
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion conducting multiblock poly(arylene ether sulfone)s containing hydrophilic segments densely functionalized with quaternary ammonium groups†
Eva Annika
Weiber
,
David
Meis‡
and
Patric
Jannasch
*
Department of Chemistry, Polymer and Material Chemistry, Lund University, P. O. Box 124, Lund SE-221 00, Sweden. E-mail: patric.jannasch@chem.lu.se
First published on 5th January 2015
Abstract
We have prepared poly(arylene ether sulfone) multiblock copolymers with cationic blocks containing single dioxyphenylene rings functionalized with two, three or four quaternary ammonium (QA) groups in order to investigate the influence of the ionic concentration and distribution on the anionic conductivity. Precursor blocks were prepared by polycondensation of 4,4′-difluorodiphenyl sulfone and either di-, tri- or tetramethylhydroquinone. Subsequently, these blocks were combined with precursor blocks prepared from 4,4′-dichlorodiphenyl sulfone and bisphenol A to form alternating multiblock copolymers with different block ratios. The benzylic methyl groups of the hydroquinone monomer units were then fully brominated using N-bromosuccinimide. Quaternization with trimethylamine gave multiblock copolymers with extremely high ion exchange capacities (IECs) of the hydrophilic blocks, i.e., 3.2, 4.9, and 5.8 meq. g−1, respectively, in the Br− form. X-ray scattering and atomic force microscopy of the anion exchange membranes (AEMs) showed a distinct nanophase separation of the blocks. At a given IEC, the ionic conductivity was found to decrease with increasing number of QA groups per dioxyphenylene ring, probably because of limited ionic dissociation resulting from the close proximity of the QA groups. Thus, at a similar IEC, the conductivity of a block copolymer with tetra-functionalized rings reached the same level of conductivity as a corresponding polymer with randomly distributed QA groups, whereas a block copolymer with di-functionalized rings exceeded the conductivity of the latter polymer by a factor 4.2, despite a lower water uptake. These findings strongly highlight the importance of controlling and optimizing the local ionic concentration and distribution for highly anion conductive AEMs based on block copolymers.
1 Introduction
Durable and efficient anion exchange membranes (AEMs) are enabling materials for new electrochemical systems for energy storage and conversion such as flow batteries,1 fuel cells2 and electrolysers.3 The expected benefits of electro-catalysis in systems based on liquid alkaline electrolytes (e.g., aqueous KOH) employed in alkaline fuel cells include the use of non-precious metal catalysts and a wider choice of fuels.2 Yet, the use of these liquid electrolytes brings considerable complications such as electrolyte leakage, corrosion, and reactions with carbon dioxide leading to the formation of insoluble carbonates that block the electrodes. This has evoked a great interest to instead use AEMs based on polymers functionalized with organic cations such as quaternary ammonium (QA) and imidazolium.2,4–6 Because the cation is immobilized in the polymer structure, the disadvantages of a liquid electrolyte are largely avoided which decreases the complexity of the system and dramatically increases the power density. However, the inherently low anionic conductivity in relation to proton exchange membranes (PEMs) as well as the chemical stability of the organic cationic groups, which are sensitive towards nucleophilic attack by the hydroxide anion, need to be improved before practical use.5Over the last decade, an extensive research effort has been directed toward developing PEMs based on sulfonated hydrocarbon polymers to overcome the drawbacks of the Nafion membrane for use in PEM fuel cells.7,8 One of the most efficient synthetic strategies to enhance the proton conductivity of PEMs is to concentrate the sulfonic acid groups to specific sites or segments in the polymeric structure to induce a higher level of organization of the ionic phase domains in the membrane.8–10 This has for example been achieved by directing the sulfonic acid groups to side chains11–14 and by precisely sequencing the sulfonic acids along the polymer backbone.15,16 However, the most successful results have been achieved with block copolymers having very high concentrations of sulfonic acid attached to one of the blocks.17,18 Because of the immiscibility of the ionic and non-ionic blocks, these copolymers characteristically self-assemble to form various well-ordered phase structures on the nanometer level, thus forming a separate and highly sulfonated phase for proton transport.9 Block copolymer PEMs typically show a higher level of conductivity than statistical copolymers at low water contents because the ionic groups are locally concentrated to nanosized domains which are able to retain a higher level of percolation. Block copolymers for use as AEMs are novel materials which are still far less studied. For example, it is still not known if the synthetic strategies used to obtain highly conductive block copolymer PEMs can be employed also in the case of block copolymer AEMs. A number of research groups have reported on improved ionic conductivity and reduced swelling of multiblock AEMs compared to membranes based on random copolymers at the same ionic content.19–21 At the same water content and ion exchange capacity (IEC), Watanabe et al. observed a higher hydroxide conductivity (42 mS cm−1) of a multiblock copolymer compared with a random copolymer (9 mS cm−1) under immersed conditions at 60 °C.22 Beckham and co-workers reported a generally higher hydroxide conductivity of multiblock copolymers compared to random copolymers at the same IEC, however not in all cases.20 The block ratios had to be optimized in order to obtain the higher performance of the multiblock copolymer. Up until now, the effect of the local ionic concentration on the conductivity of block copolymer AEMs has not been systematically investigated.
We have previously prepared and studied a series of statistical poly(arylene ether sulfone)s (PAESs) with QA groups attached to single dioxyphenylene rings along the backbone.23 Small-angle X-ray scattering (SAXS) of AEMs based on these copolymers revealed extensive ionic clustering, promoted by an increasing local density of QA. At moderate IECs, we found the conductivity of the statistical copolymers with strictly di-functionalized rings to be considerably higher than that of a corresponding randomly functionalized polymer, despite a higher water uptake of the latter. The ionic conductivity then decreased with increasing local ionic concentration.
The scope of the present work was to study structure property relationships of alternating hydrophilic–hydrophobic multiblock copolymers in order to specifically investigate the influence of the ionic concentration and distribution in the hydrophilic block on AEM properties. Consequently, we have prepared PAES multiblock copolymers with hydrophilic blocks containing dioxyphenylene units carrying two, three or four QA groups. The multiblock copolymers were synthesized by coupling of precursor blocks based on methylhydroquinones (MHQs) and bisphenol A, respectively, followed by selective benzylic bromination and quaternization. This gave block copolymers with cationic blocks having very high IEC values, i.e., 3.2, 4.9 and 5.8 meq. g−1, respectively. To our knowledge, multiblock copolymer AEMs with such high local ionic concentrations on the length scale of the phase separated block copolymer domains have not been studied previously. The morphology, thermal properties, water uptake and ionic conductivity of the multiblock AEMs were evaluated and compared with corresponding statistical copolymers.
