
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of cyclic polymers
Rebecca J.
Williams
,
Andrew P.
Dove
* and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: a.p.dove@warwick.ac.uk; r.k.o-reilly@warwick.ac.uk
First published on 11th March 2015
Abstract
The self-assembly of block copolymers in solution is an expansive area of research as a consequence of the significant potential the resulting soft nanostructures possess in numerous applications (e.g. drug delivery, imaging and catalysis), as well as our desire to mimic nature's nanostructures (e.g. viruses and proteins). Of the various factors that affect self-assembly behaviour, the effect of polymer architecture is relatively unexplored despite the successful synthesis of a range of non-linear amphiphilic polymers. Indeed, recent synthetic breakthroughs have allowed the preparation of well-defined, high purity amphiphilic cyclic polymers and as a result the self-assembly of cyclic polymers is an area of increasing interest. This review will discuss the self-assembly of cyclic block copolymers, in addition to more complex cyclic architectures, as well as providing a comparison to the self-assembly of equivalent linear systems to elucidate the effect of cyclization on self-assembly.
 Andrew P. Dove, Rebecca J. Williams, Rachel K. O'Reilly | Rebecca Williams graduated with a MChem degree from the University of Warwick in 2010 having undertaken research projects in the laboratories of Dr Ross A. Hatton and Prof. Rachel K. O'Reilly. She subsequently completed her PhD under the joint supervision of Prof. Rachel K. O'Reilly and Prof. Andrew P. Dove investigating the synthesis and self-assembly of functional degradable poly(carbonate)s. She is currently a postdoctoral researcher in the O'Reilly group studying the effects of polymer architecture on self-assembly. Rachel O'Reilly is currently a professor and an ERC consolidator grant awardee in the Department of Chemistry at the University of Warwick. She started an independent career in 2005 with the award of a Royal Society Dorothy Hodgkin Fellowship at the University of Cambridge. In 2009 she moved to Warwick with the award of an EPSRC career acceleration fellowship and in 2012 was promoted to full professor. In 2012 she won the IUPAC-Samsung young polymer scientist award and the RSC Hickinbottom medal. In 2013 she was awarded the ACS Mark Young Scholar award and was elected as an ACS POLY fellow. Her research focuses on bridging the interface between creative synthetic, polymer and catalysis chemistry, to allow for the development of materials that are of significant importance in medical, materials and nanoscience applications. Andrew Dove is currently a full professor in the Department of Chemistry at the University of Warwick. Andrew graduated from the University of York with a MChem degree in 1999. His subsequent PhD studies were conducted under the supervision of Prof. Vernon C. Gibson FRS at Imperial College, London, focused on metal catalysed co-ordination insertion polymerisation. Andrew undertook postdoctoral research first under the guidance of Prof. Robert M. Waymouth at Stanford University, California and then as a CIPMA postdoctoral fellow at IBM, San Jose, California under the supervision of Dr James L. Hedrick and Prof. Robert M. Waymouth. Andrew returned to the UK to take up a RCUK Fellowship in Nanotechnology in September 2005 before being appointed as an Assistant Professor in September 2006, an Associate Professor in September 2009 and subsequently as a Full Professor in June 2014. Andrew was awarded the Macro Group UK Young Researcher Medal in 2009 and the RSC Gibson-Fawcett Award in 2014. |
Introduction
The self-assembly of amphiphilic molecules is fundamental in nature and everyday life; the membranes of living cells are comprised of self-assembled phospholipids and countless consumer products contain self-assembled surfactants acting as detergents, emulsifiers and foaming agents. Consequently, the solution self-assembly of amphiphilic molecules is an area of significant research.1–5 A small molecule amphiphile consists of a hydrophobic tail and a hydrophilic head group. Amphiphiles self-assemble in selective solvents to minimize unfavourable hydrophobic–hydrophilic interactions and the resulting morphology of the self-assembly is determined by the packing parameter, p = ν/aolc, where ν is the volume of the hydrophobic tail, ao is the contact area of the hydrophilic head group and lc is the length of the hydrophobic tail. Spherical micelles are favoured when p < 1/3, cylindrical micelles are favoured when 1/3 < p < 1/2 and vesicles when 1/2 < p < 1.6The development of living and controlled polymerization techniques has allowed the preparation of well-defined amphiphilic polymers that will undergo analogous self-assembly in a selective solvent to yield well-defined aggregates. These self-assembled polymeric aggregates exhibit greater stability than small molecule aggregates as a result of their superior mechanical and physical properties7 and consequently polymeric self-assemblies have been utilized as catalytic nanoreactors,8–10 drug delivery vehicles11–18 and molecular imaging agents.19,20 Among the possible architectures of amphiphilic polymers, linear block copolymer systems are by far the most studied and as a result the self-assembly of linear block copolymers is well established and has been extensively reviewed.11,18,21–24
A wide range of aggregate morphologies are accessible via the self-assembly of linear block copolymers,25 including spherical and cylindrical micelles, vesicles and lamellae, as well as morphologies that possess greater complexity, such as multi-compartment micelles,26,27 helical micelles28 and multi-lamellar “onion” vesicles.29 The resulting morphology of a block copolymer aggregate is determined by three factors which govern the free energy of the system: the degree of stretching of the core forming block, the interfacial tension between the core and the solvated corona and the repulsive interactions of the corona chains.11,21 Consequently, the morphology of polymeric assemblies can be influenced by a wide range of variables that affect these three factors, including polymer composition, concentration, water content, assembly technique and the presence of additives. Furthermore, block copolymer assemblies may be defined as either thermodynamically stable or kinetically frozen, depending on the mobility of the constituent polymer chains with respect to unimer exchange and the employed method of assembly.30,31
Polymer architecture is also known to influence self-assembly behaviour, however, in contrast to the self-assembly of linear block copolymers, reports of the self-assembly of non-linear amphiphilic polymers are limited.32–39 The major advancement of controlled and living polymerization techniques has enabled the preparation of a range of well-defined non-linear polymer architectures, including star,33,40–42 graft,43–47 branched48–51 and cyclic.52–56 Of these architectures, cyclic polymers are perhaps the least explored as a consequence of the difficulties encountered during both their synthesis and purification. However, despite these difficulties cyclic polymers are of significant interest as a result of the unique properties they exhibit in comparison to analogous linear polymers.54,57
Interest in the solution self-assembly of cyclic polymers began in the mid 1990s,58 not long after initial investigations into the solution self-assembly of linear block copolymers. However, as a consequence of the synthetic difficulties encountered in the preparation of well-defined, high purity cyclic polymers, this area of research remained comparatively limited. Recent developments in the preparation of cyclic polymers55 have allowed these synthetic difficulties to be overcome and the self-assembly of cyclic polymers has received increasing attention. This review will begin with a brief introduction to the synthesis of cyclic polymers and the unique properties that cyclic polymers exhibit in comparison to their linear counterparts. An overview of the limited but growing field of cyclic polymer self-assembly will follow in an attempt to elucidate the effect of cyclization on polymer aggregation. This overview will discuss the assembly of amphiphilic cyclic block copolymers, in addition to more complex amphiphilic cyclic architectures, highlighting topological differences observed in comparison to the self-assembly of equivalent linear systems where appropriate.
