
Impact of dendritic polymers on nanomaterials
R.
Soleyman
a and
M.
Adeli
*bc
aPolymer Science and Technology Division, Research Institute of Petroleum Industry (RIPI), Tehran, Iran
bDepartment of Chemistry, Faculty of Science, Lorestan University, Khorramabad, Iran. E-mail: mohadeli@yahoo.com; m.aadeli@fu-berlin.de
cInstitut für Chemie und Biochemie, Freie Universität Berlin, Germany
First published on 2nd October 2014
Abstract
For many years scientists have employed dendritic polymers (dendrimers and hyperbranched polymers) in association with other nanomaterials (such as graphene, carbon nanotubes, proteins and peptides, as well as metallic nanoparticles) to synthesize hybrid nanomaterials with improved biocompatibility, biodegradability, functionality, physicochemical properties and the capability of carrying other molecules. However, more recent studies demonstrate that one of the less noticed effects and newly observed facets of dendritic polymers is their role in changing the structure (shape, size and sheet multiplicity) of the obtained hybrid nanomaterials, upon covalent and noncovalent interactions. In this review, we intend to have a more specialized look at these reports and discuss the ‘whys’ and ‘hows’ of this phenomenon.
 Rouhollah Soleyman | Rouhollah Soleyman was born in 1983 in Karaj (Iran). He obtained all of his academic degrees (BSc, MSc and PhD) in Chemistry from Sharif University of Technology (2002–2012). Now, he is senior researcher at the polymer science and technology division of the Research Institute of Petroleum Industry (RIPI). His research interests include synthesis and application of polymers, nanoparticles and hybrid nanomaterials. |
 Mohsen Adeli | Mohsen Adeli received his undergraduate degree from Lorestan University (Iran) in 1996. He obtained his MS and PhD degrees from Tabriz University. After joining Lorestan University in 2005, he added a postdoctoral stage from the Institute for Nanoscience and Nanotechnology of Sharif University of Technology in 2007. He was promoted to the rank of associate and full professor in 2010 and 2013, respectively. Mohsen Adeli was awarded the Razi Prize (2011) and prize for the most developed professor in chemistry (2012) administered by the Ministry of Health and Medical Education of Iran and Iran Nanotechnology Initiative Council, respectively. He joined the Free University of Berlin as a guest professor in 2014. |
1. Introduction
Common polymer science is mainly based on linear polymers. However, because the properties of branched macromolecules are quite different from conventional polymers, in the last three decades dendritic polymers have found a superior place in polymer technology. Dendritic structures introduce a variety of benefits compared to other architectural forms of polymers, including monodispersity, nanoscale size and host–guest potential and high ability as molecular shuttles, which make their passage easier through biological barriers such as vascular endothelial cells. Because of these outstanding characteristics of dendritic polymers, their applications in drug and gene delivery,1–11 cancer diagnosis and therapy,1,12–18 stimuli-responsive nanocarriers,19,20 catalysis,21,22 self-assembly,23,24 nanomedicine development,25–27 and biomedical applications28–30 have been repeatedly reviewed. Moreover, various methods for the synthesis and functionalization of different types of dendritic polymers have been studied in several reviews and papers.22,31–36 In addition to these, in a special review, the merits of chemotherapeutic drugs-dendrimer covalent conjugation have been reported compared to noncovalent encapsulation methods.37 Moreover, in 2012, glycodendrimers as functional antigens and antitumor vaccines have been properly reviewed.38In the current review, we have focused on the effect of dendritic polymers on the structure of the prepared hybrid nanomaterials of graphene, carbon nanotubes (CNTs), metallic nanoparticles and proteins/peptides via covalent–noncovalent interactions. Indeed, we have revealed the newly observed facets of dendritic polymers as transformers.
2. Dendritic polymers
Dendritic polymers are mostly globular macromolecules, which have potential that cannot be concurrently found in other materials. These polymers are almost the latest member of the polymer family, whose name originates from the Greek words ‘dendron’ meaning tree and ‘meros’ meaning part. In 1978, the first report on the synthesis of these nanomaterials was published by Vogtle et al. in the synthesis of branched polypropylene-amine ‘cascade molecules’. In the middle of the 1980s, Tomalia et al.39 (Dow chemical company) and Newkome et al.40 in parallel synthetic investigations reported the synthesis of monodisperse poly(amidoamine) (PAMAM) ‘dendrimers’ and poly(ether-amide) ‘arborols’, respectively. Thereafter, dendronization has always been considered as a useful synthetic strategy for the preparation of multifunctional materials.41–44 Nowadays, dendritic polymers have extensively attracted scientists attention because of their unique merits in biomedicine and biomedical backgrounds.2.1. Structure
Dendritic polymers are a series of macromolecules, which synthetically have a core accompanied by side branches. The core may be a small organic molecule with the ability to form several functional groups, or another macromolecule with a high functionality. On the whole, from the structural viewpoint, dendritic polymers can be classified in two major groups: monodisperse frameworks such as dendrimers and dendrons, and polydisperse frameworks including hyperbranched polymers, dendrigrafts and linear-dendritic polymers (Fig. 1).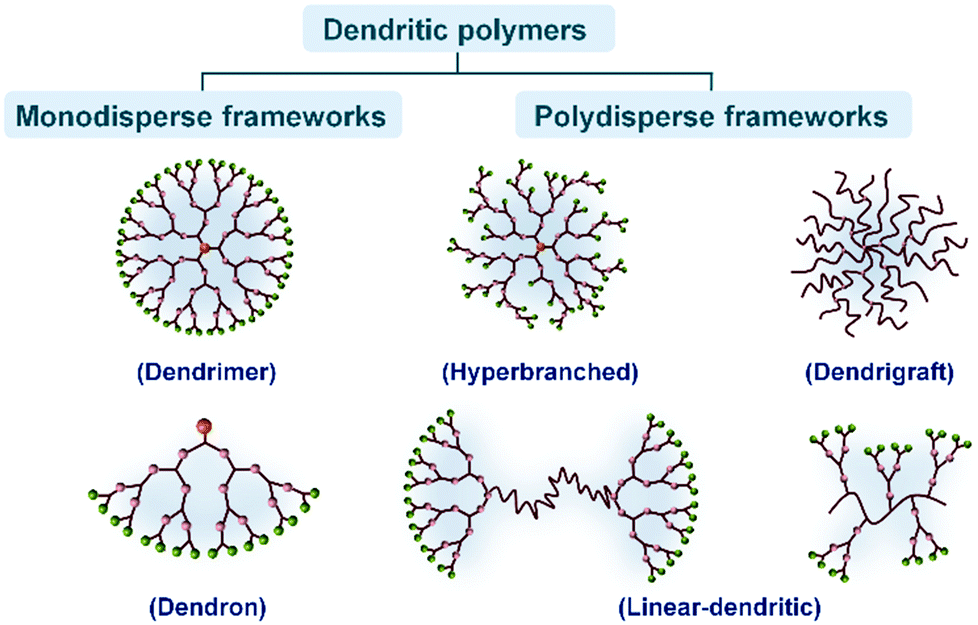 | ||
| Fig. 1 Schematic classification of the dendritic polymers including two major categories and their subgroups. | ||
Among all dendritic polymers, dendrimers are the most important and most studied family. Dendrimers are defined as globular, monodisperse polymers with three-dimensional architectures, in which all the bonds radially emerge from a central focal point (core) with repeating units that each contribute to a branching point in the dendrimer (Fig. 2a).45 Dendrimers possess three distinguishable architectural components: an interior core, interior layers (generations) composed of repeating units radially attached to the interior core, and exterior (terminal functionality) attached to the outermost interior generation46 (Fig. 2b). The higher-generation dendrimers, because of their globular structure, occupy a smaller hydrodynamic volume compared to the corresponding linear polymers. The dendritic scaffold may contain different types of functional groups such as secondary or tertiary amines in the case of polyamidoamine that enable the entrapment of drug molecules via hydrogen bonding, ionic bonding or hydrophobic interactions. The outer layer possesses a defined number of reactive functional groups that can be modified to influence the valency, solubility, tissue binding, and pharmacokinetics (e.g. PEG, PEGylation, acetylation, glycosylation, and amino acid functionalization) or biodistribution properties of the dendrimer (Fig. 2c). In addition, the surface reactive groups may also be used to covalently attach the drug molecules via labile chemical linkages (ester, amide, hydrazone, peptide, disulfide spacer.). Using these approaches, dendrimers can be constructed with precise control over several generations and surface functionalities, producing structures with typically high monodispersity when compared to the synthesis of traditional polymers, which typically produces statistical mixtures of products.
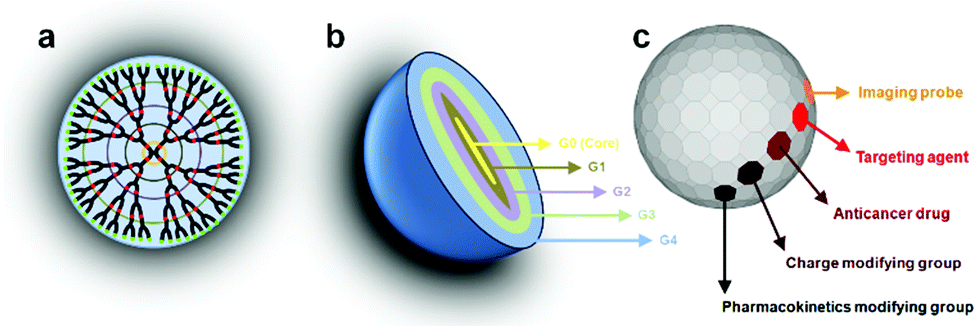 | ||
| Fig. 2 (a, b) Representation of the layers or generations (G) of a dendrimer. (c) The surface may be composed of any appropriate functional group. | ||
Although dendrimers have a unique size and symmetric structure with branching units without any structural defects, the tedious stepwise procedure for dendrimers often results in expensive products with limited availability, and therefore, results in a restricted use for large-scale industrial applications. Unlike dendrimers, hyperbranched and dendritic polymers are often easy to synthesize in a large-scale single-step synthetic process, and therefore, are considered to be potential alternatives to perfectly branched dendrimers in the field of engineering materials.47–49 However, the one-step procedures used for the preparation of hyperbranched polymers lead to uncontrolled statistical growth. Consequently, the resulting structures are imperfect and polydisperse. Furthermore, unlike dendrimers, the control over layers or generations, as well as over molecular weights deteriorates.
2.2. Toxicity
Although all the toxicity issues of dendrimers and solutions for their prevention have been recently summarized in a review paper,50 in this study, we briefly mention some key points. The toxicity of dendrimers in biological systems is generally characterized by hemolytic toxicity, cytotoxicity and hematological toxicity. Generally, the in vivo toxicity of dendrimers is attributed to the surface cationic charge interactions of dendrimers with negatively charged biological membranes, which can result in membrane disruption via pore formation, membrane thinning and erosion. To overcome or minimize this toxicity two major strategies can be employed: (i) the design and synthesis of biocompatible dendrimers via employing a biodegradable core and branching units or utilizing intermediates of various metabolic pathways and (ii) masking of the peripheral cationic charge of dendrimers by the neutralization of charge, for example PEGylation, acetylation, carbohydrate and peptide conjugation, or by introducing negative charge such as half-generation dendrimers. It has been shown that neutral and negatively charged dendrimers do not interact with the biological environment, and hence, are compatible for clinical applications. Moreover, it has been reported that hydrophobically derivatized hyperbranched polyglycerol (PG) can be a suitable substitute for human serum albumin.513. Surface modification of nanomaterials with dendritic polymers
The prefix “nano”, derived from the Greek “nanos”, signifying “dwarf”, is becoming increasingly common in scientific literature. Nano is now a popular label for modern science, and several nano-words have recently appeared in dictionaries, including nanometer, nanoscale, nanoscience, nanotechnology, nanostructure, nanotube, nanowire, and nanorobot.52 Nanostructured materials are the core components of nanotechnology, providing basic building blocks for fabricating complex devices with desired functions. Owing to their inherent quantum size and shape effects, nanomaterials have several important applications in electronics, optoelectronics, information processing, catalysis, biomedical science, environmental science, energy conversion and storage, advanced defence technologies, and various other fields.53 In the past years, numerous novel hybrid nanomaterials have been developed for biomedical applications.54 Among the polymer materials employed for surface modification of hybrid nanomaterials, dendritic polymers have obtained significant interest because of their interesting architecture and special chemical and physical properties.55,56 Dendrimers and hyperbranched polymers exhibit a highly branched, non-entangled architecture and numerous terminal groups. They exhibit considerably lower melt and solution viscosities and higher solubility in comparison with their linear analogues. Hence, dendrimers or hyperbranched polymers-functionalized nanomaterials even with a low functionalization degree are easier to solubilize. The lower viscosity of the functionalized nanomaterials in a solution or the molten state will favour the manipulation and processing of the nanohybrids. Furthermore, dendrimers and hyperbranched polymers can be advantageously used as functional surface and interfacial materials. The numerous terminal groups facilitate further modification and can be covalently modified with some linear polymers or a broad range of functional groups such as fluorophores, electroactive groups, perfluorinated moieties, and dyes. On the other hand, they can be used to entrap or stabilize various metal, metal oxide and semiconductor nanoparticles. Therefore, dendrimers and hyperbranched polymers are ideal materials for the modification of nanomaterials.4. Changing the structure of nanomaterials by dendritic polymers
The main goal of this review is to study the whys and hows of the impact of dendritic polymers on changing the structure of other nanomaterials. For example, it has been shown that dendritic polymers are able to result in direct graphite delamination, the formation of CNTs and graphene nanocapsules, prevention of amyloid formation and metallic nanoparticles size control. Here, as a major part of the paper, we extensively review the impact of dendritic polymers on graphenes, CNTs, proteins/peptides and metallic nanoparticles.4.1. Graphene
Graphene as a newly emerged carbon material has attracted much attention because of its outstanding properties and a wide range of fascinating applications. However, the mass production problem has restricted its extensive use in industry. The reduction from graphene oxide has been considered as one of the potential procedures for mass-scalable preparation. However, it suffers from restacking of the final graphene sheets after reduction because of the strong intersheet interactions. Recently, the utilization of dendritic polymers for edge-functionalization of graphene has attracted considerable attention. Because of their high solubility in solvents and natural bulky nanostructures, dendritic polymers are able to effectively prevent restacking of graphene sheets and provide a solution for the abovementioned problem. Although there are numerous reports and review papers in the literature about the synthesis and applications of nanomaterials based on graphene in biomedical and biomedicine areas including drug delivery,57–63 cancer therapy,62,64–68 non-viral-based gene therapy,69,70 tissue engineering,48 cellular imaging,61,71 and biosensors,62,72–74 its unique nature and inimitable properties have forced scientists in all fields to accelerate their researches on this ‘magic’ material.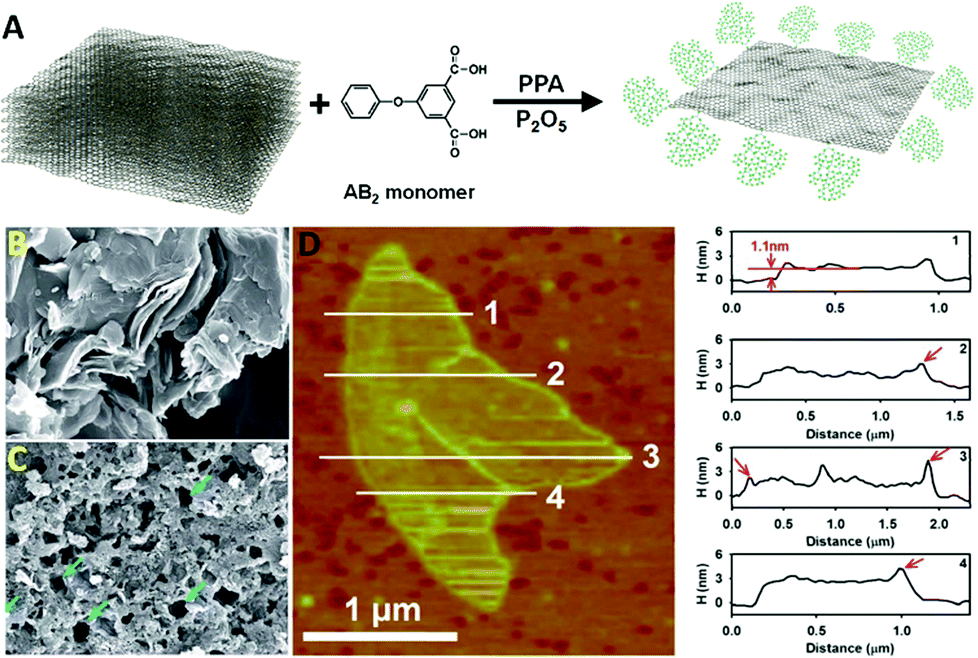 | ||
| Fig. 3 (A) Schematic representation of the direct delamination of graphite; SEM images of (B) pristine graphite and (C) HPEK-g-graphite; (D) AFM image (left) of HPEK-g-graphite with topographic height profiles (right). Reproduced from ref. 75 with permission from the Royal Society of Chemistry. | ||
The enzymatic functionalization of graphene in certain sites and with special patterns has been already reported.81 Because of the selectivity of enzyme pocket and also the topology of graphene sheets; different factors dominate the pattern of the functionalization and properties of the final hybrid nanomaterials. For example the functionalization of graphene oxide by polyethylene glycol mediated by Novozyme leads to hybrid nanomaterials possessing PEG on their surface. However, the same method for the functionalization of graphene but using citric acid as a substrate leads to functionalized hybrid nanomaterials having hyperbranched polycitric acid in circular shapes on their surface.76
In another report, a remarkable dendrimer size effect on the solubility and bulk electrical conductivity of dendronized graphene has been extensively described.77 In this work, a one-step covalent functionalization and simultaneous reduction of graphene oxide (GO) with dendritic anilines (DnPhNH2) has resulted in dendronized graphenes, named DnG, where n is the generation number of the dendritic structure having a value of 0–3 (Fig. 4). It is well known that the reduction from GO is one of the potential method for the mass-scalable preparation of graphene. However, as mentioned earlier, it is faced with the problem of graphene sheets restacking after reduction. Surprisingly, Li et al.77 have found that the graphene dendronization process not only hampers the restacking of the final graphene sheets and significantly increases their dispersity in a variety of organic solvents, but also interestingly reinforces the bulk electrical conductivity with an appropriate dendritic structure. The recovery of a honey-like crystalline lattice and few-layer characteristics of these dendronized graphenes have been revealed by the TEM technique. As shown in Fig. 4, most of the sheets are transparent and have a silk-like wrinkle. In some cases, folded layers were observed. The well-defined six-fold symmetry patterns in normal-incidence selected-area electron diffraction (SAED) for all the samples are similar to that of liquid-exfoliated graphene, demonstrating that the obtained dendronized carbon nanosheets have good crystallinity. These observations have demonstrated that most of the oxygen moieties have been removed during dendronization and the crystalline lattice of graphene has been re-established. Furthermore, the restacking of graphene sheets in cases of D0G has occurred, while that was rare for D2G and D3G, illustrating that a large dendritic wedge can efficiently prevent the agglomeration of carbon sheets. In addition, the AFM images of D1G, D2G, and D3G have shown a film thickness of 1.1, 1.5, and 2.1 nm, respectively. These values are greater than the literature values for single-layer GO, but in the range of chemically modified graphenes.78–81 The increment in thickness as compared with GO is reasonable when taking the bulky size of the dendritic wedges into consideration. However, AFM of D0G has shown aggregated objects in place of single flat sheets. This contrast demonstrates the importance of bulky dendritic wedges in suppressing the re-aggregation of the carbon sheets after reduction. As a great achievement of this study, the dispersity has shown a marked dependence on the size of the dendritic structure, in which the larger dendritic substituents afford better dispersity. Surprisingly, dendronization with an appropriate size of dendritic structure does not reduce, but significantly enhances the bulk electrical conducting capability. Furthermore, another additional bonus of this dendronization is that the final graphene products can be easily separated and purified from the unreacted starting materials, reaction media, and other impurities, simply by filtration and washing. Moreover, the final dendronized graphene products are solid powders and can be redispersed in any desired organic solvents. This feature is highly appealing for bulk production because it is beneficial for storage and transportation. The applications of these highly conductive and easily dispersed dendronized graphenes in reinforced composites and optoelectronic devices are worthy of further investigation.77
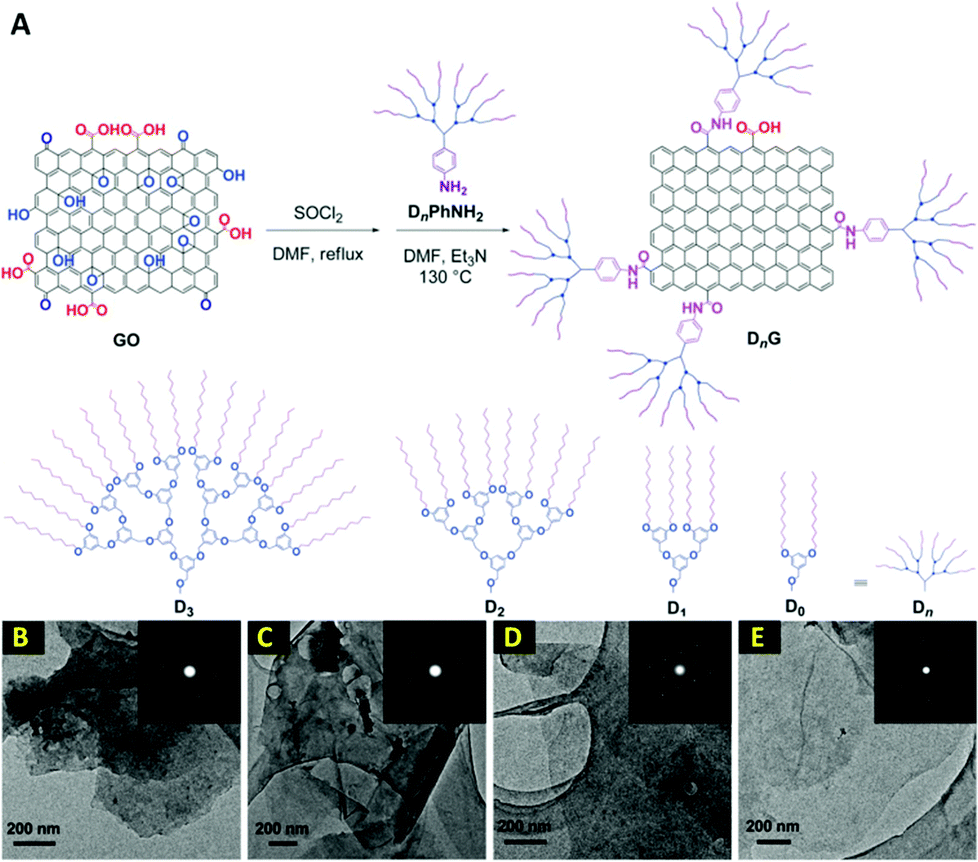 | ||
| Fig. 4 (A) Schematic description of the synthesis of dendronized graphenes; (B–E) TEM images of D0G, D1G, D2G, and D3G, respectively. Insets show SAED patterns. Reproduced from ref. 77 with permission from the Royal Society of Chemistry. | ||
The functionalization of graphene by biopolymers and proteins such as albumin to produce anticancer drug delivery systems has been reported. The conjugation of albumin onto the surface of graphene by a PEG linker leads to globular hybrid nanomaterials, which are able to transfer anticancer drugs, such as paclitaxel, both by the graphene surface and the albumin cavity.82
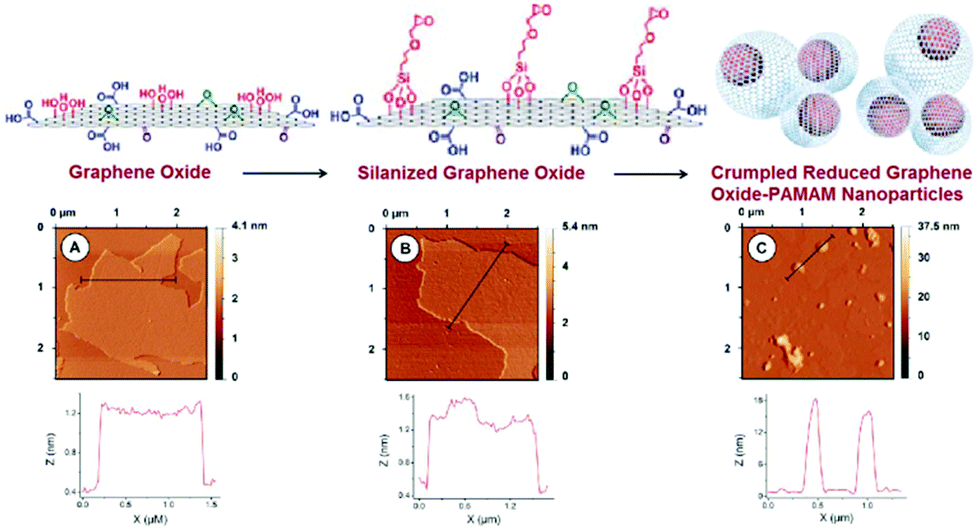 | ||
| Fig. 5 Schematic display of the steps involved in the preparation of PAMAM-Sil-rGO. AFM and section analysis for (A) GO, (B) Sil-GO and (C) PAMAM-Sil-rGO. Reproduced from ref. 83 with permission from the Royal Society of Chemistry. | ||
The shape of these nanomaterials varied from compact nanoparticles to less ordered structures, in which pendant and wrinkled graphene sheets were observed in some cases. In addition, some partially creased graphene sheets were also detected. The graphene nanoparticles showed high polydispersity with a diameter distribution ranging from 100 nm to 850 nm, and with a peak size of 420 nm. Moreover, the morphology of the nanoparticles was also studied by the FESEM technique. The crumpled morphology of the PAMAM-Sil-rGO nanoparticles was confirmed, but it was also revealed that they can be formed either by crumpling of a single graphene sheet or by the association of several of these nanomaterials. In some cases, it seems that a single graphene sheet can be involved in the formation of two or more nanoparticles yielding larger and more complex condensed structures. This fact can contribute to the high size polydispersity exhibited by the crumpled nanostructures. It should also be noted that graphene oxide prepared by Hummers method is a highly polydisperse material, with sheets ranging from a hundred nanometers to several micrometers in size. Thus, the production of crumpled nanostructures with a great variety of diameters can be expected.83
In addition, in one of our newest researches, we have selectively functionalized graphene sheets from the edges by hyperbranched polyglycerol (HPG) having a polycyclic focal point.84 Surprisingly, edge-functionalized graphenes have the capability of forming self-assembled nanocapsules in aqueous solutions, because of the hydrophilicity of their functionalized edges and hydrophobicity of their flat surface. These nanocapsules are able to efficiently deliver hydrophobic molecules such as anticancer drugs, e.g. doxorubicin (DOX). For the preparation of our considered HPG-based dendritic polymer, we arranged a set of reactions for our macromolecular architecture. On the basis of the plan, first, a tricyclic molecule was synthesized by a nucleophilic substitution reaction between cyanuric chloride and naphthol. Then, the third chlorine substitute of the triazine moiety of 2-chloro-4,6-dinaphthoxy-1,3,5-triazine was replaced by diethanolamine and 2-diethanolamino-4,6-dinaphthoxy-1,3,5-triazine (DDT) was obtained (Fig. 6A). Then, DDT was used as a macroinitiator in the anionic ring-opening polymerization of glycidol and DDT-g-HPG (DDTP) was synthesized. As can be seen from Fig. 6, by changing the angle between naphthol(s) and triazine rings, many conformations for the aromatic focal point of DDTP are possible, when it is interacting with graphene sheets. However two extreme cases are B and C, which are shown in Fig. 6. All other cases lie between these two cases. Actually, case B explains the sandwiching of the graphene edges by the polymer, whereas case C shows the stacking of the focal point of DDTP onto the surface of graphene. Additionally, we have visually indicated that HPG with a bidentate aromatic segment in its focal point, DDTP, is able to interact with graphene edges and form self-assembled nanostructures (Fig. 6D). Because of the hydrophobicity of the flat surface of the edge-functionalized graphenes and hydrophilicity of their edges, they changed their conformation from the extended to the closed state and formed nanocapsules in aqueous solutions. Spectroscopy and microscopy evaluations showed that the average size of nanocapsules is 300 nm. The aqueous solutions of nanocapsules and those with encapsulated doxorubicin were stable at room temperature for several weeks.72 In addition, we have demonstrated that DDTP-G-DOX is globular and its size is around 400 nm. TEM images not only confirm the functionalization of graphene by DDTP from the edges, but also display a new conformation for edge-functionalized graphenes in aqueous solutions.84
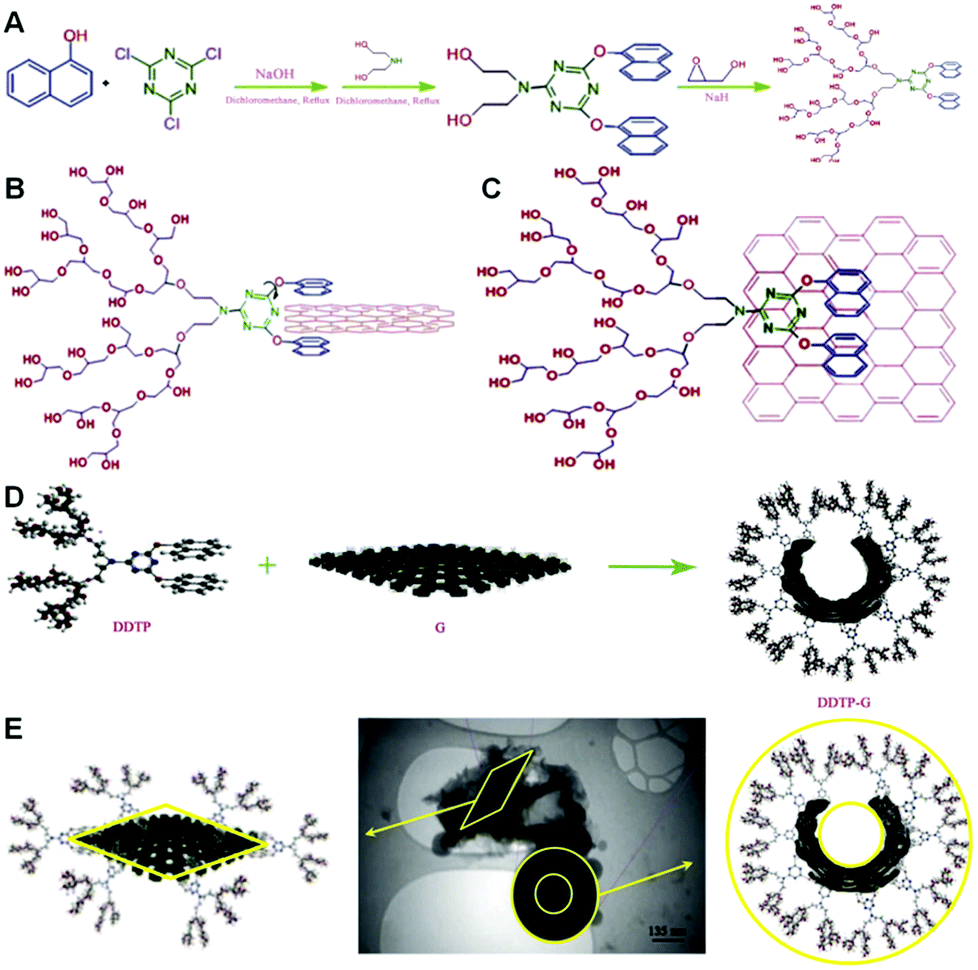 | ||
| Fig. 6 (A) Schematic representation of the synthesis of DDTP, (B and C) schematic representation of two extreme conformations for naphthol rings in the focal point of DDTP when it is interacting with the surface of graphene, (D) interactions between graphene sheets and DDTP to produce globular shapes, and (E) TEM image of the nanocapsules with a hydrophobic core and hydrophilic shell and hydrophobic graphene sheet. Reprinted from ref. 84 Copyright (2013) with permission from Elsevier. | ||
Moreover, hyperbranched polymeric ionic liquids (hyper-PILs) having C18 aliphatic chains with onion-like topology have been synthesized and employed for dispersing functionalized graphene (FG) in toluene.85 FG, prepared by the thermal reduction of graphite oxide, is readily dispersed in polar solvents by high-pressure homogenization, and mainly forms single FG nanosheets.86 In the presence of C18-hyper-PILs, stable FG dispersions have been formed in nonpolar media, such as toluene, whereas the dispersions of FG in pure toluene undergo rapid sedimentation. In fact, the interactions between the C18-hyper-PILs dendritic polymer and FG nanosheets have resulted in a hybrid nanomaterial, which is highly soluble in nonpolar solvents.
In addition to the interactions between graphene sheets and dendrimers, the driving forces of conformational changes in single-layer graphene oxide have interestingly been studied. It has been indicated that single-layer graphene oxide (SLGO) in the wet state is dried from the wet state, and in the dry state exhibits very different textural characteristics that are governed by the chemical environment, at particular pH, as well as its oxygen-containing functional groups and their surface charge. At basic pH, SLGO sheets tend to fold owing to the electrostatic repulsion between negatively charged deprotonated acidic groups, and SLGO suspensions are stable, whereas at low pH, SLGO suspensions are destabilized owing to stronger intersheet interactions. Upon drying, SLGO sheets form dense aggregates with complex structure and porosity, and low BET surface area. Depending on the geometric size of SLGO sheets and the drying conditions, they can unfold or remain folded because of the hydrogen bonding between polar surface functional groups and dispersion interactions between nonpolar graphene fragments. Therefore, a careful selection of the chemical environment and the surface functionality and reactivity of single-layer graphene oxide sheets is required when tailoring SLGO for specific applications.87 Overall, it is well known that SLGO sheets undergo morphological changes depending on the pH of the system and this may account for their restricted chemical reactivity. They can also capture nanoparticles through layering and enveloping when the pH is changed, demonstrating potential usefulness in drug delivery or waste-material capture.88
4.2. Carbon nanotubes (CNTs)
In our research group, we have extensively grafted hyperbranched PCA and PG as highly biocompatible polymers on the MWCNTs surface to produce nanomedicines.96,98,100,109–111 In another research, we synthesized MWCNT-g-PCA-PTX as a highly efficient anticancer drug delivery system. Surprisingly, during the functionalization of MWCNTs by PCA, the conformation of the nanotubes changed from a linear toward a circular type because of secondary interactions between PCA macromolecules (Fig. 7).99 This system is one of the most surprising, and the first system observed by our group in which CNTs cannot be found in their extended conformation and the drug delivery system is in the form of nanoparticles. TEM and AFM images show that functionalized CNTs are agglomerated to form bigger nanoparticles. Because CNTs are functionalized by PCA branches that have numerous carboxyl functional groups on their surfaces, hydrogen bonds between the peripheral functional groups of these branches are the major driving force and the main secondary interaction to make globular shapes from CNTs.
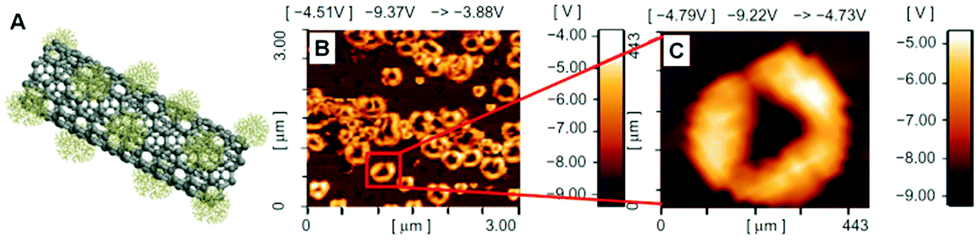 | ||
| Fig. 7 (A) Scheme of MWCNT-g-PCA and (B, C) phase contrast AFM images of MWCNT-g-PCA-PTX donut-shaped conjugates. Reprinted from ref. 99 Copyright (2011) with permission from Dove Press. | ||
Next, the cytotoxicity of MWCNT-g-PCA-PTX nanomaterials against SKOV3 ovarian cancer cells and A549 lung cancer cells has been evaluated. The results of cytotoxicity assays demonstrated that MWCNT-g-PCA-PTX exhibits a higher cytotoxic effect compared with unconjugated paclitaxel, which can be attributed to increased cell penetration.
Moreover, pristine MWCNTs were opened and functionalized by a treatment with acid and HPG was covalently grafted onto their surface by the “grafting from” approach based on the anionic ROP of glycidol monomers. TEM images show that PG is grown onto the tip of carbon nanotubes more than other regions where more hydroxyl functional groups, initiators for the polymerization of glycidol, are created upon treatment with acid. When the surface of carbon nanotubes is covered by polymers, especially in the case of the “grafting from” method in which the density of polymerization onto the surface of CNTs is higher, the physicochemical properties of carbon nanotubes and their interactions with biological systems are dominated by polymers. In this work the death of cells incubated with MWCNT-g-HPG and other interactions with cells have been investigated and it can be found that polyglycerol directly affects all these interactions. In vitro cytotoxicity tests and hemolysis assays did not show any adverse effects on HT1080 cells and red blood cells. The functionalization of CNTs with HPG decreases their in vitro cytotoxicity and makes them promising nanomaterials in nanomedicines.96 Although different analyses and other observations in several works show that polymers attached onto the surface of CNTs dominate their biological properties, carbon nanotubes retain their own properties in these systems and actually the properties of these systems originate from all segments, but with different priorities. For example, in the case of MWCNT-g-HPG, toxicity is controlled by the polyglycerol shell, whereas carbon nanotubes have a large effect on the rate of transfer of the system from the cell membrane such that most of the system is transferred from the cell membrane in one hour.
Here, the linear-dendritic copolymer is fully hydrophilic despite of other works in which at least one of the polymer segments should be hydrophobic. Therefore, the types of secondary interactions between the linear-dendritic copolymers and CNTs were investigated by different thermal analysis and spectroscopy methods. It has been found that hydrogen bonding between the functional groups of linear-dendritic copolymers and hydroxyl groups created on the surface of CNTs, or on the surface of iron oxide nanoparticles attached to the surface of CNTs, is a type of effective secondary interaction between these two segments. Spectroscopy investigations have proved that interactions between the π system of carbonyl groups of linear-dendritic copolymers and CNTs are another type of interactions, which attach copolymers to CNTs.
An effective method to incorporate cadmium selenide (CdSe) nanoparticles with different capping agents onto the surface of f-MWCNTs has been developed. In this work, CdSe NPs with negative and positive surface charges were synthesized using thioglycolic acid (TGA) and gelatin capping agents, respectively. Then, they were deposited onto the surface of MWCNTs using linear-dendritic ABA-type block copolymers containing polyethylene glycol as B blocks and poly(citric acid) as A blocks, PCA-PEG-PCA. This method led to hybrid nanomaterials consisting of well-dispersed CdSe NPs on the surface of MWCNTs, whereas the deposition of CdSe NPs onto the surface of MWCNTs without linear-dendritic copolymer intermediates, directly, resulted in the agglomerates of CdSe NPs in some areas on their surface.115
For the first time, we proved that polymers not only raise the functionality, biocompatibility and water solubility of CNTs but also are able to dramatically change the CNTs conformations. Different spectroscopy methods, such as NMR, IR and DLS, together with microscopy images proved that there are strong interactions between CNTs and linear-dendritic copolymers. It was clear that polar interactions are among the most effective driving forces at changing the conformation of CNTs. It means effective secondary interactions among polymers lead to changes in CNTs morphology.96,97,116 It has been observed that interactions between polyglycerol-poly(ethylene glycol)-polyglycerol (PG-PEG-PG) ABA linear-dendritic copolymer and CNTs lead to the conformation alteration of CNTs from the extended toward the closed state because of liposome-like nanocapsules formation (Fig. 8). On the other hand, DLS experiments showed that the size of CNTs dramatically decreased upon interactions with linear-dendritic copolymers, which again proves the changes in the conformation of CNTs from extended toward globular. Because one of the proposed reasons for the carcinogenicity of CNTs is their long lengths and rigid structures, flexible liposome-like nanocapsules prepared by this strategy could be safer and far from the asbestos-like physicochemical properties of CNTs. In this research, the biocompatibility of liposome-like nanocapsules and their ability to load and deliver anticancer drugs such as doxorubicin (DOX) have been evaluated and it has been proved that their loading capacity is more than two grams of drug to one gram of liposome-like nanocapsules.117
 | ||
| Fig. 8 (a) schematic representation of noncovalent interactions between linear-dendritic copolymers and surface of CNTs that lead to new hybrid nanomaterials with improved properties (b) AFM image of PG-PEG-PG/MWCNT liposome-like nanocapsules containing encapsulated DOX molecules. The highlighted object in the top-left part of the image, by a black line, shows the MWCNTs. Reprinted from ref. 114 with permission from the Royal Society of Chemistry. | ||
PG-PEG-PG ABA linear-dendritic copolymer is a fully hydrophilic compound and consists of a backbone with C–O bonds and peripheral hydroxyl functional groups. Therefore, interactions between the copolymer and the π system of CNTs or hydrophobic interactions between these two segments should not be significant factors in this case. Hydrogen bonds between hydroxyl functional groups of CNTs and the PG-PEG-PG ABA linear-dendritic copolymer are the main driving force to not only attach the copolymer onto the surface of CNTs but also change the conformation of CNTs toward a globular shape.118
In one of our recent works, noncovalent interactions between poly (amidoamine)-poly(ethylene glycol)-poly(amidoamine) (PAMAM-PEG-PAMAM) ABA type linear-dendritic copolymers and CNTs and changes in the conformation of CNTs upon these interactions have been extensively investigated. The modification of the end functional groups of these copolymers leads to a hybrid nanomaterial containing CNTs with a copolymer shell in which CNTs retain their rigid structure. It has been shown that π–π stacking dominates interactions between the linear-dendritic copolymer and CNTs in this case, such that in the defect sites stacked copolymer was not observed at all. However, when PAMAM-PEG-PAMAM was not modified, π–π interactions between the carbonyl functional groups of the copolymer and the π system of CNTs were the most effective factor in attaching the copolymer onto the surface of CNTs, while the secondary interactions between the end amino functional groups of the dendritic block of the copolymer force CNTs to change their conformation from a strand type to a packed form (Fig. 9).119
 | ||
| Fig. 9 Molecular structure of (a) PAMAM-PEG-PAMAM ABA-type linear-dendritic copolymers and (b) PAMAM-PEG-PAMAM linear-dendritic copolymers modified by 4,6-diphenoxy-1,3,5-triazine. (c and d) schematic interaction of macromolecule ‘a’ and macromolecule ‘b’, respectively, (e and g) AFM and TEM images of CNTs interacting with macromolecule ‘a’ and (f and h) AFM and TEM images of CNTs interacting with macromolecule ‘b’. Reprinted from ref. 119 with permission from the Royal Society of Chemistry. | ||
However, by changing the conditions, secondary interactions lead to other types of hybrid nanomaterials with extremely different topology. For example interactions between PAMAM-PEG-PAMAM linear-dendritic copolymers modified by 4,6-diphenoxy-1,3,5-triazine with carbon nanotubes result in supramolecular ultra-long carbon nanotubes, which are suggested as promising candidates to avoid the asbestos-like health risk of this type of nanomaterials.120
It has been demonstrated that methylated β-cyclodextrins (CDs) modified by a triazole group, itself substituted by highly hydrophilic moieties, such as sulfonate or hydroxyl groups, can constitute a new class of MWCNT dispersing agents via noncovalent interactions.121 TEM captured images of CDTSO3Na-MWCNT have also displayed a coil structure of carbon nanotubes, which can be correlated to the secondary molecular interactions among CDTSO3Na segments.
Moreover, recently, PAMAM dendrimer has been used as a crosslinker to produce magnetic carbon nanotube aerogels.122 For this synthesis, acid-treated MWCNT, amino-terminated generation 5 PAMAM dendrimer, gluconolactone (GDL) and magnetic Fe3O4 colloidal suspension have been used. With the help of the hydrolyzed product of the GDL to decrease the pH value of the mixture, in situ protonated PAMAM dendrimer was connected with carboxyl groups attached to different MWCNTs via electrostatic interactions to generate aerogels after either supercritical CO2 drying or freeze-drying.
4.3. Nanoparticles
In one of the rarest reports related to the impact of dendritic structures on nanoparticles, Yan et al. have described a strategy for the synthesis of gold nanoparticles (Au-NPs), for which the size and surface reactivity of the particles are controlled by the molecular sizes of dendritic arenethiols (DAT) as capping agents. This strategy exploits two important attributes of the DAT molecules: one involving the utilization of the umbrella-like structure with a single thiol as the anchorage handle and the other involving the rib with an expandable dendritic structure as a spacing-tunable cap. The synthesis of Au-NPs with controllable sizes below 10 nm has been demonstrated using different sizes of DAT in both one-phase and two-phase solutions. The unique structural properties of the dendritic arenethiol capping molecules not only provide the ability to control the size growth of Au-NPs but also a ready surface exchange reaction for surface derivatization and interparticle assembly. The viability of this strategy for the control of size and surface reactivity has been demonstrated by results from structural and morphological characterizations of the nanoparticles and assemblies (Fig. 10).123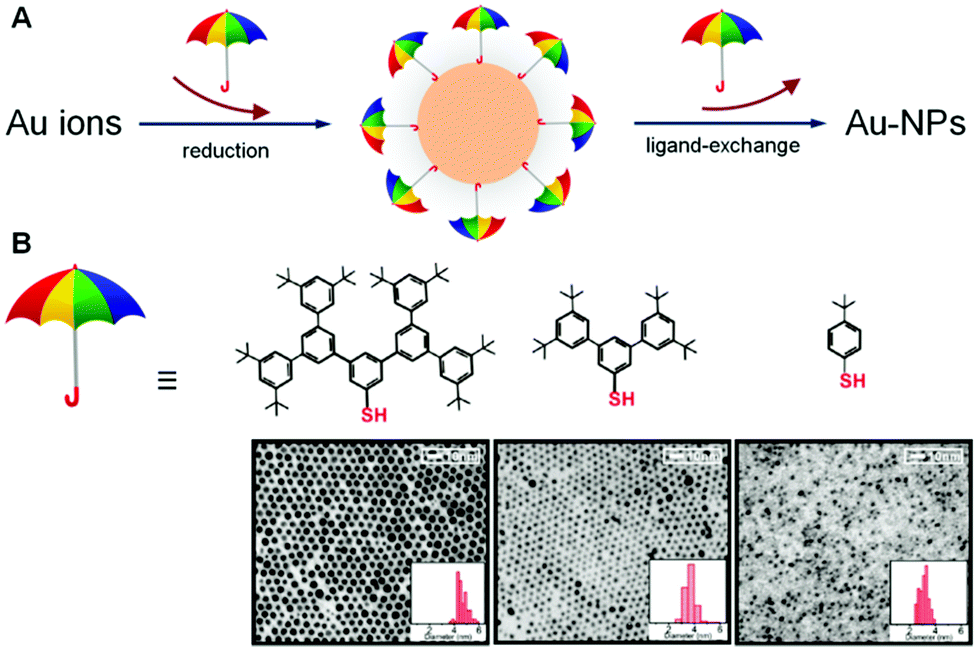 | ||
| Fig. 10 (A) Illustration of the exploitation of umbrella-like DAT molecules by the utilization of the structure with a single thiol as the anchorage handle and the rib with an expandable dendritic structure as a spacing-tunable cap for tuning the nanoparticle sizes and surface reactivity, (B) structures of the dendritic arenethiols and their corresponding TEM micrographs and size distributions for DAT-capped Au-NPs (synthesized by two-phase method) after ligand exchange with decanethiols. Reproduced from ref. 123 with permission from the Royal Society of Chemistry. | ||
The combined impact of these results has demonstrated the feasibility of exploiting the molecular sizes of dendritic arenethiols (DAT) as capping agents for the control of the size and surface reactivity of Au nanoparticles. This type of controllability was achieved by the unique umbrella-like structure of DATs with a single thiol as the anchorage handle and the rib having an expandable dendritic structure as a spacing-tunable cap. This structure was shown to effectively tune the surface reactivity, as evidenced by the easy replacement of the DAT molecules by a ligand having a stronger binding ability to the gold surface than DAT. One important aspect of this structural character for size control is the dependence of the binding strength of DATs to gold particles on the “umbrella rib” size, which leads to the capability of controlling the particle sizes. Another important aspect is the combination of the interparticle weak interactions and voids that facilitate the surface reactivity and interparticle assembly by molecular linkers.
Molecular self-assembly of quantum dots and metal nanoparticles mediated by polyrotaxanes to produce new nano-objects has also been previously observed by our groups.124–126
Furthermore, a simple and effective non-lithographic method for the preparation of a novel organization of noble metal nanoparticles into horseshoe-like nanostructures via self-assembly has been studied.127 The adsorption of Au nanoparticles stabilized with the dendrimer 1,2,3,4,5,6-hexakis[(3′,5′-bis(benzyloxy)benzyl)sulfanylmethyl] benzene (S6G1) on hydrophilic surfaces has resulted in the formation of spatially correlated droplet aggregates. The annealing of Au/S6G1 in thin films has caused amalgamated droplets to form arrays of horseshoe-like nanostructures with an average size of 250 nm and an average height of 13 nm (Fig. 11). The mobility and the manner in which the semi-capped Au nanoparticles are distributed on the hydrophilic substrate are believed to be the promoters that control the growth of the nucleation to create the horseshoe-like structures. Atomic force microscopy (AFM) measurements demonstrated the changes in height and size of the nanoparticles before and after the annealing process. Oxygen plasma etching has been used to remove the S6G1 dendrimer to reveal the orientation of the Au nanocrystals in the nanostructure matrix. The results of this research have been comparable to the results obtained by Nolte and co-workers128 on the formation of the ring-like structures of porphyrin molecules.
 | ||
| Fig. 11 (A) Schematic representation of the mechanism of the formation of the horseshoe-like structures during the annealing process. The affinity of the dendrimer molecules to each other and the surface energy of the Au nanoparticles determine the structure formation, (B) AFM height image of Au/S6G1 on the Si/SiO2 substrate after annealing at 140 °C. Insets: 3D horseshoe structures and higher magnification image of a cross-section of the horseshoe structure. Reproduced from ref. 127 with permission from the American Chemical Society. | ||
Moreover, PCA-PEG-PCA linear-dendritic copolymers have been used as capping and reducing agents to produce gold nanoparticles. Because there are two blocks in the structure of copolymers, which can interact with the surface of the prepared gold nanoparticles, necklace-type gold nanoparticles could be obtained by this procedure.129 In addition, it has been indicated that host–guest interactions between linear polymers or alkyl chains and dendritic polymers with cyclodextrin moieties can result in globular soft nanoparticles with interesting properties. For example polyglycerol (PG) with a cyclodextrin in its focal point (CD-PG) associates with alkyl chains with a guest molecule in their focal point to produce nanoparticles as pH- and thermosensitive anticancer drug delivery systems.130 Host–guest interactions between the phenyl rings of polystyrene and CD-PG also lead to soft globular nanoparticles.131
5. Impact on proteins and peptides
The formation and accumulation of amyloid fibrils is a pathological feature of more than two dozen clinical syndromes. Thus, the inhibition of fibril assembly is a potential therapeutic strategy in neurodegenerative disorders such as prion and Alzheimer's diseases. It has been proved that dendrimers are able to interact with amyloids as anti-amyloidogenic agents. Amyloid fibrils are formed from a large number of different proteins and peptides that share the property of adopting a crossed β-sheet conformation under particular conditions.132 PAMAM, PPI and phosphorus dendrimers, as well as glycodendrimers have been shown to be able to modulate amyloid formation.In 2006, the impact of three generations (3rd, 4th, and 5th) of PAMAM dendrimers on their activity against amyloid fibril aggregation was investigated. In that research, Klajnert et al.133 reported the anti-amyloidogenic effect of PAMAM dendrimers on amyloid formation of the prion peptide PrP (185–208) and the Alzheimer's peptide Aβ (1–28). The results have shown that the higher the dendrimer generation, the larger the degree of inhibition of amyloid formation and the more effective are dendrimers in disrupting the already existing fibrils. In addition, a hypothesis on a dendrimer–peptide interaction mechanism has been suggested based on the dendrimers’ molecular structure and TEM images.133
Recently, PPI dendrimers decorated with maltose (Mal) as glycodendrimers with low intrinsic toxicity have demonstrated great capacity to interfere with Alzheimer's amyloid peptide Aβ (1–40) fibrilization. It has been indicated that the interactions between PPI-Mal glycodendrimers and peptides are generation-dependent. PPI-G5-Mal blocks amyloid fibril formation generating granular non-fibrillar amorphous aggregates, whereas PPI-G4-Mal generates clumped fibrils at low dendrimer–peptide ratios and amorphous aggregates at high ratios. Both PPI-G4-Mal and PPI-G5-Mal are found to be nontoxic to PC12 and SH-SY5Y neuronal cell lines. PPI-G4-Mal reduces amyloid toxicity by clumping fibrils together, whereas amorphous aggregates are toxic to PC12 cells. The results have shown that glycodendrimers are promising nontoxic nanomaterials in the search for anti-amyloidogenic agents (Fig. 12).134
 | ||
| Fig. 12 Graphic schematically representing the morphology of the different dendrimer-amyloid aggregated species and the interactions that can explain their formation. (A) Nucleation-dependent polymerization process of Aβ (1–40): monomeric peptide (◊) assembles as lower- and higher-molecular-weight peptide oligomers (○) (rapid formation under our experimental conditions); prefibrillar structures form by combination of peptide oligomers (○○○) and finally convert into fibrils (□□). (B) In the presence of PPI-G4-Mal dendrimer at low dendrimer–peptide ratios, the interaction of the dendrimer with the monomeric peptide does not prevent the formation of prefibrillar and fibrillar structures but the fibrils are clumped, possibly glued together by the dendrimer. (C) PPI-G4-Mal at high dendrimer–peptide ratios does prevent the formation of prefibrillar structures by interfering with oligomer formation (the dendrimer can interfere by interacting with the peptide monomers, small oligomers and big oligomers) forming dendrimer–peptide oligomer complexes that combine in the form of granular non-fibrillar amorphous aggregates. (D) PPI-G5-Mal is able to alter the peptide oligomeric structures (probably due to its capacity to form a higher number of hydrogen bonds per dendrimer) even at low dendrimer–peptide ratios, impeding in this way the formation of prefibrillar structures and leading to the formation of amorphous aggregates. Reproduced from ref. 137 with permission from the American Chemical Society. | ||
Moreover, two structurally distinct maltose shell-modified 5th generation (G5) poly(propylene imine) (PPI) glycodendrimers fluorescently labeled (a) with open maltose shell, cationically charged G5-PPI-OS and (b) with dense maltose shell and nearly neutral G5-PPI-DS, have been tested in relation to several melanoma cell lines. It has been found that three melanoma cell lines internalize the G5-PPI-DS structure more efficiently than non-tumoral HEK297 T cells. Furthermore, the internalization pathways of G5-PPI-OS and G5-PPI-DS are characteristic for each tumour cell phenotype and include more than one mechanism. As a general trend, large amounts of both G5-PPI-OS and G5-PPI-DS are internalized on a cholesterol-dependent pathway in MJS primary melanoma cells and on non-conventional pathways in SK28 metastatic melanoma cells. G5-PPI-OS, temporarily retained at the plasma membrane in both cell lines, is internalized slower in the metastatic than in the primary phenotype. Unlike G5-PPI-OS, G5-PPI-DS is immediately endocytosed in both cell lines. The unconventional internalization pathway and trafficking, exclusively used by G5-PPI-DS in metastatic cells, is described at the molecular level. The decay kinetics of fluorescently labeled G5-PPI-OS and G5-PPI-DS is distinct in the two cellular phenotypes.
Both cationic and neutral maltose G5-PPI glycodendrimeric structures represent molecules based on which design of new formulations for therapy and/or diagnosis of melanoma can be further developed.135
Moreover, the impact of heparin (a highly sulfated glycosaminoglycan, which is widely used as an injectable anticoagulant) and PAMAM-G3 dendrimers on the aggregation of two amyloid peptides related to Alzheimer's and prion diseases have interestingly been studied. The obtained data have clearly shown that the Aβ (1–28) and PrP (185–208) aggregation processes have been individually enhanced by heparin and PAMAM-G3 dendrimers, which modulate the aggregation process by affecting the nucleation rate at low concentrations and the elongation rate at high concentrations.136 It is well known that the inhibition of fibril assembly is a potential therapeutic strategy in prion diseases. on the basis of this, the effect of cationic phosphorus dendrimers on the aggregation process of the prion peptide PrP (185–208) has been studied. The results have shown that the fourth generation of phosphorus dendrimers are able to clearly interfere with the PrP (185–208) aggregation process by both slowing down the formation of aggregates (by causing a decrease in the nucleation rate) and by lowering the final amount of amyloid fibrils, a common hallmark of conformational diseases.137
Furthermore, the effect of pH on the amyloid inhibiting activity of PPI dendrimers at the molecular level has also been evaluated. As mentioned earlier, the formation of amyloid plaques is a key pathological event in neurodegenerative disorders, such as prion and Alzheimer's diseases. The ionization state of acidic and alkaline residues in prion and Alzheimer's peptides plays an important role in interactions between dendrimers and peptides, which can result in an enhancement or a decrease in the peptide amyloidogenicity. Our results can help to establish the conditions for which dendrimers are effective inhibitors. These conditions are different for PrP (185–208) and Aβ (1–28) and strongly depend on the amino acid sequence. The studies show that dendrimers can serve as a tool to study the mechanism of formation of amyloid-like structures and to generate useful knowledge for the design of compounds with therapeutic utility.138
It has been demonstrated that electron paramagnetic resonance (EPR) is a useful approach to monitor interactions between dendrimers and proteins. Spin-probe and spin-label techniques were used to study the interactions of the Aβ (1–28) peptide involved in Alzheimer's disease and the PrP (106–126) peptide suspected to be preferentially involved in spongiform encephalopathies with three different types of PAMAM dendrimers. A computer-aided EPR analysis of a positively charged and a neutral spin probe was performed by comparing the pure dendrimer and peptide systems with the dendrimer–peptide ones. Moreover, spin-labeled PAMAM dendrimers were used to test the interactions. The results show the interactions between dendrimer and peptide monomer to be stronger for Aβ (1–28) than for PrP (106–126). PAMAM dendrimers perturb the aggregation of the peptides more than PPI dendrimers. The Alzheimer's peptide Aβ (1–28) and prion peptide PrP (185–208) when exposed to destabilizing factors (for instance adding heparin and lowering pH) form fibrils. In a preceding study using fluorescence, it was revealed that the dendrimers are able to prevent peptide aggregation. A probable mechanism is that the dendrimers interact with peptide monomers, and therefore, inhibit their capability of growing into fibrils. Then, to understand the mechanism of inhibition of the aggregation process, we have studied the peptide–dendrimer interactions using EPR. In this report, aggregate-free (without heparin and at neutral pH) Aβ (1–28) and PrP (106–126) (this sequence is suspected to be preferentially involved in spongiform encephalopathies) were allowed to interact with the three different types of dendrimers: PAMAM dendrimers (5th and 6th generations), PPI dendrimers (G3) and P-dendrimers (G4). Both spin-probe (the neutral TOH and the positively charged CAT1 spin probes were used) and spin-label (the PAMAM dendrimers were labeled with a nitroxide radical) techniques were used to monitor the peptide–dendrimer interactions. The computer-aided analysis of the EPR spectra at 255 K provided information on the type and strength of interactions occurring in the different systems. The EPR analysis showed that Aβ (1–28) has a dipolar interaction with the dendrimers preventing the separation of the probes in the form of aggregates at 255 K. Part of the probes interact with the peptide, but this interaction is perturbed by the addition of the dendrimers. Another part of the probes are free, captured in the hydration layer at the dendrimer–peptide interface. The mobility and the relative amount of the interacting and the free component, obtained from spectral computation, change as a function of the dendrimer type, indicating a stronger interaction with PAMAM dendrimers, which are therefore suspected to function better as peptide aggregation scavengers with respect to the other dendrimers. Furthermore, the use of spin-labeled PAMAM dendrimers supported the finding of dipolar interactions between these dendrimers and Aβ (1–28). The interactions of the dendrimers with PrP (106–126) are weaker than with Aβ (1–28). TOH showed stronger interactions than CAT1, indicating that the interactions arise from polar and low-polar groups. In this case too, the PAMAM dendrimers showed a relatively stronger binding with this peptide. The labeled dendrimers also show a weaker interaction of the dendrimers with PrP (106–126) than with Aβ (1–28).139
Recently, the kinetics of amyloid and prion fibril formation in the absence and presence of dense-shell sugar-decorated dendrimers has been investigated. PPI dendrimers at generation 5 (G5) with a dense shell of maltose and maltotriose units have been employed for this assessment. Thioflavin T (ThT) fluorescence assay and circular dichroism (CD) experiments indicated that fibril formation is enhanced at low dendrimer concentrations, while it is prevented at relatively high dendrimer concentrations. EPR analysis not only has demonstrated this behavior, but also provided detailed information on the mechanism of fibril formation and on the different behavior of the differently decorated dendrimers. EPR results also indicate that the perturbations caused by PPI (G5)-maltose are more effective on PrP (185–208) than on Aβ (1–28), while PPI (G5)-maltotriose is less effective towards PrP (185–208) in both promoting aggregation and preventing it by changing the dendrimer concentration. These results provide useful information about the mechanism and interactions, which regulate the ability of macromolecules like dendrimers to promote, prevent or cure neurodegenerative diseases.140
6. Conclusion
In summary, this review reveals the new face of dendritic polymers as agents to change the size, shape or sheet multiplicity of other nanomaterials. Indeed, this paper has reviewed primary covalent–noncovalent interactions between dendritic polymers and other nanomaterials and also secondary noncovalent interactions among dendritic polymers, which can result in modern nanostructures with improved characteristics.Because in most cases secondary interactions are driving forces to change the physicochemical properties of nanomaterials, in designing a dendritic polymer to modify nanomaterials by covalent or noncovalent approaches these types of interactions should be taken into account. This could be achieved by the modification of the functional groups of dendritic polymers or conjugation of other molecules, such as linear polymers, onto their backbone.
Finally, the consideration of all possible ways of interaction of dendritic polymers with graphene, CNTs, proteins/peptides and nanoparticles leds us to gain better knowledge in the design of novel hybrid nanomaterials.
Acknowledgements
Authors would like to thank Iran National Science Foundation: INSF to support this work financially.References
- M. Adeli, R. Soleyman, Z. Beiranvand and F. Madani, Chem. Soc. Rev., 2013, 42, 5231 RSC.
- C. Dufes, I. F. Uchegbu and A. G. Schatzlein, Adv. Drug Delivery Rev., 2005, 57, 2177 CrossRef CAS PubMed.
- C. M. Paleos, D. Tsiourvas and Z. Sideratou, Mol. Pharmaceutics, 2007, 4, 169 CrossRef CAS PubMed.
- M. Sun, A. Fan, Zh. Wang and Y. Zhao, Soft Matter, 2012, 8, 4301 RSC.
- A. K. Patri, I. J. Majoros and J. R. Baker, Curr. Opin. Chem. Biol., 2002, 6, 466 CrossRef CAS.
- B. K. Nanjwade, H. M. Bechra, G. K. Derkar, F. V. Manvi and V. K. Nanjwade, Eur. J. Pharm. Sci., 2009, 38, 185 CrossRef CAS PubMed.
- S. Svenson, Eur. J. Pharm.. Biopharm., 2009, 71, 445 CrossRef CAS PubMed.
- M. Liu and J. M. J. Frechet, Pharm. Sci. Technol. Today, 1999, 2, 393 CrossRef CAS.
- G. M. Soliman, A. Sharma, D. Maysinger and A. Kakkar, Chem. Commun., 2011, 47, 9572 RSC.
- C. Li, Adv. Drug Delivery Rev., 2002, 54, 695 CrossRef CAS.
- S. El Kazzouli, S. Mignani, M. Bousmina and J.-P. Majoral, New J. Chem., 2012, 36, 227 RSC.
- J. H. Park, S. Lee, J.-H. Kim, K. Park, K. Kim and I. C. Kwon, Prog. Polym. Sci., 2008, 33, 113 CrossRef PubMed.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2004, 56, 1649 CrossRef CAS PubMed.
- E. Blanco, A. Hsiao, A. P. Mann, M. G. Landry, F. Meric-Bernstam and M. Ferrari, Cancer Sci., 2011, 102, 1247 CrossRef CAS PubMed.
- S.-G. Sampathkumar and K. J. Yarema, in Dendrimers in Cancer Treatment and Diagnosis, Nanotechnologies for the Life Sciences, ed. C. S. S. R. Kumar, WILEY-VCH, Weinheim, 2007, Vol. 7, pp. 1–44 Search PubMed.
- Y. Cheng, L. Zhao, Y. Li and T. Xu, Chem. Soc. Rev., 2011, 40, 2673 RSC.
- S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141 CrossRef CAS PubMed.
- R. K. Tekade, P. V. Kumar and N. K. Jain, Chem. Rev., 2009, 109, 49 CrossRef CAS PubMed.
- M. Calderón, M. A. Quadir, M. Strumia and R. Haag, Biochimie, 2010, 92, 1242 CrossRef PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866 CrossRef CAS PubMed.
- C. Liang and J. M. J. Fréchet, Prog. Polym. Sci., 2005, 30, 385 CrossRef CAS PubMed.
- K. Inoue, Prog. Polym. Sci., 2000, 25, 453 CrossRef CAS.
- F. Vogtle, S. Gestermann, R. Hesse, H. Schwierz and B. Windisch, Prog. Polym. Sci., 2000, 25, 987 CrossRef CAS.
- D. K. Smith, A. R. Hirst, C. S. Love, J. G. Hardy, S. V. Brignell and B. Huang, Prog. Polym. Sci., 2005, 30, 220 CrossRef CAS PubMed.
- J. Khandare, M. Calderón, N. M. Dagiaa and R. Haag, Chem. Soc. Rev., 2012, 41, 2824 RSC.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171 CrossRef CAS PubMed.
- S. Gupta, R. Tyagi, V. S. Parmar, S. K. Sharma and R. Haag, Polymer, 2012, 53, 3053 CrossRef CAS PubMed.
- A. Sousa-Herves, R. Riguera and E. Fernandez-Megia, New J. Chem., 2012, 36, 205 RSC.
- M. A. Quadir and R. Haag, J. Controlled Release, 2012, 161, 484 CrossRef CAS PubMed.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190 CrossRef PubMed.
- H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325 CrossRef CAS PubMed.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS PubMed.
- M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593 RSC.
- G. R. Newkome and C. D. Shreiner, Polymer, 2008, 49, 1 CrossRef CAS PubMed.
- M. Adeli, in Hybrid nanostructures in cancer therapy, Nova publisher, 2012, pp. 1–204 Search PubMed.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294 CrossRef CAS PubMed.
- L. M. Kaminskas, V. M. McLeod, C. J. H. Porter and B. J. Boyd, Mol. Pharmaceutics, 2012, 9, 355 CrossRef CAS PubMed.
- T. C. Shiao and R. Roy, New J. Chem., 2012, 36, 324 RSC.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117 CrossRef CAS.
- G. R. Newkome, Z. Q. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003 CrossRef CAS.
- J. Paez, M. Martinelli, V. Brunetti and M. Strumia, Polymers, 2012, 4, 355 CrossRef PubMed.
- B. Zhang, R. Wepf, M. Kroger, A. Halperin and A. D. Schluter, Macromolecules, 2011, 44, 6785 CrossRef CAS.
- S. Fornera, T. E. Balmer, B. Zhang, A. D. Schluter and P. Walde, Macromol. Biosci., 2011, 11, 1052 CrossRef CAS PubMed.
- J. C. Cuggino, M. Calderon, C. I. Alvarez, M. C. Strumia, K. N. Silva, E. K. Penott-Chang and A. J. Muller, J. Colloid Interface Sci., 2011, 357, 147 CrossRef CAS PubMed.
- J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4782 CrossRef PubMed.
- H. Namazi and M. Adeli, Eur. Polym. J., 2003, 39, 1491 CrossRef CAS.
- D. K. Smith, A. R. Hirst, C. S. Love, J. G. Hardy, S. V. Brignell and B. Huang, Prog. Polym. Sci., 2005, 30, 220 CrossRef CAS PubMed.
- M. Seiler, Chem. Eng. Technol., 2002, 25, 3 CrossRef.
- D. K. Smith, Chem. Commun., 2006, 34 RSC.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Int. J. Pharm., 2010, 394, 122 CrossRef CAS PubMed.
- R. K. Kainthan, J. Janzen, J. N. Kizhakkedathu, D. V. Devine and D. E. Brooks, Biomaterials, 2008, 29, 1693 CrossRef CAS PubMed.
- C. Buzea, I. I. Pacheco and K. Robbie, Biointerphases, 2007, 2, 17 CrossRef.
- Y. Yin and D. Talapin, Chem. Soc. Rev., 2013, 42, 2484 RSC.
- Z. F. Gang and L. W. Shi, Sci. China Chem., 2011, 54, 28 Search PubMed.
- J. T. Sun, C. Y. Hong and C. Y. Pan, Polym. Chem., 2011, 2, 998 RSC.
- K. Kono, Polym. J., 2012, 44, 531 CrossRef CAS.
- A. R. Maity, A. Chakraborty, A. Mondal and N. R. Jana, Nanoscale, 2014, 6, 2752 RSC.
- H.-W. Yang, C.-Y. Huang, C.-W. Lin, H.-L. Liu, C.-W. Huang, S.-S. Liao, P.-Y. Chen, Y.-J. Lu, K.-C. Wei and C.-C. M. Ma, Biomaterials, 2014, 35, 6534 CrossRef CAS PubMed.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243 CrossRef CAS PubMed.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2014, 173, 75 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203 CrossRef CAS PubMed.
- L. Feng and Z. Liu, Nanomedicine, 2011, 6, 317 CrossRef CAS PubMed.
- Y. Pan, N. G. Sahoo and L. Li, Expert Opin. Drug Delivery, 2012, 9, 1365 CrossRef CAS PubMed.
- X. Wang and W. Zhang, Carbon, 2014, 67, 795 CrossRef PubMed.
- X. C. Qin, Z. Y. Guo, Z. M. Liu, W. Zhang, M. M. Wan and B. W. Yang, J. Photochem. Photobiol., B, 2013, 120, 156 CrossRef CAS PubMed.
- J.-L. Li, X.-L. Hou, H.-C. Bao, L. Sun, B. Tang, J. Wang, X.-G. Wang and M. Gu, J. Biomed. Mater. Res., Part A, 2014, 102, 2181 CrossRef PubMed.
- X. Yang, Y. Wang, X. Huang, Y. Ma, Y. Huang, R. Yang, H. Duan and Y. Chen, J. Mater. Chem., 2011, 21, 3448 RSC.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876 CrossRef CAS PubMed.
- L. Feng, S. Zhang and Z. Liu, Nanoscale, 2011, 3, 1252 RSC.
- H. Shen, L. Zhang, M. Liu and Z. Zhang, Theranostics, 2012, 2, 283 CrossRef CAS PubMed.
- P. S. Wate, S. S. Banerjee, A. Jalota-Badhwar, R. R. Mascarenhas, K. R. Zope, J. Khandare and R. D. K. Misra, Nanotechnology, 2012, 23, 415101 CrossRef PubMed.
- K. Hasan, M. H. Asif, O. Nur and M. Willander, J. Biosens. Bioelectron., 2012, 3, 1 Search PubMed.
- Y. Wang, Z. Li, J. Wang, J. Li and Y. Lin, Trends Biotechnol., 2011, 29, 205 CrossRef CAS PubMed.
- X. Dong, Q. Long, J. Wang, M. B. Chan-Park, Y. Huang, W. Huang and P. Chen, Nanoscale, 2011, 3, 5156 RSC.
- I.-Y. Jeon, H.-J. Choi, S.-Y. Bae, D. W. Chang and J.-B. Baek, J. Mater. Chem., 2011, 21, 7820 RSC.
- D. M. Maleki, M. Adeli, A. Kakanejadifard, S. Movahedi and F. Bani, Polymer, 2013, 54, 4802 CrossRef PubMed.
- F.-G. Zhao and W.-S. Li, J. Mater. Chem., 2012, 22, 3082 RSC.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720 CrossRef CAS PubMed.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201 CrossRef CAS PubMed.
- L. M. Veca, F. Lu, M. J. Meziani, L. Cao, P. Zhang, G. Qi, L. Qu, M. Shrestha and Y.-P. Sun, Chem. Commun., 2009, 14, 2565 RSC.
- D. Wang, G. Ye, X. Wang and X. Wang, Adv. Mater., 2011, 23, 1122 CrossRef CAS PubMed.
- S. Jokar, A. Pourjavadi and M. Adeli, RSC Adv., 2014, 4, 33001 RSC.
- E. Araque, R. Villalonga, M. Gamella, P. Martinez-Ruiz, J. Reviejo and J. M. Pingarron, J. Mater. Chem. B, 2013, 1, 2289 RSC.
- S. Movahedi, M. Adeli, A. Kakanejadi Fard, M. Maleki, M. Sadeghizadeh and F. Bani, Polymer, 2013, 54, 2917–2925 CrossRef CAS PubMed.
- F. Schuler, B. Kerscher, F. Beckert, R. Thomann and R. Mulhaupt, Angew. Chem., Int. Ed., 2013, 52, 455 CrossRef PubMed.
- F. J. Tolle, M. Fabritius and R. Mulhaupt, Adv. Funct. Mater., 2012, 22, 1136 CrossRef.
- R. L. D. Whitby, V. M. Gunko, A. Korobeinyk, R. Busquets, A. B. C. Kristina Laslo, J. Skubiszewska-Zieba, R. Leboda, E. Tombac, I. Y. Toth, K. Kovacs and S. V. Mikhalovsky, ACS Nano, 2012, 6, 3967 CrossRef CAS PubMed.
- R. L. D. Whitby, A. Korobeinyk, V. M. Gunko, R. Busquets, A. B. Cundy, K. Laslo, J. Skubiszewska-Zieba, R. Leboda, E. Tombac, I. Y. Toth, K. Kovacs and S. V. Mikhalovsky, Chem. Commun., 2011, 47, 9645 RSC.
- L. R. Gu, T. Elkin, X. P. Jiang, H. P. Li, Y. Lin, L. W. Qu, T. R. J. Tzeng, R. Joseph and Y. P. Sun, Chem. Commun., 2005, 874 RSC.
- L. Gu, P. G. Luo, H. Wang, M. J. Meziani, Y. Lin, L. M. Veca, L. Cao, F. Lu, X. Wang, R. A. Quinn, W. Wang, P. Zhang, S. Lacher and Y. P. Sun, Biomacromolecules, 2008, 9, 2408 CrossRef CAS PubMed.
- E. Murugan and G. Vimala, J. Colloid Interface Sci., 2011, 357, 354 CrossRef CAS PubMed.
- K. T. Al-Jamal, F. M. Toma, A. Yilmazer, H. Ali-Boucetta, A. Nunes, M. A. Herrero, B. Tian, A. Eddaoudi, W. T. Al-Jamal, A. Bianco, M. Prato and K. Kostarelos, FASEB J., 2010, 24, 4354 CrossRef CAS PubMed.
- B. Zhang, Q. Chen, H. Tang, Q. Xie, M. Ma, L. Tan, Y. Zhang and S. Yao, Colloids Surf., B, 2010, 80, 18–25 CrossRef CAS PubMed.
- W. Qin, K. Yang, H. Tang, L. Tan, Q. Xie, M. Ma, Y. Zhang and S. Yao, Colloids Surf., B, 2011, 84, 206–213 CrossRef CAS PubMed.
- L. Tao, G. Chen, G. Mantovani, S. York and D. M. Haddleton, Chem. Commun., 2006, 4949 RSC.
- M. Adeli, N. Mirab, M. S. Alavidjeh, Z. Sobhani and F. Atyabi, Polymer, 2009, 50, 3528 CrossRef CAS PubMed.
- M. Adeli, N. Mirab and F. Zabihi, Nanotechnology, 2009, 20, 485603 CrossRef PubMed.
- M. Adeli, A. Bahari and H. Hekmatara, Nano, 2008, 3, 37–44 CrossRef CAS.
- Z. Sohhani, M. Adeli, R. Dinarvand, M. Ghahremani and F. Atyabi, Int. J. Nanomed., 2011, 6, 705 Search PubMed.
- M. Adeli, E. Mehdipour and M. Bavadi, J. Appl. Polym. Sci., 2010, 116, 2188 CAS.
- C. H. Xue, R. J. Zhou, M. M. Shi, Y. Gao, G. Wu, X. B. Zhang, H. Z. Chen and M. Wang, Nanotechnology, 2008, 19, 325605 CrossRef PubMed.
- G. J. Sohn, H. J. Choi, I. Y. Jeon, D. W. Chang, L. Dai and J. B. Baek, ACS Nano, 2012, 6, 6345 CrossRef CAS PubMed.
- L. Xu, Z. Ye, Q. Cui and Z. Gu, Macromol. Chem. Phys., 2009, 210, 2194 CrossRef CAS.
- L. He, J. Sun, X. Wang, L. Yao, J. Li, R. Song, Y. Hao, Y. He and W. Huang, J. Colloid Interface Sci., 2011, 363, 122 CrossRef CAS PubMed.
- T. Ogoshi, T. Saito, T. Yamagishi and Y. Nakamoto, Carbon, 2009, 47, 117 CrossRef CAS PubMed.
- K. Awasthi, D. P. Singh, S. Singh, D. Dash and O. N. Srivastava, New Carbon Mater., 2009, 24, 301 CrossRef CAS.
- A. Star and J. F. Stoddart, Macromolecules, 2002, 35, 7516 CrossRef CAS.
- C. Gao, S. Muthukrishnan, W. Li, J. Yuan, Y. Xu and A. H. E. Muller, Macromolecules, 2007, 40, 1803 CrossRef CAS.
- A. T. Naeini, M. Adeli and M. Vossoughi, Nanomedicine, 2010, 6, 556 CrossRef CAS PubMed.
- N. Sarlak, M. Adeli, M. Karimi, M. Bordbare and M. A. Farahmandnejad, J. Mol. Liq., 2013, 180, 39 CrossRef CAS PubMed.
- A. Bahari, H. Hekmatara, R. Sepahvand and M. Adeli, Nano, 2009, 4, 217 CrossRef CAS.
- M. Adeli, F. Hakimpoor, M. Ashiri, R. Kabiri and M. Bavadi, Soft Matter, 2011, 7, 4062 RSC.
- E. Mehdipoor, M. Adeli, M. Bavadi, P. Sasanpour, B. Rashidian and M. Kalantari, J. Mater. Chem., 2011, 21, 15456 RSC.
- M. Adeli, S. Beyranvand and M. Hamid, J. Mater. Chem., 2012, 22, 6947 RSC.
- M. Adeli, A. Bahari and S. M. Hosseini, J. Nanostruct. Polym. Nanocomp., 2011, 7, 52 Search PubMed.
- M. Eskandari, S. H. Hosseini, M. Adeli and A. Pourjavadi, Iran. Polym. J., 2014, 23, 387 CrossRef CAS.
- M. Adeli, M. S. Alavidjeh and E. Mohammadifar, Iran. Polym. J., 2014, 23, 195 CrossRef CAS PubMed.
- M. Adeli, M. Ashiri, B. Khodadadi Chegeni and P. Sasanpour, J. Iran. Chem. Soc., 2013, 10, 701 CrossRef CAS PubMed.
- M. Adeli, S. Beyranvand and R. Kabiri, Polym. Chem., 2013, 4, 669 RSC.
- M. Adeli, E. Mehdipour and S. Beyranvand, New J. Chem., 2013, 37, 1871 RSC.
- B. Leger, S. Menuel, D. Landy, J.-F. Blach, E. Monflier and A. Ponchel, Chem. Commun., 2010, 46, 7382 RSC.
- X. Zhang, L. Chen, T. Yuan, H. Huang, Z. Sui, R. Du, X. Li, Y. Lu and Q. Li, Mater. Horiz., 2014, 1, 232 RSC.
- H. Yan, C. Wong, A. R. Chianese, J. Luo, L. Wang, J. Yin and C-J. Zhong, Chem. Mater., 2010, 22, 5918 CrossRef CAS.
- F. Hosseini, A. Panahifar, M. Adeli, H. Amiri, A. Lascialfari, F. Orsini, M. R. Doschak and M. Mahmoudi, Colloids Surf., B, 2013, 103, 652 CrossRef CAS PubMed.
- M. Adeli, R. S. Sarabi, R. Yadollahi Farsi, M. Mahmoudi and M. Kalantari, J. Mater. Chem., 2011, 21, 18686 RSC.
- M. Adeli, M. Kalantari, M. Parsamanesh, E. Sadeghi and M. Mahmoudi, J. Nanomed. Nanotechnol., 2011, 7, 806 CrossRef CAS PubMed.
- A. Fahmi, A. DAleo, R. M. Williams, L. De Cola, N. Gindy and F. Vögtle, Langmuir, 2007, 23, 7831–7835 CrossRef CAS PubMed.
- P. H. Albertus, J. Schenning, B. Fransicus, G. Benneker, P. Hubertus, M. Geurts, X. Y. Liu and R. J. M. Nolte, J. Am. Chem. Soc., 1996, 118, 8549 CrossRef.
- A. Tavakoli Naeini, M. Adeli and M. Vossoughi, Eur. Polym. J., 2010, 46, 165 CrossRef CAS PubMed.
- M. Adeli, A. Kakanejadifard, F. Abedi, B. Kodadadi Chegeni and F. Bani, J. Nanomed. Nanotechnol., 2013, 9, 1203 CrossRef CAS PubMed.
- A. Pourjavadi, M. Adeli and M. Yazdi, New J. Chem., 2013, 37, 295 RSC.
- A. Lorenzo And and B. A. Yankner, Ann. N. Y. Acad. Sci., 1996, 17, 89 CrossRef PubMed.
- B. Klajnert, M. Cortijo-Arellano, J. Cladera and M. Bryszewska, Biochem. Biophys.. Res. Commun., 2006, 345, 21–28 CrossRef CAS PubMed.
- O. Klementieva, N. Benseny-Cases, A. Gella, D. Appelhans, B. Voit and J. Cladera, Biomacromolecules, 2011, 12, 3903 CrossRef CAS PubMed.
- A. Filimon, L. E. Sima, D. Appelhans, B. Voit and G. Negroiu, Curr. Med. Chem., 2012, 19, 4955 CrossRef CAS.
- B. Klajnert, M. Cortijo-Arellano, M. Bryszewska and J. Cladera, Biochem. Biophys. Res. Commun., 2006, 339, 577 CrossRef CAS PubMed.
- B. Klajnert, M. Cortijo-Arellano, J. Cladera, J. P. Majoral, A. M. Caminade and M. Bryszewska, Biochem. Biophys. Res. Commun., 2007, 364, 20 CrossRef CAS PubMed.
- B. Klajnert, J. Cladera and M. Bryszewska, Biomacromolecules, 2006, 7, 2186 CrossRef CAS PubMed.
- B. Klajnert, M. Cangiotti, S. Calici, J. P. Majoral, A. M. Caminade, J. Cladera, M. Bryszewska and M. F. Ottaviani, Macromol. Biosci., 2007, 7, 1065 CrossRef CAS PubMed.
- M. F. Ottaviani, M. Cangiotti, L. Fiorani, A. Fattori, T. Wasiak, D. Appelhans and B. Klajnert, Curr. Med. Chem., 2012, 19, 5907 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |