
A new oligo(hexafluoropropylene oxide)-b-oligo(ethylene oxide) diblock surfactant obtained by radical reactions†
Jiří
Lapčík
ab,
Olinda
Gimello
b,
Vincent
Ladmiral
b,
Chadron Mark
Friesen
*c and
Bruno
Ameduri
*b
aDepartment of Organic Chemistry, Institute of Chemical Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic
bIngenierie et Architectures Macromoleculaires, Institut Charles Gerhardt, Ecole Nationale Supérieure de Chimie de Montpellier (UMR5253-CNRS), 8, rue de l'Ecole Normale, 34296 Montpellier Cedex 5, France
cDepartment of Chemistry, Trinity Western University, Langley, British Columbia V2Y 1Y1, Canada. E-mail: chad.friesen@twu.ca; bruno.ameduri@enscm.fr
First published on 29th August 2014
Abstract
The synthesis and characterization of a new oligo(hexafluoropropylene oxide)-b-oligo(ethylene oxide), oligo(HFPO)-b-oligo(PEG), diblock co-oligomer are presented. First, the model reactions dealing with the radical addition of 1-iodoperfluorohexane (C6F13I) onto allyl alcohol and allyl-O-PEG-OCH3 were optimized in terms of the choice of the initiator (azobisisobutyronitrile [AIBN], tert-butylperoxypivalate [TBBPi], and benzoyl peroxide [BPO]) and of the solvent, temperature, and time. Allyl-O-PEG-OCH3 was obtained from the etherification of ω-hydroxy-PEG with allyl bromide. End-capping of oligo(HFPO) with PEG was successfully achieved by the radical addition of 1-iodoperfluoropropyl-2-oligo(hexafluoropropylene oxide) [oligo(HFPO)-CF(CF3)CF2I] onto allyl-O-PEG-OCH3 using the best conditions of the model reactions. Although TBPPi failed and led to oligo(HFPO)-isobutyl iodide, AIBN and BPO yielded oligo(HFPO)-CH2CHICH2-oligo(PEG). The selective reduction of the latter compound led to oligo(HFPO)-b-oligo(PEG) in 77% yield, the surface tension properties of which were compared to those of commercially available ammonium perfluorooctanoate (APFO) and perfluorooctanoic acid (PFOA). Its critical micelle concentration was 0.04 g mol−1. All models, intermediates, and diblock co-oligomers were characterized by 1H, 19F, and 13C NMR spectroscopy as well as matrix assisted laser desorption ionization (MALDI) and atmospheric pressure solids analysis probe (ASAP) time-of-flight mass spectrometry (TOF-MS).
1. Introduction
Polyfluorinated compounds (PFCs) are useful chemicals involved in a wide range of products. Among them, molecules that bear both fluorinated hydrophobic moieties and hydrophilic parts, called “surfactants”,1–4 are valuable compounds. Surfactants are being used in more than 200 applications5–10 ranging from the protection of surfaces (textile, paper, carpets, masonry, metal, and leather), as stimulating fluids for oil recovery, fire-fighting foam, skin protection from chemical agents, soil and stain-repellents, plane hydraulic fluids,1,11 paints, lubricants, electroplating, photographic emulsifiers, pressure sensitive additives, pharmaceuticals, and insecticides, or are involved in cosmetic formulations. Perfluorooctanoic acid (PFOA), for example, is frequently used as a surfactant in the aqueous media (co)polymerization of hydrophobic monomers (e.g. fluorinated monomers5,12 PFOA and perfluorooctane sulfonic acid (PFOS) are the most used fluorosurfactants). Their surface tensions and critical micelle concentrations are very low.1,2 They feature both pronounced hydrophobicity and oleophobicity and display high chemical and thermal resistance. They can be synthesized either by electrochemical fluorination (ECF)13 or by telomerization.14 The former process leads to ca. 70% straight chain PFOS along with 30% branched and cyclic isomers.15 PFOA is being used in fast food packaging (≥300 μg kg−1) owing to its water and oil repellencies.4bTelomerization reactions14 are straightforward and the most representative example is the radical telomerization of tetrafluoroethylene (produced industrially by various chemical industries) that generated telomers containing a hydrophobic perfluorinated CnF2n+1 end-group (where n is an even number).5,14 D'Eon and Mabury16 reported the fluorotelomer production values from 1970 to 2020. Telomeric alcohols have been widely used for polymers and surface coatings with an estimated annual production of ca. 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t per year in 2004.7 In spite of its remarkable properties and wide range of applications, PFOS was listed in Annex B of the Stockholm convention as a persistent organic pollutant in 2009.17 As research has demonstrated that many of the long-chain PFCs are toxic, persistent, and bioaccumulative, government and regulatory agencies have been working toward agreements and regulations to limit the production of some PFCs.10,18 These three severe health and environmental issues arise from the high stability of the perfluorinated chains which are not degraded either by enzymes or metabolic processes.19 In addition, PFCs bioaccumulate in the food chains and have long half-lives in human blood:16 3.6, 5.4,20–22 and 8.5 years for PFOA, PFOS and perfluorohexane sulfonate, respectively.23 Other severe limitations are the foetal development toxicity, the immunotoxicity, and the effects on thyroidal hormones.24 PFOS itself is quite stable in the environment, with no known natural mechanism of degradation.24 Attempts to degrade PFOA and PFOS were suggested by Parsons et al.22 PFOS remains the predominant PFC found in all living species, tissues, and locations analyzed around the world.25 PFOA and PFOS accumulations in marine mammals (e.g., >1200 ng g−1 in liver) are common all over the Northern hemisphere.11,16,26 In a recent review, Fromme et al.27 evaluated potential PFC exposures from indoor and outdoor air, house dust, drinking water, and food28 and concluded that median uptakes of PFOS and PFOA were of the order of 2–3 ng kg−1 per day, with food being responsible for greater than 90% of this exposure.29 However, with the wide variety of foods consumed and the difficulty in establishing sensitive analytical methods that accurately measure contaminants, there is still a great deal of uncertainty about the role of food as an exposure route.30 An increasing number of studies have suggested that fish from contaminated water bodies may be the prevailing source of exposure to PFOS and possibly other long-chain perfluorocarbon acids (PFCAs).26,31 In addition, the consumption of contaminated drinking water15 has been linked with an increased surfactant amount in blood, as reviewed by Lindstrom et al.15
000 t per year in 2004.7 In spite of its remarkable properties and wide range of applications, PFOS was listed in Annex B of the Stockholm convention as a persistent organic pollutant in 2009.17 As research has demonstrated that many of the long-chain PFCs are toxic, persistent, and bioaccumulative, government and regulatory agencies have been working toward agreements and regulations to limit the production of some PFCs.10,18 These three severe health and environmental issues arise from the high stability of the perfluorinated chains which are not degraded either by enzymes or metabolic processes.19 In addition, PFCs bioaccumulate in the food chains and have long half-lives in human blood:16 3.6, 5.4,20–22 and 8.5 years for PFOA, PFOS and perfluorohexane sulfonate, respectively.23 Other severe limitations are the foetal development toxicity, the immunotoxicity, and the effects on thyroidal hormones.24 PFOS itself is quite stable in the environment, with no known natural mechanism of degradation.24 Attempts to degrade PFOA and PFOS were suggested by Parsons et al.22 PFOS remains the predominant PFC found in all living species, tissues, and locations analyzed around the world.25 PFOA and PFOS accumulations in marine mammals (e.g., >1200 ng g−1 in liver) are common all over the Northern hemisphere.11,16,26 In a recent review, Fromme et al.27 evaluated potential PFC exposures from indoor and outdoor air, house dust, drinking water, and food28 and concluded that median uptakes of PFOS and PFOA were of the order of 2–3 ng kg−1 per day, with food being responsible for greater than 90% of this exposure.29 However, with the wide variety of foods consumed and the difficulty in establishing sensitive analytical methods that accurately measure contaminants, there is still a great deal of uncertainty about the role of food as an exposure route.30 An increasing number of studies have suggested that fish from contaminated water bodies may be the prevailing source of exposure to PFOS and possibly other long-chain perfluorocarbon acids (PFCAs).26,31 In addition, the consumption of contaminated drinking water15 has been linked with an increased surfactant amount in blood, as reviewed by Lindstrom et al.15
Indeed, in 2006, the European Union set out a ban on the use of PFOS in a number of goods. However, PFCs are still produced and released into the environment16 and have been quoted as the “PCBs of the XXIst century”. In the same time, EPA launched the 2010/2015 PFOA Stewardship Program32 to reduce emissions and residual content of PFOA and long-chain PFCAs by 95% by 2010 with the goal to eliminate long chain PFCs by 2015. While there has been some success with voluntary controls for some PFCs,32 limited incentive for companies to join these voluntary agreements was noted. Hence, a growing interest in the synthesis of short perfluoroalkyl chain surfactants was noted,33–36 especially since the 3M company developed perfluorobutane sulfonyl compounds.37–39
Various strategies to synthesize potentially non-bio accumulable alternatives to PFOA have been reported:40 (1) chemicals bearing either a CF3O or (CF3)2N end-group,41 (2) compounds produced from small perfluorinated chains with non-ionic oligoethylene oxide or carbohydrate,36,42 (3) carboxylate gemini surfactants,43,44 (4) vinylidene fluoride (VDF) telomers with short 1-iodoperfluoroalkane where methylene groups may act as “weak” degradable points,45,46 (5) 3,3,3-trifluoropropene (TFP) telomers from either 1-iodoperfluoroalkanes or other chain transfer agents,47 (6) VDF and TFP cotelomers,48 and (7) compounds derived from oligo(hexafluoropropylene oxide), oligo(HFPO).49 Interestingly, these perfluorooligoethers were shown to be nontoxic and nonbioaccumulable50 and thus offer further opportunities to synthesize more environmentally friendly fluorinated surfactants. For example, Li et al.51 reported the synthesis of oligo(HFPO)-CO2-PEG from the esterification of trimer(HFPO)C(O)F with HO-PEG leading to a surfactant that possesses a critical micelle concentration of 0.6 g L−1. However, the ester group induces limitations in some applications due to their stability and polarity. The objective of this article is thus to overcome this issue. It describes the use of longer oligo(HFPO)-based novel surfactants. Oligo(HFPO) iodide was reacted under radical conditions with ω-unsaturated oligo(ethylene oxide) to yield the desired oligo(HFPO)-based surfactants. The study of their surface tension properties is also presented.
2. Results and discussion
The purpose of this work was to explore conditions that were favorable in converting 1-iodo perfluorooligoalkane ether, specifically oligo(hexafluoropropylene oxide) [oligo(HFPO)] primary iodide, into diblock surfactants using polyethylene glycol (PEG) as the hydrophilic sequence. This oligo(HFPO)-b-oligo(EG) diblock surfactant was achieved by the radical addition of [oligo(HFPO)] primary iodide onto allyl-PEG followed by a selective reduction of the iodine atom (Scheme 1).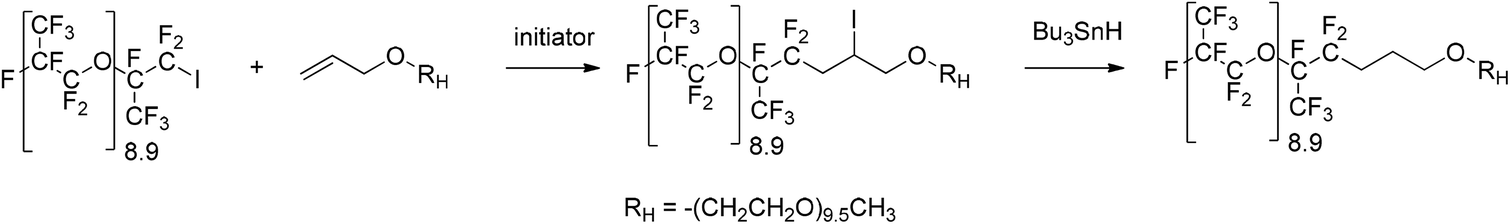 | ||
| Scheme 1 Synthesis of oligo(hexafluoropropylene oxide)-b-polyethylene glycol, oligo(HFPO)-b-PEG diblock surfactant by radical addition of [oligo(HFPO)] primary iodide onto allyl PEG followed by a selective reduction of the iodine atom. | ||
Earlier work had demonstrated that the synthesis of an oligomeric diblock was possible with aromatics and oligo(HFPO)-CF(CF3)CF2I.52 Other studies reported that perfluoroalkyl iodides can be added onto alkenes in high yields53 under radical conditions, as well as act as efficient chain transfer agents in iodine transfer copolymerization of vinylidene fluoride (VDF) and hexafluoropropylene (HFP).54 The radical addition process was considered the best route to form a diblock of perfluoroalkyl or 1-iodo perfluorooligoalkane ether with polyethylene glycol (PEG).9 Indeed, other types of linkages such as esters or amides are likely to hydrolyze over time and thus prone to degradation. The synthesis of the target amphiphilic diblock proceeded in two steps: (i) the introduction of an allylic ether on the hydroxyl end-group of monomethyl ether polyethylene glycol via Williamson ether syntheses (Scheme 2), and (ii) the radical addition of the iodide precursor onto the allylic moiety. Depending on the conditions used, a reduction step was achieved.