2 Experimental
2.1 Materials
2,3-Dimethylhydroquinone (2MHQ, 97%, Acros), trimethylhydroquinone (3MHQ, 97%, Acros), tetramethylhydroquinone (4MHQ, 95%, TCI) and bisphenol A (BPA, 97%, Acros) were recrystallized from toluene. 4,4′-Difluorophenyl sulfone (DFDPS, Fluorochem) and 4,4′-dichlorodiphenyl sulfone (DCDPS, 97%, Acros) were recrystallized from N,N-dimethylformamide and K2CO3 (99%, Acros) was dried at 120 °C prior to synthesis. N-Bromosuccinimide (NBS, 99%, Acros), azobisisobutyronitrile (AIBN, 98%, Acros), trimethylamine (7.3 M aqueous solution, Acros), toluene (HPLC grade, Honeywell), N,N-dimethylacetamide (DMAc, 99+%, Acros), 2-propanol (IPA, HPLC grade, Honeywell), 1,2-dichlorobenzene (99+%, Fluka Analytical) and N-methyl-2-pyrrolidinone (NMP, reagent grade, Acros) were all used as received.2.2 Precursor block synthesis
Four F-terminated PAES precursor blocks with number average molecular weights (Mns) in the range 9.7–18 kg mol−1 were synthesized via K2CO3 mediated polycondensations of DFDPS and 2MHQ, 3MHQ or 4MHQ as shown in Scheme 1. A precise stoichiometric excess of DFDPS was employed to control the molecular weight and chain end functionality of the resulting precursor blocks. Here follows a typical procedure, the synthesis of a precursor block with the target molecular weight of 7 kg mol−1. DFDPS (4.07 g, 16.0 mmol), 3MHQ (2.35 g, 15.4 mmol), K2CO3 (10.4 g, 78 mmol), DMAc (62 mL) and toluene (62 mL) were added to a two-neck round-bottom flask. The flask was equipped with a nitrogen inlet, a Dean-Stark trap and a condenser. First, the reaction mixture was dehydrated during 4 h at 130 °C under nitrogen flow. Then toluene was removed from the Dean-Stark trap and the temperature was increased to 160 °C. After 20 h of reaction, the product was precipitated by pouring the dark viscous reaction mixture into IPA. The precipitate was purified by repeated washing in IPA and water. The precursor block obtained had Mn = 9.7 kg mol−1 as evaluated by 1H NMR spectroscopy. Four OH-terminated precursor blocks with Mn = 10–18 kg mol−1 were synthesized by the same procedure but instead using DCDPS and a precise excess of BPA, as seen in Scheme 1. The F- and OH-terminated precursor blocks were designated ol-m-Fa and ol-n-OH respectively, where m and n corresponded to the number of repeating units in the precursor blocks, as evaluated by 1H NMR spectroscopy, and a to the number of methyl groups in the MHQ (2, 3 or 4). Structural data of the precursor blocks are summarized in Table 1.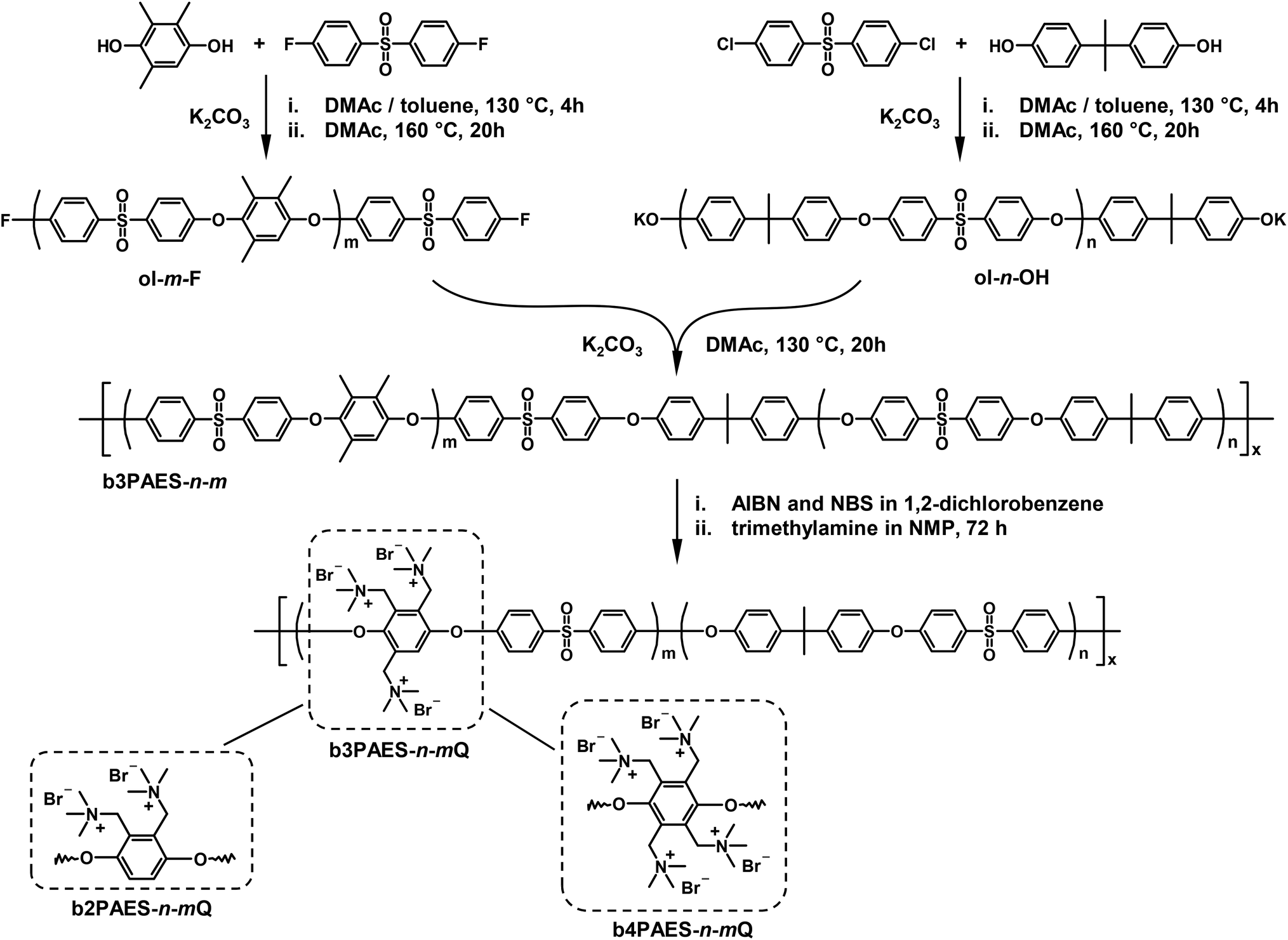 | ||
| Scheme 1 Synthetic pathway to the b3PAES-n-mQ multiblock copolymers by precursor block preparation via polycondensation, block copolymer formation via coupling, and selective benzylic bromination and quaternization. The b2PAES-n-mQ and b4PAES-n-mQ multiblock copolymers were prepared using the same method, but by using 2MHQ and 4MHQ, respectively, instead of 3MHQ. | ||
| Precursor block | M n,NMR (kg mol−1) | Number of repeating unitsa | M n, SEC (kg mol−1) | PDIb,c | T g (°C) |
|---|---|---|---|---|---|
| a Determined from 1H NMR data. b Evaluated by SEC using polystyrene standards. c Polydispersity index, Mw/Mn. d Determined by DSC at a heating rate of 10 °C min−1; ns = not soluble in chloroform, not analysed. | |||||
| ol-23-OH | 10.4 | 23 | 19 | 1.5 | 186 |
| ol-26-OH | 11.7 | 26 | 22 | 1.5 | 187 |
| ol-33-OH | 14.6 | 33 | 23 | 1.6 | 188 |
| ol-42-OH | 18.8 | 42 | 29 | 1.5 | 192 |
| ol-34-F2 | 10.8 | 34 | ns | ns | 197 |
| ol-26-F3 | 9.7 | 26 | ns | ns | 253 |
| ol-49-F3 | 18.2 | 49 | ns | ns | 237 |
| ol-20-F4 | 7.8 | 20 | ns | ns | 269 |
2.3 Block copolymer synthesis and bromination
A series of PAES multiblock copolymers with varying block lengths of the two precursor blocks were synthesized by coupling selected F- and OH-terminated precursor blocks as depicted in Scheme 1. The synthesis of the multiblock copolymer based on of the precursor block ol-26-F3 and ol-23-OH will serve as an example. F- (1.17 g, 0.1 mmol) and OH-terminated (0.95 g, 0.1 mmol) precursor blocks were mixed with K2CO3 (0.17 g, 1.2 mmol) and dissolved in 20 mL DMAc in a dry 50 mL two-neck round-bottom flask equipped with a condenser under nitrogen flow. The coupling reaction was performed at 130 °C during 20 h. This is a lower temperature than previously reported for such reactions19,24,25 and was chosen in order to suppress transetherification. To isolate the product, the viscous mixture was poured into IPA, followed by washing in IPA and water, and drying at 50 °C in a vacuum oven over night. The multiblock copolymers were designated as baPAES-n-m.The blocks containing the MHQ residues were selectively benzylbrominated via a radical route with NBS as the brominating agent and AIBN as the radical source. A typical synthesis procedure for the benzylbromination was as follows. The copolymer (0.8 g, 0.4 mmol) was dissolved in 20 mL of 1,2-dichlorobenzene and flushed with a nitrogen flow for 1 h. Next, NBS (1.2 g, 6.8 mmol) and AIBN (0.03 g, 0.2 mmol) were added and the reaction mixture was stirred at 120 °C. After 2 h, a second batch of NBS (1.2 g, 6.8 mmol) and AIBN (0.03 g, 0.2 mmol) was added, and the reaction was stopped after a total reaction time of 4 h. The mixture was poured into IPA and the brominated copolymer was purified by washing in IPA. The product was re-dissolved in chloroform, precipitated and again washed with IPA. Finally, the product was dried at 50 °C in the vacuum oven.
2.4 Quaternization and membrane preparation
The multiblock copolymers were quaternized by adding a 50 wt% aqueous solution of trimethylamine to a 5 wt% solution of the benzylbrominated copolymers in NMP. After 72 h, the quaternized polymers were precipitated and washed in IPA followed by drying in vacuo at room temperature.AEMs with a thickness in the range 30–70 μm were cast from solutions of the quaternized copolymers (5 wt%) in NMP. The solution was poured into a Petri dish and placed in a casting oven at 80 °C over night. The quaternized membranes were designated as baPAES-n-m-Q.
2.5 Structural characterization
All samples were analysed by 1H NMR spectroscopy using a Bruker DRX400 spectrometer at 400.13 MHz using CDCl3 (δ = 7.26 ppm), DMSO-d6 (δ = 2.50 ppm) or DMF-d7 (δ = 8.01, 2.93, 2.73 ppm) as solvents. The multiblock copolymers were also analyzed by FTIR spectroscopy (ESI, Fig. S1†). Size-exclusion chromatography (SEC) was employed to determine the molecular weight and polydispersity index (PDI, MwMn−1) of the polymer samples. Samples were dissolved in chloroform and passed through a series of three Shodex columns (KF-805, -804, and -802.5) and a refractive index detector at room temperature. The elution rate was 1 ml min−1. Four low-polydispersity polystyrene (PS) standard samples (Mn = 650 kg mol−1 from Water Associates, 96 and 30 kg mol−1 from Polymer Laboratories, and 3.18 kg mol−1 from Agilent Technologies) were used for calibration.2.6 Thermal analysis
The glass transition temperature (Tg) of the polymers was evaluated by differential scanning calorimetry (DSC) using a TA instruments Q2000 calorimeter at a heating rate of 10 °C min−1. The samples were subjected to a heating–cooling–heating cycle between 50 and 300 °C, and the Tg was determined from the thermogram of the second heating ramp. Thermogravimetric analysis (TGA) was employed in order to study the thermal degradation of the polymers, and a TA instruments Q500 analyser was used for this purpose. The thermal degradation was evaluated under nitrogen at a heating rate of 10 °C min−1 up to 600 °C. Prior to this heating ramp the samples were dried at 150 °C for 10 min. The degradation temperature (Td,95) was determined at the point where 95% of the sample remained.2.7 Morphological characterization
The AEMs were analysed by SAXS in the q range between 0.048 and 6.7 nm−1. Dry membranes in the Br− form were placed in a solid sample stage in a SAXSLAB SAXS instrument, from JJ X-ray Systems ApS (Denmark) equipped with a Pilatus detector. The scattering experiments were performed using Cu Kα radiation with a wavelength (l) of 1.542 Å generated within a high brilliance micro focus sealed tube with shaped multilayer optics operating at 50 kV and 60 mA. The wave vector (q) was calculated as: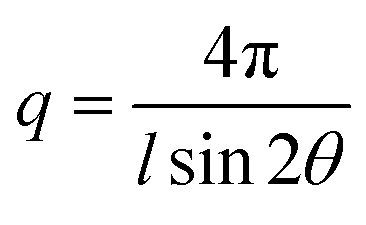 | (1) |
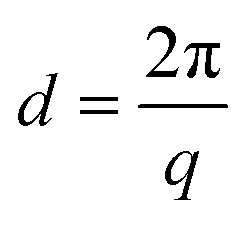 | (2) |
The surface of the membranes was analysed by atom force microscopy (AFM) in tapping mode under ambient air. An XE-100 Park System was used to carry out the measurement at a scanning frequency of 1 Hz. No image processing was performed.
2.8 Membrane properties
Mohr titrations were employed to evaluate the IEC of the AEMs in the Br− form. The membranes were dried (vacuum oven, 50 °C, 24 h), weighed and then immersed in 0.2 M aq. AgNO3 (25.00 mL) during 48 h. The resulting solution was titrated with 0.01 M aq. AgNO3 to determine the concentration of Br− ions, using 0.1 N aq. K2CrO4 as colorimetric indicator.The water uptake of the AEMs in the Br− form was evaluated at 20, 40, 60 and 80 °C. The membranes were placed in water and allowed to equilibrate at each temperature. Equilibrated membranes were gently wiped with tissue paper to remove water on the surface and weighed to determine the mass in the fully hydrated state (W′). The weight of the dry membrane (W) was determined after drying in a vacuum oven at 50 °C for 24 h. The water uptake (WU%) was then calculated as:
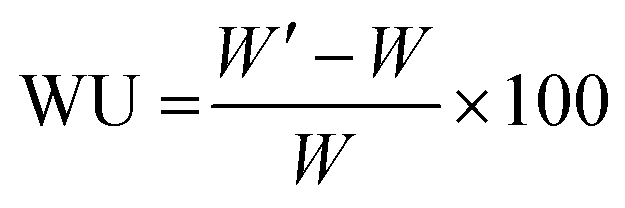 | (3) |
From the water uptake data the number of water molecules per ionic group (hydration number [λ]) was calculated using:
 | (4) |
The swelling ratio of the AEMs was evaluated by measuring the length (l) of a rectangular piece of the membranes dried in a vacuum oven at 50 °C for 24 h. The pieces were then immersed in water for 24 h and the length (l′) of the hydrated membranes were measured. The swelling in the length direction (Δl (%)) was calculated as:
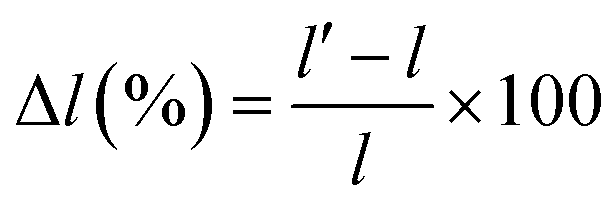 | (5) |
The Br− conductivity of fully hydrated membranes was determined by electrochemical impedance spectroscopy (EIS) in the temperature range −20–80 °C using a Novocontrol high-resolution dielectric analyser V 1.01S in the frequency range 10−1–107 Hz at 50 mV. A sealed two-probe cell was used for the evaluation and the membranes were immersed in deionized water during the measurement. The Br− conductivity was also evaluated as a function of relative humidity using a Gamry potentiostat/galvanostat/ZRA in combination with a four-probe Fumatech MK3 conductivity cell. The measurements were performed in the frequency range 10−1–105 Hz at 80 °C between 30 and 90% RH.
3 Results and discussion
The preparation of aromatic polymers using MHQ monomers gives a unique possibility to introduce precisely one, two, three, or four functional groups on single phenylene rings along the polymer backbone by exploiting the benzylic methyl groups as reactive sites.23 In the present work we have used this synthetic strategy to prepare multiblock copolymers with segments based on 2-, 3-, and 4MHQ monomers, respectively. Aiming at quantitative functionalization of the benzylic sites with QA groups, we targeted block copolymers having hydrophilic blocks with precisely controlled ionic distributions and very high local ions concentrations in the polymer structure.3.1 Multiblock copolymer synthesis
Precursor blocks were synthesized via K2CO3 mediated nucleophilic aromatic substitution reactions of either DFDPS and MHQs or DCDPS and BPA as shown in Scheme 1. The molecular weight and chain end functionality of the fluorine and hydroxyl terminated precursors were controlled by using an excess of DFDPS and BPA, respectively. Representative 1H NMR spectra of the precursor blocks are collected in Fig. 1. The molecular weight of the fluorine terminated precursor blocks was calculated from the ratios of the integrated signals e at 6.75 ppm, assigned to the aryl proton of the 3MHQ residues, and signal c′ at 7.10 ppm, which was ascribed to the chain end protons in meta position to the sulfone group (Fig. 1a). The Mn values of the hydroxy terminated precursor blocks were calculated from the ratio between the integrals of signal b and e′ assigned to the proton in ortho position to the sulfone bridge and the proton in the BPA chain end group, respectively. In all, four OH-terminated precursor blocks were prepared based on BPA with Mn ranging from 10.4 to 18.8 kg mol−1, and four F-terminated precursors were prepared based on the MHQs with Mn = 7.8 to 18.2 kg mol−1 (Table 1). | ||
| Fig. 1 Representative 1H NMR spectra of the PAES precursor blocks: fluorine terminated ol-26-F3 (a), and phenolate terminated ol-23-OH (b). | ||
Attempts were also made to determine the molecular weights of the precursor blocks that were soluble in chloroform by SEC using PS standards (Table 1). As seen, these values were far above the ones calculated from the NMR data, which may be explained by the much higher molecular stiffness of the polysulfone chains in relation to PS, leading to larger hydrodynamic radii of the former polymers at a given Mn. Still, both techniques gave the same order of increasing block length.
Seven multiblock copolymers with different block molecular weights and ratios were prepared by K2CO3 mediated coupling reactions of equimolar amounts of hydroxy and fluorine terminated precursor blocks (Scheme 1). Previously, the coupling of similar blocks to form multiblock copolymers has been performed at temperatures up to 180 °C.19,24–25 However, to lower the risk of transetherification reactions, the coupling was performed 130 °C in the present case.
A typical 1H NMR spectrum of a multiblock copolymer is shown in Fig. 2a. The signals from the end groups of the precursor blocks were absent in all the spectra of the copolymers, indicating an efficient coupling reaction and a high copolymer molecular weight. The molar ratio between the two blocks in the block copolymers was calculated from the 1H NMR data as m(m + n)−1, where m and n are the number of repeating units in the fluorine and hydroxy terminated precursor block, respectively (Fig. 2). The values presented in Table 2 coincide reasonably well with the theoretical ones calculated on basis of the Mn values of the precursor blocks, again confirming an efficient coupling reaction. The copolymers formed mechanically strong transparent films upon casting from 5 wt% NMP solutions, which indicated that sufficiently high molecular weights were reached. Yet again, SEC analysis using PS standards proved inaccurate and showed very low Mn values, in some cases even lower than the value of the precursor blocks evaluated previously.
 | ||
| Fig. 2 Representative 1H NMR spectra of multiblock copolymers before modification, (a) b3PAES-26-26, after benzylic bromination, (b) b3PAES-26-26Br, and after quaternization, (c) b3PAES-26-26Q. | ||
| Sample | nMHQ based precursora (kg mol−1) | BPA based precursora (kg mol−1) | m/(m + n) theoretical | m/(m + n)NMRa | M n (kg mol−1) | PDIb,c | T g (°C) | T d,95 (°C) | Degree of brominationa (%) | T d,95Br , (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Determined by SEC using polystyrene standards. c PDI: polydispersity index. d Evaluated by DSC at a heating rate of 10 °C min−1. e Measured under N2 by TGA. f T d,95 of the brominated derivative. | ||||||||||
| b2PAES-23-34 | 10.8 | 10.4 | 0.60 | 0.53 | 34 | 2.0 | 205 | 415 | ∼100 | 289 |
| b3PAES-23-26 | 9.6 | 10.4 | 0.53 | 0.50 | 25 | 1.6 | 222 | 384 | 98 | 254 |
| b3PAES-26-26 | 9.6 | 11.7 | 0.50 | 0.47 | 30 | 1.6 | 216 | 404 | 96 | 261 |
| b3PAES-33-26 | 9.6 | 14.6 | 0.44 | 0.38 | 26 | 1.6 | 216 | 456 | 94 | 260 |
| b3PAES-42-26 | 9.6 | 18.8 | 0.38 | 0.32 | 75 | 2.1 | 214 | 394 | 92 | 262 |
| b3PAES-42-49 | 18.2 | 18.8 | 0.54 | 0.49 | 42 | 1.7 | 219 | 398 | 92 | 276 |
| b4PAES-23-20 | 7.8 | 10.4 | 0.47 | 0.42 | 29 | 1.6 | 227 | 398 | 93 | 270 |
| b4PAES-42-20 | 7.8 | 18.8 | 0.32 | 0.27 | 41 | 1.6 | 213 | 464 | 94 | 276 |
3.2 Bromination and quaternization
The benzylic positions of the blocks containing the MHQ residues were selectively brominated using NBS with AIBN as the radical source. By employing a 500% excess of NBS, between 92 and ∼100% of the benzylic positions were brominated (Table 2). Initially the reaction was evaluated in DMF, chlorobenzene and 1,2-dichlorobenzene. The outcome showed that the latter was the solvent of choice since precipitation and gelation was observed in the other two cases. A typical 1H NMR spectrum of a benzylbrominated multiblock copolymer (baPAES-m-nBr) is shown in Fig. 2b. As seen, new peaks appeared between 4.0 and 4.8 ppm assigned to the methylene protons of the bromomethyl groups. Concurrently, the signal from the methyl protons at 2 ppm virtually disappeared to further confirm the successful bromination of the benzylic positions. The degree of bromination was calculated by integrating and comparing the signals of the –CH2Br protons (b) and the residual –CH3 protons from the MHQ monomer residues.The benzylbrominated copolymers were quaternized in NMP at room temperature using an excess of trimethylamine. Care was taken not to heat the quaternized copolymers during the subsequent purification step in order to avoid crosslinking before membrane casting. The resulting baPAES-n-mQ multiblock copolymers were not soluble in chloroform and the 1H NMR spectra were thus collected using DMSO-d6. As seen in Fig. 2c, the signals were considerably broadened after quaternization, partly due to the change of solvent and partly to the character of the cationic copolymers. The signal at approximately 3.1 ppm (h) was assigned to the QA (–CH3) groups indicating the successful quaternization.
Membranes with a thickness of approximately 50 μm were cast at 80 °C from NMP solutions of the quaternized copolymers. Mohr titrations of the membranes further confirmed successful quaternization since the experimentally determined IEC values of the AEMs corresponded well with the theoretical ones (Table 3).
| AEM | IECtargeta (meq. g−1) | IECNMR (meq. g−1) | IECtitr (meq. g−1) | Content of ionic blocksb (wt%) | T d,95 (°C) | Water uptakec (wt%) | λ | Δld (%) | σ 20 °C (mS cm−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Target IEC in the Br− form. b Determined by 1H NMR. c Measured by TGA under N2. d Fully hydrated membranes at 20 °C. | |||||||||
| b2PAES-23-34Q | 2.2 | 2.0 | 2.1 | 62 | 269 | 42 | 11 | 9 | 8.5 |
| b3PAES-23-26Q | 2.6 | 2.5 | 2.6 | 64 | 240 | 55 | 12 | 4 | 5.3 |
| b3PAES-26-26Q | 2.5 | 2.3 | 2.5 | 61 | 246 | 61 | 13 | 5 | 9.8 |
| b3PAES-33-26Q | 2.2 | 2.0 | 2.2 | 52 | 233 | 28 | 7 | 5 | 5.2 |
| b3PAES-42-26Q | 2.0 | 1.8 | 1.9 | 45 | 236 | 37 | 11 | 3 | 4.5 |
| b3PAES-42-49Q | 2.6 | 2.5 | 2.5 | 63 | 260 | 81 | 18 | 8 | 6.1 |
| b4PAES-23-20Q | 2.8 | 2.6 | 2.6 | 60 | 249 | 113 | 24 | 14 | 13 |
| b4PAES-42-20Q | 2.2 | 1.9 | 2.1 | 40 | 276 | 41 | 11 | 2 | 1.7 |
3.3 Membrane morphology
The average separation length between inhomogeneities in a membrane (d) can be evaluated by SAXS and is derived from the position of the scattering maximum (qmax) via Braggs law.26 The multiblock copolymer AEMs in the dry state showed quite broad scattering peaks with qmax in the range 0.19–0.27 nm−1 (Fig. 3). This corresponded to characteristic separation lengths of 23–33 nm. The magnitude of the separation lengths indicated that the scattering was linked to the microphase separation of the blocks and not to the formation of the kind of ion clusters usually observed in statistical copolymers which gives smaller separation lengths.23 In general, the influence of the molecular parameters of ionic multiblock copolymers on membrane morphology is complex due to the solvent casting process, the rather high polydispersity of the blocks, and the ionic and strongly polar nature of the hydrophilic blocks. In the present case, there was no apparent correlation between the block length and the characteristic separation length as usually found for non-ionic block copolymers consisting of low polydispersity blocks.9 The absence of higher order peaks indicated a low long-range order in these materials.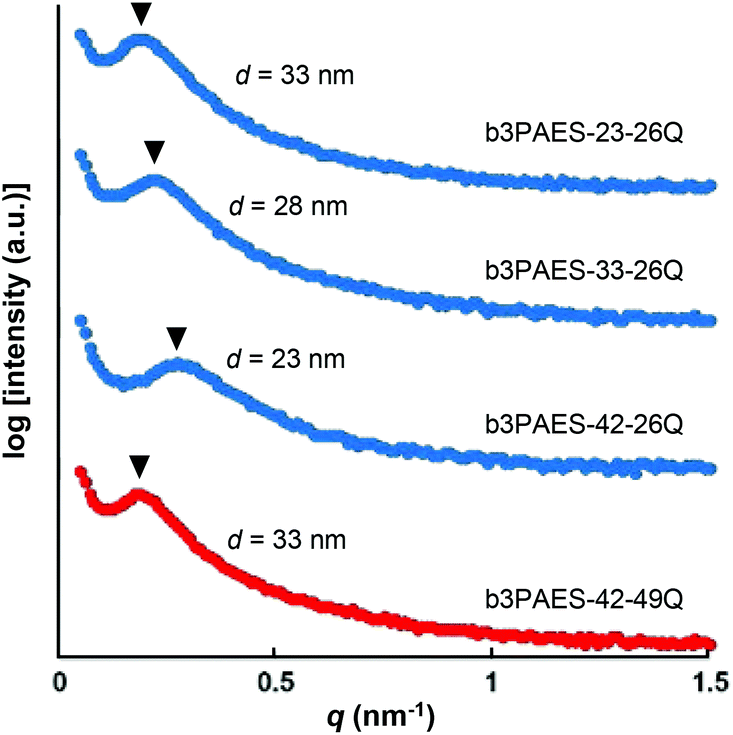 | ||
| Fig. 3 SAXS data of nominally dry multiblock copolymer membranes. The data have been shifted vertically for clarity and the arrowheads indicate qmax. | ||
AFM phase images of multiblock copolymer AEMs before modification showed a smooth and featureless surface (Fig. 4a). This was expected since these block copolymers consisted of two blocks with high Tgs which gave rise to two hard phases. On the other hand, the AFM phase images of the membranes after quaternization (Fig. 4b) indicated a phase separation between the hard hydrophobic phase (bright regions) and the softer hydrophilic phase (dark regions). Accordingly, the corresponding amplitude images show a much rougher surface for the AEM than for the unmodified copolymer (Fig. 4d and c, respectively). The estimated distance between the hydrophilic regions from the image in Fig. 4b is approximately 30 nm which is in the same range as the d value of 33 nm evaluated by SAXS. This indicated that similar repeating distances were present at the surface and in the bulk of the material, and furthermore supported the argument that the peaks observed in the scattering experiment correspond to the distance between the hydrophilic phase domains. The present domain distances were in the same range as those previously observed by AFM of sulfonated hydrophilic–hydrophobic multiblock copolymer membranes with similar block sizes.27
 | ||
| Fig. 4 AFM phase images of; (a) b3PAES-23-26 and (b) of b3PAES-23-26Q, and the corresponding amplitude images of; (c) b3PAES-23-26 and (d) b3PAES-23-26Q. | ||
3.4 Thermal properties
The thermal decomposition of the multiblock copolymers was studied by TGA under nitrogen atmosphere at a heating rate of 10 °C min−1. Prior to the measurements the samples were held at 150 °C for 10 min in order to remove any remaining water. The non-modified multiblock copolymers b2PAES-n-m, b3PAES-n-m and b4PAES-n-m possessed a high thermal stability with Td,95 between 384 and 464 °C. In Fig. 5 the TGA trace of b3PAES-33-26 shows a slight tendency for degradation in two steps (all TGA traces are found in Fig. S2†).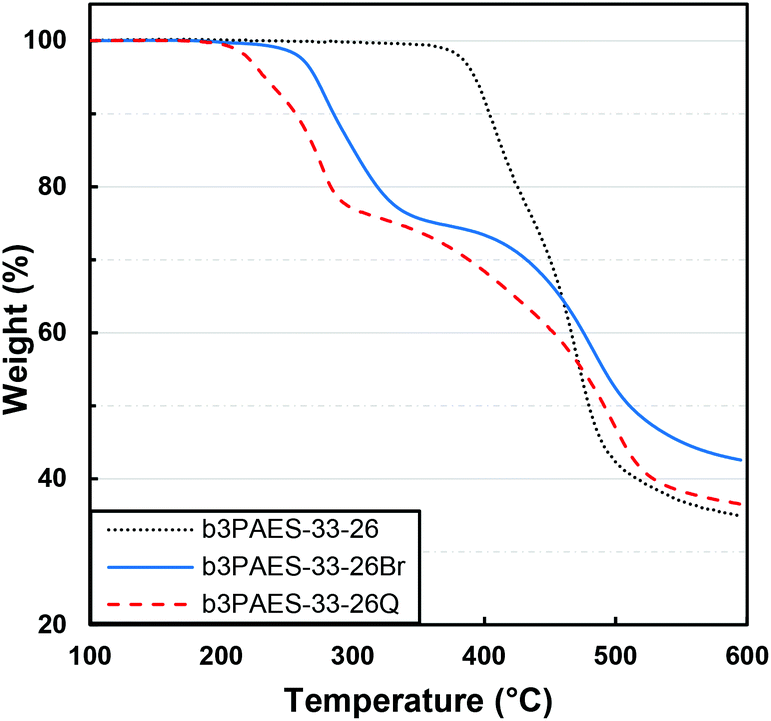 | ||
| Fig. 5 TGA traces of b3PAES-33-26 in the unmodified, benzylbrominated and quaternized form. | ||
The benzylbrominated multiblock copolymers degraded at a significantly lower temperature with Td,95 in the range between 254 and 289 °C. As seen in Fig. 5, these copolymers degraded in two steps, where the first most likely was due to loss of the brominated groups. The observed weight loss between 245 and 345 °C correlated with the wt% of CH2Br groups in the copolymer, as evaluated by NMR spectroscopy (see Fig. S3†). The second degradation step coincided with that of the unmodified multiblock copolymer, and was presumably due to decomposition of the copolymer backbone. The quaternized copolymers also degraded in two distinct steps (Fig. 5). The first degradation step started approximately 10–30 °C below that of the corresponding brominated copolymers.
The Tgs of the precursor blocks and the multiblock copolymers prior to modification were evaluated by DSC and the data are collected in Table 1 and 2, respectively. The Tg values of the 2MHQ, 3MHQ and 4MHQ based precursor blocks increased with the number of substituted methyl groups and were higher than the values for the BPA containing precursor blocks. This is presumably due to the stiffening effect of the benzylic methyl groups and the rather flexible linkage of the BPA residues. The Tg of the BPA containing precursor blocks increased slightly with increasing Mn as expected. On the other hand, the 3MHQ containing precursor blocks showed a rather unexpected raise in Tg with decreasing Mn. After coupling, the multiblock copolymers all displayed a single Tg around 210 °C. This indicated most likely that the blocks were at least partly miscible which may further explain the featureless AFM images of the multiblock copolymers prior to modification (Fig. 4a and c). After the quaternization, the AEMs showed no Tg below the decomposition temperature.
3.5 Water uptake
Some research groups have reported a high stability of PAESs under the strongly nucleophilic and basic conditions caused by OH−.28–30 However, other groups have reported stability issues including backbone degradation.31–34 By 1H NMR analysis before and after storage in an alkaline solution (1 M aq. NaOH, 40 °C, 7 days), we observed a substantial loss (∼80%) of QA functionality of an AEM based on a statistical PAES with a similar structure as the present multiblock copolymers (ESI, Fig. S4†). This observation clearly confirmed the poor stability of the AEMs in the presence of OH− and made further testing under alkaline conditions superfluous. Consequently, we studied the water uptake and ionic conductivity of the membranes in the Br− form to establish fundamental structure–property relationships without the need to consider possible effects of degradation. We anticipate that the transport data is still relevant because of the general ion conduction mechanism.The water uptake of the AEMs in the Br− form was measured under fully hydrated conditions as a function of temperature (Fig. 6). As expected, increasing temperatures led to increasing water uptake of most of the membranes. The b3PAES-n-26Q series, with a constant length of the quaternized block, showed the lowest water uptake with a trend of increasing water uptake as a function of IEC. Membranes b3PAES-26-26Q and b3PAES-42-49Q both had an IEC of 2.5 meq. g−1, but contain different block lengths. As seen, the latter membrane with the longer blocks clearly showed a higher water uptake (81 vs. 61 wt% at 20 °C). This finding was in line with most published work regarding sulfonated block copolymers. However, the opposite tendency has previously been reported for AEMs.22,25 From Fig. 6 it is clear that the membranes with the tetra-QA functionalized units took up more water than the corresponding membranes having tri-QA functionalized units at similar IEC. The reason may be the presence of a more distinct hydrophilic phase which allows the accommodation of large amounts of water. Membrane b4PAES-23-20Q had a very high IEC (2.6 meq. g−1) and ion concentration which led to an excessive water uptake. The hydration number (λ), i.e., the number of water molecules per QA group, was calculated from the water uptake data (Fig. S5†). As seen in Table 3, λ was found to be in the range 7–24 at room temperature with the high-IEC membranes displaying the highest values. The swelling ratio in the plane of the membrane was measured at room temperature (Table 3). For all membranes except for b4PAES-23-20Q the swelling was found to be moderate, in the range Δl = 2–14%. No freezing water was detected by DSC in any of the AEMs after full hydration and cooling at 5 °C min−1 to −60 °C.
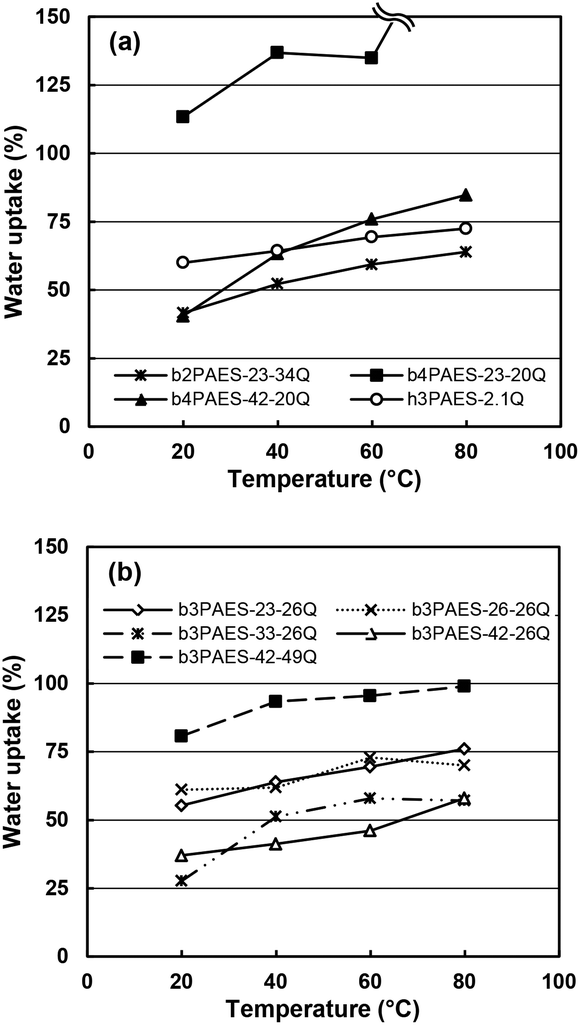 | ||
| Fig. 6 Water uptake data of the b2PAES-n-mQ, b4PAES-n-mQ (a), and the b3PAES-n-mQ (b) AEMs in the Br− form as a function of temperature under fully hydrated (immersed) conditions. The data is provided in two plots for clarity. | ||
3.6 Ion conductivity
The ionic conductivity of the AEMs in the Br− form was evaluated by EIS under fully hydrated conditions (Fig. 7a). A steep increase of the conductivity was observed when the temperature was raised from −20 to 20 °C due to the melting of ice. The b3PAES-n-26Q series showed increasing ion conductivity with increasing IEC. This can be correlated to the higher water uptake of the membranes with higher ionic content. Comparing the ionic conductivities of membranes b3PAES-42-49Q and b3PAES-26-26Q with similar IEC, the copolymer with longer blocks had a slightly lower conductivity. This may partly be due to a dilution effect caused by the higher water uptake of the former membrane. The copolymer AEM with the highest water uptake, b4PAES-23-20Q, also displayed the highest ion conductivity. However, because of the high water content this membrane was not as mechanically stable as the others. | ||
| Fig. 7 Br− conductivity of the AEMs in the fully hydrated (immersed) state as a function of temperature (a), as a function of water uptake in the temperature range 20–80 °C (b), and as a function of relative humidity at 80 °C (c). | ||
When comparing the Br− conductivity of b4PAES-42-20Q, b3PAES-33-26Q and b2PAES-23-34Q with IECs of 2.1, 2.2, and 2.1 meq. g−1, respectively, it is obvious that the Br− conductivity increased with decreasing local density of ionic groups. The water uptake of these membranes was 41, 28 and 42 wt%, respectively, at 20 °C. The most probable explanation for the trend in the conductivity is that counter-ion condensation increased when the QA groups were placed in very close proximity.35 This reduced the number of available charge carriers and subsequently the ion conductivity. The observation may also be partly explained by differences in morphology and degree of percolation since the ionic blocks of these multiblock copolymers constituted 40, 52 and 62 wt%, respectively (Table 3). Fig. 7b shows the ionic conductivity as a function of water uptake, and also indicates a higher conductivity of the b2PAES-23-34Q membranes in relation to the b3PAES-n-mQ and b4PAES-n-mQ AEMs at a given water uptake. In comparison, the Br− conductivity of a commercial AEM [FAA-3, FuMaTec GmbH, IEC = 2.1 meq. g−1 (Cl− form)] has been reported to reach 4–7 × 10−3 S cm−1 at water volume fractions between 0.4 and 0.7 at 25 °C.35
The Br− conductivity of membranes b3PAES-42-49Q, b3PAES-26-26Q and b2PAES-23-34Q was also evaluated as a function of relative humidity at 80 °C (Fig. 7c). Considering the former two AEMs, both with an IEC of 2.5 meq. g−1, the membrane with smallest block size (b3PAES-26-26Q) reached the highest ion conductivity in the entire investigated humidity range. This was also the case under fully hydrated conditions, indicating that the water absorbed by the membrane with smaller block sizes provided more efficient ion transport. The b2PAES-23-34Q membrane had a lower conductivity than the b3PAES-26-26Q membrane under reduced relative humidity, despite a comparable conductivity under fully immersed conditions. Probably, the latter membrane's slightly higher IEC and higher local ion concentration promoted the conductivity at low water contents.
3.7 Block versus statistical copolymers for AEMs
In order to clarify possible advantages of the multiblock copolymer architecture for AEMs, the properties need to be compared with more accessible copolymers. Hence, our previous data on corresponding statistical copolymers based on the same QA functionalized monomer units will serve as a suitable basis for comparison.23Fig. 8a shows schematically PAESs with different distributions of QA groups, including a randomly functionalized homopolymer (1), statistical copolymers containing di-, tri- and tetra-functionalized units (2–4), and multiblock copolymers containing segments with di-, tri- and tetra-functionalized units (5–7). The Br− conductivity and water uptake of AEMs based on these different polymers, all within a narrow IEC range (2.1–2.2 meq. g−1), are summarized in Fig. 8b and c. As seen, the randomly functionalized polymer displayed a comparatively low conductivity, despite a high water content. This membrane did not show any ionic clustering and hence the QA groups and the water can be expected to be dispersed throughout the membrane, leading to the low conductivity.23 When the QA groups were arranged in pairs in a statistical copolymer, they efficiently formed clusters and the conductivity increased considerably. Increasing the local ionic concentration by introducing tri- and tetra-functionalized sites resulted in a gradual decrease in the conductivity.23 The reason was most probably a decreased ionic dissociation because of the close proximity of QA groups in the copolymer structure, leading to a decrease in the charge carrier concentration.35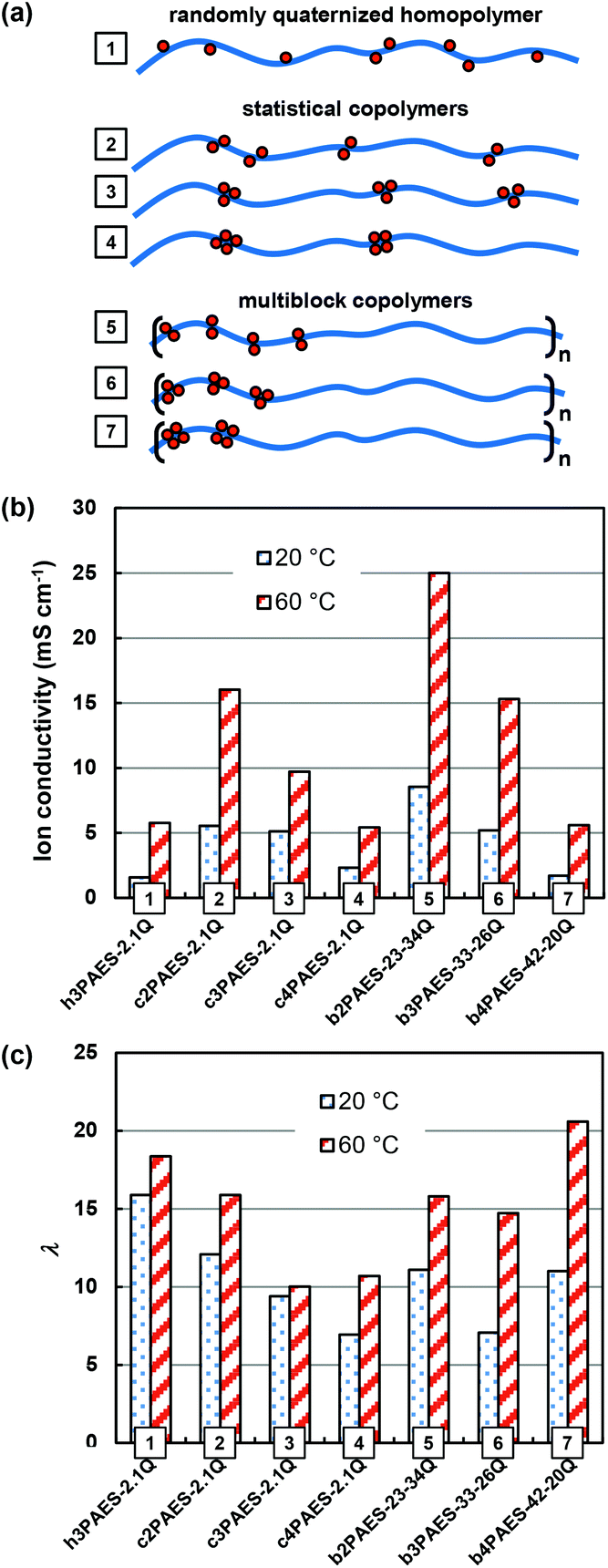 | ||
| Fig. 8 Schematic structures (a) showing the ionic distribution of a homopolymer randomly functionalized with QA groups (1), as well as statistical (2–4) and multiblock (5–7) copolymers with di-, tri-, and tetra-QA functionalized units. Comparative data of the corresponding Br− conductivity (b) and hydration number (c) data at 20 and 60 °C of the different PAES AEMs with the various ionic distributions (1–7) within a narrow IEC range (1.9–2.2 meq. g−1). Data for the non-block copolymers were taken from ref. 23. | ||
As already mentioned above, the multiblock copolymers of the present study showed a similar trend as the statistical copolymers with the conductivity decreasing with increasing local ionic concentration. The reason was most probably linked to decreasing ion dissociation, but may also result from a less favourable morphology of the densely functionalized block copolymers since the ionic block length, and hence the volume fraction, of the ionic blocks decreased with the local ionic concentration at a given IEC (Fig. 8a). The multiblock copolymers containing di-, and tri-QA functionalized units (b2PAES-23-34Q and b3PAES-33-26Q, respectively) showed a higher conductivity in comparison with the corresponding statistical membranes by a factor of 1.6. However, the statistical and multiblock copolymers with tetra-QA functionalized units, c4PAES-2.1Q and b4PAES-42-20Q, both had the same conductivity – at the same level as the randomly functionalized polymer. The multiblock copolymer with di-functionalized units showed a higher conductivity than the randomly functionalized polymer by a factor 4.2. This finding agrees very well with the results found by Watanabe et al. when comparing the OH− conductivity of a randomly QA functionalized polymer with a multiblock copolymer.22
The comparison of the ionic conductivities of statistical and multiblock copolymer AEMs strongly underscores the importance of the local distribution of the ionic groups in the copolymer structures. Still, if the ionic distribution and concentration are optimized, a block copolymer structure seems to reach a higher ionic conductivity. This is certainly true when comparing with a homopolymer with randomly distributed ionic groups. Critical factors for the conductivity are ion cluster formation which facilitates conductivity and counter-ion condensation which has a negative effect. Both these factors are favoured by increasing local ion concentrations.
4 Conclusions
Multiblock copolymers having hydrophilic blocks with dense and controlled distributions of QA groups were prepared by utilizing MHQ monomers substituted with two, three or four methyl groups. After precursor block formation and multiblock copolymer preparation, the benzylbromination and quaternization were selective and nearly complete. SAXS and AFM analysis verified that the block copolymer AEMs were phase separated to contain nanophases with very high local QA concentrations. The anionic conductivity of these membranes was found to decrease with increasing local ionic concentration, which is in contrast to the results normally found for the proton conductivity of sulfonated multiblock copolymer PEMs. The explanation is most probably that an increasing proximity of the QA groups induces a reduced ion-dissociation, resulting in a decreasing charge carrier concentration. Still, at a similar IEC the multiblock copolymer with di-substituted MHQ residues showed a markedly higher conductivity when compared with a polymer having randomly placed ionic groups. The present results strongly emphasize the importance of the local ionic distribution and concentration of AEMs based on multiblock copolymers if the conductivity is to surpass that of more easily accessible statistical copolymers.Acknowledgements
We thank the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning for financial support. We are also grateful to Marc Obiols-Rabasa for assistance with SAXS measurements and data treatment, and to Zhecheng Shao for acquiring the AFM images.Notes and references
- D. Y. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, ACS Appl. Mater. Interfaces, 2013, 5, 7559 CAS.
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187 CrossRef CAS.
- Y. J. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054 CrossRef CAS PubMed.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1727 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1 CrossRef CAS PubMed.
- G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521 CrossRef CAS PubMed.
- G. Maier and J. Meier-Haack, Adv. Polym. Sci., 2008, 216, 1 CAS.
- Y. A. Elabd and M. A. Hickner, Macromolecules, 2011, 44, 1 CrossRef CAS.
- Y. Yang and S. Holdcroft, Fuel Cells, 2005, 5, 171 CrossRef CAS.
- K. Matsumoto, T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 1161 CrossRef CAS.
- O. Savard, T. J. Peckham, Y. Yang and S. Holdcroft, Polymer, 2008, 49, 4949 CrossRef CAS PubMed.
- Y. Chang, G. F. Brunello, J. Fuller, M. L. Disabb-Miller, M. E. Hawley, Y. S. Kim, M. A. Hickner, S. S. Jang and C. Bae, Polym. Chem., 2013, 4, 272 RSC.
- J. H. Pang, S. N. Feng, Y. Y. Yu, H. B. Zhang and Z. H. Jiang, Polym. Chem., 2014, 5, 1477 RSC.
- E. P. Jutemar and P. Jannasch, J. Membr. Sci., 2010, 351, 87 CrossRef CAS PubMed.
- X.-F. Li, F. P. V. Paoloni, E. A. Weiber, Z. H. Jiang and P. Jannasch, Macromolecules, 2012, 45, 1447 CrossRef CAS.
- Z. Zhu, N. M. Walsby, H. M. Colquhoun, D. Thompsett and E. Petrucco, Fuel Cells, 2009, 9, 305 CrossRef CAS.
- M. S. Jung, T.-H. Kim, Y. J. Yoon, C. G. Kang, D. M. Yu, J. Y. Lee, H.-J. Kim and Y. T. Hong, J. Membr. Sci., 2014, 459, 72–85 CrossRef CAS PubMed.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2684 CrossRef CAS.
- Z. Zhao, J. H. Wang, S. H. Li and S. B. Zhang, J. Power Sources, 2011, 196, 4445 CrossRef CAS PubMed.
- D.-Y. Park, P. A. Kohl and H. W. Beckham, J. Phys. Chem. C, 2013, 117, 15468 CAS.
- M. A. Hossain, Y. Lim, S. Lee, H. Jang, S. Choi, Y. Jeon, J. Lim and W. G. Kim, Int. J. Hydrogen Energy, 2014, 39, 2731 CrossRef CAS PubMed.
- M. Tanaka, K. Fukasawa, E. Nishino, S. Yamaguchi, K. Yamada, H. Tanaka, B. Bae, K. Miyatake and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 10646 CrossRef CAS PubMed.
- E. A. Weiber and P. Jannasch, ChemSusChem, 2014, 7, 2621 CrossRef CAS PubMed.
- A. H. N. Rao, R. L. Thankamony, H.-J. Kim, S. Nam and T.-H. Kim, Polymer, 2013, 54, 111 CrossRef CAS PubMed.
- X. Li, Y. Yu, Q. Liu and Y. Meng, J. Membr. Sci., 2013, 436, 202 CrossRef CAS PubMed.
- G. Gebel and O. Diat, Fuel Cells, 2005, 5, 261 CrossRef CAS.
- S. Takamuku and P. Jannasch, Adv. Energy Mater., 2012, 2, 129 CrossRef CAS.
- J. H. Wang, J. Wang, S. H. Li and S. B. Zhang, J. Membr. Sci., 2011, 368, 246 CrossRef CAS PubMed.
- A. H. N. Rao, H.-J. Kim, S. Nam and T.-H. Kim, Polymer, 2013, 54, 6918 CrossRef CAS PubMed.
- J. Pan, C. Chen, Y. Li, L. Wang, L. S. Tan, G. W. Li, X. Tang, L. Xiao, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2014, 7, 354 CAS.
- D. Chen and M. A. Hickner, ACS Appl. Mater. Interfaces, 2012, 4, 5775 CAS.
- J. Miyake, K. Fukasawa, M. Watanabe and K. Miyatake, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 383 CrossRef CAS.
- S. A. Nuñez and M. A. Hickner, ACS Macro Lett., 2013, 2, 49 CrossRef.
- N. Li, Q. Zhang, C. Y. Wang, Y. M. Lee and M. D. Guiver, Macromolecules, 2012, 45, 2411 CrossRef CAS.
- M. G. Marino, J. P. Melchior, A. Wohlfarth and K. D. Kreuer, J. Membr. Sci., 2014, 464, 61 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: FTIR spectra, all TGA traces, the correlation between the Br content determined by 1H NMR and TGA, evaluation of AEM stability under alkaline conditions, and the hydration number of the AEMs as a function of temperature. See DOI: 10.1039/c4py01588f |
| ‡ Present address: Department of Chemistry, University of Hamburg, Martin-Luther-King Platz 6, 20146 Hamburg, Germany |
| This journal is © The Royal Society of Chemistry 2015 |