
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly thermally stable hydrogels derived from monolayered two-dimensional supramolecular polymers†
Tian-You
Zhou
a,
Qiao-Yan
Qi
a,
Qiao-Ling
Zhao
a,
Jie
Fu
a,
Yi
Liu
b,
Zhi
Ma
a and
Xin
Zhao
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, China. E-mail: xzhao@mail.sioc.ac.cn
bThe Molecular Foundry, Lawrence Berkeley National Laboratory, One Cyclotron Road, MS 67R6110, Berkeley, CA 94720, USA
First published on 19th February 2015
Abstract
It has been predicted that the properties of materials are dramatically influenced if their structures are confined to two-dimensional (2D) space. A representative example is graphene. However, for synthetic 2D materials, such influences have rarely been demonstrated. In this work, a rare example of how a 2D monolayer structure can impact the properties of bulk materials has been demonstrated by the construction of 2D supramolecular polymers (SPs) and their utilization in the fabrication of hydrogels. Maintaining the intrinsic 2D structures, the as-prepared hydrogels exhibited exceptional thermal stabilities (>180 °C), as revealed by an inversion test and a variable-temperature rheological study. The microstructures and morphologies of the 2D SPs have been extensively characterized by NMR spectroscopy, dynamic light scattering, small-angel X-ray scattering, transmission electron microscopy and atomic force microscopy. Furthermore, molecular dynamic simulations were also performed to shed light on the formation mechanism of the hydrogels.
Introduction
The recent boom in research interest on graphene1 has spurred great research interest on two dimensional (2D) synthetic materials due to the specific atom arrangement, large surface area and extremely high aspect ratio of 2D structures.2 Due to the substantial synthetic challenges, much effort has been devoted towards the controlled synthesis of isolated, layered 2D structures, broadly known as 2D polymers. Several 2D polymers have been constructed via chemical synthesis3 or self-assembly4 over the past two years, some of which possess impressive atomic structural precision within individual layers. Despite these great synthetic advances, the emergent material properties derived from the unique 2D structures of such polymers still remain to be demonstrated.Hydrogels, a family of jelly-like soft materials fabricated from the gelation of water by specific molecules, have been extensively studied in the past few decades because of their widespread applications in biomaterials,5 chemosensors,6 and separation technology.7 A general mechanism for the formation of hydrogels is the immobilization of water molecules in three-dimensional (3D) networks, which are created by the entanglement of linear polymers or nanofibers self-assembled from small molecules. The thermal stabilities of hydrogels are usually quite low, especially for hydrogels fabricated from the self-assembly of small molecules. This prevents them from being used at elevated temperatures.8
In this article, we demonstrate a rare example of how a 2D monolayer structure can impact the properties of bulk materials by the construction of 2D supramolecular polymers (SPs) and their utilization in the formation of hydrogels with exceptional thermal stabilities (Scheme 1). These hydrogels with intrinsic 2D periodical structures exhibit extremely high thermal stabilities. The gel phase and elasticity are maintained even at 180 °C, significantly above the boiling point of water, demonstrating that the thermal stability of a hydrogel could be significantly enhanced by taking advantage of the unique features of a 2D structure. The building blocks for the construction of 2D SPs were designed by incorporating three phenyl-pyridinium (PP) units into the para-positions of a triphenylamine skeleton (1 and 2, Scheme 1). The dimerization of PP units can be considerably enhanced by the encapsulation of two PP units in the cavity of a CB[8] molecule,9 an approach employing the potent cucurbit[8]uril (CB[8])-based host–guest chemistry.10 As revealed by our previous work,9 the two PP units should adopt a head-to-tail arrangement in the cavity of CB[8]. This advantage removes the requirement to introduce steric groups into the skeleton of the monomer, which was necessary for the first example of a 2D SP reported by us recently.4a In that system dimerization of the whole monomer was observed as a result of the head-to-head arrangement of the 4,4′-bipyridin-1-ium (BP) units in the cavity of CB[8] when steric groups were not introduced. DFT calculations revealed that these triphenylamine-based building blocks adopted a C3 symmetric conformation with a coplanar arrangement of the three N-aryl bonds (Fig. S1, ESI†), which could facilitate the formation of extended 2D networks with hexagonal pores.
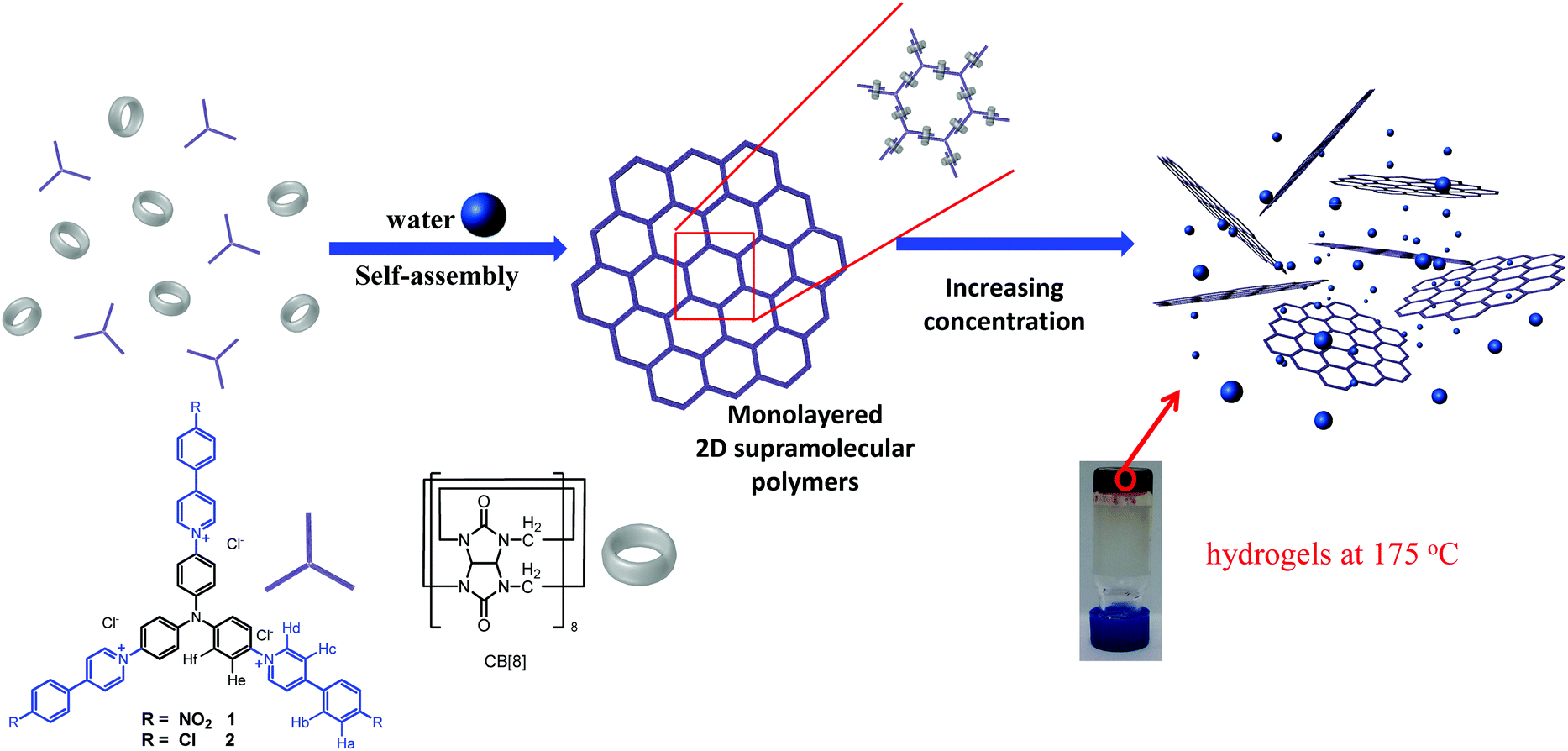 | ||
| Scheme 1 Structures of building blocks 1, 2 and CB[8], and cartoon representations of the formation of 2D SPs and hydrogels. | ||
Results and discussion
Characterization of the assembled structures in solution phase
The binding between building block 1 and CB[8] was first investigated by 1H NMR titration experiments (Fig. 1). Following the addition of CB[8] into a solution of 1 in D2O, the intensities of the protons (Ha–Hd) of the PP units decreased while the signals corresponding to the protons (He and Hf) of the triphenyl amine segment remained almost unchanged. At the same time, a new set of signals grew in the range of 9.1–6.8 ppm. This new set of signals is attributed to the encapsulated PP units in the cavity of CB[8], for which the rate of interchange between encapsulated and non-encapsulated 1 was slower than the NMR time scale.11 The binding stoichiometry between 1 and CB[8] was revealed to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 by a Job's plot (Fig. S2, ESI†). This was also reflected by the 1H NMR titration experiments: no further changes in the signals were observed after the ratio of 1 to CB[8] reached 2:3. The dimerization of the PP units in the cavity of CB[8] was further corroborated by 2D 1H NMR nuclear Overhauser effect spectroscopy (NOESY). Through-space correlations were observed between Ha and Hd, and Hb and Hd (Fig. S3, ESI†), clearly indicating a head-to-tail arrangement of two PP units in the cavity of one CB[8] molecule. Driven by such host–guest interactions, the formation of extended honeycomb-like 2D networks could be expected as a result of the precise direction control offered by the rigid skeleton of the building blocks. Compound 2 exhibited similar complexation behavior as compound 1 when mixed with CB[8] (Fig. S4–S6, ESI†).
:3 by a Job's plot (Fig. S2, ESI†). This was also reflected by the 1H NMR titration experiments: no further changes in the signals were observed after the ratio of 1 to CB[8] reached 2:3. The dimerization of the PP units in the cavity of CB[8] was further corroborated by 2D 1H NMR nuclear Overhauser effect spectroscopy (NOESY). Through-space correlations were observed between Ha and Hd, and Hb and Hd (Fig. S3, ESI†), clearly indicating a head-to-tail arrangement of two PP units in the cavity of one CB[8] molecule. Driven by such host–guest interactions, the formation of extended honeycomb-like 2D networks could be expected as a result of the precise direction control offered by the rigid skeleton of the building blocks. Compound 2 exhibited similar complexation behavior as compound 1 when mixed with CB[8] (Fig. S4–S6, ESI†).
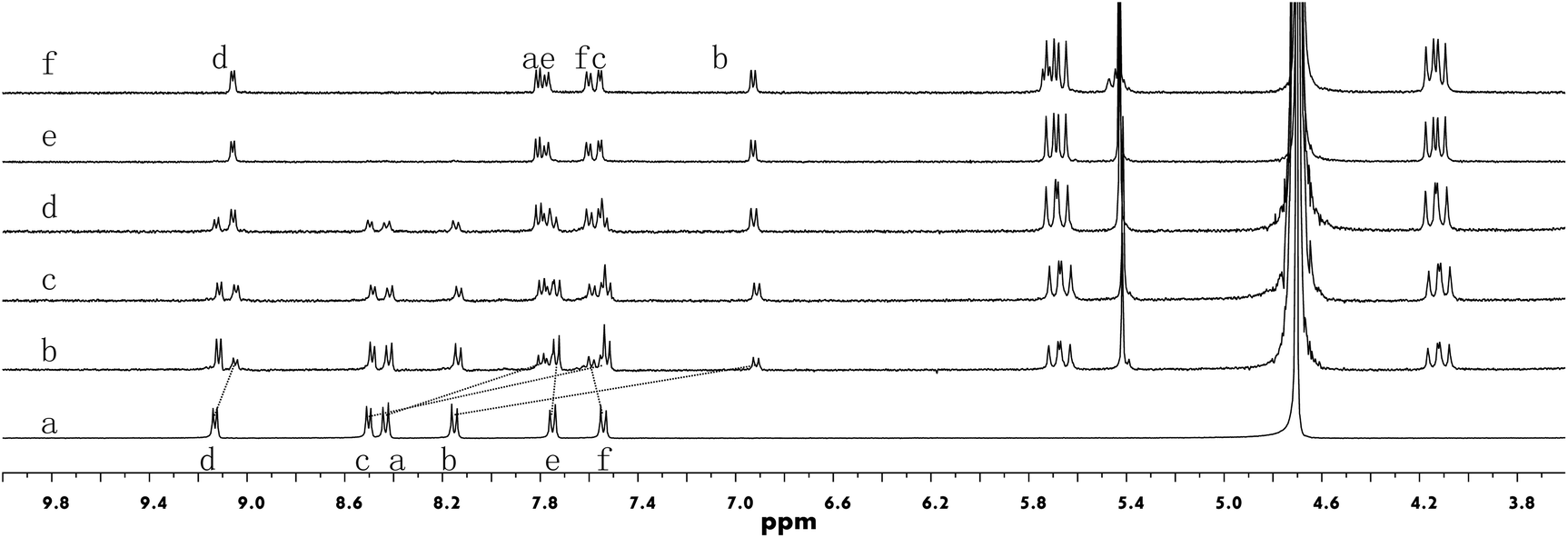 | ||
| Fig. 1 1H NMR spectra (500 MHz) of (a) 1 (0.5 mM), (b) 1 + CB[8] (0.3 equiv.), (c) 1 + CB[8] (0.75 equiv.), (d) 1 + CB[8] (1.1 equiv.), (e) 1 + CB[8] (1.5 equiv.), and (f) 1 + CB[8] (1.8 equiv.) in D2O at 25 °C. | ||
The formation of polymeric structures in solution was proven by dynamic light scattering (DLS). With the increase of concentration, the hydrodynamic diameter (Dh) of the mixture of 1 and CB[8] (2:3) in water increased sharply within the low concentration range (lower than 0.05 mM) and then became much more smooth and finally reached a plateau value around 1100 nm at higher concentrations (Fig. S7, ESI†). The mixture of 2 and CB[8] also gave a similar trend (Fig. S8, ESI†). These results suggest that supramolecular polymers formed even at very low concentrations and reached equilibrium at high concentrations. This is a typical feature of self-assembled systems which are under thermodynamic control.
The molecular weights of the supramolecular polymers were estimated by static light scattering (SLS) experiments. It was also found that the molecular weights increased monotonically with the concentrations of the stoichiometric (2:3) mixtures of [1 + CB[8]] and [2 + CB[8]] (Fig. S9, ESI†). At the highest concentration recorded (0.45 mM for 1 and 2), the molecular weights were estimated to be 1.36 × 106 and 8.5 × 105 g mol−1, corresponding to degrees of polymerization of 462 and 292 for the 2D polymers fabricated from 1 and 2, respectively.
The formation of highly thermally stable hydrogels
When the concentrations of the self-assembled systems were further increased, formation of hydrogels was observed (Fig. S10–11, ESI†). The critical gelation concentrations of the hydrogels were estimated to be ca. 2.0 mM (6.5 wt%) for 1–CB[8] and 2.5 mM (7.8 wt%) for 2–CB[8], respectively. Notably, these hydrogels exhibited extremely high thermal stabilities. The gel state could still be maintained even at 175 °C for the hydrogel fabricated from 1 and CB[8] under an ambient atmosphere in a sealed tube. Furthermore, no hydrogel collapse was observed after the as-prepared hydrogel was sonicated for 10 minutes. The hydrogel fabricated from 2 and CB[8] also exhibited a comparable high thermal stability. The high thermal stabilities of the supramolecular structures in solution were also confirmed by variable-temperature 1H NMR studies. When the temperature was increased from 25 to 75 °C, no observable changes were detected for the chemical shifts and the shape of the signals in the SP spectra, suggesting the 2D polymeric structures were maintained over this temperature range (Fig. S12–13, ESI†). Although 1H NMR experiments at higher temperatures were not conducted on account of pressure buildup in the NMR tube, the SPs are expected to remain stable in solution at temperatures well above 75 °C. In addition, the apparent binding constants (Ka) were determined by fitting the data obtained from UV-vis titration experiments at 25 and 75 °C. Both of the SPs exhibited high binding strengths. At 25 °C, 1–CB[8] had a Ka value of 1.68 (±0.18) × 1012 M−2 and for 2–CB[8] the value was 6.75 (±0.55) × 1012 M−2. As the temperature increased to 75 °C, the Ka value for 1–CB[8] remained almost unchanged (1.37 (±0.05) × 1012 M−2), while the value for 2–CB[8] slightly decreased to 6.08 (±0.85) × 1011 M−2, indicating again the high thermal stabilities of the SPs (Fig. S14, ESI†).In order to get some insights to the properties of the hydrogels, the rheological properties of the hydrogels were first investigated at 25 and 80 °C under an ambient atmosphere.12 At the two temperatures, both hydrogels exhibited higher storage modulus (G′) values than loss modulus (G′′) values over the whole scanning frequency range recorded (Fig. S15, ESI†). This result clearly indicates that these hydrogels exhibited typical elastic properties and the elasticity was maintained at elevated temperatures. Temperature-dependent rheological experiments were further performed to get a deeper insight into the thermal stabilities of the hydrogels. These experiments were carried out in a sealed nitrogen atmosphere under 50 bar pressure to avoid the evaporation of water. The following observations confirmed the high thermal stabilities of both hydrogels: (i) the storage modulus values of the hydrogels were always higher than their loss modulus values over the whole temperature range recorded (30–180 °C); (ii) the values of both the storage modulus and loss modulus of the hydrogels remained nearly constant over the whole temperature range (Fig. 2).
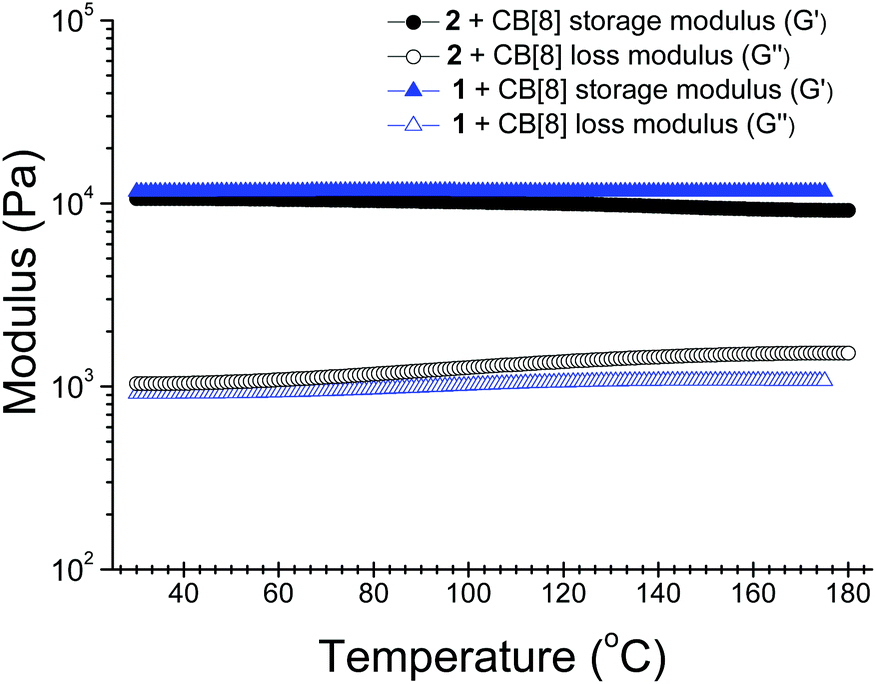 | ||
| Fig. 2 Temperature-dependent storage modulus (G′) and loss modulus (G′′) for the hydrogels fabricated from CB[8] and 1 or 2 (50 bar, 1 Hz; 2 °C min−1). The concentrations used were 16.4 wt% for 1–CB[8] and 15.6 wt% for 2–CB[8]. The molar ratio for CB[8] to 1 or 2 was 2:3. | ||
The morphologies of the xerogels after water was evaporated were examined by scanning electron microscopy (SEM). Layered flakes were observed, consistent with the postulation that the hydrogels were derived from 2D sheets (Fig. S16, ESI†). A polarized optical microscopy (POM) study on the xerogels revealed birefringence textures corresponding to crystalline features (Fig. S17, ESI†), suggestive of well-ordered internal microstructures in these hydrogels. The hydrogel was regenerated when water was introduced into the xerogel fabricated from 1 and CB[8], indicating re-hydration of the dehydrated 2D SP structures.
Evidence for the formation of single-layer 2D sheets
The extremely high thermal stabilities of these hydrogels are attributed to their distinctive formation mechanism, which is proposed to be driven by the interactions between water molecules and the extraordinarily large surface area of the 2D polymeric structures. The mechanism is markedly different from the conventional hydrogels reported previously, which were usually generated through 3D entangling of linear fiber-like structures. In order to investigate the formation of 2D supramolecular polymer layers, a variety of techniques have been used to characterize the structures of the materials formed through the self-assembly of CB[8] and 1 or 2 in water. The morphologies of the self-assembled structures were first characterized by transmission electron microscopy (TEM). As can be seen in Fig. 3a, sheet-like structures can be observed for the sample prepared from [1 + CB[8] (2:3)]. The contrast between the sheets and the TEM substrate (copper grid-supported carbon film) was very low, consistent with the ultra thin thickness of single layer sheets. Larger area sheets with lateral sizes up to several micrometers were also observed at higher concentrations (Fig. 3b). Similar TEM results were also obtained for the sample prepared from [2 + CB[8] (2:3)] (Fig. S18, ESI†). Most notably, honeycomb units with a pore size of around 3.6 nm could be observed by high resolution (HR)-TEM (Fig. 3c). This gives direct evidence for the formation of the proposed 2D network. Since the thicknesses of the sheets could not be estimated from TEM, atomic force microscopy (AFM) was employed to characterize the sheet-like assemblies. Flat and uniform sheets were observed by AFM measurements, with thicknesses of 2.02 and 2.21 nm for the SPs fabricated from 1 and 2 with CB[8], respectively (Fig. 3d and S19, ESI†). These values are very close to the theoretical thicknesses of the expected monolayered 2D sheets determined by the outer diameter of CB[8] (Fig. 3e).13
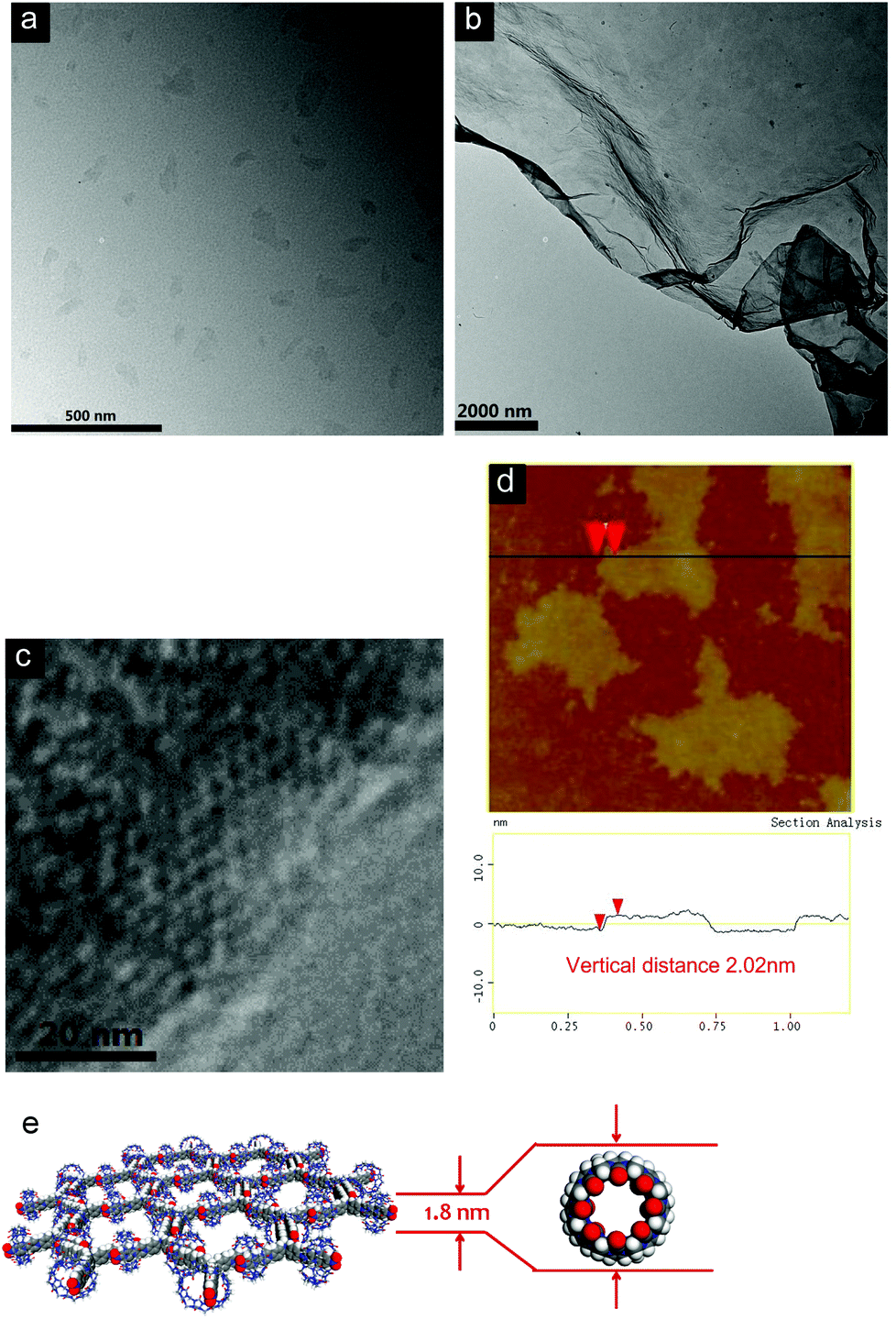 | ||
| Fig. 3 TEM images of samples (a) 1 (0.05 mM) and CB[8] (0.075 mM) and (b) 1 (0.25 mM) and CB[8] (0.375 mM). (c) HR-TEM image of the sheet, indicating its honeycomb-like microstructure. (d) Tapping-mode AFM image and cross-section analysis of the sample of [1 + CB[8] (2:3)]. (e) Theoretical thickness of the monolayered 2D SPs. The concentration was 0.05 mM for 1. | ||
Evidence for long-range ordered 2D periodicity in the 2D SPs
A key criterion for a true 2D polymer is the existence of long-range ordered periodicity in two dimensions. In order to prove that the sheets generated by the self-assembly of CB[8] and 1 or 2 did hold periodic 2D polymeric structures, small angle X-ray scattering (SAXS) experiments were carried out. For the sample fabricated from 1 and CB[8],14 a scattering peak corresponding to a d spacing of 3.61 nm (100) was observed (Fig. 4a). Moreover, a (200) scattering peak was also observed at a d spacing of 1.81 nm, further confirming the existence of a periodic size of 3.61 nm. In the case of the sample prepared from 2 and CB[8], scattering peaks corresponding to (100) and (200) were observed at 3.49 and 1.73 nm, respectively (Fig. 4c). Semiempirical calculations at PM3 level revealed that the theoretical pore diameter of the expected honeycomb structure is 3.60 nm for both of the SPs fabricated from the co-assembly of CB[8] with 1 or 2. The experimentally obtained values agree well with the theoretical ones, corroborating the formation of the expected 2D networks with well-ordered internal microstructures. These scattering peaks could still be observed when the samples were heated to 85 °C (Fig. 4b and d), suggesting that the 2D networks remained intact at elevated temperatures. This again indicates the high thermal stabilities of these 2D supramolecular polymers.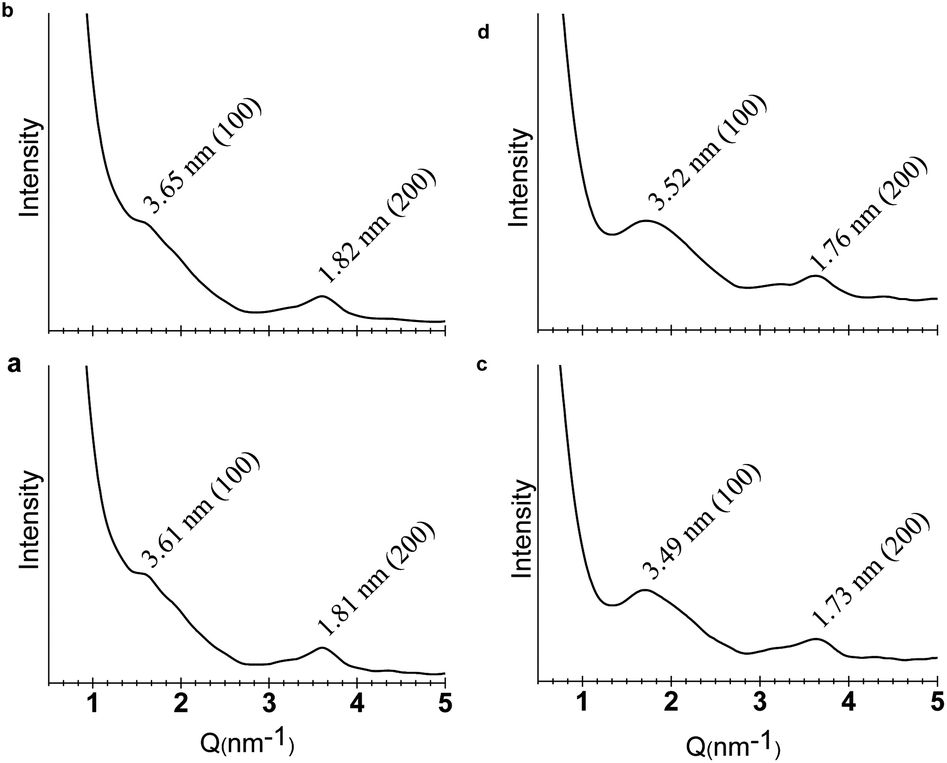 | ||
| Fig. 4 SAXS patterns of the sample prepared from a hydrogel of 1 and CB[8] (13.0 wt%) in water (pH = 7) at (a) 25 °C and (b) 85 °C. SAXS patterns of the sample prepared from a hydrogel of 2 and CB[8] (9.36 wt%) in water (pH = 7) at (c) 25 °C and (d) 85 °C. | ||
In order to shed light on the formation mechanism of the hydrogels, molecular dynamic simulations were performed for the hydrogel formed from 1 and CB[8] (Fig. 5, see ESI for more details†). A snapshot of a (4 × 4) segment of the 2D layer revealed that water molecules were mainly distributed near the skeleton of the honeycomb structure. In contrast, the pore centers had the lowest distribution density of water molecules. This result suggests that the 2D supramolecular polymers have a high affinity for water molecules, which may be ascribed to the formation of hydrogen bonds between the O and N atoms of CB[8] and the NO2 groups of the PP units (for building block 1). Through these hydrogen-bonding interactions, a huge number of water molecules could be localized on the surfaces of the monolayered sheets, which further extended to produce hydrogels. In this context, the high thermal stability of the hydrogel could be attributed to the extraordinarily large surface areas of the 2D structures, which provide a maximum number of interaction sites for the formation of hydrogen bonds. Compared to 2–CB[8], 1–CB[8] exhibited a lower critical gelation concentration and higher molecular weights and storage modulus. This could be attributed to the additional hydrogen-bonding sites provided by the nitro group of compound 1.
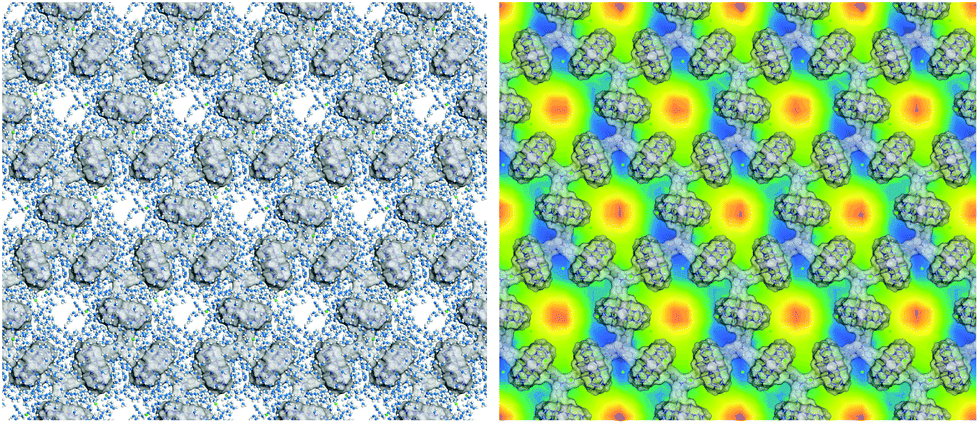 | ||
| Fig. 5 Left: a snapshot of the simulated hydrogel formed from 1 and CB[8]. The skeleton is illustrated by a Connolly surface for clarity. The oxygen atoms of H2O are represented by the blue-colored particles, the hydrogen atoms are represented by the gray-colored particles and Cl− ions are represented by the green-colored particles. Right: the statistical slice of the density field of H2O to display the water distribution on the 2D structure. The blue color shows the relatively high density of H2O and the red color shows the relatively low density of H2O. This indicates that the water molecules are more distributed near the skeleton of the 2D SP. | ||
Conclusions
In conclusion, the significant impact of a unique 2D structure on the performance of soft materials has been demonstrated. Different from traditional hydrogels, which are usually formed by the 3D entanglement of flexible linear structures, the hydrogels reported here were generated from monolayered 2D supramolecular polymers without any flexible side chains. Thanks to the extraordinarily large surface areas of the 2D structures, a maximum number of interaction sites is provided to immobilize water molecules on the surfaces and thus the thermal stabilities of the hydrogels are enhanced significantly. Currently the research on synthetic 2D polymers is still at a very early stage. Much attention has been paid to the construction and characterization of 2D structures, while their properties and applications are awaiting further exploration. The highly thermostable hydrogels demonstrated in this work reveal that the unique features of 2D structures could be essential for the fabrication of unconventional functional materials. As this work takes advantage of only one feature of 2D structures, i.e. the large surface area, we believe that the other features of this type of structure will also endow 2D polymer-based materials with unique properties and functions. For example, the long-range ordered periodicity in 2D space could be exploited for the construction of well-ordered arrays of sensors to achieve ultrahigh sensitivity. This is currently under investigation in our laboratory.Acknowledgements
We thank the National Natural Science Foundation of China (nos. 21172249 and 91127007) for financial support. Y.L. acknowledges the support from the Molecular Foundry, Lawrence Berkeley National Laboratory, supported by the Office of Science, Office of Basic Energy Sciences, Scientific User Facilities Division, of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231. We also thank Prof. Zhan-Ting Li and Mr Jia Tian (Fudan University) for their help in preparing the manuscript and Prof. Li-Zhu Wu (Technical Institute of Physics and Chemistry) for her helpful advice.Notes and references
- (a) K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed; (b) K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef CAS PubMed; (c) M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS PubMed.
- (a) J. Sakamoto, J. van Heijst, O. Lukin and A. D. Schlüter, Angew. Chem., Int. Ed., 2009, 48, 1030 CrossRef CAS PubMed; (b) R. Mas-Ballesté, C. Gómez-Navarro, J. Gómez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20 RSC.
- (a) T.-Y. Zhou, F. Lin, Z.-T. Li and X. Zhao, Macromolecules, 2013, 46, 7745 CrossRef CAS; (b) P. Kissel, R. Erni, W. B. Schweizer, M. D. Rossell, B. T. King, T. Bauer, S. Gotzinger, A. D. Schlüter and J. Sakamoto, Nat. Chem., 2012, 4, 287 CrossRef CAS PubMed; (c) P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774 CrossRef CAS PubMed; (d) M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779 CrossRef CAS PubMed; (e) K. Baek, G. Yun, Y. Kim, D. Kim, R. Hota, I. Hwang, D. Xu, Y. H. Ko, G. H. Gu, J. H. Suh, C. G. Park, B. J. Sung and K. Kim, J. Am. Chem. Soc., 2013, 135, 6523 CrossRef CAS PubMed.
- (a) K.-D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C.-H. Sue, T.-Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z.-T. Li, J. Am. Chem. Soc., 2013, 135, 17913 CrossRef CAS PubMed; (b) L. Zhang, T.-Y. Zhou, J. Tian, H. Wang, D.-W. Zhang, X. Zhao and Z.-T. Li, Polym. Chem., 2014, 5, 4715 RSC; (c) X. Zhang, C.-B. Nie, T.-Y. Zhou, Q.-Y. Qi, J. Fu, X.-Z. Wang, L. Dai, Y. Chen and X. Zhao, Polym. Chem., 2015, 6, 1923 RSC.
- (a) O. P. Oommen, S. Wang, M. Kisiel, M. Sloff, J. Hilborn and O. P. Varghese, Adv. Funct. Mater., 2013, 23, 1273 CrossRef CAS; (b) N. Annabi, K. Tsang, S. M. Mithieux, M. Nikkhah, A. Ameri, A. Khademhosseini and A. S. Weiss, Adv. Funct. Mater., 2013, 23, 4950 CrossRef CAS PubMed; (c) Y. Liu and M. B. Chan-Park, Biomaterials, 2009, 30, 196 CrossRef PubMed.
- (a) D. Zhai, B. Liu, Y. Shi, L. Pan, Y. Wang, W. Li, R. Zhang and G. Yu, ACS Nano, 2013, 7, 3540 CrossRef CAS PubMed; (b) D. Pussak, D. Ponader, S. Mosca, S. V. Ruiz, L. Hartmann and S. Schmidt, Angew. Chem., Int. Ed., 2013, 52, 6084 CrossRef CAS PubMed; (c) S. Bhowmik, B. N. Ghosh, V. Marjomaki and K. Rissanen, J. Am. Chem. Soc., 2014, 136, 5543 CrossRef CAS PubMed.
- (a) Y. Liu, S. Yu, H. Wu, Y. Li, S. Wang, Z. Tian and Z. Jiang, J. Membr. Sci., 2014, 469, 198 CrossRef CAS PubMed; (b) F. Lu, Y. Chen, N. Liu, Y. Cao, L. Xu, Y. Wei and L. Feng, RSC Adv., 2014, 4, 32544 RSC.
- (a) M. Guo, L. Pitet, H. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969 CrossRef CAS PubMed; (b) Z. Lv, L. Chang, X. Long, J. Liu, Y. Xiang, J. Liu, J. Liu, H. Deng, L. Deng and A. Dong, Carbohydr. Polym., 2014, 108, 26 CrossRef CAS PubMed; (c) F. Lv, L. Mao and T. Liu, Mater. Sci. Eng., C, 2014, 43, 221 CrossRef CAS PubMed; (d) G. Azevedo, B. Domingues, H. Abreu, I. Sousa-Pinto, G. Feio and L. Hilliou, Carbohydr. Polym., 2014, 108, 140 Search PubMed.
- Y. Zhang, T.-Y. Zhou, K.-D. Zhang, J.-L. Dai, Y.-Y. Zhu and X. Zhao, Chem. – Asian. J., 2014, 9, 1530 CrossRef CAS PubMed.
- (a) C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574 CrossRef CAS PubMed; (b) Y. H. Ko, Y. Kim, H. Kim and K. Kim, Chem. – Asian. J., 2011, 6, 652 CrossRef CAS PubMed; (c) Z.-J. Ding, H.-Y. Zhang, L.-H. Wang, F. Ding and Y. Liu, Org. Lett., 2011, 13, 856 CrossRef CAS PubMed; (d) Q. Wang, Y. Chen and Y. Liu, Polym. Chem., 2013, 4, 4192 RSC; (e) W. S. Jeon, H. J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828 RSC; (f) A. Y. Ziganshina, Y. H. Ko, W. S. Jeon and K. Kim, Chem. Commun., 2004, 806 RSC; (g) Y. Lan, Y. Wu, A. Karas and O. A. Scherman, Angew. Chem., Int. Ed., 2014, 53, 2166 CrossRef CAS PubMed; (h) Z. Huang, L. Yang, Y. Liu, Z. Wang, O. A. Scherman and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5351 CrossRef CAS PubMed; (i) J. R. McKee, E. A. Appel, J. Seitsonen, E. Kontturi, O. A. Scherman and O. Ikkala, Adv. Funct. Mater., 2014, 24, 2706 CrossRef CAS; (j) F. Biedermann, I. Ross and O. A. Scherman, Polym. Chem., 2014, 5, 5375 RSC; (k) J. del Barrio, P. N. Horton, D. Lairez, G. O. Lloyd, C. Toprakcioglu and O. A. Scherman, J. Am. Chem. Soc., 2013, 135, 11760 CrossRef CAS PubMed; (l) F. Biedermann, M. Vendruscolo, O. A. Scherman, A. De Simone and W. M. Nau, J. Am. Chem. Soc., 2013, 135, 14879 CrossRef CAS PubMed; (m) H. Qian, D.-S. Guo and Y. Liu, Chem. – Eur. J., 2012, 18, 5087 CrossRef CAS PubMed; (n) C. Yang, C. Ke, W. T. Liang, G. Fukuhara, T. Mori, Y. Liu and Y. Inoue, J. Am. Chem. Soc., 2011, 133, 13786 CrossRef CAS PubMed; (o) H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Acc. Chem. Res., 2014, 47, 2106 CrossRef CAS PubMed; (p) H. Yang, Z. Ma, Z. Wang and X. Zhang, Polym. Chem., 2014, 5, 1471 RSC; (q) F. Lin, T.-G. Zhan, T.-Y. Zhou, K.-D. Zhang, G.-Y. Li, J. Wu and X. Zhao, Chem. Commun., 2014, 50, 7982 RSC.
- Z.-J. Zhang, H.-Y. Zhang, L. Chen and Y. Liu, J. Org. Chem., 2011, 76, 8270 CrossRef CAS PubMed.
- (a) G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697 RSC; (b) X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, Adv. Mater., 2012, 24, 362 CrossRef CAS PubMed; (c) J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686 RSC.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267 RSC.
- In order to inject the samples into the sample tube of SAXS, partially gelated samples were prepared for SAXS experiment by dispersing the as-prepared hydrogels in water.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterizations of compounds, 1H NMR 2D NOESY spectra, variable-temperature 1H NMR spectra, SEM images, POM images, pictures of the hydrogels, additional rheological experiments, 1H NMR titration spectra and TEM images. See DOI: 10.1039/c5py00072f |
| This journal is © The Royal Society of Chemistry 2015 |