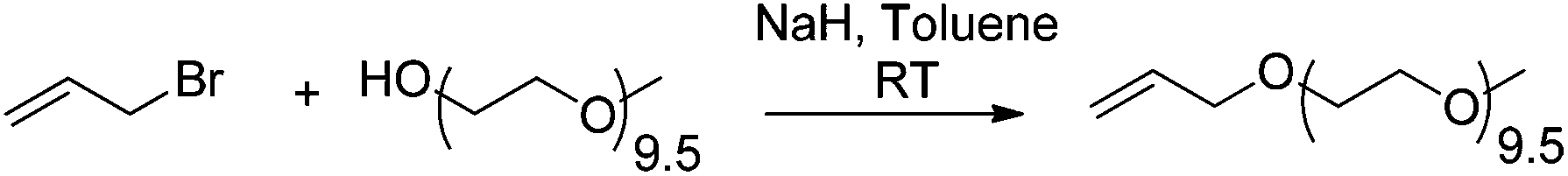 | ||
| Scheme 2 Synthesis of allyl oligo(ethylene oxide) allyl-O-PEG-OCH3 from the etherification of HO-PEG-OCH3 with allyl bromide. | ||
The yield of allyl-O-PEG-OCH3 was found to be ca. 65%. The 1H NMR spectrum (Fig. S1†) displays the expected multiplets assigned to the three ethylenic protons centered at 5.11, 5.20, and 5.85 ppm while the signals assigned to the methylene groups adjacent to oxygen atoms in the oligo(ethylene oxide) chain and characteristic of the CH3O end-group are located at 3.96 ppm and a singlet is centered at 3.31 ppm, respectively. The 13C NMR spectrum (Fig. S2†) confirms its structure from the signals centered at 136 and 116, 70.7, and 59 ppm assigned to both ethylenic carbon atoms, methylenes in ethylene oxide units, and the –OCH3 end-group, respectively. The synthetic strategy for this work relies on the radical addition of 1-iodoperfluoroalkane or 1-iodoperfluoroalkylether [oligo(HFPO)-CF(CF3)CF2I] onto an alkene compound such as the above allyl-O-PEG-OCH3.
To explore the appropriate and best conditions to prepare oligo(HFPO)-b-oligo(PEG) diblock co-oligomers in high yield using radical initiators, a systematic study was undertaken. This article reports the study of five systems: (1) short 1-iodoperfluoroalkane (C6F13I) reacting with a model alkene [e.g., allyl alcohol], (2) small perfluorinated alkyl iodides reacting with a polymeric alkene [e.g., allyl-O-PEG-OCH3], (3) polymeric fluorinated iodides [oligo(HFPO)-CF(CF3)CF2I] with a simple alkene [e.g., allyl alcohol], (4) polymeric fluorinated iodides with a polymeric alkene, and (5) small molecule and polymeric fluorinated iodides reacting strictly with three radical initiators: azobisisobutyronitrile [AIBN], tert-butylperoxypivalate [TBBPi], and benzoyl peroxide [BPO], to observe potential by-product formation.
2.1. Radical addition of 1-iodoperfluorohexane (C6F13I) onto unsaturated alkenes (allyl alcohol and allyl ether, allyl-O-PEG-OCH3)
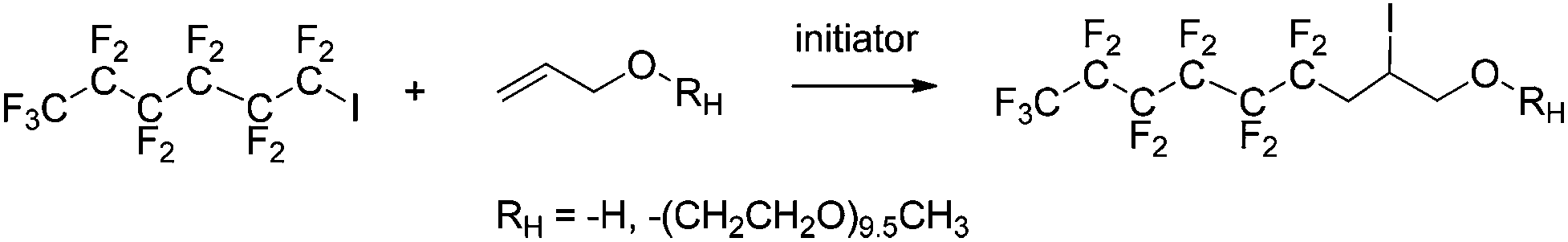 | ||
| Scheme 3 Radical reaction of 1-iodoperfluorohexane with allyl alcohol and allyl-O-PEG-OCH3 (initiated by azobisisobutyronitrile AIBN, tert-butylperoxypivalate, TBBPi, and benzoyl peroxide, BPO). | ||
The reaction temperatures were first adjusted so that the half-lives of the initiators were approximately one hour (Table 1). The reactions were initially monitored by gas chromatography/mass spectrometry (GC/MS) to determine reaction times. After the reaction and removal of traces of unreacted reactants, the total product mixture was characterized by 1H 19F, and 13C NMR spectroscopy to determine the reaction yield. In all cases, C6F13I conversion was higher than 93%. The first 1H NMR spectrum (Fig. S3†) did not display any signals in the ca. 5.5–7.0 ppm range assigned to the ethylenic protons of allyl alcohol (Fig. S4†) but instead showed complex systems at 2.95 and 2.65 ppm and 4.3 ppm attributed to C6F13CH2– and –CHI–, respectively. The 19F-NMR spectrum (Fig. S5†) showed the high field shift of the signal assigned initially to –CF2I (−59 ppm, Fig. S6†) and then to –CF2CH2– (−112 ppm) as a characteristic AB system. All other signals such as the CF3– centered at −83 ppm, and those in the −123 to −128 ppm range assigned to the (CF2)n–, remained relatively unchanged compared to those of the spectrum of C6F13I. Finally, the 13C NMR spectrum (Fig. S7†) exhibited a doublet of triplets, and both singlets were centered at 37.18, 67.8, and 20.4 ppm assigned to the methylene groups in –CF2CH2–, –CH2OH, and the methine –CHI– group, respectively. This 13C NMR spectrum did not show any signal assigned to –CF2I at 110 pm (Fig. S8†) for the potentially remaining C6F13I and displayed a minimal amount of unreacted allyl alcohol (Fig. S9†). Electron impact mass spectrometry (Fig. S10†) was also useful in detecting the formation of the iodhydrin characterized by the molecular ion at m/z = 504 (M+ ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6F13CH2CHICH2OH), and characteristic fragments at m/z = 377 (M − I) and m/z = 357 (M − I − HF).
C6F13CH2CHICH2OH), and characteristic fragments at m/z = 377 (M − I) and m/z = 357 (M − I − HF).
| Rxn # | Allyl compound | C6F13I (mmol) | Allyl (mmol) | Initiator (mmol) | Initiator | t (h) | T (°C) | Conv. C6F13Id (%) | Yield (%) 19F-NMR |
|---|---|---|---|---|---|---|---|---|---|
| Initiator for reactions 1–6 was only added once at the beginning of the reaction.a Ref. 56.b Ref. 57.c Ref. 9a; AIBN, TBPPi, and BPO stand for azobisisobutyronitrile, tert-butylperoxypivalate, and benzoyl peroxide, respectively.d Assessed from 19F NMR. | |||||||||
| 1a | Allyl alcohol | 1.150 | 1.875 | 0.152 | TBPPI | 2 | 75 | >99 | >99 |
| 2a,b | Allyl alcohol | 1.120 | 1.484 | 0.023 | AIBN | 4 | 90 | 93 | 92 |
| 3 | Allyl alcohol | 1.120 | 1.680 | 1.008 | BPO | 8 | 90 | 100 | 0 |
| 4 | Allyl-O-PEG-OCH3 | 0.359 | 0.417 | 0.217 | TBPPI | 4 | 90 | 84 | 69 |
| 5c | Allyl-O-PEG-OCH3 | 0.209 | 0.417 | 0.006 | AIBN | 4 | 90 | 51 | 37 |
| 6 | Allyl-O-PEG-OCH3 | 0.093 | 0.417 | 0.310 | BPO | 8 | 90 | 100 | 0 |
However, it is worth noting that conversion of the alkyl iodide does not necessarily equate to product yield. In all cases, complete conversion of the iodide took place within 8 to 9 h. Both TBPPI and AIBN were suitable in yielding the desired iodhydrin adduct. In contrast, benzoyl peroxide did not yield any product (yield = 0%, Table 1).
2.2. Radical addition of 1-iodoperfluoropropyl-2-oligo(hexafluoropropylene oxide) onto unsaturated alkenes (allyl alcohol and ether)
 | ||
| Scheme 4 Radical reaction of 1-iodoperfluoropropyl-2-oligo(hexafluoro-propylene oxide) onto allyl alcohol and allyl-O-PEG-OCH3 (initiated by AIBN, TBPPI, and BPO). | ||
The progress of the reaction was monitored by the decrease of the starting iodide (Fig. S15†) and the formation of the product (Fig. S16†) using gas chromatography/mass spectrometry (GC/MS). The spectrum showed that 38 to 72 hours were necessary to reach the desired yields (>80%). The 1H NMR spectrum of the iodhydrin based on oligo(HFPO) (Fig. S17 or S18†) exhibited the characteristic signals for –CHI– group at 4.31 and 4.15 ppm resulting from the –CF2CHaHb– at 2.84 and 2.54 ppm, respectively. The respective methylene in –CH2OH and the –OH are noted at 3.66 and 3.99 ppm, respectively (Fig. S18†). The 19F-NMR spectra (Fig. 1A & B) show the absence of the characteristic triplet of doublets centered at −58 ppm (Fig. 1A and S19†) assigned to the CF2I end-group in oligo(HFPO)-CF(CF3)CF2I. Instead, they show a high field shifted signal centered at −112 ppm and resulting from a complex AB system containing two stereocenters (Fig. 1B and S20†). The 13C-NMR spectrum (Fig. S21†) exhibits the absence of the triplet of doublets at 91.3 ppm, which is normally present in the iodide (Fig. S22†). Typical signals of oligo(HFPO)-CF(CF3)CF2 were observed in the 124 ppm to 98 ppm range, while those of iodhydrin can be noted at 66.32, 36.52 and 18 ppm attributed to the –CH2OH, –CF2CH2–, and –CHI groups, respectively.
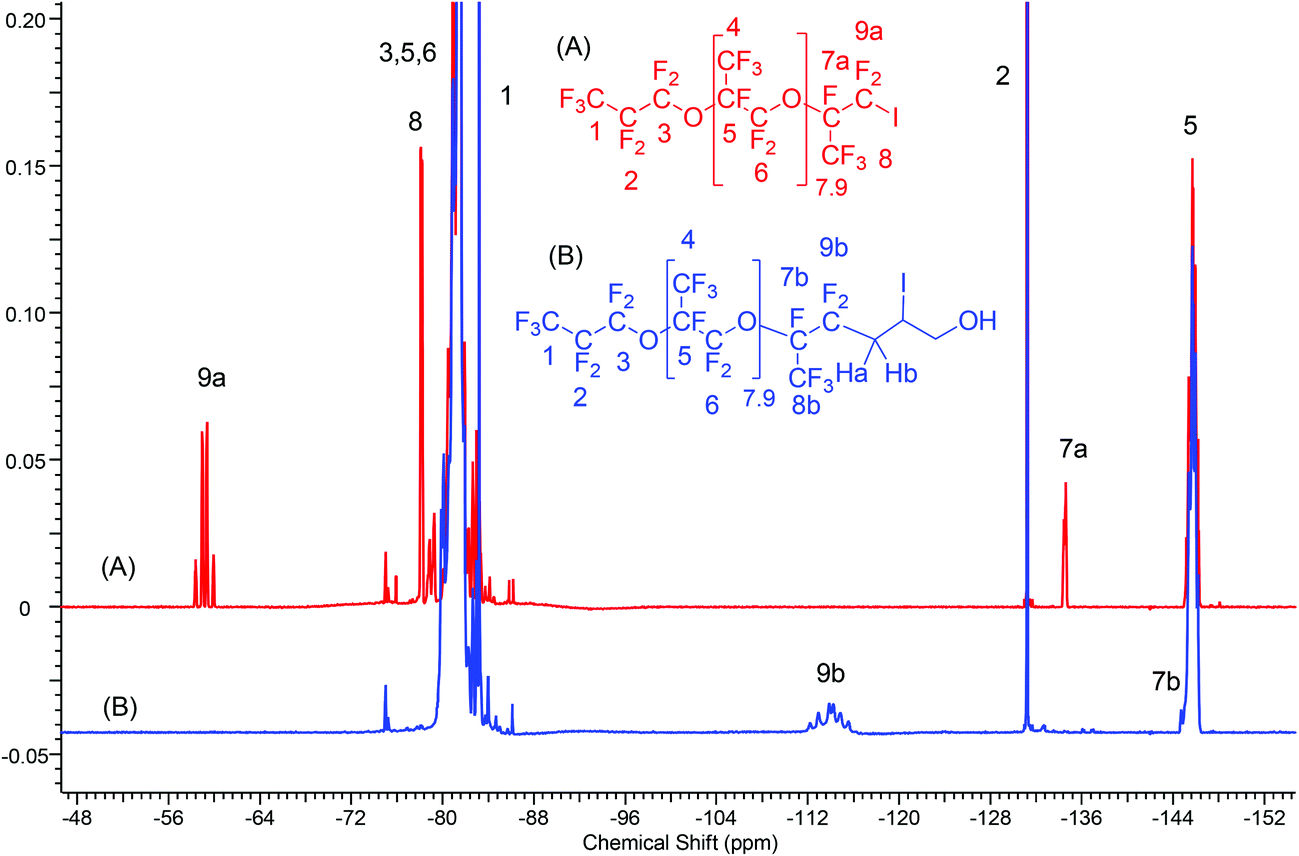 | ||
| Fig. 1 19F-NMR spectra of 1-iodo-2-oligo(hexafluoropropylene oxide)perfluoropropane(F[CF(CF3)CF2O]8.9CF(CF3)CF2I) (A) and F[CF(CF3)CF2O]8.9CH2CHICH2OH (B). | ||
The atmospheric pressure solids analysis probe (ASAP) time-of-flight mass spectrometry (TOF-MS) spectrum in negative ion mode for oligo(HFPO)-CF(CF3)CF2I (Fig. S23†) displays one distribution between 2400 and 4200 m/z corresponding to oligo(HFPO)-based polymers as evidenced by the m/z difference between two consecutive oligomeric peaks (Δm/z = 166 Da) which corresponds to the HFPO repeat unit mass. This distribution is attributed to the deprotonation molecular ion of (F[CF(CF3)CF2O]nCF(CF3)CF2I + CH3OH–H)– when methanol is present in the probe. In addition, fragments of F[CF(CF3)CF2O]nCF(CF3)O– were observed between 184 and 1845 m/z. In negative ion mode, the MALDI-TOF-MS spectrum of oligo(HFPO)-CF(CF3)CF2I (Fig. S24†) shows one distribution between 2300 and 4400 m/z. This distribution corresponds to the deprotonation of oligo(HFPO)-I with formic acid (F[CF(CF3)CF2O]n CF(CF3)CF2 I + HCOOH–H)–. The repeat units Δm/z = 166 Da confirm the presence of oligo(HFPO). In the ASAP-TOF-MS spectrum of F[CF(CF3)CF2O]n CF(CF3)CF2CH2CHICH2OH iodhydrin (Fig. S25†) in positive ion mode a main distribution of the M+ = (F[CF(CF3)CF2O]n CF(CF3)CF2CH2CHICH2)+ radical cation was observed. The loss of the OH group is possible in this ionization mode. A second distribution corresponding to the radical cation M+ with acetonitrile from the probe and a few fragments that may arise from the main compound were also observed. The MALDI-TOF-MS spectrum of F[CF(CF3)CF2O]nCF(CF3)CF2CH2CHICH2OH iodhydrin (Fig. S26†) highlights the positive ion of F[CF(CF3)CF2O]nCF(CF3)CF2CH2CHICH2OH. One distribution was detected between 1400 and 3000 m/z. This corresponds to (F[CF(CF3)CF2O]nCFCF3CF2CH2CHICH2O + Li)+ lithium adduct. The repeat units of 166 m/z indicate that oligo(HFPO) was present, whereas several fragmentations of this compound were observed between 400 and 3000 m/z.
The reaction of oligo(HFPO)-CF(CF3)CF2I with allyl alcohol carried out on the same scale (0.56 mmol) initiated by TBPPI (0.5 mole ratio to the iodide) was almost twice faster than AIBN (0.6 mole ratio to the iodide). It is believed that the rate of the reaction was increased with a higher amount of formed free radicals. TBPPI produces tBu˙ while t-BuO˙ initially generated also released CH3˙ at more elevated temperatures.58 In contrast, AIBN only produces two (CH3)2(CN)C˙. Once again, BPO43 did not form any of the desired iodhydrin although it produces two radicals as well: Ph˙ or PhC(O)O˙ (Table 2).
| Rxn # | Allyl compound | HFPO-I (mmol) | Allyl (mmol) | Initiator (mmol) | Initiator | t (h) | T (°C) | Conv. HFPO-I (%) | 1 HEC (%) | Yield (%) 19F-NMR |
|---|---|---|---|---|---|---|---|---|---|---|
| a Additions beyond the first amount of initiator – (7) 6 additions, (8) 18 additions, (9) 16 additions, (10) 7 additions, (11) 17 additions, (12) 35 additions, (13) 0 addition, (14) 1 addition, (15) 19 additions, (16) 15 additions, (17) 20 additions, (18) 86 additions, (19) 1 addition, (20) 5 additions; calculated using 19F NMR. | ||||||||||
| 7 | Allyl alcohol | 0.564 | 5.279 | 0.080 | TBPPI | 48 | 75 | 100 | 2.3 | 88 |
| 8 | 0.564 | 16.357 | 0.284 | 38 | 90 | 94.5 | 4 | 80 | ||
| 9 | 5.640 | 30.120 | 7.293 | 32 | 75 | 73 | 2.5 | 0 | ||
| 10 | Allyl alcohol | 1.147 | 12.162 | 0.180 | AIBN | 64 | 90 | 100 | 1.12 | 86 |
| 11 | 0.564 | 1.510 | 0.203 | 72 | 90 | 100 | 3 | 86 | ||
| 12 | 5.640 | 112.861 | 4.056 | 72 | 90 | 100 | 2.5 | 89 | ||
| 13 | Allyl alcohol | 0.564 | 1.119 | 0.845 | BPO | 8 | 90 | 68 | 2.7 | 0 |
| 14 | 0.564 | 2.256 | 3.619 | 8 | 90 | 95.5 | 6.5 | 0 | ||
| 15 | Allyl-O-PEG(450)-OCH3 | 0.564 | 0.862 | 0.230 | TBPPI | 80 | 75 | 96.5 | 8.1 | 0 |
| 16 | 5.640 | 17.239 | 18.365 | 64 | 75 | 99.0 | 4 | 0 | ||
| 17 | Allyl-O-PEG(450)-OCH3 | 0.564 | 2.586 | 2.326 | AIBN | 80 | 90 | 100 | 25 | 48 |
| 18 | 5.640 | 23.015 | 23.997 | 172 | 90 | 100 | 24 | 50 | ||
| 19 | Allyl-O-PEG(450)-OCH3 | 0.564 | 2.761 | 2.343 | BPO | 16 | 90 | 100 | 6 | 71 |
| 20 | 5.640 | 11.280 | 23.618 | 48 | 90 | 97.0 | 19 | 51 | ||
The reactions with allyl alcohol are difficult to reproduce due to the biphase nature of the reaction media, the reactants being immiscible, and it is expected that the reaction most likely occurred at the interface of both liquid phases (Table 2, RXNs 9 and 10). Because of such a non-homogeneous medium, a situation may be created in which the radicals react more easily with oligo(HFPO)-CF(CF3)CF2I. The abstraction of the iodine atom in oligo(HFPO)-CF(CF3)CF2I from the radical initiators leads to the oligo(HFPO)-CF(CF3)CF2˙ macroradical that may recombine with itself. But this observation was not noted in any of the MALDI-TOF-MS spectra.59 However, this macroradical could trap a radical initiator fragment or add onto an aromatic decomposition product. The product corresponding to the latter hypothesis was detected by 19F NMR spectroscopy (Fig. 2) as evidenced by the presence of the broad signal centered at −116.3 ppm. This signal is in close agreement with the oligo(HFPO)-CF(CF3)CF2-C6H5 structure previously reported.43 The difluoromethylene attached to benzene gives a complex multiplet from −110.76 to −113.67 ppm; an upfield shift to −116 ppm is probably with an activated group such as iodine attached to the benzene ring. The 1H-NMR spectrum (Fig. S27†) does indicate the presence of aromatic compounds in the mixture ranging from 7.5 to 8.5 ppm.
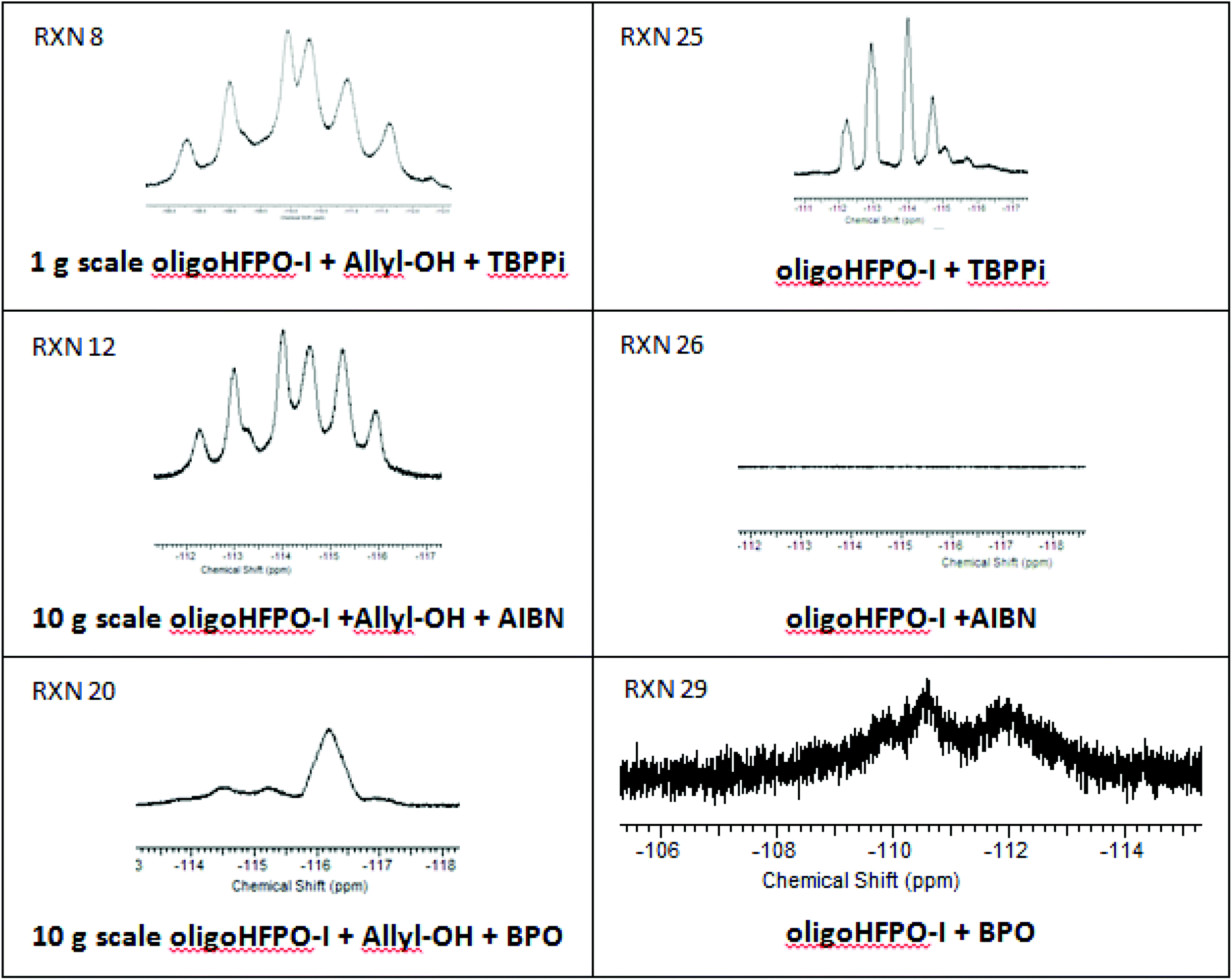 | ||
| Fig. 2 Comparison of the 19F-NMR expansions of 19F NMR spectra the reaction of oligo(HFPO)-CF(CF3)CF2I with the initiator (TBPPi, AIBN, BPO) (right column) and initiator with allyl alcohol (left column). | ||
| Rxn # | Initiator type | Catalyst/Solvent | 1 HEC (%) | Conv. 1 iodide (%) |
|---|---|---|---|---|
| a 1 HEC = C6F13H or oligo(HFPO)-CF(CF3)CF2H. | ||||
| 1, 4, 7–9, 15–16 | TBPPI | None | 2.3–8.1 | 94.5–100 |
| 3, 6, 13–14, 19–20 | BPO | Cu(OAc)2 | 2.7–19 | 68–100 |
| 2, 5, 10, 12, 17–18 | AIBN | None | 1.12–25 | 100 |
| 11 | AIBN in CHCl3 | None | 3 | 100 |
Fig. 3 compares the 1H NMR spectra of allyl-PEG-OCH3 (Fig. 3A) with the resulting iodinated diblock co-oligomer (Fig. 3B or S28†). It is noted that the ethylenic protons initially located at 5.11, 5.20, and 5.85 ppm have disappeared and are replaced by an AB system and two multiplets centered at 2.55 and 3.05 ppm, 3.79, and 4.36 ppm assigned to methylene groups in –CF2CHaHb, –CH2OH, and –CHI–, respectively.