Synthesis of cyclic polymers
The existence of cyclic polymers has been long established in nature with the discovery of circular DNA59 and in synthetic chemistry as cyclic contaminants in step-growth polymerizations. Indeed, original synthetic methods to prepare cyclic polymers were based upon the ring-chain equilibrium of poly(dimethylsiloxane),60,61 where cyclic species were separated from linear polymers through laborious fractional precipitations and preparative size exclusion chromatography (SEC).62 Despite the obvious limitations of this method, it allowed the first investigation of cyclic polymer properties, verifying the unique behaviour of cyclic topologies.Recent synthetic breakthroughs have since allowed the preparation of well-defined cyclic polymers in the absence of linear impurities, as well as a diverse range of more complex cyclic architectures. There are now two main approaches to prepare cyclic polymers; ring-closure63–65 and ring-expansion66 (Fig. 1). The synthesis of cyclic polymers has been extensively reviewed,52–55,67 therefore here only an overview of these techniques will be given.
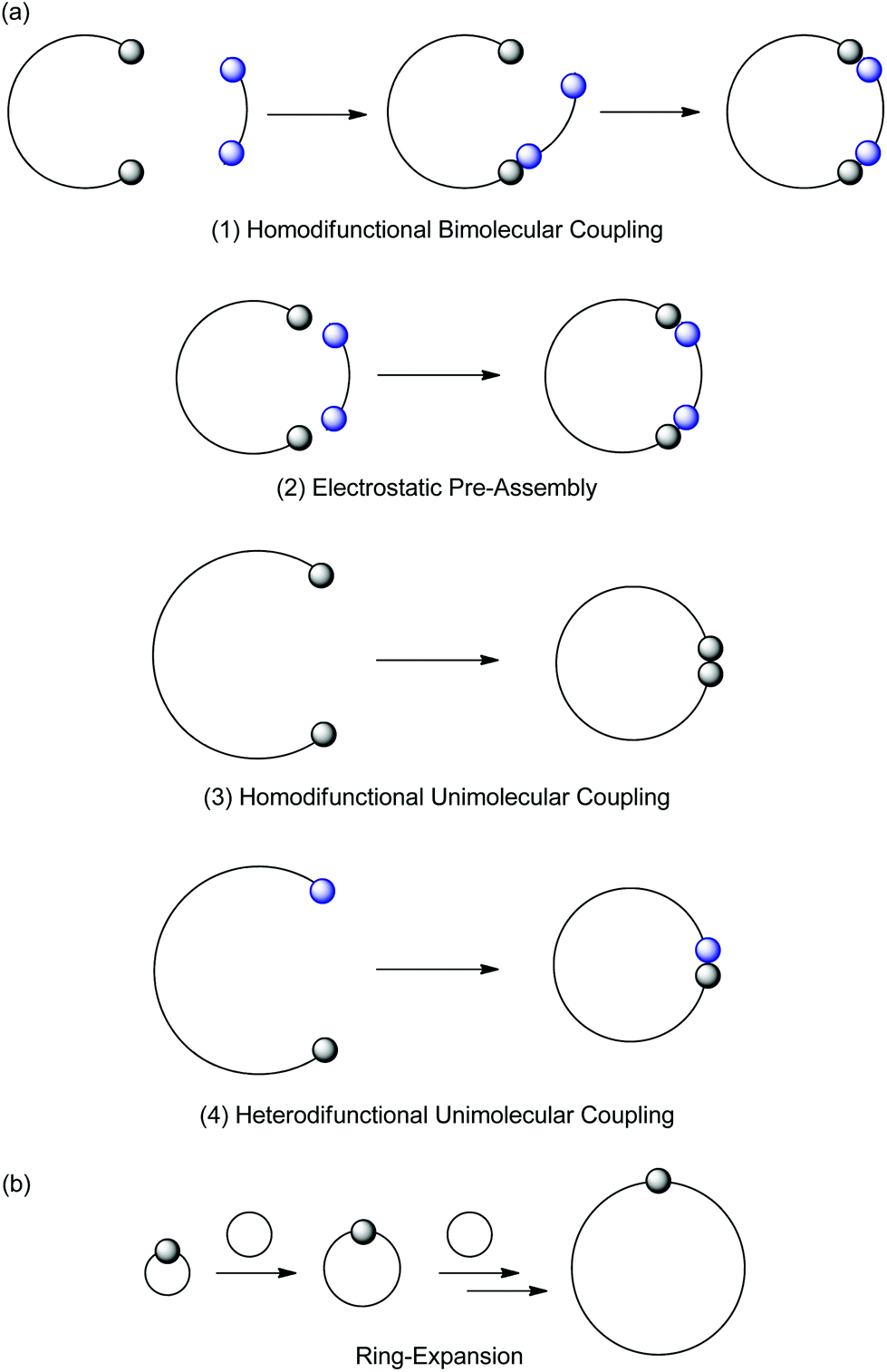 | ||
| Fig. 1 Synthesis of cyclic polymers via ring-closure (a) and ring-expansion techniques (b). | ||
Ring-closure techniques involve the coupling of the reactive chain ends of a linear polymer to yield a cyclic polymer (Fig. 1(a)). Ring-closure can be achieved through the bimolecular homodifunctional coupling of a linear polymer with a small molecule linker (Fig. 1(a)-1 and (a)-2) or the unimolecular homodifunctional (Fig. 1(a)-3) or heterodifunctional coupling of a linear polymer (Fig. 1(a)-4). The development of living and controlled polymerization techniques has allowed the preparation of polymers with high chain end functionality, enabling the success of these techniques. Furthermore, in all ring-closure techniques, the use of highly efficient coupling reactions is crucial to ensure high purity cyclic polymers.
