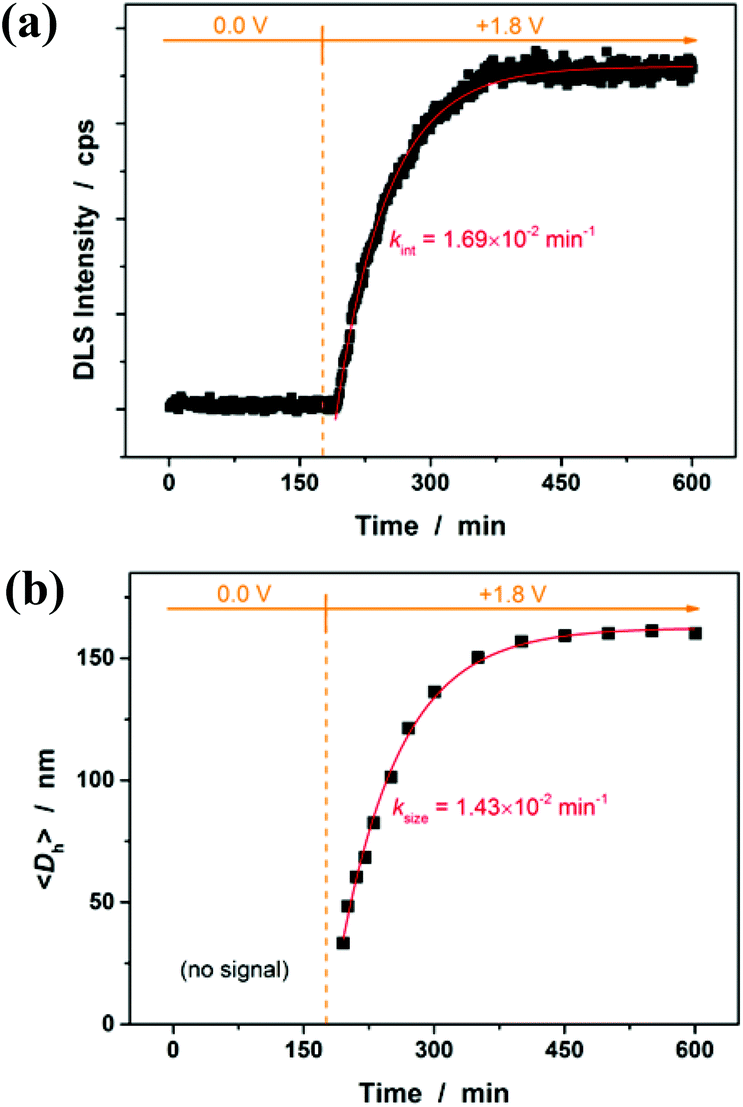
Then, the microgel synthesis was conducted using a potentiostat at +1.8 V versus Ag/AgCl and at room temperature. The whole synthesis process was monitored using in situ DLS. As shown in Fig. 3a, two stages can be observed after addition of the monomer and the crosslinker to the reaction medium: (i) DLS intensity remained nearly the same 3 h before applying the potential, as well as the first ca. 10 min after applying the potential, and then (ii) increased gradually and reached stability within the reaction time of 3 h. According to the light scattering theory41,48 and the formation mechanism proposed for polymer microgels that were synthesized with chemicals as initiators,6–10 our observation is interpreted as follows: without applying the potential, the formation of polymer particles can be readily ignored; after the radical polymerization was electrochemically initiated by applying the potential, the copolymer fragments of NIPAM and MBAAm would be formed at the early stage of reaction to yield very tiny nuclei; with the polymerization reaction proceeding, more copolymer fragments were added to the initially formed nuclei, leading to a continuous growth in size of the gel particles until the reaction was completed.6,9,49–51 Based on the evolution of DLS intensity, the apparent rate constant kint of 1.69 × 10−2 min−1 was derived from the fitting of the second-stage of the time-dependent DLS intensity with an exponential growth. The kinetic evolution was further substantiated by in situ DLS measurement of the average hydrodynamic diameter, <Dh>, versus reaction time (ksize = 1.43 × 10−2 min−1; Fig. 3b). At the end of the polymerization, the light-blue colour (owing to the Tyndall effect) was observed in the dispersion. In IR spectra (see Fig. S1 in the ESI†), the characteristic bands of C–H vibrations of –CH(CH3)2 at 1385 cm−1 and 1367 cm−1 of NIPAM units were recorded, which confirmed the structure of the purified microgels. TEM images shown in Fig. 4 display a typically spherical shape of the obtained PNIPAM microgels. A remark has to be made concerning the particle size measured from TEM images: the particle size measured by this technique is somewhat unreliable as the microgels may have a tendency to shrink, flatten and spread on the TEM grid during the sample preparation. This can also lead to a larger apparent polydispersity; individual microgels are not expected to interact with the substrate in a homogeneous fashion.52 It is for these reasons that we perform particle sizing via DLS, a less perturbing method.
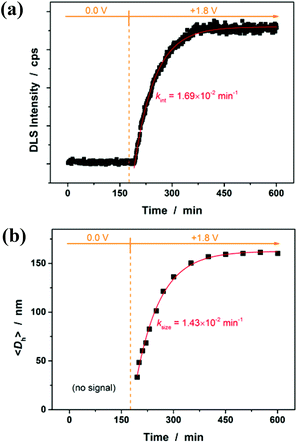 |
| | Fig. 3 Change in (a) the DLS intensity in the reaction medium and (b) the average hydrodynamic diameter, <Dh>, of PNIPAM microgels with polymerization time. Solid lines: 1st-order kinetic fits. | |
 |
| | Fig. 4 Typical TEM images of PNIPAM microgels harvested after 3 hours’ reaction. | |
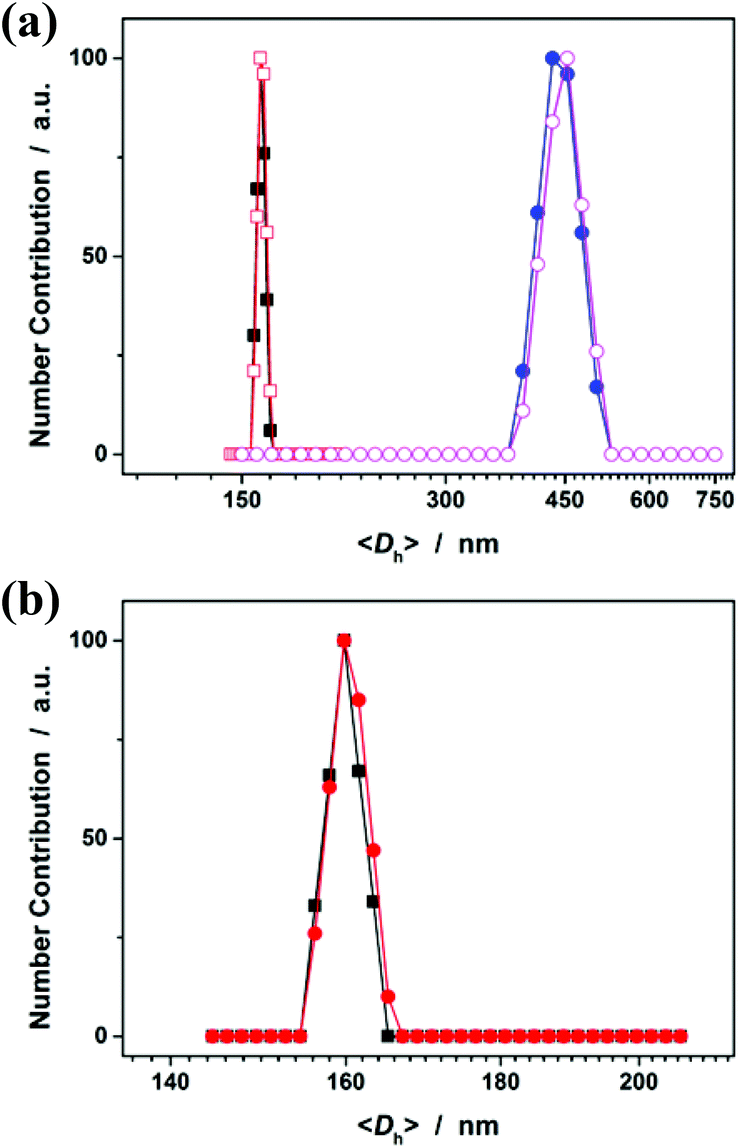
Fig. 5a shows DLS size distribution of PNIPAM microgels harvested after 3 hours’ reaction. Only a single peak with a narrow size distribution range from 156 to 165 nm is observed and the <Dh> was determined to be 160 nm (see Fig. S2 in the ESI† for a screenshot). The microgels can be well reproducible from batch to batch, with a considerably high yield of ≥98%. The microgels can undergo temperature-responsive volume phase transitions (Fig. S3 in the ESI†), similar to those microgels reported in previous studies that were synthesized using persulfates as initiators.1 Meanwhile, an experiment (serve as a control) was also conducted following the procedures for the synthesis of the presented PNIPAM microgels, except that the crosslinker MBAAm was not added. Surprisingly, polymer particles (we denoted these polymer particles as PNIPAM particles, so as to distinguish between them and the ones in the presence of MBAAm) can also be obtained. From Fig. 5a, it can be seen that the <Dh> of PNIPAM particles (442 nm) is much larger than that of PNIPAM microgels, suggesting that the presence of the crosslinker MBAAm in the synthesis further solidifies and stabilizes the microgels; this is also supported by the fact that after the dialysis against water, the light-blue solution was retained for PNIPAM microgels (these microgels showed good stability, with the size distribution kept nearly the same before and after dialysis, or even before and after 2 months’ storage at room temperature; Fig. 5b), while the solution would became completely colorless for PNIPAM particles. If the reaction time was prolonged, no remarkable change in the <Dh> of the final product was observed for both PNIPAM microgels and PNIPAM particles (Fig. 5a). It should be noted that no macroscopic gelation occurred in all experiments, standing in vivid contrast against the synthesis with the same monomer/crosslinker concentration via conventional free radical polymerization wherein macroscopic gelation occurs under the same conditions. This is one of the advantages of the electrochemical approach developed herein.
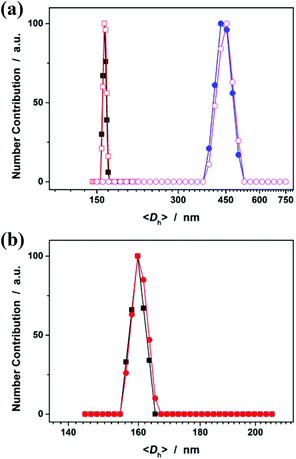 |
| | Fig. 5 (a) DLS size distribution of PNIPAM microgels (■, □; in the presence of MBAAm in the synthesis) and PNIPAM particles (●, ○; in the absence of MBAAm in the synthesis; as a control) harvested after 3 hours’ (solid symbols) or 24 hours’ (open symbols) reaction, and before being purified by dialysis. (b) The size distribution of PNIPAM microgels harvested after 3 hours’ reaction and further being purified by 3 days’ dialysis (■), and after 2 months’ storage (●). | |
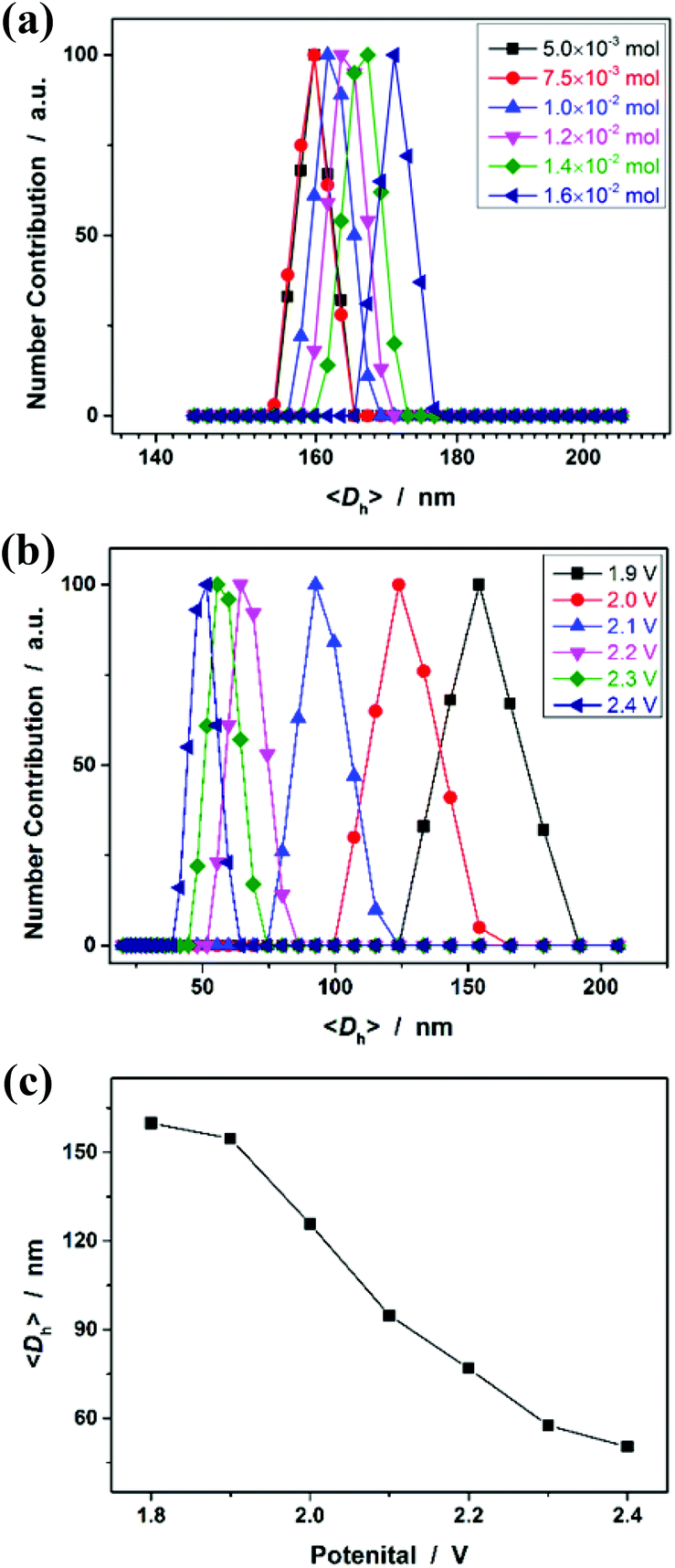
As polymer networks, which are limited in micro-/nanoscale size, are targeted to enable the harvest of microgels, a further challenge lies in understanding fundamentally the experiments that allow avoiding the formation of macroscopic networks, i.e., macroscopic gelation. Targeting the chemical synthesis of microgels through polymerization of monomers, the main approaches reported previously in the literature rely on the control of the distance between growing polymer chains,53 which typically is achieved by two conditions: (a) conducting the experiments in a highly diluted homogeneous solution (i.e., decreasing the monomer concentration; in order to limit intermolecular crosslinking and increasing the probability of intramolecular crosslinking), or (b) applying heterogeneous polymerization processes (in dispersed systems, such as emulsion, miniemulsion, microemulsion, precipitation, stabilizer-aided dispersion, and suspension systems) where the polymerization is performed in a confined nanometric/micrometric space (in order to confine the crosslinking to intraparticle rather than interparticle).6–11 As for our cases, however, our observations cannot be explained perfectly by this principle, since our experiments can be readily extended to the reaction systems containing quite high monomer concentrations (Fig. 6a), and no precipitation was observed in all those systems without the aid of stabilizers. Alternatively, we note that Stansbury and co-workers reported the chemical synthesis of polymer microgels, at high monomer concentrations, based on the use of substantial amounts of chain transfer agents, which are likely to be required to controllably limit the length of the propagating chains such that bridging between growing microgel regions can be eliminated.54 Inspired by this work, it is speculated that the electrochemical synthesis to obtain microgels instead of macrogels, even at high monomer concentrations, might rely on limitation of the primary chain length. The electrochemical process may generate a high concentration of radical intermediates during micro-gelation and thus efficiently induce intramolecular crosslinking and the termination of polymerization, i.e., a coupling reaction of microgels, to inhibit the propagation of linear polymer chains. As it appears that limitation of the primary chain length could be achieved using large amounts of chemical additives as the chain transfer agents or initiators,54–56 a change in the applied potential should be able to vary the size of PNIPAM microgels; it is reasonable that the radical concentration will increase at elevated potentials.21,22Fig. 6b shows the DLS size distribution of the microgels harvested after 3 hours’ reaction at different applied potentials. Indeed, the higher the potential, the smaller is the <Dh> of the microgels. Therefore, these results can not only provide direct experimental proof for the hypothesis for the formation of the microgels, but also foreshadow a facile route to tune the size of the microgels.
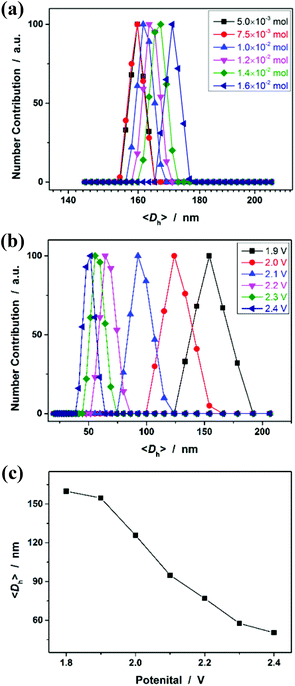 |
| | Fig. 6 (a) DLS size distribution of PNIPAM microgels synthesized with different feeding amounts of NIPAM, where the mole ratio of NIPAM/MBAAm was fixed at 1/0.05. (b) DLS size distribution of the microgels synthesized with different applied potentials, where the feeding amounts of NIPAM (1.0 × 10−3 mol) and MBAAm (5.0 × 10−5 mol) were fixed. (c) The <Dh> as a function of the applied potential. All microgels were harvested after 3 hours’ reaction. | |
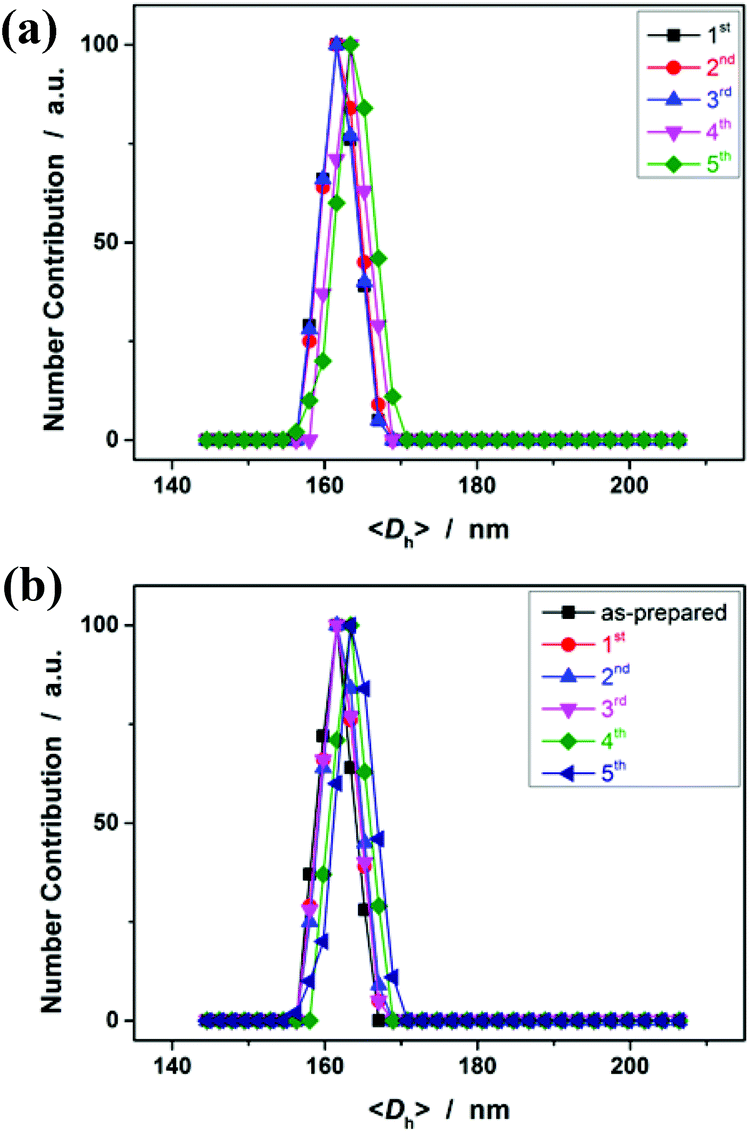
Having demonstrated the feasibility of electrochemical synthesis of PNIPAM microgels, we then examined the reusability of the reaction medium. After full conversion of the monomers within the reaction time of 3 h, the reaction medium was recovered simply by centrifugation, and was reused for the next cycle of synthesis where only the monomer and the crosslinker were added (no more NaClO4 or CH3CN was added). The recycling experiments clearly revealed the robustness of the approach (conducted at +1.8 V versus Ag/AgCl for 3 h; Fig. 7a). In more than 5 recycling experiments, no significant change of the size (both the <Dh> and DLS size distribution) of the obtained microgels was observed, due to the fact that the concentration of the formed C-localized radicals and thus the size of the microgels mainly depended on the reactant concentration (Fig. 6a) and the applied potential (Fig. 6b), as was discussed above. While the reaction of the [ClO4]˙ radicals with the monomer/crosslinker might also contribute to the formation of C-localized radicals, it should not be a prerequisite in the synthesis (see Fig. S4 in the ESI† for cyclic voltammograms of NIPAM and MBAAm in CH3CN). The role of NaClO4 is possibly to improve the conductivity of the reaction medium. In another series of recycling experiments with pure CH3CN as the reaction medium, we found that the obtained microgels (<Dh> ≈ 162 nm; see Fig. 7b) have nearly the same size as those synthesized in the presence of NaClO4 (Fig. 7a). Therefore, the electrochemical approach is easy to handle and facilitates robust stability for the synthesis of polymer microgels.
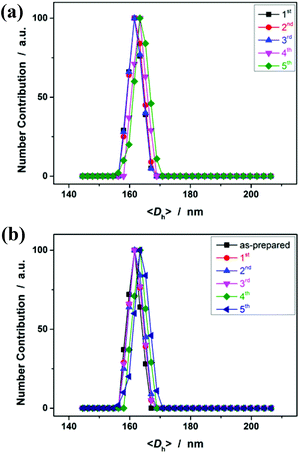 |
| | Fig. 7 (a) A comparison of DLS size distribution of PNIPAM microgels during five cycles of synthesis using the recycled reaction medium. (b) DLS size distribution of PNIPAM microgels synthesized in the absence of NaClO4. All microgels were harvested after 3 hours’ reaction. | |
In the next step, we conducted additionally two series of experiments, using AAm and AA, the two commonly used monomers, as examples, to demonstrate the feasibility of extending the presented electrochemical approach for synthesizing microgels of other polymers. It is known that AAm, AA and their corresponding polymers are usually referred to be highly hydrophilic. Although several methods have been exploited to synthesize polymer microgels of such highly hydrophilic polymers, the microgels reported previously in the literature are typically synthesized based on the preformed polymers.38–40,57–59 To the best of our knowledge, sub-micron or even nanoscale sized microgels of those polymers have not been synthesized through polymerization of the corresponding monomers directly. As discussed above, the electrochemical approach for the synthesis of polymer microgels essentially means initiation by the electron transfer processes which occur at the electrodes, in that by controlling the applied potential it is possible to control the generation of initiating species. In this respect, in principle, this approach should be general.
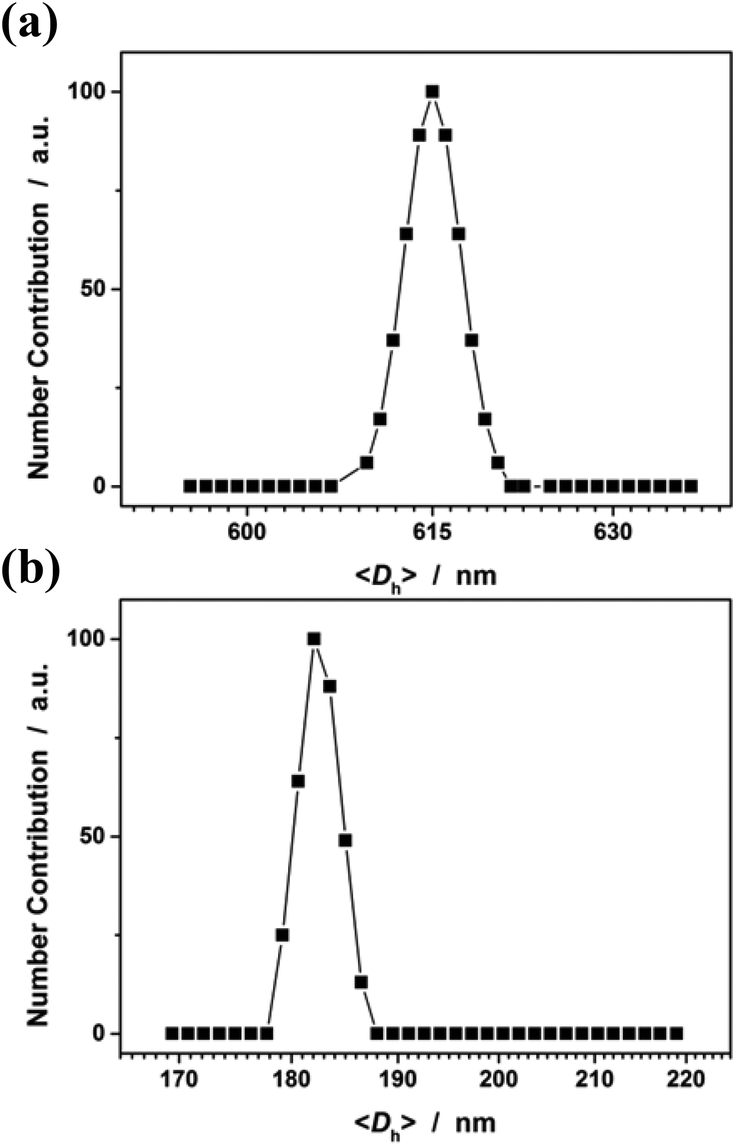
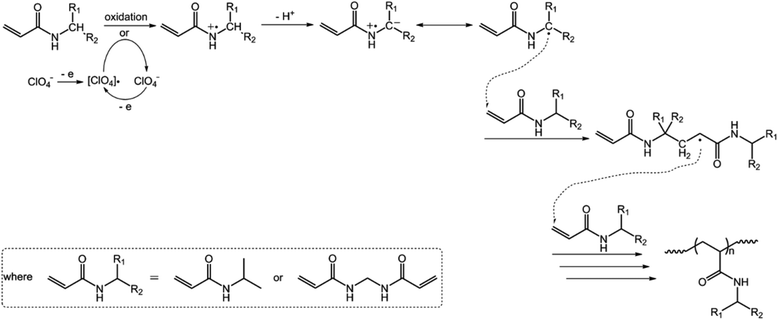
In the first series of experiments, we would like to synthesize PAAm microgels. Similarly to the anodic oxidation of NIPAM, if applying a suitable potential above the electrochemical oxidation waves (the oxidation reaction starts to develop on AAm at about +1.4 V versus Ag/AgCl; Fig. 8), the anodic oxidation of AAm would lead to the initial formation directly of the N-localized radicals;43–47 whilst, in contrast to the N-localized radicals of NIPAM, the N-localized radicals of AAm could initiate polymerization on attacking the vinyl groups of the reactants immediately.23,24 The electrochemical-initiated polymerization mechanism is depicted in Scheme 2. We thus anticipate that it is possible to synthesize PAAm microgels following the presented electrochemical approach. The synthesis was conducted using a potentiostat at +2.3 V versus Ag/AgCl. As expected, microgels of spherical shape (Fig. 9) can be obtained. In DLS (Fig. 10; see Fig. S5 in the ESI† for a screenshot), only a single peak with a narrow size distribution (the polydispersity index μ2/<Γ>2 ≤ 0.005) is observed and the <Dh> was determined to be 615 nm. The structure of the purified microgels was also confirmed by IR analysis (Fig. S6 in the ESI†), where the characteristic bands of amide I at 1667 cm−1 and amide II at 1617 cm−1 of AAm units were recorded. Moreover, PAAm microgels of smaller sizes were synthesized at higher applied potentials (+2.5 and +2.6 V; see Fig. S7 in the ESI† for TEM images, and Fig. S8† for DLS data).
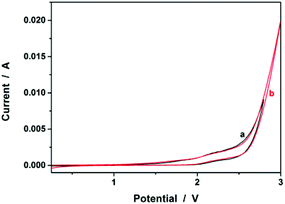 |
| | Fig. 8 Typical cyclic voltammograms of (a) AAm (1.0 × 10−3 mol) and (b) AA (1.0 × 10−3 mol) in the reaction medium (a solution of 0.1 M NaClO4 in CH3CN). | |
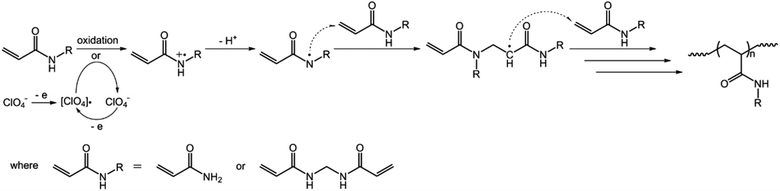 |
| | Scheme 2 A proposed electrochemical-initiated polymerization mechanism for the electrochemical synthesis of PAAm microgels. | |
 |
| | Fig. 9 TEM images of (a, b) PAAm microgels, and (c, d) PAA microgels. All microgels were synthesized using a potentiostat at +2.3 V versus Ag/AgCl, and harvested after 3 hours’ reaction. | |
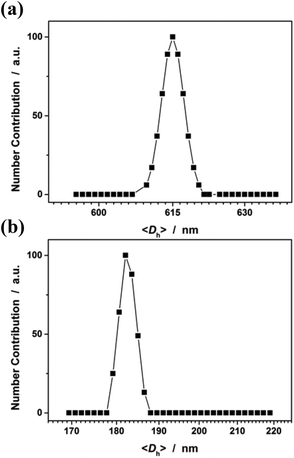 |
| | Fig. 10 DLS size distribution of (a) PAAm microgels and (b) PAA microgels. All microgels were synthesized using a potentiostat at +2.3 V versus Ag/AgCl, and harvested after 3 hours’ reaction. | |
In the second experiment, we synthesized PAA microgels directly through polymerization of the AA monomer in the presence of the crosslinker MBAAm. The synthesis was conducted using a potentiostat at +2.3 V versus Ag/AgCl, a suitable potential above the electrochemical oxidation waves (the oxidation reaction starts developing on AA at about +1.5 V versus Ag/AgCl; Fig. 8). The electrochemical-initiated polymerization of AA was taken because of the well-known Kolbe electrolysis reaction.21,22 In the Kolbe electrolysis, the carboxylic acid is oxidized to form O-localized radicals. As schematically depicted in Scheme 3, the O-localized radicals could initiate polymerization on attacking the vinyl groups of the monomer/crosslinker, making the system enter into the polymerization process.23,24 TEM images in Fig. 9c and d show a spherical shape of the microgels harvested after 3 hours’ reaction. In Fig. 10b (see Fig. S9 in the ESI† for a screenshot), DLS size distribution shows only a single peak for the narrowly distributed microgels, with the polydispersity index μ2/<Γ>2 ≤ 0.005 and <Dh> ≈ 182 nm. The structure of the purified microgels was confirmed by IR analysis (Fig. S10 in the ESI†), where the characteristic bands of the carboxylic group at 1721 cm−1 of AA units were recorded. Therefore, these results provide further support for the feasibility of extending the presented electrochemical approach for synthesizing microgels of polymers other than PNIPAM.
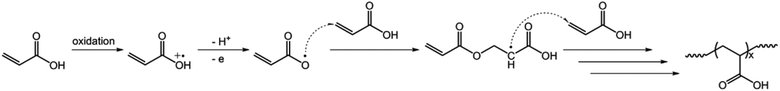 |
| | Scheme 3 A proposed mechanism for electrochemical polymerization of AA in the formation of PAA microgels. | |
Conclusions
We reported the electrochemical synthesis of nearly monodisperse PNIPAM microgels via polymerization of the NIPAM monomer in the presence of the crosslinker MBAAm, in a three-neck round-bottom flask equipped with a three-electrode system. Such an electrochemical approach essentially means initiation by the electron transfer processes that occur at the electrodes upon applying a suitable potential above the electrochemical oxidation waves of the reactants, in that by controlling the applied potential it is possible to control the generation of free radicals and/or other reactive species. The apparent rate constant was determined to be 1.69 × 10−2 min−1 on the basis of the evolution of DLS intensity or 1.43 × 10−2 min−1 based on the <Dh>. The underlying formation mechanism for the electrochemical-initiated polymerization and crosslinking reactions to reach microgels instead of macrogels, even at high monomer concentrations (as high as 0.8 mol L−1), is possibly due to the limitation of the primary chain length such that bridging between growing microgel regions can be eliminated. The microgel size can be tuned by varying the applied potential, with the smaller size being achieved at the higher applied potential. The reaction medium can be recycled and reused directly without a notable impact on the next cycle of synthesis. Moreover, this electrochemical approach can be extended to synthesize microgels of other polymers, such as poly(acrylamide) and poly(acrylic acid) (as the additional models) that are difficult to be reduced to sub-micron or even nano in size directly through polymerization of the corresponding monomers by other approaches. It is anticipated that this may open up exciting new possibilities for developing novel functional polymer microgels with huge potential in a wide range of fields.
Notes and references
- C. Wu and S. Q. Zhou, Macromolecules, 1997, 30, 574 CrossRef CAS.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1 CrossRef CAS.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174 CrossRef CAS PubMed.
- A. Döring, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391 RSC.
- S. Saxena, C. E. Hansen and L. A. Lyon, Acc. Chem. Res., 2014, 47, 2426 CrossRef CAS PubMed.
- G. Z. Zhang, A. Z. Niu, S. F. Peng, M. Jiang, Y. F. Tu, M. Li and C. Wu, Acc. Chem. Res., 2001, 34, 249 CrossRef CAS PubMed.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747 CrossRef CAS PubMed.
- A. Pich and W. Richtering, Adv. Polym. Sci., 2010, 234, 1 CrossRef CAS.
- N. Sanson and J. Rieger, Polym. Chem., 2010, 1, 965 RSC.
- S. Seiffert, Angew. Chem., Int. Ed., 2013, 52, 11462 CrossRef CAS PubMed.
- F. Krahl and K. F. Arndt, Adv. Polym. Sci., 2010, 234, 95 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- N. Welsch, M. Ballauff and Y. Lu, Adv. Polym. Sci., 2011, 234, 129 CrossRef.
- W. T. Wu, T. Zhou and S. Q. Zhou, Chem. Mater., 2009, 21, 2851 CrossRef CAS.
- T. Terashima, A. Nomura, M. Ito, M. Ouchi and M. Sawamoto, Angew. Chem., Int. Ed., 2011, 50, 7892 CrossRef CAS PubMed.
- Q. S. Wu, H. Cheng, A. P. Chang, X. Bai, F. Lu and W. T. Wu, Chem. Commun., 2014, 50, 14217 RSC.
- W. Schnabel and U. Borgward, Makromol. Chem., 1969, 123(1), 73 CrossRef CAS PubMed.
- Q. D. Chen, X. H. Shen and H. C. Gao, Chin. J. Polym. Sci., 2005, 23(6), 643 CrossRef CAS.
- Q. Yuan, L. B. Yang, M. Z. Wang, H. Wang, X. P. Ge and X. W. Ge, Langmuir, 2009, 25, 2729 CrossRef CAS PubMed.
- A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623 CrossRef CAS.
- H. J. Schäfer, Top. Curr. Chem., 1990, 152, 91 CrossRef.
- N. Yamazaki, Adv. Polym. Sci., 1969, 6, 377 CrossRef CAS.
- O. F. Olaj, Makromol. Chem., Macromol. Symp., 1987, 8, 235 CrossRef CAS PubMed.
- K. Gurunathan, A. Vadivel Murugan, R. Marimuthu, U. P. Mulik and D. P. Amalnerkar, Mater. Chem. Phys., 1999, 61, 173 CrossRef CAS.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81 CrossRef CAS PubMed.
- N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391 CrossRef CAS PubMed.
- C. S. Lee and J. P. Bell, J. Mater. Sci., 1995, 30, 3827 CrossRef CAS.
- G. Yildiz, H. Çatalgil-Giz and F. Kadirgan, J. Appl. Electrochem., 2000, 30, 71 CrossRef CAS.
- N. Baute, C. Jérôme, L. Martinot, M. Mertens, V. M. Geskin, R. Lazzaroni, J.-L. Brédas and R. Jérôme, Eur. J. Inorg. Chem., 2001, 2001(5), 1097 CrossRef.
- S. L. Cram, G. M. Spinks, G. G. Wallace and H. R. Brown, J. Appl. Polym. Sci., 2003, 87, 765 CrossRef CAS PubMed.
- J. Reuber, H. Reinhardt and D. Johannsmann, Langmuir, 2006, 22, 3362 CrossRef CAS PubMed.
- J. Bünsow, M. Mänz, P. Vana and D. Johannsmann, Macromol. Chem. Phys., 2010, 211, 761 CrossRef PubMed.
- K. Kaniewska, M. Karbarz and Z. Stojek, Electrochim. Acta, 2015 DOI:10.1016/j.electacta.2015.02.196.
- J. Kan, Y. Jiang and Y. Zhang, Mater. Chem. Phys., 2007, 102, 260 CrossRef CAS PubMed.
- R. Ganesan, S. Shanmugam and A. Gedanken, Synth. Met., 2008, 158, 848 CrossRef CAS PubMed.
- J. J. Richardson, H. Ejima, S. L. Lçrcher, K. Liang, P. Senn, J. W. Cui and F. Caruso, Angew. Chem., Int. Ed., 2013, 52, 6455 CrossRef CAS PubMed.
- H. Bysell and M. Malmsten, Langmuir, 2006, 22, 5476 CrossRef CAS PubMed.
- H. Bysell, P. Hansson and M. Malmsten, J. Colloid Interface Sci., 2008, 323, 60 CrossRef CAS PubMed.
- H. Bysell, A. Schmidtchen and M. Malmsten, Biomacromolecules, 2009, 10, 2162 CrossRef CAS PubMed.
-
B. Chu, Laser Light Scattering, Academic Press, New York, 2nd edn, 1991 Search PubMed.
- J. Heinze, Angew. Chem., Int. Ed. Engl., 1984, 23, 831 CrossRef PubMed.
- H. C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2008, 130, 13542 CrossRef CAS PubMed.
- H. C. Xu and K. D. Moeller, Org. Lett., 2010, 12, 1720 CrossRef CAS PubMed.
- H. C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2010, 132, 2839 CrossRef CAS PubMed.
- H. C. Xu and K. D. Moeller, Angew. Chem., Int. Ed., 2010, 49, 8004 CrossRef CAS PubMed.
- J. M. Campbell, H. C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2012, 134, 18338 CrossRef CAS PubMed.
- D. J. Gan and L. A. Lyon, J. Am. Chem. Soc., 2001, 123, 7511 CrossRef CAS PubMed.
- G. Z. Zhang, F. M. Winnik and C. Wu, Phys. Rev. Lett., 2003, 90, 035506 CrossRef.
- W. T. Wu, T. Zhou, A. Berliner, P. Banerjee and S. Q. Zhou, Chem. Mater., 2010, 22, 1966 CrossRef CAS.
- L. X. Li, A. P. Chang, Y. M. Hu, L. Y. Zhang and W. T. Wu, Chem. Commun., 2013, 49, 6534 RSC.
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317 CrossRef CAS PubMed.
- J. W. Stansbury, M. Trujillo-Lemon, X. Ding and S. M. Newman, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2006, 47, 825 CAS.
- O. Okay and W. Funke, Macromolecules, 1990, 23, 2623 CrossRef CAS.
- L. Pille and D. H. Solomon, Macromol. Chem. Phys., 1994, 195, 2477 CrossRef CAS PubMed.
- L. Montanari, R. Scotti and T. P. Lockhart, Macromolecules, 1994, 27, 3341 CrossRef CAS.
- C. L. Dona, D. W. Green and G. P. Willhite, J. Appl. Polym. Sci., 1997, 64, 1381 CrossRef CAS.
- F. G. Chen, H. J. Wang, C. F. Zhu, L. L. Ren and J. Li, J. Appl. Polym. Sci., 2004, 94, 1375 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra (Fig. S1, S6, S10), screenshots of DLS size distribution (Fig. S2, S5, S8, S9), temperature-dependent <Dh> of PNIPAM microgels (Fig. S3), cyclic voltammograms recorded in CH3CN (Fig. S4), and TEM images (Fig. S7). See DOI: 10.1039/c5py00365b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence