DOI:
10.1039/C5PY00188A
(Paper)
Polym. Chem., 2015,
6, 3024-3030
Phosphonated furan-functionalized poly(ethylene oxide)s using orthogonal click chemistries: synthesis and Diels–Alder reactivity†
Received
5th February 2015
, Accepted 2nd March 2015
First published on 4th March 2015
Abstract
The synthesis and the reactivity in Diels–Alder and retro Diels–Alder (DA/rDA) reactions of a series of novel phosphonated furan-functionalized PEO monomethyl ethers were investigated. Dimethylphosphonate-terminated furan-functionalized PEO monomethyl ethers and their phosphonic acid-terminated derivatives have been successfully prepared by using a combination of click copper-catalyzed 1,3-dipolar cycloaddition and Kabachnik–Fields reactions. Influence of both the substitution pattern of the furan ring and the solvent onto the DA/rDA process were investigated. It was found that the 3-substituted furan is the more reactive and that water facilitates both the DA and the rDA reactions, while maintaining the polymeric structure intact. The results demonstrate the potential of such structures for dynamic covalent applications and controlled drug delivery systems such as thermoreversible linkage of biological entities onto metallic nanoparticles.
Introduction
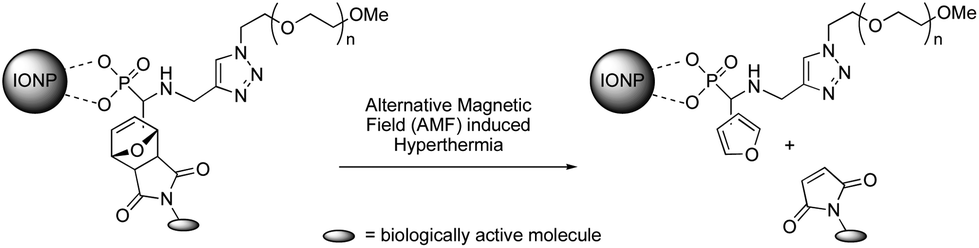
The Diels–Alder (DA) reaction is a well-known thermoreversible [4 + 2] cycloaddition reaction between diene and alkene (dienophiles) derivatives.1,2 Because of its thermal reversibility, in addition to high yields and superior selectivity under mild (aqueous) conditions, this cycloaddition reaction is one of the most attractive members of the click chemistry family.3–7 DA reactions involving furan as a diene to form oxanorbornenes have attracted much attention in polymer chemistry, particularly in providing new materials and products,8–15 including bioconjugates.16–19 Recently, the reversible nature of the DA reaction has been exploited for the dynamic covalent synthesis of organic materials.20,21 We have relied on this methodology to prepare novel functional iron oxide magnetic nanoparticles (IONPs) that show unprecedented hyperthermia-induced drug release by magnetically stimulated retro Diels–Alder (rDA) process.22 Our strategy is based on a new versatile multifunctional ligand incorporating a phosphonic acid group, which strongly binds onto the iron oxide surface of the IONPs, and two orthogonal clickable (alkyne and furan) groups. The alkyne moiety is used for installing via copper-catalyzed azide–alkyne cycloaddition (CuAAC) an azido-end-functionalized hydrophilic poly(ethylene oxide) (PEO) that affords water-dispersibility and stability, anti-fouling, and biocompatibility. The furan ring acts as a thermoreversible linker for a biologically active molecule via thermally reversible DA chemistry (Scheme 1). We have demonstrated that upon alternating magnetic field (AMF) exposure, sufficient local energy is brought in close proximity of the cycloadduct to initiate the rDA reaction without the need for heating the solution at the elevated temperature usually required for such a process.22 Those functional IONPs have thus the potential to improve hyperthermia therapies by expanding the range of polymers and drugs that can be used.
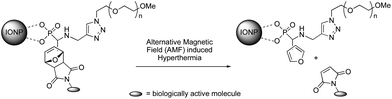 |
| | Scheme 1 The concept of magnetically stimulated rDA reaction using IONPs. | |
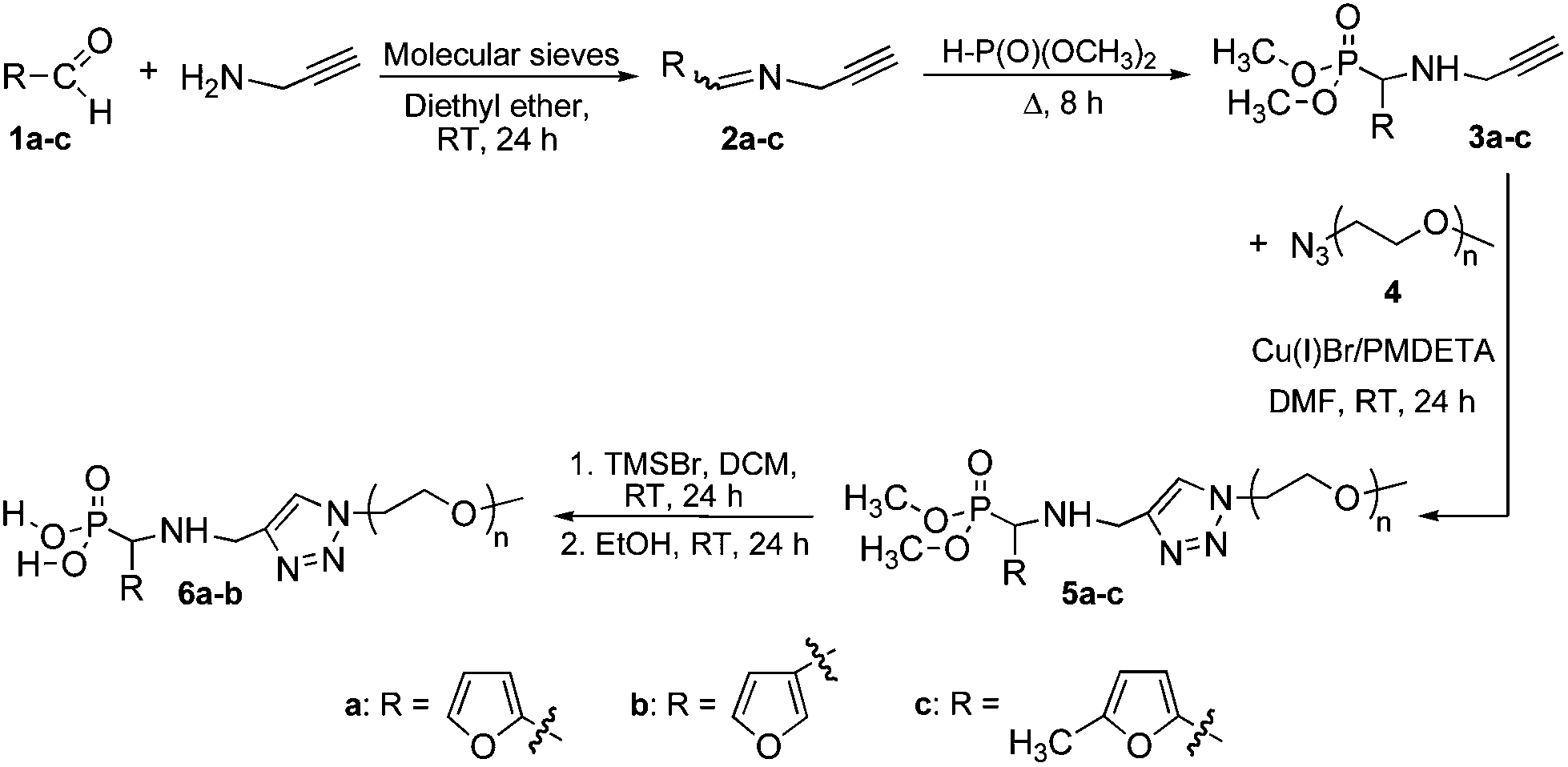
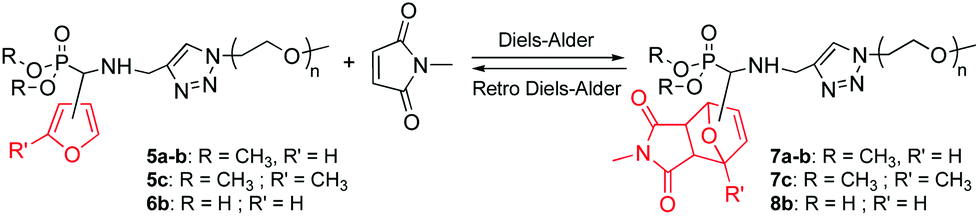
Herein, we report on the synthesis and reactivity in DA/rDA processes of a series of new phosphonated furan-functionalized PEO monomethyl ethers that are potential candidates for a wide range of uses, including stabilization and dispersion of metallic nanoparticles. In order to study the influence of the furan substitution pattern, we first synthesized dimethylphosphonate-terminated furan-functionalized PEO monomethyl ethers 5a–c and their phosphonic acid-terminated homologues 6a–b according to our reported procedure (Scheme 2)22,23 combining the click CuAAC3 and the Kabachnik–Fields24–27 reaction that is rarely utilized in polymer chemistry.23,28–32 Next we looked into the role of the furan substitution position onto the DA/rDA reactivity of the prepared furan-functionalized phosphonated PEOs by using N-methylmaleimide as a model dienophile.
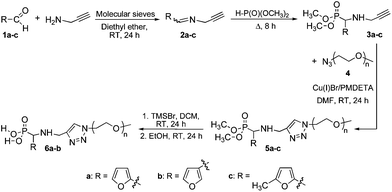 |
| | Scheme 2 Synthesis of phosphonated furan-functionalized PEO monomethyl ethers. | |
Experimental
Materials
Dimethylphosphonate-terminated furan-functionalized poly(ethylene oxide) (PEO) monomethyl ethers 5a–c and their phosphonic acid-terminated homologues 6a–b were synthesized according to the literature22,23 and the whole procedure as well as the characterization are included in the ESI.† All other chemicals were purchased from commercial sources and used without further purification.
General characterization
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 400 spectrometer operating at 400.16 MHz for 1H, 100.62 MHz for 13C, and 161.96 MHz for 31P using either deuterated chloroform (CDCl3), deuterated dimethyl sulfoxide (DMSO-d6), deuterium oxide (D2O) or deuterated 1,1,2,2-tetrachloroethane (TCE-d2) as the solvent. 31P NMR spectra were proton decoupled. Diastereoisomers ratios were calculated from the peak integration area of a quantitative 1H decoupled 31P NMR spectrum acquired by a 1D sequence with inverse gated coupling (zgig) and a relaxation delay D1 = 30 s. 1H and 13C NMR spectra were referenced to tetramethylsilane signals while 31P NMR chemical shifts were referenced to 85% phosphoric acid as an external reference, with positive shift values downfield from the reference. Coupling constants and chemical shifts are reported in hertz and in parts per million (ppm), respectively. Fourier transform infra-red (FT-IR) spectra were recorded using a Nicolet avatar 370 DTGS spectrometer in transmittance mode. High resolution mass spectra (HR-MS) were recorded on a Waters-Micromass® GCT Premier™ (GC, CI+, methane) using a HP 6890 GC apparatus equipped with a chromatographic column of 25 m, diameter 250 μm, thickness 0.25 μm. The sample was warmed at a temperature of 40 °C for 5 min and then further heated at a heating rate of 10 °C min−1 up to 220 °C.
General procedure for the Diels–Alder reaction between dimethylphosphonate-terminated furan-functionalized PEO monomethyl ethers 5a–c or phosphonic acid-terminated furan-functionalized PEO monomethyl ether 6b and N-methylmaleimide model compound
The following protocol was used for reactions carried out in deuterated chloroform (CDCl3) reported in Table 1. Dimethylphosphonate-terminated furan-functionalized PEO derivatives 5a–b (0.7 g; 0.315 mmol) and the desired quantity of N-methylmaleimide (0.315, 1.575 or 6.3 mmol; 1, 5 or 20 equivalents) were introduced in a 10 mL round-bottom flask equipped with a magnetic stirrer and a reflux condenser. The reaction mixture was subsequently dissolved in 3 mL of CDCl3. When a homogeneous solution was obtained, the 10 mL round-bottom flask was immersed in an oil bath preset at 40 °C to allow the reaction to proceed (initial reaction time, t = 0). Samples were taken out during the reaction to monitor the conversion of the Diels–Alder (DA) reaction by 1H NMR spectroscopy by comparing the peaks areas of the bridgehead protons of the oxanorbornene cycloadduct at δ = 5.13–5.39 ppm and the methylene protons linked to the triazole ring at δ = 4.52 ppm. For the DA reactions carried out in a NMR tube in situ (Fig. 1, 2 & Table 2), the following formulations were used: 5b (0.1 g; 0.045 mmol), N-methylmaleimide (0.9 mmol) and deuterated solvent: CDCl3, water (D2O) or dimethyl sulfoxide (DMSO-d6) (0.4 mL). The NMR tube was immersed in an oil bath preset at 25 or 40 °C to allow the reaction to proceed (initial reaction time, t = 0). NMR spectra were carried out periodically to monitor the conversion of the DA reaction by 1H NMR spectroscopy. The DA reaction from the phosphonic acid-terminated furan-functionalized PEO monomethyl ether 6b was carried out in a NMR tube in situ using the following formulation: 6b (0.045 mmol), N-methylmaleimide (0.9 mmol) and D2O (0.4 mL).
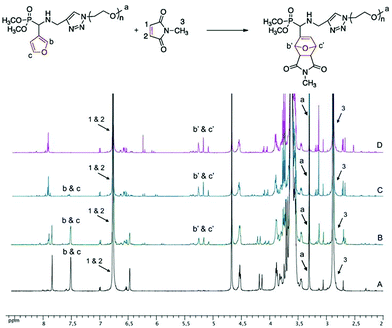 |
| | Fig. 1 Overlay of 1H NMR spectra of the reaction mixture of 5b and N-methylmaleimide for a [N-methylmaleimide]/[5b] molar ratio of 20 in D2O at 40 °C recorded for a reaction time of (A) t = 0, (B) t = 3 h, (C) t = 1 day and (D) t = 3 days. | |
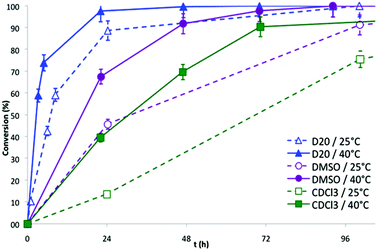 |
| | Fig. 2 Conversion of the DA reaction between 5b and N-methylmaleimide in different solvents at 25 °C and 40 °C versus time. | |
Table 1 Influence of the furan position on the DA reaction between 5a–c and N-methylmaleimide at 40 °C during 5 days
| Run |
Dimethylphosphonate-terminated PEO |
Solvent |
[N-Methylmaleimide]0/[5a–c]0 |
Conv.a (%) |
|
Determined by 1H NMR spectroscopy by comparing the peak areas of the CH–O groups of oxanorbornene at δ = 5.13–5.39 ppm and the methylene protons of the PEO in α of the triazole ring at δ = 4.52 ppm.
|
| 1 |
5a
|
CDCl3 |
1 |
0 |
| 2 |
5b
|
CDCl3 |
1 |
10 |
| 3 |
5a
|
CDCl3 |
5 |
0 |
| 4 |
5b
|
CDCl3 |
5 |
13 |
| 5 |
5a
|
CDCl3 |
20 |
0 |
| 6 |
5b
|
CDCl3 |
20 |
82 |
| 7 |
5a
|
DMF |
20 |
18 |
| 8 |
5b
|
DMF |
20 |
98 |
| 9 |
5c
|
DMF |
20 |
50 |
Table 2 Conversion of the DA reaction between furan-functionalized PEO 5b or 6b and N-methylmaleimide for a [N-methylmaleimide]/[5b or 6b] molar ratio of 20 after a reaction time of 24 h
| Run |
Furan-functionalized PEO |
Solvent |
Temperature (°C) |
Conv.a (%) |
|
Determined by 1H NMR spectroscopy by comparing the peak areas of the CH–O groups in the furan ring at δ = 7.40–7.60 ppm and the CH3 end-group of PEO at δ = 3.38 ppm.
|
| 1 |
5b
|
CDCl3 |
25 |
14 |
| 2 |
5b
|
DMSO-d6 |
25 |
46 |
| 3 |
5b
|
D2O |
25 |
89 |
| 4 |
6b
|
D2O |
25 |
47 |
| 5 |
5b
|
CDCl3 |
40 |
59 |
| 6 |
5b
|
DMSO-d6 |
40 |
68 |
| 7 |
5b
|
D2O |
40 |
98 |
| 8 |
6b
|
D2O |
40 |
90 |
For reactions carried out in non-deuterated solvent N,N-dimethylformamide (DMF) reported in Table 1, dimethylphosphonate-terminated furan-functionalized PEO derivatives 5a–c (0.2 g; 0.09 mmol) and N-methylmaleimide (1.8 mmol; 20 equivalents) were introduced in a 25 mL round-bottom flask equipped with a magnetic stirrer and a reflux condenser. The reaction mixture was subsequently dissolved in 5 mL of DMF. When a homogeneous solution was obtained, the 25 mL round-bottom flask was immersed in an oil bath preset at 40 °C to allow the reaction to proceed (initial reaction time, t = 0). At the end of the reaction, the resulting mixture was concentrated under vacuum and precipitated into diethyl ether. The yellow powder was subsequently dried under vacuum at ambient temperature. Conversions were determined from 1H NMR spectra by comparing the peaks areas of the bridgehead protons of the oxanorbornene cycloadduct at δ = 5.13–5.39 ppm and the methylene protons linked to the triazole ring at δ = 4.52 ppm.
Dimethylphosphonate-terminated oxanorbornene-functionalized PEO monomethyl ether 7a.
[N-Methylmaleimide]/[5a] = 20 (Table 1, run 7); conv.: 18%. 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.62 (s, 1H, triazole); 6.28 (d, J = 6.22 Hz, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); 5.23 (m, 1H, CH–O); 4.52 (m, 2H, Ntriazole–CH2–CH2–O); 4.23 (d, 1H, J = 21.43 Hz, CHP); 3.94 (m, 2H, Ntriazole–CH2–CH2–O); 3.87 (m, 6H, P(O)O–CH3); 3.78–3.51 (m, 172H, CH2–CH2–O); 3.46 (t, J = 4.60 Hz, 2H, NH–CH2); 3.38 (s, 3H, O–CH3); 3.21 (m, 2H, CHCO); 2.98 (s, 3H, CH3–N); 2.32 (s, 1H, NH). 31P NMR (CDCl3, 161.96 MHz), δ (ppm): 27.69; 26.72; 25.48; 24.37.
C); 5.23 (m, 1H, CH–O); 4.52 (m, 2H, Ntriazole–CH2–CH2–O); 4.23 (d, 1H, J = 21.43 Hz, CHP); 3.94 (m, 2H, Ntriazole–CH2–CH2–O); 3.87 (m, 6H, P(O)O–CH3); 3.78–3.51 (m, 172H, CH2–CH2–O); 3.46 (t, J = 4.60 Hz, 2H, NH–CH2); 3.38 (s, 3H, O–CH3); 3.21 (m, 2H, CHCO); 2.98 (s, 3H, CH3–N); 2.32 (s, 1H, NH). 31P NMR (CDCl3, 161.96 MHz), δ (ppm): 27.69; 26.72; 25.48; 24.37.
Dimethylphosphonate-terminated oxanorbornene-functionalized PEO monomethyl ether 7b.
[N-Methylmaleimide]/[5b] = 20 (Table 1, run 8); conv.: 98%. 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.66 (s, 1H, triazole); 6.46 (d, J = 6.17 Hz, 1H, CHC); 5.23 (m, 2H, CH–O); 4.53 (t, J = 4.18 Hz, 2H, Ntriazole–CH2–CH2–O); 4.04 (m, 1H, CHP); 3.95 (t, J = 5.64 Hz, 2H, Ntriazole–CH2–CH2–O); 3.86 (m, 6H, P(O)O–CH3); 3.78–3.52 (m, 172H, CH2–CH2–O); 3.46 (t, J = 4.87 Hz, 2H, NH–CH2); 3.38 (s, 3H, O–CH3); 3.26 (d, J = 6.95 Hz, 1H, CHCO); 3.22 (d, J = 5.95 Hz, 1H, CHCO); 2.97 (s, 3H, CH3–N); 2.52 (s, 1H, NH). 13C NMR (CDCl3, 100.62 MHz), δ (ppm): 176.37 (CO); 146.80 (CCH); 145.13 (CC–Ntriazole); 133.57 (CH–O); 133.45 (CH–O); 123.18 (CC–Ntriazole); 122.89 (CHC); 81.65 (CH3–N); 71.90 (CH2–O–CH3); 70.53 (–CH2–O); 69.41 (Ntriazole–CH2–CH2–O); 58.99 (CH3–O); 53.44 (P(O)O–CH3); 52.56 (CHP); 50.23 (Ntriazole–CH2–CH2–O); 48.51 (CHCO); 42.88 (N–CH2). 31P NMR (CDCl3, 161.96 MHz), δ (ppm): 24.43; 24.38; 23.35; 23.19. FT-IR (ν cm−1): 2892 (νC–H); 1700 (νCO); 1468 (νCC triazole); 1241 (νPO).
Dimethylphosphonate-terminated oxanorbornene-functionalized PEO monomethyl ether 7c.
[N-Methylmaleimide]/[5c] = 20 (Table 1, run 9); conv.: 50%. 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.63 (s, 1H, triazole); 6.40 (t, J = 6.72 Hz, 1H, C(CH3)–CHC); 6.11 (t, J = 6.72 Hz, 1H, CHCH–C(CH3)); 4.53 (t, J = 8.66 Hz, 2H, Ntriazole–CH2–CH2–O); 4.16 (d, J = 15.60 Hz, 1H, CHP); 3.95 (t, J = 13.56 Hz, 2H, Ntriazole–CH2–CH2–O); 3.86 (m, 6H, P(O)O–CH3); 3.84–3.51 (m, 172H, CH2–CH2–O); 3.46 (t, J = 6.24 Hz, 2H, NH–CH2); 3.38 (s, 3H, O–CH3); 3.12–2.95 (m, 2H, CH–CO); 2.79 (s, 3H, CH3–N); 2.31 (s, 3H, C(O)CH3); 2.18 (s, 1H, NH). 31P NMR (CDCl3, 161.96 MHz), δ (ppm): 28.20; 26.98; 25.78; 24.48.
Phosphonic acid-terminated oxanorbornene-functionalized PEO monomethyl ether 8b.
[N-Methylmaleimide]/[6b] = 20 (Table 3, run 5); conv.: 94%. 1H NMR (DMSO-d6, 400 MHz), δ (ppm): 8.23 (s, 1H, triazole); 6.73 (s, 1H, CHC); 5.22 (m, 2H, CH–O); 4.58 (t, J = 5.25 Hz, 2H, Ntriazole–CH2–CH2–O); 4.38 (m, 1H, CHP); 3.84 (t, J = 5.98 Hz, 2H, Ntriazole–CH2–CH2–O); 3.77–3.36 (m, 172H, CH2–CH2–O); 3.35 (t, J = 5.38 Hz, 2H, NH–CH2); 3.24 (s, 3H, O–CH3); 3.05 (m, 2H, CHCO); 2.86 (s, 3H, CH3–N); 2.51 (s, 1H, NH). 31P NMR (DMSO-d6, 161.96 MHz), δ (ppm): 10.96; 10.00; 9.77; 9.06. FT-IR (ν cm−1): 3368 (νOH); 2881 (νC–H); 1698 (νCO); 1656 (νCC); 1466 (νCC triazole); 1241 (νPO).
Table 3 Conversion of the rDA reaction with 7b and 8b at different temperatures and solvents
| Run |
Oxanorbornene-functionalized-PEO |
Solvent |
Temperature (°C) |
Reaction time (h) |
Conv.a (%) |
|
Determined by 1H NMR spectroscopy by comparing the peak areas of the CH–O groups of oxanorbornene at δ = 5.13–5.39 ppm and the methylene protons of the PEO in α of the triazole ring at δ = 4.52 ppm.
Degradation of the product in case of a longer reaction time.
Degradation of the product.
|
| 1 |
7b
|
DMSO-d6 |
110 |
12b |
86 |
| 2 |
7b
|
TCE-d2 |
110 |
12b |
90 |
| 3 |
8b
|
DMSO-d6 |
110 |
1c |
— |
| 4 |
8b
|
DMSO-d6 |
80 |
24 |
30 |
| 5 |
8b
|
TCE-d2 |
110 |
6b |
94 |
| 6 |
8b
|
D2O |
80 |
48 |
71 |
General procedure of the retro Diels–Alder reaction from dimethylphosphonate-terminated oxanorbornene-functionalized PEO monomethyl ether 7b or phosphonic acid-terminated oxanorbornene-functionalized PEO monomethyl ether 8b
Dimethylphosphonate-terminated or phosphonic acid-terminated oxanorbornene-functionalized PEO derivatives 7b and 8b (0.05 g; 0.0225 mmol) was introduced in a NMR tube. The reaction mixture was subsequently dissolved in 0.4 mL of deuterated solvent: DMSO-d6, D2O or 1,1,2,2-tetrachloroethane (TCE-d2). When a homogeneous solution was obtained, the NMR tube was immersed in an oil bath preset at 80 or 110 °C to allow the reaction to proceed (initial reaction time, t = 0). NMR spectra were carried out periodically to monitor the conversion using 1H NMR spectroscopy by comparing the peaks areas of the bridgehead protons of the oxanorbornene cycloadduct at δ = 5.13–5.39 ppm and the methylene protons linked to the triazole ring at δ = 4.52 ppm.
Results and discussion
Synthesis of phosphonated furan-functionalized PEO monomethyl ethers
A series of dimethylphosphonate and phosphonic acid-terminated furan-functionalized poly(ethylene oxide) (PEO) monomethyl ethers (5a–c and 6a–b, respectively) was synthesized according to a strategy we have previously developed,22,23 combining the click CuAAC3 and the Kabachnik–Fields24–27 reactions (Scheme 2). The detailed procedure is described in the ESI.† Briefly, the Schiff base (2a–c), issued from reaction between aldehyde (1a–c) and N-propargylamine was reacted with dimethyl hydrogenophosphonate to afford the expected α-aminophosphonate (3a–c). The aminophosphonate was then engaged in a typical click coupling reaction with azido-terminated PEO monomethyl ether 2000 to afford the dimethylphosphonate-terminated furan-functionalized PEO monomethyl ether (5a–c), which was subsequently converted into the phosphonic acid homologue (6a–b) by dealkylation. All the structures were confirmed by Fourier transform-infra red (FT-IR), 1H, 13C, 31P nuclear magnetic resonance (NMR) spectroscopy, and mass spectrometry (see ESI†).
Reactivity of the furan functionality in DA reaction
The suitability of dimethylphosphonate-terminated furan-functionalized PEO monomethyl ethers (5a–c) and the phosphonic acid homologue (6b) for the DA reaction was investigated using N-methylmaleimide as the dienophile according to Scheme 3.
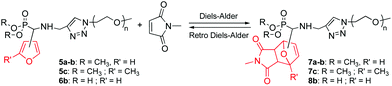 |
| | Scheme 3 DA and rDA reactions between furan-functionalized PEO monomethyl ethers and N-methylmaleimide. | |
When 5a–b were heated in deuterated chloroform (CDCl3) at 40 °C for 5 days with a [N-methylmaleimide]/[5a–b] molar ratio of 1 to 5 (runs 1–4, Table 1), only minimal amounts, up to 13%, could be detected using the typical peaks of the CH–O groups of oxanorbornene at δ = 5.13–5.39 ppm. However, when [N-methylmaleimide]/[5a–b] molar ratio was increased to 20, conversion increased up to 82% (run 6, Table 1). Furthermore, when N,N-dimethylformamide (DMF) was used as the solvent, a near complete conversion to the oxanorbornene adduct was observed from 5b (run 8, Table 1). Moreover, comparative experiments in DMF show the remarkable influence of the furan group according to the position of the substitution (2-substituted one 5avs. 3-substituted one 5b) and the presence of a methyl group (5c, from commercially available 5-methyl-2-furaldehyde). Indeed, the order of reactivity with respect to DA reaction was observed as: 3-substituted furan 5b > 2,5-disubstituted furan 5c > 2-substituted furan 5a (runs 7–9, Table 1). It seems that the furan side chain can have steric effects that may impact the progress of the reaction. In order for the DA reaction to occur, the furan moiety must be able to form a bridgehead; thus the rigidity of 2-substituted furan versus 3-substituted furan may also contribute to reduce the reactivity.33 The positive influence of the methyl group in 5-position of the 2-substituted furan relative to the non-substituted one can be attributed to the electron-donating inductive effect of the methyl group as it is well-known that DA cycloadditions involving electron-rich dienes and electron-poor dienophiles proceed more favorably.34
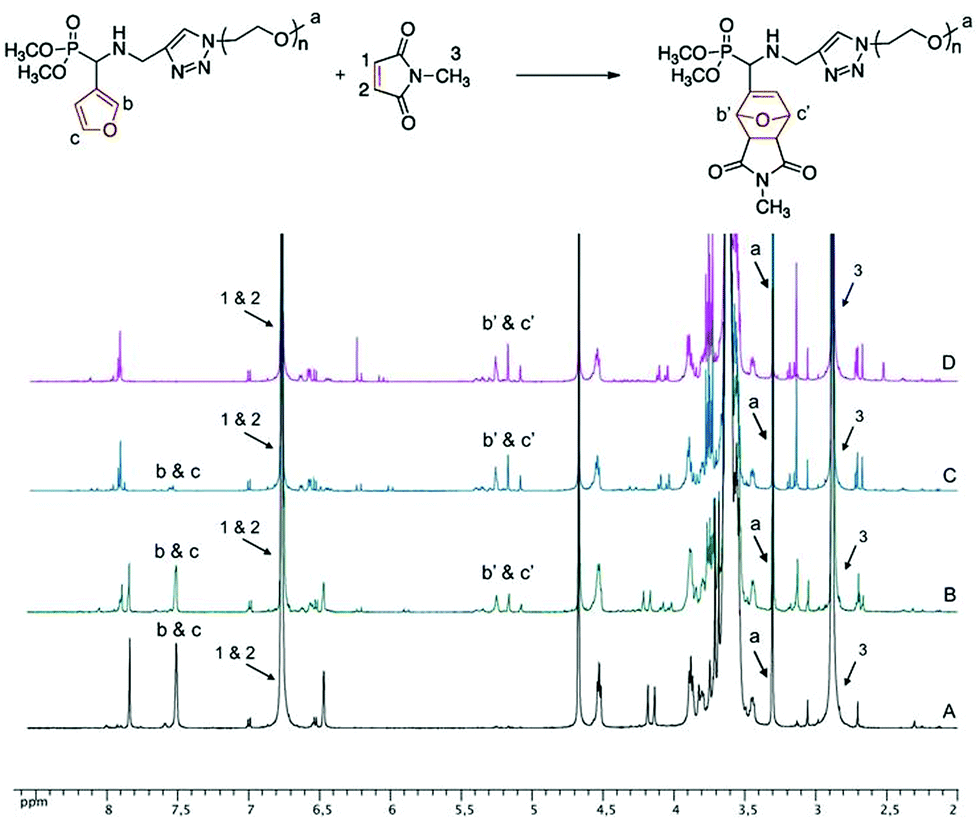
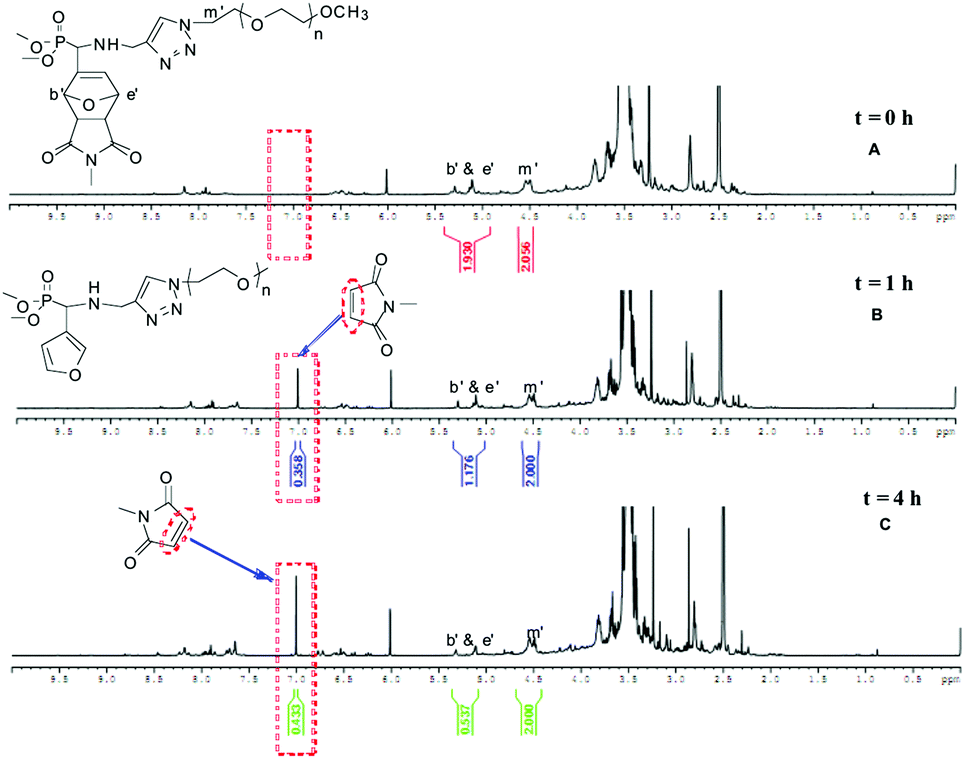
The effect of solvent media on the conversion of the DA reaction has been examined. The reaction between 5b and N-methylmaleimide in deuterated solvents (CDCl3, DMSO-d6, D2O) at 25 and 40 °C has been monitored using 1H NMR spectroscopy. For example, Fig. 1 shows an overlay of 1H NMR spectra of the reaction mixture of 5b and N-methylmaleimide in D2O at 40 °C at different intervals. Progress of the reaction is indicated by the disappearance of the signal of CH groups linked to the oxygen in the furan ring at δ = 7.40–7.60 ppm (labeled b & c in Fig. 1) and the concomitant appearance of the signal of the bridgehead protons of the oxanorbornene cycloadduct at δ = 5.00–5.50 ppm (labeled b′ & c′ in Fig. 1). The conversion of the DA reaction was calculated by comparing the integration areas of the CH3 end-group of PEO at 3.38 ppm (labeled a in Fig. 1) and of CH groups linked to the oxygen in the furan ring at δ = 7.40–7.60 ppm (labeled b & c in Fig. 1).
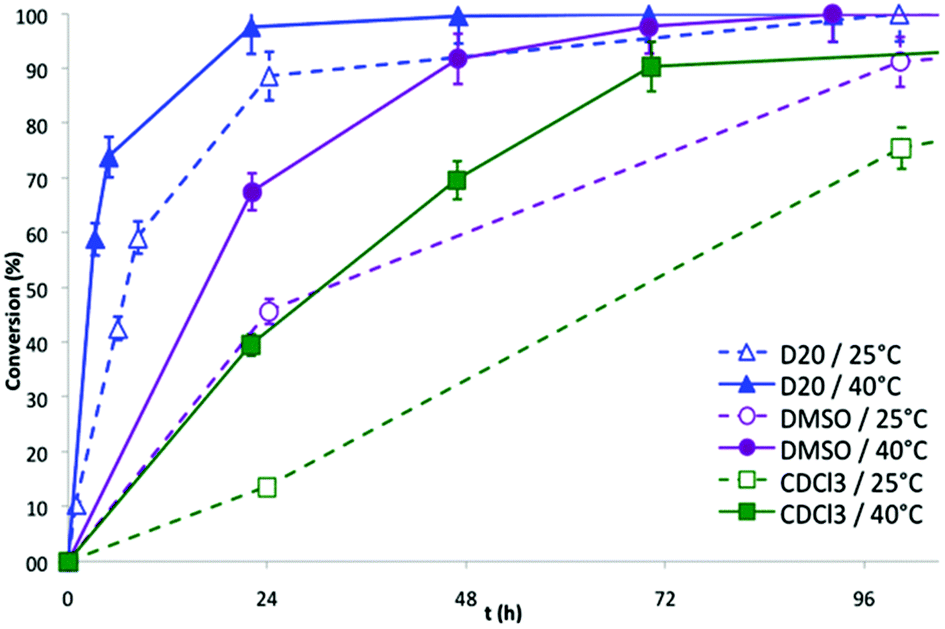
The series of NMR spectra obtained were then used to generate Fig. 2, which shows the conversion of the DA reaction according to the solvent and the temperature versus time. The data suggest that the reaction proceeds more efficiently in water (open and black triangles in Fig. 2, 89% and 98% conversion in 24 h at 25 °C and 40 °C, respectively). Lower yields of the resulting dimethylphosphonate-terminated oxanorbornene-functionalized PEO monomethyl ether 7b were observed when less polar organic solvents, namely DMSO (open and black circles in Fig. 2, 46% and 68% conversion in 24 h at 25 °C and 40 °C, respectively) and chloroform (open and black squares in Fig. 2, 14% and 59% conversion in 24 h at 25 °C and 40 °C, respectively) were used.
The acceleration of the DA reaction in water solution is in accordance with previous studies involving non-polymeric cycloreactants33,35–38 and can been ascribed to enforced hydrophobic interactions between the cycloreactants and hydrogen-bonding interactions between the dipolarophile and the solvent, both stabilizing the transition state.37 The reactivity of the phosphonic acid homologue 6b showed a less pronounced water-induced acceleration as compared to that of 5b (run 3 vs. 4 & run 7 vs. 8, Table 2), probably due to the presence of the strong hydrophilic character of the phosphonic acid moiety near the diene, which restricts the stabilizing hydrophobic interactions with the dienophiles in the transition state.
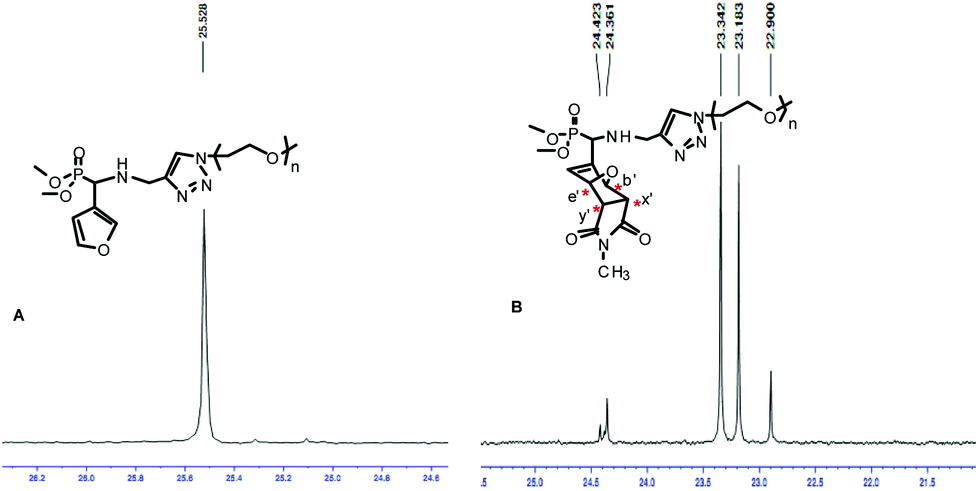
The 31P NMR spectra of the oxanorbornene-functionalized PEO monomethyl ether cycloadducts 7b (Fig. 3) and 8b (Fig. S9B in the ESI†) indicated the presence of four diastereoisomers corresponding to the four possible DA cycloadducts resulting from the endo/exo and facial approaches, in a 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :29:12:6 ratio obtained from the peak integration area of the quantitative 31P NMR spectrum of 7b (Fig. S10 in the ESI†) acquired by a 1D sequence with inverse gated decoupling (zgig sequence that allows quantitative determination of the diastereoisomers ratios by suppression of nuclear overhauser effect). Based on the assumption that this thermal DA reaction is endo-selective (as commonly reported from related non-polymeric reactants) and proceeds with low facial control in respect of the stereogenicity of the chiral (racemic) diene,39 the two major isomers detected on the 31P NMR at δ = 23.34 and 23.18 ppm could be attributed to endo isomers. On the statement of an overall (global) endo selectivity better than 4 to 1, and within the context of dynamic covalent chemistry, it should be noted that the predominant formation of the endo isomers is of particular interest since it has been shown that the rDA of endo DA-adducts most often takes place at 20–30 K lower temperatures than that of the corresponding exo DA-adducts.40
:29:12:6 ratio obtained from the peak integration area of the quantitative 31P NMR spectrum of 7b (Fig. S10 in the ESI†) acquired by a 1D sequence with inverse gated decoupling (zgig sequence that allows quantitative determination of the diastereoisomers ratios by suppression of nuclear overhauser effect). Based on the assumption that this thermal DA reaction is endo-selective (as commonly reported from related non-polymeric reactants) and proceeds with low facial control in respect of the stereogenicity of the chiral (racemic) diene,39 the two major isomers detected on the 31P NMR at δ = 23.34 and 23.18 ppm could be attributed to endo isomers. On the statement of an overall (global) endo selectivity better than 4 to 1, and within the context of dynamic covalent chemistry, it should be noted that the predominant formation of the endo isomers is of particular interest since it has been shown that the rDA of endo DA-adducts most often takes place at 20–30 K lower temperatures than that of the corresponding exo DA-adducts.40
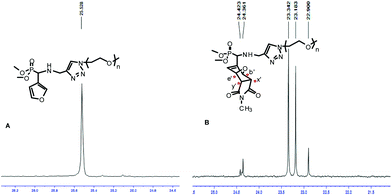 |
| | Fig. 3
1H decoupled 31P NMR spectra of (A) 6b and (B) 7b; solvent: CDCl3. | |
Retro Diels–Alder reaction
DA reactions can be reversed, via the retro-DA (rDA) reaction, typically at temperatures above 120 °C resulting in the original diene and the dienophile.41,42 The feasibility of the rDA reaction was investigated with 7b and 8b in different solvents (Table 3). The rDA reaction was followed by 1H NMR spectroscopy by comparing the peak areas of the CH–O groups of oxanorbornene at δ = 5.13–5.39 ppm and the methylene protons of the PEO in α of the triazole ring at δ = 4.52 ppm. A typical experiment is shown in Fig. 4, where the initial spectrum shows the oxanorbornene cycloadduct 7b in DMSO-d6 (see Fig. S11 in the ESI† for the oxanorbornene cycloadduct 8b). The middle and third spectra show the situation reached at 110 °C after 1 h and 4 h, corresponding to 41% and 73% conversion, respectively. The rDA reaction of the dimethylphosphonate-terminated oxanorbornene-functionalized PEO monomethyl ether 7b proceeded in a quasi-quantitative way at 110 °C within 12 h in both organic solvents DMSO-d6 and deuterated 1,1,2,2-tetrachloroethane (TCE-d2) tested (runs 1 & 2, Table 3), indicating no influence of the polarity of the organic solvent.43 Similar results were obtained with the phosphonic acid-terminated oxanorbornene-functionalized PEO monomethyl ether 8b for the rDA reaction carried out in TCE-d2 (run 5, Table 3). When DMSO-d6 was used as the solvent (runs 3 & 4, Table 3), the reaction had to be performed at 80 °C instead of 110 °C to prevent the degradation of 8b, resulting in a decrease of the conversion, as the temperature is not high enough to shift heavily the equilibrium of the reversible DA reaction to the predominant reversion to the precursors.41 Unlike what is observed with TCE-d2 and DMSO-d6 at 110 °C (runs 3 & 5, Table 3), the reaction carried out in D2O proceeded in 71% conversion at 80 °C after 48 h while preserving the integrity of the polymer (run 6, Table 3), allowing their utilization as controlled delivery systems in aqueous media.
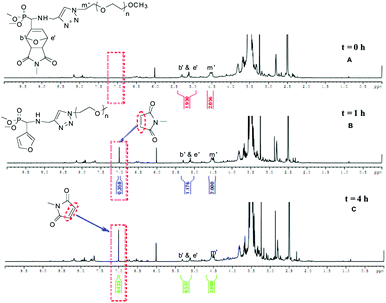 |
| | Fig. 4
1H NMR spectra of the rDA reaction of 7b in DMSO-d6 at 110 °C for (A) t = 0 h, (B) t = 1 h et (C) t = 4 h. | |
Conclusions
The present work investigated the synthesis and the reactivity in the DA/rDA process of a series of novel phosphonated furan-functionalized PEO monomethyl ethers. Various dimethylphosphonate-terminated furan-functionalized PEO monomethyl ethers (5a–c) and their phosphonic acid-terminated homologues (6a–b) have been successfully obtained using a combination of click CuAAC and Kabachnik–Fields reactions, starting from commercially available 2-furaldehyde, 3-furaldehyde, and 5-methyl-2-furaldehyde. Comparative experiments in DMF as the solvent have shown that the substitution of the furan diene greatly affects the reactivity during the DA reaction with N-methylmaleimide, the 3-substituted furan being the more reactive. The influence of the solvent on the DA/rDA process has shown that water facilitates both the DA and the rDA reactions, while maintaining the integrity of the polymer structure. These results have important implications in the area of controlled drug delivery systems and demonstrate that such phosphonated furan-functionalized POE have great potential in dynamic covalent applications, especially as thermolabile coatings for metallic nanoparticles.
Acknowledgements
We thank Emmanuelle Mebold, Patricia Gangnery, Amélie Durand, and Corentin Jacquemmoz for MALDI-TOF mass spectrometry, high resolution mass spectrometry (HR-MS), and 1H nuclear magnetic resonance (NMR) analyses.
Notes and references
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannaki, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- J. A. Funel and S. Abele, Angew. Chem., Int. Ed., 2013, 52, 3822–3863 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- G. K. Such, A. P. R. Johnston, K. Liang and F. Caruso, Prog. Polym. Sci., 2012, 37, 985–1003 CrossRef CAS.
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed.
- N. Zydziak, B. Yameen and C. Barner-Kowollik, Polym. Chem., 2013, 4, 4072–4086 RSC.
- Y.-L. Liu and T.-W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- A. Gandini, Polym. Chem., 2010, 1, 245–251 RSC.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- D. Le, V. Montembault, J.-C. Soutif, M. Rutnakornpituk and L. Fontaine, Macromolecules, 2010, 43, 5611–5617 CrossRef CAS.
- G. Morandi, G. Mantovani, V. Montembault, D. M. Haddleton and L. Fontaine, New J. Chem., 2007, 31, 1826–1829 RSC.
- V. Lapinte, L. Fontaine, V. Montembault, I. Campistron and D. Reyx, J. Mol. Catal. A: Chem., 2002, 190, 117–129 CrossRef CAS.
- M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825–839 CrossRef CAS PubMed.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- M. F. Debets, S. S. Van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. Van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- J. M. Palomo, Eur. J. Org. Chem., 2010, 6303–6314 CrossRef CAS.
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- T. T. T. N'Guyen, H. T. T. Duong, J. Basuki, V. Montembault, S. Pascual, C. Guibert, J. Fresnais, C. Boyer, M. R. Whittaker, T. P. Davis and L. Fontaine, Angew. Chem., Int. Ed., 2013, 52, 14152–14156 CrossRef PubMed.
- T. T. T. N'Guyen, K. Oussadi, V. Montembault and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 415–423 CrossRef.
- M. I. Kabachnik and T. Y. Medved, Dokl. Akad. Nauk SSSR, 1952, 83, 689–692 (
Chem. Abstr.
, 1953
, 47
, 2724b
) CAS.
- E. K. Fields, J. Am. Chem. Soc., 1952, 74, 1528–1531 CrossRef CAS.
- N. S. Zefirov and E. D. Matveeva, ARKIVOC, 2008, 1–17 CAS.
- G. Keglevich and E. Balint, Molecules, 2012, 17, 12821–12835 CrossRef CAS PubMed.
- L. Ménard, L. Fontaine and J.-C. Brosse, React. Polym., 1994, 23, 201–212 CrossRef.
- R. Kakuchi and P. Theato, ACS Macro Lett., 2014, 3, 329–332 CrossRef CAS.
- Y. Zhang, Y. Zhao, B. Yang, C. Zhu, Y. Wei and L. Tao, Polym. Chem., 2014, 5, 1857–1862 RSC.
- N. Illy, G. Couture, R. Auvergne, S. Caillol, G. David and B. Boutevin, RSC Adv., 2014, 4, 24042–24052 RSC.
- C.-O. Turrin, A. Hameau and A.-M. Caminade, Synthesis, 2012, 1628–1630 CrossRef CAS.
- K. C. Koehler, A. Durackova, C. J. Kloxin and C. N. Bowman, AIChE J., 2012, 58, 3545–3552 CrossRef CAS.
- R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994–8002 CrossRef CAS PubMed.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- R. Breslow, U. Maitra and D. Rideout, Tetrahedron Lett., 1983, 24, 1901–1904 CrossRef CAS.
- S. Otto and J. F. B. N. Engberts, Pure Appl. Chem., 2000, 72, 1365–1372 CrossRef CAS , and references cited.
- A. Lubineau and E. Meyer, Tetrahedron, 1988, 44, 6065–6070 CrossRef CAS.
- A. Whiting, Adv. Asymmetric. Synth., 1996, 126–145 CAS.
- J. Canadell, H. Fischer, G. De With and R. A. T. M. Van Bethem, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 43456–43467 CrossRef.
- H. Kwart and K. King, Chem. Rev., 1968, 68, 415–447 CrossRef CAS.
- B. Rickborn, Org. React., 1998, 52, 1–393 CrossRef CAS.
- H.-L. Wei, Z. Yang, H.-J. Chu, J. Zhu, Z.-C. Li and J.-S. Cui, Polymer, 2010, 51, 1694–1702 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and NMR spectra. See DOI: 10.1039/c5py00188a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a