In a bimolecular ring-closure strategy (Fig. 1(a)-1), the linear polymer first undergoes an intermolecular reaction with the small molecule linker, forming an intermediate species which then undergoes intramolecular cyclization. Reactions are performed at high dilution or pseudo-high dilution to avoid reaction of the intermediate species with another polymer chain, however the concentration of reactants must be sufficiently high for the first step of this method to be effective. Furthermore, precise 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the linear polymer and small molecule linker is needed. To overcome the limitations of bimolecular ring-closure, electrostatic interactions between the linear polymer chain ends and small molecule linker can be used to template cyclization (Fig. 1(a)-2).68 In contrast, for unimolecular ring-closure techniques (Fig. 1(a)-3 and (a)-4), high dilution alone is required to suppress polymer–polymer coupling side reactions, as such this method has been highly successful in the preparation of well-defined cyclic polymers and is generally favoured over bimolecular ring-closure.
:1 stoichiometry of the linear polymer and small molecule linker is needed. To overcome the limitations of bimolecular ring-closure, electrostatic interactions between the linear polymer chain ends and small molecule linker can be used to template cyclization (Fig. 1(a)-2).68 In contrast, for unimolecular ring-closure techniques (Fig. 1(a)-3 and (a)-4), high dilution alone is required to suppress polymer–polymer coupling side reactions, as such this method has been highly successful in the preparation of well-defined cyclic polymers and is generally favoured over bimolecular ring-closure.
Ring-expansion techniques involve the successive insertion of a cyclic monomer into a cyclic catalyst,69 initiator66 or propagating species70 (Fig. 1(b)). Ring-expansion techniques do not require high dilution and therefore afford cyclic polymers in considerably higher yields than ring-closure techniques. However, careful catalyst design is required to ensure the formation of high molecular weight cyclic polymers with low dispersities and to ensure elimination of the catalyst from the final polymer.
The advances made in cyclic polymer synthesis, controlled polymerization techniques and highly efficient “click” coupling reactions,71 have also allowed for the preparation of a diverse range of cyclic polymer topologies including tadpole,72 jellyfish,73 sun-shaped,74 theta-shaped,75 figure-of-eight76 and other multi-cyclic topologies.68
Topological effects
Cyclic polymers possess many unique physical properties in comparison to their linear polymer analogues in both solution and bulk.52,54,56,57 These differences provide opportunities for exploitation in many applications, as well as increasing our fundamental understanding of structure–property relationships. Cyclic polymers possess smaller hydrodynamic volumes77,78 and radii of gyration79,80 in comparison to their linear counterparts as a consequence of the more confined conformation of cyclic polymer chains. This difference has been exploited in the characterization of cyclic polymers by SEC analysis, where cyclic polymers exhibit longer retention times and therefore lower apparent molecular weights than the equivalent linear polymers of the same molecular weight.As a consequence of their smaller hydrodynamic volume and lack of chain ends, cyclic polymers exhibit significantly higher critical entanglement molecular weights than analogous linear polymers. Similarly, the solution viscosities and melt viscosities of cyclic polymers are lower than the equivalent linear polymers.79,81 Interestingly the melt viscosities of blends of cyclic and linear polymers are higher than either component, as a consequence of the threading of linear chains through cyclic polymer chains.82
The glass transition temperatures (Tg) of analogous linear and cyclic polymers exhibit very different trends.83,84 For low and medium molecular weight polymers (101–103 kDa), cyclic polymers exhibit higher Tg values than their linear counterparts as a consequence of the different mobilities of cyclic and linear polymers. Cyclic polymers are inherently less mobile than linear polymers because of their confined nature, smaller volumes and lack of chain ends. For high molecular weight polymers (>103 kDa), the effect of end-groups becomes negligible and cyclic and linear polymers possess the same value of Tg. Furthermore, because of a lack of polymer chain ends, cyclic polymers show very little dependence of Tg on molecular weight and exhibit values of Tg similar to those of high molecular weight linear polymer (Tg(∞)). The melting transition temperatures (Tm) of cyclic and linear polymers exhibit similar trends in behaviour.
Cyclic and linear polymers are known to exhibit different modes of diffusion.85 The accepted mode of diffusion for linear polymers is a reptation mechanism;86,87 this process is governed by the mobility of the polymer chain ends. As cyclic polymers do not possess chain ends, diffusion must occur by a different mechanism, however this precise mode of diffusion has yet to be elucidated.
The unique structural and physical properties of cyclic polymers have been exploited in a variety of applications. Hawker and coworkers recently reported the use of cyclic diblock copolymers to prepare thin films for lithography applications, where the reduced volume of the cyclic polymer allowed a 30% decrease in domain spacing, compared to the corresponding linear diblock copolymer (Fig. 2).88 Zhang et al. prepared cyclic polymer based gels via a combination of ring-opening metathesis polymerization and thiol–ene chemistry. The gels prepared from cyclic polymers were found to exhibit markedly different swelling and mechanical properties in comparison to the equivalent gels comprised of linear polymers.89 In addition, Szoka and coworkers reported that cyclic polymers exhibit longer in vivo circulation times and higher tumour accumulation compared to linear analogues.90
 | ||
| Fig. 2 AFM height images of cyclic and linear PS-b-PEO thin films (scale bar = 250 nm). Reprinted with permission from ref. 88. Copyright 2012 American Chemical Society. | ||
Self-assembly of cyclic block copolymers
The earliest reported investigations into the effect of polymer cyclization on self-assembly were undertaken by Booth and coworkers, studying cyclic diblock copolymers comprised of either poly(ethylene oxide)-b-poly(butylene oxide) (cyclic-PEOx-b-PBOy) or poly(ethylene oxide)-b-poly(propylene oxide) (cyclic-PEOx-b-PPOy).58,91,92 The self-assembly behaviour of the cyclic diblock copolymers was compared with the self-assembly of linear triblock copolymers and linear diblock copolymers of equivalent block composition. The authors reported similar aggregation behaviour for the cyclic diblock and linear triblock copolymers, with both polymers forming micellar assemblies, with comparable values of hydrodynamic radii (Rh) and critical micelle concentrations (cmc). However, one notable difference was observed when comparing the aggregation numbers (Nagg) of the cyclic diblock and linear triblock assemblies, with values of Nagg consistently higher for the cyclic diblock micelles, suggestive of a more dense assembly (cyclic-PEO42-b-PBO8Rh = 4.4 nm, Nagg = 16, PEO21-b-PBO8-b-PEO21Rh = 4.0 nm Nagg = 6). When comparing the cyclic diblock copolymers with the linear diblock copolymers, a more distinct difference in self-assembly behaviour was observed. The linear diblock copolymer assemblies consistently exhibited significantly larger values of Rh and Nagg and lower cmc values (PEO41-b-PBO8Rh = 7.1 nm, Nagg = 44, cmc = 0.3 g L−1), compared to both the cyclic diblock and linear triblock systems.The findings of these initial studies can be explained by considering the respective conformation of the three polymer architectures in a micellar state (Fig. 3). The cyclic diblock and linear triblock copolymers are entropically disfavoured because each polymer chain has two block junctions located at the solvent-core interface, in comparison to linear diblock copolymers which possess only one block junction. To this end, the cmc values for cyclic diblock and linear triblock copolymers are expected to be higher than the equivalent linear diblock assemblies. The relative size of the resulting assemblies will also be influenced by the conformation of the different architectures. As the core-forming block of the linear diblock copolymer assembly is not required to loop and can stretch without restriction, the value of Rh for a linear diblock micellar assembly is expected to be larger than that of equivalent cyclic diblock or linear triblock assemblies. Furthermore, as a consequence of their unrestricted structure, allowing better packing, linear diblock copolymer micelles are expected to be denser than micelles comprised of the equivalent cyclic diblock or linear triblock. In addition, because cyclic polymers possess smaller hydrodynamic volumes than linear polymers, the assembly of the cyclic diblock copolymer may also be smaller than the assembly of the linear triblock copolymer. Thus the observed respective particle sizes of cyclic diblock, linear diblock and linear triblock copolymers are a balance between their hydrodynamic volume, conformation and relative stretching and packing abilities.
 | ||
| Fig. 3 Chain conformations of cyclic diblock, linear diblock and linear triblock copolymers in a micellar state. | ||
Subsequent studies by other research groups have also compared the self-assembly of cyclic diblock and linear diblock copolymers and reported similar findings. Ge et al. studied the self-assembly of cyclic poly(2-(2-methoxy-ethoxy) ethyl methacrylate)-b-poly(oligo(ethylene glycol) methyl ether methacrylate) (cyclic-PMEO2MA35-b-POEGMA12) and cyclic poly(2-(dimethylamino) ethyl methacrylate)-b-poly(2-(diethylamino) ethyl methacrylate) (cyclic-PDMAEMA110-b-PDEAEMA89) in comparison to linear diblock copolymers of the same composition.93 For both polymer systems, the cyclic diblock assemblies exhibited smaller hydrodynamic radii and higher cmc values than the equivalent linear diblock copolymer (PMEO2MA35-b-POEGMA12 cyclic: Rh = 24 nm, cmc = 1.39 × 10−2 g L−1, linear: Rh = 34 nm, cmc = 1.02 × 10−2 g L−1, PDMAEMA105-b-PDEAEMA90 cyclic: Rh = 25 nm, cmc = 9.7 × 10−3 g L−1, linear: Rh = 42 nm, cmc = 7.9 × 10−3 g L−1). Additionally, Zhang et al. observed that the hydrodynamic diameter of cyclic poly(ethylene glycol)-b-poly(ε-caprolactone) (PEGx-b-PCLy) micelles was approximately half that of linear PEGx-b-PCLy micelles (cyclic Dh = 15 nm, linear Dh = 27 nm) (Fig. 4).94 Meanwhile, Hadjichristidis and coworkers reported a significantly larger aggregation number and hydrodynamic radius for aggregates of linear poly(styrene)-b-poly(butadiene) (PS28-b-PBd22) in DMF (Rh = 30 nm, Nagg = 1050), a selective solvent for PS, in comparison to cyclic PS28-b-PBd22 (Rh = 23 nm, Nagg = 450), which was also smaller than assemblies of the equivalent linear triblock PS14-b-PBd22-b-PS14 (Rh = 26 nm).95
 | ||
| Fig. 4 (Top) Cyclization and self-assembly of PEG-b-PCL. (Bottom) Light scattering data (number distributions) for cyclic and linear PCL-b-PEG. Adapted with permission from ref. 94. Copyright 2013 American Chemical Society. | ||
Isono et al. reported the self-assembly of cyclic poly(decyl glycidyl ether)51-b-poly(2-(2-(2-methoxyethoxy)ethoxy ethyl glycidyl ether)50 and the equivalent linear diblock copolymer.96 The cmc of the cyclic diblock copolymer assemblies was observed to be higher than that of the linear diblock copolymer (cyclic cmc = 1.8 × 10−3 g L−1, linear cmc = 1.4 × 10−3 g L−1), but in contrast to previous examples, the cyclic diblock copolymer assemblies were observed to be larger than the assemblies of the linear diblock (cyclic Dh = 166 nm, linear Dh = 122 nm). However, considering the fully extended chain lengths of the linear and cyclic copolymers, these assemblies cannot be classical core–shell micelles and indeed further analysis of the assemblies by transmission electron microscopy (TEM) revealed large spherical compound structures. The increased complexity of these aggregates makes it harder to elucidate the effect of cyclization on the particle dimensions. However, the observed difference in particle size may result from the reduced packing ability of cyclic polymer chains within the compound micelle compared to linear polymers, resulting in a greater value of Dh for the cyclic diblock assembly.
Yamamoto and Tezuka compared the self-assembly behaviour of cyclic poly(butyl acrylate)-b-poly(ethylene oxide) (PBA12-b-PEO59) with respect to the precursor linear triblock PBA6-b-PEO59-b-PBA6.97 Upon micellization the hydrophilic block of the linear triblock copolymer is looped and in contrast to previous studies the linear triblock assembly is conformationally restricted in the corona and not the core. The cyclic diblock and linear triblock assemblies displayed comparable values of Dh and cmc (cyclic Dh = 20 nm, cmc = 0.14 g L−1, linear Dh = 20 nm, cmc = 0.13 g L−1). However, significantly different thermal stabilities were exhibited by the cyclic and linear assemblies, with the micelles comprised of cyclic diblock copolymer displaying cloud points >40 °C higher than the micelles comprised of the linear triblock copolymer. The lower thermal stability of the linear triblock assemblies was attributed to the occurrence of inter-micelle bridging via dangling polymer chains in combination with dehydration, resulting in agglomeration at lower temperatures (Fig. 5). In comparison, the cyclic polymer chains cannot form inter-micelle bridges and agglomeration will only occur as a consequence of polymer dehydration, resulting in a higher transition temperature. Through coassembly of the cyclic and linear polymers, micelles with tuneable cloud point temperatures were observed. The same group has also reported that micelles of cyclic PBAx-b-PEOy and cyclic poly(methyl acrylate)-b-poly(ethylene oxide) (PMAx-b-PEOy) exhibit greater robustness in response to salt additives (NaCl and MgSO4), i.e. the cyclic diblock assemblies displayed higher salting-out concentrations in comparison to their linear PBAx/2-b-PEOy-b-PBAx/2 and PMAx/2-b-PEOy-b-PMAx/2 counterparts.98 The greater thermal and salt stabilities of the cyclic diblock assemblies were exploited through their use as catalytic nanoreactors in reactions that required elevated temperatures and high salt concentrations.
 | ||
| Fig. 5 Modes of temperature induced agglomeration for cyclic diblock PBA-b-PEO and linear triblock PBA-b-PEO-b-PBA flower-like micelles. Reprinted with permission from ref. 97. Copyright 2010 American Chemical Society. | ||
Yamamoto and Ree have subsequently reported a detailed small angle X-ray scattering (SAXS) investigation that highlighted subtle structural differences between assemblies of cyclic PBA10-b-PEO69 and linear PBA5-b-PEO69-b-PBA5.99 Both micelles were found to exhibit a core-fuzzy-shell structure, however the core and corona of the cyclic diblock copolymer micelle were more compact than the linear triblock copolymer micelle, as a result of the greater confinement and smaller effective volume of cyclic polymers.
The work highlighted so far has focused on the aggregation of cyclic block copolymers that possess a longer hydrophilic block relative to the hydrophobic block or comparable hydrophilic and hydrophobic block lengths; such polymers assemble to form so-called “star-like” micelles. Borsali and coworkers reported the self-assembly behaviour of cyclic poly(styrene)-b-poly(isoprene) (PSx-b-PIy) copolymers, that possess a significantly longer core-forming PS block than the corona-forming PI block; these assemblies are commonly referred to as “crew-cut”.100–106 In contrast to the “star-like” assemblies discussed above, these “crew-cut” assemblies exhibited much greater structural and morphological differences with respect to their linear PSx-b-PIy analogues. The linear PSx-b-PIy copolymers were observed to form spherical micelles of consistent size and low dispersities, regardless of polymer concentration, temperature or solvent choice (various n-alkanes selective for the PI block). However, the morphology of the cyclic PSx-b-PIy copolymers was found to change dramatically as these parameters were varied and a transition from spherical flower-like micelles to giant worm-like micelles was observed (Fig. 6). As was discussed in previous examples, the contrasting self-assembly behaviour between the cyclic and linear polymers was attributed in part to the looped nature of the core block of the cyclic polymer, restricting the packing of the core and resulting in a greater number of unfavourable PS-solvent interactions. However, as a consequence of the large solvophobic PS block in these “crew-cut” assemblies, this effect is more pronounced compared to examples of “star-like” micelles. To minimize unfavourable PS-solvent interactions, the flower-like micelles of the cyclic diblock copolymer cohere forming more energetically favourable worm-like micelles. The transition from flower-like micelles to worm-like micelles is more pronounced as polymer concentration and temperature are increased, as the probability of cohesive collisions increases. Furthermore, as the solvent quality for PS is reduced (n-pentane < n-heptane < n-decane), the driving force for cohesion is greater. Conversely, without the restrictive loop structure the spherical micelles of the linear polymer are more energetically favourable than the equivalent cyclic assemblies and possess no driving force for cohesion.
 | ||
| Fig. 6 (Top) Contrasting self-assembly behaviour of linear and cyclic PS-b-PI. Reprinted with permission from ref. 102. Copyright 2003 American Chemical Society. (Bottom) (A) Cryo-TEM image of linear PS-b-PI, (B) cryo-TEM image of cyclic PS-b-PI, (C) AFM image of linear PS-b-PI, (D) AFM image of cyclic PS-b-PI. Reprinted with permission from ref. 100. Copyright 2003 American Chemical Society. | ||
The effect of cyclization on the aggregation behaviour of a crystallization-driven self-assembly107 has also been investigated. Cyclic and linear amphiphilic diblock polypeptoids, poly(N-decylglycine)10-b-poly(N-methylglycine)105 were found to initially form spherical micelles upon self-assembly in methanol, a selective solvent for the poly(N-methylglycine) block.108 However, a morphological transition from spheres to cylindrical micelles was observed by cryo-TEM for both cyclic and linear polymers as the core-forming poly(N-decylglycine) block crystallized over time. This morphological transition was observed to take approximately twice as long for the cyclic diblock polypeptoid (15 days) compared to the linear diblock polypeptoid (8 days). This difference in behaviour was attributed to retarded crystallization of the cyclic diblock polypeptoid as a consequence of the restricted conformation of cyclic polymers. The resulting cylindrical micelles comprised of cyclic and linear polypeptoids exhibited similar dimensions by cryo-TEM (cyclic average core diameter = 12.2 nm, linear average core diameter = 12.6 nm).
Self-assembly of complex cyclic architectures
In addition to the self-assembly of amphiphilic cyclic diblock copolymers, some examples of the self-assembly of more complex amphiphilic cyclic architectures have been reported. Wan et al. reported the self-assembly of an amphiphilic tadpole-shaped polymer; where a tadpole-shaped polymer consists of a cyclic polymer attached to a linear polymer chain.109 The ring of the tadpole consisted of hydrophilic poly(N-isopropylacrylamide), whereas the linear tail consisted of hydrophobic poly(ε-caprolactone) ((cyclic-PNiPAm45)-b-PCL60). When the self-assembly behaviour of the amphiphilic tadpole was compared with that of the equivalent linear diblock assembly, the tadpole-shaped polymer was reported to form slightly larger assemblies than the linear diblock copolymer (tadpole Rh = 70 nm, linear Rh = 62 nm). This suggested that the incorporation of cycles in the tadpole-shaped polymer hindered packing of the polymer during aggregation, resulting in larger, less compact particles. Such a hypothesis could be confirmed by determination of Nagg for the tadpole-shaped and linear polymers. As the values of Rh for both the tadpole-shaped polymer and linear polymer were larger than the maximum polymer chain length, the particles were ascribed to large compound structures. The viability of these tadpole and linear assemblies as drug carriers was investigated by loading the particles with doxorubicin hydrochloride and monitoring the subsequent release of the drug. The assemblies consisting of the tadpole-shaped polymer were found to exhibit faster release profiles, further indicating that the tadpole assembly was less compact than the equivalent linear system.In direct contrast, when Isono et al. studied the self-assembly of tadpole-shaped polymers with a hydrophilic ring (poly(2-(2-(2-methoxyethoxy)ethoxy ethyl glycidyl ether)49) and hydrophobic tail (poly(decyl glycidyl ether)51), the tadpole assemblies displayed significantly smaller solution diameters than the equivalent linear assemblies (tadpole Dh = 83 nm, linear Dh = 123 nm).96 These aggregates were also reported to be large compound structures.
The self-assembly behaviour of tadpole-shaped polymers comprised of a hydrophobic ring and a hydrophilic tail has also been studied. Dong et al. prepared tadpole-shaped polymers with a poly(styrene) ring and a poly(ethylene oxide) tail ((cyclic-PS68)-b-PEO45).110 Subsequent self-assembly afforded vesicles with an average solution diameter, Dh, of 160 nm, whereas vesicles prepared from the analogous linear PS65-b-PEO45 copolymer displayed a smaller solution diameter, Dh, of 70 nm. This difference in particle size was further confirmed by TEM analysis (Fig. 7).
 | ||
| Fig. 7 TEM images of (a) linear PS-b-PEO and (b) tadpole PS-b-PEO. Reprinted with permission from ref. 110. Copyright 2009 American Chemical Society. | ||
Lonsdale and Monteiro compared the self-assembly behaviour of different tadpole architectures comprised of hydrophobic poly(styrene) rings and hydrophilic poly(acrylic acid) (PAA) tails.111 Depending on the block length of the PAA tails, either micelles or vesicles were formed during self-assembly (Fig. 8). Assemblies that possessed two PAA tails but only one PS ring (C in Fig. 8) formed the smallest structures with the lowest values of Nagg as a consequence of the relatively low hydrophobic volume of these assemblies. Conversely, as a consequence of increased hydrophobic volume, assemblies with only one PAA tail but two PS rings (B in Fig. 8) formed the largest structures and exhibited the largest values of Nagg. The greater restriction of two polymer rings per chain may also hinder the packing of the hydrophobic block in comparison to tadpole-shaped polymers with only one ring, contributing to the larger size of this assembly.
 | ||
| Fig. 8 TEM images of PS-b-PAA tadpole-shaped polymers with varying architectures. Reprinted with permission from ref. 111. Copyright 2011 Wiley Periodicals, Inc. | ||
Similarly, when Fan et al. studied the self-assembly of an amphiphilic figure-of-eight shaped polymer in comparison to its precursor, a 4-armed amphiphilic star polymer, the assembly of the figure-of-eight polymer exhibited a significantly larger solution diameter (figure-of-eight Dh = 42 nm, star Dh = 18 nm).112 The 4-armed star polymer consisted of two PS arms and two PEO arms, whereas both rings in the figure-of-eight shaped polymer possessed a diblock PSx-b-PEOy structure. The conformation of the figure-of-eight shaped polymer is extremely restricted upon aggregation, which greatly limits its ability to pack during self-assembly and results in a considerably larger micelle size compared to the star copolymer system. Furthermore, micelles of the figure-of-eight polymer possess three core-solvent junctions reducing their entropic favourability, whereas micelles comprised of the star copolymer possess only one core-solvent junction.
Meanwhile, Wang et al. studied the self-assembly of figure-of-eight shaped cyclic-(PSx-b-PAAy)2 in comparison to linear-(PSx-b-PAAy)2 (Fig. 9).113 Through variation of PS and PAA block lengths either conventional core–shell micelles ((PS8-b-PAA26)2) or large compound structures ((PS14-b-PAA17)2) were formed. In both cases the figure-of-eight polymer formed larger structures that possessed lower values of Nagg compared to the equivalent linear polymer (figure-of-eight Rh = 46 nm, Nagg = 250, linear Rh = 35 nm, Nagg = 367), providing further evidence of the loose nature of aggregates prepared from figure-of-eight shaped polymers.
 | ||
| Fig. 9 Self-assembly of linear and cyclic-(PS-b-PAA)2.Reprinted with permission from ref. 112. Copyright 2014 American Chemical Society. | ||
The self-assembly of jellyfish-shaped amphiphilic polymers has also been briefly investigated. Cai et al. prepared jellyfish-shaped polymers with a hydrophobic block copolymer ring comprised of PCL and poly(pentafluorostyrene) (PPFS), with hydrophilic PEG side arms attached to the latter block, yielding cyclic-PCLx-b-(PPFSy-g-PEGz).114 Upon self-assembly in water, spherical micelles with a diameter of 50–60 nm were observed by scanning electron microscopy (SEM). However, no comparison with an equivalent linear structure was reported. In another example, Coulembier et al. prepared jellyfish-shaped polymers with a cyclic poly(L-lactide) (PLLA40) backbone and three poly(methyl methacrylate) (PMMA) side arms.73 When a solution of the polymer in THF was deposited on a mica substrate and analyzed by atomic force microscopy (AFM), short cylindrical structures, toroids and other structures were observed. The height and width of the cylinders and toroids corresponded to the diameter of the PLLA ring, suggesting the jellyfish assembled in a cofacial manner.
In a similar manner, Schappacher and Deffieux prepared well-defined polymeric nanotubes via the self-assembly of densely grafted, high molecular weight cyclic brush copolymers.115 The cyclic polymer backbone consisted of poly(chloroethyl vinyl ether)1000 that had been grafted with a mixture of randomly distributed PS170 and PI50 arms (Fig. 10). The cyclic brush copolymers were found to self-assemble in n-heptane, a selective solvent for PI, to afford nanotubes with a diameter of ca. 100 nm and length of up to 700 nm. The diameter of the assemblies corresponded to the diameter of the cyclic brushes, again suggesting self-assembly occurred in a cofacial manner between cyclic brush copolymers. These last two examples highlight the significant and unique self-assembly behaviour of polymers that possess a cyclic architecture, where these particular examples of self-assembly are impossible to achieve with polymers that possess a linear architecture.
 | ||
| Fig. 10 (Top) Preparation of PS and PS/PI cyclic brush copolymers. (Bottom) AFM images of PS/PI nanotubes (E) topological images, (F) phase images, inset: reverse mode, purple = PS, green = PI. From ref. 115. Reprinted with permission from AAAS. | ||
Conclusions
As a consequence of improved synthetic methods, enabling the preparation of well-defined, high purity cyclic polymers, the self-assembly of amphiphilic cyclic polymers has received increasing attention in recent years. By comparing the aggregation of cyclic polymers with equivalent linear polymers, we can determine the effect of cyclization on self-assembly and increase our understanding of structure–property relationships. Indeed, the examples discussed in this review have highlighted the profound effect cyclization can have on particle dimensions, stability, and morphology, as well as the packing of polymer chains within the assembly. In general, cyclic diblock copolymers form smaller, entropically disfavoured aggregates in comparison to linear diblock copolymers, as a consequence of the confined and looped nature of cyclic polymers, resulting in a greater number of unfavourable core-solvent junctions. The self-assembly of cyclic diblock copolymers is however similar to that of linear triblock copolymers, which are also required to loop upon aggregation. For the self-assembly of more complex cyclic topologies, assemblies of cyclic polymers are often larger than the equivalent linear polymer assembly as a consequence of poor polymer packing. Furthermore, the cyclization of amphiphilic polymers can lead to unique self-assembly behaviour that cannot be achieved through the self-assembly of linear polymers or can impart improved properties to the resulting nanostructures, for example, greater thermal stability and robustness towards salt additives.Notes and references
- H. F. Eicke, Pure Appl. Chem., 1980, 52, 1349–1357 CrossRef CAS.
- P. D. I. Fletcher, Curr. Opin. Colloid Interface Sci., 1996, 1, 101–106 CrossRef CAS.
- H. Hoffmann, Adv. Mater., 1994, 6, 116–129 CrossRef CAS.
- S. P. Moulik, Curr. Sci., 1996, 71, 368–376 CAS.
- P. Schurtenberger, Curr. Opin. Colloid Interface Sci., 1996, 1, 773–778 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- P. Cotanda, N. Petzetakis and R. K. O'Reilly, MRS Commun., 2012, 2, 119–126 CrossRef CAS.
- A. Lu and R. K. O'Reilly, Curr. Opin. Biotechnol., 2013, 24, 639–645 CrossRef CAS PubMed.
- D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomed. Nanotechnol. Biol. Med., 2010, 6, 714–729 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- P. V. Pawar, S. V. Gohil, J. P. Jain and N. Kumar, Polym. Chem., 2013, 4, 3160–3176 RSC.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- V. S. Trubetskoy, Adv. Drug Delivery Rev., 1999, 37, 81–88 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- K. Nakashima and P. Bahadur, Adv. Colloid Interface Sci., 2006, 123–126, 75–96 CrossRef CAS PubMed.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2011, 45, 2–19 CrossRef.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- S. Zhong, H. Cui, Z. Chen, K. L. Wooley and D. J. Pochan, Soft Matter, 2008, 4, 90–93 RSC.
- H. Shen and A. Eisenberg, Angew. Chem., Int. Ed., 2000, 39, 3310–3312 CrossRef CAS.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- T. Nicolai, O. Colombani and C. Chassenieux, Soft Matter, 2010, 6, 3111–3118 RSC.
- Z. Ge and S. Liu, Macromol. Rapid Commun., 2009, 30, 1523–1532 CrossRef CAS PubMed.
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171–1185 RSC.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed.
- D. K. Smith, Chem. Commun., 2006, 34–44 RSC.
- D. K. Smith, A. R. Hirst, C. S. Love, J. G. Hardy, S. V. Brignell and B. Huang, Prog. Polym. Sci., 2005, 30, 220–293 CrossRef CAS PubMed.
- G. M. Soliman, A. Sharma, D. Maysinger and A. Kakkar, Chem. Commun., 2011, 47, 9572–9587 RSC.
- Y. Zhou, W. Huang, J. Liu, X. Zhu and D. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS PubMed.
- Y. Zhou and D. Yan, Chem. Commun., 2009, 1172–1188 RSC.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS PubMed.
- Y. Deng, S. Zhang, G. Lu and X. Huang, Polym. Chem., 2013, 4, 1289–1299 RSC.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS PubMed.
- C. Feng, Y. Li, D. Yang, J. Hu, X. Zhang and X. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS PubMed.
- D. Uhrig and J. Mays, Polym. Chem., 2011, 2, 69–76 RSC.
- M. Zhang and A. H. E. Müller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3461–3481 CrossRef CAS.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef CAS.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- R. M. England and S. Rimmer, Polym. Chem., 2010, 1, 1533–1544 RSC.
- K. Inoue, Prog. Polym. Sci., 2000, 25, 453–571 CrossRef CAS.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- K. Endo, in New Frontiers in Polymer Synthesis, ed. S. Kobayashi, Springer, Berlin Heidelberg, 2008, vol. 217, pp. 121–183 Search PubMed.
- Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097 CrossRef CAS.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- Y. Tezuka, Topological Polymer Chemistry: Progress of Cyclic Polymers in Syntheses, Properties and Functions, World Scientific Publishing Co. Pte. Ltd, 2013 Search PubMed.
- T. Yamamoto and Y. Tezuka, Polym. Chem., 2011, 2, 1930–1941 RSC.
- G.-E. Yu, Z. Yang, D. Attwood, C. Price and C. Booth, Macromolecules, 1996, 29, 8479–8486 CrossRef CAS.
- D. Freifelder, A. K. Kleinschmidt and R. L. Sinsheimer, Science, 1964, 146, 254–255 CAS.
- J. F. Brown and G. M. J. Slusarczuk, J. Am. Chem. Soc., 1965, 87, 931–932 CrossRef.
- D. W. Scott, J. Am. Chem. Soc., 1946, 68, 2294–2298 CrossRef CAS.
- K. Dodgson, D. Sympson and J. A. Semlyen, Polymer, 1978, 19, 1285–1289 CrossRef CAS.
- D. Geiser and H. Höcker, Macromolecules, 1980, 13, 653–656 CrossRef CAS.
- G. Hild, A. Kohler and P. Rempp, Eur. Polym. J., 1980, 16, 525–527 CrossRef CAS.
- B. Vollmert and J. Huang, Makromol. Chem. Rapid Commun., 1980, 1, 333–339 CrossRef CAS.
- H. R. Kricheldorf and S.-R. Lee, Macromolecules, 1995, 28, 6718–6725 CrossRef CAS.
- J. N. Hoskins and S. M. Grayson, Polym. Chem., 2011, 2, 289–299 RSC.
- H. Oike, H. Imaizumi, T. Mouri, Y. Yoshioka, A. Uchibori and Y. Tezuka, J. Am. Chem. Soc., 2000, 122, 9592–9599 CrossRef CAS.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS PubMed.
- D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 2627–2630 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Kubo, T. Hayashi, H. Kobayashi and T. Itoh, Macromolecules, 1998, 31, 1053–1057 CrossRef CAS.
- O. Coulembier, S. Moins, J. De Winter, P. Gerbaux, P. Leclère, R. Lazzaroni and P. Dubois, Macromolecules, 2010, 43, 575–579 CrossRef CAS.
- M. Schappacher, C. Billaud, C. Paulo and A. Deffieux, Macromol. Chem. Phys., 1999, 200, 2377–2386 CrossRef CAS.
- Y. Tezuka, A. Tsuchitani, Y. Yoshioka and H. Oike, Macromolecules, 2002, 36, 65–70 CrossRef.
- M. Antonietti and K. J. Fölsch, Makromol. Chem. Rapid Commun., 1988, 9, 423–430 CrossRef CAS.
- G. Hadziioannou, P. M. Cotts, G. ten Brinke, C. C. Han, P. Lutz, C. Strazielle, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 493–497 CrossRef CAS.
- M. Duval, P. Lutz and C. Strazielle, Makromol. Chem. Rapid Commun., 1985, 6, 71–76 CrossRef CAS.
- J. Roovers, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 1117–1126 CrossRef CAS.
- M. Ragnetti, D. Geiser, H. Höcker and R. C. Oberthür, Makromol. Chem., 1985, 186, 1701–1709 CrossRef CAS.
- G. B. McKenna, G. Hadziioannou, P. Lutz, G. Hild, C. Strazielle, C. Straupe, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 498–512 CrossRef CAS.
- D. J. Orrah, J. A. Semlyen and S. B. Ross-Murphy, Polymer, 1988, 29, 1455–1458 CrossRef CAS.
- J. Roovers, Macromolecules, 1985, 18, 1359–1361 CrossRef CAS.
- P. G. Santangelo, C. M. Roland, T. Chang, D. Cho and J. Roovers, Macromolecules, 2001, 34, 9002–9005 CrossRef CAS.
- S. Habuchi, N. Satoh, T. Yamamoto, Y. Tezuka and M. Vacha, Angew. Chem., Int. Ed., 2010, 49, 1418–1421 CrossRef CAS PubMed.
- T. Perkins, D. Smith and S. Chu, Science, 1994, 264, 819–822 CAS.
- J. Käs, H. Strey and E. Sackmann, Nature, 1994, 368, 226–229 CrossRef PubMed.
- J. E. Poelma, K. Ono, D. Miyajima, T. Aida, K. Satoh and C. J. Hawker, ACS Nano, 2012, 6, 10845–10854 CAS.
- K. Zhang, M. A. Lackey, J. Cui and G. N. Tew, J. Am. Chem. Soc., 2011, 133, 4140–4148 CrossRef CAS PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS PubMed.
- G.-E. Yu, C. A. Garrett, S.-M. Mai, H. Altinok, D. Attwood, C. Price and C. Booth, Langmuir, 1998, 14, 2278–2285 CrossRef CAS.
- G.-E. Yu, Z.-K. Zhou, D. Attwood, C. Price, C. Booth, P. C. Griffiths and P. Stilbs, J. Chem. Soc., Faraday Trans., 1996, 92, 5021–5028 RSC.
- Z. Ge, Y. Zhou, J. Xu, H. Liu, D. Chen and S. Liu, J. Am. Chem. Soc., 2009, 131, 1628–1629 CrossRef CAS PubMed.
- B. Zhang, H. Zhang, Y. Li, J. N. Hoskins and S. M. Grayson, ACS Macro Lett., 2013, 2, 845–848 CrossRef CAS.
- H. Iatrou, N. Hadjichristidis, G. Meier, H. Frielinghaus and M. Monkenbusch, Macromolecules, 2002, 35, 5426–5437 CrossRef CAS.
- T. Isono, Y. Satoh, K. Miyachi, Y. Chen, S. Sato, K. Tajima, T. Satoh and T. Kakuchi, Macromolecules, 2014, 47, 2853–2863 CrossRef CAS.
- S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251–10253 CrossRef CAS PubMed.
- S. Honda, T. Yamamoto and Y. Tezuka, Nat. Commun., 2013, 4, 1574 CrossRef PubMed.
- K. Heo, Y. Y. Kim, Y. Kitazawa, M. Kim, K. S. Jin, T. Yamamoto and M. Ree, ACS Macro Lett., 2014, 3, 233–239 CrossRef CAS.
- R. Borsali, E. Minatti, J.-L. Putaux, M. Schappacher, A. Deffieux, P. Viville, R. Lazzaroni and T. Narayanan, Langmuir, 2002, 19, 6–9 CrossRef.
- E. Minatti, R. Borsali, M. Schappacher, A. Deffieux, V. Soldi, T. Narayanan and J.-L. Putaux, Macromol. Rapid Commun., 2002, 23, 978–982 CrossRef CAS.
- E. Minatti, P. Viville, R. Borsali, M. Schappacher, A. Deffieux and R. Lazzaroni, Macromolecules, 2003, 36, 4125–4133 CrossRef CAS.
- N. Ouarti, P. Viville, R. Lazzaroni, E. Minatti, M. Schappacher, A. Deffieux and R. Borsali, Langmuir, 2005, 21, 1180–1186 CrossRef CAS PubMed.
- N. Ouarti, P. Viville, R. Lazzaroni, E. Minatti, M. Schappacher, A. Deffieux, J.-L. Putaux and R. Borsali, Langmuir, 2005, 21, 9085–9090 CrossRef CAS PubMed.
- J.-L. Putaux, E. Minatti, C. Lefebvre, R. Borsali, M. Schappacher and A. Deffieux, Faraday Discuss., 2005, 128, 163–178 RSC.
- M. Schappacher and A. Deffieux, Macromol. Chem. Phys., 2002, 203, 2463–2469 CrossRef CAS.
- W.-N. He and J.-T. Xu, Prog. Polym. Sci., 2012, 37, 1350–1400 CrossRef CAS PubMed.
- C.-U. Lee, T. P. Smart, L. Guo, T. H. Epps and D. Zhang, Macromolecules, 2011, 44, 9574–9585 CrossRef CAS PubMed.
- X. Wan, T. Liu and S. Liu, Biomacromolecules, 2011, 12, 1146–1154 CrossRef CAS PubMed.
- Y.-Q. Dong, Y.-Y. Tong, B.-T. Dong, F.-S. Du and Z.-C. Li, Macromolecules, 2009, 42, 2940–2948 CrossRef CAS.
- D. E. Lonsdale and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4603–4612 CrossRef CAS.
- X. Fan, B. Huang, G. Wang and J. Huang, Macromolecules, 2012, 45, 3779–3786 CrossRef CAS.
- X. Wang, L. Li, X. Ye and C. Wu, Macromolecules, 2014, 47, 2487–2495 CrossRef CAS.
- T. Cai, W. J. Yang, K.-G. Neoh and E.-T. Kang, Polym. Chem., 2012, 3, 1061–1068 RSC.
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512–1515 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |