
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignocellulosic biomass: a sustainable platform for the production of bio-based chemicals and polymers
Furkan H.
Isikgor
a and
C. Remzi
Becer
*b
aDepartment of Chemistry, Boğaziçi University, Bebek, 34342 İstanbul, Turkey
bSchool of Engineering and Materials Science, Queen Mary University of London, Mile End Road, E1 4NS London, UK. E-mail: r.becer@qmul.ac.uk; Tel: +44 (0)20 78826534
First published on 5th May 2015
Abstract
The demand for petroleum dependent chemicals and materials has been increasing despite the dwindling of their fossil resources. As the dead-end of petroleum based industry has started to appear, today's modern society has to implement alternative energy and valuable chemical resources immediately. Owing to the importance of lignocellulosic biomass being the most abundant and bio-renewable biomass on earth, this critical review provides insights into the potential of lignocellulosic biomass as an alternative platform to fossil resources. In this context, over 200 value-added compounds, which can be derived from lignocellulosic biomass by various treatment methods, are presented with their references. Lignocellulosic biomass based polymers and their commercial importance are also reported mainly in the frame of these compounds. This review article aims to draw the map of lignocellulosic biomass derived chemicals and their synthetic polymers, and to reveal the scope of this map in today's modern chemical and polymer industry.
 Furkan H. Isikgor | Furkan H. Isikgor received his Master of Science degree from the Department of Chemistry at Boğaziçi (Bosphorus) University in Turkey. His research covered synthesis, HPLC and NMR studies on axially chiral pyridine compounds as potential bidentate N,N′-ligands. In 2013, he was an academic visitor in the group of Dr Remzi Becer, University of Warwick, UK, and he worked on the synthesis of glycopolymers. Currently, he is a Ph.D. candidate in the Department of Materials Science and Engineering at National University of Singapore. His research focuses mainly on high performance perovskite solar cells. |
 C. Remzi Becer | Remzi Becer received his BSc and MSc degrees in Chemistry at Istanbul Technical University. He completed his PhD in 2009 under the guidance of Prof. Ulrich Schubert at Eindhoven University of Technology. He received a Marie Curie IntraEuropean Fellowship in 2009 and joined the group of Prof. David Haddleton, University of Warwick. He was awarded a Science City Interdisciplinary Senior Research Fellowship in 2011 to start up his independent research group at the University of Warwick. Since 2013, he has been appointed as a Senior Lecturer in the School of Engineering and Materials Science at Queen Mary University of London. |
1. Introduction
Modern industrial polymerization technologies make it possible to produce versatile polymers with highly tunable properties and a broad range of applications. Depending on request, today's polymers can be stiff or soft, transparent or opaque, conducting or insulating, permeable or impermeable, and stable or degradable. Some indispensable and irreplaceable applications of them include high strength fibers, composites, construction materials, light weight engineering plastics, coatings, adhesives, packaging materials, microelectronics and novel materials for biomedical applications such as drug delivery systems, implants, membranes for artificial kidneys and water purification, dental fillings, wound dressing and artificial hearts. No other class of materials can have such diverse properties and versatile applicability. This means that modern life would be impossible without polymeric materials since they provide high quality of life for all humankind.1–3On the other hand, industrial production of a wide range of chemicals and synthetic polymers heavily relies on fossil resources.4 Dwindling of these resources together with their frightening environmental effects, such as global warming and littering problems, have started to threaten the future of the polymer industry. In the early part of the 19th century, Henry Ford suggested that the implementation of a bio-based economy is a logical and necessary option for the growth of any civilization. This implementation was postponed because oil has always been cheaper than any other commodity products. However, the competitive price advantage of fossil fuels during the last century has disappeared.5 After crossing the oil production peak, the dwindling of fossil resources will further boost the oil price and this situation will drastically impact the cost-effectiveness and competiveness of polymers. More importantly, mass consumption of petroleum based materials leaves devastating environmental problems, which are lethal threats to human beings. Growing concerns regarding these issues have inevitably started to force our society to demand sustainable and green products. The European Union has already approved laws for the reduction of environmentally abusive materials and started to put greater efforts for finding eco-friendly materials based on natural resources. Hence, alternative solutions are sought to develop sustainable polymers from renewable natural resources for decreasing the current dependence on fossil resources and fixing the production rate of CO2 to its consumption rate.6,7
Biomass and biomass derived materials have been pointed out to be one of the most promising alternatives.8,9 These materials are generated from available atmospheric CO2, water and sunlight through biological photosynthesis. Therefore, biomass has been considered to be the only sustainable source of organic carbon in earth and the perfect equivalent to petroleum for the production of fuels and fine chemicals with net zero carbon emission.10,11 In this context, lignocellulosic biomass, which is the most abundant and bio-renewable biomass on earth,10 has critical importance. Many studies have shown that lignocellulosic biomass holds enormous potential for sustainable production of chemicals and fuels. Besides, it is a renewable feedstock in abundance and is available worldwide.12,13 Lignocellulosic biomass has been projected as an abundant carbon-neutral renewable source, which can decrease CO2 emissions and atmospheric pollution. Thus, it is a promising alternative to limit crude oil, which can be utilized to produce biofuels, biomolecules and biomaterials.14–16 Furthermore, the major component of lignocellulosic biomass, cellulose, is considered to be the strongest potential candidate for the substitution of petroleum-based polymers owing to its eco-friendly properties like renewability, bio-compatibility and bio-degradability.7
Sustainability of the production of fuels and chemicals from biomass, on the other hand, has been greatly debated. As an example, there are critical concerns regarding the sustainability of current production of bioethanol, which relies on starch and sugar crops. The limited supply of such crops can lead to competition with food production.17 Lignocellulosic feedstocks have crucial advantages over other biomass supplies in this manner because they are the non-edible portion of the plant and therefore they do not interfere with food supplies.18 Moreover, forestry, agricultural and agro-industrial lignocellulosic wastes are accumulated every year in large quantities. Disposal of these wastes to the soil or landfill causes serious environmental problems; however, they could be utilized for the production of a number of value-added products.19 From the economic point of view, lignocellulosic biomass can be produced quickly and at lower cost than other agriculturally important biofuel feedstocks such as corn starch, soybeans and sugar cane. It is also significantly cheaper than crude oil.20
On the other hand, the development of the conversion of lignocellulosic biomass to fine chemicals and polymers still remains a big challenge.10 Lignocellulose has evolved to resist degradation. This inherent property of lignocellulosic materials makes them resistant to enzymatic and chemical degradation.16 For changing the physical and chemical properties of the lignocellulosic matrix, pretreatment of lignocellulosic biomass, which is an expensive procedure with respect to cost and energy, is essential.21 Although lignocellulosic materials are abundant and usually low-priced, the crucial challenge in converting lignocellulosic biomass is to produce value-added chemicals at high selectivities and yields at economical cost.22 Extensive research is currently being undertaken all over the world to address this problem.23 Biorefinery and biofuel technologies are developed to refine biomass in analogy to petrochemistry for producing renewable oil and green monomers.24 In addition, the number of biorefinery-related pilot and demonstration plants has been increasing.25 For instance, Lignol, Verenium and Mascoma are promising companies, which aim to undertake the development of biorefining technologies for the production of advanced biofuels, biochemicals and biomaterials from non-food cellulosic biomass feedstocks.
In this review article, we report the ongoing activities in the field of lignocellulosic biomass for the production of value-added chemicals and polymers that can be utilized to replace petroleum-based products. After a description of the structure and sources of lignocellulosic biomass, different pre- and post-treatment methods for the degradation of lignocellulosic biomass into its components are summarized. Over 200 value-added compounds, which can be derived from lignocellulosic biomass using various treatment methods, are presented with their references. Finally, detailed overviews of the polymers that can be produced mainly from these compounds are given. Current research studies and commercial product examples of these polymers are also explained in order to reveal their indispensable need in our modern society.
2. Structure and sources of lignocellulosic biomass
Lignocellulosic biomass is mainly composed of three polymers: cellulose, hemicellulose and lignin together with small amounts of other components like acetyl groups, minerals and phenolic substituents (Fig. 1). Depending on the type of lignocellulosic biomass, these polymers are organized into complex non-uniform three-dimensional structures to different degrees and varying relative composition. Lignocellulose has evolved to resist degradation and this robustness or recalcitrance of lignocellulose stems from the crystallinity of cellulose, hydrophobicity of lignin, and encapsulation of cellulose by the lignin–hemicellulose matrix.16,26,27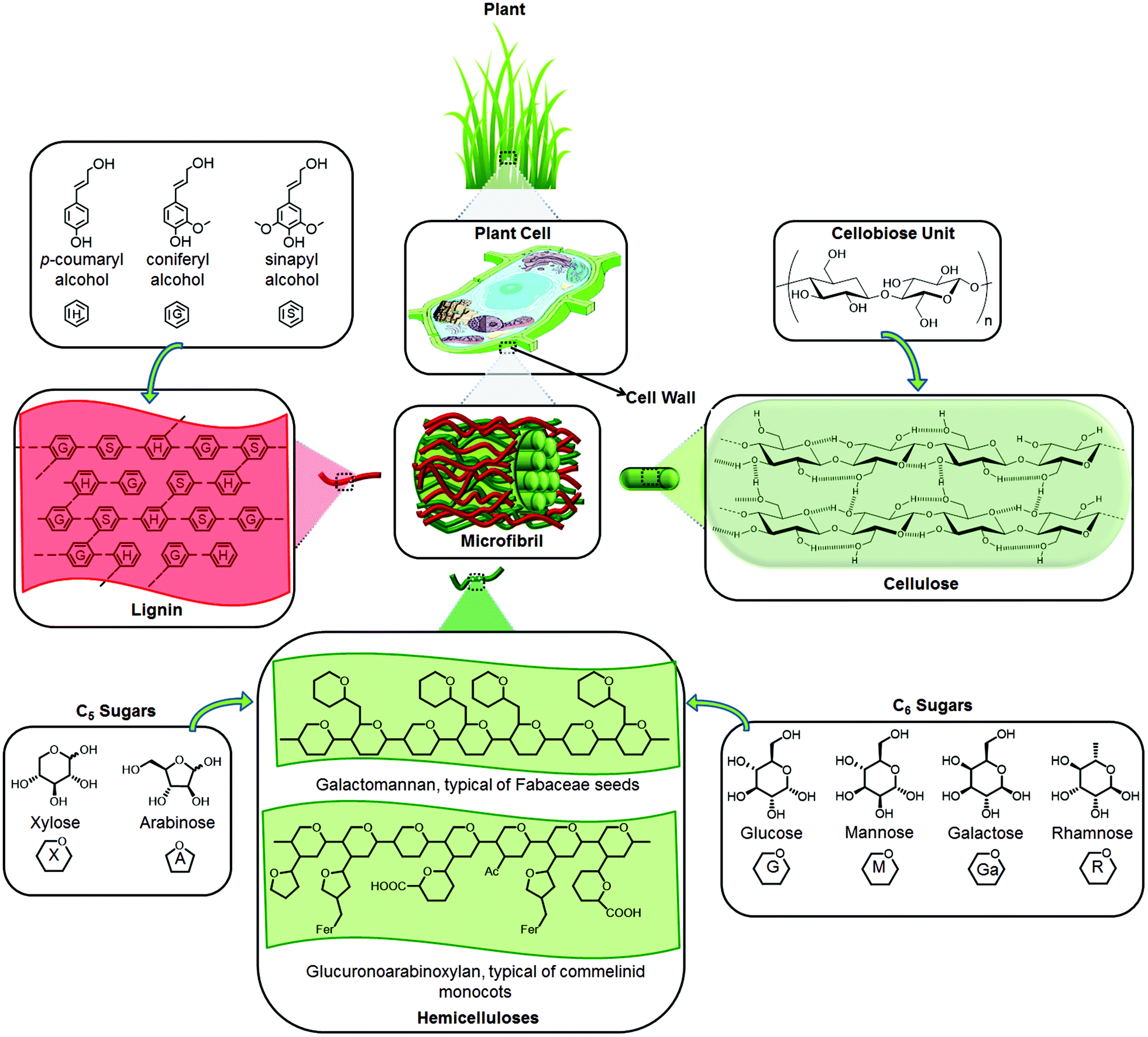 | ||
| Fig. 1 The main components and structure of lignocellulose. “Gl” represents glucuronic acid and “Fer” represents esterification with ferulic acid, which is characteristic of xylans in commelinid monocots.28 | ||
The major component of lignocellulosic biomass is cellulose. Unlike glucose in other glucan polymers, the repeating unit of the cellulose chain is the disaccharide cellobiose. Its structure consists of extensive intramolecular and intermolecular hydrogen bonding networks, which tightly binds the glucose units (Fig. 1). Since about half of the organic carbon in the biosphere is present in the form of cellulose, the conversion of cellulose into fuels and valuable chemicals has paramount importance.10,12,13
Hemicellulose is the second most abundant polymer. Unlike cellulose, hemicellulose has a random and amorphous structure, which is composed of several heteropolymers including xylan, galactomannan, glucuronoxylan, arabinoxylan, glucomannan and xyloglucan (Fig. 1). Hemicelluloses differ in composition too; hardwood hemicelluloses contain mostly xylans, whereas softwood hemicelluloses contain mostly glucomannans. The heteropolymers of hemicellulose are composed of different 5- and 6-carbon monosaccharide units: pentoses (xylose, arabinose), hexoses (mannose, glucose, galactose) and acetylated sugars. Hemicelluloses are imbedded in the plant cell walls to form a complex network of bonds that provide structural strength by linking cellulose fibres into microfibrils and cross-linking with lignin (Fig. 1).26,28
Finally, lignin is a three-dimensional polymer of phenylpropanoid units. It functions as the cellular glue which provides compressive strength to the plant tissue and the individual fibres, stiffness to the cell wall and resistance against insects and pathogens.29 The oxidative coupling of three different phenylpropane building blocks: monolignols: p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol, forms the structure of lignin. The corresponding phenylpropanoid monomeric units in the lignin polymer are identified as p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units, respectively (Fig. 1).30
Cellulose, hemicellulose and lignin are not uniformly distributed within the cell walls. The structure and the quantity of these plant cell wall components vary according to species, tissues and maturity of the plant cell wall.16 Generally, lignocellulosic biomass consists of 35–50% cellulose, 20–35% hemicellulose, and 10–25% lignin. Proteins, oils, and ash make up the remaining fraction.21Table 1 summarizes particular types of lignocellulosic biomass and their chemical composition.17,31
| Lignocellulosic biomass | Cellulose (%) | Hemicellulose (%) | Lignin (%) | |
|---|---|---|---|---|
| Hardwood | Poplar | 50.8–53.3 | 26.2–28.7 | 15.5–16.3 |
| Oak | 40.4 | 35.9 | 24.1 | |
| Eucalyptus | 54.1 | 18.4 | 21.5 | |
| Softwood | Pine | 42.0–50.0 | 24.0–27.0 | 20.0 |
| Douglas fir | 44.0 | 11.0 | 27.0 | |
| Spruce | 45.5 | 22.9 | 27.9 | |
| Agricultural waste | Wheat Straw | 35.0–39.0 | 23.0–30.0 | 12.0–16.0 |
| Barley Hull | 34.0 | 36.0 | 13.8–19.0 | |
| Barley Straw | 36.0–43.0 | 24.0–33.0 | 6.3–9.8 | |
| Rice Straw | 29.2–34.7 | 23.0–25.9 | 17.0–19.0 | |
| Rice Husks | 28.7–35.6 | 12.0–29.3 | 15.4–20.0 | |
| Oat Straw | 31.0–35.0 | 20.0–26.0 | 10.0–15.0 | |
| Ray Straw | 36.2–47.0 | 19.0–24.5 | 9.9–24.0 | |
| Corn Cobs | 33.7–41.2 | 31.9–36.0 | 6.1–15.9 | |
| Corn Stalks | 35.0–39.6 | 16.8–35.0 | 7.0–18.4 | |
| Sugarcane Bagasse | 25.0–45.0 | 28.0–32.0 | 15.0–25.0 | |
| Sorghum Straw | 32.0–35.0 | 24.0–27.0 | 15.0–21.0 | |
| Grasses | Grasses | 25.0–40.0 | 25.0–50.0 | 10.0–30.0 |
| Switchgrass | 35.0–40.0 | 25.0–30.0 | 15.0–20.0 | |
3. Lignocellulosic biomass treatment methods
One of the most important goals of lignocellulosic biomass refining is to fractionate lignocellulose into its three major components: cellulose, hemicelluloses and lignin. Single step treatment methods, like pyrolysis, are not efficient. Although they render lower costs, deconstruction of the lignocellulosic biomass takes place since these methods generally rely on high temperatures. It is highly inconvenient and difficult to separate the targeted chemicals and fuels via single step methods because the produced bio-oil consists of a mixture of hundreds of compounds. For downstream and efficient separations, additional costs and various pretreatment methods are required. The application of the pretreatment methods changes the natural binding characteristics of lignocellulosic materials by modifying the supramolecular structure of the cellulose–hemicellulose–lignin matrix. Hence, the pretreatment of lignocellulosic biomass, prior to other treatment methods, is an essential step in order to increase cellulose and hemicellulose accessibility and biodegradability for enzymatic or chemical action.16,22Pretreatment methods are divided into different categories such as mechanical, chemical, physicochemical and biological methods or various combinations of these.16 Various pretreatment options were reported to fractionate, solubilize, hydrolyse, and separate cellulose, hemicellulose, and lignin components.21 Some of them include milling, irradiation, microwave, steam explosion, ammonia fiber explosion (AFEX), supercritical CO2 and its explosion, SO2, alkaline hydrolysis, liquid hot-water pretreatment, organosolv processes, wet oxidation, ozonolysis, dilute- and concentrated-acid hydrolyses, and biological pretreatments.19,21 The common goal of these methods is to reduce the biomass in size and open its physical structure. Each of these methods has been reported to have distinct advantages and disadvantages.
Through research and development, pretreatment of lignocellulosic biomass has great potential for the improvement of efficiency and lowering the cost of production.32 The integration of various biomass pretreatment methods with other processes like enzymatic saccharification, detoxification, fermentation of the hydrolysates, and recovery of products will greatly reduce the overall cost of using lignocellulose for practical purposes.21 Hence, the future achievements of lignocellulosic conversion at a commercial scale are expected to depend on the improvements in pretreatment technologies, cellulolytic enzyme producing microorganisms, fullest exploitation of biomass components and process integration.33
4. Valuable chemicals from lignocellulosic biomass
Lignocellulosic biomass has higher amount of oxygen and lower fractions of hydrogen and carbon with respect to petroleum resources. Owing to this compositional variety, more classes of products can be obtained from lignocellulosic biorefineries than petroleum based ones. Nevertheless, a relatively large range of processing technologies is needed for the treatment of lignocellulosic biomass. In fact, most of these technologies are still at the pre-commercial stage.25,34 Nowadays, only production of bio-ethanol from biomass feedstocks is well established and it is turning out to be a mature technology. Fermentation of glucose to lactic acid is also established on the market and it is commercially available.25,35Higher oxygen content in biofuels reduces the heat content of the products and prevents their blending with existing fossil fuels. Therefore, in terms of transportation fuels and chemicals, lignocellulosic biomass needs to be depolymerized and deoxygenated. For the production of other value-added chemicals, the presence of oxygen often provides valuable physical and chemical properties to the product. Thus, the production process requires much less deoxygenation.25,36 Oxygenation or deoxygenation of biomass feedstocks results in completely different products. Besides these, various sources of lignocellulosic biomass need to be considered separately since they have different compositions of cellulose, hemicellulose and lignin. Against all odds, the depolymerization process of the lignocellulosic biomass is a common goal for all different feedstocks for the production of all types of chemicals.
4.1. C5 and C6 sugar production from lignocellulosic biomass
The first platform chemicals in the biorefinery can be sugar compounds obtained from non-food biomass.37 Efficient release of the C5 and C6 sugars (Fig. 1) with lower energy consumption has critical importance because the generation of further degradation products depends on that step.Glucose is the sugar degradation product of cellulose. The depolymerization of hemicellulose, on the other hand, results in the formation of both glucose as well as the other five (xylose, arabinose) and six (mannose, galactose, rhamnose) membered sugars (Fig. 1). According to Zviely, concentrated HCl-driven hydrolysis is currently the most powerful and industrially proven technology for the conversion of lignocellulosic biomass to low-cost fermentable sugars.38 However, recovery of the acid still remains a key limitation to any concentrated acid hydrolysis. Continued research is needed for addressing the use and separation of mineral acids, increasing the concentration of product streams and improving product separations. In this respect, a roadmap was presented by Wettstein et al. to address current challenges and future prospects of lignocellulosic biomass conversion to sugars, fine chemicals and fuels.39
4.2. Lignocellulosic sugar and lignin derivable chemicals
Fig. 2–5 depict the map of lignocellulosic sugars and lignin derivable chemicals under 16 platforms with their references. These chemicals can be produced via biological or chemical conversions. The 16 building blocks can subsequently be converted to a number of valuable chemicals or materials. The chemistry of these conversions was described in detail in the corresponding references. As such, the report of the US Department of Energy (DOE) describes twelve sugar derivable building block chemicals, which can be transformed into new families of useful molecules. These C5 and C6 sugar derived platform chemicals include 1,4-diacids (succinic acid, fumaric acid, malic acid), 2,5-furan dicarboxylic acid (2,5-FDCA), 3-hydroxy propionic acid (3-HPA), aspartic acid, glucaric acid, glutamic acid, itaconic acid, levulinic acid, 3-hydroxybutyrolactone (3-HBL), glycerol, sorbitol and xylitol/arabinitol.40 Gallezot has also reviewed the synthesis of chemicals by conversion of platform molecules obtained by depolymerisation and fermentation of biopolymers.41 Successful catalytic conversion of these building blocks into intermediates, specialties and fine chemicals was examined in detail. Unlike the DOE report, Gallezot has considered 5-hydroxymethyl furfural (5-HMF) as a separate building block because its derivatives, such as 2,5-FDCA, were identified as very promising chemical intermediates. In the same review, xylose and furfural were considered as a C5 platform. A variety of chemicals that can be obtained from that platform were also presented.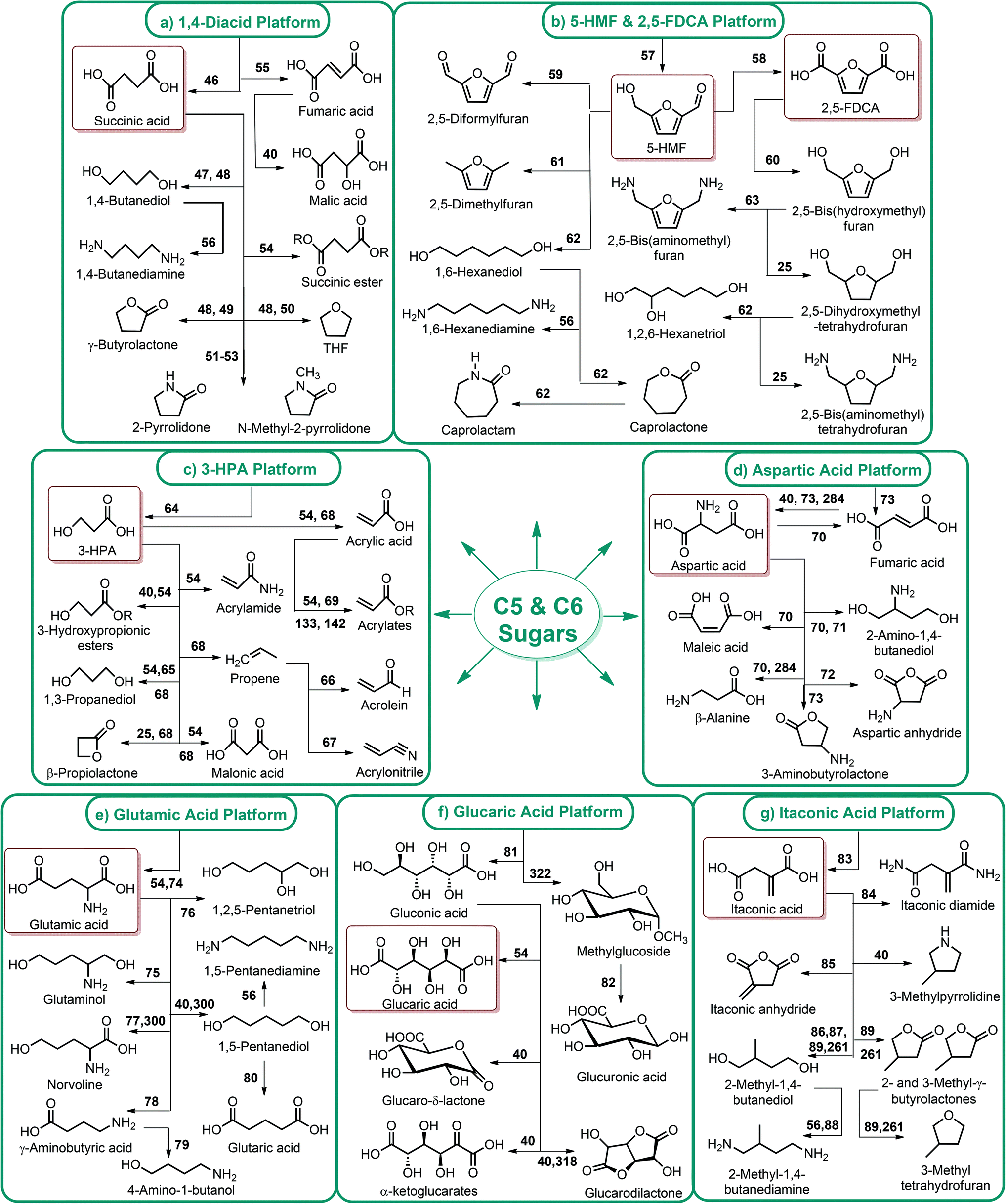 | ||
| Fig. 2 (a) 1,4-diacid, (b) 5-HMF and 2,5-FDCA, (c) 3-HPA, (d) aspartic acid, (e) glutamic acid, (f) glucaric acid, and (g) itaconic acid platform chemicals. | ||
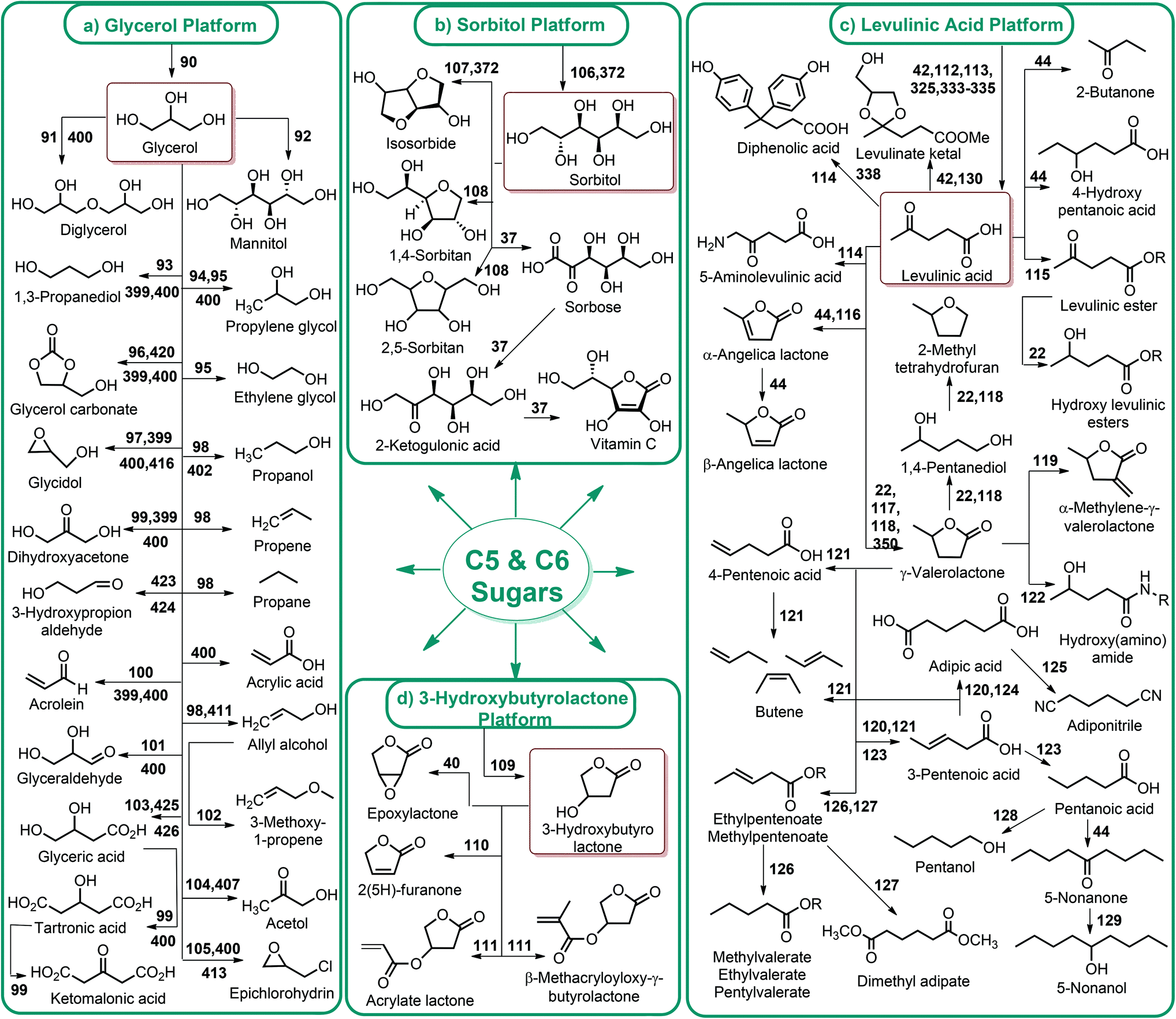 | ||
| Fig. 3 Glycerol, sorbitol, 3-hydroxybutyrolactone and levulinic acid platform chemicals. | ||
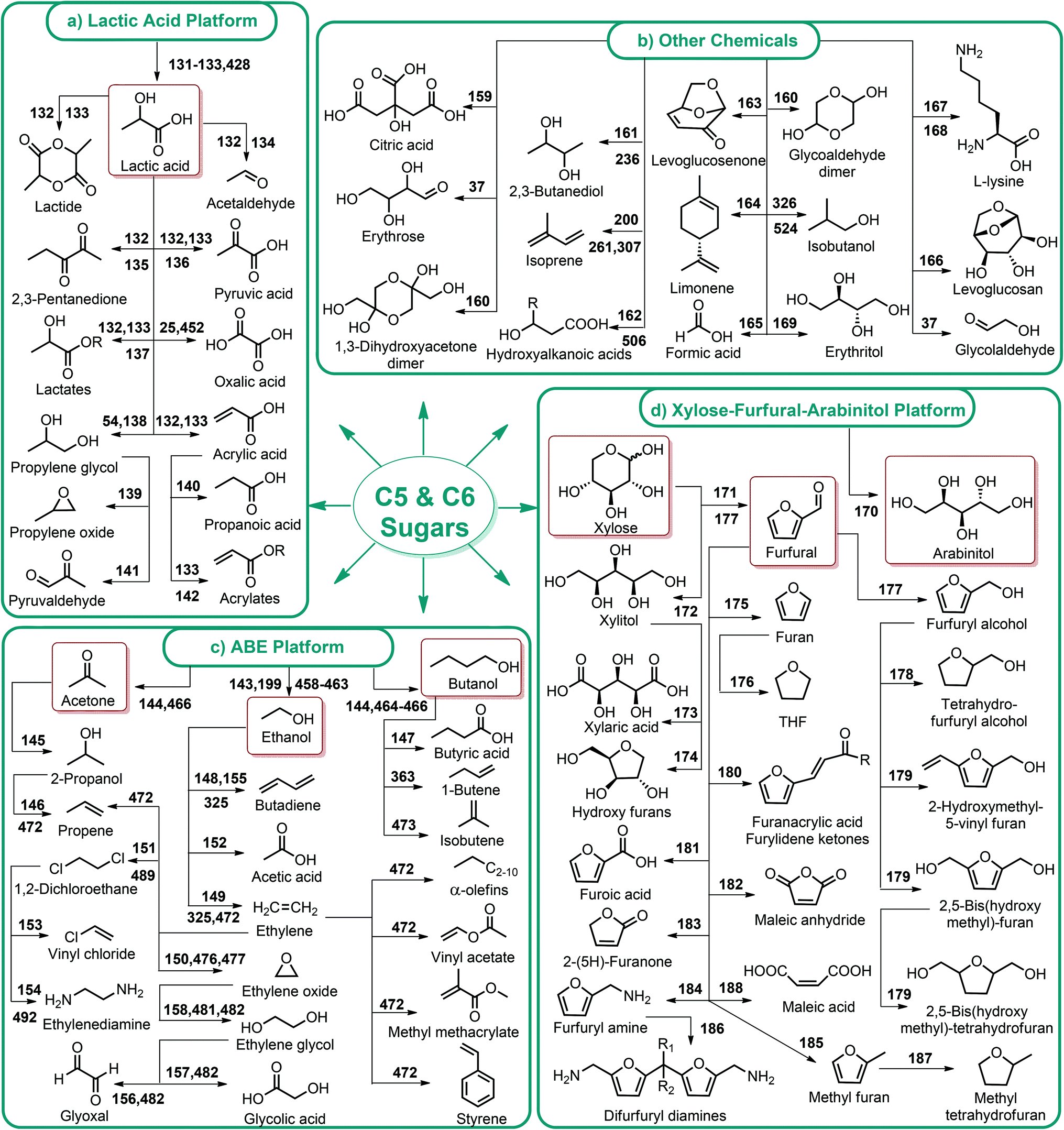 | ||
| Fig. 4 Lactic acid, ABE, xylose–furfural–arabinitol platforms and other lignocellulosic chemicals. | ||
 | ||
| Fig. 5 Lignin derived chemicals. | ||
Since the original DOE report, considerable progress in biobased product development has been made. As an example, ethanol was omitted from DOE's original list because it was categorized as a supercommodity chemical due to its expected high production volume. On the other hand, Bozell and Petersen revisited that report and presented an updated group of candidate structures. They included ethanol as a platform chemical because bio-based ethanol and related alcohols such as bio-butanol are promising precursors to the corresponding olefins via dehydration.42 In this way, acetone can also be considered as a platform chemical because its conversion products include important monomeric chemicals. The acetone, butanol and ethanol (ABE) fermentation process was largely studied by the scientific and industrial communities.43 Their conversion products such as ethylene, ethylene glycol, butadiene, propene and vinyl chloride have made a great impact on polymer chemistry. Lactic acid was also not indicated as a building block in the DOE report. However, lactic acid is the most widely occurring carboxylic acid in nature. Owing to its biofunctionality, it can be converted into a variety of reaction compounds such as acetaldehyde, acrylic acid, propanoic acid, 2,3-pentanedione and dilactide.44
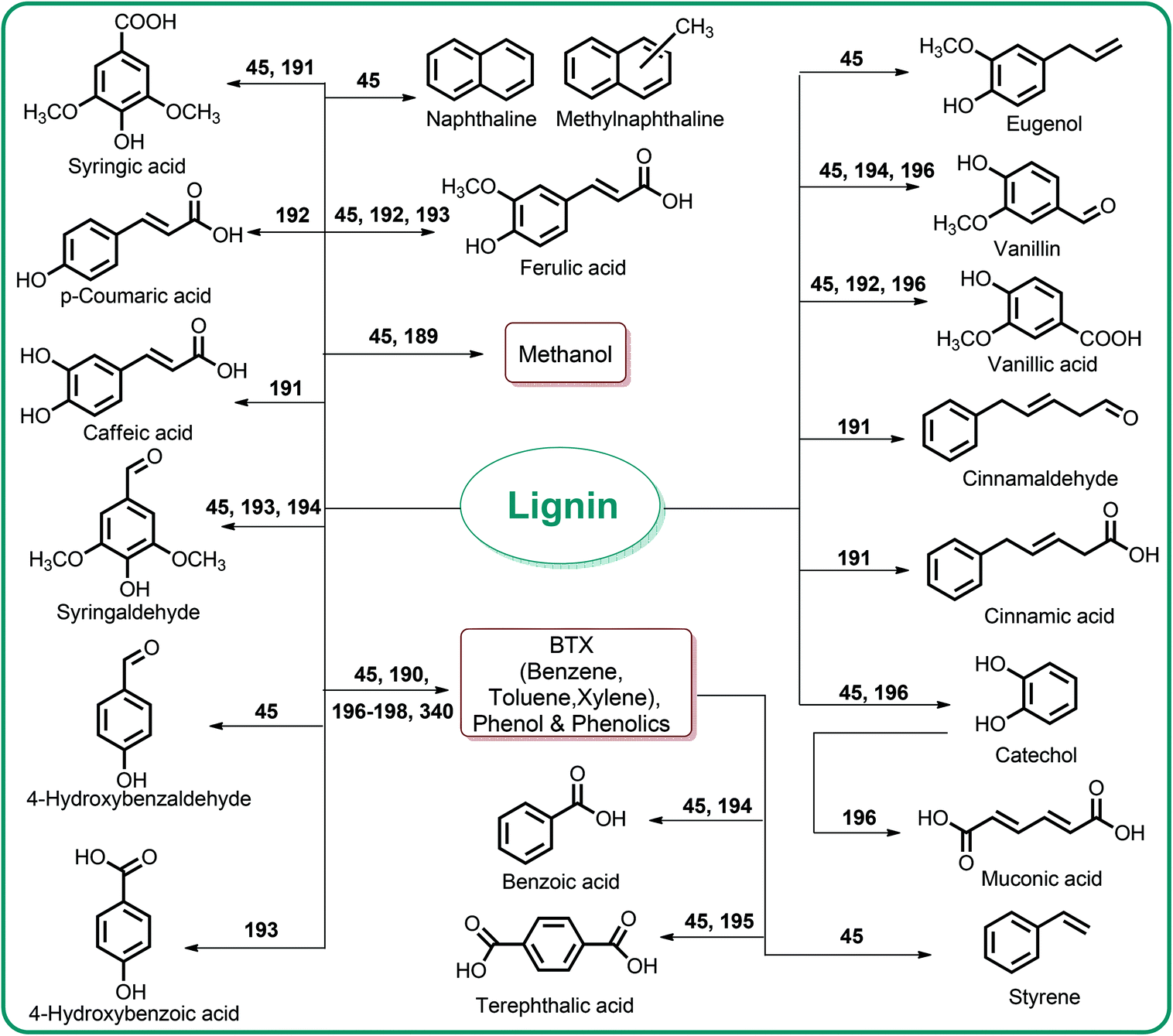
Lignin conversion also has promising potential. The unique structure and chemical properties of lignin allow the production of a wide variety of chemicals, particularly aromatic compounds. Hence, lignin can be considered to be the major aromatic resource of the bio-based economy. Different approaches and strategies that have been reported for catalytic lignin conversion were comprehensively reviewed by Zakzeski et al.45,46–200
5. Sustainable polymers from lignocellulosic biomass
There are three main routes for the production of polymeric materials from lignocellulosic biomass. The least common route is to revaluate of the waste stream of the biorefinery or paper mill, which consists of lignin, cellulose, monomeric sugars and various extractives. The second route includes the cell wall polymers themselves in which cellulose, hemicellulosic polysaccharides and lignin are isolated, processed, and converted to end products.201 It can be more convenient to directly make use of these readily available bio-polymers because the process of polymerization is done perfectly by nature itself. But the challenge for lignocellulosic biomass remains in the efficient separation of cellulose, hemicellulose and lignin from each other. In this respect, lignocellulosic biomass related research studies are also focused on that particular concept. Recently, promising studies on the isolation of cellulose, hemicellulose and lignin from various lignocellulosic biomass resources have been reported.202–205 Once isolated, cellulose, hemicellulose and lignin can be converted and/or incorporated into a wide range of materials. The production strategies and specific applications of functionalized polymers of cellulose, hemicellulose and lignin have already been highlighted in the recent literature.201,206,207 That is why this review article focuses on the third route and in fact the most important route which relies on the deconstruction of the cell wall polysaccharides into monomeric hexose and pentose sugars. These sugars are then converted into a wide range of value-added chemicals and bio-based polymers.5.1. Sugar containing polymers
Lignocellulosic biomass can become a major feedstock for sugar containing polymers by providing C5 (xylose, arabinose) and C6 (glucose, mannose, galactose, rhamnose) monosaccharides (Fig. 1), and their many functionalized derivatives including glucaro-δ-lactone, methylglucoside and glucuronic acid. These sugars and their derivatives can either be incorporated in the polymer backbone or be used as pendant groups. The latter type is identified as glycopolymers: synthetic macromolecules with pendant carbohydrate moieties (Fig. 6a).208 Such polymers have gained special interest since they are promising in mimicking structural and functional responsibilities of glycoproteins.209 Their multivalent character, which is displayed by the large number of repeating carbohydrate units, can represent “the cluster glycoside effect”: a high affinity derived from multivalency in oligosaccharide ligands. The cluster glycoside effect greatly enhances carbohydrate–lectin binding events.210,211 Therefore, different glycopolymer architectures have been developed with a specific effort to design novel drug and gene delivery systems. Current research on glycopolymeric drugs mainly focuses on millions of people affecting diseases such as influenza hemagglutinin and neuraminidase inhibitors, HIV, and Alzheimer's disease. However, the majority of these studies are still at the preliminary level and extensive research is required for clinical trials of glycopolymer-based drugs.212,213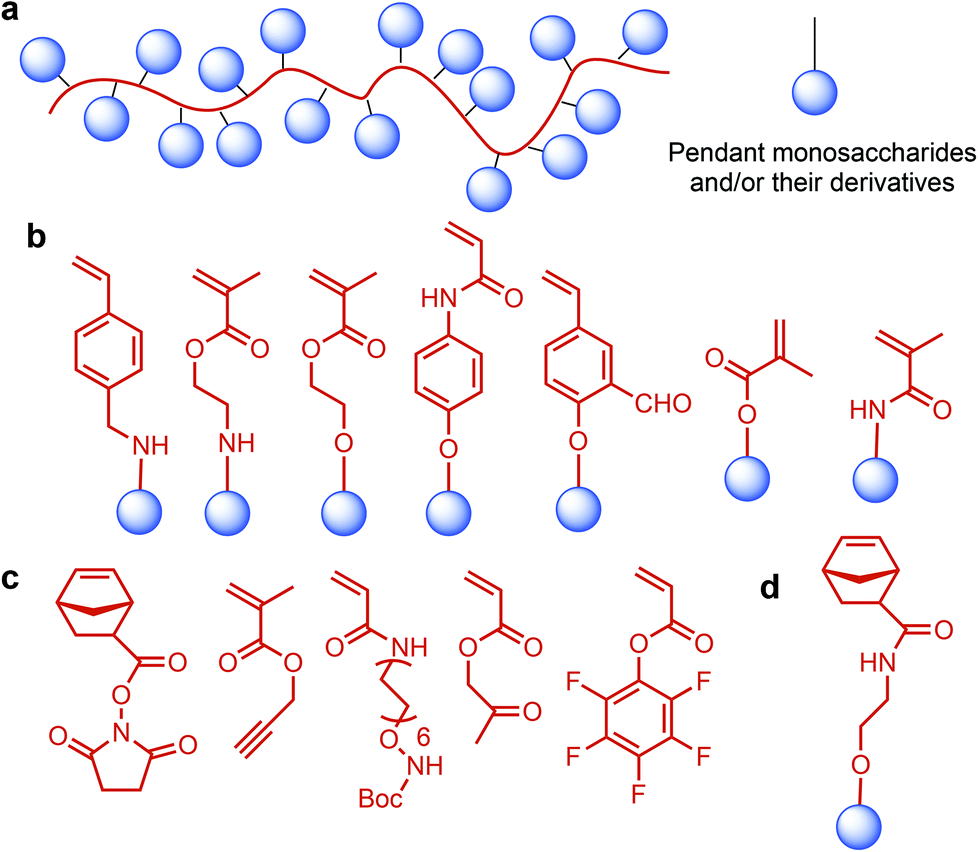 | ||
| Fig. 6 (a) Glycopolymer structure. Monomer structures of glycopolymers used for living polymerization: monomers for (b) LRP, (c) polymerization with the saccharide-reactive group, (d) ROMP. | ||
Recent advances in polymer chemistry have enabled the living polymerization of glycopolymers having various architectures.214 In that respect, Miura reviewed the design and synthesis of glycopolymer incorporating systems such as block copolymers, micelles and particles, star polymers, bioconjugated polymers, hyperbranched polymers, and polymer brushes. For the synthesis of such systems, even when the monomer structure of the glycopolymer is bulky, various living radical polymerization (LRP) techniques, such as nitroxide-mediated polymerization (NMP), atom-transfer radical polymerization (ATRP), and reversible addition-fragment chain transfer (RAFT) polymerization, have proven to be very promising.215–224 Ring-opening metathesis polymerization (ROMP) and polymerization with the saccharide-reactive group approaches also provide a living manner. For these living polymerization methods, several monomer structures of glycopolymers were developed (Fig. 6b and c).225
Controlled/living polymerization of glycopolymers with a combination of click chemistry has attracted attention over the last few years.226–232 Controlled polymerization can give well-defined and fine-tuned glycopolymers. In addition, click chemistry can provide highly efficient and facile click of monosaccharide units to the monomer itself or to the polymer backbone. Slavin et al. reviewed the combination of different polymerization techniques, i.e. RAFT polymerization, NMP, ATRP, cobalt catalyzed chain transfer polymerization (CCCTP) and ring opening polymerization (ROP), with selected click reactions including copper catalyzed azide–alkyne cycloaddition (CuAAC), p-fluoro–thiol click, thiol–ene click, thio–halogen click, and thiol–yne click.226 Three different click chemistry and controlled polymerization combining synthetic routes have been developed so far. The first route is called the glycomonomer approach. In this strategy, carbohydrate-bearing monomers can be polymerized to yield glycopolymers. The second route is based on post-polymerization modifications of clickable polymers with carbohydrates. In the third route, glycopolymers are synthesized via a one-pot process with a simultaneous click reaction and living radical polymerization. Examples of these three synthetic routes to glycopolymers using ATRP and copper catalyzed azide–alkyne cycloaddition (CuAAC) reactions are summarized in Fig. 7.226
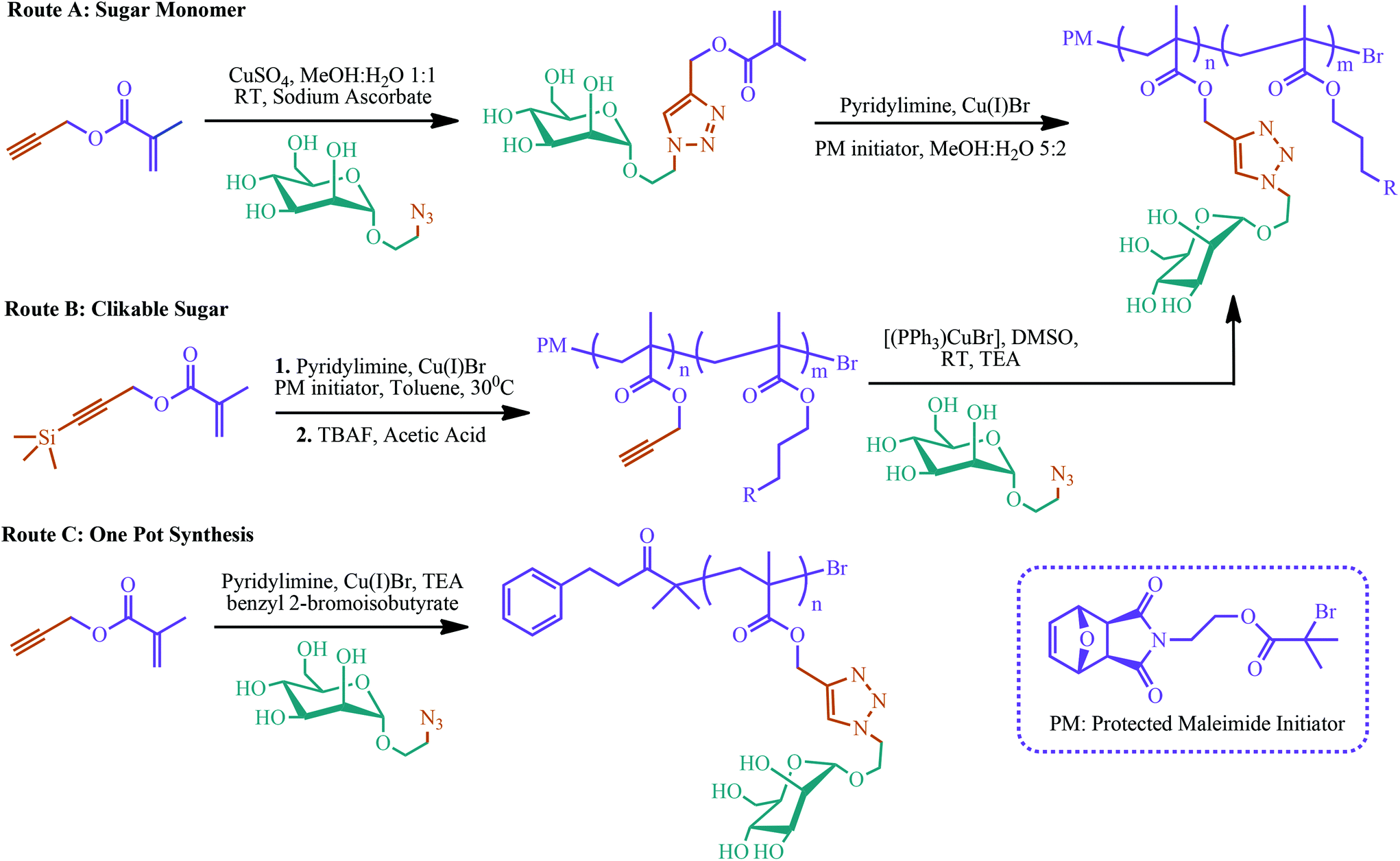 | ||
| Fig. 7 Various synthetic routes to glycopolymers using ATRP and CuAAC employed by Slavin et al. Adapted from ref. 226. | ||
Apart from providing C5 and C6 monosaccharides, lignocellulosic biomass is also a huge feedstock for linear derivatives of these monosaccharides. Lignocellulosic biomass derivable alditols (isosorbide, erythritol, mannitol, sorbitol, xylitol, arabinitol), aldaric acids (glucaric acid, xylaric acid, α-ketoglucarates), and aldonic acids (gluconic acid, 2-ketogluconic acid) (Fig. 2–4) can offer outstanding advantages for the preparation of bio-based polymers. These linear carbohydrate-based polymers typically display enhanced hydrophilicity. They are more prone to be non-toxic and biodegradable than petrochemical-based polycondensates. Therefore, they offer a wide range of applications in food packaging and medical devices. The –OH and –COOH rich functionality of the linear carbohydrates makes them particularly useful in polycondensation reactions. However, group protection and activation procedures are generally required before their polycondensation process.233,234 Munoz-Guerra reported that linear polycondensates, such as polyesters, polyamides, polycarbonates, polyureas, polyanhydrides, and polyurethanes, are obtained by the end group functionalization and/or blocking or removal of the secondary –OH groups (Fig. 8). A variety of polycondensates have been derived from tetroses, pentoses and hexoses. Their structures have been examined, and some of their more relevant properties comparatively evaluated.234 Apart from polycondensation reactions, linear carbohydrates are also employed as pendant groups. In this respect, the common strategy is to anchor a linear sugar derivative to a polymerizable double bond through ester, amide, ether and C–C linkages. As an example, Narain et al. described the polymerization of a C–C connected monomer, namely 4-vinylphenyl-δ-gluco(d-manno)hexitol, which is derived from δ-gluconolactone.235
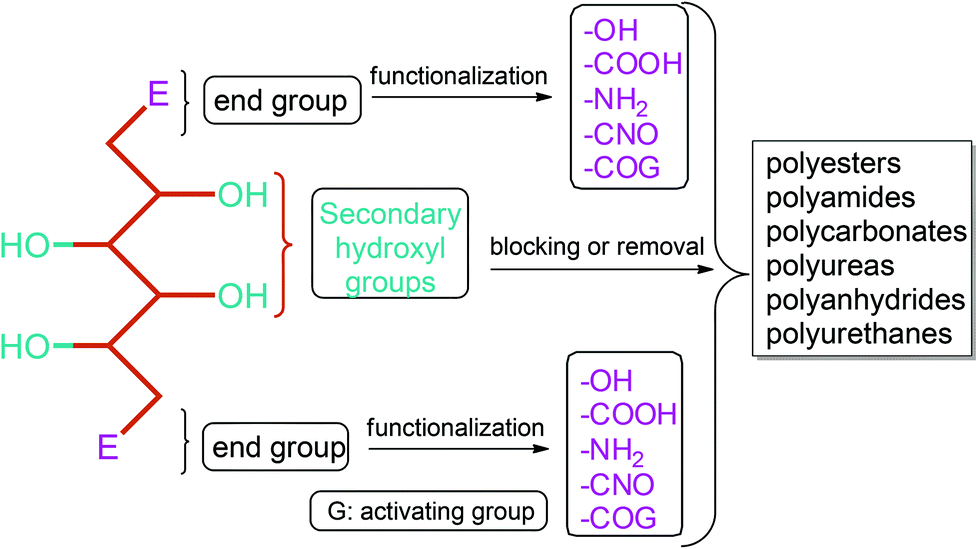 | ||
| Fig. 8 Linear polycondensates from carbohydrates. Adapted from ref. 234. | ||
For the production of commodity materials, protection/deprotection as well as further functionalization processes of sugar monomers are required. These processes add up to the manufacture costs and thus decrease the cost competitiveness of sugar containing polymers. Hence, polymer research in this field is much more directed on specialty products instead of commodity materials. Particularly, controlled/living polymerization of sugar monomers with the help of click chemistry has the highest contribution to the synthesis of sugar containing specialty polymers. Nonetheless, extensive research is required since the majority of these studies are at the preliminary level for bio-applications.
5.2. 1,4-Diacid platform based polymers
As the microbial production of succinic acid (Fig. 2a) is turning out to be a mature technology,46 several companies have started to develop renewable polymers from succinic acid and its derivatives. As an example, NatureWorks and BioAmber recently formed an alliance (AmberWorks) to investigate the production of completely renewable polyester copolymers of succinic acid and 1,4-butanediol (1,4-BDO).236 Various polyamides (PA), polyesters (PE), and poly(ester amide)s (PEA) can be produced via the condensation reaction of succinic acid or succinic acid diesters with diamines or diols. In terms of polyamides, the literature generally describes polyamides which are either on the basis of 1,4-butanediamine (PA 4n) or succinic acid (PA n4).237 Certain types of succinic acid based polyesters, particularly poly(alkylene succinate)s (PAS), can also be manufactured. Poly(ethylene succinate) (PES), poly(propylene succinate) (PPS) and poly(butylene succinate) (PBS) are the most studied polyesters of succinic acid. From these sets of polyesters, PES and PBS are successfully commercialized due to their relatively high melting temperatures, controllable biodegradation rate and good processability. They have polyethylene (PE) like crystallization behavior, and their elongation at break and tensile strength are comparable with those of polypropylene (PP) and low-density polyethylene (LDPE).238 PBS and its copolymers have been receiving special interest in today's polymer market.239 Commercially available products of PBS are presented in Table 2, which summarizes the most important commercial products of the 1,4-diacid platform, their large scale manufacturers and specific application areas.| Polymer structures | Product names and manufacturers | Major applications |
|---|---|---|
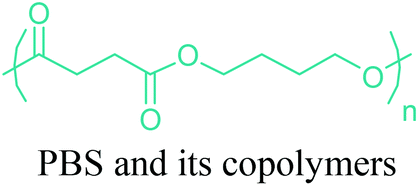
|
GS Pla® (Mitsubishi Chemical), Bionolle® (Showa), Lunare SE® (Nippon Shokubai), Skygreen® (SK Chemical) etc. | Disposable goods, films (compost bag, shopping bag, packaging film etc.), agriculture or horticulture (mulch film, plant pot, rope etc.), fishing gear (fishing net, fishing trap, fishing line etc.), containers (tray, food containers, bottles etc.) |
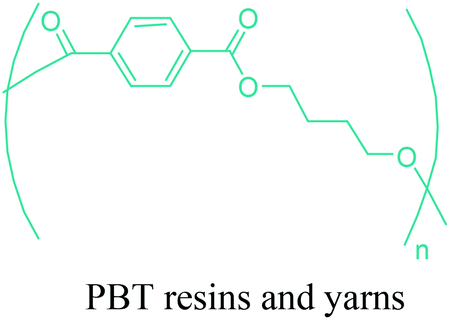
|
Arnite® (DSM), Crastin® (DuPont), Ultradur® (BASF), Advanite™ (SASA), Celanex® (Ticona), Toraycon® (Toray), Valox® (SABIC) etc. | Automotive parts, electrical (as an insulator in the electrical and electronic industries), footwear, recreation equipment, appliances, furniture etc. |
| PBT yarn: sportswear, underwear, outerwear, car interior textiles etc. | ||
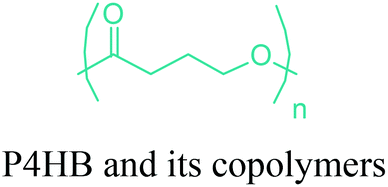
|
PHA4400 (Tepha Inc.) | Medical applications and devices: cardiovascular, wound healing, orthopedic, drug delivery and tissue engineering. |
| Ex: tissue engineered heart valve, vascular grafts, stents, patches, sutures etc. | ||
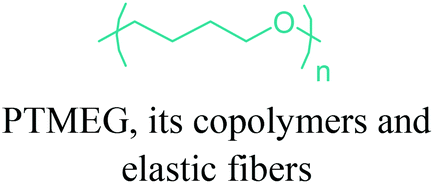
|
Terethane (Invista), (PolyTHF) BASF | Stretchable fabrics, artificial leather, apparel and clothing, compression garments, home furnishing. |
|
Stanyl (DSM Engineering Plastics) | Electrical & electronics, outdoor power equipment, automotive |
|
EcoPaXX™ (DSM Engineering Plastics) |
Through hydrogenation of succinic acid, 1,4-BDO,47,48 γ-butyrolactone (GBL)48,49 and THF48,50 are obtained (Fig. 2a). Luque et al. proved that the selectivity of the formation of these products can be tuned by the suitable choice of the catalyst.48 The production of polybutylene terephthalate (PBT) (Table 2) is the second largest use of 1,4-BDO. Recently, there are over 50 companies engaged in the production of PBT. Among these companies, Toray has recently manufactured a partially bio-based PBT using 1,4-BDO made with Genomatica's bio-based process technology. PBT exhibits excellent thermal stability, high strength, high chemical resistance and good durability. It is also easily made into yarn which has a natural stretch similar to Lycra®. These characteristics of PBT polyester resins and yarns lead to its many uses. A great deal of effort has also been devoted to further broaden its current applications by changing its crystallinity and crystallization rate.240
In terms of ring opening polymerization (ROP), poly(4-hydroxybutyrate) (P4HB) (Table 2) is produced from GBL. However, the polymerization reaction gives low molecular weight products.241 Nonetheless, the copolymers of GBL with L-lactide, glycolide, β-propiolactone, δ-valerolactone and ε-caprolactone were reported. It was stated that the use of GBL as a monomer for the synthesis of biodegradable polymer materials offers distinct advantages.242,243 P4HB polyester belongs to the polyhydroxyalkanoates (PHAs) family (Section 5.16). So, another and more efficient production route for the production of P4HB is the fermentation process, in which a much higher molecular weight, strong and flexible plastic can be derived. For instance, Tepha Inc. currently produces PHA4400, the commercial product of P4HB, for medical applications using a proprietary transgenic fermentation process.244 Unlike GBL, the ROP of THF has attracted much more attention. The resulting linear polymer of the ring opening process is poly(tetramethylene oxide) (PTMO). The most important difunctional derivative of PTMO is PTMEG (Table 2), an α,ω-dihydroxy-terminated polymer. PTMEG has turned out to be a large-scale commercial product and the main use of this polymer is to make thermoplastic elastomers.245 In this manner, it can be either reacted with diisocyanates to produce thermoplastic polyurethane elastomers or reacted with diacids or their derivatives to produce thermoplastic polyester elastomers. Thermoplastic elastomers contain two different types of blocks or chain segments to form a hard and a soft segment.246 In particular, PTMEG is often applied as polyol because compared to other soft segments, it renders low temperature flexibility, low content of extractable substances, microbial resistance and hydrolytic stability.247 For instance, the main use of PTMEG is to make spandex (Elastane, Lycra), a highly elastic synthetic fiber. PTMEG based spandex is typically produced by first making a prepolymer of PTMEG and methylene diphenyl diisocyanate (MDI). Later on, the prepolymer is extended via a diamine extension process in which ethylenediamine is often preferred.248 Other examples of PTMEG based commercial thermoplastic elastomers include Arnitel (DSM), Hytrel (DuPont) and Pibiflex (SOFTER). Common applications of these products are summarized in Table 2.
Through reductive amination of succinic acid by using amine, ammonium or ammonia and optionally alcohol, 2-pyrrolidone can be synthesized (Fig. 2a).51 2-Pyrrolidone can undergo an anionic ROP. The corresponding polymer, PA-4, has been known to be difficult to synthesize and process due to its thermal instability. Its melting and decomposition points are very close and when melted it reverts to its thermodynamically stable monomer.249 Although it has not been commercially produced due to its insufficient thermal stability, PA-4 membranes show interesting dialysis properties which are comparable to those of commercial cellophane and cellulose acetate membranes.238
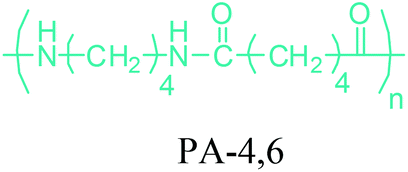
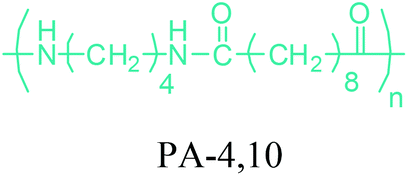
Either by employing metal catalysts or biocatalytic methods, 1,4-BDO can be transformed to its diamine product: 1,4-butanediamine (putrescine) (Fig. 2a). In the presence of metal catalysts, the primary alcohol firstly gives the corresponding carbonyl compound and hydrogen and then the imine product. Subsequent hydrogenation of the imine leads to the desired amination product.56 Putrescine has been extensively used in the production of polyamides. DSM Engineering Plastics currently uses putrescine to produce PA-4,6 and PA-4,10 (Table 2), which have been marketed as Stanyl™ and EcoPaXX™, by using adipic acid and sebacic acids as co-monomers, respectively.236 These polyamides have led to the emergence of novel technologies. For example, EcoPaXX™ has started to be used in the engine cover of the Mercedes Benz A-Class engine. Moreover, Bauser, an injection molding company located in Wehingen, Germany, has successfully introduced a production process for automotive gears using Stanyl™. PA-4I (I: isophthalic acid), PA-4T (T: terephthalic acid), and PA-4,2 are the other polymers of putrescine which were studied.250
In addition to succinic acid, the other diacid members of this platform, fumaric and malic acids, can be obtained through fermentation which overproduces C4 diacids from Krebs cycle pathways (Fig. 2a).40,55 Homopolymers of these diacids, either alone or in combination with other polymers, have been used for general biomedical applications. Unlike succinic acid, having a double bond in its structure can allow fumaric acid to form cross-linked, degradable polymer networks with tunable material properties. Among them, poly(propylene fumarate) (PPF) has been widely investigated because PPF-based polymers can be crosslinked in situ to form solid polymer networks for injectable applications. Recently, Kasper et al. reported a novel protocol for the synthesis of PPF by a two-step reaction of diethyl fumarate and propylene glycol for its potential utilization in the fabrication of orthopedic implants, scaffolds for tissue engineering, controlled bioactive factor delivery systems and cell transplantation vehicles.251 By providing high water solubility, poly(malic acid) was specifically studied for the design of targeted drug delivery systems.252
Recently, bio-succinic acid costs higher than its petroleum based counterpart. Although this is the case, microbial production of succinic acid has been attracting a lot of interest mainly due to three factors: (i) the estimated $115.2 million market value of succinic acid in 2013 is expected to reach $1.1 billion by 2020.253 So, bio-based feedstocks are needed to compensate this demand. (ii) The succinic acid produced through traditional chemical routes using petroleum-based starting materials cannot give high purity and yield without the usage of high temperature, high pressure, and catalysts.254 On the other hand, microbial succinic acid production provides milder and environmental friendly conditions as well as high purity and yields. (iii) As the microbial production of succinic acid becomes a mature technology, the current bio-succinic acid production costs will further decrease. It is highly expected that petroleum-based succinic acid will lose its cost advantage in the near future. Microbial production of succinic acid also opens new opportunities to bio-based production of 1,4-BDO, THF, putrescine, and polymers of these such as PBS, PBT, PTMEG, PA-4,6 and PA-4,10. Hence, bio-versions of these products will expand together with the bio-succinic market.
5.3. 5-HMF & 2,5-FDCA platform based polymers
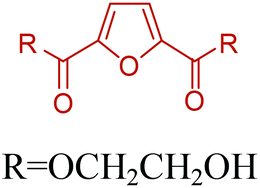
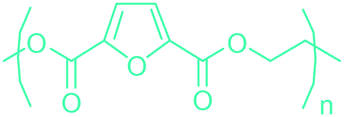
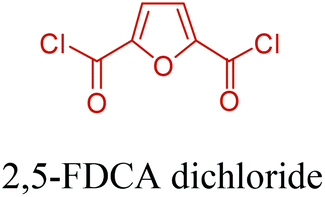
5-HMF can easily be converted to various 2,5-disubstituted furan derivatives as shown in Fig. 2b. These compounds have been called the “sleeping giants” of renewable intermediate chemicals for having enormous market potential.255 In fact, the “sleeping giants” have already woken-up, and started to claim their place in today's market. As an example, Avantium has developed 100% bio-based polyethylene-furanoate (PEF) (Table 3) bottles, fibers and films under the brand name of YXY by using plant sugar derived 2,5-FDCA.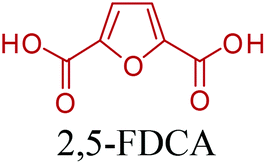
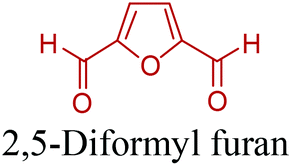
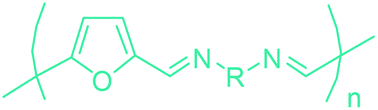
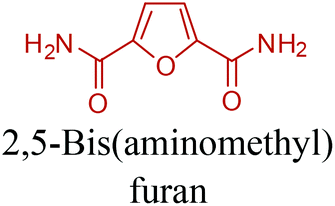
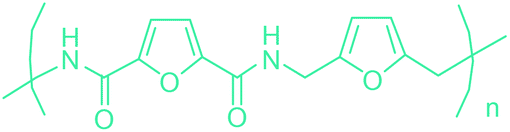
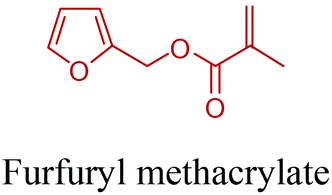
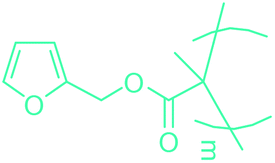
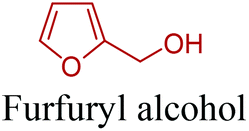
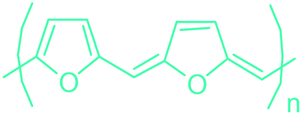
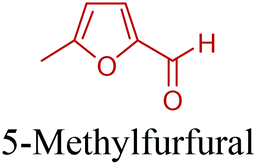
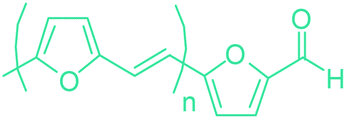
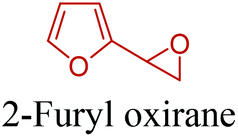
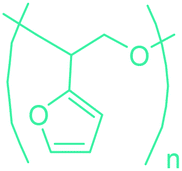
5-HMF has caused its derivatives to become potential building blocks for step-growth polymers. In addition to this, the development of the vinyl polymers inexpensively from biomass-derived 5-HMF can introduce new substituents for commodity polymers. For instance, Yoshida et al. synthesized novel biomass-based vinyl polymers from 5-HMF. They efficiently converted HMF or its methylated derivative, 5-(methoxymethyl)furfural (5-MMF), to the vinyl monomers by the Wittig reaction in a solid-liquid phase transfer process, followed by free radical polymerization (Fig. 9).256
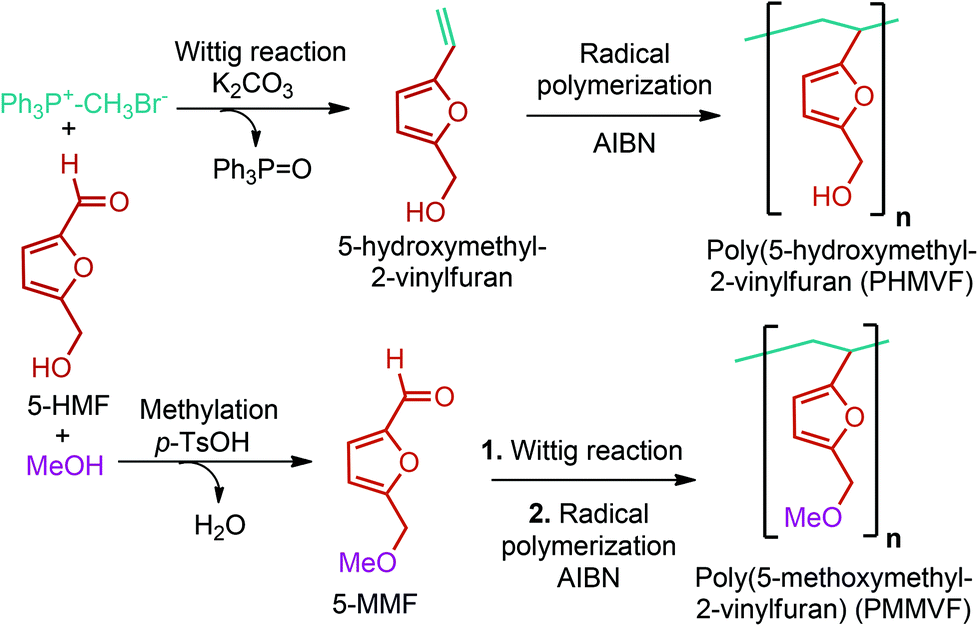 | ||
| Fig. 9 Synthesis of PHMVF and PMMVF. Adapted from ref. 256. | ||
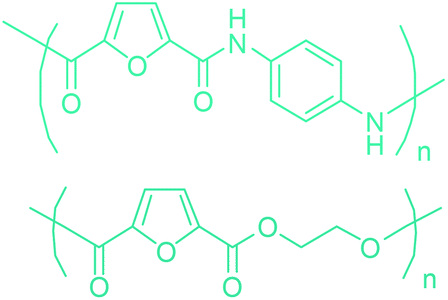
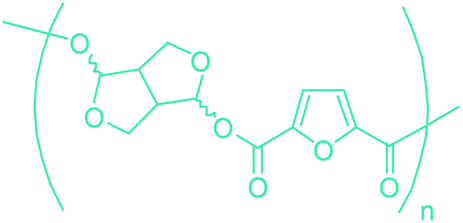
Table 3 summarizes some of the most important furanoic homo- and copolymers mainly in the frame of furan ring based monomers (Fig. 2b and 4d). The presented furan-based polymers can offer distinct advantages and special features. As such, the aromatic nature of the furan ring allows the synthesis of conjugated polymers, especially for optoelectronic applications. Recent progress in the synthesis, characterization, and physical properties of 5-HMF derived furanoic polymers including poly-Schiff-bases, polyesters, polyamides, polyurethanes, polybenzoimidazoles and polyoxadiazoles was reviewed in detail by Amarasekara.260 Also, Gandini discussed a wide range of furan-monomers and their corresponding polymers.257 Amongst these monomers, particular attention has been recently paid to 2,5-FDCA. The oxidation of HMF to FDCA was mainly catalyzed by Pt, Pd, and Au-based catalysts. The polyamide, which is produced via polycondensation of 2–5-FDCA and p-phenylenediamine (PPD), can render high strength by forming many interchain hydrogen bonds as in the case of its entirely aromatic counterpart Kevlar.257 But more importantly, 2,5-FDCA shows comparable properties to terephthalic acid (TA) in a range of polyesters. Hence, Avantium has been developing next generation 2,5-FDCA based bioplastics, called “YXY building blocks”, to replace oil-based polyesters. For instance, Avantium aims to replace oil-based PET with its furanoic counterpart: polyethylene furanoate (PEF) (Table 3). For PEF to become a successful replacement polymer, it has to compete with PET in terms of price and performance as well as deliver a better environmental footprint. Regarding the physical properties, PEF has a higher glass transition temperature (Tg), heat deflection temperature (HDT) and tensile strength but lower melting temperature (Tm) and elongation to break than PET. PEF bottles exhibit superior barrier properties than PET: for both H2O and CO2 more than two times and for O2 six times better. Also, recyclability of PEF should be excellent. It was shown that both mechanical and chemical recycling of PEF is feasible. Moreover, PEF production can reduce the non-renewable energy usage and greenhouse gas emissions by 40–50% and 45–55%, respectively, compared to PET. It is expected that the price of 2,5-FDCA will drop below €1000 once its production capacity exceeds 300 kt per year and then it will be competitive with purified terephthalic acid (TA) produced at the same scale. All these findings suggest that PEF can compete with PET in terms of price and performance, particularly in bottling applications, and it can provide a significantly better environmental footprint.258,263
Recently, one of the most important motivations to study furan-derived monomers is their utilization in Diels–Alder (DA) reactions for new polymeric materials. Both the DA and retro-DA reactions between furan and maleimide, which are straightforward and not affected by side reactions, have been well investigated over the past several years (Fig. 10). The DA reaction can be employed to produce both linear and branching polymers by a simple modification of the number of maleimide funtionalities.261
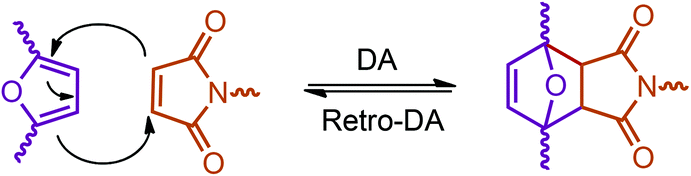 | ||
| Fig. 10 DA and retro-DA reactions between furan and maleimide. | ||
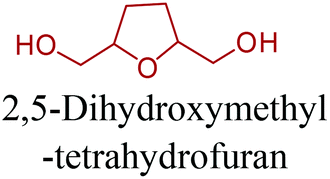
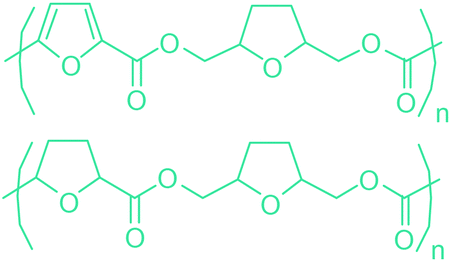
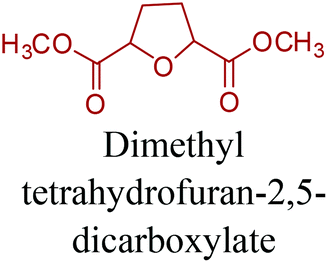
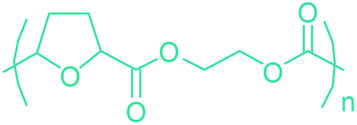
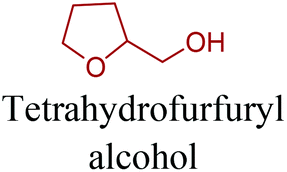
Sequentially, dihydrofuran and/or tetrahydrofuran based monomers can be obtained via hydrogenation of furan ring based compounds as shown in Fig. 2b and 4d. The resulting new monomers’ behaviors are quite dissimilar to that of their furan precursors because the aromatic nature of the furan ring is now broken down in the process. Through the step-growth polymerization method, THF ring bearing polyesters264 and polyamides265 are reported and some of their literature examples are summarized in Table 4. In terms of ROP, another derivative of THF, tetrahydrofurfuryl alcohol, can give cationic-ROP for the production of its homo- or hyperbranced-polymer architectures.266,267
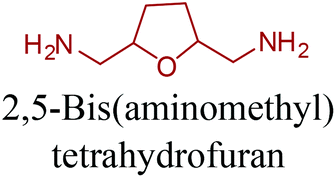
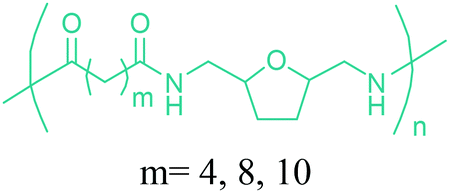
Buntara et al. proposed four different reaction pathways for the conversion of 5-HMF into 1,6-hexanediol (1,6-HDO). These pathways include: (i) the direct hydrogenation of 5-HMF to 1,6-HDO. (ii) A two-step process through 2,5-THF-dimethanol (THFDM) intermediate. (iii) A three-step synthesis through THFDM and 1,2,6-hexanetriol (1,2,6-HT) intermediates, respectively. (iv) A four-step synthesis via THFDM, 1,2,6-HT, and tetrahydro-2H-pyran-2-ylmethanol intermediates.62 The first route is much more desirable since it avoids the usage of intermediate steps and chemicals. Once obtained, the double diamination of 1,6-HDO yields 1,6-hexanediamine (1,6-HDA) (Fig. 11).56 The conversion of 5-HMF to 1,6-HDO, and subsequently to 1,6-HDA is particularly important since these products have a last-born and very large polymer market. Successful commercialization of their polymers mainly depends on their fairly long hydrocarbon chain, which provides hardness and flexibility to their incorporating polymers, and high reactivity of their terminal alcohol or amine groups. Ube Industries Ltd currently produces 1,6-HDO based polycarbonatediols under the trade name of Eternacoll® (Fig. 11), particularly for the production of polyurethanes as coatings, elastomers and adhesives.269 In the case of 1,6-HDA, more than 95% of it is reacted with adipic acid for the manufacture of PA-6,6.270,271 Recently, the world's largest producer of this polyamide has been Invista and this polyamide is commercialized as Torzen® for general use in industrial, textile, and automotive applications (Fig. 11). 1,6-HDA can be effectively converted to its diisocyanate derivative, hexamethylene-diisocyanate (HDI), by employing a phosgene-free synthetic approach.272 The resulting new monomer is especially useful for the synthesis of polyurethanes (PU).
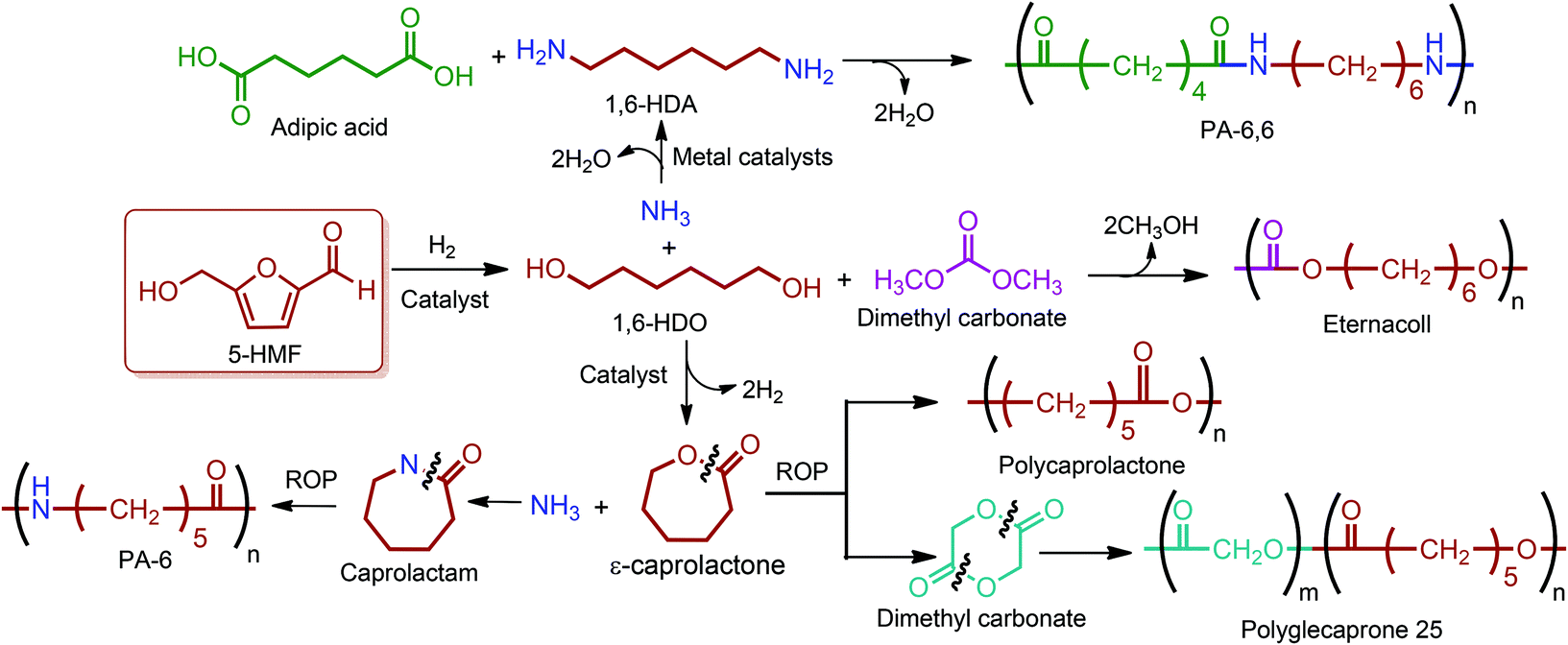 | ||
| Fig. 11 Commercial polymer products of 1,6-hexanediol, 1,6-hexanediamine, ε-caprolactone and caprolactam. | ||
5-HMF derived 1,6-HDO can be alternatively oxidized to ε-caprolactone. In this process, 1,6-HDO is oxidized to its corresponding monoaldehyde, which cyclizes spontaneously to the lactol, and then the dehydrogenation of the lactol yields ε-caprolactone. Further amination of ε-caprolactone gives caprolactam (Fig. 11), which is the monomer for PA-6, a widely used synthetic polymer with an annual production of about 4 million tons. This 5-HMF-based bio-route may become an alternative to the traditional caprolactam synthesis process. The bio-route has only four steps; on the other hand, the traditional caprolactam process requires six steps starting from benzene and ammonia.62 In this respect, the bio-route is more feasible but still requires process development in terms of product yields and cost competitiveness.
Although the great majority of ε-CL has been used for the commercial production of caprolactam, it is also converted to its commercially available homopolymer, polycaprolactone (PCL), via the ROP of it using a variety of anionic, cationic and co-ordination catalysts. Perstorp (CAPA), Dow Chemical (Tone) and Daicel (Celgreen) are the major producers of this polyester. Woodruff and Hutmacher have reviewed the important applications of PCL as a biomaterial over the last two decades with a particular focus on medical devices, drug delivery and tissue engineering.273 Another significant industrial application of ε-CL is its utilization in the production of polyglecaprone 25, the copolymer of glycolide and ε-CL. This copolymer is formed into filaments for use as sutures and has been trademarked by Ethicon as Monocryl®.274 For caprolactam, approximately 90% of it is processed to PA-6 filament and fiber (Perlon), and most of the remaining 10% is used for the manufacture of plastics.275 This aliphatic polyamide is trademarked as Capron®, Ultramid® and Nylatron®. It is exceptionally useful whenever high strength and stiffness are required. The applications and properties of PA-6 and PA-6,6 are described in detail by James E. Mark in “Polymer Data Handbook”.270 Also, important commercial polymers of the 5-HMF & 2,5-FDCA platform are depicted in Fig. 11.
5.4. 3-HPA platform based polymers
3-HPA is a key building block for the production of high value products such as acrylic monomers, 1,3-propanediol (PDO), propene, malonic acid, β-propiolactone, and 3-hydroxypropionic esters (Fig. 2c).68 It can be chemically produced (i) through oxidation of PDO in the presence of a Pd-containing supported catalyst, (ii) through oxidation of 3-hydroxypropionaldehyde in the presence of a Pd-containing supported catalyst, or (iii) through hydration of acrylic acid in the presence of acid catalysts. However, these chemical routes are too costly and, thus, a biosynthetic pathway is required for large scale production of 3-HPA. In this consideration, cheap sugar feedstocks should be employed to obtain 3-HPA in a commercially efficient way. Even so, no known organisms can produce 3-HPA as a major metabolic end product from sugars. Hence, large scale production of 3-HPA can be realized through fermentation of sugars by employing genetically modified microorganisms. The process requires an improved microbial biocatalyst as well as a reduction in the production cost.64,683-HPA and its various derivatives, including 3-hydroxypropanal, β-propiolactone or its oligomers and esters of 3-HPA, can be used for the production of 1,3-propanediol (1,3-PDO) through the hydrogenation process (Fig. 2c).54,65,68 In fact, direct production of PDO through the fermentation of sugars is more feasible and, thus, the current industry on large scale production of 1,3-PDO has been shifting to this direction. As such, DuPont and Tate & Lyle have recently formed a joint venture for the production of bio-PDO from fermentation of corn sugar by using a genetically modified strain of E. coli.276 It was claimed that the bio-PDO process results in 40% less energy consumption and 20% reduction of greenhouse gas emissions. Based on the corn derived bio-PDO, DuPont has commercialized the Sorona® family of poly(trimethylene terephthalate) (PTT), the polyester of 1,3-propanediol and terephthalic acid (or dimethyl terephthalate).277
3-HPA can be directly converted to acrylic acid by thermal dehydration at reduced pressure using homogeneous acid catalysts in the presence of a copper powder. This method provides yields of acrylic acid of around 80%. Higher yields are attained when heterogeneous catalysts, such as NaH2PO4 supported on silica gel, are employed.54 As an alternative route, 3-HPA can be converted to propene through a dehydration reaction coupled with a decarboxylation process (Fig. 12). But this reaction is yet to be realized. Once propene is obtained, the vapour phase oxidation of propene in the presence of oxygen to acrylic acid through acrolein intermediate is the most widely accepted commercial process. So, as shown in Fig. 2c, the dehydration of 3-HPA, with or without further modification of its carboxylic unit, can open a completely new window to the world of acrylic monomers and their corresponding polymeric materials.54,66–69,133,142 The realization of technology in the end will depend either on (i) the cost competitive production of 3-HPA and its efficient dehydration to acrylic monomers, or (ii) the cost competitive production of 3-HPA derived bio-propene with respect to the petroleum derived propene.
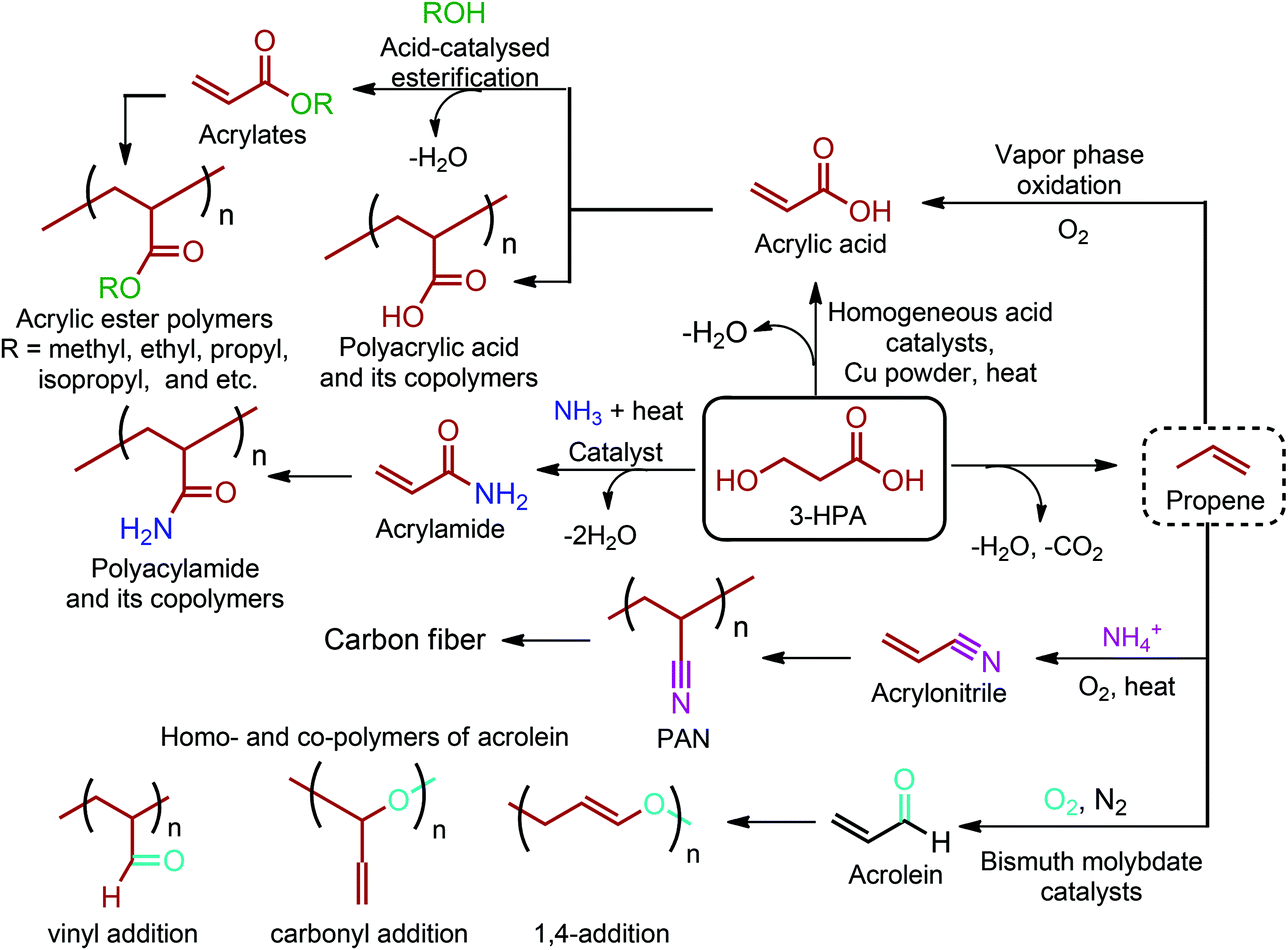 | ||
| Fig. 12 Industrially important 3-HPA derivable acrylic polymers. | ||
Production of acrylic monomers starting from 3-HPA is quite important owing to the giant market size of acrylic polymers. Almost all commercially produced acrylic polymers contain acrylic acid and/or methacrylic acid to some extent. Hence, homo- or copolymers of acrylic (Fig. 12) and methacrylic acids have a myriad of applications. Many companies have been involved in the production of these polymers. For polyacrylic acid, some of them include The Dow Chemical Company (Acrysol™, Acumer™, Acusol™, and Dualite™), AkzoNobel (Alcogum®, Alcosperse®, and Aquatreat®), Lubrizol (Carbopol®) and BASF (Sokalan®). Swift summarized common applications of polyacrylic acid and polymethacrylic acid under their specific types including emulsion, water-soluble, alkali-soluble, gel, block and graft polymers.270,278 In terms of acrylates, methyl acrylate and ethyl acrylate were the first produced derivatives of acrylic acid. The physical properties of acrylic ester polymers (Fig. 12) greatly depend on their R ester groups. For instance, the glass-transition range can vary widely among the acrylic ester polymers from −65 °C for 2-ethylhexyl acrylate (R = C4H9) to 103 °C for acrylic acid (R = H). Since the R side-chain group conveys such a wide range of properties, acrylic ester polymers are used in various applications varying from paints to adhesives, concrete modifiers and thickeners. More detailed information regarding many different acrylic esters and their corresponding polymers can be found in Slone's article.279
Acrylamides are also produced from 3-HPA by simply heating mixtures of 3-HPA and an amine without or with a catalyst which enhances the rate of the dehydration reaction (Fig. 12).54 Although acrylamide is toxic, its homopolymer, polyacrylamide, is harmless and it has a wide range of applicability. Huang et al. have extensively reviewed a very large number of applications of acrylamide-containing polymers. Some major applications of these polymers include flocculants in water treatment, paper manufacture, mining, oil recovery, absorbents, and gels for electrophoresis.270,280
Acrylonitrile and acrolein are the other two industrially important acrylic derivatives of 3-HPA. These monomers can be obtained from 3-HPA derived bio-propene (Fig. 12). Acrylonitrile is produced commercially via SOHIO's catalytic vapor-phase propylene ammoxidation process in which propylene, ammonia, and air are reacted in the presence of a solid catalyst at 400–510 °C.67 On the other hand, propene is selectively oxidized to acrolein in the presence of bismuth molybdate catalysts and nitric acid.66
Acrylonitrile has unalterable status in today's fiber industry. It is the major monomer of DuPont's first acrylic fiber, Orlon. Moreover, polyacrylonitrile (PAN) fibers are the chemical precursor of high-quality carbon fiber. Carbon fibers are uniquely qualified for use in both high-tech and common daily applications since they have the inherent combination of high strength, high stiffness and light weight.281 Wu stated that acrylonitrile easily copolymerizes with electron-donor monomers and there are over 800 acrylonitrile copolymers that have been registered. Some of the important copolymers include styrene-acrylonitrile (SAN), acrylonitrile-butadiene-styrene (ABS), acrylonitrile-styrene-acrylate (ASA), acrylonitrile-butadiene (ANB) and acrylonitrile–methyl acrylate (AN/MA).270,282
Acrolein can polymerize in three different ways: by 1,2-addition at the vinyl group or at the carbonyl group or by 1,4-addition as a conjugated diene across the carbonyl and α,β unsaturation (Fig. 12). Acrolein homo- and co-polymers generally have been used as biocidal and biostatic agents. Moreover, a commercial product having both pendant aldehyde and carboxylic acid groups is obtained by oxidative co-polymerization of acrylonitrile with acrylic acid. The material has the Degussa trade name POC and it is particularly useful as a sequestering agent for water treatment. Schulz reviewed radical, anionic and cationic polymerizations of acrolein and more detailed information on acrolein as a monomer and its polymers can be found in this article.283
5.5. Aspartic acid platform based polymers
Large scale production of aspartic acid has recently been realized from ammonia and fumaric acid by employing immobilized aspartase from E. coli or suspended cells of Brevibacterium bravum. Its direct production from sugars is more desirable for cutting-off intermediate steps. However, this is not possible with the current technology because a favorable equilibrium of the aspartase-catalyzed reaction requires toxic concentrations of intracellular ammonia and fumaric acid.284 Thus, bio-based fumaric acid is required to industrially produce aspartic acid from renewable feedstocks.As a chemical building block, aspartic acid leads to the formation of many valuable compounds including fumaric acid, maleic acid, 2-amino-1,4-butanediol, β-alanine, aspartic anhydride and amino-γ-butyrolactone (Fig. 2d).40,70–73,284 Also, aspartic acid based polymers, poly(amino acids) with free carboxylic groups, have attained the greatest commercial success in terms of water soluble biodegradable polymers. Poly(aspartic acid)s (PASA) can be produced by different routes. Thermal polyaspartate (TPA) production is the simplest and oldest approach. In this process, powdered L-aspartic acid is firstly heated to high enough temperature, at least 185 °C, to initiate a condensation reaction. Then, the temperature is increased to 225 °C and maintained at that temperature until at least 80% conversion of L-aspartic acid to polysuccinimide (PSI) is achieved. The subsequent step includes the alkaline hydrolysis of the intermediate PSI polymer to give the resulting TPA polymer (Fig. 13). TPAs are functionally equivalent to poly(acrylic acid) and highly linear TPAs are fully biodegradable. These distinctive features of TPAs make them one of the best target polymers for use in three global markets: performance chemicals, diapers, and agriculture, which have a global market potential of $20 billion.285,286
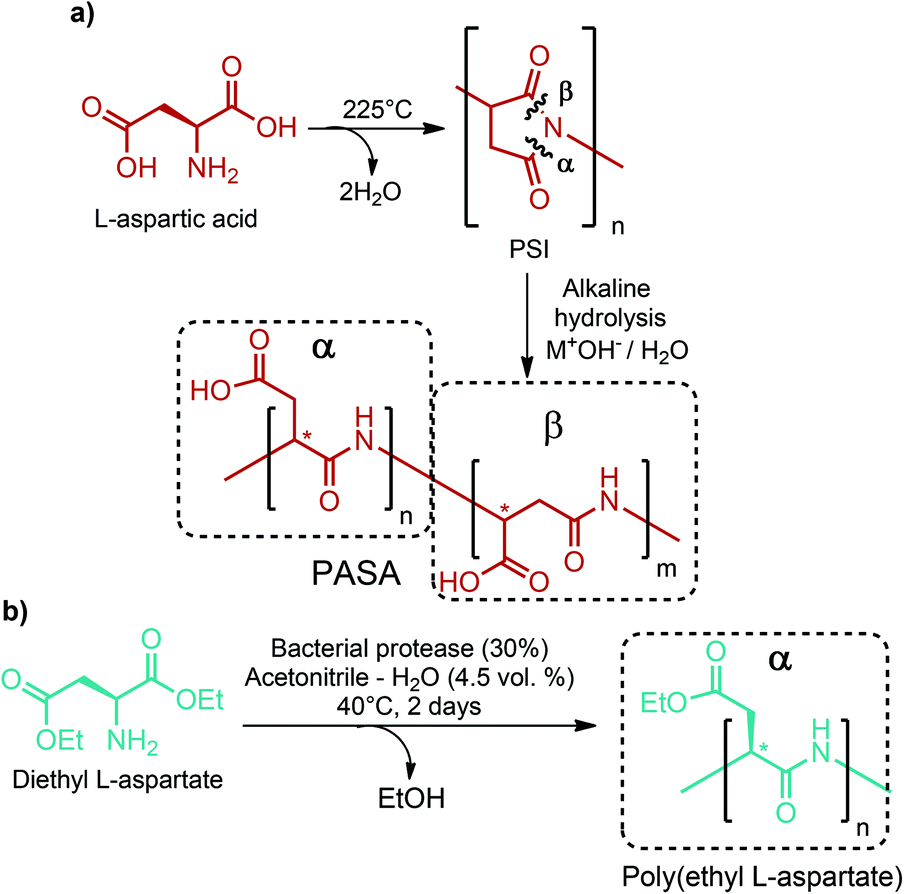 | ||
| Fig. 13 PASAs obtained via (a) the TPA and (b) the bacterial protease methods. | ||
There are certain disadvantages of the TPA process. First of all, a copolymer is always obtained at the end of the polymerization. In addition, nearly complete racemization occurs during the thermal polycondensation reaction and the alkaline hydrolysis may proceed at both carbonyls of the PSI. Hence, the resulting copolymer not only consists of D- and L-isomers, but may also contain α- or β-peptide bonds in the main chain (Fig. 13).286 Control over the repeating unit isomers, D- or L-PASA with pure α- or β-links, can be achieved by enzymatic preparation of PASA. For instance, Soeda et al. polymerized diethyl L-aspartate by employing a bacterial protease from Bacillus subtilis to yield only α-linked poly(ethyl L-aspartate) having an Mw of up to 3700. The best polymerization result was obtained using 30% protease Bacillus subtilis in acetonitrile containing 4.5 volume percent of water at 40 °C for 2 days (Fig. 13).287 Although the enzymatic preparation route can give only D- or L-PASA with pure α- or β-links, it is not a suitable approach for large scale production. The TPA process is much cheaper and more suitable for commercial manufacture of PASA. Nonetheless, the enzymatic process is quite useful for specialty polymer applications in which the isomeric purity of PASA really matters.
PSI has also been used as a polymer precursor for the production of polyaspartamide (PAA) based polymers apart from being an intermediate polymer for the production of commercial TPA. Recent literature studies on PAA derived polymers mainly focus on their use as drug carriers and stimuli-responsive polymeric materials.288,289 As a recent example, Xu et al. synthesized a novel polyaspartamide derivative composed of partially tetraethylenepentamine grafted poly(aspartic acid) (PASP-pg-TEPA) nanoparticles as a potential intracellular delivery system (Fig. 14).290
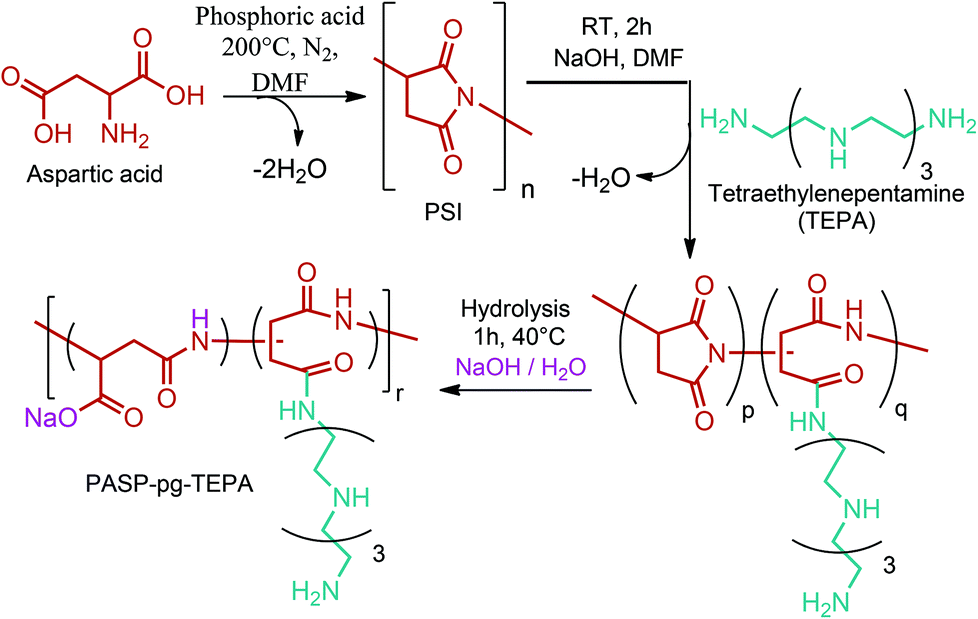 | ||
| Fig. 14 Synthesis of the zwitterionic polyaspartamide derivative composed of PASP-pg-TEPA. Adapted from ref. 290. | ||
α-Decarboxylation of aspartic acid results in the formation of β-alanine (Fig. 2d).70,284 On the other hand, it is highly difficult to achieve this reaction through chemical methods because the β-carboxyl group of aspartic acid can also undergo a decarboxylation reaction. The perfect conversion can be realized using E. coliL-aspartate α-decarboxylase. However, this enzymatic method still requires process development mainly due to the occurring enzyme inactivation during the catalysis.284 Solution of this problem is critical for large scale enzymatic synthesis of β-alanine. Once cost competitive β-alanine is industrially available, it may open new opportunities for the production of nitrogen containing base chemicals such as acrylamide and acrylonitrile. Besides, the direct condensation of β-alanine, or its esters, is used for the preparation of PA-3 (poly(β-alanine)), which is used as a stabilizer for polyoxymethylene, and polyacetal resin. It is also an excellent formaldehyde scavenger.270 Apart from these, Liu et al. have revealed the intriguing biological properties of PA-3. They introduced a new family of PA-3 polymers (poly-β-peptides) and these polymers were shown to display significant and selective toxicity toward Candida albicans, the most common fungal pathogen among humans.291 In another study, they reported that PA-3 backbone based polymers selectively support in vitro culture of endothelial cells but do not support the culture of smooth muscle cells or fibroblasts. There are very few reported synthetic material examples which can display selective cell growth. Hence, these findings of Liu et al. on selective encouragement of the growth of specific cell types are very valuable for the engineering of complex tissues.292
Amino acid-based polymers of aspartic acid and β-alanine, and also other lignocellulosic biomass derived amino acids such as glutamic acid, γ-benzyl glutamate, norvoline, γ-aminobutyric acid (Section 5.6), 5-aminolevulinic acid (Section 5.9) and lysine (Section 5.18), may offer many different advantages. These polymers can be modified further to introduce new functions such as imaging and molecular targeting. Their degradation products are expected to be non-toxic and readily excreted from the body. Drugs can be chemically bonded to these polymers. Moreover, these polymers can enhance biological properties like cell migration, adhesion and biodegradability, and improve mechanical and thermal properties. Three major applications of them include drug delivery, gene delivery and tissue engineering. Various types of polymerization techniques, particularly different polycondensation and ROP reactions, can be employed to produce polymers consisting of amino acid moieties in the main chain. Besides, radical, anionic and cationic polymerizations are generally used to obtain polymers consisting of amino acid moieties in the side chain. On the basis of these techniques, it has been reviewed that polyamides, polyesters, polyurethanes, polyacetylenes, polymethacrylamides, polyanhydrides, polyphosphazines, polysulfides, poly(amide-ester-imide)s, poly(amide-imide)s, poly(amide-imide-urethane)s, poly(ester-amide)s, poly(ester-amide-sulfide)s, poly(ester-imide)s and poly(imide-urethane)s are produced with different architectures. Amino acid based polymers were reviewed in detail in the recent literature.293–296 Thus, no further detail is provided in this article. Nonetheless, the design of novel functional amino acid based monomers and their corresponding polymerization methods are briefly described in Fig. 15.296 Although the figure covers α-amino acid derived monomers and their polymerization methods, it can be extended to β-, γ, δ-amino acids such as β-alanine, γ-benzyl glutamate, γ-aminobutyric acid (Section 5.6), and 5-aminolevulinic acid (Section 5.9).
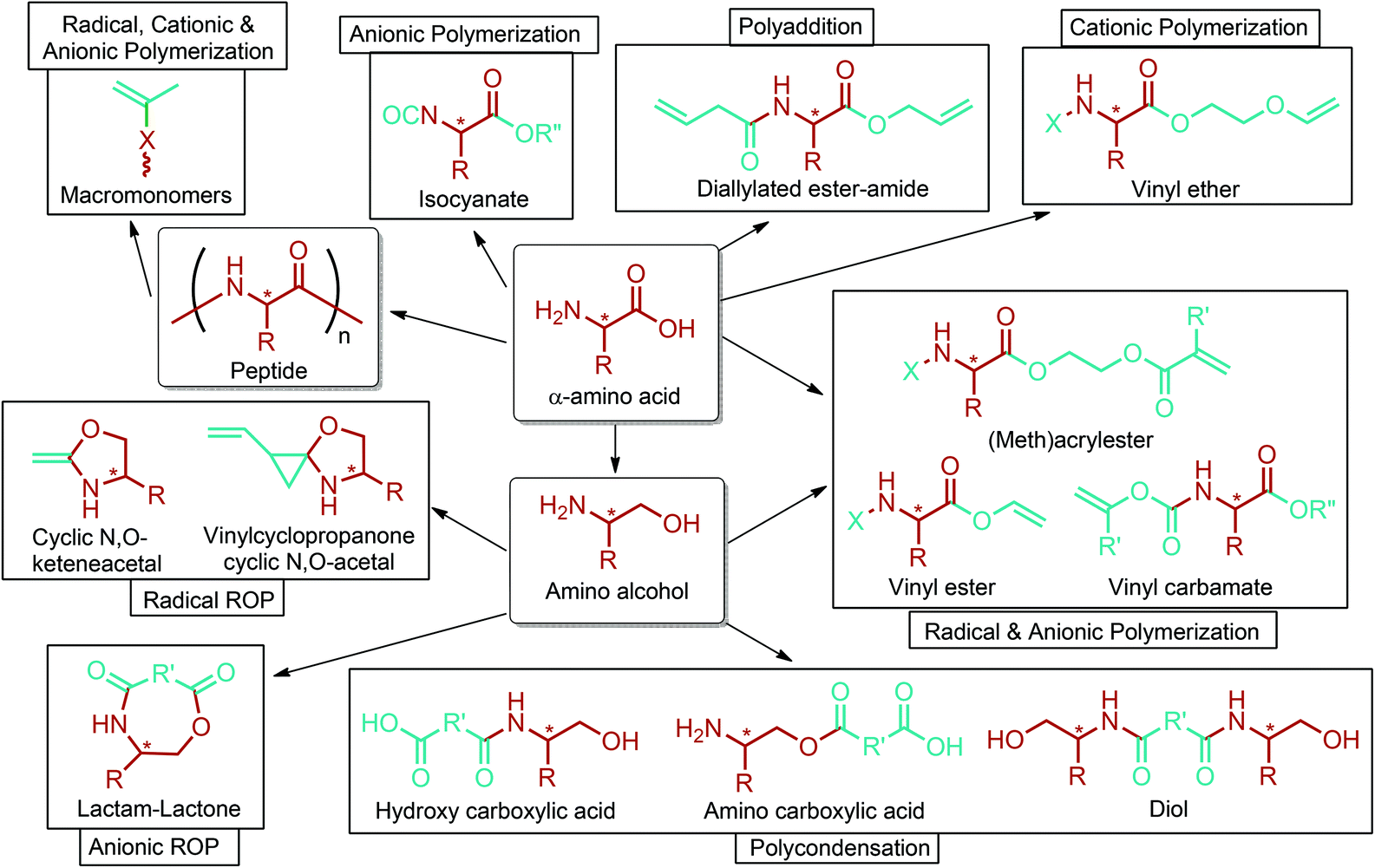 | ||
| Fig. 15 The design of novel functional monomers based on amino acids and their corresponding polymerization methods. Adapted from ref. 296. | ||
5.6. Glutamic acid platform based polymers
Currently, the global production capacity of glutamic acid is more than 200 kt per year and most of this production has been achieved through fermentation. Various species of Brevibacterium and Corynebacterium can produce glutamic acid from different cheap carbon sources such as glucose, ethanol and glycerol. Although glutamic acid is produced from these cheap feedstocks, there are certain limitations in its large scale production process. The main limitation stems from its complex processing which consists of a number of downstream treatment schemes like precipitation, conventional filtration, acidification, carbon adsorption and evaporation. All these treatments are essential to obtain high purity glutamic acid but they greatly increase the production cost in the end. Thus, a new process is required to produce glutamic acid in a more eco-friendly and economical manner. In this consideration, a membrane-based processing is envisioned to eliminate the need for separate purification units and to reduce the overall production cost.74 Thus, a shift from the current complex processing to a membrane-based technology might be observed in the glutamic acid market in the near future.Industrial production of glutamic acid is quite important for the production of the C5 compounds (Fig. 2e) and their corresponding polymers. So far, the most successful commercial polymer form of glutamic acid is poly-γ-glutamic acid (γ-PGA), an anionic biopolymer formed via the gamma-amide linkage. γ-PGA is isolated particularly from various strains of B. licheniformis and B. subtilis. It can be produced from a variety of carbon sources such as glutamic acid, glucose, fructose, sucrose, citrate and glycerol. The proposed pathway for γ-PGA synthesis from L-glutamic acid in B. subtilis IFO 3335 is shown in Fig. 16a. The recent pilot trial findings of Zhu et al. have suggested that the environmentally friendly and efficient production of γ-PGA can also be achieved using the low-cost and renewable lignocellulosic biomass.297 γ-PGA is water-soluble, edible, biodegradable, biocompatible and non-toxic for humans and the environment. Hence, γ-PGA and its derivatives have been of interest in the past few years, especially in food, cosmetics, medicine, and water treatment industries. A wide range of unique applications of them in these industries were reviewed by Shih and Van.298
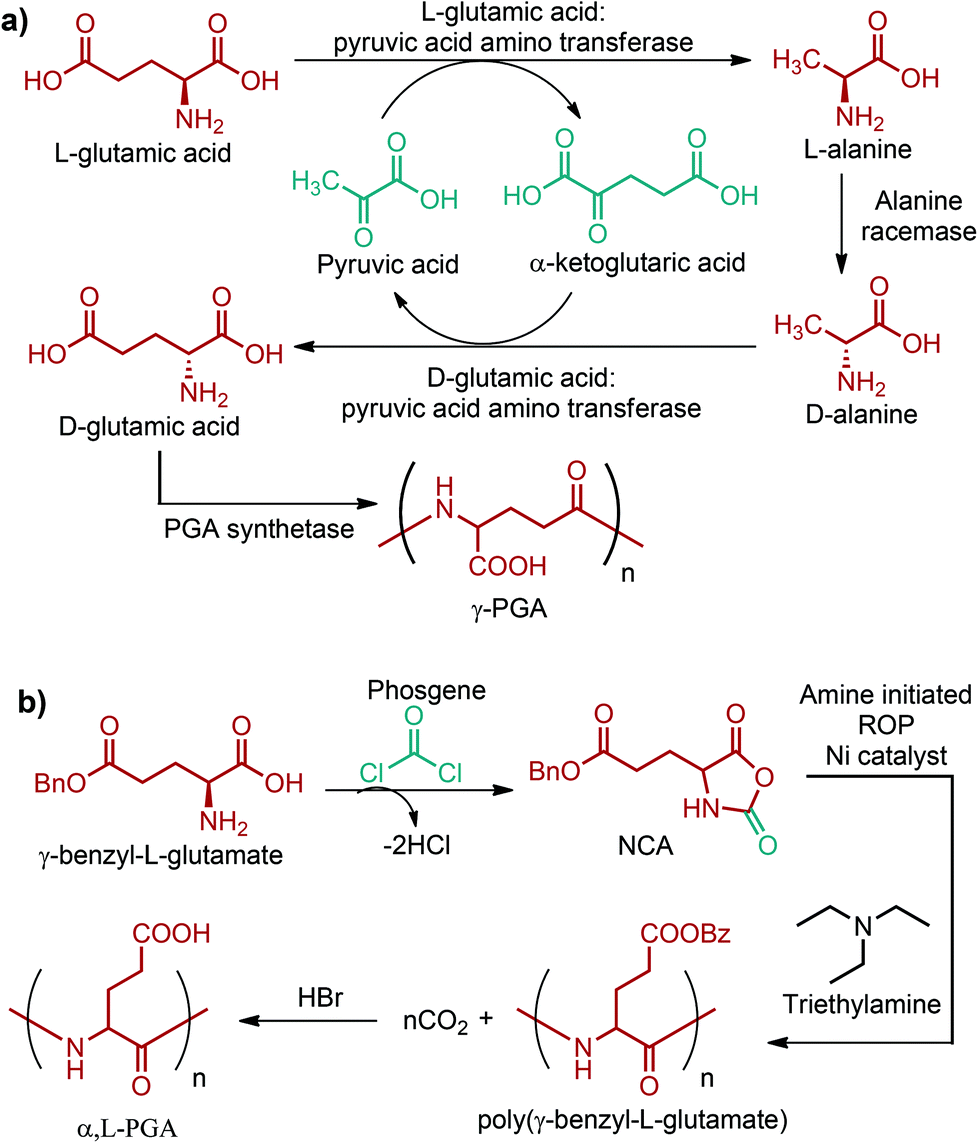 | ||
| Fig. 16 (a) Proposed pathway for γ-PGA synthesis from L-glutamic acid in B. subtilis IFO 3335 (the pathway starting from glucose to L-glutamic acid was not indicated). (b) Reaction scheme for the synthesis of α,L-PGA. | ||
A structurally different polymer of glutamic acid is poly-α,L-glutamic acid (α,L-PGA). It is generally synthesized starting from poly(γ-benzyl-L-glutamate) (PBLG). At first, γ-benzyl-L-glutamate is reacted with a carbonyl dichloride compound, such as phosphene, to give the corresponding NCA derivative. Later on, the triethylamine-initiated polymerization of the NCA of γ-benzyl-L-glutamate yields poly(γ-benzyl-L-glutamate). Finally, removing the benzyl protecting group of poly(γ-benzyl-L-glutamate) with the use of HBr in the presence of CO2 results in the formation of α,L-PGA (Fig. 16b).299 On the other hand, a better method, most probably a microbial method, is necessary because the above-mentioned route requires the synthesis of the intermediate NCA compound as well as the usage of highly toxic carbonyl dichloride compounds, and protection/deprotection steps. α,L-PGA shows the general characteristics of γ-PGA and, additionally, its carboxylic group provides functionality for drug attachment. Thus, anticancer drug conjugates of α,L-PGA have been studied extensively. α,L-PGA based anti-cancer drug conjugates were reviewed in detail by Li.299
Norvoline and γ-aminobutyric acid (GABA) are the other two amino acids of this platform in addition to glutamic acid (Fig. 2e).77,78,300 Through chemical methods, it is quite difficult to control selective reduction of γ-carboxyl or selective decarboxylation of α-carboxyl groups of glutamic acid for the production of norvoline and GABA, respectively. Hence, biosynthetic routes are more convenient for the production of these amino acids from glutamic acid. As such, the glutamate decarboxylase catalyzed reaction of glutamic acid to GABA is superior to conventional chemical methods owing to its higher catalytic efficiency and environmental compatibility and milder reaction conditions.78 Norvoline, GABA and other lignocellulosic biomass derivable monomers are quite useful for the synthesis of novel biodegradable and bio-compatible amino acid containing polymers. As an example, Rodríguez-Galán et al. studied poly(ester amide)s derived from glycolic acid and GABA or β-alanine for non-coated monofilament and flexible sutures.301 GABA or β-alanine were specifically chosen as co-monomers because they can enhance enzymatic degradation and improve intermolecular interactions of their incorporating polymers. Moreover, they can bring functionalizable groups where molecules with a pharmacological activity could be attached. Although GABA and β-alanine were the only studied amino acids, the study can be extended to other amino acids as shown in Fig. 17. Amino acid derived monomers and their corresponding polymerization techniques are high-lightened in more detail in Section 5.5.
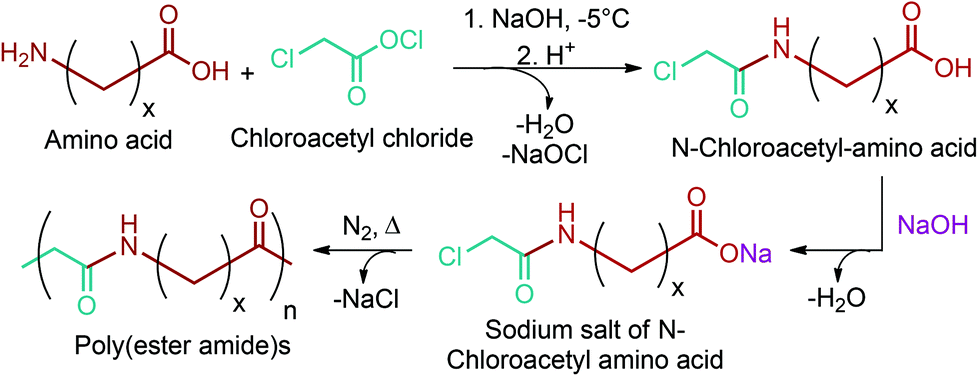 | ||
| Fig. 17 Synthesis of poly(ester amide)s based on glycolic acid and amino acids for suture applications. | ||
GABA can be effectively reduced to 4-amino-1-butanol with a conversion of more than 99.9% and a yield of 91.9% through a hydrogenation process over a Rh-MoOx/SiO2 heterogeneous catalyst (Fig. 2e).79 4-amino-1-butanol monomer has started to be widely used for the production of poly(α- or β-amino ester)s. Recently, there has been a great increase in the number of publications related to poly(amino ester)s, especially after the promising results of these polymers in gene delivery applications. For instance, Li et al. stated that cationic poly(β-amino ester)s have superior transfection efficiency and less toxicity in comparison with commercial reagents such as Lipofectamine 2000, a transfection reagent produced by Invitrogen.302 Many 4-amino-1-butanol containing poly(amino ester) systems have been reported to deliver DNA, siRNA and proteins for gene therapy applications.302–305 Different libraries of structurally related poly(amino ester)s can be produced by the systematic modification of the polymer backbone, side chain, and end group. Sunshine et al.306 introduced a new library of 320 poly(β-amino ester)s by implementing this method (Fig. 18). In the process, the primary amino alcohols were reacted with diacrylates by Michael addition to yield the corresponding diacrylate-terminated base polymers. The reaction was carried out in the presence of excess diacrylate with no solvent stirring at 90 °C for 24 h. Subsequently, the base polymers were dissolved in DMSO and then allowed to react with an end-capping amine at RT for 24 h. As can be seen from the figure, other lignocellulosic biomass derivable alkanolamines such as 1,3-propanolamin and 1,5-pentanolamin, which are reduction products of β-alanine and 5-aminolevulinic acid, respectively, can also be employed for the introduction of novel poly(amino ester) libraries.304,306
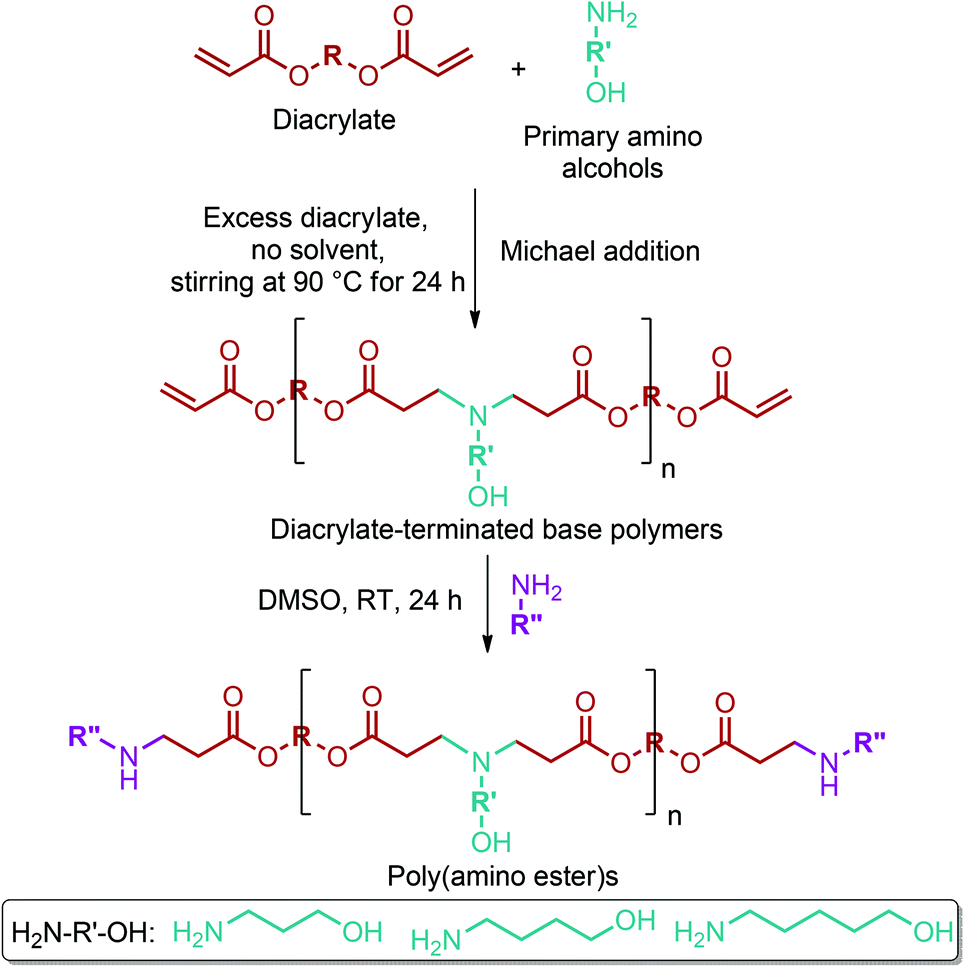 | ||
| Fig. 18 Synthesis of the poly(β-amino ester) library. Adapted from ref. 306. | ||
By providing amine functionality together with a carboxyl, or an alcohol group, glutamic acid platform chemicals can boost up the manufacture of amino acid based polymeric materials once their commercial production routes are optimized. In addition, their conversion to di-functional products such as 1,5-pentanediamine (or cavaderine), glutaric acid and 1,5-pentanediol (1,5-PDO) (Fig. 2e) opens a completely new window to the industry of polyamides and polyesters. Among these chemicals, cadaverine can be produced by the diamination process of glutamic acid derived 1,5-PDO,56 or as a different route, it can be effectively exported from E. coli or C. glutamicum cells as the decarboxylation product of L-lysine.236 Various PA-5 series can be produced by combining cadaverine with diverse diacids. For example, PA-5,10, PA-5,5 and PA-5,4 are obtained by polymerizing cadaverine with sebacic acid, glutaric acid and succinic acid, respectively (Fig. 19).307 According to Kind and Wittmann, polycondensation of microbial biosynthesis derived cadaverine with these bio-blocks provides completely bio-based PAs. Cadaverine incorporating PAs possess desirable properties such as high melting points and low water absorption. Hence, they are proposed to be bio-based alternatives to several petroleum-based PAs.308 In fact, Carothers invented PA-5,10 even before PA-6,6;309 a keystone in today's polymer industry. However, PA-5,10 is more expensive to make even though it has superior properties. So, industrial shifts from conventional PAs to bio-based PA-5,10 products might be observed in the future depending on the cheaper and more efficient production of cadaverine from biomass resources than its petroleum-based production routes.
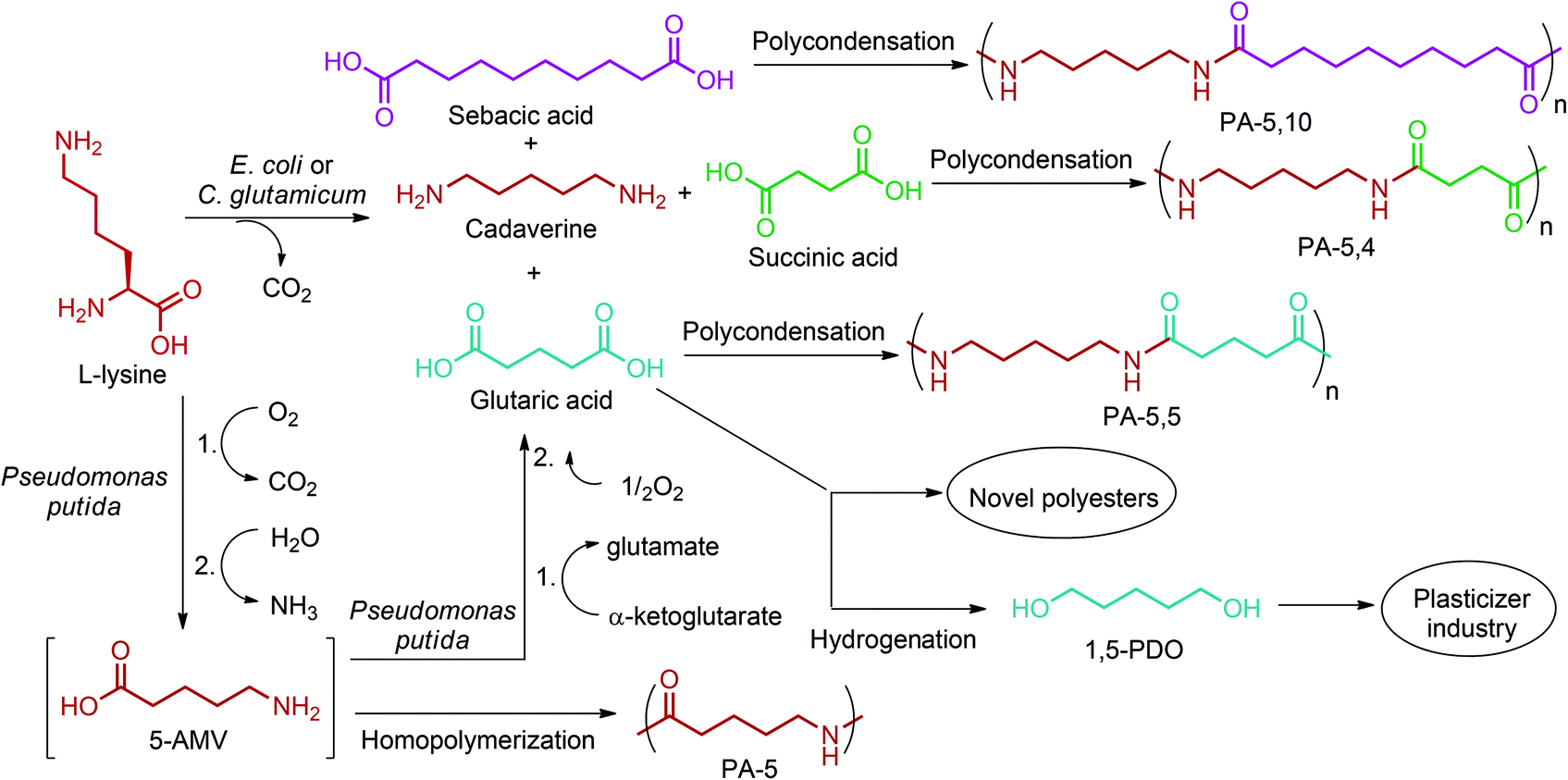 | ||
| Fig. 19 Glutamic acid platform based PAs and PEs. | ||
Like cadaverine, glutaric acid (Fig. 2e) is also a natural L-lysine degradation product. Pseudomonas putida uses L-lysine as the sole carbon and nitrogen source to produce glutaric acid by having 5-aminovaleric acid (5-AMV) as the key intermediate.310 Thus, reconstruction of the 5-AMV pathway in a L-lysine over-producing host can bring renewable routes to PA-5 and PA-5,5 polymers (Fig. 19). PA-5, being a homopolymer of 5-AMV, possesses similar properties to PA-4,6 and could be a suitable substitute.236 PA-5 based products have also been commercially manufactured by Lipo Chemicals Company under the trade name of Orgasol Caresse® for skin care and sun care applications.311
Glutamic acid platform can provide bio-based monomers for PAs as shown in Fig. 19. In addition, glutaric acid can be extensively employed in the polyester industry. Numerous polyesters polyols composed of about 50–70% by weight of glutaric acid were patented by Altounian and Grenier.312 More importantly, its hydrogenation product, 1,5-PDO (Fig. 2e),300 has already been used as a common plasticizer in today's modern polymer industry owing to its fairly long carbon chain.276 Flynn and Torres recently patented bio-derived 1,5-PDO plasticizer for biopolymers, which improves the flexibility of the polymers while not adversely affecting their modulus.313
5.7. Glucaric acid platform based polymers
The main contribution of a glucaric acid platform (Fig. 2f) to polymer chemistry would be in the design and synthesis of novel sugar containing polymers. In terms of linear monosaccharide derivatives, D-gluconic acid has an estimated market size of 60 kt per year. It is usually produced by the enzymatic oxidation of D-glucose, which is facilitated by the enzyme glucose oxidase and glucose dehydrogenase.81D-gluconic acid is considered to be an interesting source of comonomer to synthesize functionalized polyesters for biomedical and pharmaceutical applications. Marcincinova-Benabdillah et al. reported novel heterocyclic 1,4-dioxane-2,5-dione monomers derived from D-gluconic acid through selective protection of the 3-, 4-, 5-, and 6-hydroxyl groups. In the process, δ-gluconolactone is reacted with methoxypropane and then treated with NaOH and HCl, respectively, to yield 3,4:5,6-di-O-isopropylidene gluconic acid. Further reaction of the protected gluconic acid with bromoacetyl chloride in the presence of triethylamine, and subsequently with NaHCO3 gives the corresponding cyclic dilactone. Finally, the ROP of the cyclic dilactone with lactide yields novel degradable polyesters (Fig. 20) with higher glass transition temperatures than poly(lactic acid-co-glycolic acid) polymers, which are routinely used for sutures, bone prostheses, and drug delivery systems.314 However, the polyesters are not suitable for large scale production because several laborious and costly steps are required to synthesize the dilactone monomer. | ||
| Fig. 20 The versatile utilization of glucaric acid platform based monomers in production novel polymers. | ||
As another linear monosaccharide derivative of this platform, glucaric acid can be obtained by oxidation of the primary hydroxy group of gluconic acid on Pt/C catalysts (Fig. 2f).54 The recent literature on polymers of glucaric acid is mainly based on poly(D-glucaramidoamine)s (PGAAs) (Fig. 20), efficient and degradable gene delivery vehicles that consist of three main functionalities: carbohydrate groups, secondary amines, and amide bonds. As a promising example, Liu et al. synthesized a series of new PGAAs by reacting esterified D-glucaric acid comonomer with the amine-containing comonomers: diethylenetriamine, triethylenetetramine, tetraethylenepentamine, and pentaethylenehexamine, at room temperature in methanol. The PGAAs were proven to exhibit high gene delivery efficiency without toxicity with BHK-21 cells.315 More detailed information on PGAA and other carbohydrate-based nucleic acid delivery vehicles was reviewed by Ingle et al.316 By applying the same synthetic approach, the polycondensation of esterified glucaric acid, Nobes et al. synthesized and tested a number of poly(glucaramides), hydroxylated PAs (Fig. 20), for applications requiring water resistance. In the study, the poly(glucaramides) were used as an additive in fiber-reinforced pressed panels. The water resistance of the panels increased dramatically with increasing amounts of poly(glucaramide) in the panels.317 Another route for producing hydroxylated PAs is ring-opening polyaddition of glucaric acid derived glucarodilactone318 (Fig. 2f) with several alkylenediamines. Hashimoto et al. synthesized various hydroxylated PAs through this route even at room temperature and with no catalyst.319 The same group also carried out the polyaddition of glucarodilactone with hexamethylene diisocyanate and methyl (S)-2,6-diisocyanatocaproate to obtain the corresponding polyurethanes (Fig. 20) by using dibutyltin dilaurate as a catalyst.320 Very recently, Gallagher et al. reported the synthesis of a new glucarodilactone based dimethacrylate monomer, glucarodilactone methacrylate (GDMA). Thermally initiated free radical polymerization of GDMA in the bulk yields a highly cross-linked thermoset network. The thermoset polymer is reported to have comparable mechanical properties with respect to commercially available stiff poly(dimethacrylates).321
For cyclic monosaccharide derivatives, their versatile usage as a monomer or as an initiator has been reported. Such a compound is methylglucoside, which can be obtained through the methanolysis of cellulose in methanol in the presence of an acid catalyst at 200 °C.322 However, the yield of this process is low (between 40 and 50%) and thus a process development is required to further increase the yield (Fig. 2f). Park et al. utilized β-methylglucoside for the chemoenzymatic synthesis of sugar-containing biocompatible hydrogels (Fig. 20) by a two step reaction. In the first step, lipase-catalyzed esterification of β-methylglucoside was performed with acrylic acid, methacrylic acid, vinyl acrylate or vinyl methacrylate. In the subsequent step, the esterified β-methylglucoside monomers were used to synthesize poly(β-methylglucoside acrylate) and poly(β-methylglucoside methacrylate) by free-radical polymerization of the monomers with and without ethylene glycol dimethacrylate (EGDMA) as a crosslinker. The results of the study suggest that the obtained polymers, not crosslinked with EGDMA, are highly biocompatible and they are readily available for various biomedical applications.323 A different utilization of β-methylglucoside was reported by Suriano et al. They synthesized amphiphilic A3B mikto-arm copolymers by using a t-butyl-diphenyl silyl-based methylglucoside derivative. The methylglucoside derivative was used as an initiator for the polymerization of ε-CL to obtain a star-shaped poly(ε-caprolactone) macroinitiator (Fig. 20). ATRP of the macroinitiator in the presence of galactose methacrylate allowed the formation of A3B mikto-arm copolymers with different compositions and molecular weights. Finally, selective deprotection of sugar protecting groups generated amphiphilic mikto-arm copolymers. Such polymeric micelles offer the advantage to trap drugs, such as anticancer agents, e.g., paclitaxel and doxorubicin, in their hydrophobic core.324
Fig. 20 shows the versatile utilization of glucaric acid platform based monomers in the production of novel polyesters, polyurethanes, hydrogels, hydroxylated PAs, and novel initiators. As shown in the figure, the applicability of polymers of a glucaric acid platform is quite versatile. Their research and industrial utilization can extend from composites to cosmetic and biomedical materials. Since glucaric acid platform consists of monosaccharides and their derivatives, more detailed information regarding the incorporation of sugars and their derivatives either in a polymer backbone or as pendant groups is provided in Section 5.1.
5.8. Itaconic acid platform based polymers
Itaconic acid (ITA) (Fig. 2g) is one of the most attractive monomers for producing bio-based polymers by providing two functional acid groups as well as a vinyl functionality. It is produced industrially via fermentation of carbohydrates such as glucose by fungi and its recent market volume is about 15 kt per year. Chemical synthesis of ITA starting from different compounds, like citric acid, is also known but it is not economically and ecologically convenient for large scale production. According to Yao and Tang, ITA may become a replacement monomer for petrochemical-based acrylic or methacrylic acid owing to its similarity in structure.261,325 Current production of petroleum-based acrylic and methacrylic acid requires several chemical synthesis steps. In this consideration, ITA has process and ecological advantages since it is a direct fermentation product of cheap carbohydrates. On the other hand, its market penetration requires a cost competitive production with respect to other acrylic monomers.ITA has already been used in the manufacture of synthetic fibers, coatings, adhesives, thickeners and binders.261 For instance, it is primarily used as a comonomer in styrene butadiene polymers to provide dye receptivity in the fiber industry. Moreover, its homopolymer, polyitaconic acid (PIA), is commercially available. Itaconix Corporation, USA, produces linear PIA partially neutralized with sodium salt for everyday applications (Fig. 21).326 The company uses bio-based ITA which is produced by fermentation of carbohydrates using Aspergillus terreus. Hence, 100% sustainable polymers from bio-based itaconic acid are already in the market since all of the carbon comes from renewable biomass resources. PIA has also been envisioned as a replacement polymer to polyacrylic acid. For this to happen, its current production cost (∼$3 per kg) must reach below $1.5 per kg and this can be achieved via an integrated production infrastructure starting from carbohydrates to the final PIA polymer.327 Versatile utilization of ITA based polymers in resin, lattice, fibre, detergent and cleaner industries is summarized by Willke and Vorlop.328
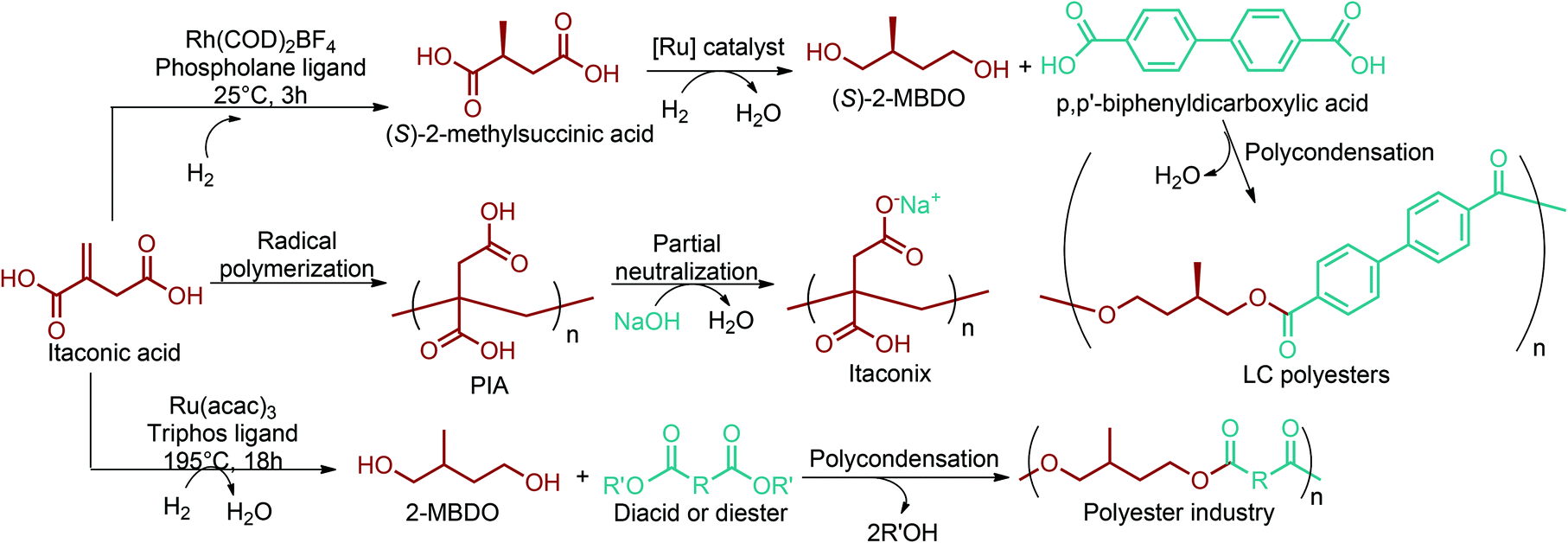 | ||
| Fig. 21 Itaconic acid and 2-MBDO based polymers. | ||
Apart from being a valuable monomer itself, ITA also serves as a parent molecule for the production of other monomers (Fig. 2g). 2-methyl-1,4-butanediol (2-MBDO) is a promising example. It can be obtained through ruthenium-catalyzed hydrogenation of ITA in the presence of triphos ligand at 195 °C. The reaction proceeds with excellent yields of up to 93%.89 2-MBDO can have a great contribution to the current polyester industry owing to its structural resemblance to 1,4-butanediol. Its usage in the production of polyesters still awaits exploration. Besides, selective hydrogenation of ITA to R- or S-2-MBDO has significance because enantiomerically pure 2-MDBO is a promising monomer for the synthesis of liquid crystalline (LC) polyesters. In this consideration, Almena et al. showed that the vinyl group of ITA can be effectively reduced with an enantiomeric excess (ee) of 97% with the help of a chiral phospholane ligand and a Rh(COD)2BF4 catalyst.86 Further reduction of the two carboxyl groups can give chiral 2-MBDO compounds. Recently, liquid crystalline (LC) polyesters of (S)-2-MBDO with p,p′-biphenyldicarboxylic acid or p,p′-biphenyldicarboxylate (Fig. 21) have been attracting interest. As an example, Uchimura et al. incorporated 9,10-diphenylanthracene moieties into the LC polyesters consisting of (S)-2-MBDO, p,p′-biphenyldicarboxylate and 1,6-hexanediol units. The circular dichroism spectra showed that the polymer with 1 mol% of the anthracene moiety exhibited the chiral smectic C phase. On the other hand, only a smectic A phase was formed in the other polymers.329
Upon a dehydration reaction under acidic conditions and at high temperatures, carbohydrate derived ITA can be converted to itaconic anhydride (ITAnh). However, a faster synthesis process, which can proceed under milder conditions, is needed for this conversion. Such a process was reported by Robert et al.85 They showed that reacting ITA with dimethyl dicarbonate in the presence of chromium(III)-based salen complex perfectly gives ITAnh with 100% product yield (Fig. 22a). This conversion is quite valuable because it allows the formation of an ITAnh monomer starting from carbohydrates in just two steps. ITAnh can be employed both in ROP and radical polymerization reactions owing to its cyclic anhydride and vinyl functionalities, respectively. It is one of the unique monomers for designing cyclic anhydride copolymers, which can exhibit biodegradable characteristics and are derived from renewable resources. Such copolymers have been drawing attention because they can be further modified for target applications. As an example, Shang et al. synthesized comb-like random copolymers of ITA and stearyl methacrylate (SM) by radical polymerization. These copolymers were then easily converted to ionomers (Na, Ca or Zn carboxylates) via the partial neutralization of the copolymers (Fig. 22b). Both of the copolymers and their ionomers showed crystallinity of stearyl side-chains, by having a melting point of about 30 °C. Since the melting point is just below human body temperature (37 °C), these materials were suggested to be potential candidates for biomaterials such as scaffolds, drug delivery systems, and prosthetics.330 As another approach, ITA can be first utilized in polycondensation reactions, and then, the remaining vinyl functionality of its ring-opened copolymers can be further used in post-functionalization reactions. For instance, Okuda et al. performed the Sn-catalyzed ROP of ITA with poly(L-lactic acid) (PLA) to produce ITA-PLA macromonomers. The intact vinyl functionality of the macromonomers was then radically copolymerized with n-butyl methacrylate (BMA), n-butyl acrylate (BA), methyl methacrylate (MMA), and ethyl methacrylate (EMA) to efficiently give the corresponding graft copolymers (Fig. 22c).331
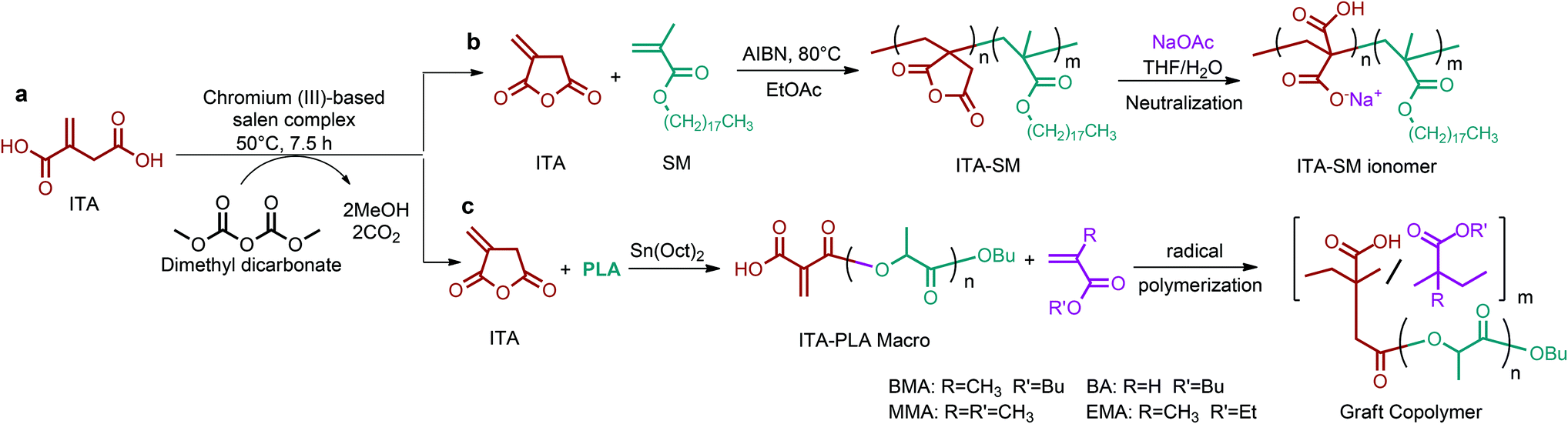 | ||
| Fig. 22 (a) Dehydration of ITA to ITAnh. (b) Synthesis of ITA-SM copolymers and their conversion to the ITA-SM ionomer. (c) Synthesis of ITA-PLA graft copolymers. | ||
Hydrogenation of the vinyl group of itaconic acid leads to the formation of hydroxyl acids. Then, intramolecular esterification of the hydroxyl acids yields two regioisomeric monomers: 2-methyl butyrolactone (2-MGBL) and 3-methyl butyrolactone (3-MGBL) (Fig. 2g). Polymerization of these butyrolactones has been rarely investigated because γ-lactones (5-membered) are thermodynamically stable. Nonetheless, it was stated that these monomers are worthy of more investigation.261 Further reduction and dehydration of these lactones result in the formation of 3-methyltetrahydrofuran (3-MTHF) (Fig. 2g). 3-MTHF has a THF like cationic polymerization; however, its comparatively low value of activation entropy prevents its polymerization above 4 °C.261,332 Literature reports on the polymerization of the other derivatives of ITA (Fig. 2g): itaconic diamide, 2-methyl-1,4-butanediamine and 3-methylpyrrolidine, are scarce. Depending on the large scale production of ITA from lignocellulosic biomass, these derivatives can have a larger contribution to polymer research and industry.
5.9. Levulinic acid platform based polymers
Amongst the lignocellulosic biomass derived monomers, levulinic acid (LevA) (Fig. 3c) has critical importance because its large scale production from lignocellulosic biomass has already been achieved. For instance, Maine BioProducts announced the “Biofine Process” for the commercial production of LevA. The process is based on acid catalyzed dehydration of lignocellulosic feedstocks in a two-stage process.325 Moreover, Avantium's YXY technology has developed a tuneable process to convert plant-based carbohydrates into high purity methyl levulinate, which can be easily converted to LevA through a straightforward process.333 Although these processes for the first time provide cost competitive and high volume access to levulinics, the current LevA prices remaining in between 5–8 $ per kg must be further decreased to 1 $ per kg or below. Thereupon, the realization of LevA as a giant commodity chemical can be achieved. For further reduction of the LevA prices, its direct production from cheap cellulosic biomass feedstocks is more convenient and this issue is under intense investigation.42,325,334,335LevA is the precursor of the levulinic family and it branches to many valuable new compounds having novel applications (Fig. 3c). Hence, it is a significant precursor to many pharmaceuticals, plasticizers, and additives.336 Various companies recently launched the production of novel bio-polymers based on LevA or its derivatives with a motivation towards producing cost competitive and sustainable polymers. As an encouraging example, Segetis recently launched the “Levulinic Ketal Technology” to investigate the direct utilization of LevA based ketals in polyurethane and thermoplastic applications.325 In the process, esters of LevA, as degradation products of carbohydrates, are combined with alcohols derived from vegetable oils for the synthesis of levulinic ketals (Fig. 23). Leibig et al. evaluated over 25 levulinic ketal derivatives in terms of their plasticizer performance in PVC. A number of levulinic ketals exhibited superior performance in comparison with the commercial phthalate plasticizers.337 In addition to giving rise to the development of novel polymer materials, levulinic monomers have the potential for substitution of petrochemical building blocks. For instance, there is growing concern regarding the sustainability of bisphenol A (BPA) in consumer products and food containers since it has a pseudo-hormonal effect on the body.338 The Food and Drug Administration (FDA) has already ended its authorization of the use of BPA in certain products.339 LevA derived diphenolic acid (DPA) is considered to be a sustainable replacement for BPA. It can be easily produced by reacting levulinic acid with two moles of phenol at 100 °C under acid conditions (Fig. 23).114,338 Both of these starting chemicals can be produced from lignocellulosic biomass.112,113,340 So, petroleum based BPA can be replaced with fully renewable and sustainable DPA, depending on its cheaper production from lignocellulosic biomass. DPA has also been attracting interest in the synthesis of poly(arylene ether ketone) (PAEK) and poly(ether ether ketone) (PEEK) based membranes for fuel cell applications. In a recent study, Zhou and Kim produced sulfonated PAEK (SPAEK) by polymerizing DPA with 4,4′-difluorobenzophenone and 5,5′-carbonylbis(2-fluoro benzenesulfonate) in the presence of DMSO and K2CO3. The intact carboxylic groups of the polymerized DPA served as pendant moieties so that they were then converted to unsaturated groups to obtain a series of cross-linked SPAEK (CSPAEK) membranes. The CSPAEK membrane with 80% sulfonation and 20% cross-linking degree exhibited much lower methanol permeability and a comparable proton conductivity with respect to its commercial competitor, Nafion® 117.341 Moreover, DPA is quite a useful monomer in the synthesis of hyperbranched polymers by providing tri-functionality: having two phenol and a carboxylic acid groups. Foix et al. made use of this characteristic of DPA. They prepared a new hyperbranched–linear–hyperbranched polymer in a one pot process via polycondensation of DPA with poly(ethylene glycol) using DCC as a coupling agent and 4-(N,N-dimethylamino)pyridinium-p-toluenesulfonate as a catalyst (Fig. 23).342
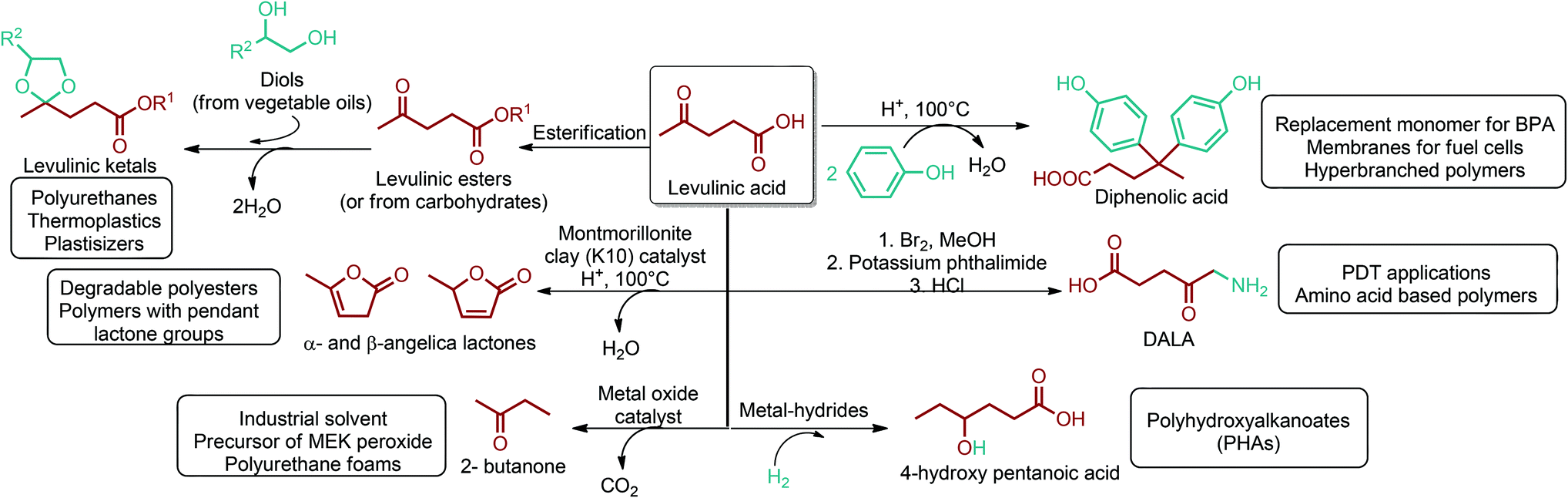 | ||
| Fig. 23 Synthesis and common applications of direct derivatives of LevA. | ||
In addition to DPA and levulinic ketals, LevA can be directly converted to levulinic esters,115 5-aminolevulinic acid (δ-aminolevulinic acid, DALA),114 angelica lactones (ANLs),44,116 2-butanone,44 4-hydroxy pentanoic acid or its esters,22,44 and gamma-valerolactone (GVL)22,117,118 (Fig. 3c). Common applications of these direct derivatives of LevA are summarized in Fig. 23.
Amongst these derivatives, synthesis of DALA requires the formation of a C–N bond at the C5 carbon. Although various amination methods have been successful on the laboratory scale,114 a process development is required for its industrial production. Particularly, the laborious amination processes based on the bromination (Fig. 23) must be replaced with a low cost and facile method. DALA is frequently used as an agent for photodynamic therapy. Fotinos et al. reviewed the use of DALA derivatives in photodynamic therapy (PDT) and fluorescence photodetection (FD) from a chemical, a biochemical and a pharmaceutical point of view. Two derivatives of DALA: methylaminolevulinate and hexylaminolevulinate, are marketed under the trade names of Metvix and Hexvix, respectively.343 Macromolecules based on DALA were also reported for PDT applications. Battah and co-workers synthesized novel DALA containing well-defined dendritic molecules, in which, the DALA moieties were attached to the periphery by ester linkages which are hydrolysable under cellular conditions.344,345 As it is an amino acid itself, DALA can provide novel opportunities in amino acid based polymers. Such polymers were described in more detail in Section 5.5.
As they are dehydration products of LevA (Fig. 23), ANLs can potentially provide a route for producing functionalized aliphatic polyesters from renewable resources. In that respect, Chen et al. prepared a degradable polymer from α-ANL. The resulting polyester exhibited good degradability under light or acidic/basic circumstances owing to the presence of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.346 Hirabayashi and Yokota utilized β-ANL in a different manner. They carried out radical copolymerization of β-ANL with styrene in order to obtain polymers containing lactone units in the backbone chains.347
C bond.346 Hirabayashi and Yokota utilized β-ANL in a different manner. They carried out radical copolymerization of β-ANL with styrene in order to obtain polymers containing lactone units in the backbone chains.347
2-Butanone (methyl ethyl ketone, MEK) (Fig. 23) is commonly used as an industrial solvent. Although it is the precursor of MEK peroxide, a commonly used peroxide in the polymer industry,348 its direct utilization as a monomer is scarce. Nonetheless, Glowacz-Czerwonka obtained polyurethane foams from melamine solution in reactive solvents based on MEK and 4,4′-diphenylmethane diisocyanate.349
4-Hydroxypentanoic acid (4-hydroxyvaleric acid) (Fig. 23) is a monomer belonging to the hydroxyalkanoates family. It can be polymerized into polyhydroxyalkanoates (PHAs) by chemical and/or biological methods. Detailed scrutiny of PHAs based on lignocellulosic biomass is provided in Section 5.16.
LevA can be hydrogenated to GVL by employing either homogeneous or heterogeneous catalysts. So far, the best results were obtained with noble metal-based catalysts, particularly with Ru-based catalysts. However, the utilization of expensive noble metal-based catalysts prevents scaling-up of the GVL production process. Replacement of noble metals by more widely available cheap metals as well as recovery and recycling of non-precious metal-based catalysts are important issues in preventing GVL to become a commodity chemical. The challenges associated with industrial production of GVL were highlighted in detail by Wright and Palkovits.350 Conversion of LevA to GVL holds great potential because GVL is a valuable fuel and green solvent as well as it is a precursor for other value-added chemicals as shown in Fig. 3c.22,117,118 Alonso et al. reviewed the upgrading of lignocellulose derived GVL to various chemicals and fuels, including polymers, fuel additives, and jet fuels.22 Jedlinski et al. synthesized a homopolymer of GVL with alkali metal alkoxides and alkali metal supramolecular complexes based initiator systems to obtain polymeric materials with potential biodegradability.351 Its copolymerization with various monomers, including diglycidyl ether of BPA,352,353 ε-CL,354 β-butyrolactone,355 and L-lactide,356 were also studied (Fig. 24). Over basic catalysts, gas phase catalytic condensation of formaldehyde with GVL yields α-methylene-γ-valerolactone (MeGVL). There are several problems regarding this chemistry. First of all, it is very difficult to handle formaldehyde in the gas phase because it polymerizes very rapidly. MeGVL is also a reactive monomer so that it polymerizes on standing. Fortunately, dissolving GVL in formalin (37 wt% aqueous solution) works well and a MeGVL yield of more than 95% is obtained using barium-based catalysts supported on silica.119 MeGVL is a new and an attractive acrylic monomer that imparts high thermal stability to polymers.119 Naturally renewable MeGVL has been attracting interest in exploring the prospects of substituting the petroleum-based methacrylate monomers for specialty chemicals production. The cyclic ring in MeGVL imparts significant enhancements such as enhanced resistance to heat, solvent and scratch in the materials properties of its polmers.357 For instance, Tg of poly-MeGVL (Fig. 24) is about 200 °C, which is about 100 °C higher than that of its acyclic counterpart, PMMA. However, the absence of an economically attractive catalytic process prevents the commercial development of MeGVL.119 Particularly, its synthesis through the LA-GVL-MeGVL pathway must be optimized accordingly for its large scale production. Achieving this pathway in one pot would be more desirable for cutting-off extra synthesis steps and thus the production costs.
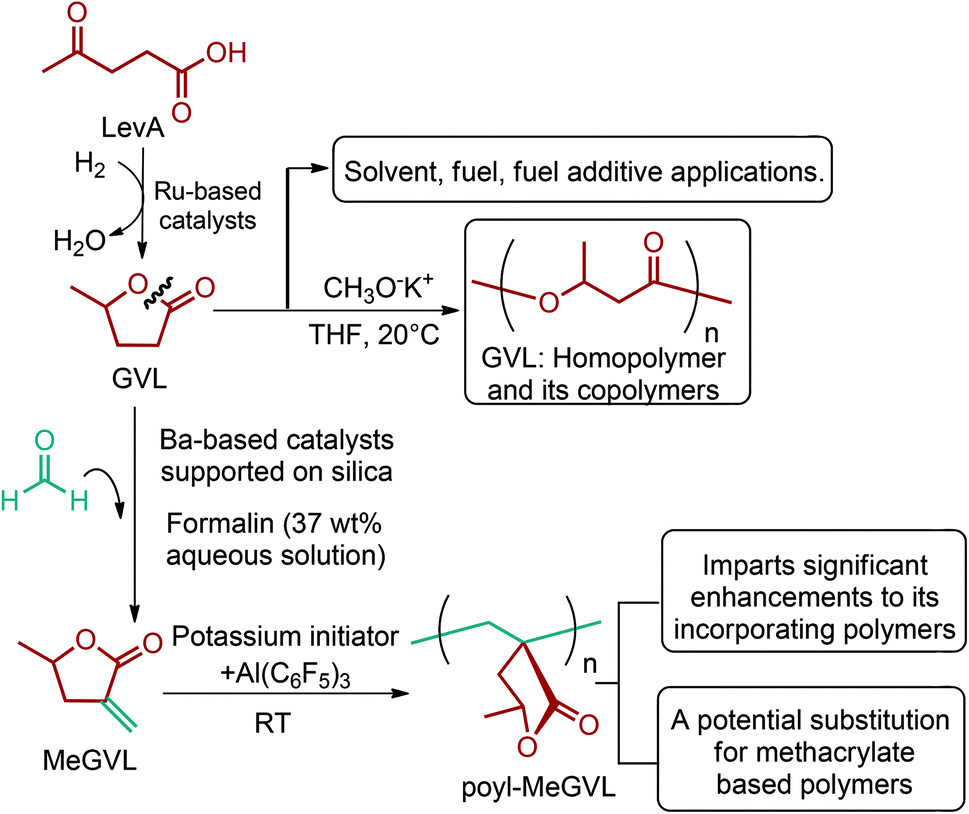 | ||
| Fig. 24 Applications of GVL and MeGVL. | ||
Ring-opening of GVL is a new versatile route to produce polymer precursors with relatively high molecular weights (Fig. 3c and 25).358 In the presence of an amine, the GVL ring can be opened to give the corresponding γ-hydroxy(amino)amide compounds. Chalid et al. synthesized novel GVL derived γ-hydroxy-amide monomers through the addition of various amino compounds. Yields of the final products ranged between 22 and 95%. It was shown that sterically less hindered amine compounds can effectively result in ring opening of GVL even without the use of a catalyst and a solvent. Hence, GVL ring opening by amines, which do not have steric hindrance around their nucleophilic nitrogen centre, is a promising pathway for the production of novel monomers.122 This new family of monomers can be employed to produce polymers such as polyethers or polyurethanes.22 For instance, a GVL/1,2-aminoethanol adduct was polymerized with 2,4-toluene-di-isocyanate (TDI) at 140 °C by using TEA as the catalyst and DMA as the solvent. A polymer with a molecular weight of 156 kDa was produced in 97% yield.358 Also, the hydrogenation GVL can be performed at 40 °C to yield 1,4-pentanediol (1,4-PDO) over an inexpensive Cu catalyst, which was calcinated in the presence of H2, with a selectivity of over 98% at complete conversion.22 Although polycondensation of 1,4-PDO is rarely reported, it has a similar structure to 1,4-BDO so that it can be largely employed in the synthesis of polymers. As such, Rothen-Weinhold et al. used 1,4-PDO in the synthesis of poly(ortho ester)s, hydrophobic and bioerodable polymers that have been investigated for pharmaceutical use. In the study, poly(ortho ester)s, prepared from 3,9-diethylidene-2,4,8,10-tetraoxaspiro[5.5]un decane, 1,4-PDO and 1,6-hexanediol glycolide, were used as agents for bovine serum albumin (BSA) protein delivery.359,360 Acid catalyzed ring opening of GVL using a SiO2/Al2O3 catalyst at 225–375 °C results in the formation of an isomeric mixture of pentenoic acids.120,121,123 Amongst these isomers, 4-pentenoic acid (4-PA) can be utilized in both radical and condensation reactions owing to its dual functionality (vinyl and carboxylic acid groups). An et al. employed this characteristic of 4-PA in the production of UV-induced thiol–ene crosslinked films. In the study, well-controlled pendant hydroxyl containing copolymers of 2-hydroxyethyl methacrylate (HEMA) and methyl methacrylate (MMA) were synthesized in the presence of the benzyl α-bromoisobutyrate initiator. The pendant –OH groups were then converted to pendant enes by their coupling with the carboxyl group of 4-PA. Under UV radiation, the resulting pendant ene groups can undergo thiol–ene polyaddition reactions with polythiols to form crosslinked films with a uniform network.361 Lange et al. produced another ring-opened derivative of GVL, methyl pentenoate, in more than 95% yield via a transesterification method.127 The process basically relies on the large boiling point difference between GVL (207 °C) and methyl pentenoate (127 °C). In the process, methanol was slowly fed into the reaction flask containing GVL and para-toluene sulfonic acid (pTSA) as the catalyst while boiling the reaction medium at 200 °C. A mixture of methyl pentenoate, methanol and water was distilled off continuously, and then condensed at 90 °C at the top of a rectification column. Finally, methyl pentenoate was collected over consecutive distillate fractions. Methyl pentenoate is considered to be a valuable PA precursor because it can be converted to ε-CL, caprolactam, or adipic acid by hydroformylation, hydrocyanation, or hydroxycarbonylation reactions, respectively. So, commercialization of the methyl pentenoate production through the LevA-GVL-methyl pentenoate pathway may open new opportunities for the production of conventional PAs (Fig. 25).22,362
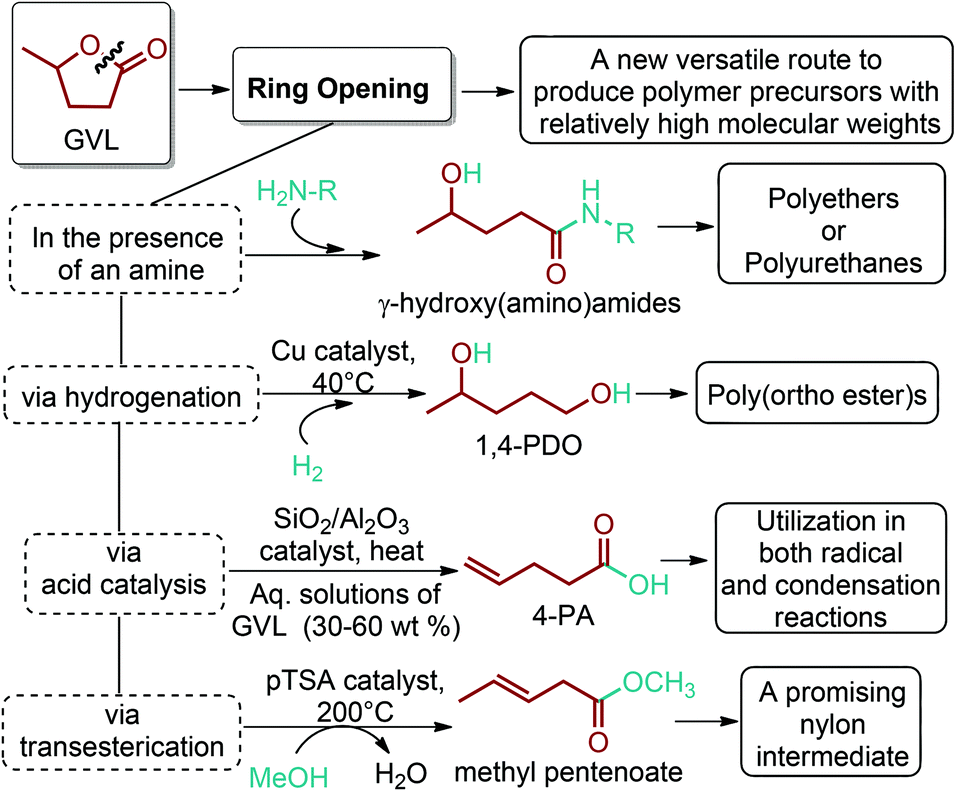 | ||
| Fig. 25 Ring opening of GVL as a new versatile route to produce polymer precursors. | ||
The levulinic acid platform can also open novel synthetic routes for the production of giant monomers such as butene and adipic acid (ADA) (Fig. 3c). In this consideration, decarboxylation of GVL derived pentenoic acids on a SiO2/Al2O3 catalyst at 375 °C results in the formation of butene isomers, including 1-butene (1-B).121 In fact, GVL ring opening on acid catalysts at high temperatures first gives an isomeric mixture of pentenoic acid, and subsequently, an isomeric mixture of butenes. So, selective synthesis of 1-B starting from GVL is not feasible under these conditions. A better route for the production of 1-B would be its synthesis from n-butanol. Cobalt and The Naval Air Warfare Center recently teamed up for scaling up and optimizing the dehydration chemistry for the conversion of non-food feedstock based bio n-butanol to 1-B.363 Polymerization of 1-B by using supported Ziegler-Natta catalysts results in its high molecular weight homopolymer, poly(1-butene) (PB-1). PB-1 exhibits excellent creep properties and its major applications are summarized in Fig. 26.270 1-B is also used as a comonomer in the production of sine qua non polymers of today's modern society such as linear low density polyethylene (LLDPE), LLDPEs based on the metallocene catalyst technology (mLLDPE), very low density polyethylene (VLDPE) and isotactic polypropylene (i-PP).270,364,365 Kraton Polymers Company employs 1-butene in the production of linear styrene–(ethylene–butylene)–styrene triblock copolymer series under the trade name of Kraton G1600 SEBS (Fig. 26).270
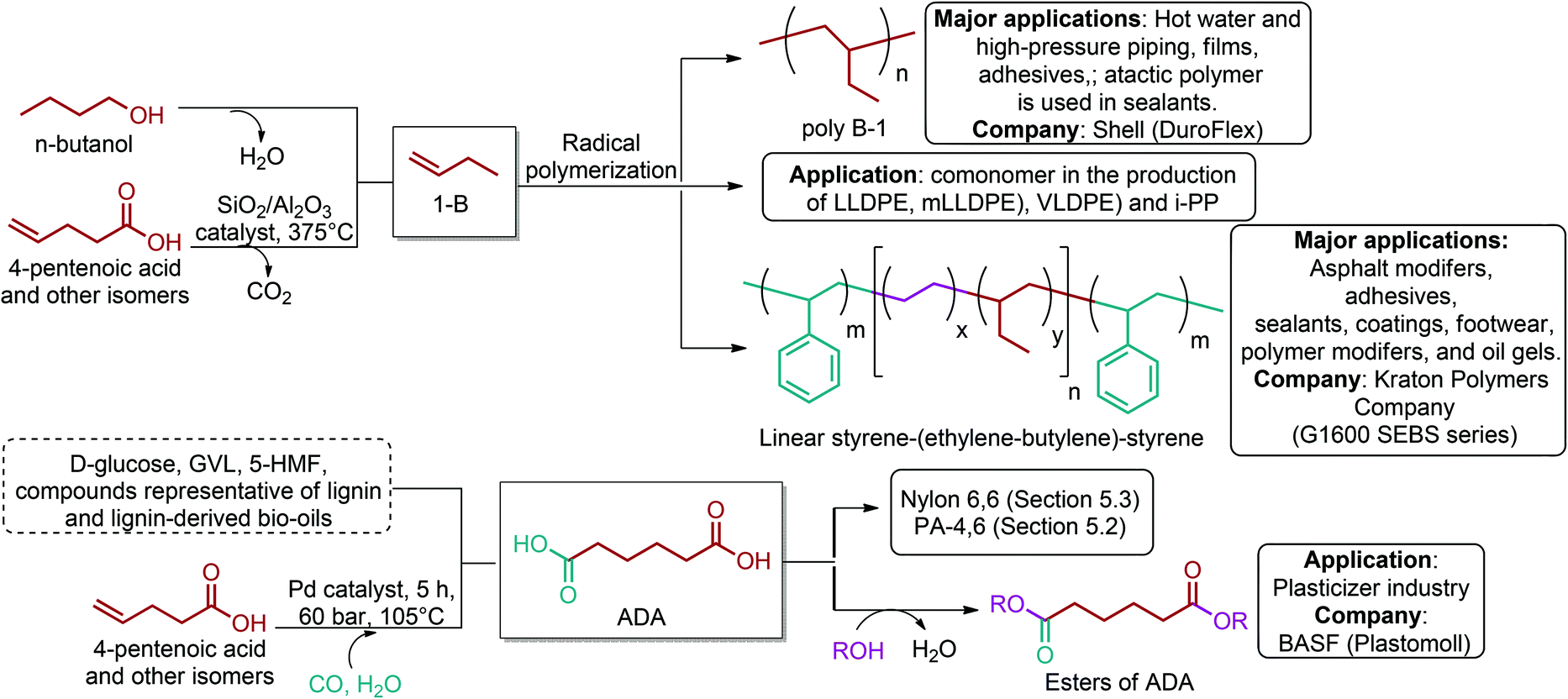 | ||
| Fig. 26 Major applications of 1-B and ADA. | ||
GVL derived pentenoic acids can be carbonylated to produce ADA over a palladium catalyst for 5 h under 60 bar of CO at 105 °C (Fig. 26). However, poor yields of 22–48% are obtained. For a further improvement of the reaction yields, more information on the kinetics and the thermodynamic constraints of the process is required.124 In fact, there are many other possibilities for the production of ADA. There is a growing research interest in both industry and academia in the development of bio- and chemocatalytic routes for ADA from other biorefinery building blocks such as 5-HMF, D-glucose, compounds representative of lignin, and lignin-derived bio-oils. Van de Vyver and Roman-Leshkov stated that the production of ADA particularly from lignocellulosic biomass-derived chemicals could provide an even more sustainable route, instead of its petroleum-derived production route.124 In the end, these bio-based routes for ADA production have to be cost-competitive with its current industrial manufacture process, which is based on catalytic oxidation of a mixture of cyclohexanol and cyclohexanone with nitric acid.
The global market size of ADA is projected to be 6 billion pounds by 2017.124 Since it has such a huge market, many start-up companies such as Rennovia, Verdezyne, BioAmber, Celexion, and Genomatica have been engaged in developing bio-based routes to produce ADA. Some of them have already reached advanced pilot or demonstration scales. Especially, Rennovia's process was suggested to be highly cost competitive with the conventional DuPont/Invista cyclohexane-based oxidation process.124 The ultimate motivation of these companies for the synthesis of bio-ADA is to produce 100% bio-based PAs. In this consideration, ADA is a critical monomer. About 85% of the total produced ADA is used for the production of PA-6,6.366 Another important commercial polymer of ADA is PA-4,6. After the polyamides, esters and polyesters constitute the second most important class of ADA derivatives.367 Esters of ADA can be obtained from alkylpenteonates,127 or as another route, ADA can be readily reacted with alcohols to give either its mono- or diesters.367 Moderately long-chain esters of ADA find large applicability as plasticizers, particularly for poly(vinyl chloride) (PVC).367 As an example, BASF manufactures and markets di-2-ethylhexyl adipate and di-isononyl adipate derivatives of ADA under the trade name of Plastomoll® (Fig. 26).368
5.10. 3-Hydroxybutyrolactone platform based polymers
3-Hydroxybutyrolactone (3-HBL) is a versatile chiral building block (Fig. 3d) having applications in the synthesis of a variety of pharmaceuticals, polymers and solvents. Enantiopure 3-HBL is a significant precursor for chiral drugs such as the cholesterol-reducing statins like Lipitor® (Pfizer) and Crestor® (AstraZeneca), antibiotics such as carbapenems and linezolid (Zyvox®), and the anti-hyperlipidemic medication Zetia®. Both enantiomers of 3-HBL can be used in the synthesis of L-carnitine, the nutritional supplement. Moreover, the functional groups of enantiopure 3-HBL can be derivatized to yield chiral building blocks such as lactones, THFs, amides, nitriles, epoxides, and solvents.40,109,369,370Owing to its versatile applicability, the U.S. Department of Energy proposed 3-HBL as a top value-added chemical from biomass. Even so, there is not a facile and low-cost chemical pathway for its large scale synthesis. For instance, the commercial synthesis of (S)-3-HBL is performed through a continuous chemical synthesis process using high pressure hydrogenation of L-malic acid over a Ru-based catalyst. However, the process consists of expensive catalysts and purification steps as well as hazardous processing conditions. The other various chemical and chemoenzymatic synthesis routes of 3-HBL also consist of multiple steps and expensive catalysts. That is why, 3-HBL currently has an expensive wholesale cost of ∼$450 per kg and this alleviates 3-HBL from being a commodity chemical.109
Since the chemical synthesis of 3-HBL is messy, special attention should be given to its biological production routes. Even so, there are no known naturally occurring biosynthetic pathways for 3-HBL. Here, the design of a novel pathway through microbial engineering holds great importance. Recently, Martin et al. presented such a promising platform pathway for 3-hydroxyacid synthesis, which provides a biosynthetic route to 3-HBL, for the first time. The pathway results in the complete biological production of 3-HBL and its hydrolysed form, 3,4-dihydroxybutyric acid (3,4-DHBA), in recombinant Escherichia coli by using glucose and glycolic acid as feedstocks. It was also stated that direct production of 3,4-DHBA/3-HBL from glucose requires integration of the endogenous glyoxylate shunt with the 3,4-DHBA/3-HBL pathway. Engineering of this integration was described in detail in their recent articles.109,369
Once large scale production of 3-HBL via a biosynthetic route is achieved, it can be converted to a variety of fine chemicals by employing mainly three different chemistries: (i) functionalization of the –OH group, (ii) dehydration of the –OH group, and (iii) ring opening of the cyclic ester. Particularly, functionalization of the –OH group of 3-HBL can open new windows to novel acrylic monomers. As such, Murata et al. patented a process for converting 3-HBL to β-(meth)acryloyloxy-γ-butyrolactones (B(M)AL-GBLs) by reacting 3-HBL with (meth)acrylic acid chloride, (meth)acrylic acid or (meth)acrylic ester in a simple and safe manner (Fig. 27). These acrylic derivatives of 3-HBL are stated to be useful as a constituent monomer of paints, adhesives, sticking-agents, and resins for ink. Despite their expected use for various purposes, they have never been produced industrially due to a high risk of explosion in the chemical synthesis of their parent molecule, 3-HBL.111 But now, as previously mentioned, achievements in the production of 3-HBL via fermentation routes can solve this problem.109,369 B(M)AL-GBLs have been finding applicability particularly in resist compositions and patterning processes.371 As a different synthetic route, the –OH group of 3-HBL also provides functionality for its dehydration to the corresponding unsaturated lactone derivative, 2(5H)-furanone. The dehydration reaction can be easily performed in the presence of an acid, such as polyphosphoric acid, and under vacuum distillation (160 °C).110 Once obtained, 2(5H)-furanone can undergo a facile reaction with a primary amine by a Michael-type addition to give α,β-amino-gamma-lactone, which subsequently polymerizes to yield a polyamide with pendant R groups. In the end, many different types of polyamides can be produced starting from 3-HBL depending on the structure of the side R group (Fig. 27).268
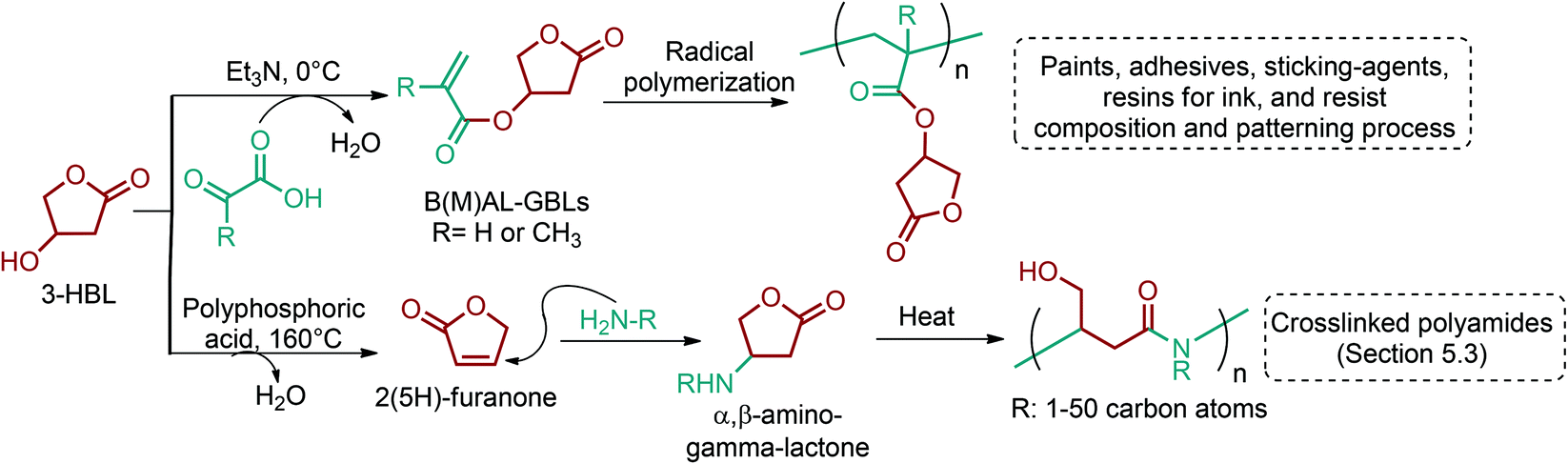 | ||
| Fig. 27 Polymer applications of 3-HBL derived B(M)AL-GBLs and 2(5H)-furanone. | ||
5.11. Sorbitol platform based polymers
Amongst similar polyols, sorbitol (SBL) (Fig. 3b) is the least costly and the most commonly used sugar alcohol. It has the biggest market share and it is extensively used as sweetener, thickener, humectants, excipient, dispersant in food, cosmetic, toothpaste and other related industries. Owing to its large market volume, many companies have been involved in its commercial production. Recently, Roquette Freres has been the biggest SBL producer and shares over 70% of the total market volume together with Cargil and SPI Polyols.372,373 Currently, no technical development is essentially needed for the use of SBL as a building block because its production is commercially practiced by several companies and the yields demonstrated are about 99%. It is suggested that the only necessary change requires a shift from the batch process to the continuous process.40 SBL's industrial production begins with raw materials such as corn, cassava and wheat. Through enzymatic hydrolysis, these raw materials are first converted to glucose, and then the hydrogenation of glucose at 403–423 K with H2 pressure ranging from 4.0 to 12.0 MPa provides the production of SBL. Also, there is a growing interest in one-pot conversion of biomass into SBL.106,372 Very recently, Zhu et al. reported efficient hydrogenolysis of cellulose into SBL in one-pot in the presence of a bifunctional, sulfonic acid-functionalized silica-supported ruthenium catalyst. When the reaction was performed at 150 °C for 10 h, a maximum yield of 61.2% was obtained.374 Such direct conversions of raw materials into SBL are more desirable for further decreasing current production prices of SBL. Nonetheless, cheap catalysts must be employed instead of noble metal-based catalysts and the reaction yields must be ∼99%. In this consideration, research studies should be directed at the development of cheap and recyclable multifunctional catalyst systems for the one-pot synthesis of SBL.SBL is an outstanding building block for achieving sustainable energy supply, chemicals and polymer production. It is industrially converted to vitamin C (ascorbic acid) by fermentation with or without chemical processes through sorbose and 2-ketogluconic acid as intermediates.37 This process consumes almost 15% of world SBL production. As a different chemistry, 2-fold dehydration of SBL in the presence of a sulfated copper oxide catalyst at 200 °C results in the formation of isosorbide (IS).107 In this process, cyclodehydration of SBL first forms 1,4-sorbitan and 3,6-sorbitan intermediates, and afterwards the dehydration of these intermediates yields IS (Fig. 3b). One-step conversion of lignocellulose feeds to IS is also possible with designed reaction systems.37,372 In a recent study, Op de Beeck et al. reported the catalytic production of IS from a diverse range of lignocellulosic biomass feedstocks by combining the heteropolyacid, H4SiW12O40, with the redox catalyst, commercial Ru on carbon.375 Apart from these, SBL hydrogenolysis with multifunctional catalysts can constitute a major route for the synthesis of lower alcohols such as glycerol, propylene glycol, ethylene glycol, ethanol and methanol. It is noteworthy that these lower alcohols have enormous market volumes and they can be further utilized to obtain many other high value-added products. Hence, SBL hydrolysis is expected to be a major research field in which the development of cheap and recyclable multifunctional catalysts as well as mild and facile reactions deserves priority. Repeated cycling of dehydration and hydrogenation reactions can also open up a new field in SBL chemistry, particularly for the production of H2 and alkanes. Detailed information regarding the transformation of SBL to biofuels, in the frame of chemical and industrial considerations, is provided in the review article of Vilcocq et al.37,372,376
Polymer production from SBL or its derivatives constitutes another important building block consideration. Copolymerization of SBL with other glycols would create a major opportunity in the unsaturated polyester resin market.40 Roquette recently introduced sorbitol-based polymer clarifiers under the trade name of Disorbene®, a bis(3,4-dimethylbenzylidene) sorbitol (bis-DMBS) product.377 Bis-DMBS can be produced by heating 3,4-dimethyl benzaldehyde and D-sorbitol in the presence of a Brønsted acid catalyst (Fig. 28).378 As it is a linear monosaccharide, SBL can potentially be employed for the production of a wide range of polymer families through its selective functionalization (Fig. 8 – Section 5.1). The most recent research studies (Table 5) reveal that SBL can be employed in a variety of polymerization reactions. Hence, SBL-based polymers can have a wide range of applicability, ranging from biodegradable polymers for everyday applications to specialty products including biocomposites and biomedicines.
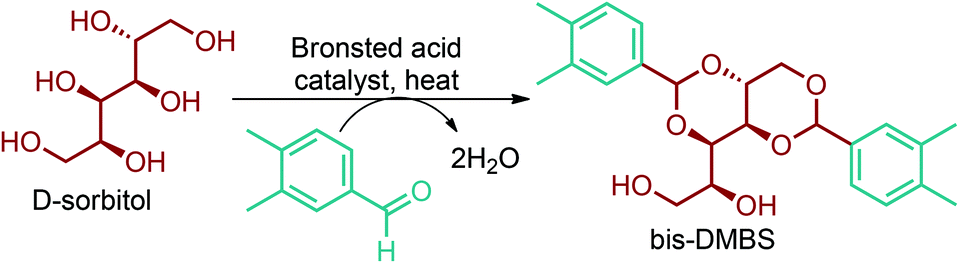 | ||
| Fig. 28 Synthesis of SBL derived bis-DMBS for polymer clarifier applications. | ||
| Monomers | Method | Polymer structure | (Potential) applications | Ref. |
|---|---|---|---|---|
| SBL, divinyl adipate | Enzymatic polycondensation (Candida antarctica) |
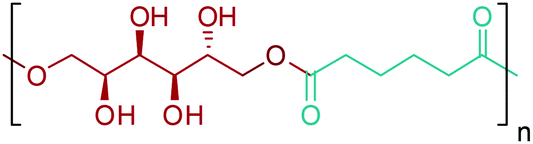
|
Self-assembled nanostructures, super-soft elastomers, biomedicine | 379 |
| SBL, 1,2,3,6-tetrahydro phthalic anhydride, adipic acid and diethylene glycol | Melt condensation |
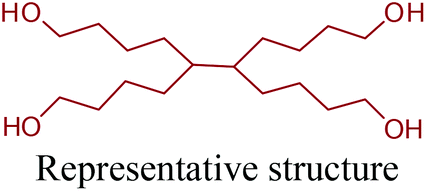
|
Polyurethane coatings (after reacting with polyisocyanates) | 380 |
| SBL polyglycidyl ether, quercetin or phenol novolac | Thermal polymerization and then, compression molding | The structure was not provided. | Biocomposites | 381 |
| SBL dimethacrylate, polyethylenimine | Michael addition |
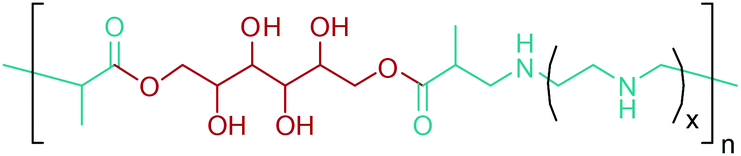
|
Gene carrier, osmotically active transporter | 382 |
| SBL, citric acid (or tartaric acid), sebacic acid | Catalyst-free melt condensation |
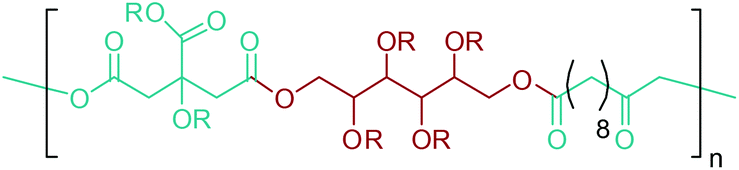
|
Biodegradable polymers for various applications | 383 |
| SBL methacrylate | Surface-initiated polymerization |
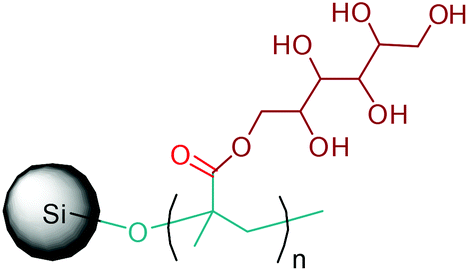
|
Hydrophilic interaction chromatography | 384 |
| SBL, L-aspartic acid | Thermal polycondensation (no catalyst) |
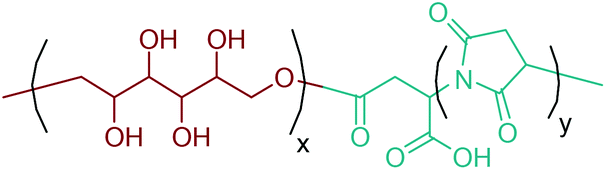
|
Biocompatible gels, absorbents, controlled release matrices | 385 |
As a selective dehydration product of SBL, IS (Fig. 29) imparts superior properties to its incorporating polymers by providing rigidity and chiral centers.372 For instance, unlike the conventional PET having a Tg of 353 K, poly(ethylene-co-isosorbide)terephthalate (PEIT) (Fig. 29) can show a much higher Tg (up to 470 K) by increasing the ratio of IS to ethylene glycol. Hence, PEIT polyesters are quite useful for hot-fill bottles. Among aliphatic polyesters, poly(isosorbide oxalate) (Fig. 29) also has a remarkably high Tg of 445 K. Moreover, it can exhibit good biodegradable properties.37,326 Recently, Naves et al. reported enzymatic copolymerization of IS with diethyladipate and different unsaturated diesters including diethyl itaconate, diethyl fumarate, diethyl glutaconate, and diethyl hydromuconate.386 The study is specifically important for providing insights into unsaturated polyester production from fully renewable monomers. Another IS incorporating sustainable polyester production was reported by Gioia et al. The sustainability stems from a novel method which combines the chemical recycling of poly(ethylene terephthalate) with the use of IS and succinic acid derived from renewable resources.387 Other IS based polymer families can also exhibit unique characteristics. As such, poly(isosorbide carbonate) (PIC, Fig. 29) is highly transparent, heat-resistant and water-tolerant. As it is a bio-based and harmless plastic, PIC is identified as a promising alternative to the polycarbonate derived from BPA. Hence, the commercial production of PEIT and PIC can boost up in the future since their petroleum derived counterparts, PET and PC, have an annual consumption capacity of about 5 × 107 and 3.5 × 106 t, respectively. Owing to the current interest in IS based polymers, various companies have been involved in the commercialization of IS based polymeric materials. Mitsubishi and Teijin chemical companies launched mass production of PIC in 2012 and 2011, respectively. Roquette also constructed a plant in 2007 for the manufacture of polymer-grade IS. The company also produces IS diesters, as phthalate-free plasticizers, under the trade name Polysorb® ID (Fig. 29). The diesters are produced as substitutes for conventional petroleum-based plasticisers such as dioctyl phthalate and particularly for the production of a flexible polyvinyl chloride. Many other kinds of IS polymers have been studied in recent years and detailed information about them was provided in the review articles of Fenouillot et al. and Rose and Palkovits.37,388,389
 | ||
| Fig. 29 Common polymers based on SBL dehydration compounds; IS and sorbitan. | ||
SBL derived sorbitans108 (Fig. 3b) could also potentially be employed in polyester production.325 Besides, esterification of sorbitan with fatty acid methyl esters, followed by PEG-ylation, results in commercially available polysorbates (Fig. 29). These products are commonly sold as emulsifiers and solubilizers in the cosmetic, pharmaceutical, and food industries under different trade names such as Tween, Span, Arlacel and Crill.37,390
5.12. Glycerol platform based polymers
Microbial production of GLY has been known for about 150 years. Significant improvements have been made in the GLY production using osmotolerant yeasts on a commercial scale, particularly in China. Microbial fermentation has been supplying more than 12% of the country's needs. More detailed information regarding GLY production by microbial fermentation was provided in the review article of Wang et al.90 On the other hand, the fast growth in the biodiesel industry has led to overproduction of GLY worldwide. About 90% of GLY has been recently produced as a by-product in the conversion of fats and oils to fatty acids or fatty acid methyl esters for biodiesel. That is why the other production routes of GLY such as fermentation of sugar or hydrogenation of carbohydrates are not industrially important.391 Instead of this, the main consideration has currently been focused on the valorization of overproduced GLY. Both industry and academia have been putting a lot of effort into the conversion of GLY to other commodity chemicals and polymers. So, the global demand for GLY as a platform chemical is also expected to increase in parallel with its production capacity. In fact, this is confirmed by a recent report published by Transparency Market Research. According to the report, the global demand for glycerol (GLY) was 1995.5 kilotons in 2011. This demand is expected to reach 3060.4 kilotons, growing at a compound annual growth rate (CAGR) of 6.3% from 2012 to 2018. In terms of revenues, the demand is predicted to reach $2.1 billion by 2018.392There are more than 2000 different applications of GLY which arise from its unique properties. Its main application areas include cosmetics, pharmaceuticals, food, tobacco, plastics and resins. About 20% of the totally produced GLY has been utilized in plastic and resin applications.391 GLY is commonly used as a polyol for the production of alkyd resins: synthetic resins made from polyhydric alcohols and polybasic acids, and modified with resins, fatty oils, or fatty acids. The largest use of alkyd resins is their utilization as binders for surface coatings. Alkyd resins are the most versatile of coating binders and thus they can be extensively used in all major categories of coatings. Other uses of alkyds include ink binders, caulks, adhesives, and plasticizers.393 As the main by-product of biodiesel, crude GLY is a promising and abundant feedstock for industrial microbiology. Without any pretreatment step, it can be used to produce polyhydroxyalkanoates with an economically competitive overall production process.394 Many different copolymers of GLY including poly(glycerol sebacate), poly(glycerol methacrylate)s and poly(glycerol-co-ε-caprolactone) were reported for various applications in the literature. It is outside the scope of this article to summarize countless GLY based polymeric materials and their applications. Detailed scrutiny is provided in review articles on polyglycerols (PGs).395–398
Research efforts have been directed to making use of the large surplus of GLY by introducing a number of selective processes for its conversion into commercially value-added products (Fig. 3a). Hence, GLY is considered to be a platform-central raw material in the future chemical industry.399,400 Particularly, its hydrogenolysis opens an alternative route to the industrial production of its diol derivatives: 1,3-PDO, propylene glycol and ethylene glycol,93–95,399,400 which are indispensible monomers of today's modern industry. Selective hydrogenolysis of GLY and the resulting diol yields are highly dependent on the catalyst used. Nakagawa et al. reported that conventional-unmodified catalysts generally result in the formation of propylene glycol as the main product, whereas Pt–WO3 catalysts on Al-based supports and Ir–ReOx/SiO2 catalysts specifically favor the production of 1,3-PDO.93 Further hydrogenolysis of GLY is also critically important since the resulting products are bio-based fuels: 1-propanol (1-PO) and propane.98 As a common fuel, production of propanol is currently too expensive.401 Its production routes from lignocellulosic biomass might decrease its current market price. As a promising study, Zhu et al. converted biomass-derived GLY to biopropanols in one step over bi-functional catalysts. In particular, the conversion of GLY to 1-PO in the presence of a Pt–HSiW catalyst supported over ZrO2 is quite promising. The reaction proceeded with a conversion of 99.7% and a selectivity of 80.0%.402 However, the above-mentioned chemical transformations of GLY employ expensive noble-metal catalysts. For industrial applicability, research studies should be directed to cheap and recyclable catalyst systems. Apart from these, Deng and Fong reported the direct conversion of untreated lignocellulosic biomass to 1-PO by using an engineered strain of the actino bacterium, Thermobifidafusca.403 Such direct conversions of raw lignocellulosic biomass to target compounds are more desirable. Of course, commercialization of such shortcuts will depend on the production costs and efficiencies.
As a different way of valorization, GLY can be converted to its short length oligomers (Fig. 3a). GLY-derived oligomers have GLY like applications so that they are also widely used in cosmetics, food industry, and in polymer and plastics industries. An important GLY oligomer is its dimer, diglycerol (DGLY). Heterogeneous catalysts exhibit better selectivities for oligomerization of GLY to DGLY. As such, Amberlyst catalysts provide DGLY selectivities up to 85%; however; the conversion of GLY remains very low (35–40%). Hence, development of heterogeneous catalysts with very high GLY conversion efficiencies is essential for selective oligomerization of GLY.91,400 Once selectively obtained, DGLY can be extensively used in the manufacture of polyurethanes and polyesters. Its industrial applications include the production of plasticizer in polyvinyl alcohol films or starch-based biodegradable thermoplastic compositions.404 Satoh et al. have recently employed DGLY in a different manner. They produced a novel monomer, difurfurylidene diglycerol (DFDG), by the acetalization reaction of DGLY and furfural in the presence of an Amberlyst 15 catalyst at 75 °C under a reduced pressure of 100 mbar for 48 h. The new DFDG monomer can be fully bio-based since both DGLY and furfural are bio-derivable compounds. A bio-based linear polyimide with Mw of 5400 was then obtained as a result of Diels–Alder (DA) polymerization of DFDG with 4,40-bismaleimidodiphenylmethane (BMI) at 60 °C for 48 h (Fig. 30a). The polymaleimide is expected to be environmentally benign having feedstock recovery, thermo-responsive and remendable features.405
 | ||
| Fig. 30 (a) Thermo-reversible DA polymerization of DFDG and BMI. (b) Production of mannitol based PExManxyT copolyesters. | ||
Production of hexitols from GLY is also possible. For instance, under aerobic conditions, Khan et al. reported the production of mannitol (Fig. 3a) from GLY by employing resting cells of Candida magnoliae.92 As shown in Fig. 8, a large variety of polymers can be obtained through selective functionalization of hexitols. As a recent example, Lavilla et al. produced a novel carbohydrate-based bicyclic diol: 2,4:3,5-di-O-methylene-D-mannitol (Manx), by internal acetalization of D-mannitol. Manx monomer was then reacted in the melt with ethylene glycol and dimethylterephthalate for the production of random PExManxyT copolyesters (Fig. 30b). The produced copolyesters have similar thermal stability, higher Tg and low crystallizability with respect to PET. These findings suggest that Manx, as a bio-based comonomer, is suitable for obtaining amorphous PET products with enhanced glass-transition temperature. It can be used in polymer applications requiring thermal stability and transparency.406 More detailed information regarding the utilization of hexitols for the preparation of bio-based polymers was provided in Section 5.1.
GLY and its derivatives, such as hydroxyacetone (acetol)104 (Fig. 3a), can be valorized as potential platform compounds in the synthesis of renewable diesel or jet fuel.407,408 For the first time, Li et al. obtained diesel or jet fuel range branched alkanes by the hydroxyalkylation–alkylation (HAA) of 2-methylfuran (2-MF) with lignocellulose derived hydroxyacetone. The hydroxyacetone pathway showed higher HAA reactivity and diesel yield over the previous acetone route owing to the electron-withdrawing effect of the hydroxyl group.407 Another GLY derivable hydroxy ketone compound is dihydroxyacetone (DHA) (Fig. 3a).99,399,400 DHA is industrially produced by microbial fermentation of GLY over Gluconobacter oxydans. It is an intermediate in the metabolism of glucose in humans as well as an FDA approved compound for topical use as the active ingredient in sunless tanning lotions. DHA-based polymers are expected to have advantageous biomaterial applications since DHA degradation products can enter the normal metabolic pathway. Owing to this motivation, Putnam and Zelikin patented polycarbonates, poly(acetal carbonate)s, poly(spiroacetal)s, polyesters and polyurethanes by using chemically protected DHA and/or its dimers.409 In solution, monomeric DHA is in equilibrium with its hemiacetal dimer. Locking monomeric DHA in the dimer form allows the preparation of poly(carbonate acetal)s. Alternatively, through the conversion of the C2 carbonyl into a dimethoxy acetal, DHA can be converted to 2,2-dimethoxypropane-1,3-diol carbonate (TMC(OMe)2). The bulk ROP of TMC(OMe)2 in the presence of stannous octanoate (Sn(oct)2) results in the formation of polycarbonates (Fig. 31).410
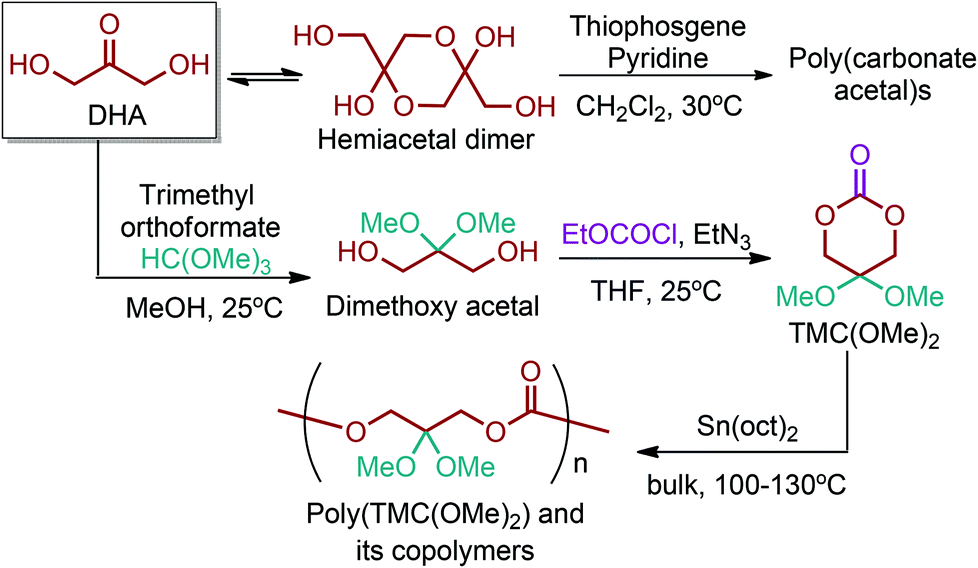 | ||
| Fig. 31 Conversion of DHA to poly(carbonate)s and poly(carbonate acetal)s. Adapted from ref. 410. | ||
Selective dehydration of GLY affords the production of extensively used vinyl monomers such as acrylic acid, acrolein, propene, allyl alcohol and 3-methoxy-1-propene (Fig. 3a). Acrolein is commercially produced by oxidation of petroleum-derived propene with a Bi/Mo mixed oxide catalyst at temperatures above 300 °C. Large scale production of acrylic acid also starts from propene. The process involves a two-step reactor system and metal oxide catalysts, in which propene is first oxidized to acrolein as the intermediate, and then further oxidation of acrolein yields acrylic acid (Fig. 32). As shown in Section 5.4 – Fig. 12, bio-based production of acrolein and acrylic acid can be realized starting from 3-HPA as the feedstock. In the process, 3-HPA can be converted to propene through a dehydration reaction coupled with a decarboxylation process. Conversion of propene to acrolein and/or acrylic acid has already been commercially practiced as mentioned above. On the other hand, the conversion of 3-HPA to propene is yet to be realized. Besides, no known organisms can produce 3-HPA as a major metabolic end product from sugars. These issues currently alleviate 3-HPA from being a platform chemical for large scale production of propene and acrylic monomers. In this consideration, GLY, as a cheap byproduct of biodiesel production, may become a better alternative. As such, acrolein can be produced from GLY via the dehydration of GLY on acidic solid catalysts. In the process, full conversion of GLY to acrolein is achieved by passing a mixture of GLY–H2O gases at 250–340 °C over an acidic solid catalyst (Fig. 32). Recently, the usage of sub- and supercritical water as the reaction media and a biocatalysis pathway employing Lactobacillus reuteri have also been investigated.399,400 Nonetheless, these acrolein production routes from GLY do not satisfy the criteria of an economical process, and thus, they were not commercialized so far.399 The GLY-based synthesis methods should be cost-competitive in comparison with the current propene-based process. Hence, a process development is required for further reducing the GLY-derived acrolein production costs. Once obtained, GLY-derived acrolein can be further oxidized to acrylic acid by employing the current industrial process (Fig. 32). It is also possible to produce propene through the GLY–acrolein–allyl alcohol–propanol–propene pathway.98 However, this process is quite cumbersome and it does not seem to be a cost-competitive route for the production of bio-propene. Common polymers of acrylics and propene were described in Sections 5.4 and 5.14, respectively. So, no further details are provided in this section.
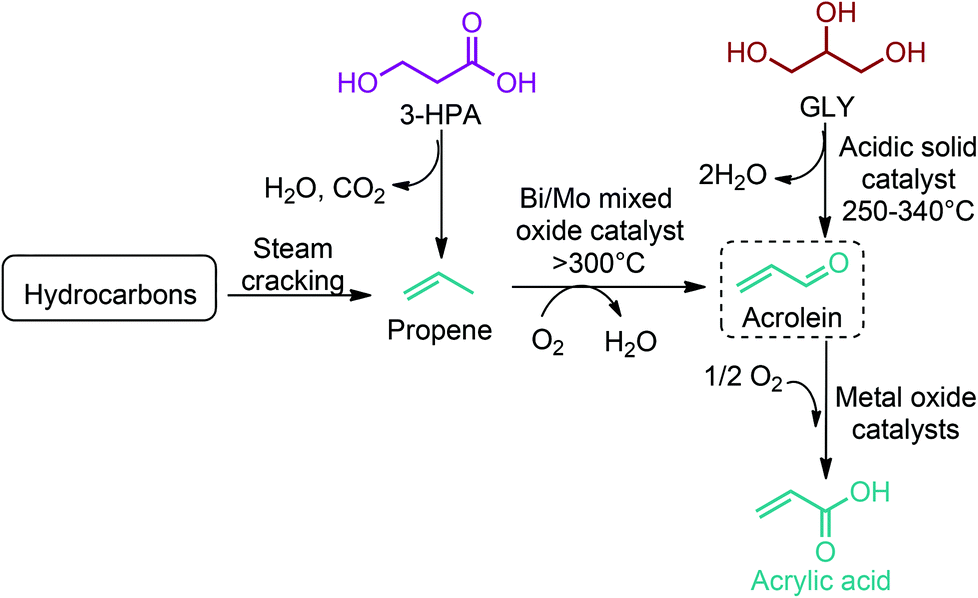 | ||
| Fig. 32 Production pathways of propene, acrolein, and acrylic acid from different feedstocks. | ||
Liu et al. have shown that using iron oxide as a catalyst first catalyzes the dehydration of GLY to acrolein and, consecutively, the selective hydrogenation of acrolein to allyl alcohol (AlOH). The catalyst results in full conversion of GLY but AlOH yields remain between 20 and 25%.411 Although the yields are low, the study provides promising insights for direct conversion of GLY to AlOH. GLY-derived AlOH can be copolymerized with other monomers. As an example, in the presence of oxygen, it is copolymerized with styrene. AlOH based copolymers are used as an intermediate for the production of flame-resistant materials, or as a nematocide, fungicide, or preservative. It was reported that its condensation with methyl glucoside polyethers, and subsequent bromination and addition of isocyanates, yields flame-resistant polyurethane foams. Apart from these, AlOH is also a building block for the synthesis of polymerizable allyl esters and ethers. Allyl esters are generally produced by reacting AlOH with the free acids, acid anhydrides, or acid chlorides in the presence of pTsOH. On the other hand, allyl ethers are obtained when AlOH is heated with monoalkanols in the presence of mineral acids (Fig. 33). Detailed information regarding polymeric applications of AlOH derived allyl esters and allyl ethers was provided by Krähling et al.412
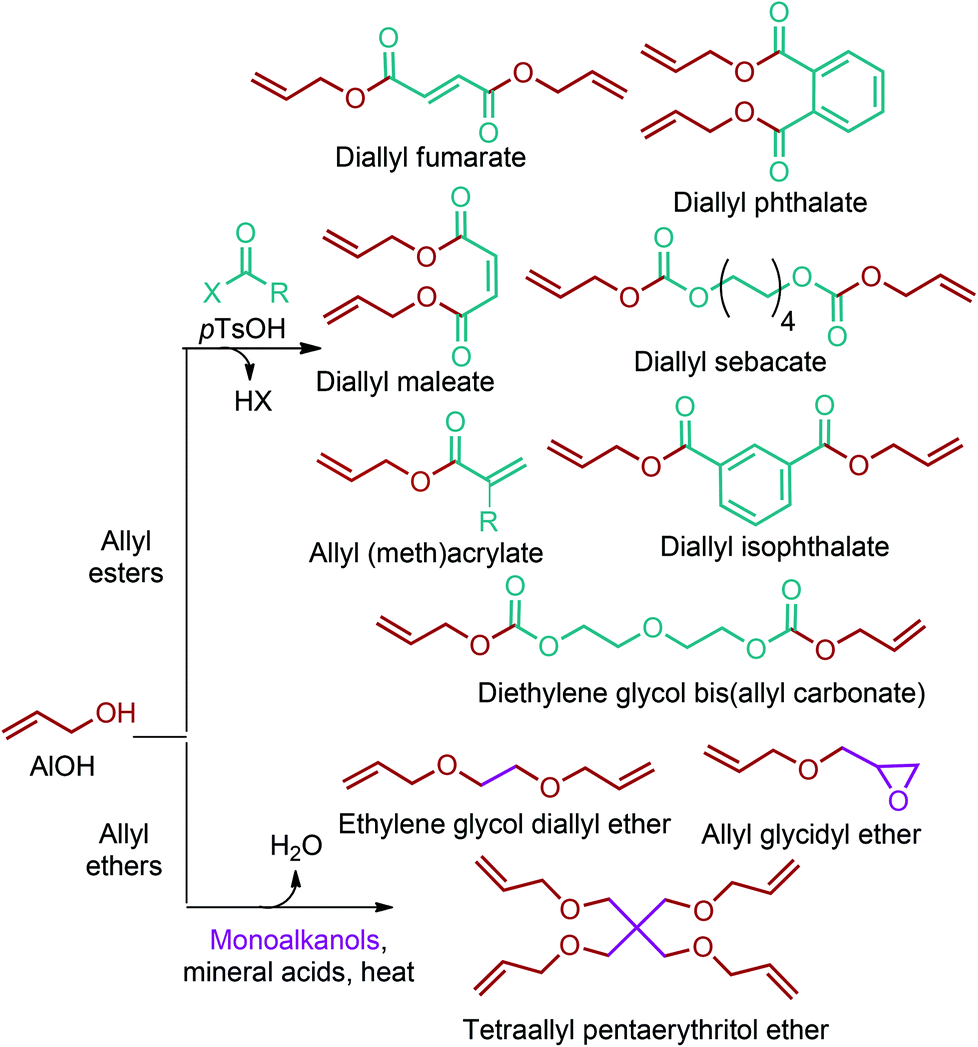 | ||
| Fig. 33 AlOH derived allyl ester and allyl ether monomers. | ||
Ring formation reactions can be performed on GLY for the production of industrially important cyclic monomers such as epichlorohydrin (ECH), glycidol and glycerol carbonate (GC) (Fig. 3a). Through the use of GLY as a renewable feedstock, the Dow Chemical Company has announced a significant improvement in the production of ECH by introducing a solventless, glycerin to epichlorohydrin (GTE), process, which proceeds in two steps.105 In the first step, GLY is hydrochlorinated to dichlorohydrins in the presence of a carboxylic acid catalyst under pressure at 120 °C. Subsequently, the ring closure reaction of the dichlorohydrins in the presence of a base yields ECH (Fig. 34). This process produces only one equivalent of waste chloride. On the other hand, the dominant commercial route employing propene as the starting compound retains only one of the four employed chlorine atoms in the final ECH product.105 This shows that the GLY-based process is more efficient in terms of atom economy. Besides, the two step GTE route is less laborious in comparison with the propene-based process (3 steps). Owing to these advantages, a shift from the dominant process to the GLY-based process might be observed in the near future. As such, Solvay Chemicals has also launched a GLY-based Epicerol® technology for the production of bio-based ECH in its new production plant in China.413 As a chemical intermediate, ECH has a wide range of applications (Fig. 34). It is primarily used in the manufacture of epoxy resins. The most familiar epoxy resin, bisphenol A diglycidyl ether (BADGE), is produced by condensing ECH with BPA. ECH is also homopolymerized or copolymerized with other monomers to form elastomers. ECH based elastomers exhibit excellent physical properties and are resistant to oxygen, weather, fuel, oil and ozone. The reaction of ECH with alcohols, alcoholates, or the sodium salts of fatty acids gives products which are used as vinyl polymer plasticizers. It is used in the manufacture of ion-exchange resins, polyamines and polyquaternary ammonium salts for water treatment applications. Many ECH-based surface-active agents are produced by reacting ECH with a polyamine. Other main application areas of ECH include textile, paper, agricultural and pharmaceutical industries.414,415
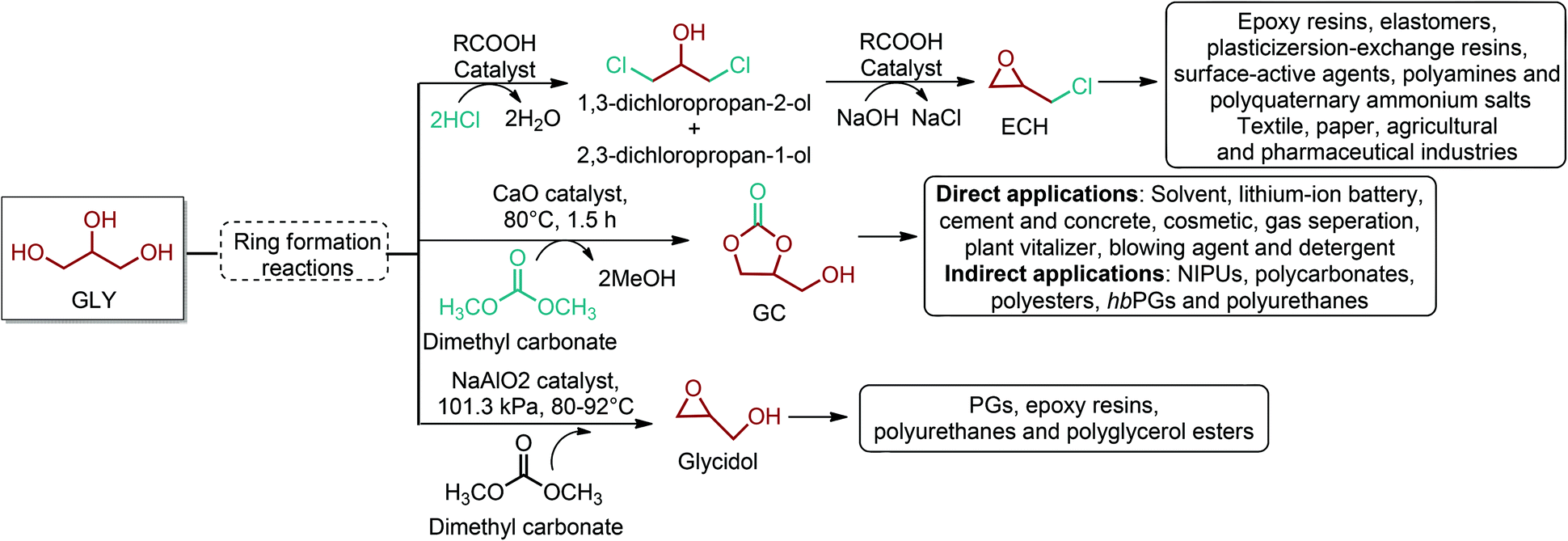 | ||
| Fig. 34 Common applications of GLY derived ECH, GC and glycidol. | ||
Another GLY derivable epoxide is glycidol (Fig. 3a). Commercially, glycidol is produced through epoxidation of AlOH or reaction of 3-chloro-1,2-propanediol with bases. However, both of the processes suffer from drawbacks mainly including: (i) petroleum-based raw materials which are not sustainable, (ii) multistep synthesis which decreases the synthetic efficiency and thus increases the production cost, and (iii) production of a large amount of waste liquid and salt.416 So, a bio-based process development for large scale production of glycidol is necessary. Such a process was recently reported by Bai et al.416 They demonstrated the one-pot synthesis of glycidol from GLY and dimethyl carbonate (DMC) in the presence of a reusable and recoverable NaAlO2 catalyst under pressure (101.3 kPa) at 80–92 °C. The selectivity of glycidol and the conversion of GLY were reported to reach 80.7% and 94.7%, respectively. As a different route, glycidol can be synthesized from the decarboxylation of GC. Zeolithe A was proved to be a very efficient catalyst for this reaction by providing a yield of 86% and a purity of 99% at a temperature of 180 °C and a pressure of 35 mbar.399,400 These developments can potentially provide large scale synthesis of bio-glycidol starting from GLY in a two step process (Fig. 34). Once obtained on a large scale, bio-glycidol can have a variety of industrial uses owing to its bifunctionality, having both epoxide and alcohol functional groups. Like GLY, glycidol can be polymerized into PGs. Sunder et al. described in detail the synthesis of hyperbranched PGs (hbPGs) through the reaction of glycidol with trimethylolpropane.417,418 For applications ranging from cosmetics to controlled drug release, a variety of polyglycerols were commercialized. For instance, Hyperpolymers Company (Germany) has been providing small-scale production hbPGs with different molecular weights and special derivatives. Besides these, glycidol is a high-value compound in the production of epoxy resins, polyurethanes and polyglycerol esters (PGEs).54,399,400 Esters of glycidol are also useful monomers. Recently, Geschwind and Frey reported the poly(glycerol carbonate)s which were synthesized from the polymerization of the glycidyl ethers: ethoxy ethyl glycidyl ether or benzyl glycidyl ether, with CO2. In the study, an innovative route for the synthesis of poly(1,2-glycerol carbonate) was presented.419
A different GLY derivable important cyclic monomer is GC. By taking industrial feasibility into account, Ochoa-Gómez et al. stated that the synthesis of GC starting from GLY and/or CO2-derivatives is the most attractive. Thus, the most suitable industrial process seems to be the transesterification of GLY with dimethyl carbonate in the presence of uncalcined CaO as a catalyst. This process is highly promising because (i) CaO is very cheap and widely available, (ii) the reaction proceeds with a 100% GLY conversion and a GC yield of 95%, and (iii) the reaction is complete in just 1.5 h at 80 °C.96 Many other synthesis routes of GC by considering its reactivity and applications were discussed in detail by Sonnati et al.420 Over the last 20 years, GC has attracted much interest because it is a very important compound in valorization of waste GLY. Owing to its wide reactivity, it has many direct and indirect applications (Fig. 34). Unlike other cyclic carbonates, the reactivity of GC stem not only from the dioxolane ring but also from the pendant hydroxyl moiety. The wide reactivity of GC opens numerous opportunities for its utilization as a raw material for the synthesis of industrially important chemical intermediates such as glycidol and ECH, as well as polymers such as non-isocyanate polyurethanes (NIPUs), polycarbonates, polyesters, hbPGs and polyurethanes. These indirect applications of GC hold enormous potential in manufacturing useful materials such as coatings, adhesives, foams, and lubricants.96,420–422
GLY oxidation creates new opportunities for the synthesis of value-added aldehyde and acid compounds (Fig. 3a). Kim et al. demonstrated that the electrochemical dehydrogenation process can be used to oxidize glycerol to glyceraldehyde (GAD).101 Another GLY derivable value-added aldehyde is 3-hydroxypropionaldehyde (3-HPAl), which can function as a precursor for monomers such as 3-HPA, 1,3-PDO, acrolein and acrylic acid. Furthermore, it is transformed into polymeric derivatives and resins. 3-HPAl is produced through both bacterial fermentation and traditional chemistry. It was stated that biotechnological production has several advantages with respect to the chemical process.423 In a recent study, Krauter et al. reported very high 3-HPAl concentration and productivities from glycerol by employing Lactobacillus reuteri. In the study, 138 g L−1 glycerol was converted into 108 g L−1 3-HPAl with an overall productivity of 21.6 g L−1 h−1 in a single fed-batch biotransformation at 45 °C.424 Further oxidation of GAD results in the formation of glyceric acid (GLA)103 and, subsequently, tartronic acid (TTA).99,400 The oxidation of both primary and secondary alcohol groups, on the other hand, gives the highly functionalized compound ketomalonic (mesoxalic) acid (KMA).99 GLA has both medicinal and industrial values.103 However, it is very expensive and thus it has not been commercially produced so far. Nonetheless, it can be abundantly produced from GLY by employing acetic acid bacteria425 and this pathway may open new opportunities for its mass production. The biotechnological production of GLA and its possible applications were reviewed by Habe et al.426 GLA acts as an ABB′-type trifunctional monomer since it has one –COOH and two –OH groups. Hence, various GLA based polymers having different structures such as linear, hyperbranched, and network structures can be obtained through suitable polymerization reactions. Fukuoka et al. reported a novel hyperbranched poly(lactic acid) by polymerizing lactide in the presence of GLA. The produced branched polymer could potentially be used as a bio-based modifier for poly(lactic acid)s.425 Catalytic oxidation of TTA in water over a BiPt/C catalyst first results in the formation of KMA and then in the polymerization of the produced KMA to its corresponding polyether: poly(ketomalonate) (PKM). Kimura stated that the decarboxylation of PKM can be easily performed and this proves that PKM has pendant carbon dioxide groups. When completely decarboxylated, poly(oxymethylene) is produced from PKM.427
5.13. Lactic acid platform based polymers
Lactic acid (LA) (Fig. 4a) is the most widely studied carboxylic acid from natural resources and it has achieved extensive success in commercialization. Industrially, LA is mainly produced via the fermentation of glucose and sucrose by lactic acid bacteria. Through the fermentation process, the global production of LA is around 350 kt per year and it is expected that there will be a substantial growth in the next decade.261 Since microbial production of LA has been extensively reviewed in the recent literature,131–133,428 it has not been described in detail here. However, it is noteworthy that the production of enantiopure L-lactic acid or D-lactic acid depends on the microbial strain used during the fermentation process. This is highly significant because LA stereo-chemistry greatly controls relevant physical properties of its final products.236 According to Abdel-Rahman et al.,131 it is very expensive when sugars, such as glucose, sucrose, and starch, are used as the feedstock for LA production. Hence, lignocellulosic biomass can have a great contribution to the expected substantial growth of LA production by considering its great availability, sustainability and low cost compared to refined sugars. Despite these advantages, the commercial use of lignocellulose for LA production is still problematic. The main problem stems from the costly pretreatment and enzymatic hydrolysis of lignocellulosic biomass for the production of fermentative sugars (Section 3). The pretreatment is a sine qua non process in which the structure of lignocellulosic biomass is broken down to separate cellulose and hemicelluloses from lignin. Hence, highly efficient and cheap pretreatment technologies need to be sought. On the other hand, the costly enzymatic hydrolysis step for depolymerising cellulose and hemicelluloses to fermentative sugars can be by-passed. In this consideration, the development of genetically modified lactic acid bacteria, which can directly ferment cellulose and/or xylan, is a necessary option.131 Fortunately, the commercial production of LA from lignocellulosic biomass does not seem to be too far away. As an encouraging example, the industrial biotechnology company Direvo has recently fermented lactic acid on a pilot scale by introducing a consolidated bioprocessing technology. The consolidated process allows the conversion of lignocelluloses in a single step without having to add enzymes.429LA is primarily used for the production of biodegradable polymers, and in food and beverage sectors. These sectors are followed by pharmaceuticals and personal care products.430 In the polymer industry, LA is mainly consumed in production of polylactic acid (PLA). There are two major chemical ways to prepare polylactic acid (PLA): polycondensation of LA or ROP of its cyclic dimer, lactide (Fig. 35). For the preparation of high molecular weight PLA, the ROP is more widely used. It is difficult to obtain high molecular weight PLA via polycondensation due to water formation during the reaction. ROP proceeds in the presence of catalysts and it allows the control of molecular weight as well as stereotacticity of PLA in comparison with polycondensation.261,431 Several distinct forms of PLA exist due to the chiral nature of LA. Polymerization of racemates results in the formation of amorphous poly-DL-lactide (PDLLA). On the other hand, poly-L-lactide (PLLA), the product resulting from polymerization of L,L-lactide, exhibits high crystallinity (37%), a glass transition temperature between 60 and 65 °C, and a melting temperature of around 175 °C. Hence, the degree of crystallinity depends mostly on a defined ratio of D-and L-enantiomers and it determines most physical properties of the final products.325
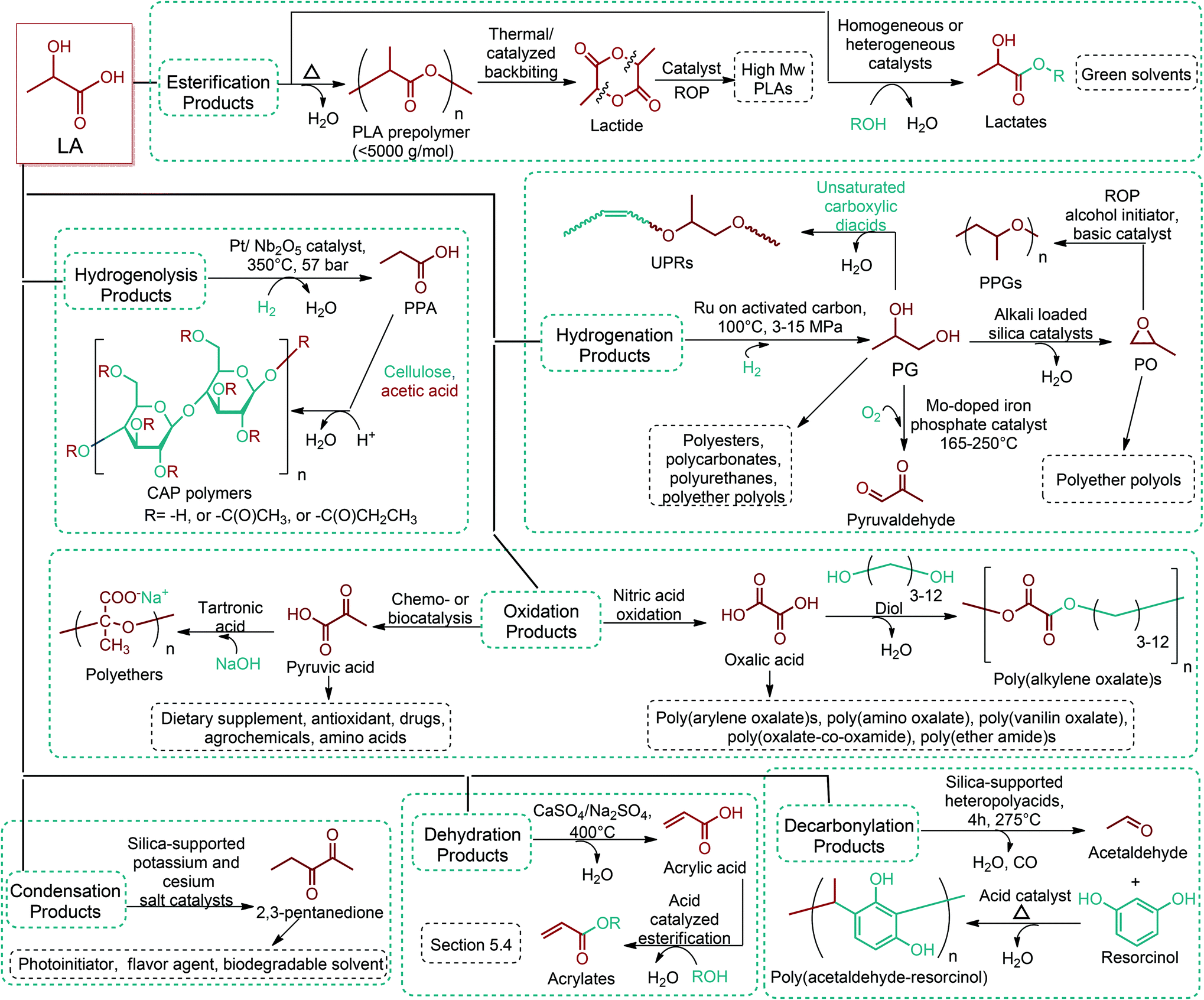 | ||
| Fig. 35 LA as a platform compound for the production of fine chemicals and polymers. | ||
Owing to its low price and availability in the market, PLA has one of the highest potential amongst the other biodegradable polyesters.261,326 Its market volume is projected to exceed $4840.1 million by 2019.430 Thus, many companies have been involved in its commercial production (Table 6). NatureWorks is currently the major supplier of PLA under the brand name of Ingeo. There are other PLA manufacturers in the USA, Europe, China and Japan. They have been developing various grades of PLA suitable for different industrial sectors.325,431 Until the late 1980s, the applicability of PLAs outside the medical field was restricted by high production costs. Later on, major breakthroughs in the process technology decreased the production costs and this allowed the commercial-scale production of biodegradable polymers from LA for nonmedical applications.285 Currently, the produced PLA is mainly used in the packaging market, which is projected to be valued at $994.9 million by 2019. The textile segment accounts for the second-largest share in the PLA market and it is followed by agriculture, electronics, automobile and other segments.430 PLA as a biodegradable and thermoplastic polyester has the potential to replace traditional polymers such as PET, PS, and PC for various applications. However, there are still many challenges to be addressed. Tg and brittleness are the most noticeable weakness of PLA and there are many research groups actively involved in overcoming and addressing solutions to these problems.261,431 Toughening of PLA by blending it with a variety of materials is a commonly employed approach in this consideration.432 However, a completely renewable and biodegradable toughening agent should be used to preserve the renewability and biodegradability of the final product. Since PLA based polymers and their applications are extensively reviewed in the literature, it was not described in detail here. Detailed information regarding synthesis and production, thermal–chemical–mechanical–rheological properties, degradation and stability, and applications of PLAs is provided in the comprehensive books and review articles.433–438
| Company | Brand name | Location |
|---|---|---|
| NatureWorks LLC | Ingeo | USA |
| Purac | Purasorb | Netherlands |
| Futerro | Futerro | Belgium |
| Tate & Lyle | Hycail | Netherlands |
| Synbra | Biofoam | Netherlands |
| Toyobo | Vylocol | Japan |
| Teijin | Biofront | Japan |
| Uhde Inventa-Fischer | Germany | |
| Hiusan Biosciences | Hisun | China |
LA is transformed to other valuable chemicals via esterification, hydrogenolysis, dehydration, decarbonylation, oxidation, and reduction processes (Fig. 4a and 35). The leading esterification products of LA are lactide132,133 and PLAs. Its esterification leads to the formation of lactates too, depending on the reaction conditions. A wide range of alkyl lactates (linear esters), which exhibit unique solvation properties, are easily produced from LA using either homogeneous or heterogeneous catalysts. The esterification of LA has been investigated with different alcohols including methanol, ethanol, 2-propanol, isobutanol, n-butanol and benzyl alcohol. The most prominent example is ethyl lactate. This short chain ester is amongst the most promising green solvents owing to its high boiling point, low vapour pressure, low surface tension and renewable origin.132,133,137
The dehydration and reduction of LA result in the formation of giant monomers: acrylic acid (Section 5.4) and propylene glycol, respectively.54,132,133,138 In 1958, LA was for the first time converted to acrylic acid via a direct dehydration reaction. A maximum yield of 68% of acrylic acid was obtained over a CaSO4/Na2SO4 heterogeneous catalyst system at 400 °C. Thus far, no important progress has been reported so that the acrylic acid yields generally remained below ∼70%.132,133 Mainly due to this reason, LA-derived acrylic acid production has not been put into commercial practice. Finding the right heterogeneous catalyst seems to be the key challenge since the main consideration is to increase the acrylic acid yield. In this respect, Dusselier et al. suggested the use of alkali modified zeolities which can eliminate formation of the side products including CO2, acetaldehyde and coke.133 The LA derived AA can be further functionalized. It is converted to acrylates with the help of efficient, cheap and reusable catalysts (Section 5.4). A different way of producing acrylates is their direct production from lactates through the dehydration reaction.133,142 The production of AA or its esters starting from LA might become an industrially feasible technology depending on its cost competitiveness in comparison with the current propene process. Acrylic polymers and their applications were described in detail in Section 5.4.
Hydrogenolysis of the secondary alcohol group of LA generates propanoic acid (PPA). The reaction can take place over a bifunctional Pt/Nb2O5 catalyst at 350 °C and 57 bar pressure. In the process, LA is first dehydrated on the acid support and, subsequently, hydrogenated to PPA on the metal sites.140 Nonetheless, the reaction yields a very low amount of PPA (18%), and thus, a process development is required. Particularly, instead of expensive Pt-based catalysts, the development of bifunctional, cheap and recyclable catalysts which can provide high LA conversions as well as high PPA yields should be considered. Once obtained on a large scale, LA-based PPA can be employed in the production of cellulose–acetate–propionate (CAP) polymers (Fig. 35). Different formulations of CAP polymers are currently produced by Eastman Company for printing ink and nail care applications.439 In addition, CAP has been studied as composite, adsorbent, membrane and coating material for a wide range of applications.440–443
Corma et al. stated that the direct hydrogenation of LA or lactates to propylene glycol (PG) can be an alternative green route to the current petroleum-based process, which is based on hydroperoxidation chemistry or the chlorohydrin process involving the use of hypochlorous acid.54 Particularly, ruthenium on activated carbon was reported to be an effective catalyst to perform the hydrogenation process by providing 95% LA conversion and PG selectivities higher than 90%.54 Nevertheless, the industrial manufacture of bio-PG is currently focused on glycerol mainly due to the large and cheap surplus of glycerol in the market, which is obtained as a byproduct in the production of biodiesel from vegetable oils (Section 5.12). Companies such as Dow Chemicals, Cargill/Ashland, ADM, and Senergy have been producing bio-based PG from glycerol.444 Annually, 1.5 million tons of PG are manufactured by employing the above-mentioned petroleum-based process.37 Such a high production capacity of PG simply stems from its large applicability. Nearly 45% of PG is consumed in the manufacture of unsaturated polyester resins (UPRs) (Fig. 35), in which it is reacted with saturated and unsaturated carboxylic diacids such as isophthalic acid and maleic anhydride. The resulting resins are dissolved in styrene or another polymerizable monomer, which is combined by using filler, a chopped glass, a peroxide polymerization initiator, and other additives. Then, they are cured to give a hard, cross-linked, thermoset composite. The end-use applications for such UPRs include laminates, automotive plastics, fiber glass boats, construction, coatings, art objects, insulation, electrical components, pipes and tanks. Polymeric applications of PG are not limited to polyesters, but they also consist of polycarbonates, polyurethanes and polyether polyols.270,325,445 PG has many other diverse applications since it is conferred as “Generally Regarded As Safe” (GRAS) by the FDA. It is commonly used as a cosmetic and food additive, an antifreeze agent, a brake fluid, a lubricant, and an aircraft de-icing fluid.37,445 Besides, PG is employed as an intermediate for the production of other valuable compounds including propylene oxide and pyruvaldehyde (Fig. 4a and 35).139,141 Between 60% and 70% of the total volume of PO is used in the manufacture of polyether polyols. The Dow Chemical Company is the world's largest producer of PO and polyether polyols. Commercial polyether polyols are based on di-, tri-, or polyhydric alcohols which are usually copolymerized with PO/ethylene oxide. They are mainly consumed in the production of polyurethanes. Such polyurethanes can exhibit a wide range of hardness, rigidity, and density characteristics. Polyols with molecular masses higher than 3000 are used for the manufacture of flexible polyurethane foams for applications requiring flexibility such as furniture and automobile seating, bedding, and carpet underlay. On the other hand, polyols with lower molecular masses lead to the formation of rigid foams for applications such as thermal insulation.446,447 A different PO based polymer family is polypropylene glycols (PPGs) (Fig. 35). They are produced by ROP of PO in the presence of an alcohol initiator and a basic catalyst. A variety of PPGs can be obtained by using different alcohol initiators and/or by copolymerizing PO with other monomers. Depending on the functionality of the initiator (i.e. mono-, di-, or multi-functional initiators), PPGs having different architectures such as linear, branched, or even linear-dendritic hybrid systems can be obtained for diverse applications.446,448–451
Other considerable transformations of LA include its decarbonylation to acetaldehyde, condensation to 2,3-pentanedione, and oxidation to pyruvic and oxalic acids (Fig. 4a and 35).25,132–136,452 Regarding common applications of these fine chemicals, acetaldehyde is used in the manufacture of novolak resins of resorcinol (Fig. 35). Acetaldehyde modified novolaks exhibit certain characteristics, i.e. special solubility or compatibility properties in rubber compounding applications, which may not be provided by straight resorcinol-formaldehyde resins.453 2,3-Pentanedione has applications as a flavor agent, a biodegradable solvent, a photoinitiator, and an intermediate for other high-value compounds.132 As an oxidation product of LA, pyruvic acid has been increasingly used as a dietary supplement and an antioxidant, as well as a precursor for the synthesis of drugs, amino acids and agrochemicals.133 Moreover, this alpha-keto acid can be anionically polymerized to form the corresponding polyether (Fig. 35).454 Very recently, Boamah et al. employed pyruvic acid in a different manner. They prepared pyruvic acid modified low molecular weight chitosans as potential lead adsorbent materials.455 The other oxidation product of LA, oxalic acid, can be used in polycondensation reactions. Nonetheless, its diester456 or dichloride (oxalyl chloride)457 derivatives are more commonly used instead of oxalic acid in the literature since they provide more facile polycondensation reactions.
5.14. Acetone–butanol–ethanol (ABE) platform based polymers
Biomass based fuels currently seem to be the most suitable alternative for the production of renewable, sustainable, and economically viable fuels such as bio-ethanol. In this context, lignocellulosic biomass resources have certain advantages. First of all, lignocellulosic biomass is the most abundant sustainable raw material worldwide and it has widespread availability. Furthermore, it has non-competitiveness with food supplies and high mitigation effects on GHG emissions. Owing to these characteristics, various companies have been engaged in the commercial production of bio-ethanol from lignocellulosic biomass resources (Table 7). Although the production of bio-ethanol from lignocellulosic sugars is now a commercialized technology, the process of conversion is more complicated than that of starch based biofuels. This mainly stems from the rigid and complex molecular polymeric structure of lignocellulosic biomass. Hence, the production of lignocellulosic ethanol is still a challenge with many opportunities for progress. The current status and challenges as well as the future prospects of bio-ethanol production from lignocellulosic biomass were not comprehensively presented in this article since the detailed scrutiny was provided in the corresponding books and review articles.143,199,458–463 Renewed attention has also been paid to butanol and acetone production from lignocellulose through the acetone–butanol–ethanol (ABE) fermentation process (Fig. 4c). In fact, butanol as a higher alcohol is a more promising gasoline substitute compared to ethanol. That is why the production of bio-butanol has been receiving great interest from both small biofuel start-ups and large oil and chemical companies such as British Petroleum, Chevron, DuPont and DSM.144,464–466| Company | Plant location | Feedstock |
|---|---|---|
| Abengoa | USA | Corn stover, wheat straw, milo (sorghum) stubble, switchgrass, |
| Abengoa | Spain | Wheat straw, cereal |
| Agroethanol AB | Sweden | Wheat |
| ALICO, Inc. | USA | Yard and citrus wastes |
| American Process Inc. | USA | Woody biomass |
| BioEthanol Japan | Japan | Wood construction waste |
| BioFuels Energy Corp. | USA | Grass and tree trimmings |
| BlueFire Ethanol | USA | Green waste |
| Borregaard Industries Ltd | Norway | Wood |
| British Sugar | England | Sugar beet |
| Broin jointly with US DoE, DuPont and Novozymes | USA | Corn fiber and stover |
| Colusa Biomass Energy Corporation | USA | Rice straw and hulls |
| China Resources Alcohol Corporation | China | Corn stover |
| DINS Sakai | Japan | Waste construction wood |
| DuPont-BP Biofuels | England | Sugar beets |
| ICM Inc. | USA | Corn stover, switchgrass |
| Inbicon | Denmark | Wheat straw |
| Iogen | USA | Wheat straw, barley straw, corn stover, switchgrass and rice straw |
| Iogen | Canada | Wheat, oat and barley straw |
| Lignol | Canada | Softwood and hardwood |
| Mascoma | USA | Paper sludge, wood chips, switch grass and corn stover |
| Pacific Ethanol Inc. | USA | Wheat straw, corn cob, woody biomass |
| Poet | USA | Corn fiber, corn stover |
| Range Fuels | USA | Timber and forest residue |
| Sekab | Sweden | Forestry products |
| Tereos | France | Sugar beet, wheat and sugar cane |
| Verenium | USA | Sugarcane bagasse and specially bred energy cane |
| Western Biomass | USA | Ponderosa pine wood chips, waste |
ABE compounds are promising precursors to many value-added chemicals (Fig. 4c). They are converted to other value-added chemicals through different chemical processes such as oxidation, reduction, dehydration, and chlorination reactions. For instance, Ozkar and Finke developed a system for room temperature and acid-assisted hydrogenation of acetone to 2-propanol in the presence of Ir(0) nanoclusters. The system exhibits exceptional activity by providing up to 100% selectivity at 100% conversion, and up to 188![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 total turnover catalytic lifetime.145 In terms of oxidation processes, ethanol and butanol can be selectively converted into acetic152 and butyric acids,147 respectively. Acetic acid can also be produced through fermentation of C5 and C6 sugars. As an example, ZeaChem Inc. has recently achieved a commercially scalable acetogenic process for the fermentation of the cellulosic sugars to acetic acid without CO2 as a by-product.326 Cheung et al. pointed out that the global production of acetic acid was approximately 10.6 × 106 tons per annum in 2008. More than 65% of this production is converted into vinyl acetate or cellulose based polymers (Fig. 36).467 For the preparation of vinyl acetate monomer (VAM), acetic acid is reacted with ethylene and oxygen either in the liquid phase in the presence of Pd/Cu, or in the gas phase on heterogeneous catalysts containing palladium.468 It should be noted that raw materials for VAM, ethylene and acetic acid, are nowadays based on fossil resources. As shown by Amann and Minge, this could be switched to a bio-ethanol based renewable, sustainable and CO2-neutral production process.469 The polymerization of VAM can be carried out by employing different types of standard polymerization techniques like emulsion, suspension and solution polymerization. Through these techniques, VAM is used to produce poly(vinyl acetate) (PVAc) as well as a wide range of PVAc copolymers. Particularly, ethylene–vinyl acetate copolymers are one of the most important classes. Such polymers are abbreviated as EVA (high ethylene and low vinyl acetate content) or conversely VAE depending on the ethylene and VAM content. Around 30% of the produced VAM is converted to poly(vinyl alcohol) (PVA) through the saponification or transesterification process of PVAc. The remaining 20% is valorized in other ways, such as in the production of poly(vinyl butyral) (PVB). The major manufacturers of VAM and VAM-based polymers include, but are not limited to, BP, Wacker Chemie, Celanese Chemicals, Dow Chemical Corp., DuPont, Millenium, Kuraray, Nippon Gohsei, Showa Denko, Dairen Chemical, and Shanghai Petrochemical.469 In addition to VAM-based polymers, acetic acid is also utilized in the synthesis of cellulose based polymers, predominantly cellulose acetate. Detailed scrutiny regarding properties, applications, and degradation behaviors of cellulose acetate-based materials was presented by Fischer et al.470 and Puls et al.471 Major applications of the above-mentioned acetic acid derived polymers are summarized in Fig. 36.270,468–471
000 total turnover catalytic lifetime.145 In terms of oxidation processes, ethanol and butanol can be selectively converted into acetic152 and butyric acids,147 respectively. Acetic acid can also be produced through fermentation of C5 and C6 sugars. As an example, ZeaChem Inc. has recently achieved a commercially scalable acetogenic process for the fermentation of the cellulosic sugars to acetic acid without CO2 as a by-product.326 Cheung et al. pointed out that the global production of acetic acid was approximately 10.6 × 106 tons per annum in 2008. More than 65% of this production is converted into vinyl acetate or cellulose based polymers (Fig. 36).467 For the preparation of vinyl acetate monomer (VAM), acetic acid is reacted with ethylene and oxygen either in the liquid phase in the presence of Pd/Cu, or in the gas phase on heterogeneous catalysts containing palladium.468 It should be noted that raw materials for VAM, ethylene and acetic acid, are nowadays based on fossil resources. As shown by Amann and Minge, this could be switched to a bio-ethanol based renewable, sustainable and CO2-neutral production process.469 The polymerization of VAM can be carried out by employing different types of standard polymerization techniques like emulsion, suspension and solution polymerization. Through these techniques, VAM is used to produce poly(vinyl acetate) (PVAc) as well as a wide range of PVAc copolymers. Particularly, ethylene–vinyl acetate copolymers are one of the most important classes. Such polymers are abbreviated as EVA (high ethylene and low vinyl acetate content) or conversely VAE depending on the ethylene and VAM content. Around 30% of the produced VAM is converted to poly(vinyl alcohol) (PVA) through the saponification or transesterification process of PVAc. The remaining 20% is valorized in other ways, such as in the production of poly(vinyl butyral) (PVB). The major manufacturers of VAM and VAM-based polymers include, but are not limited to, BP, Wacker Chemie, Celanese Chemicals, Dow Chemical Corp., DuPont, Millenium, Kuraray, Nippon Gohsei, Showa Denko, Dairen Chemical, and Shanghai Petrochemical.469 In addition to VAM-based polymers, acetic acid is also utilized in the synthesis of cellulose based polymers, predominantly cellulose acetate. Detailed scrutiny regarding properties, applications, and degradation behaviors of cellulose acetate-based materials was presented by Fischer et al.470 and Puls et al.471 Major applications of the above-mentioned acetic acid derived polymers are summarized in Fig. 36.270,468–471
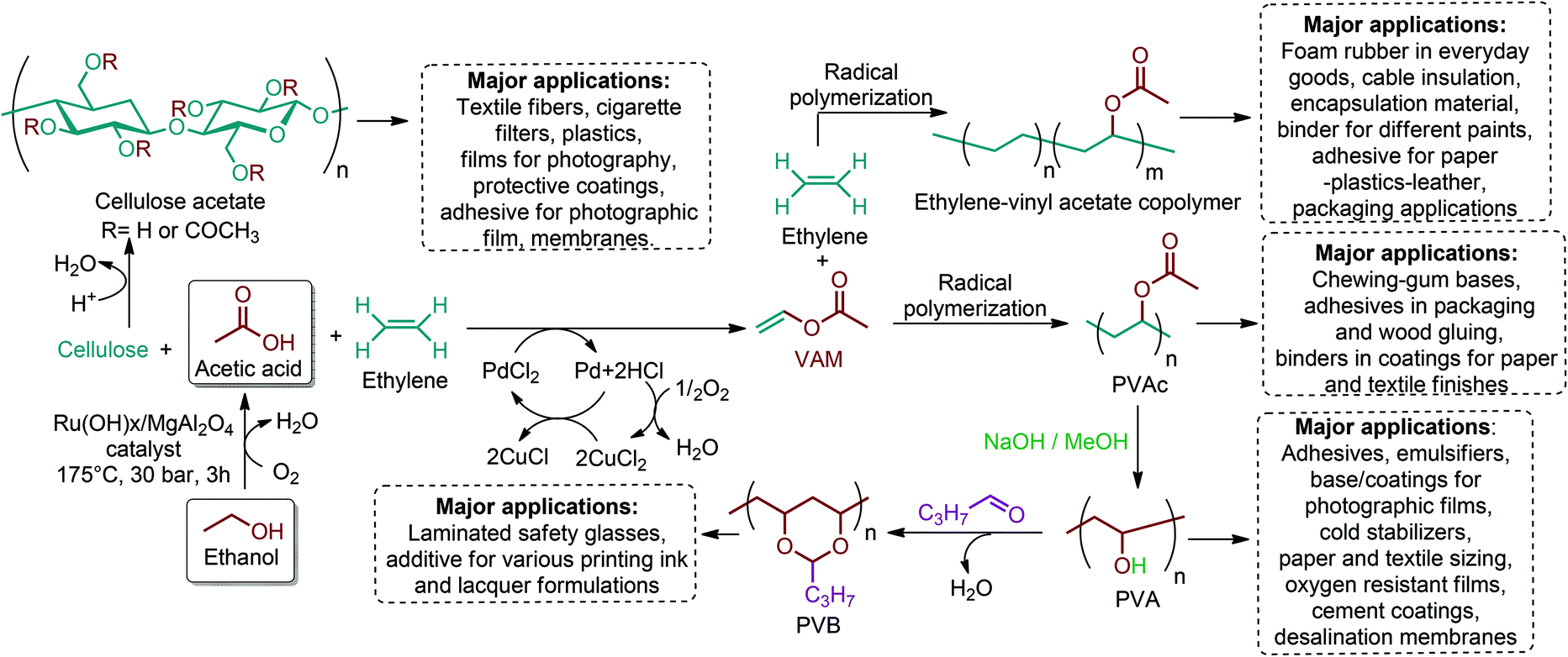 | ||
| Fig. 36 Acetic acid based commercial polymers. | ||
Without a doubt, the most important process that can be performed on ABE alcohols is dehydration. Giant monomers of today's polymer industry such as ethylene,149,325,472 propylene,146,472 butadiene,148 1-butene,363 and isobutene473 have been produced through dehydration of ethanol, butanol, and 2-propanol as shown in Fig. 4c.
Bio-derived ethanol has been particularly dehydrated to ethylene. In fact, in the early 20th century, ethylene was mainly produced from ethanol. Later on, the process was shifted to the current petrochemical route in which ethylene is exclusively produced via steam cracking of hydrocarbons at 750–950 °C. The main reason for this shift was the unbeatable cheap price of oil. Nonetheless, due to the current cost-competitive production of bio-ethanol and public interest in polymeric products derived from renewable resources, several companies like Dow, Braskem and Solvay have engaged in bioethanol-to-ethylene projects. The ethanol dehydration reaction was recently conducted in the vapor phase through fixed bed or fluidized bed reactors over solid acid catalysts like alumina or silica-alumina. Over these catalysts and at around 400 °C, full conversion of ethanol and 99.9% selectivity of ethylene could be achieved.325 This conversion is quite valuable because ethylene dominates the petrochemical market by being the largest petroleum chemical. In 2010, the worldwide production capacity of ethylene was approximately 160 × 109 lbs per year. To meet the demand for the enormous quantity of ethylene consumed worldwide, large quantities of renewable materials are needed. This, in fact, is another factor for the current shift towards the dehydration of the bio-ethanol process.325,472 As an intermediate chemical, ethylene is predominately used in the manufacture of many most important commodity polymers like high- and low-density polyethylene, poly(vinyl chloride), polystyrene and poly(ethylene terephthalate). These polymers make up almost two thirds of the current plastics market. Polyethylene (PE), the world's most widely used plastic, is produced by polymerization of ethylene under pressure and temperature, and in the presence of a catalyst. Ethylene can be either used as a monomer for high-density polyethylene (HDPE) or as a comonomer for linear low density polyethylene (LLDPE) (Fig. 37). Moreover, it can be copolymerized with a wide range of monomers such as propylene, styrene, vinyl acetate, and acrylic acid to manufacture different materials with the desired properties. With increases in oil prices, microbial PE or green PE is now available in the market owing to the commercial production of bio-ethylene. Braskem is the first and largest producer of bio-PE with 52% market share, followed by Dow Chemical and Solvay with market shares of 12% and 10%, respectively. Bio-PE has exactly the same chemical, physical, and mechanical properties as petrochemical polyethylene, which means that all the applications of current fossil-based PE can be replaced by bio-PE. Currently, bio-PE is widely used in engineering, agriculture, packaging, and many day-to-day commodity applications due to its low price and good performance.261,270,326,431,472,474,475
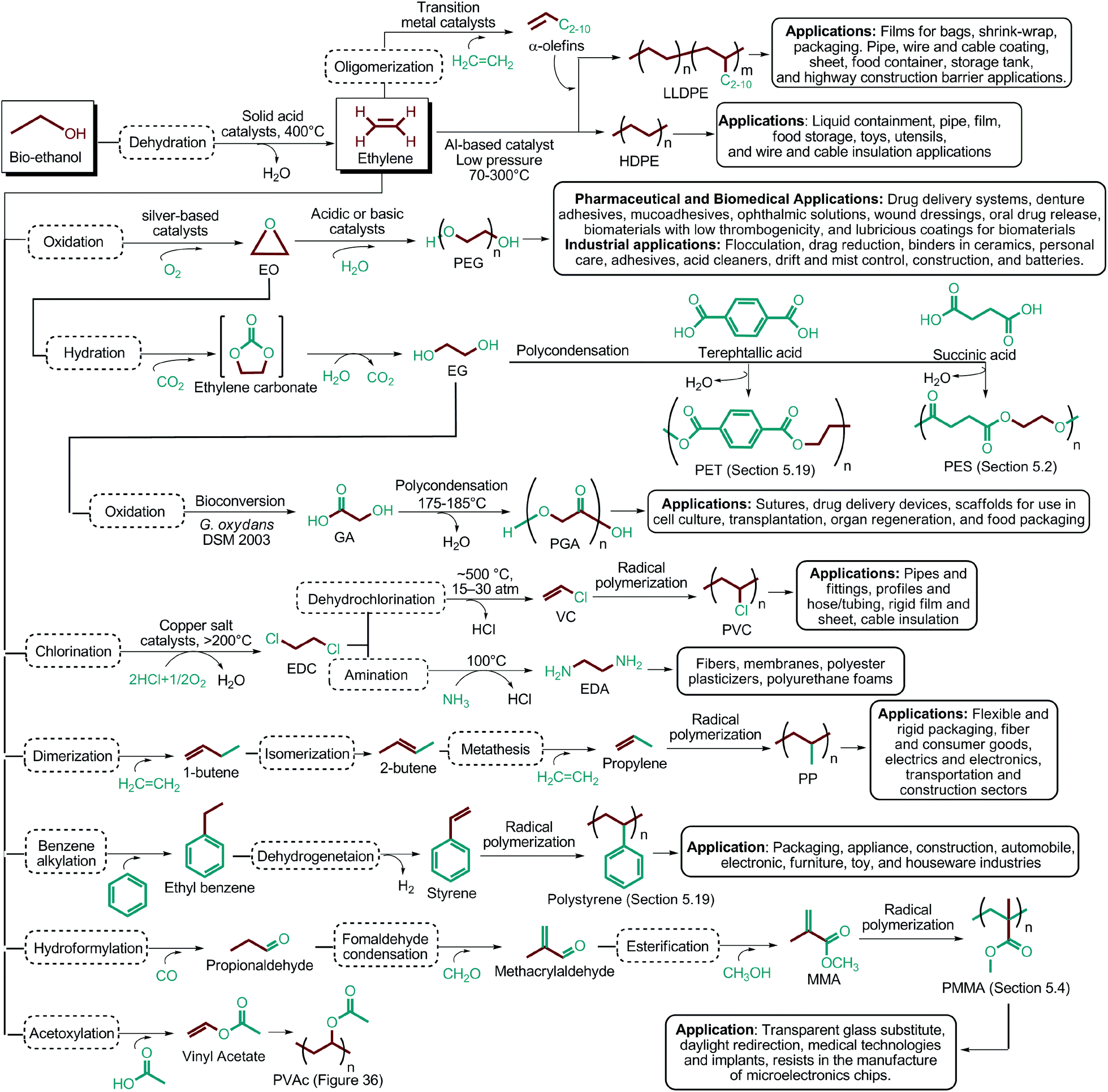 | ||
| Fig. 37 Bio-ethanol derived ethylene as a platform chemical for synthesis of commodity monomers and their corresponding polymers. | ||
Ethylene is also a valuable precursor for the production of other related commodity compounds. Once bio-ethylene is obtained, no process development is required for its conversion to ethylene oxide (EO) and ethylene glycol (EG). Ethylene has already been commercially oxidized to ethylene oxide (EO), which is then hydrolysed to produce ethylene glycol (EG) (Fig. 4c). In 2011, the worldwide demand for EO was estimated to be higher than $28000 million. From 2013 to 2018, this demand is expected to grow at a CAGR of over 6%, and pass $40000 million in 2018. Currently, the global EO demand has been supplied by the vapor-phase oxidation of ethylene over silver-based catalysts. Several companies such as Dow Chemical Company, Shell, SABIC, Scientific Design, Japan Catalytic, BASF and China Chemical have been implementing this technology. However, depending on whether pure oxygen or air is used for oxidation, differences may exist in their technological details.150,476,477 The produced EO is utilized as a decent disinfectant, a sterilizing agent, and a fumigant. Its ROP can proceed anionically or cationically but both of them generally give lower molecular weight products. For the production of extremely high molecular weight polymers, the process must involve a coordinate anionic reaction, in which EO is coordinated with a metal atom of the catalyst and subsequently attacked by an anion.478 Its major polymer derivative is polyethylene glycol (PEG), also known as polyethylene oxide (PEO) (Fig. 37). PEGs exhibit very high solubility, low toxicity, unique solution rheology, complexation with organic acids, low ash content, and thermoplasticity.478 It is feasible to produce various PEG copolymers having different architectures such as linear, branched, star shaped, and comb-like PEGs. Furthermore, highly diverse PEG-based polymers or polymeric systems can be developed via the PEGylation method; the process of covalent attachment of one or more PEG chains to another molecule. For instance, various commercially available polysorbates have been produced through PEG-ylation of sorbitan, which is followed by acylation with fatty acids (Section 5.11).37,390,479 Owing to these characteristics, PEGs have myriad applications ranging from industrial to medical uses (Fig. 37).478 It is noteworthy that PEG is the most used polymer in the field of polymer-based drug delivery. There are many PEG-stabilized drug delivery systems in the market, which have gained regulatory approval from the US and/or the EU. The overwhelming utilization of PEG in biomedical applications is highlighted and discussed delicately in the review article of Knop et al.480
Approximately 60% of the global production of EO is converted to EG (Fig. 4c) via the hydration of ethylene oxidation technology. Currently, this is the major industrial process for the production of EG.481,482 The process simply involves non-catalytic liquid-phase hydration of EO. However, co-products such as diethylene glycol (DEG) and triethylene glycol (TEG) form together with EG. In order to ensure higher EG selectivities (roughly 90%), a large excess of water (20–25 mol water per mol EO) is generally used. This necessary operation, on the other hand, increases capital investment for unit operations.158,482 For the purpose of further increasing the EG selectivity as well as decreasing the production costs, Shell Global Solutions has implemented OMEGA, “Only MEG Advantage” technology. In the OMEGA process, EO is reacted with CO2 to produce ethylene carbonate, which is then hydrolysed to yield EG and CO2. This robust operation gives over 99% EG yield without having a considerable by-product and it also uses nearly 30% less wastewater and 20% less steam.483,484 Although these are different industrial approaches for the manufacture of EG, maximization of the EG yield and selectivity is a standard practice because EG is the most important one amongst the above-mentioned by-products. In 2010, its worldwide consumption capacity was pointed out to be about 20 million metric tons. Such a giant consumption capacity simply stems from its large applicability in diverse industrial areas such as energy, plastics, automobiles, and chemicals.158,482 Approximately 50% of the EG is utilized as an antifreeze. It is also consumed as a solvent, a coolant, a heat transfer fluid, a hydrate inhibitor and a fuel cell component. Besides its direct applications, nearly 40% of EG is directed into the polymer industry (Fig. 37), particularly into the manufacture of polyethylene terephthalate (PET) polyesters for fiber and bottling applications.481,482 Another commercially available EG based polyester, poly(ethylene succinate), was described in Section 5.2. Apart from the EG based polyesters, it is also feasible to convert EG into other types of polymers, like polyurethanes (PU). As a recent example, Paşahan et al. prepared PU films by reacting EG with glucose and diphenylmethane diisocyanate. The PU films were then used to design a novel polymer electrode for the detection of epinephrine. The PU film containing 5% glucose by weight can be used as a membrane for EP detection in the presence of a high concentration of ascorbic acid.485
EG is also a fine chemical intermediate. It can be completely reduced to generate H2-rich fuel gases or selectively oxidized to glycolic acid (GA) or glyoxal (Fig. 4c).156,157,482 Partial oxidation of EG to GA can be performed on gold-based catalysts in aqueous solutions.482 However, gold-based catalysts are too expensive, and it is difficult to control the oxidation process so that further oxidation results in the formation of oxalic acid. Since the selective oxidation is perfectly done in microbial species, the bioconversion route could be a better option. As such, Wei et al. introduced an integrated bioprocess for the GA production from EG by employing G. oxydans DSM 2003. In the integrated process, simultaneous separation of GA was achieved using anion ion-exchange resins to eliminate the effect of end product inhibition.486 EG-derived GA can be polycondensed to form its homopolymer, poly(glycolic acid) (PGA) (Fig. 37). This is the simplest process to synthesize PGA; however; it gives a low Mwt product. Nonetheless, high Mwt PGA is obtained by ROP of a cyclic dimer of GA, glycolide.487 PGA is a low-cost tough fibre forming polymer but it is also biodegradable mainly by simple hydrolysis, which initially limited its use. Later on, this disadvantage of PGA turned out to be an advantage. Davis & Geck used PGA to develop the first synthetic absorbable structure, which was marketed under the trade name of Dexon. Nowadays, PGAs are routinely used for suture manufacture. PGA based commercial sutures are produced by either copolymerization of PGA with other monomers, such as lactic acid (Vicryl®, Polysorb®), ε-caprolactone (Monocryl®), and trimethylcarbonate (Maxon®), or a mixture of monomers, like ε-caprolactone and trimethylcarbonate (Monosyn®) or p-dioxanone, and trimethylcarbonate (Bisyn®). Major applications of PGAs are not limited to sutures but also cover drug delivery devices, scaffolds for use in cell culture, transplantation, organ regeneration, and food packaging applications. For instance, Kureha Corporation commercialized PGA as a film having good gas barrier properties for food packaging.37,270,301 The other selective oxidation product of EG, glyoxal, is used as a cross-linking agent in the polymer industry. It has been employed to crosslink functionalized polymers such as cellulose, polyacrylamides, poly(vinylalcohol), keratin, and other polycondensates.488
Chlorination of ethylene to 1,2-dichloroethane, or ethylene dichloride (EDC), has also been industrially practiced. EDC currently belongs to those chemicals with the highest production rates since it is the starting material for the production of vinyl chloride (VC) and subsequently of poly(vinyl chloride) (PVC) (Fig. 4c and 37). The conversion of EDC to VC process is carried out through either direct or oxychlorination methods, in which chlorine or hydrogen chloride is used, respectively. Industrially, both the methods are practiced together and in parallel because most EDC plants are connected to vinyl chloride (VC) production units. The hydrogen chloride generated from VC production is re-used in the oxychlorination of ethylene.151,153,489 In 2012, approximately 37.4 million tons of PVC was produced from VC. With such a huge production capacity, PVC is the third most widely produced plastic after polyethylene and polypropylene. The top manufacturers are Shin-Etsu Chemical, Formosa Plastics, Solvay, LG Chem, ChemChina, Ineos and OxyVinyls.490 Among these companies, Solvay is the leader in the production of bio-PVC, in which the company employs bio-ethylene as the starting material.325,431 Depending on the intended end use, each PVC company produces a range of PVC polymers varying in molecular mass and morphology. Moreover, commercial PVC is never used alone. Heat stabilizers, lubricants, plasticizers, fillers, and other additives are always mixed with PVC in order to make its processing possible. These additives consequently influence physical and mechanical properties of PVC. For instance, unplasticized (rigid) and plasticized (flexible) PVC can have very distinct applications.491 The most important application areas of PVC were reported to be pipes and fittings (42%), profiles and hose/tubing (18%), rigid film and sheet (17%) as well as cable (8%).490 Although there are myriad applications of PVC in these areas, its usage in daily life is still debated due to the carcinogenic nature of VC monomer. Strict limits have been set for the quantity of residual VC in PVC polymers, which are produced for food contact applications.491 Hence, replacement of PVC with its alternative and more reliable polymers is more desirable.
Another commercial derivative of EDC is ethylenediamine (EDA). This diamine is mainly produced by reacting EDC with aqueous or liquid NH3 at about 100 °C (Fig. 4c).154,492 Major application sectors of EDA include detergents, resins, crop protection agents, paper chemicals, lubricants, and pharmaceuticals. Having diamine functionality makes EDA a useful monomer in different polycondensation reactions (Fig. 37). For example, EDA-polyester condensates, which are hydroxymethylated with formaldehyde, can be used as plasticizers. As nitrogen containing polyol components, condensates of EDA, epoxides, and urea were proposed for the production of polyurethane foams. Also, the incorporation of EDA into diisocyanate–polyester prepolymers forms useful polymers for the manufacture of elastic polyurethane fibers.492 In the recent literature, EDA cored poly(amidoamine) (PAMAM) has emerged as an alternative monomer in the synthesis of polyamide thin film composite (TFC) membranes owing to its dendritic and hydrophilic nature.493 Furthermore, research studies were also reported on plasma polymerized EDA (PPEDA) films for various applications.494
The oligomerization of ethylene is a common method for the synthesis of alpha-olefins, such as 1-butene, 1-hexene, and other higher alpha-olefins (Fig. 4c). A wide variety of catalysts based on nickel, chromium, iron, cobalt, and aluminum can be employed in that respect. As previously mentioned in this section, α-olefins are predominantly copolymerized with ethylene for the commercial production of LLDPE (Fig. 37).472 Vinyl acetate, methyl methacrylate (MMA), and styrene are the other ethylene derivable giant monomers. Amongst these monomers, vinyl acetate is obtained via the reaction of ethylene with acetic acid, and it is mainly consumed in the production of PVAc and PVA (Fig. 36). MMA is currently commercially produced by condensation of acetone and hydrogen cyanide (ACH process). However, hydrogen cyanide is highly toxic and the ACH process results in the formation of large quantities of ammonium bisulphate as a corrosive byproduct. Thus, many alternative routes have been put into practice for replacing the problematic ACH process. As a promising method, BASF employs ethylene, carbon monoxide and formaldehyde as raw materials (Fig. 37).495 In this respect, a sustainable and renewable route for the synthesis MMA could be realized once petroleum based ethylene is replaced with bio-ethanol derived ethylene. As the other giant monomer, styrene is produced on the industrial scale via the dehydrogenation of ethyl benzene, which in turn was prepared by alkylation of benzene with ethylene. Hence, the combination of lignin derived benzene (Section 5.19) with bio-ethanol derived ethylene could potentially provide a sustainable pathway for mass production of styrene.472 As shown in Fig. 37, the development of bio-based routes for the production of MMA and styrene is quite important because these monomers are particularly converted into their homopolymers, which have myriad applications.270
After ethylene, propylene is the second most demanded product in the petrochemical industry.261 Mathers pointed out that the high demand for propylene production would be alleviated from its petroleum-based industrial route to biomass resources.472 In that respect, bio-based alcohols have a flaming potential as key intermediates in the synthesis of bio-propylene. For instance, Braskem has been planning to produce propylene from bio-ethanol. The process involves bio-ethanol conversion to ethylene, and then to propylene through dehydration, dimerization, isomerization, and metathesis processes (Fig. 37). Other strategies might include fermentation of biomass to yield 2-propanol, butanol and methanol, and then conversion of these alcohols to bio-propylene (Fig. 4c). Furthermore, fermentation and direct cracking of cellulose, lignin, and sugar for propylene production have been actively pursued in industry.146,261,472 About two thirds of the globally produced propylene is consumed in the manufacture of polypropylene (PP) (Fig. 37).472 According to the new market study of Ceresana, the worldwide PP production capacity is about 62 million tonnes and this is expected to increase more than 23.5 million tonnes by 2019. Commercial PP is primarily consumed in flexible and rigid packaging, which accounts for more than 50% of the demand. This is followed by fiber and consumer goods (12% each), electrics and electronics as well as the transportation and construction (6% each) sectors.496 Several kinds of PPs, such as atactic PP, elastomeric PP, isotactic PP and syndiotactic PP, are available in the market under different trade names and each of them has their unique applications.270
5.15. Xylose/furfural/arabinitol platform based polymers
Xylose and arabinose, being C5 sugars of lignocellulosic biomass, led to the production of many value added chemicals as shown in Fig. 4d. Ring-opened derivatives of these sugars, such as xylitol, arabinitol, and xylaric acid,170,172,173 are useful intermediates for the synthesis of various kinds of linear polycondensates (Section 5.1). For instance, Zamora et al. synthesized and characterized new polyesters based on L-arabinitol and xylitol as PET and poly(butylene terephthalate) (PBT) analogs.497 In another study, García-Martín et al. reported a new series of AABB-type polyamides derived from L-arabinitol and xylitol.498 Very recently, Pohjanlehto et al. employed xylaric acid in the preparation of a series of lignin-xylaric acid polyurethanes. The polyurethanes were prepared through the network-forming reaction between lignin and esterified xylaric acid. In the reaction, MDI, PEG, and di-n-butyltin dilaurate were used as a coupling agent, a compatibilizer and a catalyst, respectively. The content of bio-based starting materials in the polyurethane products was reported to be as high as 35%.499Industrially, most of the xylose is consumed in the production of furfural through its dehydration using mineral acids as homogeneous catalysts. Besides, many studies have been reported to search new sustainable ways for furfural production by employing heterogeneous acid catalysts and different extracting techniques as well as by tuning the temperature, pressure and solvent.171,177 This conversion holds important industrial opportunities because xylose derived furfural can be further transformed to furan and many other furan derivatives such as furfuryl alcohol, hydroxy furans, furoic acid, 2(5H)-furanone, furfuryl amine, difurfuryl diamines, furanacrylic acid, furylidene ketones, methyl furan, 2-hydroxymethyl-5-vinyl furan and 2,5-bis(hydroxymethyl) furan (Fig. 4d).174,175,177,179–181,183–186 Moreover, the corresponding reductions of the parent furan ring of these products result in the formation of THF and THF derivatives including 2-methyltetrahydrofuran, tetrahydrofurfuryl alcohol and 2,5-bis(hydroxymethyl) tetrahydrofuran (Fig. 4d).176,178,179,187 Common polymers of these furfural derived compounds were discussed in Section 5.3.
In addition to furans and THFs, maleic anhydride and maleic acid are the other two furfural derivable industrially important compounds (Fig. 4d).182,188 MA is mostly converted to fumaric acid, which is often preferred instead of MA to produce polyesters and copolymers. Nonetheless, small amounts of MA are utilized for maleinate resins and for copolymers.500 Unlike MA, MAnh is a major monomer of the current polymer industry. Particularly, it is employed in the manufacture of glass-reinforced or unreinforced unsaturated polyester resins (UPRs). Moreover, MAnh is used in myriad applications through its copolymerization with other molecules having vinyl functionality. Typical copolymers of MAnh and their end uses are summarized in Table 8.500–502
| MAnh-based polymers | Major applications |
|---|---|
| UPRs and alkyd resins | Myriad applications in the marine, construction, corrosion, transportation and electrical industries |
| MAnh–styrene copolymers | Engineering plastics, paper treatment, floor polishes, emulsifiers, protective colloids, antisoil agents, dispersants, stabilizing agent, adhesives, detergents, cosmetics, polymer–protein conjugates |
| MAnh – acrylic acid copolymers | Detergent industry |
| MAnh – diisobutylene copolymers | Dispersing agent |
| MAnh – butadiene copolymers | Sizing agent |
| MAnh – C18 α-olefin copolymers | Emulsification agent and paper coating |
| Polyaspartic acid | Section 5.5 |
5.16. Polyhydroxyalkanoates
Polyhydroxyalkanoates (PHAs) are a family of naturally-occurring diverse biopolyesters produced by various microorganisms.431,503,504 As shown in Fig. 38, each monomer unit of PHAs harbors a side chain R group which is generally a saturated alkyl group. Although it is less common, the R group can also take the form of unsaturated alkyl groups, branched alkyl groups, and substituted alkyl groups. PHAs be classified as either short- (3–5 carbon atoms), medium- (6–14 carbon atoms), or long-chain length depending on the total number of carbon atoms within a PHA monomer.504,505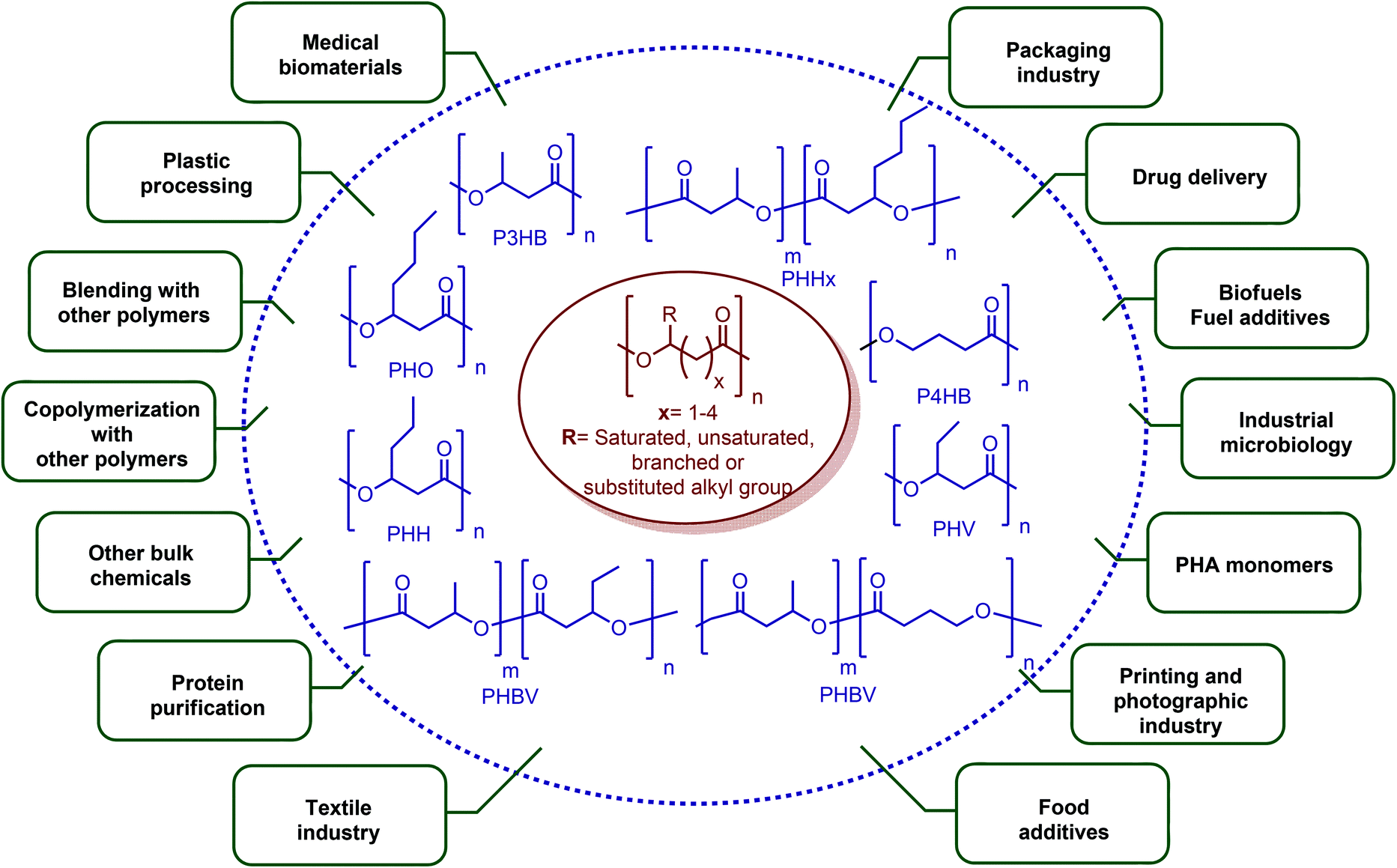 | ||
| Fig. 38 General structure, specific examples and common applications of PHAs. | ||
PHAs are produced by large scale microbial production. Chen and Patel pointed out that over 30% of soil inhabiting bacteria can synthesize PHA. Moreover, many bacteria species having habitats in activated sludge, in high sea, and in extreme environments are also capable of producing PHA in their cell environment.506 In a nutshell, the microbial production process mainly involves strain development, shake flask optimization, lab and pilot fermentor studies, and finally industrial scale-up. Various factors, such as the growth rate of the bacteria, time duration for reaching high final cell density, the final cell density, PHA percentage in cell dry weight, substrate to product transformation efficiency, price of substrates, and robust extraction and purification of PHA, are critical for the efficient microbial production of PHAs.503 Depending on the specific PHA required, the PHA feedstocks can include cellulosics, vegetable oils, organic waste, municipal solid waste, and fatty acids.431 Amongst these feedstocks, the main contribution of lignocellulosic biomass would be to supply glucose as a carbon source for PHA fermentation. The fermentation of glucose has been well studied and thus it is commonly employed both in the laboratory and industry for PHA production.507 In addition to glucose, other lignocellulosic monomer sugars, such as xylose, arabinose, mannose, galactose and rhamnose, and some lignocellulose derivable compounds, such as glycerol, lactic acid and levulinic acid, can be employed as PHA feedstocks.507–509 Furthermore, it is possible to produce PHAs directly from cellulose.510 Detailed scrutiny regarding recent trends and future perspectives of microbial PHA production from different feedstocks, including lignocellulosic biomass, are provided in the comprehensive books and articles.503–505,507–509,511,512
PHAs are completely biodegradable, biocompatible and thermoplastic polymers. They are enantiomerically pure, non-toxic, water-insoluble, inert and stable in air. Moreover, they exhibit piezoelectricity, and they have good processability as well as high structural diversity. Their thermal and mechanical properties depend on their composition so that the Tg of the polymers varies from −40 °C to 5 °C, and the melting temperatures range from 50 °C to 180 °C.431,505 They can be blended with synthetic or natural polymers for tuning their physical properties and biodegradability. PHAs having longer side chains are elastomeric; on the other hand, short side chain PHAs show similar characteristics of polypropylene.285 Owing to these characteristics, many companies (Table 9) have engaged in commercial manufacture of PHA polymers431,503 with a specific motivation towards producing low cost and 100% bio-polymers which can replace conventional polymers like polyethylene and polypropylene in certain applications.
| Company | Brand name | Country | PHA type | Applications |
|---|---|---|---|---|
| Metabolix | Mirel | USA | Several PHA | Packaging |
| TEPHA | TephaFLEX, TephaELAST | USA | Several PHA | Medical bio-implants |
| PHB Industrial S/A | Biocycle | Brazil | PHB | Raw materials |
| Mitsubishi Gas Chemicals | Biogreen | Japan | PHB | Packaging |
| Tianjin Green Bio-Sciences | GreenBio | China | P3HB4HB | Raw materials and packaging |
| Tianan Biological Materials | Enmat | China | PHBV, PHB | Thermoplastics, fiber, nonwovens |
| Bio-On | Minerv | Italy | PHA (unclear) | Raw materials |
| P&G | Nodax | USA | PHBH | Packaging |
| ICI (1980s to 1990s) | Biopol | UK | PHBV | Packaging |
| Biomer | Biomer | Germany | PHB | Packaging and drug delivery |
| BASF | Germany | PHB, PHBV | Blending with Ecoflex | |
| ADM | USA | Several PHA | Raw materials | |
| Meridian | USA | Several PHA | Raw materials | |
| Kaneka (with P&G) | Aonilex | Japan | Several PHA | Packaging |
| Biomatera | Biomatera | Canada | Several PHA | Packaging and biomedical products |
| PolyFerm | VersaMer | Canada | mcl PHAs | Bioplastic materials |
| Zhejiang Tian An | China | PHBV | Raw materials | |
| Jiangmen Biotech Ctr | China | PHBHHx | Raw materials |
Depending on the bacterial species and growth conditions, it is possible to produce homopolymers, random copolymers, and block copolymers of PHA.506 Polyhydroxybutyrate (PHB), poly(3-hydroxybutyrate-co-3-hydroxyvalerate (PHBV), poly(3-hydroxybutyrate-co-4-hydroxybutyrate) (P3HB4HB), poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHBHHx) and medium-chain length (mcl) PHAs are successfully commercialized so far.503 Some specific examples of PHB include poly-3-hydroxybutyrate (P3HB), poly-4-hydroxybutyrate (P4HB), polyhydroxyvalerate (PHV), polyhydroxyhexanoate (PHH) and polyhydroxyoctanoate (PHO) (Fig. 38, Table 9).
PHAs and their related technologies have industrial importance ranging from fermentation, materials, energy to medical fields. As shown in Table 9, recent commercial applications of PHAs mainly include packaging, fiber and nonwoven production, blending with other conventional polymers, biomedical products and drug delivery systems. In the packaging industry, PHA based materials are suitable for short term usage due to their biodegradability. P&G, Biomers and Metabolix have developed PHA based packaging films generally for shopping bags, containers and paper coatings, disposable items, cosmetic containers and cups, and compostable bags and lids.503 PHA can also be processed into fibers like conventional PAs.503 Thermoplastic processing of PHB was already optimized for the production of fibers which have physical properties suitable for the production of scaffolds or for other medical applications.513 It is convenient to blend PHA with other polymers. As such, BASF has been blending PHAs with its commercially available product Ecoflex503 to further improve the use of the product. In the literature, blending different kinds of PHAs among themselves or blending PHAs with other low cost materials, such as cellulose and starch, has also been reported to tune their physical properties and biodegradability.503,514,515 PHAs and their copolymers are widely preferred as biomedical materials. Biomedical applications of PHAs include but are not limited to sutures, suture fasteners, meniscus repair devices, rivets, bone plates, surgical mesh, repair patches, cardiovascular patches, tissue repair patches, and stem cell growth.431 Tepha Inc. specializes in manufacturing PHA based biomedical materials. It markets P4HB for medical application under the name of PHA4400 (Section 5.2).503 Furthermore, PHAs have become candidates for use as drug carriers owing to their biocompatibility and controlled degradability.431 So far, only PHB and PHBV have been studied for controlled drug release. Hence, this application area still remains to be exploited.503
Apart from the above-mentioned commercial uses, applications of PHAs have been rapidly expanding (Fig. 38). Advancements in PHA research have greatly contributed to the progress of industrial microbiology. In this context, the performances of industrial microbial strains can be improved by the PHA synthesis operon which is utilized as a metabolic regulator or a resistance enhancer.503,516,517 As another important point, PHAs are degraded into its constituent monomers. These monomers can be produced via various routes including chemical synthesis, acidic hydrolysis of PHA, and in vitro and in vivo enzymatic depolymerization of PHA.506 So far, more than 150 PHA monomers have been identified.431 PHA derived monomers can be utilized as chiral building blocks for the synthesis of novel polymers, particularly for chiral polyesters.506 In addition, chemical modifications, which cannot be easily achieved by bioconversion processes, can add valuable attributes to PHAs.518 For example, through transformation of PHAs into PHA diols, block copolymerization of PHAs with other polymers becomes feasible.503 PHAs are also suitable candidates for the production of other bulk chemicals, such as heat sensitive adhesives, latex, smart gels,503 biofuels or fuel additives,503,519 and healthy food additives.503 Moreover, PHA granule binding proteins are used to purify recombinant proteins and stained PHAs can find applications in the printing and photographic industries.503,520,521
5.17. Rubber polymers
The huge market volume of rubber has been substantially expanding particularly due to the growth of the automotive industry and the growing demand for tyres.522 Hence, the market has been experiencing a tightening supply of both natural and synthetic rubber. In this context, the production of isoprene, isobutene, and butadiene, the three most important monomers for the manufacture of renewable rubber, would create a very positive impact in the field.261 In fact, the three largest tyre manufacturers: Goodyear, Michelin, and Bridgestone, have already been engaged in the production of renewable rubber from biomass resources. Recently, the collaboration of Genencor and Goodyear has resulted in the development of a microbial process for bio-based isoprene production. By adopting genes from Enterococcus faecalis, Methanosarcina mazei, S. cerevisiae, and Populus alba, the companies synthetically constructed a pathway towards isoprene production in E. coli. The successful metabolic engineering of the isoprene pathway has resulted in the production of 60 g of isoprene per liter of sugar.261,307 Moreover, Michelin teamed up with Amyris, and Bridgestone announced that they are working with Ajinomoto for developing isoprene-based tyres.523Lignocellulosic biomass can also play a very active role in the development of the bio-based rubber industry (Fig. 39). Recently, Zhang et al. patented methods for the production of bio-isoprene from lignocellulosic biomass by using a genetically engineered strain of saprophytic bacterium.200 Another interesting possibility concerns the conversion of lignocellulosic bio-ethanol (Section 5.14) into butadiene.148,325 In this consideration, two possible routes have been reported. The first route is to transform ethanol to butadiene at 400–450 °C in the presence of metal oxide catalysts. Alternatively, ethanol and acetaldehyde can be converted at 325–350 °C. These processes are even used on the industrial scale in Eastern Europe, China, and India.325 Furthermore, cellulose derived sugars can be fermented to give isobutanol and then the dehydration of isobutanol over alumina catalysts at 250–400 °C results in the formation of isobutene.524,525 Gevo has been producing bio-based isobutanol from sugars by fermentation through its Integrated Fermentation Technology (GIFT), which combines genetically engineered yeast with a continuous separation process.326 As an alternative method, lignocellulosic biomass derived bio-butanol can be selectively dehydrated into isobutene in one step over zeolite catalysts.473
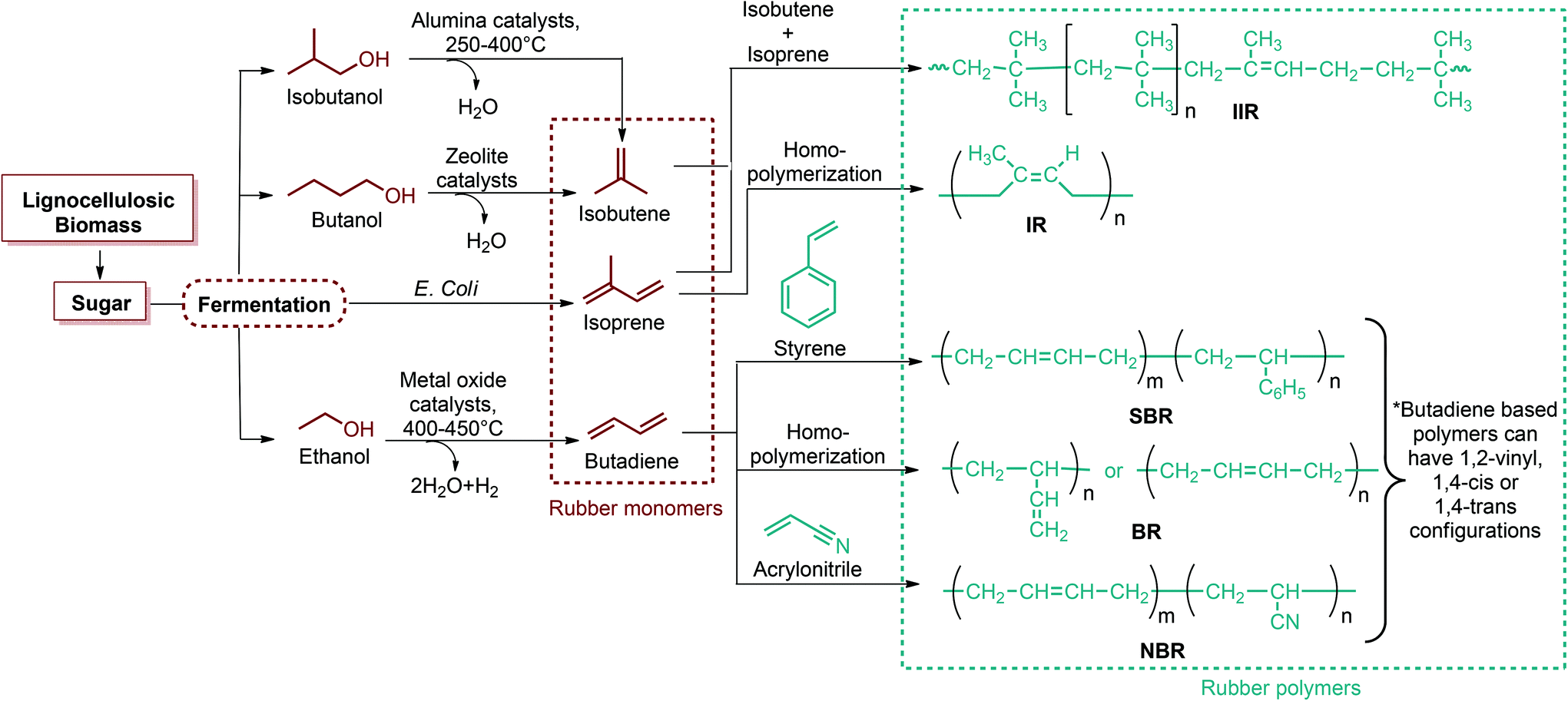 | ||
| Fig. 39 Lignocellulosic biomass derivable rubber monomers and polymers. | ||
About two thirds of the total rubber consumed worldwide is synthetic and one third is natural. Primarily owing to its heat and mineral oil resistance, synthetic rubber (SR) is superior to natural rubber (NR).526 Hence, many different synthetic rubbers having various monomer compositions have been developed (Fig. 39). In 2002, the worldwide SR capacity was 11.2 × 106 t. 43% of this production was belonging to styrene-butadiene rubber (SBR). Other important butadiene based rubber polymers such as polybutadiene, or butadiene rubber (BR), and acrylonitrile-butadiene rubber (NBR) occupied 26% and 4% of the worldwide production capacity, respectively. There are different types of polybutadiene, which include 1,2-polybutadiene, cis-1,4-polybutadiene, and trans-1,4-polybutadiene. These structurally different polybutadienes are prepared by using different catalysts.270,526 Homopolymer of isoprene is another important rubber polymer. It has mainly been extracted from rubber tree (NR) or it has also been manufactured synthetically (IR).307,527 Higher 1,4-cis configuration of polyisoprene most closely exhibits the properties of natural rubber. Hence, a nearly pure cis-1,4 structure allows the production of synthetic natural rubber (IR) (Fig. 39).528 This is achieved by coordination, anionic, free-radical, or cationic polymerization of isoprene through the use of various catalysts.270 Another widely used rubber polymer is butyl rubber (IIR), a copolymer of isobutylene and a small amount of isoprene.529 Halley and Dorgan stated that butyl rubber will soon be available from renewable resources.524 The usefulness of butyl rubber is greatly extended by halogenation, which provides higher vulcanization rates and improves the compatibility with highly unsaturated elastomers. In this consideration, butyl rubber can be either chlorinated or brominated (Fig. 39).529
5.18. Other lignocellulosic biomass derivable polymers
More classes of final chemicals can be produced from lignocellulosic feedstocks than petroleum resources owing to their compositional variety.25 Although industrially important chemicals are presented under different platforms (Fig. 2–5), it is beyond the scope of this review article to cover all of the lignocellulosic biomass derivable compounds. Nonetheless, some of the important ones are presented in Fig. 4b.Citric acid can be obtained through aerobic fermentation of lignocellulosic glucose with A. niger. Its main applications include food, beverages, pharmaceuticals, detergents, buffering and chelating agents.159 It is used as a multifunctional monomer to react with different aliphatic diols to give biodegradable polyester elastomers. Tran et al. reviewed various other citric acid based elastomers, including crosslinked urethane-doped polyesters (CUPEs), poly(alkylene maleate citrates) (PAMCs), poly (xylitol-cocitrate) (PXC) and poly(poly(ethylene glycol) maleate citrate) (PPEGMC).530
Another interesting monomer is limonene and it can be produced from citrus wastes. Particularly, the wastes from the orange juice industry can be valorized in the production of this terpene derivative.1 For removal of limonene from citrus wastes, Pourbafrani et al. reported a dilute acid hydrolysis process at 150 °C for 6 min. In the process, the hydrolysis, followed by explosive pressure reduction (flashing), resulted in a drastic decrease of limonene in the hydrolysates. 99% of the limonene content can be removed from citrus wastes by this method. The removed limonene can then be recovered through its condensation process.164 Once removed from citrus wastes, limonene can be widely employed in the synthesis of polymers (Fig. 40).261 For instance, Singh and Kamal have reported radical homopolymerization of limonene in the presence of benzoyl peroxide as an initiator at 85 °C in xylene. The resulting polylimonene has a weight-average molecular of 43000 g mol−1 and a Tg of 116 °C.531 Copolymerization of limonene can also be carried out with different monomers including styrene532 and N-phenylmaleimide.533 Furthermore, limonene is oxidized to form mono- as well as di-functional epoxides.54
 | ||
| Fig. 40 Limonene based polymers. | ||
Byrne et al. performed catalytic copolymerization of limonene monoxide with carbon dioxide. The resulting thermoplastic polylimonene carbonates show polystyrene like properties.1,534 Instead of carbon dioxide, limonene monoxide can alternatively be copolymerized with dicarboxylic acid anhydrides, such as succinic anhydride, to give limonene-based polyesters.1,85 Limonene dioxide, the difunctional epoxide derivative of limonene, can be further converted into limonene dicarbonates. This process enables chemical fixation of 34 wt% carbon dioxide. Curing of this novel limonene-derived monomer with polyfunctional amines, such as citric aminoamides, yields a wide variety of crosslinked green polyurethanes without requiring the use of isocyanates (Fig. 40).1,535
Erythritol can be produced by several osmophilic yeasts such as Aureobasidium, Candida, Moniliella, Pichia, Pseudozyma, Trigonopsis, Trichosporon, Tri-chosporonoides, and Yarrowia.169 Having –OH functionality allows erythritol to be used in polycondensation reactions. Barrett et al. produced various soft elastomers based on the polycondensation of erythritol with a dicarboxylic acid, including glutaric, adipic, pimelic, suberic, azelaic, sebacic, dodecanedioic, and tetradecanedioic acids. The elastomers exhibit a wide range of physical and mechanical properties. Young's modulus, ultimate tensile stress, and rupture strain values of the elastomers are in between 0.08–80.37 MPa, 0.14–16.65 MPa, and 22–466%, respectively.536
2,3-Butanediol (2,3-BDO) is produced as a natural fermentation product of many microorganisms, including many species of Klebsiella, Bacillus, and lactic acid bacteria.161,236 LanzaTech has been developing 2,3-BDO as a platform chemical for the production of bio-based butadiene monomer.236 Besides, several studies were reported on the copolymerization of 2,3-BDO with 2,5-FDCA and other FDCA derivatives in the recent literature. The common motivation of these studies is to produce fully bio-based polyesters.258,537,538
Another notable compound is L-lysine, an essential amino acid for humans. In 2005, about 1.5 million tons of L-lysine were produced by employing Gram-positive Corynebacterium glutamicum.167,168 The most well known polymer of L-lysine is its homopolyamide, ε-polylysine (ε-PL). Naturally occurring ε-PL is nontoxic, water soluble, biodegradable and edible. Furthermore, it exhibits excellent antimicrobial activity and heat stability. Owing to these characteristics, ε-PL has been attracting interest especially in food, medicine and electronics industries. Its derivatives also offer a wide range of applications including emulsifying agents, dietary agents, biodegradable fibers, highly water absorbable hydrogels, drug carriers, anticancer agent enhancers, and biochip coatings. The production and medical applications of ε-PL are elegantly reviewed by Shukla et al.539 In addition, the design of novel functional monomers based on amino acids and their corresponding polymerization methods is described in Section 5.5.
Lastly, levoglucosan is another important lignocellulosic biomass derivable compound. It is obtained through fast pyrolysis of lignocellulosic biomass.166 Ring-opening multibranching polymerization of levoglucosan, and other anhydro sugars, results in the formation of hyperbranched carbohydrate polymers. Satoh and Kakuchi have reported levoglucosan incorporating water-soluble hyperbranched carbohydrate polymers with controlled molecular weights and narrow polydispersities. Such polymer architectures are expected to have useful applications in medical and medicinal fields.540
5.19. Lignin derivable polymers
Lignin, which can be found in all vascular plants, is the second most abundant natural polymer coming after cellulose. Even so, it has received little attention regarding its valorization. For instance, approximately 50 million tons of lignin are available worldwide from pulping processes but only about 2% of this lignin is used commericially. The remaining 98% is not isolated but burned on site. Borregaard Lignotech, Westvaco and Tembec are currently the major producers of isolated lignin. These companies have been producing around 1.1 million tonnes of isolated lignin per year either through the acid sulphite or the kraft pulping processes.45,506,541Lignin is considered as the major aromatic resource of the bio-based economy. A wide variety of bulk and fine chemicals, particularly aromatic compounds, as well as fuels can be obtained from lignin. Lignin derivable compounds are reviewed in detail by Zakzeski et al.,45 Huber et al.,9,542 Bozell et al.543 and Stöcker.24 Representatives of lignin derivable aromatic compounds are also provided in Fig. 5. In terms of the polymer industry, the enormous quantity of lignin renders excellent opportunities for the production of aromatic monomers and polymers. However, the full potential of lignin for commodity polymers has recently been underutilized mainly due to the difficulty in obtaining aromatic chemicals from lignin.472 Hence, new technological developments are required to break down the chemical structure of lignin into commodity chemicals such as benzene, toluene, xylene, phenols, hydroxybenzoic acids as well as coniferyl, sinapyl, and p-coumaryl compounds (Fig. 41). According to Mathers, this difficulty could be overcome by employing either heterogeneous or homogeneous catalysts, which can break the complicated network of C–O and C–C bonds in lignin without leaving substantial amounts of tar or gasification.472
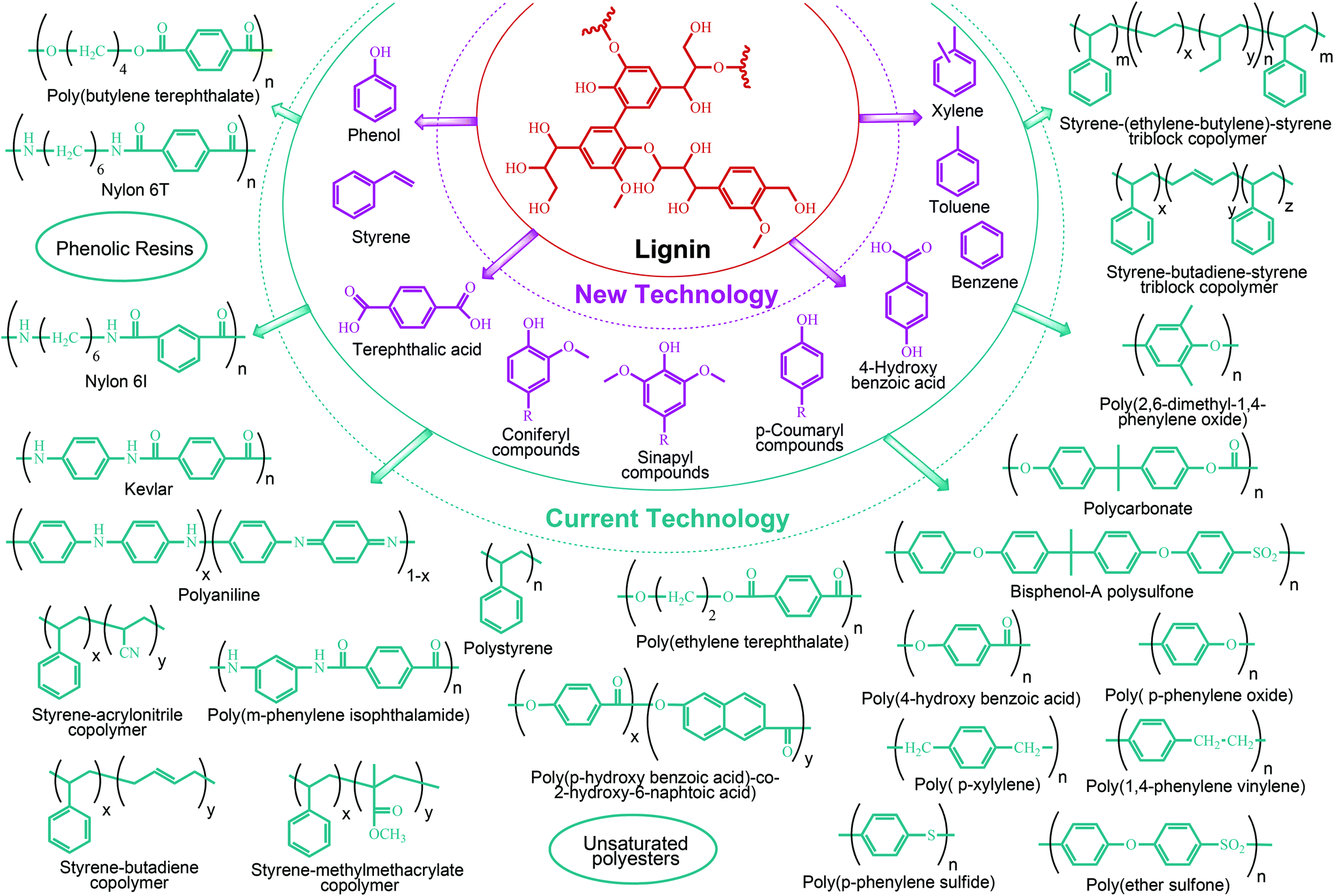 | ||
| Fig. 41 Valuable aromatic monomers and polymers obtainable from lignin via development and integration of new and current technologies. | ||
Once lignin is isolated and degraded into aromatic compounds, the subsequent processes do not require too much improvement. There is already a mature technology for the conversion of these compounds into commodity monomers and polymers. As shown in Fig. 41, the combination of the new and current technologies would provide novel opportunities for the conversion of lignin to commodity polymers like polyethylene terephthallate (PET), polystyrene, Kevlar, unsaturated polyesters, polyaniline and many more aromatic polymers. Since common applications and properties of these polymers are well documented in the literature,270 they are not reviewed here in detail. Nonethless, it should be noted that alternatives to petroleum-based aromatic polymers could be fully realized by valorization of lignin. For instance, two key components of PET: ethylene glycol and p-terephthalic acid, must be produced from renewable biomass for the production of fully bio-based PET. The Coca Cola Company has been using bioethanol-derived bio-ethylene to generate a ca. 30% plant-based PET. It is also necessary to use bio-based p-xylene as the raw material for p-terephthalic acid to produce 100% plant-based PET. Pepsi Cola has been producing 100% plant-based green PET bottles by employing C5 and/or C6 sugars derived bio-terephthallic acid.544 The processes for the production of bio-terephthallic acid from C5/C6 sugars require intermediate steps and compounds. Hence, direct conversion of lignin to terephthalic acid may become more feasible depending on the development of new lignin valorization technologies.
6. Conclusion
Industrial production of a wide range of chemicals and synthetic polymers heavily relies on fossil resources despite their dwindling resources and frightening environmental effects. Hence, alternative sources are sought to produce valuable chemicals and polymers from renewable natural resources for decreasing the current dependence on fossil resources as well as alleviating their corresponding environmental threats. In this context, lignocellulosic biomass, being the most abundant bio-renewable biomass on earth, has critical importance mainly owing to its worldwide avaliability and non-competitiveness with food supplies. Besides, it is significantly cheaper than crude oil and it can be produced quickly and at lower cost than other agriculturally important feedstocks. Thus, lignocellulosic biomass is considered as an ideal renewable feedstock to produce biofuels, commodity chemicals and polymers by exhibiting great economic significance and environmental friendliness. Owing to these characteristics, ongoing research and industrial activities in the field of lignocellulosic biomass for the production of commodity chemicals and polymers were reviewed. Initially, the structure and sources of lignocellulosic biomass were described and, subsequently, different pre- and post-treatment methods for the degradation of lignocellulosic biomass into its components were summarized. More than 200 lignocellulosic biomass derivable value-added compounds were presented with their references. Finally, the scope of the polymers that can be produced mainly in the frame of these compounds was depicted to reveal the potential of lignocellulosic biomass as an alternative platform to petroleum resources. As shown throughout this article, developments in the valorization of lignocellulosic biomass still remains a big challenge together with many opportunities. Thus, extensive research is currently being undertaken all over the world to convert lignocellulosic biomass to value-added chemicals and polymers at high selectivities, and yields at economical costs. One of the most important goals is to fractionate lignocellulose into its three major components; cellulose, hemicelluloses and lignin. Various pretreatment approaches have been developed to increase the accessibility and biodegradability of these components for enzymatic or chemical action. Once these components are isolated, target compounds can be obtained through either chemocatalytic or microbial production processes. That is why future developments in the valorization of lignocellulosic biomass are directly correlated to improvements in the fields of chemical and microbial synthesis. Owing to the recent advancements in these fields, the number and diversity of lignocellulosic biomass based commodity and specialty chemicals have been rapidly increasing. Furthermore, biorefinery and biofuel technologies have been developed to refine lignocellulosic biomass in analogy to petrochemistry for producing green fuels, chemicals and polymers. The number of biorefinery-related pilot and demonstration plants has been increasing. As such, Lignol, Verenium and Mascoma are promising companies which aim to undertake the development of biorefining technologies for the production of advanced biofuels, biochemicals and biomaterials from non-food cellulosic biomass feedstocks. The world's largest chemical firms including DuPont, BASF, SABIC, Dow Chemical, LyondellBasell, and Mitsubishi Chemical have also been actively engaged in valorization of lignocellulosic biomass. Bio-production of various platform chemicals such as ethanol, butanol, lactic acid, levulinic acid, sorbitol, glycerol, 1,3-propanediol, itaconic acid, succinic acid, and 2,5-FDCA has already been achieved. Through research and development, many more lignocellulosic biomass derivable chemicals still await the realization of their commercial production. This, of course, includes a number of monomer building blocks, which are utilized to produce many conventional and novel polymers. So, current commodity polymers are expected to be replaced by their bio-derived counterparts in the near future. For instance, the applications of petroleum derived polyethylene can be easily reproduced by bio-polyethylene. It was shown that bio-polyethylene has exactly the same chemical, physical, and mechanical properties as petrochemical polyethylene. As a different future prospect, existing conventional polymers may also be replaced by their new bio-based alternatives. As such, Avantium's 100% bio-based polyethylene-furanoate (PEF) can potentially replace PET in certain applications. However, the ultimate commercial success of bioplastics will depend on three factors: economics, performance, and environmental factors. The first factor seems to be more significant since bio-polymers have been proven to exhibit similar performance and more environmental friendliness in comparison with their petroleum-based counterparts. Economical considerations can be improved via continued research and development as well as government and private sector investment. Fortunately, the recent trends as mentioned throughout this article suggest that we are on the path of establishing a worldwide bio-based economy, and lignocellulosic biomass may have a great contribution in this context.Notes and references
- R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef.
- A. L. Andrady, Plastics and the Environment, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003 Search PubMed.
- R. Mülhaupt, Angew. Chem., Int. Ed., 2004, 43, 1054–1063 CrossRef PubMed.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- L. A. Lucia, BioResources, 2008, 3, 981–982 Search PubMed.
- K. G. Satyanarayana, G. G. C. Arizaga and F. Wypych, Prog. Polym. Sci., 2009, 34, 982–1021 CrossRef CAS.
- Y. Ahn, S. H. Lee, H. J. Kim, Y.-H. Yang, J. H. Hong, Y.-H. Kim and H. Kim, Carbohydr. Polym., 2012, 88, 395–398 CrossRef CAS.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- C. Somerville, H. Youngs, C. Taylor, S. C. Davis and S. P. Long, Science, 2010, 329, 790–792 CrossRef CAS PubMed.
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Energy Environ. Sci., 2011, 4, 793 CAS.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J. P. Mikkola, Ind. Crops Prod., 2010, 32, 175–201 CrossRef.
- N. Sun, H. Rodriguez, M. Rahman and R. D. Rogers, Chem. Commun., 2011, 47, 1405–1421 RSC.
- A. Barakat, H. de Vries and X. Rouau, Bioresour. Technol., 2013, 134, 362–373 CrossRef CAS PubMed.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- M. J. Taherzadeh and K. Karimi, Int. J. Mol. Sci., 2008, 9, 1621–1651 CrossRef CAS PubMed.
- G. W. Huber, Breaking the Chemical and Engineering Barriers to Lignocellulosic Biofuels: Next Generation Hydrocarbon Biorefineries, National Science Foundation, 2008 Search PubMed.
- B. C. Saha, Enzymes as Biocatalysts for Conversion of Lignocellulosic Biomass to Fermentable Sugars, in Handbook of Industrial Biocatalysis, ed. C. T. Hou, CRC Press, 2005 Search PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC.
- J. Holm and U. Lassi, in Ionic Liquids: Applications and Perspectives, ed. A. Kokorin, InTech, 2011 Search PubMed.
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef PubMed.
- F. Cherubini and A. H. Strømman, Biofuels, Bioprod. Biorefin., 2011, 5, 548–561 CrossRef CAS.
- V. B. Agbor, N. Cicek, R. Sparling, A. Berlin and D. B. Levin, Biotechnol. Adv., 2011, 29, 675–685 CrossRef CAS PubMed.
- S. Morais, E. Morag, Y. Barak, D. Goldman, Y. Hadar, R. Lamed, Y. Shoham, D. B. Wilson and E. A. Bayer, mBio, 2012, 3 Search PubMed.
- H. V. Scheller and P. Ulvskov, Annu. Rev. Plant Biol., 2010, 61, 263–289 CrossRef CAS PubMed.
- E. M. Rubin, Nature, 2008, 454, 841–845 CrossRef CAS PubMed.
- A. M. Abdel-Hamid, J. O. Solbiati and I. K. Cann, Adv. Appl. Microbiol., 2013, 82, 1–28 CAS.
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed.
- S. S. da Silva, A. K. Chandel, S. R. Wickramasinghe and J. M. Dominguez, J. Biomed. Biotechnol., 2012, 2012, 826162 Search PubMed.
- B. E. Dale and S. Kim, Biomass Refining Global Impact – The Biobased Economy of the 21st Century, in Biorefineries – Industrial Processes and Products: Status Quo and Future Directions, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2005 Search PubMed.
- M. Altaf, M. Venkateshwar, M. Srijana and G. Reddy, J. Appl. Microbiol., 2007, 103, 372–380 CrossRef CAS PubMed.
- J.-P. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39–48 CrossRef CAS.
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740 RSC.
- M. Zviely, in Pretreatment Techniques for Biofuels and Biorefineries, ed. Z. Fang, Springer, Berlin, Heidelberg, 2013, ch. 7, pp. 133–150, DOI:10.1007/978-3-642-32735-3_7.
- S. G. Wettstein, D. M. Alonso, E. I. Gürbüz and J. A. Dumesic, Curr. Opin. Chem. Eng., 2012, 1, 218–224 CrossRef CAS.
- T. A. Werpy and G. Petersen, Top Value Added Chemicals From Biomass: I. Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U.S. Department of Energy (DOE), 2004 Search PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- T. Ezeji, N. Qureshi and H. P. Blaschek, Process Biochem., 2007, 42, 34–39 CrossRef CAS.
- J. C. Serrano-Ruiz, R. M. West and J. A. Dumesic, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 79–100 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Wang, A. Thygesen, Y. Liu, Q. Li, M. Yang, D. Dang, Z. Wang, Y. Wan, W. Lin and J. Xing, Biotechnol. Biofuels, 2013, 6, 74 CrossRef CAS PubMed.
- D. P. Minh, M. Besson, C. Pinel, P. Fuertes and C. Petitjean, Top. Catal., 2010, 53, 1270–1273 CrossRef CAS.
- R. Luque, J. H. Clark, K. Yoshida and P. L. Gai, Chem. Commun., 2009, 5305–5307, 10.1039/b911877b.
- U. G. Hong, S. Hwang, J. G. Seo, J. Lee and I. K. Song, J. Ind. Eng. Chem., 2011, 17, 316–320 CrossRef CAS.
- U. G. Hong, H. W. Park, J. Lee, S. Hwang, J. Yi and I. K. Song, Appl. Catal., A, 2012, 415–416, 141–148 CrossRef CAS.
- C. Delhomme, D. Weuster-Botz and F. E. Kühn, Green Chem., 2009, 11, 13 RSC.
- R.-H. Fischer, R. Pinkos, M. Rosch and F. Stein, US Patent20040158078, 2004 Search PubMed.
- E. J. Hollstein and W. A. Butte, US Patent3812148, 1974 Search PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- C. A. Roa Engel, A. J. Straathof, T. W. Zijlmans, W. M. van Gulik and L. A. van der Wielen, Appl. Microbiol. Biotechnol., 2008, 78, 379–389 CrossRef CAS PubMed.
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156–9159 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- B. Liu, Y. Ren and Z. Zhang, Green Chem., 2015, 17, 1610 RSC.
- C. Carlini, P. Patrono, A. M. R. Galletti, G. Sbrana and V. Zima, Appl. Catal., A, 2005, 289, 197–204 CrossRef CAS.
- T. Thananatthanachon and T. B. Rauchfuss, ChemSusChem, 2010, 3, 1139–1141 CrossRef CAS PubMed.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2010, 49, 6616–6618 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melian-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- T. Schaub, B. Buschhaus, M. K. Brinks, M. Schelwies, R. Paciello, J.-p. Melder and M. Merger, US Patent20120232292, 2012 Search PubMed.
- X. Jiang, X. Meng and M. Xian, Appl. Microbiol. Biotechnol., 2009, 82, 995–1003 CrossRef CAS PubMed.
- S. Crabtree and R. Henderson, US Patent20030153795, 2003 Search PubMed.
- K. Schuh, W. Kleist, M. Høj, V. Trouillet, P. Beato, A. D. Jensen, G. R. Patzke and J.-D. Grunwaldt, Appl. Catal., A, 2014, 482, 145–156 CrossRef CAS.
- J. F. Brazdil, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2012 Search PubMed.
- R. Rajagopal, in Sustainable Value Creation in the Fine and Speciality Chemicals Industry, John Wiley & Sons, Ltd, 2014, pp. 153–197 Search PubMed.
- E. Sert and F. S. Atalay, Ind. Eng. Chem. Res., 2012, 51, 6666–6671 CrossRef CAS.
- E. Scott, F. Peter and J. Sanders, Appl. Microbiol. Biotechnol., 2007, 75, 751–762 CrossRef CAS PubMed.
- K. Gu, L. Bi, M. Zhao, C. Wang, C. Dolan, M. C. Kao, J. B. Tok and S. Peng, Bioorg. Med. Chem., 2006, 14, 1339–1347 CrossRef CAS PubMed.
- G. Yan, Y. Wu, W. Lin and X. Zhang, Tetrahedron: Asymmetry, 2007, 18, 2643–2648 CrossRef CAS.
- M. N. Molefe, Syntheses of D,L-1,2,4-butanetriol and [epsilon]-caprolactam from D-glucose-derived Starting Materials, Michigan State University, Department of Chemistry, 2005 Search PubMed.
- R. Kumar, D. Vikramachakravarthi and P. Pal, Chem. Eng. Process., 2014, 81, 59–71 CrossRef CAS.
- J. E. Holladay, T. A. Werpy and D. S. Muzatko, Appl. Biochem. Biotechnol., 2004, 113–116, 857–869 CrossRef CAS PubMed.
- M. A. Brimble, J. H. Parka and C. M. Taylor, Tetrahedron, 2003, 59, 5861–5868 CrossRef CAS.
- H. Luesch, D. Hoffmann, J. M. Hevel, J. E. Becker, T. Golakoti and R. E. Moore, J. Org. Chem., 2003, 68, 83–91 CrossRef CAS PubMed.
- J. Huang, L. H. Mei and J. Xia, Biotechnol. Bioeng., 2007, 96, 924–931 CrossRef CAS PubMed.
- M. Tamura, R. Tamura, Y. Takeda, Y. Nakagawa and K. Tomishige, Chem. Commun., 2014, 50, 6656–6659 RSC.
- E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nat. Chem., 2013, 5, 122–125 CrossRef CAS PubMed.
- S. Ramachandran, P. Fontanille, A. Pandey and C. Larroche, Food Technol. Biotechnol., 2006, 44, 185–195 CAS.
- H. Yuan, H. Liu, Z. Du and Y. Wu, Chem. Eng. J., 2012, 207–208, 72–75 CrossRef CAS.
- M. G. Steiger, M. L. Blumhoff, D. Mattanovich and M. Sauer, Front. Microbiol., 2013, 4, 23 CAS.
- V. Menon and M. Rao, in Microorganisms in Sustainable Agriculture and Biotechnology, ed. T. Satyanarayana, B. N. Johri and A. Prakash, Springer, London, 2012 Search PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2, 586 CrossRef PubMed.
- J. Almena, A. Monsees, R. Kadyrov, T. H. Riermeier, B. Gotov, J. Holz and A. Börnerc, Adv. Synth. Catal., 2004, 346, 1263–1266 CrossRef CAS.
- H.-J. Weyer, R. Fischer, F. Merger, J. Frank, J. Henkelmann, H. Siegel and T. Ruehl, US Patent5536854, 1996 Search PubMed.
- S. Imm, S. Bahn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599–7603 CrossRef CAS PubMed.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed.
- Z. X. Wang, J. Zhuge, H. Fang and B. A. Prior, Biotechnol. Adv., 2001, 19, 201–223 CrossRef CAS PubMed.
- A. Martin, M. P. Checinski and M. Richter, Catal. Commun., 2012, 25, 130–135 CrossRef CAS.
- A. Khan, A. Bhide and R. Gadre, Bioresour. Technol., 2009, 100, 4911–4913 CrossRef CAS PubMed.
- Y. Nakagawa, M. Tamura and K. Tomishige, J. Mater. Chem. A, 2014, 2, 6688–6702 CAS.
- N. D. Kim, J. R. Park, D. S. Park, B. K. Kwak and J. Yi, Green Chem., 2012, 14, 2638 RSC.
- A.-Y. Yin, X.-Y. Guo, W.-L. Dai and K.-N. Fan, Green Chem., 2009, 11, 1514 RSC.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and M. Belsué, Org. Process Res. Dev., 2012, 16, 389–399 CrossRef.
- A. Pienaar, L. J. P. Wilson and M. Stockenhuber, US Patent20130090497, 2013 Search PubMed.
- D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schlaf, Chem. – Eur. J., 2009, 15, 10132–10143 CrossRef CAS PubMed.
- R. Ciriminna and M. Pagliaro, Adv. Synth. Catal., 2003, 345, 383–388 CrossRef CAS.
- M. Watanabe, T. Iida, Y. Aizawa, T. M. Aida and H. Inomata, Bioresour. Technol., 2007, 98, 1285–1290 CrossRef CAS PubMed.
- H. J. Kim, J. Lee, S. K. Green, G. W. Huber and W. B. Kim, ChemSusChem, 2014, 7, 1051–1054 CrossRef CAS PubMed.
- Y. Kayaki, T. Koda and T. Ikariya, J. Org. Chem., 2004, 69, 2595–2597 CrossRef CAS PubMed.
- N. Kondamudi, M. Misra, S. Banerjee, S. Mohapatra and S. Mohapatra, Appl. Catal., B, 2012, 126, 180–185 CrossRef CAS.
- C.-W. Chiu, M. A. Dasari, G. J. Suppes and W. R. Sutterlin, AIChE J., 2006, 52, 3543–3548 CrossRef CAS.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean, 2008, 36, 657–661 CAS.
- A. Negoi, I. T. Trotus, O. M. Steiner, M. Tudorache, V. Kuncser, D. Macovei, V. I. Parvulescu and S. M. Coman, ChemSusChem, 2013, 6, 2090–2094 CrossRef CAS PubMed.
- J. Xia, D. Yu, Y. Hu, B. Zou, P. Sun, H. Li and H. Huang, Catal. Commun., 2011, 12, 544–547 CrossRef CAS.
- A. Corma, S. B. Hamid, S. Iborra and A. Velty, ChemSusChem, 2008, 1, 85–90 CrossRef CAS PubMed.
- C. H. Martin, H. Dhamankar, H. C. Tseng, M. J. Sheppard, C. R. Reisch and K. L. Prather, Nat. Commun., 2013, 4, 1414 CrossRef PubMed.
- T. S. Brima, US Patent4968817, 1990 Search PubMed.
- N. Murata, K. Sakano and T. Ikemoto, US Patent6313321, 2001 Search PubMed.
- N. Ya'aini, N. A. Amin and M. Asmadi, Bioresour. Technol., 2012, 116, 58–65 CrossRef PubMed.
- D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour. Conserv. Recy., 2000, 28, 227–239 CrossRef.
- D. R. Fernandes, A. S. Rocha, E. F. Mai, C. J. A. Mota and V. Teixeira da Silva, Appl. Catal., A, 2012, 425–426, 199–204 CrossRef CAS.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854–1857 CrossRef CAS PubMed.
- L. Deng, Y. Zhao, J. Li, Y. Fu, B. Liao and Q. X. Guo, ChemSusChem, 2010, 3, 1172–1175 CrossRef CAS PubMed.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS.
- L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. T. Horváth, ACS Catal., 2014, 4, 1470–1477 CrossRef CAS.
- J. Q. Bond, D. Martin Alonso, R. M. West and J. A. Dumesic, Langmuir, 2010, 26, 16291–16298 CrossRef CAS PubMed.
- M. Chalid, H. J. Heeres and A. A. Broekhuis, J. Appl. Polym. Sci., 2012, 123, 3556–3564 CrossRef CAS.
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- S. Van de Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465 CAS.
- R. Jacquot and P. Marion, US Patent20110288324, 2011 Search PubMed.
- C. E. Chan-Thaw, M. Marelli, R. Psaro, N. Ravasio and F. Zaccheria, RSC Adv., 2013, 3, 1302 RSC.
- J. P. Lange, J. Z. Vestering and R. J. Haan, Chem. Commun., 2007, 3488–3490, 10.1039/b705782b.
- Y.-J. Zhang, W. Dayoub, G.-R. Chen and M. Lemaire, Tetrahedron, 2012, 68, 7400–7407 CrossRef CAS.
- D. M. Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2010, 12, 992 RSC.
- S. Selifonov, US Patent20080242721, 2008 Search PubMed.
- M. A. Abdel-Rahman, Y. Tashiro and K. Sonomoto, J. Biotechnol., 2011, 156, 286–301 CrossRef CAS PubMed.
- P. Mäki-Arvela, I. L. Simakova, T. Salmi and D. Y. Murzin, Chem. Rev., 2014, 114, 1909–1971 CrossRef PubMed.
- M. Dusselier, P. V. Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS.
- B. Katryniok, S. Paula and F. Dumeignil, Green Chem., 2010, 12, 1910–1913 RSC.
- G. C. Gunter, D. J. Miller and J. E. Jackson, J. Catal., 1994, 148, 252–260 CrossRef CAS.
- T. Yasukawa, W. Ninomiya, K. Ooyachi, N. Aoki and K. Mae, Ind. Eng. Chem. Res., 2011, 50, 3858–3863 CrossRef CAS.
- X. Li, Q. Lin and L. Ma, Ultrason. Sonochem., 2010, 17, 752–755 CrossRef CAS PubMed.
- L. Yu, X.-L. Du, J. Yuan, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, ChemSusChem, 2013, 6, 42–46 CrossRef CAS PubMed.
- Z. Yu, L. Xu, Y. Wei, Y. Wang, Y. He, Q. Xia, X. Zhangad and Z. Liu, Chem. Commun., 2009, 3934–3936 RSC.
- J. Carlos Serrano-Ruiz and J. A. Dumesic, Green Chem., 2009, 11, 1101 RSC.
- M. Ai, A. Motohashi and S. Abe, Appl. Catal., A, 2003, 246, 97–102 CrossRef CAS.
- H. B. Sun, R. Hua and Y. Yin, Molecules, 2006, 11, 263–271 CrossRef CAS PubMed.
- C. Weber, A. Farwick, F. Benisch, D. Brat, H. Dietz, T. Subtil and E. Boles, Appl. Microbiol. Biotechnol., 2010, 87, 1303–1315 CrossRef CAS PubMed.
- Z. Wen, M. Wu, Y. Lin, L. Yang, J. Lin and P. Cen, Microb. Cell Fact., 2014, 13, 92 CrossRef PubMed.
- S. Özkar and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 4800–4808 CrossRef PubMed.
- H. Bai, N. Su, W. Li, X. Zhang, Y. Yan, P. Li, S. Ouyang, J. Yeb and G. Xi, J. Mater. Chem. A, 2013, 1, 6125–6129 CAS.
- T. Wang, H. Shou, Y. Kou and H. Liu, Green Chem., 2009, 11, 562 RSC.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degreve, P. A. Jacobsa and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- J. Ouyang, F. Kong, G. Su, Y. Hu and Q. Song, Catal. Lett., 2009, 132, 64–74 CrossRef CAS.
- M. Ghanta, T. Ruddy, D. Fahey, D. Busch and B. Subramaniam, Ind. Eng. Chem. Res., 2013, 52, 18–29 CAS.
- P. Leduc, F. Vanney and R. Teissier, US Patent20100036180, 2010 Search PubMed.
- Y. Y. Gorbanev, S. Kegnæs, C. W. Hanning, T. W. Hansen and A. Riisager, ACS Catal., 2012, 2, 604–612 CrossRef CAS.
- R. Baran, A. Srebowata, I. I. Kaminska, D. Lomot and S. Dzwigaj, Microporous Mesoporous Mater., 2013, 180, 209–218 CrossRef CAS.
- G. Van Cauwenberge, J.-P. Melder, K. Dahmen, K. Massonne, S. Oehlenschlaeger and K. M. Exner, US Patent7626058, 2009 Search PubMed.
- M. D. Jones, Chem. Cent. J., 2014, 8, 53 CrossRef PubMed.
- O. V. Magaev, A. S. Knyazev, O. V. Vodyankina, N. V. Dorofeeva, A. N. Salanov and A. I. Boronin, Appl. Catal., A, 2008, 344, 142–149 CrossRef CAS.
- X. Gao, Z. Ma, L. Yang and J. Ma, Appl. Biochem. Biotechnol., 2014, 174, 1572–1580 CrossRef CAS PubMed.
- J. W. van Hal, J. S. Ledford and X. Zhang, Catal. Today, 2007, 123, 310–315 CrossRef CAS.
- K. Zhang, B. Zhang and S.-T. Yang, in Bioprocessing Technologies in Biorefinery for Sustainable Production of Fuels, Chemicals, and Polymers, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013, DOI:10.1002/9781118642047.ch20.
- S. Kumar and R. B. Gupta, Ind. Eng. Chem. Res., 2008, 47, 9321–9329 CrossRef CAS.
- X. J. Ji, H. Huang, J. Du, J. G. Zhu, L. J. Ren, S. Li and Z. K. Nie, Bioresour. Technol., 2009, 100, 5214–5218 CrossRef CAS PubMed.
- D. E. Agnew and B. F. Pfleger, Chem. Eng. Sci., 2013, 103, 58–67 CrossRef CAS.
- J. Zandersons, A. Zhurinsh, G. Dobele, V. Jurkjane, J. Rizhikovs, B. Spince and A. Pazhe, J. Anal. Appl. Pyrolysis, 2013, 103, 222–226 CrossRef CAS.
- M. Pourbafrani, G. Forgacs, I. S. Horvath, C. Niklasson and M. J. Taherzadeh, Bioresour. Technol., 2010, 101, 4246–4250 CrossRef CAS PubMed.
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Rena and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC.
- L. Luque, R. Westerhof, G. V. Rossum, S. Oudenhoven, S. Kersten, F. Berruti and L. Rehmann, Bioresour. Technol., 2014, 161, 20–28 CrossRef CAS PubMed.
- J. Schneider, K. Niermann and V. F. Wendisch, J. Biotechnol., 2011, 154, 191–198 CrossRef CAS PubMed.
- N. Adachi, C. Takahashi, N. Ono-Murota, R. Yamaguchi, T. Tanaka and A. Kondo, Appl. Microbiol. Biotechnol., 2013, 97, 7165–7172 CrossRef CAS PubMed.
- G. R. Ghezelbash, I. Nahvi and A. Malekpour, Appl. Biochem. Microbiol., 2014, 50, 292–296 CrossRef CAS.
- M. Kordowska-Wiater, A. Kubik-Komar and Z. Targoński, Cent. Eur. J. Biol., 2013, 8, 835–842 CAS.
- G. Marcotullio and W. De Jong, Green Chem., 2010, 12, 1739 RSC.
- H. Cheng, B. Wang, J. Lv, M. Jiang, S. Lin and Z. Deng, Microb. Cell Fact., 2011, 10, 5 CrossRef CAS PubMed.
- P. Turner, G. Mamo and E. N. Karlsson, Microb. Cell Fact., 2007, 6, 9 CrossRef PubMed.
- M. Kurszewska, E. Skorupowa, J. Madaj and A. Wiśniewski, J. Carbohydr. Chem., 2004, 23, 169–177 CrossRef CAS.
- W. Zhang, Y. Zhu, S. Niu and Y. Li, J. Mol. Catal. A: Chem., 2011, 335, 71–81 CrossRef CAS.
- K. W. Hutchenson and S. K. Sengupta, US Patent20120035378, 2012 Search PubMed.
- R. F. Perez and M. A. Fraga, Green Chem., 2014, 16, 3942–3950 RSC.
- F.-A. Khan, A. Vallat and G. Süss-Fink, Catal. Commun., 2011, 12, 1428–1431 CrossRef CAS.
- C. Moreau, M. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11–30 CrossRef CAS.
- F. W. Lichtenthaler, in Carbohydrates as Organic Raw Materials. Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2010, DOI:10.1002/14356007.n05_n07.
- M. Krystof, M. Pérez-Sánchez and P. Domínguez de María, ChemSusChem, 2013, 6, 826–830 CrossRef CAS PubMed.
- N. Alonso-Fagundez, M. L. Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef CAS PubMed.
- A. Gassama, C. Ernenwein and N. Hoffmann, Green Chem., 2010, 12, 859 RSC.
- M. A. Ayedi, Y. L. Bigot, H. Ammar, S. Abid, R. E. Gharbi and M. Delmas, Synth. Commun., 2013, 43, 2127–2133 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, J. Catal., 2011, 284, 90–101 CrossRef CAS.
- M. S. Holfinger, A. H. Conner, D. R. Holm and C. G. Hill, J. Org. Chem., 1995, 60, 1595–1598 CrossRef CAS.
- P. Biswas, J.-H. Lin, J. Kang and V. V. Guliants, Appl. Catal., A, 2014, 475, 379–385 CrossRef CAS.
- H. Guo and G. Yin, J. Phys. Chem. C, 2011, 115, 17516–17522 CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- R. N. Olcese, J. Francois, M. M. Bettahar, D. Petitjean and A. Dufour, Energy Fuels, 2013, 27, 975–984 CrossRef CAS.
- G. Garrote, J. M. Cruz, A. Moure, H. Domínguez and J. C. Parajó, Trends Food Sci. Technol., 2004, 15, 191–200 CrossRef CAS.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CrossRef CAS PubMed.
- A. K. Chandel, S. S. da Silva and O. V. Singh, in Biofuel Production-Recent Developments and Prospects, ed. M. A. d. S. Bernardes, InTech, Rijeka, Croatia, 2011, pp. 225–247, DOI:10.5772/959.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- G. G. Lavoie, US Patent20050240055, 2005 Search PubMed.
- K. Albizati and C. Tracewell, US Patent20120107886, 2012 Search PubMed.
- T. Yoshikawa, T. Yagi, S. Shinohara, T. Fukunaga, Y. Nakasaka, T. Tago and T. Masuda, Fuel Process. Technol., 2013, 108, 69–75 CrossRef CAS.
- H. G. Kim and Y. Park, Ind. Eng. Chem. Res., 2013, 52, 10059–10062 CrossRef CAS.
- Y. Hadar, in Lignocellulose Conversion: Enzymatic and Microbial Tools for Bioethanol Production, ed. V. Faraco, Springer, London, UK, 2013, pp. 21–39 Search PubMed.
- H. Zhang, M. Suvorov and S. W. Hutcheson, WO Patent2013010057, 2013 Search PubMed.
- E. Ten and W. Vermerris, Polymer, 2013, 5, 600–642 Search PubMed.
- M. M. Ibrahim, F. A. Agblevor and W. K. El-Zawawy, BioResources, 2010, 5, 397–418 CAS.
- L. Brinchi, F. Cotana, E. Fortunati and J. M. Kenny, Carbohydr. Polym., 2013, 94, 154–169 CrossRef CAS PubMed.
- H. Krawczyk, T. Persson, A. Andersson and A. S. Joensson, Food Bioprod. Process., 2008, 86, 31–36 CrossRef CAS.
- F. A. Dottori, R. A. C. Benson and R.-O. Benech, US Patent20100269990, 2010 Search PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 9963–9965 CrossRef CAS PubMed.
- S. M. Dimick, S. C. Powell, S. A. McMahon, D. N. Moothoo, J. H. Naismith and E. J. Toone, J. Am. Chem. Soc., 1999, 121, 10286–10296 CrossRef CAS.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392 RSC.
- C. R. Becer, Macromol. Rapid Commun., 2012, 33, 742–752 CrossRef CAS PubMed.
- Q. Zhang, J. Collins, A. Anastasaki, R. Wallis, D. A. Mitchell, C. R. Becer and D. M. Haddleton, Angew. Chem., Int. Ed., 2013, 52, 4435–4439 CrossRef CAS PubMed.
- G. Yilmaz and C. R. Becer, Eur. Polym. J., 2013, 49, 3046–3051 CrossRef CAS.
- Q. Zhang, L. Su, J. Collins, G. Chen, R. Wallis, D. A. Mitchell, D. M. Haddleton and C. R. Becer, J. Am. Chem. Soc., 2014, 136, 4325–4332 CrossRef CAS PubMed.
- I. Kurtulus, G. Yilmaz, M. Ucuncu, M. Emrullahoglu, C. R. Becer and V. Bulmus, Polym. Chem., 2014, 5, 1593–1604 RSC.
- J. Huang, Q. Zhang, G.-Z. Li, D. M. Haddleton, R. Wallis, D. Mitchell, A. Heise and C. R. Becer, Macromol. Rapid Commun., 2013, 34, 1542–1546 CrossRef CAS PubMed.
- Y. Gou, S. Slavin, J. Geng, L. Voorhaar, D. M. Haddleton and C. R. Becer, ACS Macro Lett., 2012, 1, 180–183 CrossRef CAS.
- K. Babiuch, D. Pretzel, T. Tolstik, A. Vollrath, S. Stanca, F. Foertsch, C. R. Becer, M. Gottschaldt, C. Biskup and U. S. Schubert, Macromol. Biosci., 2012, 12, 1190–1199 CrossRef CAS PubMed.
- N. Vinson, Y. Gou, C. R. Becer, D. M. Haddleton and M. I. Gibson, Polym. Chem., 2011, 2, 107–113 RSC.
- K. Babiuch, R. Wyrwa, K. Wagner, T. Seemann, S. Hoeppener, C. R. Becer, R. Linke, M. Gottschaldt, J. Weisser, M. Schnabelrauch and U. S. Schubert, Biomacromolecules, 2011, 12, 681–691 CrossRef CAS PubMed.
- K. Babiuch, C. R. Becer, M. Gottschaldt, J. T. Delaney, J. Weisser, B. Beer, R. Wyrwa, M. Schnabelrauch and U. S. Schubert, Macromol. Biosci., 2011, 11, 535–548 CrossRef CAS PubMed.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130–15132 CrossRef CAS PubMed.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
- Y. Miura, Polym. J., 2012, 44, 679–689 CrossRef CAS.
- S. Slavin, J. Burns, D. M. Haddleton and C. R. Becer, Eur. Polym. J., 2011, 47, 435–446 CrossRef CAS.
- E. L. Malins, S. Amabilino, G. Yilmaz, F. H. Isikgor, B. M. Gridley and C. R. Becer, Eur. Polym. J., 2015, 62, 347–351 CrossRef CAS.
- Q. Zhang, G.-Z. Li, C. R. Becer and D. M. Haddleton, Chem. Commun., 2012, 48, 8063–8065 RSC.
- C. Weber, T. Neuwirth, K. Kempe, B. Ozkahraman, E. Tamahkar, H. Mert, C. R. Becer and U. S. Schubert, Macromolecules, 2012, 45, 20–27 CrossRef CAS.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–191 RSC.
- U. Mansfeld, C. Pietsch, R. Hoogenboom, C. R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560–1598 RSC.
- C. R. Becer, K. Kokado, C. Weber, A. Can, Y. Chujo and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1278–1286 CrossRef CAS.
- J. A. Galbis and M. G. Garcia-Martin, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Oxford, UK, 2008, pp. 89–114 Search PubMed.
- S. Munoz-Guerra, High Perform. Polym., 2012, 24, 9–23 CrossRef CAS.
- R. Narain, D. Jhurry and G. Wullf, Eur. Polym. J., 2002, 38, 273–280 CrossRef CAS.
- J. Adkins, S. Pugh, R. McKenna and D. R. Nielsen, Front. Microbiol., 2012, 3, 313 Search PubMed.
- I. Bechthold, K. Bretz, S. Kabasci, R. Kopitzky and A. Springer, Chem. Eng. Technol., 2008, 31, 647–654 CrossRef CAS.
- S. Kabasci and I. Bretz, Succinic Acid: Synthesis of Biobased Polymers from Renewable Resources, in Renewable Polymers, ed. V. Mittal, John Wiley & Sons, Inc., 2011, pp. 355–379, DOI:10.1002/9781118217689.ch8.
- J. Xu and B.-H. Guo, in Plastics from Bacteria: Natural Functions and Applications, ed. G.-Q. Chen, Springer, Berlin, 2010, ch. 14, pp. 347–388, DOI:10.1007/978-3-642-03287-5_14.
- X. Xiao, Z. Zeng, W. Xue, Q. Kong and W. Zhu, Polym. Eng. Sci., 2013, 53, 482–490 CAS.
- T. Moore, R. Adhikari and P. Gunatillake, Biomaterials, 2005, 26, 3771–3782 CrossRef CAS PubMed.
- A. Duda, J. Libiszowski, J. Mosnáček and S. Penczek, Macromol. Symp., 2005, 226, 109–120 CrossRef CAS.
- A. Nakayama, N. Kawasaki, S. Aiba, Y. Maeda, I. Arvanitoyannis and N. Yamamoto, Polymer, 1998, 39, 1213–1222 CrossRef CAS.
- D. P. Martin and S. F. Williams, Biochem. Eng. J., 2003, 16, 97–105 CrossRef CAS.
- G. Pruckmayr, P. Dreyfuss and M. P. Dreyfuss, Polyethers, Tetrahydrofuran and Oxetane Polymers, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000, DOI:10.1002/0471238961.2005201816182103.a01.
- N. Stribeck, S. Fakirov, A. A. Apostolov, Z. Denchev and R. Gehrke, Macromol. Chem. Phys., 2003, 204, 1000–1013 CrossRef CAS.
- M. Basko, M. Bednarek, L. Billiet, P. Kubisa, E. Goethals and F. Du Prez, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1597–1604 CrossRef CAS.
- S. D. Seneker and B. D. Lawrey, US Patent5691441, 1997 Search PubMed.
- M. Omer, T. Kamal, H.-H. Cho, D.-K. Kim and S.-Y. Park, Macromol. Res., 2012, 20, 810–815 CrossRef CAS.
- Y. J. Kim, K. E. Yohana, H.-s. Lee and J. Kim, Ind. Eng. Chem. Res., 2012, 51, 15801–15810 CrossRef CAS.
- F. K. Kasper, K. Tanahashi, J. P. Fisher and A. G. Mikos, Nat. Protoc., 2009, 4, 518–525 CrossRef CAS PubMed.
- J. Y. Ljubimova, M. Fujita, A. V. Ljubimov, V. P. Torchilin, K. L. Black and E. Holler, Nanomedicine, 2008, 3, 247–265 CrossRef CAS PubMed.
- Global Bio Succinic Acid Market (Applications and Geography) – Size, Share, Trends, Analysis, Research, Future Demand, Scope and Forecast, 2013–2020, Allied Market Research, 2014.
- Y. Cao, R. Zhang, C. Sun, T. Cheng, Y. Liu and M. Xian, Biomed. Res. Int., 2013, 2013, 12 Search PubMed.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- N. Yoshida, N. Kasuya, N. Haga and K. Fukuda, Polym. J., 2008, 40, 1164–1169 CrossRef CAS.
- A. Gandini, Furan Monomers and their Polymers: Synthesis, Properties and Applications, in Biopolymers – New Materials for Sustainable Films and Coatings, ed. D. Plackett, John Wiley & Sons, Ltd, Chichester, UK, 2011, pp. 179–209, DOI:10.1002/9781119994312.ch9.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Green Chem., 2014, 16, 1957–1966 RSC.
- T. Xiang, X. Liu, P. Yi, M. Guo, Y. Chen, C. Wesdemiotis, J. Xu and Y. Pang, Polym. Int., 2013, 62, 1517–1523 CrossRef CAS.
- A. S. Amarasekara, 5-Hydroxymethylfurfural Based Polymers, in Renewable Polymers: Synthesis, Processing, and Technology, ed. V. Mittal, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 381–428, DOI:10.1002/9781118217689.ch9.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- M. J. González-Tejera, E. S. de la Blanca and I. Carrillo, Synth. Met., 2008, 158, 165–189 CrossRef.
- E. d. Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, in Biobased Monomers, Polymers, and Materials, American Chemical Society, 2012, ch. 1, vol. 1105, pp. 1–13 Search PubMed.
- J. A. Moore and J. E. Kelly, Macromolecules, 1978, 11, 568–573 CrossRef CAS.
- S. Jeol, US Patent20140135449, 2014 Search PubMed.
- M. Bednarek and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6484–6493 CrossRef CAS.
- T. Satoh, M. Tamaki, T. Taguchi, H. Misaka, T. H. Nguyen, R. Sakai and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2353–2365 CrossRef CAS.
- Z. H. Huang, W. F. McDonald, S. C. Wright and A. C. Taylor, US Patent6399714, 2002 Search PubMed.
- H. Ichihashi and T. Kaneko, US Patent20100249291, 2010 Search PubMed.
- J. E. Mark, The Polymer Data Handbook, Oxford University Press, Incorporated, 2009 Search PubMed.
- R. A. Smiley, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a12_629.
- D.-L. Suna, J.-Y. Luoa, R.-Y. Wena, J.-R. Deng and Z.-S. Chao, J. Hazard. Mater., 2014, 266, 167–173 CrossRef PubMed.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- J.-T. Hong, N.-S. Cho, H.-S. Yoon, T.-H. Kim, M.-S. Koh and W.-G. Kim, J. Appl. Polym. Sci., 2006, 102, 737–743 CrossRef CAS.
- J. Ritz, H. Fuchs, H. Kieczka and W. C. Moran, Caprolactam, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011, DOI:10.1002/14356007.a05_031.pub2.
- P. Werle, M. Morawietz, S. Lundmark, K. Sörensen, E. Karvinen and J. Lehtonen, Alcohols, Polyhydric, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI:10.1002/14356007.a01_305.pub2.
- S. K. Bhatia and J. V. Kurian, Biotechnol. Lett., 2008, 30, 619–623 CrossRef CAS PubMed.
- G. Swift, Acrylic (and Methacrylic) Acid Polymers, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2002, DOI:10.1002/0471440264.pst009.
- R. V. Slone, Acrylic Ester Polymers, in Encyclopedia Of Polymer Science and Technology, John Wiley & Sons, Inc., 2010, DOI:10.1002/0471238961.1921182214152201.a01.pub2.
- S.-Y. Huang, D. W. Lipp and R. S. Farinato, Acrylamide Polymers, in Encyclopedia Of Polymer Science and Technology, John Wiley & Sons, Inc., 2001, DOI:10.1002/0471238961.0103182512091616.a01.pub2.
- J. C. Chen and I. R. Harrison, Carbon, 2002, 40, 25–45 CrossRef CAS.
- M. M. Wu, Acrylonitrile and Acrylonitrile Polymers, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2001, DOI:10.1002/0471440264.pst010.
- R. C. Schulz, Acrolein Polymers, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2011, DOI:10.1002/0471440264.pst038.
- A. J. Straathof, Chem. Rev., 2014, 114, 1871–1908 CrossRef CAS PubMed.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- S. Roweton, S. J. Huang and G. Swift, J. Environ. Polym. Degr., 1997, 5, 175–181 CAS.
- Y. Soeda, K. Toshima and S. Matsumura, Biomacromolecules, 2003, 4, 196–203 CrossRef CAS PubMed.
- Y. Ma, X. Jiang and R. Zhuo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3917–3924 CrossRef CAS.
- J. R. Moon, Y. S. Jeon, M. Zrinyi and J.-H. Kim, Polym. Int., 2013, 62, 1218–1224 CAS.
- M. Xu, Y. Zhao and M. Feng, Langmuir, 2012, 28, 11310–11318 CrossRef CAS PubMed.
- R. Liu, X. Chen, Z. Hayouka, S. Chakraborty, S. P. Falk, B. Weisblum, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2013, 135, 5270–5273 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. H. Gellman and K. S. Masters, J. Am. Chem. Soc., 2013, 135, 16296–16299 CrossRef CAS PubMed.
- W. Khan, S. Muthupandian, S. Farah, N. Kumar and A. J. Domb, Macromol. Biosci., 2011, 11, 1625–1636 CrossRef CAS PubMed.
- S. Mallakpour and M. Dinari, J. Macromol. Sci., Pure Appl. Chem., 2011, 48, 644–679 CrossRef CAS.
- H. Sun, F. Meng, A. A. Dias, M. Hendriks, J. Feijen and Z. Zhong, Biomacromolecules, 2011, 12, 1937–1955 CrossRef CAS PubMed.
- F. Sanda and T. Endo, Macromol. Chem. Phys., 1999, 200, 2651–2661 CrossRef CAS.
- F. Zhu, J. Cai, Q. Zheng, X. Zhu, P. Cen and Z. Xu, J. Chem. Technol. Biotechnol., 2014, 89, 616–622 CrossRef CAS.
- I.-L. Shih and Y.-T. Van, Bioresour. Technol., 2001, 79, 207–225 CrossRef CAS PubMed.
- C. Li, Adv. Drug Delivery Rev., 2002, 54, 695–713 CrossRef CAS PubMed.
- T. Hirasawa and H. Shimizu, in Bioprocessing of Renewable Resources to Commodity Bioproducts, John Wiley & Sons, Inc., 2014, pp. 473–495, DOI:10.1002/9781118845394.ch17.
- A. Rodríguez-Galán, M. Vera, K. Jiménez, L. Franco and J. Puiggalí, Macromol. Chem. Phys., 2003, 204, 2078–2089 CrossRef.
- C. Li, S. Y. Tzeng, L. E. Tellier and J. J. Green, ACS Appl. Mater. Interfaces, 2013, 5, 5947–5953 CAS.
- S. Cohen, G. Coue, D. Beno, R. Korenstein and J. F. Engbersen, Biomaterials, 2012, 33, 614–623 CrossRef CAS PubMed.
- S. Y. Tzeng, B. P. Hung, W. L. Grayson and J. J. Green, Biomaterials, 2012, 33, 8142–8151 CrossRef CAS PubMed.
- J. Kim, J. C. Sunshine and J. J. Green, Bioconjugate Chem., 2014, 25, 43–51 CrossRef CAS PubMed.
- J. C. Sunshine, M. I. Akanda, D. Li, K. L. Kozielski and J. J. Green, Biomacromolecules, 2011, 12, 3592–3600 CrossRef CAS PubMed.
- J. W. Lee, H. U. Kim, S. Choi, J. Yi and S. Y. Lee, Curr. Opin. Biotechnol., 2011, 22, 758–767 CrossRef CAS PubMed.
- S. Kind and C. Wittmann, Appl. Microbiol. Biotechnol., 2011, 91, 1287–1296 CrossRef CAS PubMed.
- W. H. Carothers, US Patent2130523, 1938 Search PubMed.
- O. Revelles, R.-M. Wittich and J. L. Ramos, J. Bacteriol., 2007, 189, 2787–2792 CrossRef CAS PubMed.
- A. Filbry, R. Kroepke, S. Heinecke, A. Blohm and J. Nielsen, US Patent20110268674, 2011 Search PubMed.
- G. N. Altounian and D. Grenier, US Patent4716213, 1987 Search PubMed.
- A. Flynn and L. F. Torres, US Patent20090253871, 2009 Search PubMed.
- K. Marcincinova-Benabdillah, M. Boustta, J. Coudane and M. Vert, Biomacromolecules, 2001, 2, 1279–1284 CrossRef CAS PubMed.
- Y. Liu, L. Wenning, M. Lynch and T. M. Reineke, J. Am. Chem. Soc., 2004, 126, 7422–7423 CrossRef CAS PubMed.
- N. P. Ingle, B. Malone and T. M. Reineke, Trends Biotechnol., 2011, 29, 443–453 CrossRef CAS PubMed.
- G. A. R. Nobes, W. J. Orts and G. M. Glenn, Ind. Crops Prod., 2000, 12, 125–135 CrossRef CAS.
- T. C. Gehret, A. S. Frobese, J. S. Zerbe and H. K. Chenault, J. Org. Chem., 2009, 74, 8373–8376 CrossRef CAS PubMed.
- K. Hashimoto, S. Wibullucksanakul, M. Matsuura and M. Okada, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3141–3149 CrossRef CAS.
- K. Hashimoto, S. Wibullcksanakul and M. Okada, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1495–1503 CrossRef CAS.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, Macromolecules, 2014, 47, 498–505 CrossRef CAS.
- A. J. J. E. Eerhart, W. J. J. Huijgen, R. J. H. Grisel, J. C. van der Waal, E. de Jong, A. de Sousa Dias, A. P. C. Faaij and M. K. Patel, RSC Adv., 2014, 4, 3536–3549 RSC.
- D. W. Park, S. Haam, T. G. Lee, H. S. Kim and W. S. Kim, J. Biomed. Mater. Res., Part A, 2004, 71, 497–507 CrossRef PubMed.
- F. Suriano, O. Coulembier and P. Dubois, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3271–3280 CrossRef CAS.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- B. Erickson, J. E. Nelson and P. Winters, Biotechnol. J., 2012, 7, 176–185 CrossRef CAS PubMed.
- USDA, http://www.reeis.usda.gov/web/crisprojectpages/0220209-development-of-integrated-production-of-polyitaconic-acid-from-northeast-hardwood-biomass.html, Accessed April 8, 2015.
- T. Willke and K. D. Vorlop, Appl. Microbiol. Biotechnol., 2001, 56, 289–295 CrossRef CAS PubMed.
- M. Uchimura, R. Ishige, M. Shigeta, Y. Arakawa, Y. Niko, J. Watanabe and G.-I. Konishi, Res. Chem. Intermed., 2013, 39, 403–414 CrossRef CAS.
- S. Shang, S. J. Huang and R. A. Weiss, Polymer, 2011, 52, 2764–2771 CrossRef CAS.
- T. Okuda, K. Ishimoto, H. Ohara and S. Kobayashi, Macromolecules, 2012, 45, 4166–4174 CrossRef CAS.
- L. Garrido, J. Guzman and E. Riande, Macromolecules, 1981, 14, 1132–1133 CrossRef CAS.
- Avantium, http://avantium.com/yxy/products-applications/levulinics.html, Accessed November 11, 2014.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520 RSC.
- A. Mukherjee, M.-J. Dumont and V. Raghavan, Biomass Bioenergy, 2015, 72, 143–183 CrossRef CAS.
- F. D. Klingler and W. Ebertz, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- C. Leibig, B. Mullen, T. Mullen, L. Rieth and V. Badarinarayana, Polym. Prepr., 2010, 51, 762–763 CAS.
- J. Ertl, E. Cerri, M. Rizzuto and D. Caretti, AIP Conf. Proc., 2014, 1599, 326–329 CrossRef CAS.
- The U.S. Food and Drug Administration (FDA), http://www.fda.gov/food/newsevents/constituentupdates/ucm360147.htm, Accessed June 18, 2014.
- Q. Bu, H. Lei, S. Ren, L. Wang, J. Holladay, Q. Zhang, J. Tang and R. Ruan, Bioresour. Technol., 2011, 102, 7004–7007 CrossRef CAS PubMed.
- S. Zhou and D. Kim, Electrochim. Acta, 2012, 63, 238–244 CrossRef CAS.
- D. Foix, X. Ramis, M. Sangermano and A. Serra, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1133–1142 CrossRef CAS.
- N. Fotinos, M. A. Campo, F. Popowycz, R. Gurny and N. Lange, Photochem. Photobiol., 2006, 82, 994–1015 CrossRef CAS PubMed.
- S. H. Battah, C. E. Chee, H. Nakanishi, S. Gerscher, A. J. MacRobert and C. Edwards, Bioconjugate Chem., 2001, 12, 980–988 CrossRef CAS PubMed.
- E. Gillies and J. Frechet, Drug Discovery Today, 2005, 10, 35–43 CrossRef CAS PubMed.
- T. Chen, Z. Qin, Y. Qi, T. Deng, X. Ge, J. Wang and X. Hou, Polym. Chem., 2011, 2, 1190 RSC.
- T. Hirabayashi and K. Yokota, Polym. J., 1982, 14, 789–796 CrossRef CAS.
- W. Wu, G. Qian, X.-G. Zhou and W.-K. Yuan, Chem. Eng. Sci., 2007, 62, 5127–5132 CrossRef CAS.
- D. Głowacz-Czerwonka, J. Appl. Polym. Sci., 2013, 128, 3465–3472 CrossRef.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed.
- Z. Jedliński, P. Kurcok and R. W. Lenz, J. Macromol. Sci., Pure Appl. Chem., 1995, 32, 797–810 CrossRef.
- M. Arasa, R. A. Pethrick, A. Mantecón and A. Serra, Eur. Polym. J., 2010, 46, 5–13 CrossRef CAS.
- M. Arasa, X. Ramis, J. M. Salla, F. Ferrando, A. Serra and A. Mantecón, Eur. Polym. J., 2009, 45, 1282–1292 CrossRef CAS.
- W. J. Lin, J. Biomed. Mater. Res., 1999, 47, 420–423 CrossRef CAS PubMed.
- C. W. Lee, R. Urakawa and Y. Kimura, Eur. Polym. J., 1998, 34, 117–122 CrossRef CAS.
- C. Tsutsumi, T. Hara, N. Fukukawa, K. Oro, K. Hata, Y. Nakayama and T. Shiono, Green Chem., 2012, 14, 1211 RSC.
- Y. Hu, L. O. Gustafson, H. Zhu and E. Y. X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2008–2017 CrossRef CAS.
- M. Chalid, H. J. Heeres and A. A. Broekhuis, Appl. Mech. Mater., 2012, 229–231, 297–302 CrossRef.
- A. Rothen-Weinhold, K. Schwach-Abdellaoui, J. Barr, S. Y. Ng, H. R. Shen, R. Gurny and J. Heller, J. Controlled Release, 2001, 71, 31–37 CrossRef CAS PubMed.
- S. Einmahl, S. Capancioni, K. Schwach-Abdellaoui, M. Moeller, F. Behar-Cohen and R. Gurny, Adv. Drug Delivery Rev., 2001, 53, 45–73 CrossRef CAS PubMed.
- S. Y. An, J. W. Hwang, K. N. Kim, H. W. Jung, S. M. Noh and J. K. Oh, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 572–581 CrossRef CAS.
- M. Beller and A. M. Tafesh, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, VCH, Weinheim, 1996 Search PubMed.
- Cobalt Technologies, http://www.cobalttech.com/news-item/March 20, 2012.html, Accessed June 22, 2014.
- D. B. Malpass, in Introduction to Industrial Polyethylene, John Wiley & Sons, Inc., 2010, DOI:10.1002/9780470900468.ch1.
- D. B. Malpass and E. I. Band, in Introduction to Industrial Polypropylene, John Wiley & Sons, Inc., 2012, DOI:10.1002/9781118463215.ch1.
- A. Pavone, Bio-Based Adipic Acid, Report 284, Santa Clara, California, 2012 Search PubMed.
- J. P. Oppenheim and G. L. Dickerson, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2003, DOI:10.1002/0471238961.0104091604012209.a01.pub2.
- BASF, http://www.plasticizers.basf.com/portal/5/en/dt.jsp?setCursor=1_231606, Accessed June 23, 2014.
- H. Dhamankar, Y. Tarasova, C. H. Martin and K. L. Jones Prather, Metab. Eng., 2014, 25, 72–81 CrossRef CAS PubMed.
- S. H. Lee and O. J. Park, Appl. Microbiol. Biotechnol., 2009, 84, 817–828 CrossRef CAS PubMed.
- J. Hatakeyama and M. Sagehashi, US Patent20140080055, 2014 Search PubMed.
- J. Zhang, J.-b. Li, S.-B. Wu and Y. Liu, Ind. Eng. Chem. Res., 2013, 52, 11799–11815 CrossRef CAS.
- S.-H. Luo, Q.-F. Wang, J.-F. Xiong and Z.-Y. Wang, J. Polym. Res., 2012, 19, 1–9 CrossRef CAS.
- W. Zhu, H. Yang, J. Chen, C. Chen, L. Guo, H. Gan, X. Zhao and Z. Hou, Green Chem., 2014, 16, 1534 RSC.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS PubMed.
- L. Vilcocq, A. Cabiac, C. Especel, E. Guillon and D. Duprez, Oil Gas Sci. Technol. – Rev. IFP Energies nouvelles., 2013, 68, 841–860 CrossRef CAS.
- Roquette, Additives Polym., 2013, 2013, 4 Search PubMed.
- V. Gupta, S. Singh and U. Makwana, WO Patent2009128087, 2009 Search PubMed.
- M. Jbeily, T. Naolou, M. Bilal, E. Amado and J. Kressler, Polym. Int., 2014, 63, 894–901 CrossRef CAS.
- A. Anand, R. D. Kulkarni and V. V. Gite, Prog. Org. Coat., 2012, 74, 764–767 CrossRef CAS.
- M. Shibata, S. Yoshihara, M. Yashiro and Y. Ohno, J. Appl. Polym. Sci., 2013, 128, 2753–2758 CrossRef CAS.
- M. A. Islam, J. Y. Shin, J. Firdous, T. E. Park, Y. J. Choi, M. H. Cho, C. H. Yun and C. S. Cho, Biomaterials, 2012, 33, 8868–8880 CrossRef CAS PubMed.
- S. Pasupuleti and G. Madras, J. Appl. Polym. Sci., 2011, 121, 2861–2869 CrossRef CAS.
- J. Persson, P. Hemstrom and K. Irgum, J. Sep. Sci., 2008, 31, 1504–1510 CrossRef CAS PubMed.
- R. L. Shogren, K. M. Doll, J. L. Willett and G. Swift, J. Polym. Environ., 2009, 17, 103–108 CrossRef CAS.
- A. F. Naves, H. T. C. Fernandes, A. P. S. Immich and L. H. Catalani, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3881–3891 CrossRef CAS.
- C. Gioia, M. Vannini, P. Marchese, A. Minesso, R. Cavalieri, M. Colonna and A. Celli, Green Chem., 2014, 16, 1807–1815 RSC.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- H. Schiweck, A. Bär, R. Vogel, E. Schwarz, M. Kunz, C. Dusautois, A. Clement, C. Lefranc, B. Lüssem, M. Moser and S. Peters, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012, DOI:10.1002/14356007.a25_413.pub3.
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2006, DOI:10.1002/14356007.a12_477.pub2.
- Transparency Market Research, http://www.transparencymarketresearch.com/glycerol.market.html, Accessed October 25, 2014.
- F. N. Jones, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2003, DOI:10.1002/14356007.a01_409.
- R. Moita, A. Freches and P. C. Lemos, Water Res., 2014, 58, 9–20 CrossRef CAS PubMed.
- A. Thomas, S. S. Muller and H. Frey, Biomacromolecules, 2014, 15, 1935–1954 CrossRef CAS PubMed.
- C. Zhu, S. Chiu, J. P. Nakas and C. T. Nomura, J. Appl. Polym. Sci., 2013, 130, 1–13 CrossRef CAS.
- M. Calderon, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef CAS PubMed.
- R. Rai, M. Tallawi, A. Grigore and A. R. Boccaccini, Prog. Polym. Sci., 2012, 37, 1051–1078 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13 RSC.
- Bioalcohols, http://biofuel.org.uk/bioalcohols.html, Accessed July 18, 2014.
- S. Zhu, Y. Zhu, S. Hao, H. Zheng, T. Mo and Y. Li, Green Chem., 2012, 14, 2607 RSC.
- Y. Deng and S. S. Fong, Metab. Eng., 2011, 13, 570–577 CrossRef CAS PubMed.
- A. Martin and M. Richter, Eur. J. Lipid Sci. Technol., 2011, 113, 100–117 CrossRef CAS.
- H. Satoh, A. Mineshima, T. Nakamura, N. Teramoto and M. Shibata, React. Funct. Polym., 2014, 76, 49–56 CrossRef CAS.
- C. Lavilla, A. Martínez de Ilarduya, A. Alla and S. Muñoz-Guerra, Polym. Chem., 2013, 4, 282 RSC.
- G. Li, N. Li, S. Li, A. Wang, Y. Cong, X. Wanga and T. Zhang, Chem. Commun., 2013, 49, 5727–5729 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- D. A. Putnam and A. Zelikin, US Patent7659420, 2010 Search PubMed.
- M. Helou, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2011, 2, 2789–2795 RSC.
- Y. Liu, H. Tuysuz, C. J. Jia, M. Schwickardi, R. Rinaldi, A. H. Lu, W. Schmidt and F. Schuth, Chem. Commun., 2010, 46, 1238–1240 RSC.
- L. Krähling, J. Krey, G. Jakobson, J. Grolig and L. Miksche, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a01_425.
- Solvay Chemicals, Solvay to Build Bio-Based Epichlorohydrin Plant in China to Serve Largest Market in the World, Brussels, 2012 Search PubMed.
- The Dow Chemical Company, http://msdssearch.dow.com/PublishedLiteratureDOWCOM/dh_0100/0901b80380100836.pdf?filepath=/296-01301.pdf&fromPage=GetDoc, Accessed August 15, 2014.
- Solvay SA, http://www.solvaychemicals.com/Chemicals Literature Documents/Allylic_products/APP-2200-0000-W-EN_WW_.pdf, Accessed August 15, 2014.
- R. Bai, H. Zhang, F. Mei, S. Wang, T. Li, Y. Gu and G. Li, Green Chem., 2013, 15, 2929–2934 RSC.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- M. Schömer, C. Schüll and H. Frey, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 995–1019 CrossRef.
- J. Geschwind and H. Frey, Macromolecules, 2013, 46, 3280–3287 CrossRef CAS.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- G. Rokicki, P. Rakoczy, P. Parzuchowski and M. Sobiecki, Green Chem., 2005, 7, 529–539 RSC.
- M. Fleischer, H. Blattmann and R. Mülhaupt, Green Chem., 2013, 15, 934–942 RSC.
- S. Vollenweider and C. Lacroix, Appl. Microbiol. Biotechnol., 2004, 64, 16–27 CrossRef CAS PubMed.
- H. Krauter, T. Willke and K. D. Vorlop, Nat. Biotechnol., 2012, 29, 211–217 CAS.
- T. Fukuoka, H. Habe, D. Kitamoto and K. Sakaki, J. Oleo Sci., 2011, 60, 369–373 CrossRef CAS PubMed.
- H. Habe, T. Fukuoka, D. Kitamoto and K. Sakaki, Appl. Microbiol. Biotechnol., 2009, 84, 445–452 CrossRef CAS PubMed.
- H. Kimura, Polym. Adv. Technol., 2001, 12, 697–710 CrossRef CAS.
- F. A. Castillo Martinez, E. M. Balciunas, J. M. Salgado, J. M. Domínguez González, A. Converti and R. P. d. S. Oliveira, Trends Food Sci. Technol., 2013, 30, 70–83 CrossRef.
- Direvo Industrial, http://www.b2bioworld.com/en/products_services/ps00112_Direvo_lignocellulose_lactic_acid.html, Accessed August 19, 2014.
- Lactic Acid Market by Application (Biodegradable Polymer, Food & Beverage, Personal Care & Pharmaceutical) & Polylactic Acid Market by Application (Packaging, Agriculture, Automobile, Electronics, Textile) & Geography – Global Trends & Forecasts to 2019, MarketsandMarkets.
- R. P. Babu, K. O'Connor and R. Seeram, Prog. Biomater., 2013, 2, 8 CrossRef.
- M. L. Robertson, K. Chang, W. M. Gramlich and M. A. Hillmyer, Macromolecules, 2010, 43, 1807–1814 CrossRef CAS.
- L. T. Sin, A. R. Rahmat and W. A. W. A. Rahman, Polylactic Acid: PLA Biopolymer Technology and Applications, William Andrew, Inc., London, 2012 Search PubMed.
- J. Ren, Biodegradable Poly (Lactic Acid): Synthesis, Modification, Processing and Applications, Springer, New York, 2011 Search PubMed.
- S. Inkinen, M. Hakkarainen, A. C. Albertsson and A. Sodergard, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed.
- I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. Lopez-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS.
- R. M. Rasal, A. V. Janorkar and D. E. Hirt, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS.
- A. J. Lasprilla, G. A. Martinez, B. H. Lunelli, A. L. Jardini and R. M. Filho, Biotechnol. Adv., 2012, 30, 321–328 CrossRef CAS PubMed.
- Eastman Company, http://www.eastman.com/Products/Pages/ProductList.aspx?categoryName=Cellulose+Acetate+Proprionate, Accessed August 25, 2014.
- L. Z. Flores-López, J. Caloca, E. Rogel-Hernández and H. Espinoza-Gomez, Cellulose, 2014, 21, 1987–1995 CrossRef.
- G. W. Jeon, J.-E. An and Y. G. Jeong, Compos. Part B-Eng., 2012, 43, 3412–3418 CrossRef CAS.
- B. P. Kumar, S. Ramanaiah, T. M. Reddy and K. S. Reddy, Polym. Bull., 2014, 71, 125–132 CrossRef.
- X. Li, K. Y. Wang, B. Helmer and T.-S. Chung, Ind. Eng. Chem. Res., 2012, 51, 10039–10050 CrossRef CAS.
- World-wide Investments in Bio-based Chemicals, Nova Institute, 2012 Search PubMed.
- C. J. Sullivan, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a22_163.
- D. Kahlich, U. Wiechern and J. Lindner, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a22_239.
- The Dow Chemical Company, http://www.dow.com/propyleneoxide/app/ppp.htm, Accessed 28 August, 2014.
- B. Liang, R. Tong, Z. Wang, S. Guo and H. Xia, Langmuir, 2014, 30, 9524–9532 CrossRef CAS PubMed.
- L. Xu, Z. Zhang, F. Wang, D. Xie, S. Yang, T. Wang, L. Feng and C. Chu, J. Colloid Interface Sci., 2013, 393, 174–181 CrossRef CAS PubMed.
- S. Zheng, J. Y. Shin, S. Y. Song, S. J. Yu, H. Suh and I. Kim, J. Appl. Polym. Sci., 2014, 131, 40610–40620 Search PubMed.
- R. Wipf, M. Kraska, T. Spehr, J. Nieberle, H. Frey and B. Stühn, Soft Matter, 2011, 7, 10879–10888 RSC.
- J. M. Duroux and L. M. Elie, US Patent3549696, 1970 Search PubMed.
- R. B. Durairaj, Resorcinol: Chemistry, Technology and Applications, Springer, Pittsburgh, 2005 Search PubMed.
- H. Kimura, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 189–193 CrossRef CAS.
- P. O. Boamah, Q. Zhang, M. Hua, Y. Huang, Y. Liu, W. Wang and Y. Liu, Carbohydr. Polym., 2014, 110, 518–527 CrossRef CAS PubMed.
- C. R. Reddy, P. Iyengar, G. Nagendrappa and B. S. J. Prakash, Catal. Lett., 2005, 101, 87–91 CrossRef CAS.
- A. Vogel, G. Steffan, K. Mannes and V. Trescher, US Patent4341720, 1982 Search PubMed.
- S. Haghighi Mood, A. Hossein Golfeshan, M. Tabatabaei, G. Salehi Jouzani, G. H. Najafi, M. Gholami and M. Ardjmand, Renewable Sustainable Energy Rev., 2013, 27, 77–93 CrossRef CAS.
- A. W. Bhutto, K. Qureshi, K. Harijan, G. Zahedi and A. Bahadori, RSC Adv., 2014, 4, 3392–3412 RSC.
- R. Singh, A. Shukla, S. Tiwari and M. Srivastava, Renewable Sustainable Energy Rev., 2014, 32, 713–728 CrossRef CAS.
- R. R. Singhania, P. Binod and A. Pandey, in Bioprocessing Technologies in Biorefinery for Sustainable Production of Fuels, Chemicals, and Polymers, John Wiley & Sons, Inc., 2013, pp. 193–204, DOI:10.1002/9781118642047.ch11.
- A. L. Borrion, M. C. McManus and G. P. Hammond, Renewable Sustainable Energy Rev., 2012, 16, 4638–4650 CrossRef.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- J. Zhao, C. Lu, C.-C. Chen and S.-T. Yang, in Bioprocessing Technologies in Biorefinery for Sustainable Production of Fuels, Chemicals, and Polymers, John Wiley & Sons, Inc., 2013, pp. 235–262, DOI:10.1002/9781118642047.ch13.
- Y. Tashiro, T. Yoshida, T. Noguchi and K. Sonomoto, Eng. Life Sci., 2013, 13, 432–445 CrossRef CAS.
- M. Kumar, Y. Goyal, A. Sarkar and K. Gayen, Appl. Energy, 2012, 93, 193–204 CrossRef CAS.
- H. Cheung, R. S. Tanke and G. P. Torrence, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011, DOI:10.1002/14356007.a01_045.pub2.
- G. Roscher, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a27_419.
- M. Amann and O. Minge, Adv. Polym. Sci., 2012, 245, 137–172 CrossRef CAS.
- S. Fischer, K. Thümmler, B. Volkert, K. Hettrich, I. Schmidt and K. Fischer, Macromol. Symp., 2008, 262, 89–96 CrossRef CAS.
- J. Puls, S. A. Wilson and D. Hölter, J. Polym. Environ., 2011, 19, 152–165 CrossRef CAS.
- R. T. Mathers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1–15 CrossRef CAS.
- D. Zhang, S. A. I. Barri and D. Chadwick, Appl. Catal., A, 2011, 403, 1–11 CrossRef CAS.
- H. Huang, in Plastics from Bacteria, ed. G. G.-Q. Chen, Springer, Heidelberg, 2010, ch. 15, vol. 14, pp. 389–404 Search PubMed.
- A. J. Peacock, Handbook of Polyethylene: Structures: Properties, and Applications, Marcel Dekker, New York, 2000 Search PubMed.
- Ethylene Oxide Market And Ethylene Glycol Market – Global Industry Analysis, Raw Material And Consumption Trends, Size, Share And Forecast 2012–2018, Transparency Market Research, 2012.
- S. Rebsdat and D. Mayer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2001, DOI:10.1002/14356007.a10_117.
- D. M. Back and R. L. Schmitt, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2004, DOI:10.1002/0471440264.pst528.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- S. Rebsdat and D. Mayer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a10_101.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC.
- Shell Corporation, http://www.shell.com/global/products-services/solutions-for-businesses/chemicals/media-centre/factsheets/omega.html, Accessed September 11, 2014.
- Y. Li, K. Junge and M. Beller, ChemCatChem, 2013, 5, 1072–1074 CrossRef CAS.
- A. Paşahan, S. Köytepe, M. A. Cengiz and T. Seçkin, Polym. Int., 2013, 62, 246–250 CrossRef.
- G. Wei, X. Yang, W. Zhou, J. Lin and D. Wei, Biochem. Eng. J., 2009, 47, 127–131 CrossRef CAS.
- V. Singh and M. Tiwari, Int. J. Polym. Sci., 2010, 2010, 1–23 CrossRef.
- G. Mattioda and A. Blanc, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011, DOI:10.1002/14356007.a12_491.pub2.
- E.-L. Dreher, T. R. Torkelson and K. K. Beutel, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011, DOI:10.1002/14356007.o06_o01.
- Vinnolit GmbH & Co. KG, http://www.vinnolit.com/vinnolit.nsf/id/kunststoffe-developement-pvc-market-en/$file/I-PDF VINNOLIT_PE111518_PE10_13_.pdf, Accessed September 18, 2014.
- M. W. Allsopp and G. Vianello, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a21_717.
- K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a02_001.
- J. Y. Sum, A. L. Ahmad and B. S. Ooi, J. Membr. Sci., 2014, 466, 183–191 CrossRef CAS.
- H. Testrich, H. Rebl, B. Finke, F. Hempel, B. Nebe and J. Meichsner, Mater. Sci. Eng., C, 2013, 33, 3875–3880 CrossRef CAS PubMed.
- K. Nagai, Appl. Catal., A, 2001, 221, 367–377 CrossRef CAS.
- Market Study: Polypropylene, Ceresana, 2nd edn, 2014, http://www.ceresana.com/en/market-studies/plastics/polypropylene/ Search PubMed.
- F. Zamora, K. Hakkou, A. Alla, R. Marín-bernabé, M. V. de Paz, A. Martínez de Ilarduya, S. Muñoz-guerra and J. A. Galbis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5167–5179 CrossRef CAS.
- M. d. G. García-Martín, E. Benito Hernández, R. Ruiz Pérez, A. Alla, S. Muñoz-Guerra and J. A. Galbis, Macromolecules, 2004, 37, 5550–5556 CrossRef.
- H. Pohjanlehto, H. M. Setälä, D. E. Kiely and A. G. McDonald, J. Appl. Polym. Sci., 2014, 131, 39714 CrossRef.
- K. Lohbeck, H. Haferkorn, W. Fuhrmann and N. Fedtke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, DOI:10.1002/14356007.a16_053.
- T. R. Felthouse, J. C. Burnett, B. Horrell, M. J. Mummey and Y.-J. Kuo, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2001, DOI:10.1002/0471238961.1301120506051220.a01.pub2.
- B. Klumperman, Polym. Chem., 2010, 1, 558–562 RSC.
- G. Q. Chen, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- G.-Y. Tan, C.-L. Chen, L. Li, L. Ge, L. Wang, I. Razaad, Y. Li, L. Zhao, Y. Mo and J.-Y. Wang, Polymer, 2014, 6, 706–754 Search PubMed.
- Y. K. Leong, P. L. Show, C. W. Ooi, T. C. Ling and J. C. Lan, J. Biotechnol., 2014, 180, 52–65 CrossRef CAS PubMed.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- C. Du, J. Sabirova, W. Soetaert and S. Ki Carol Lin, Curr. Chem. Biol., 2012, 6, 14–25 CAS.
- J. Yu, in Bioprocessing Technologies in Biorefinery for Sustainable Production of Fuels, Chemicals, and Polymers, John Wiley & Sons, Inc., 2013, pp. 415–426, DOI:10.1002/9781118642047.ch22.
- M. Koller, A. Atlić, M. Dias, A. Reiterer and G. Braunegg, in Plastics from Bacteria, ed. G. G.-Q. Chen, Springer, Berlin, Heidelberg, 2010, ch. 5, vol. 14, pp. 85–119 Search PubMed.
- L. E. Alva Munoz and M. R. Riley, Biotechnol. Bioeng., 2008, 100, 882–888 CrossRef PubMed.
- S. J. Park, T. W. Kim, M. K. Kim, S. Y. Lee and S. C. Lim, Biotechnol. Adv., 2012, 30, 1196–1206 CrossRef CAS PubMed.
- A. M. Gumel, M. S. M. Annuar and Y. Chisti, J. Polym. Environ., 2013, 21, 580–605 CrossRef CAS.
- R. Vogel, B. Tandler, D. Voigt, D. Jehnichen, L. Haussler, L. Peitzsch and H. Brunig, Macromol. Biosci., 2007, 7, 820–828 CrossRef CAS PubMed.
- K. C. Reis, J. Pereira, A. C. Smith, C. W. P. Carvalho, N. Wellner and I. Yakimets, J. Food Eng., 2008, 89, 361–369 CrossRef CAS.
- L. Wang, W. Zhu, X. Wang, X. Chen, G.-Q. Chen and K. Xu, J. Appl. Polym. Sci., 2008, 107, 166–173 CrossRef CAS.
- J. Zhang, N. Hao and G. Q. Chen, Appl. Microbiol. Biotechnol., 2006, 71, 222–227 CrossRef CAS PubMed.
- Q. Liu, S. P. Ouyang, J. Kim and G. Q. Chen, J. Biotechnol., 2007, 132, 273–279 CrossRef CAS PubMed.
- D. Kai and X. J. Loh, ACS Sustainable Chem. Eng., 2014, 2, 106–119 CrossRef CAS.
- X. Zhang, R. Luo, Z. Wang, Y. Deng and G.-Q. Chen, Biomacromolecules, 2009, 10, 707–711 CrossRef CAS PubMed.
- K. Yamashita, M. Yamada, K. Numata and S. Taguchi, Biomacromolecules, 2006, 7, 2449–2454 CrossRef CAS PubMed.
- Z. J. Li, L. Cai, Q. Wu and G. Q. Chen, Appl. Microbiol. Biotechnol., 2009, 83, 939–947 CrossRef CAS PubMed.
- Synthetic Rubber Market - Global Industry Analysis, Size, Share, Growth, Trends And Forecast, 2012–2018, Transparency Market Research, 2012, http://www.transparencymarketresearch.com/synthetic-rubber-market.html Search PubMed.
- Issue Thirteen: “Bio-based Products”, NNFCC Market Review, 2013.
- P. J. Halley and J. R. Dorgan, MRS Bull., 2011, 36, 687–691 CrossRef CAS.
- J. Taylor, M. Jenni and M. Peters, Top. Catal., 2010, 53, 1224–1230 CrossRef CAS.
- D. Threadingham, W. Obrecht, W. Wieder, G. Wachholz and R. Engehausen, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011, DOI:10.1002/14356007.a23_239.pub5.
- D. J. Kind and T. R. Hull, Polym. Degrad. Stab., 2012, 97, 201–213 CrossRef CAS.
- IISRP: Synthetic Polyisoprene, http://iisrp.com/webpolymers/11polyisoprene.pdf, Accessed October 25, 2014.
- R. N. Webb, T. D. Shaffer and A. H. Tsou, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2003, DOI:10.1002/0471440264.pst036.
- R. T. Tran, Y. Zhang, D. Gyawali and J. Yang, Recent Pat. Biomed. Eng., 2009, 2, 216–227 CrossRef CAS.
- A. Singh and M. Kamal, J. Appl. Polym. Sci., 2012, 125, 1456–1459 CrossRef CAS.
- S. Sharma and A. K. Srivastava, Eur. Polym. J., 2004, 40, 2235–2240 CrossRef CAS.
- K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005 CrossRef CAS PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- M. Bahr, A. Bitto and R. Mulhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- D. G. Barrett, W. Luo and M. N. Yousaf, Polym. Chem., 2010, 1, 296–302 RSC.
- E. Gubbels, L. Jasinska-Walc, B. A. J. Noordover and C. E. Koning, Eur. Polym. J., 2013, 49, 3188–3198 CrossRef CAS.
- E. Gubbels, L. Jasinska-Walc and C. E. Koning, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 890–898 CrossRef CAS.
- S. C. Shukla, A. Singh, A. K. Pandey and A. Mishra, Biochem. Eng. J., 2012, 65, 70–81 CrossRef CAS.
- T. Satoh and T. Kakuchi, Macromol. Biosci., 2007, 7, 999–1009 CrossRef CAS PubMed.
- NNFCC, Renewable Chemicals Factsheet: Lignin, 2011 Search PubMed.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- J. J. Bozell, J. E. Holladay, D. Johnson and J. F. White, Top Value Added Candidates from Biomass, Volume II: Results of Screening for Potential Candidates from Biorefinery Lignin, Pacific Northwest National Laboratory, Richland, WA, 2007 Search PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
| This journal is © The Royal Society of Chemistry 2015 |