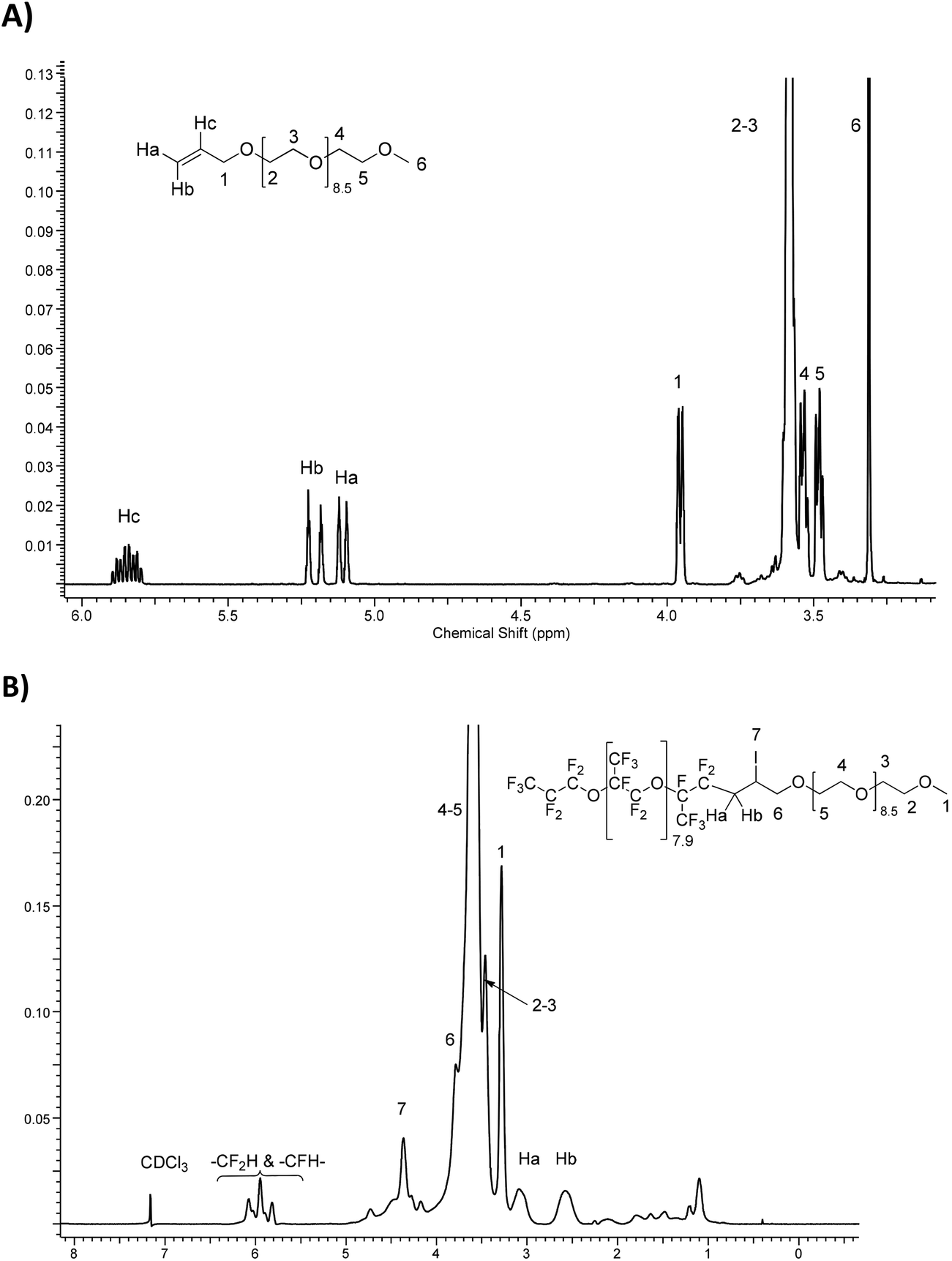 | ||
| Fig. 3
1H-NMR spectra of (A) CH2CH–CH2O(CH2CH2O)9.5CH3, allyl-PEG-OCH3 and (B) F[CF(CF3)CF2-O]8.9CF(CF3)CF2-(CH2CH2O)9.5CH3. | ||
As expected, the 19F NMR spectrum exhibits the high field shift from −60 ppm (assigned to the CF2I end-group in the iodinated precursor) to −112 to −117.5 ppm in the diblock co-oligomers (Fig. S29†).
The 13C NMR spectrum (Fig. 4 or S30†) also shows the absence of ethylenic carbon atoms at 116 and 136 ppm and the presence of fluorinated groups in the 100–125 ppm range, and of the methylene groups in PEG at 70.54 ppm.
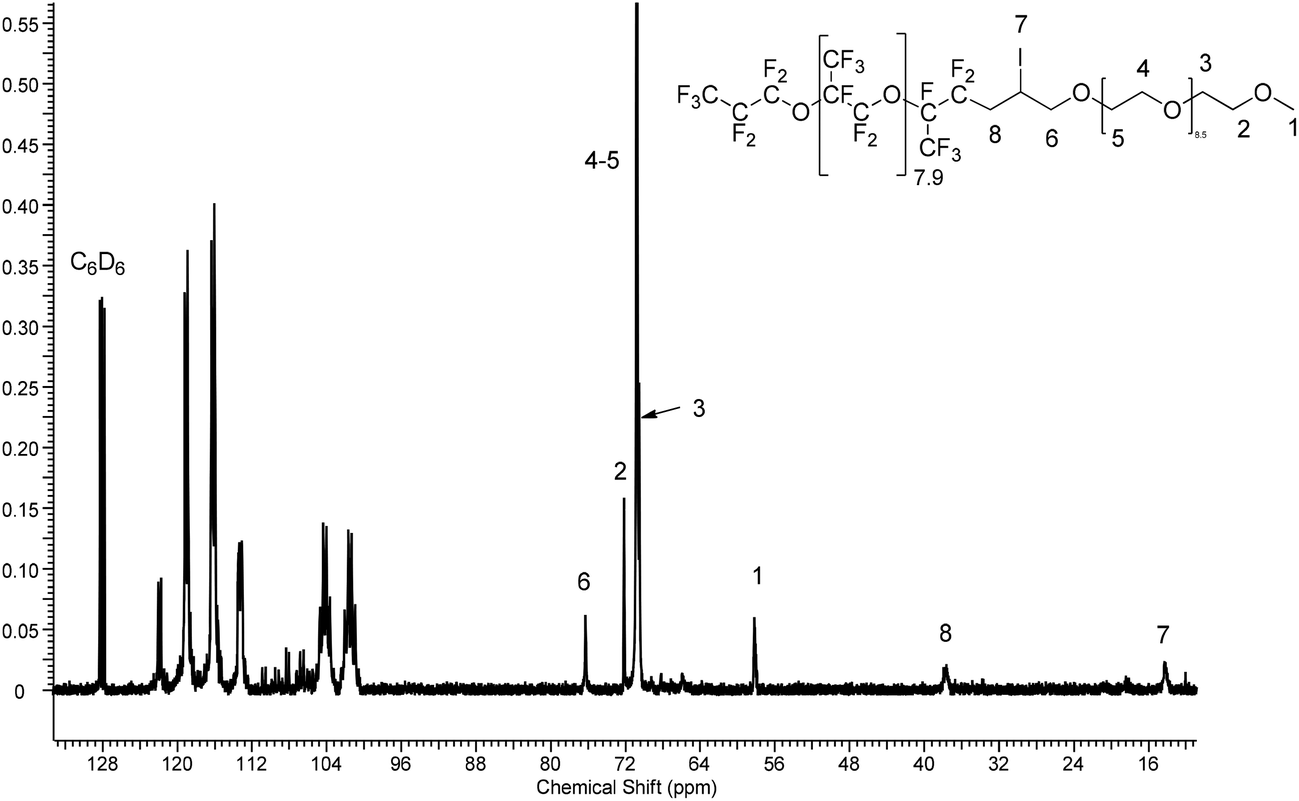 | ||
| Fig. 4 13C-NMR spectrum of F[C(CF3)CF2O]8.9CF(CF3)CF2CH2CHICH2O(CH2CH2O)9.5CH3. | ||
ASAP (Fig. 5 or S31†) and MALDI (Fig. S32†) spectra were critical for determining the coupling of the two oligomers. ASAP was more helpful in determining whether the iodo-diblock co-oligomer based on oligo(HFPO) and PEG was formed. In other experiments, it was observed that ASAP ionization caused the fragmentation of polymers.60 The desorption temperature, ranging from 50 to 650 °C, plays an important role since the high temperature may also induce a thermal degradation. The analyses using ASAP ionization of oligo(HPFO) oligomers thus led to molecular weights lower than those assessed by MALDI-TOF-MS. In ASAP ionization, the main distribution was observed between 1090 and 2700 m/z which corresponds to the F[CF(CF3)CF2O]nCF(CF3)CF2CH2CHICH2O(OCH2CH2)4CH3+ radical cation. This distribution displays a 166 m/z repeat unit as well as a 44 m/z repeat unit characteristic of HFPO and EG, respectively (Fig. 5). In positive ion mode, the MALDI-TOF-MS spectrum of F[CF(CF3)CF2O]nCF(CF3)CF2CH2CHICH2O(OCH2CH2)9.5CH3 exhibits only one distribution between 1500 and 3200 m/z for the adduct (M + Li)+ (Fig. S32†).
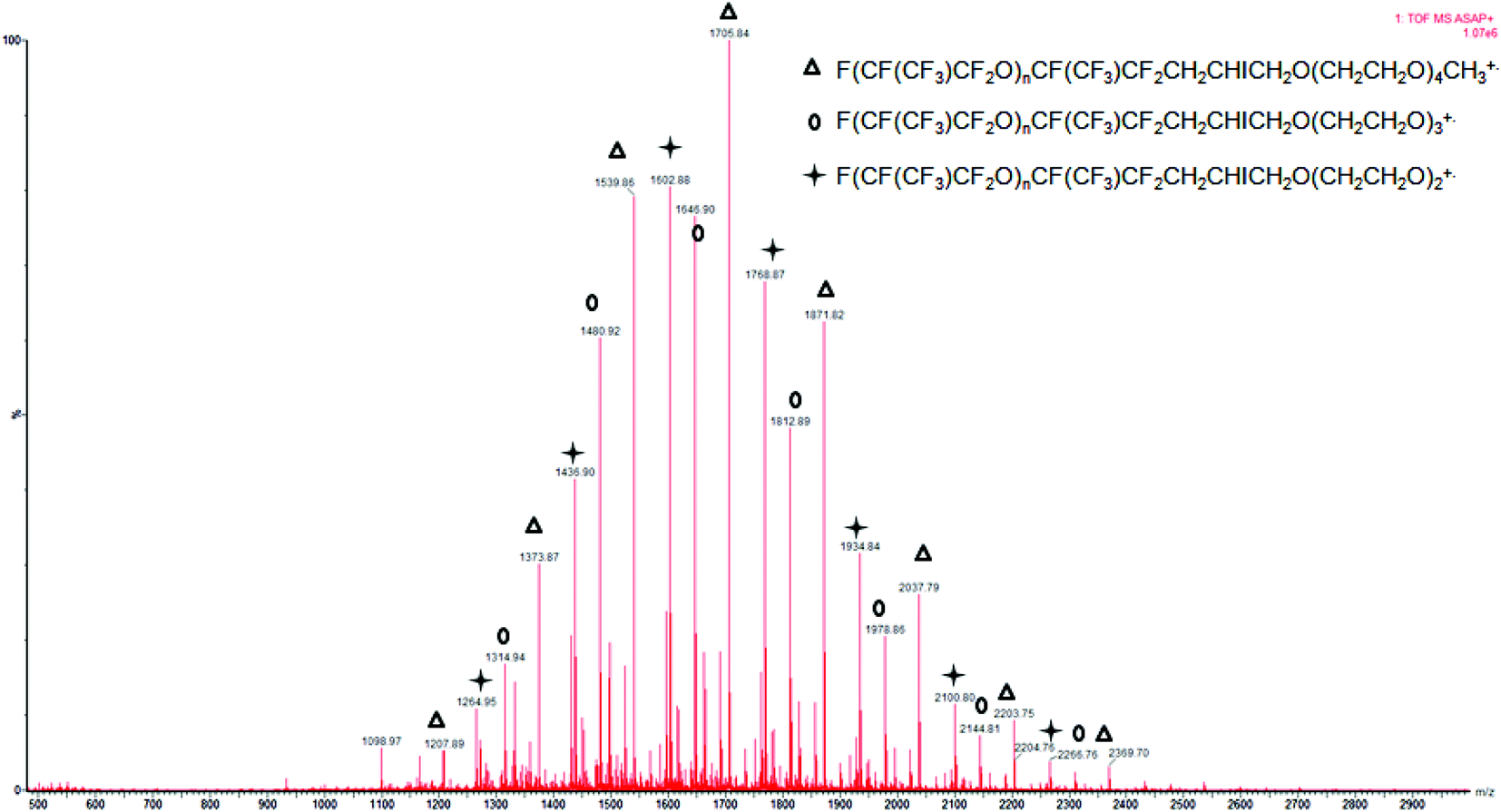 | ||
| Fig. 5 Positive mode Atmospheric pressure Solids Analysis Probe (ASAP) time-of-flight mass spectrometry (TOF-MS) spectrum of F[CF(CF3)CF2O]8.9CF(CF3)CF2CH2CHICH2O(CH2CH2O)9.5–CH3 (initiated with BPO). 166 m/z is the repeat unit for HFPO [CF(CF3)CF2O] and 44 m/z is the repeat unit for ethylene oxide (CH2CH2O). | ||
2.3. Radical reactions of 1-iodoperfluorohexane and 1-iodoperfluoro-propylene-2-oligo(hexafluoropropylene oxide) with various initiators (TBPPi, AIBN, and BPO)
In all radical reactions involving an allyl reagent, an almost complete conversion of the fluorinated iodides was observed [seen in the 19F-NMR spectra (Fig. 2 and S33–35†)]. However, this did not necessarily imply the formation of the desired product. Thus, it was worth studying the nature of radical initiators to understand their role in the production of undesired side-products. As expected, AIBN did not add onto any of the fluorinated alkyl iodides both in the case of neat addition (RXN 22 and 26, Table 4) or in the presence of sodium dithionite (RXN 28) or when chloroform was used as a solvent (RXN 23 and 27). In the presence of radicals, chloroform normally generates CCl3˙ or HCCl2˙, but the 13C-NMR spectrum did not show any chemical shifts ranging between 80 and 100 ppm.61 In contrast, TBPPI and BPO formed side-products with both fluorinated alkyl iodides, C6F13I and oligo(HFPO)-CF(CF3)CF2I. When C6F13I reacted with TBPPi (RXN 21), the 19F NMR spectrum (Fig. S33†) shows complex signals ranging between −112.3 and −113.6 ppm assigned to the by-product and a signal at −112 ppm characteristic of –CF2CF2I in the remaining C6F13I. C4F9I is known to react with aryl-compounds as evidenced by a 19F-NMR chemical signal centered at ca. −111 ppm assigned to a –CF2-aromatic group.62 The 19F-NMR spectrum of RXN 24 (Fig. S33†) displays two signals in this same area. These signals were also present in the case of radical reactions of C6F13I onto allyl compounds (Fig. S33–34†), while the other two initiators are more efficient in product formation (yield = 37 to 99%).| Rxn # | 1 iodide | Initiator type | Catalyst/Solvent | 1 HEC (%) | Conv. 1 iodide (%) |
|---|---|---|---|---|---|
| 21 | C6F13I | TBPPI | None | 0 | 92 |
| 22 | C6F13I | AIBN | None | 0 | 0 |
| 23 | C6F13I | AIBN (CHCl3) | None | 0 | 0 |
| 24 | C6F13I | BPO | Cu(OAc)2 | 0 | 100 |
| 25 | OligoHFPO-I | TBPPI | None | 4.5 | 95.5 |
| 26 | OligoHFPO-I | AIBN | None | 0 | 0 |
| 27 | OligoHFPO-I | AIBN (CHCl3) | None | 0 | 0 |
| 28 | OligoHFPO-I | AIBN | Na2S2O4 | 0 | 0 |
| 29 | OligoHFPO-I | BPO | Cu(OAc)2 | 0.5 | 7.5 |
As mentioned earlier, BPO produces aromatic side-products. The structures of these side-products were observed by GC/MS (Fig. S14†). From the reaction of 1-iodoperfluorohexane with BPO, the prevailing aromatic products were iodobenzene (4.8 min, 77 and 204 m/z), benzoic acid (7.1 min, 77, 105, 122 m/z), ortho, meta, para isomers of perfluorohexyl iodobenzene (7.9, 8.1, and 8.5 min; 126, 253, and 522 m/z, respectively) and the minor products arising from the addition of perfluorohexyl radical onto biphenyl (7.4 and 7.8 min) and benzoic acid (9.77 and 10.2 min). Therefore, it is believed that the dominant side-product in the case of the reaction between BPO and the 1-iodoperfluoroalkanes (Table 4) is an iodobenzene perfluoroadduct.
The same radical reactions were studied in the presence of oligo(HFPO)-CF(CF3)CFI. Surprisingly, this iodo compound shows a different behavior than that of C6F13I. It was noted that neither AIBN (RXN 26) nor BPO (RXN 29) led to any new signals in the −112 to −118 ppm range in the 19F-NMR spectra (Fig. S35†). In contrast, TBPPi converted all of the iodide into the undesired side-product. 13C-NMR spectroscopy (APT, Fig. S36†) and ASAP mass spectrometry (Fig. S37†) were instrumental in identifying the main side-product using TBPPi. The side-product arises from the addition of the 1-iodoperfluoroalkane onto 2-methyl-propene (Scheme 5), a decomposition product from TBPPi. This was evidenced in the 13C NMR spectrum (Fig. S36†) by the presence of the characteristic triplet (2JCF = 19 Hz) of doublets (3JCF = 8 Hz) centered at 47.5 ppm and assigned to –CH2CF2–. The other two signals which can be seen at 36.1 and 32.0 ppm are attributed to the methyl groups in iso-butyl and the tertiary carbon atom connected to the iodine atom, respectively. It is known that TBPPi generates two radicals: tert-butoxyl˙ and tert-butyl-CO2˙.58,63 The positive mode in ASAP analysis also confirms the addition of oligo(HFPO)-I onto 2-methyl-propene with the main distribution observed between 900 and 1950 m/z corresponding to the (F[CF(CF3)CF2O]nCF(CF3)CF2CH2C(CH3)2 + CH3CN)+ radical cation formed with acetonitrile in the probe.
 | ||
| Scheme 5 Conversion of poly(hexafluoropropylene oxide) primary iodide to undesired side-product using TBPPI as the radical initiator. | ||
This work carried out with the 1-iodoperfluoroalkanes and initiators provides a clear explanation for the results listed in Table 2. Most reactions were biphasic due to the immiscibility of the reactants. During the reaction with allyl alcohol and BPO in acetic acid, a dominating amount of iodobenzene was formed and could be more soluble in oligo(HFPO)-CF(CF3)CF2I or C6F13I than acetic acid. This led to a perfluoroalkyl iodide conversion into an aromatic side-product by reaction of the oligo(HFPO)-CF(CF3)CF2I- or C6F13I-derived radicals onto the iodobenzene. As for reactions with allyl-O-PEG-OCH3 initiated by BPO, the solubility of the iodobenzene in C6F13I is higher than that of the allyl-PEG. This difference in solubility may explain the failure of the reactions (yield = 0%). In the reaction involving oligo(HFPO)-CF(CF3)CF2I, allyl-O-PEG-OCH3 may have some partial solubility in the fluorinated ether which reduced the production of iodobenzene and thus minimized side-products. In summary, BPO only formed by-products with C6F13I, but not with oligo(HFPO)-CF(CF3)CF2I (at least not detectable at the scale of the reactions).
In the case of the reactions involving oligo(HFPO)-CF(CF3)CF2I and allyl-O-PEG-OCH3, if TBPPi is used as the initiator, 2-methylpropene was quickly released at 90 °C (Scheme 5) and oligo(HFPO)-CF(CF3)CF2I reacted faster with the smaller 2-methylpropene rather than with the bulky and sterically hindered allyl-O-PEG-OCH3.
2.4. Reduction of oligo(HFPO)-CH2CHICH2-oligo(PEG) using tributyltin hydride
Kaplánek et al.9a reported the formation of ω-perfluorohexyl poly(ethylene oxide) diblock co-oligomer [C6F13CH2CHICH2PEG] by the reduction of the iodine atom in the presence of zinc, NiCl2·6H2O in THF–H2O in 62% yield. The present study used tributyltin hydride which was efficient in previous studies to achieve the desired reduction.64,65 The successful reduction can be evidenced from the 1H-NMR spectrum (Fig. S38†) with the high field shift from 4.36 ppm (assigned to –CHI–, Fig. S28†) to 1.89 ppm characteristic of the central –CH2– group. The 19F-NMR spectrum (Fig. S39†) is similar to that of the iodide-containing diblock cooligomer.The 13C-NMR spectrum (Fig. S40†) exhibits the absence of the signal assigned to the methine –CHI– at 18 ppm that underwent a low field shift to 20.55 ppm for the new central methylene group. In addition, the methylene group in –CF2CH2 shifted from 37.65 to 27.80 ppm due to the reduction of the iodine atom. The yield of this reaction was 77%. The positive mode of ASAP and MALDI-TOF-MS spectra (Fig. 6 and S41–42†) also confirmed the reduction of the material and showed the connectivity of oligo(HFPO) to oligo(PEG) evidenced by the presence of 166 and 44 m/z for HFPO and ethylene oxide repeat units, respectively.
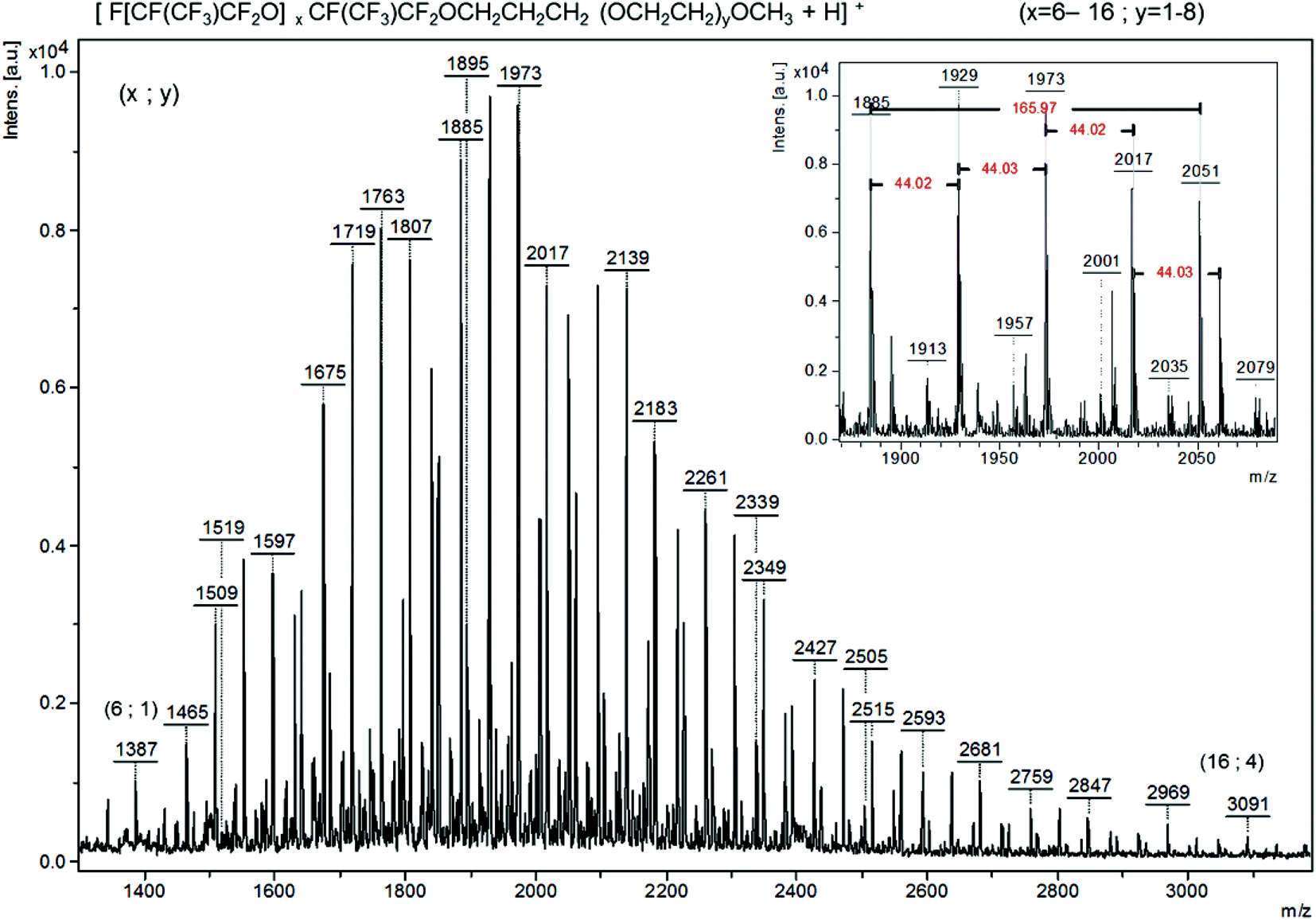 | ||
| Fig. 6 Positive ion mode MALDI-TOF-MS spectrum of oligo(HFPO)-CH2CH2CH2-oligo(PEG) (using as a matrix DCTB and LiCl as the cationizing agent), 1807 m/z is x = 8 and y = 5. The insert expansion m/z between 1850 and 2100 displays 166 and 44 m/z repeat units for HFPO [CF(CF3)CF2O] and ethylene oxide (CH2CH2O), respectively. | ||
2.5. Surface tension measurements (critical micellar concentration, CMC)
The oligo(HFPO)-b-oligo(PEG) diblock co-oligomer is as an attractive amphiphilic molecule, the surfactant behavior of which deserves to be studied. It is partially water-soluble up to ca. 0.3 g L−1. Surface tension properties of this diblock co-oligomer were assessed by tensiometry and compared to the surface tension of commercially available ammonium perfluorooctanoate (APFO). Three surfactants were measured for comparison: APFO, oligo(HFPO)-CH2CHICH2-oligo(PEG) and oligo(HFPO)-b-oligo(PEG) diblock cooligomers. APFO, a commonly used surfactant, was regarded as a reference and was measured to have a surface tension of 37.7 mN m−1 at the critical micellar concentration (CMC) of 3.77 g L−1 (Fig. S43†). Oligo(HFPO)-CH2CHICH2-oligo(PEG) and oligo(HFPO)-b-oligo(PEG) diblocks were measured to have surface tensions of 38.5 mN m−1 and 43.5 mN m−1 at the cmc of ca. 0.13 g L−1 and 0.04 g L−1, respectively. The plot of their surfaces can be seen in Fig. 7. Interestingly, it can be noted that the surface tension properties of such a diblock co-oligomeric surfactant are much better than those of APFO in terms of the amount of material required to reach the critical micellar concentration. The oligo(HFPO)-b-oligo(PEG) diblock's cmc is ca. 94 times better than that of APFO and 3 times smaller than that of oligo(HFPO)-CH2CHICH2-oligo(PEG). In comparison, the cmcs of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS) are 4.45 and 15.70 g L−1, respectively.66 At room temperature, the oligo(HFPO)-b-oligo(PEG) diblock co-oligomer is a potential surfactant, competitor to APFO.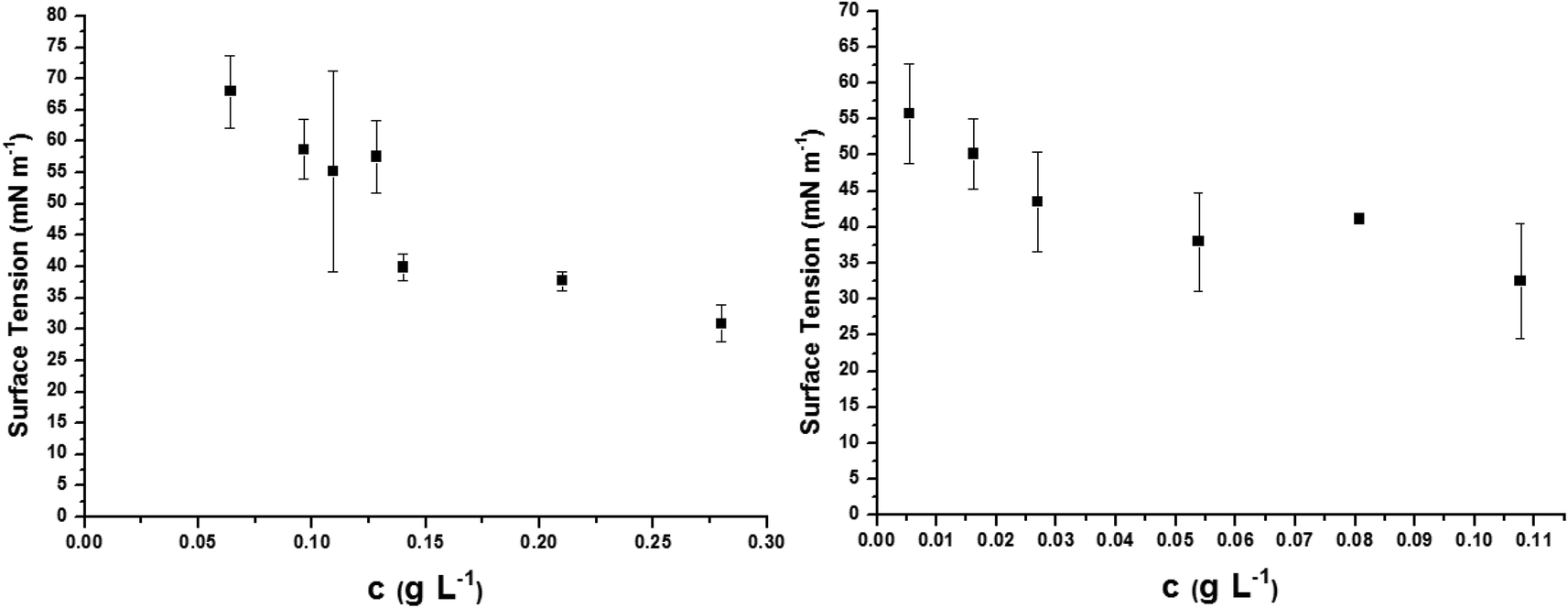 | ||
| Fig. 7 Surface tension measurements of new surfactant (left) oligo(HFPO)-CH2CHICH2-oligo(PEG) [CMC = 0.13 g L−1] (and right) oligo(HFPO)-b-oligo(PEG)[CMC = 0.04 g L−1]. Reference: APFO [CMC = 3.77 g L−1] (Fig. S42†). | ||
3. Conclusion
An original diblock co-oligomer based on oligo(hexafluoropropylene oxide) and oligo(ethylene oxide) was successfully synthesized by the radical addition of oligo(HFPO)-CF(CF3)CF2I onto an allyl-O-PEG-OCH3 derivative followed by the selective reduction of the iodine atom. Careful optimization of the experimental conditions such as the nature of the initiators (TBPPi, AIBN, or BPO), temperature, and reaction time was required to synthesize the desired diblock co-oligomer. BPO was shown to be the most efficient source of radicals with regard to both yield and reaction time. The best overall yield obtained from the oligo(HFPO)-CF(CF3)CF2I precursor was 71%. The oligo(HFPO)-b-oligo(PEG) diblock co-oligomer exhibits very interesting surfactant behavior. Indeed, the surface tension of water reached 32.5 mN m−1 for a surfactant concentration of 0.11 g L−1 and a low critical micellar concentration (0.04 g L−1 at room temperature). This newly formed diblock molecule has demonstrated relevant properties compared to those of the commercially available ammonium perfluorooctanoate (APFO). Further studies dealing with the synthesis of homologue surfactants containing various oligo(HFPO) and PEG chain lengths delving into deeper surface activity studies, absorption kinetics, and static and dynamic interfacial properties, as well as their bioaccumulation, decomposition and micellar behavior in solution, are in progress.4. Experimental details
4.1. Materials
All materials were used without further purification or drying. Krytox® primary iodide, 1-iodoperfluoropropyl-2-oligo(hexafluoropropylene oxide) (oligo(HFPO)-CF(CF3)CF2I) and Freon 113 were kindly offered by DuPont (Wilmington, USA) and iodoperfluorohexane (C6F13I, purity 95%) was generously supplied by Atofina (now Arkema, Pierre-Benite, France). Ammonium perfluoro-octanoate (Daikin Industries, Ltd), allyl alcohol and anhydrous copper(II) acetate (Fluka, 98%), tributyl stannane (Sigma Aldrich, 97%), sodium dithionite (Na2S2O4, Fisher Chemical), chloroform (Sigma Aldrich, 98%), recrystallized azobisisobutyronitrile (AIBN) in methanol (Fluka Analytical), tert-butyl peroxypivalate (TBPPi, TRIGONOX 25-C75, 75% TBPPi diluted to 63% in isodecane, Akzo Nobel), di-benzoyl peroxide (BPO) (Sigma Aldrich), polyethylene glycol methyl ether (Fluka Analytical, 500 amw), and allyl bromide (Sigma Aldrich).4.2. Analyses
:1.
4.3. Syntheses
RXN #1: A round-bottomed flask (10 mL) was charged with C6F13I (0.5135 g, 1.15 mmol), allyl alcohol (0.1089 g, 1.87 mmol) and TBPPi (0.0264 g, 0.152 mmol), heated to 75 °C, for 2 h. After elimination of unreacted reactants by a rotary evaporator, the total product mixture was dissolved in CDCl3 for NMR analysis. Yellow solid, yield by 19F-NMR = >99% yield, with >99% purity.
RXN #2: A round-bottomed flask (10 mL) was charged with C6F13I (0.501 g, 1.12 mmol), allyl alcohol (0.0667 g, 1.148 mmol), AIBN (0.0037 g, 0.0225 mmol), Na2S2O4 (0.0503 g, 0.289 mmol), and H2O (0.0663 mL), heated to 90 °C for 4 h. The product was dissolved in CDCl3. Slightly yellow solid, yield by 19F-NMR = 92% yield, with 93% purity.
RXN #3: A round-bottomed flask (10 mL) fitted with a reflux condenser was charged with C6F13I (0.501 g, 1.12 mmol), allyl alcohol (0.0976 g, 1.68 mmol), BPO (0.2442 g, 1.008 mmol), copper(II) acetate (0.0102 g, 0.055 mmol), and glacial acetic acid (0.3 mL), heated to 90 °C for 8 h. The product was dissolved in CDCl3. Yellow solid, yield by 19F-NMR = 0% yield.
4.3.1.1. Characterization. TBPPI: 1H NMR (400 MHz, CDCl3, 25 °C) (Fig. S3†): δ = 4.31 (quin, –CH2CHICH2OH, 3JHH = 6.57 Hz, 1H), 3.80, 3.74 (–CH2CHICHaHbOH, 2JHaHb = 12.13 Hz, 1H), 3.78, 3.73(d, –CH2CHICHaHbOH, 2JHbHa = 12.13 Hz, 1H) 3.01 (m, –CF2CHaHbCHI–, 1H), 2.65 (m, –CF2CHaHbCHI–, 1H), 2.95(–CH2OH, 1H); 19F NMR (376.41 MHz, CDCl3, 25 °C) (Fig. S5†): δ = −81.06 (CF3–, 3JFF = 10.33, 4JFF = 2.30 Hz, 3F), −126.36 (m, CF3CF2(CF2)4CH2–, 2F), −123.77 (m, –CF2(CF2)3CH2–, 2F), −123.05 (m, –CF2(CF2)2CH2–, 2F), −121.97 (m, –CF2CF2CH2–, 2F), −113.17, −114.20 (dm, –CF2CH2–, 2JFF = 144.68 Hz, 2F); 13C NMR (101 MHz, CDCl3, 25 °C) (Fig. S7†): δ = 118.49 (qt, CF3CF2– 1JCF = 288.34 Hz, 2JCF = 33.66 Hz), 117.70 (tt, –CF2CF2CH2–, 1JCF = 257.61 Hz, 2JCF = 32.20 Hz), 110.87 (m, CF3(CF2)4CF2–, 4C), 67.78 (s, –CH2OH, 1C), 37.18 (t, –CF2CH2CHI–, 2JCF = 20.49 Hz, 1C), 20.4 (s, –CH2CHICH2OH, 1C).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHCH2O[CH2CH2O]9.5CH3).
The same equipment as above was used and dried and the reactions were carried out under a nitrogen atmosphere. To the three neck round-bottomed flask (100 mL) containing a magnetic stirrer, sodium hydride (80% by mass) in mineral oil (0.5125 g, 17.129 mmol) was weighed, dissolved in dry toluene (5 mL) and placed in an ice bath. A mixture composed of PEG500 (5.004 g, 10.58 mmol) in 50 mL toluene (dried by distillation) was added drop-wise into the reaction medium under vigorous stirring and then stirred for 2 h at room temperature. Then, the total product mixture was cooled in an ice bath and allyl bromide (3.849 g, 31.81 mmol) was slowly added. The mixture was stirred at room temperature for 24 h. The resulting mixture was filtered over silica gel. The liquid part was dissolved in 100 mL water and extracted four times with 20 mL portions of ether. The product was dried under high vacuum (0.128 × 10−3 mbar). The product was a colourless liquid; the isolated yield was 3.5044 g, 65%.
CHCH2O[CH2CH2O]9.5CH3).
The same equipment as above was used and dried and the reactions were carried out under a nitrogen atmosphere. To the three neck round-bottomed flask (100 mL) containing a magnetic stirrer, sodium hydride (80% by mass) in mineral oil (0.5125 g, 17.129 mmol) was weighed, dissolved in dry toluene (5 mL) and placed in an ice bath. A mixture composed of PEG500 (5.004 g, 10.58 mmol) in 50 mL toluene (dried by distillation) was added drop-wise into the reaction medium under vigorous stirring and then stirred for 2 h at room temperature. Then, the total product mixture was cooled in an ice bath and allyl bromide (3.849 g, 31.81 mmol) was slowly added. The mixture was stirred at room temperature for 24 h. The resulting mixture was filtered over silica gel. The liquid part was dissolved in 100 mL water and extracted four times with 20 mL portions of ether. The product was dried under high vacuum (0.128 × 10−3 mbar). The product was a colourless liquid; the isolated yield was 3.5044 g, 65%.
4.3.2.1. Characterization. 1H NMR (400 MHz, C6D6, 25 °C) (Fig. S1†) δ = 5.85 (ddt, CHaHb
CHcCH2–, 3JHcHb(trans) = 17.34 Hz, 3JHcHa(cis) = 10.36 Hz, 3JHcH(CH2) = 5.81 Hz, 1H), 5.20 (ddt, CHaHbCHcCH2–, 2JHbHa = 1.77 Hz, 3JHbHc(trans) = 17.18 Hz, 4JHbH(CH2) = 1.77 Hz, 1H), 5.11 (dm, CHaHbCHcCH2–, 3JHaHc(cis) = 10.36 Hz, 2JHaHb = 1.77 Hz, 1H), 3.96 (dm, CHaHbCHcCH2O–, 3JH(CH2)Hc = 5.56 Hz, 2H), 3.53 (t, –OCH2CH2OCH3, 3JHH = 5.5 Hz, 2H), 3.55–3.61 (m, –CH2O–, 19 × 2H), 3.31 (s, –OCH3, 4JHH = 0.72 Hz, 3H), 3.48 (t, –CH2OCH3, 3JHH = 5.5 Hz, 2H); 13C NMR (101 MHz, C6D6, 25 °C) (Fig. S2†) δ = 135.97 (s, –CH, 1C), 116.0 (s, CH2, 1C), 72.07 (s, 1C, CH2–OCH2CHCH2, 1C), 72.07 (s, 1C, CH2–OCH3, 1C), 70.74 (s, –CH2–O, 19 × 1C), 58.65 (s, 1C, CH3).
RXN #5: C6F13I (0.093 g, 0.208 mmol), allyl-O-PEG-OCH3 (0.2113 g, 0.417 mmol), and AIBN (0.001 g, 0.0042 mmol), heated to 90 °C for 8 h. The product was dissolved in CDCl3. Brown solid, yield by 19F-NMR = 37% yield, with 51% purity.
RXN #6: A round bottom flask (50 mL) equipped with a reflux condenser contained C6F13I (0.093 g, 0.2084 mmol), allyl-O-PEG-OCH3 (0.2113 g, 0.417 mmol), benzoyl peroxide (0.101 g, 0.413 mmol), copper(II) acetate (0.0039 g, 0.0214 mmol), and glacial acetic acid (10 mL) was heated at 90 °C for 8 h. The product was dissolved in CDCl3. Dark brown solid, yield by 19F-NMR = 0% yield.
4.3.3.1. Characterization. 1H NMR (400 MHz, DMSO capillary, 25 °C) (Fig. S11†): δ = 4.35 (m, –CH2CHICH2OH, 1H), 3.80, 3.74 (–CH2CHICHaHbO–, 2JHaHb = 10.86 Hz, 1H), 3.65, 3.64 (d, –CH2CHICHaHeOH, 2JHbHa = 10.86 Hz, 1H), 3.16 (s, –OCH3, 4JHH = 0.72 Hz, 3H), 3.35 (t, –OCH2CH2OCH3, 3JHH = 5.3 Hz, 2H), 3.33 (t, –CH2OCH3, 3JHH = 6.3 Hz, 2H), 3.5–3.4 (m, –CH2O–, 19 × 2H), 3.14–3.03 (m, –CF2CHaHbCHI–, 1H), 2.70–2.59 (m, –CF2CHaHbCHI–, 1H); 19F NMR (376.41 MHz, DMSO capillary, 25 °C) (Fig. S12†): δ = −80 (t, CF3–, 3JFF = 9.4 Hz, 3F), −112.24 (d, –CFaFbCH2–, 2JFaFb = 273.06 Hz, 1F), −112.83 (d, –CFaFbCH2–, 2JFbFa = 273.06 Hz, 1F, 2F), −120.73 (m, –CF2CF2CH2–, 2F), −121.78 (m, –CF2(CF2)2CH2–, 2F), −122.62 (m, CF2(CF2)3CH2–, 2F), −125.15 (s, CF3CF2(CF2)4CH2–); impurity: −63.78 (m, CF2I, 2F), −112.59 (m, –CF2CF2I), −120.08 (m, –(CF2)2I, 2F); 13C NMR (101 MHz, CDCl3 capillary, 25 °C) (Fig. S13†): δ = −122 to −104 (m, C6F13, 6C), 76.05 (s, –CHICH2O–, 1C), 71.85 (s, –CH2OMe, 1C), 70.47 (s, –OCH2–, 19 × 1C), 70.40 (s, –OCH2CH2OMe), 58.92 (s, –OCH3, 1C), 14.39 (s, –CH2CHICH2O–, 1C), 37.34 (t, 2JCF = 20.83 Hz, –CF2CH2CHI–, 1C).
4.3.4.1. General synthetic procedure. 1-Iodo-2 oligo(hexafluoropropylene oxide) perfluoropropane (oligoHFPO-I) (M = 1773 g mol−1), allyl alcohol, and initiator were weighed in appropriate amounts. Oxygen was displaced by purging nitrogen into the reaction mixture for 10 minutes. All reactions were carried out under positive nitrogen pressure and heated using an oil bath. Initiators were added to the reaction mixtures in regular periods according to their half-lives and reaction temperatures. The progress of the reaction was monitored by GC/MS using the starting iodide as the reference. After reaction, all products were separated by an acetone extraction in the same reaction flask. The soluble side-products were first separated by decantation, followed by filtration through a Teflon® filter (2 mm porosity). The final product was dried at ca. 40 °C under vacuum (ca. 140 × 10−3 mbar) for 20 minutes.
4.3.4.2. Initiator (tert-butyl peroxypivalate, TBPPi). RXN #7: In a 10 mL round-bottomed flask, oligo(HFPO)-CF(CF3)CF2I (1.006 g, 0.564 mmol), allyl alcohol (0.0438 g, 0.754 mmol), and TBPPi (0.0020 g, 0.0113 mmol) were heated to 75 °C for 48 h. TBPPi was transferred (0.0020 g, 0.0115 mmol) every 4 h during the day for a total of six injections. Allyl alcohol (0.0438 g, 0.754 mmol) was also added 6 times. The reaction mixture was washed 3 times with 3 mL acetone and one time with 3 mL ethanol. Yellow liquid, yield by 19F-NMR = 88%, with 98% purity.
RXN #8: In a 10 mL round-bottomed flask, oligo(HFPO)-CF(CF3)CF2I (1.006 g, 0.564 mmol), allyl alcohol (0.0438 g, 0.754 mmol), and TBPPi (0.00197 g, 0.0113 mmol) were heated to 90 °C for 38 h. TBPPi was transferred (0.00263 g, 0.0151 mmol) every 4 h during the day for a total of 18 injections. Allyl alcohol (0.0503 g, 0.867 mmol) was also added 18 times. The reaction mixture was washed 3 times with 3 mL acetone and one time with 3 mL ethanol. Yellow liquid, yield by 19F-NMR = 80%, with 90.5% purity.
RXN #9: In a 25 mL round-bottomed flask, oligo(HFPO)-CF(CF3)CF2I (10.090 g, 5.694 mmol), allyl alcohol (0.438 g, 7.54 mmol), and TBPPi (0.0020 g, 0.0113 mmol) were heated to 75 °C for 32 h. TBPPi was transferred (0.0355 g, 0.204 mmol) every 4 h during the day for a total of 16 injections. Allyl alcohol (0.0820 g, 1.411 mmol) was also added 16 times. The reaction mixture was washed 3 times with 3 mL acetone and one time with 3 mL ethanol. Yield by 19F-NMR = 0%.
4.3.4.3. Initiator (azobisisobutyronitrile, AIBN). RXN #10: 1° oligo(HFPO)-CF(CF3)CF2I (2.034 g, 1.147 mmol), allyl alcohol (0.0883 g, 1.52 mmol), and AIBN (0.0037 g, 0.0226 mmol) were placed in a two necked pear-shaped flask (10 mL) and heated to 90 °C for 64 h. AIBN and allyl alcohol were both added 7 times at 4 h intervals. When the reaction was complete, extraction of by-products was carried out with acetone washings (5 × 25 mL). Yield by 19F-NMR = 86% yield, with 99% purity.
RXN #11: In a 10 mL round bottomed flask, oligo(HFPO)-CF(CF3)CF2I (1.004 g, 0.564 mmol), allyl alcohol (0.0437 g, 7.52 mmol), AIBN (20% in chloroform, AIBN (0.0019 g, 0.0116 mmol) and chloroform (0.0074 g, 0.0620 mmol) were heated to 90 °C for 72 h. AIBN in chloroform was added via a syringe 17 times every 4 h while allyl alcohol was added once after 32 h (0.044 g, 0.758 mmol). Extraction with acetone was achieved (5 times, 20 ml). Yield by 19F-NMR = 86% yield, with 97% purity.
RXN #12: In a 250 mL round bottomed flask, oligo(HFPO)-CF(CF3)CF2I (10.040 g, 5.6402 mmol), allyl alcohol (0.0437 g, 7.52 mmol), AIBN (20% in chloroform, AIBN (0.0185 g, 0.0116 mmol) and chloroform (0.074 g, 0.0620 mmol) were heated to 90 °C for 72 h. AIBN in chloroform was added via a syringe 35 times every 4 h while allyl alcohol was added one time after 32 h (0.044 g, 0.758 mmol). Extraction with acetone was achieved (5 times 20 ml). Yield by 19F-NMR = 89% yield, with 98% purity.
4.3.4.4. di-benzoyl peroxide (BPO). RXN #13: In a three-necked round-bottomed flask (50 mL) equipped with a reflux condenser, oligo(HFPO)-CF(CF3)CF2I (1.003 g, 0.564 mmol), allyl alcohol (0.065 g; 1.12 mmol), glacial acetic acid (solvent, 10 mL), copper(II) acetate (0.011 g, 0.0548 mmol), and BPO (0.273 g; 1.128 mmol) were heated to 90 °C for 8 h. The contents were cooled and the acetic acid was evaporated. A one-time extraction with water (10 mL) followed by a 4× extraction with ethanol (10 mL) achieved the final material. Yield by 19F-NMR = 0% yield.
RXN #14: In a three-necked round-bottomed flask (50 mL) equipped with a reflux condenser, oligo(HFPO)-CF(CF3)CF2I (1.003 g, 0.564 mmol), allyl alcohol (0.1310 g, 2.2559 mmol), glacial acetic acid (solvent, 10 mL), copper(II) acetate (0.005 g, 0.02738 mmol), and BPO (0.8767 g, 3.619 mmol) were heated to 90 °C for 8 h. The contents were cooled and the acetic acid was evaporated. A one-time extraction was accomplished with water (10 mL), followed by a 4× extraction with ethanol (10 mL). Yield by 19F-NMR = 0% yield.
4.3.4.5. Characterization. TBPPI: 1H NMR (400 MHz, C6D6 capillary, 25 °C) (Fig. S17†): δ = 4.4 (s, –CH2CHICH2OH, 1H), 4.21 (s, –CH2OH, 1H), 3.81 (–CH2CHICH2OH, 2H), 2.98 (m, –CF2CHaHbCHI–, 1H), 2.72 (m, –CF2CHaHbCHI–, 1H); (AIBN): 1H NMR (400 MHz, C6D6 capillary, 25 °C) (Fig. S18†): δ = 4.31, 4.25 (s, –CH2CHICH2OH, 1H), 3.99 (s, –CH2OH, 1H), 3.66 (–CH2CHICH2OH, 2H), 2.84 (m, –CF2CHaHbCHI–, 1H), 2.54 (m, –CF2CHaHbCHI–, 1H); 19F NMR (376.41 MHz, C6D6, 25 °C) (Fig. S20†): δ = −80 to −85 (m, CF(CF3)CF2O–), −80.15 (s, CF3CF2CF2O–, 3F), −81.80 (s, CF3CF2CF2O–, 2F), −110.92 (d, –CF(CF3)CFaFbCH2–, 2JFF = 262.73 Hz), −112.54 (d, –CF(CF3)CFaFbCH2–, 2JFF = 237.49 Hz), −113.99 (d, –CF(CF3)CFaFbCH2–, 2JFF = 261.58 Hz), −129.80 (s, CF3CF2CF2–, 2F), −146.80 (m, –CF(CF3)CF2–, 8.9 × 1F); 13C NMR (101 MHz, DMSO/C6D6 capillary, 25 °C) (Fig. S22†): δ = 118.0 (qd, 1JCF = 290.9, 2JCF = 28.2 Hz, –OCF(CF3)CF2–), 117.6 (qt, 1JCF = 286.15 Hz, 2JCF = 32.93 Hz, CF3CF2CF2O–, 1C), 117.5 (qd, 1JCF = 286.15, 2JCF = 34.40 Hz, CF3CF2CF2O, 1C), 114.7 (td, 1JCF = 285.74, 2JCF = 31.26 Hz, –OCF(CF3)CF2–, 8.9 × 1C), 105.2 (tsext, 1JCF = 267.03 Hz, 2JCF = 36.68 Hz, CF3CF2CF2O–, 1C), 101.8 (dsext, 1JCF = 270.7, 2JCF = 36.7 Hz, –OCF(CF3)CF2–), 66.32 (s, –CH2CHICH2OH, 1C), 36.52 (m, –CF2CH2CHICH2OH, 1C), 18.09, 17.90 (s, –CH2CHICH2OH, 1C).
4.3.5.1. Characterization56. 1H NMR (400 MHz, CDCl3): δ = 3.8 (t, 3JHH = 5.6 Hz, –CH2CH2OH), 2.3 (m, –CF2CH2CH2–), 1.9 (m, –CH2CH2CH2–); 13C NMR (101 MHz, CDCl3 capillary, 25 °C) δ = 60.8 (s, –CH2OH, 1C), 27.9 (t, 2JCF = 24.3, –CF2CH2–, 1C), 23.5 (s, –CH2CH2CH2–, 1C); the 19F-NMR is similar to that of F[CF(CF3)CF2O]8.9CF(CF3)CF2CH2CHICH2OH. ASAP-TOF-MS spectrum (positive and negative, Fig. S44–45†).
4.3.6.1. General procedure. 1-Iodo-2-oligo(hexafluoropropylene oxide) perfluoropropane (oligo(HFPO)-CF(CF3)CF2I), polyethylene glycol allyl (allyl-O-PEG-OCH3), and the initiator were weighed in appropriate amounts. Oxygen was displaced by purging nitrogen into the reaction mixture for 10 minutes. All reactions were carried out under positive nitrogen pressure and heated using an oil bath. Initiators were added to the reaction mixtures in regular periods according to their half-lives and temperatures. The progress of the reaction was monitored by GC/MS using the starting iodide as the reference. When the reaction was complete, the GC/MS showed no oligo(HFPO) iodide. After the reaction, all products were separated by acetone extraction in the same reaction flask. The soluble side-products were first separated by decantation, followed by filtration through a Teflon® filter (2 mm porosity). The final product was dried at ca. 40 °C under vacuum (ca. 140 × 10−3 mbar) for 20 minutes and resulted in a light yellow oil.
4.3.6.2. Initiator (tert-butyl peroxypivalate, TBPPi). RXN #15: In a 10 mL round-bottomed flask equipped with a reflux condenser, oligo(HFPO)-CF(CF3)CF2I (1.0 g, 0.564 mmol), allyl-O-PEG-OCH3 (0.437 g, 0.8619 mmol), and TBPPi (0.312 g, 1.13 mmol) were heated to 75 °C. TBPPi (3.202 g, 0.23 mmol) was added every 4 h for a total of 19 additions. The product was washed 5× with ethanol (10 mL). A yellow liquid was obtained but the yield by 19F-NMR spectroscopy was 0%.
RXN #16: In a 3 neck 100 mL round-bottomed flask equipped with a reflux condenser, oligo(HFPO)-CF(CF3)CF2I (10.008 g, 5.64 mmol), allyl-O-PEG-OCH3 (4.373 g, 8.61 mmol), and TBPPi (0.312 g, 1.13 mmol) were heated to 75 °C for 64 h. TBPPi (0.312 g, 1.13 mmol) was added every 4 h for a total of 15 additions. The product was washed 3 times with ethanol (20 mL). Light yellow liquid, yield by 19F-NMR = 0% yield.
4.3.6.3. Initiator (azobisisobutyronitrile, AIBN). RXN #17: In a 2 necked round-bottomed flask, oligo(HFPO)-CF(CF3)CF2I (1.005 g, 0.564 mmol), allyl-O-PEG-OCH3 (1.3110 g, 2.5858 mmol), and AIBN (0.002 g, 0.012 mmol) were heated to 90 °C for 80 h. AIBN (0.020 g, 0.12 mmol) was added 20 times every 4 h. After reaction, extractions with acetone (5 × 10 mL) were performed. A light honey brown liquid was obtained. A yield of 48% and a purity of 75 % were calculated by 19F-NMR.
RXN #18: In a two-neck round-bottomed flask equipped with a Claisen condenser and a magnetic stir bar, oligo(HFPO)-CF(CF3)CF2I (10.012 g, 5.64 mmol), allyl-O-PEG-OCH3 (3.8228 g, 7.54 mmol), AIBN (0.0185 g, 0.113 mmol) and chloroform (0.074 g, 6.197 mmol) were added. The reaction mixture was bubbled for ca. 10 minutes with nitrogen and then placed in an oil bath that was progressively heated up to 90 °C. Additional allyl-O-PEG-OCH3 (3.8228 g, 7.54 mmol) was transferred into the reaction mixture after 40 h. AIBN in chloroform (0.0185 g, 0.113 mmol, 0.074 g, 0.618 mmol) was added 73 times in total with the additions occurring every 2 h. Because complete conversion was not detected with NMR more of allyl-O-PEG-OCH3 was added (4.023 g, 7.93 mmol) and AIBN in chloroform (0.185 g, 1.13 mmol, 0.74 g, 6.197 mmol) was added 14 times in total occurring every 2 h. The product mixture was washed with 10 mL of water and then 3 times with 20 mL of acetone. The product was dried using a high-vacuum pump and filtered through a ring filter. A yellow-brown oil was obtained. The yield by 19F-NMR = 50%, with 76% purity.
4.3.6.4. Initiator (di-benzoyl peroxide (BPO)). RXN #19: In a two-neck 10 mL round-bottomed flask equipped with a reflux condenser, oligo(HFPO)-CF(CF3)CF2I (1.0007 g, 0.564 mmol), allyl-O-PEG-OCH3 (1.4003 g, 2.7613 mmol), copper(II)acetate (0.0052 g, 0.0274 mmol), glacial acetic acid (3 mL) and BPO (0.5675 g, 2.343 mmol) were added and heated to 90 °C. After the reaction, acetic acid was evaporated and washed 7 times with acetone (10 mL). Green oil, yield by 19F-NMR = 51% yield, with 88% purity.
RXN #20: In a three-neck 100 mL round-bottomed flask equipped with a reflux condenser, oligo(HFPO)-CF(CF3)CF2I (10.007 g, 5.64 mmol), allyl-O-PEG-OCH3 (4.291 g, 8.46 mmol), copper(II) acetate (0.052 g, 0.274 mmol), glacial acetic acid (3 mL), and BPO (1.641 g, 6.77 mmol) were added and then heated to 90 °C. After 8 h, additional allyl-O-PEG-OCH3 (1.431 g, 2.821 mmol) and BPO (1.192 g, 4.91 mmol) were added. The number of allyl-O-PEG-OCH3 and BPO additions was 5 times every 8 h. After the reaction the acetic acid was evaporated, and washed 7 times with acetone (10 mL). Dark green oil, yield by 19F-NMR = 71% yield, with 94% purity.
4.3.6.5. Characterization. 1H NMR (400 MHz, CDCl3 capillary, 25 °C) (Fig. S28†): δ = 4.36 (b, –CHI–, 1H), 3.79 (b, –CHICH2O, 2H), 3.59 (b, –CH2O, 19 × 1H), 3.46 (b, –CH2CH2OCH3, 4H), 3.28 (s, CH3O–, 3H), 3.09 (vb, –CF2CHaHbCHI–, 1H), 2.58 (vb, CF2CHaHbCHI–, 1H); 19F NMR (376.41 MHz, C6D6, 25 °C) (Fig. S29†): δ = −80 to −84 (CF(CF3)CF2O–), −84.04 (CF3CF2CF2O–, 3F), −82.37 (CF3CF2CF2O–, 2F), −112 to –117.5 (b, –CF(CF3)CF2CH2CHI–, 2F), −132.03 (s, CF3CF2CF2–, 2F), −146.54 (m, –CF(CF3)CF2–, 8.9 × 1F); 13C NMR (101 MHz, C6D6, 25 °C) (Fig. S30†): δ = 118.0 (qd, 1JCF = 290.9, 2JCF = 28.2 Hz –OCF(CF3)CF2–), 117.6 (qt, 1JCF = 286.15 Hz, 2JCF = 32.93 Hz, CF3CF2CF2O–, 1C), 117.5 (qd, 1JCF = 286.15, 2JCF = 34.40 Hz, CF3CF2CF2O, 1C), 114.7 (td, 1JCF = 285.74, 2JCF = 31.26 Hz, –OCF(CF3)CF2–, 8.9 × 1C), 105.2 (tsext, 1JCF = 267.03 Hz, 2JCF = 36.68 Hz, CF3CF2CF2O–, 1C), 101.8 (dsext, 1JCF = 270.7, 2JCF = 36.7 Hz, –OCF(CF3)CF2–), 76.27 (s, –CH2CHICH2O–, 1C), 72.17 (s, CH2–CH2–OMe, 2C), 70.79 (s, 19 × 1C, –CH2–O), 70.54 (s, –CH2CH2OMe, 1C), 58.65 (s, 1C, CH3), 37.65 (m, –CF2CH2CHICH2O–, 1C), 14.30 (s, –CH2CHICH2O–, 1C).
4.3.7.1. Characterization. 1H NMR (400 MHz, CDCl3 capillary, 25 °C) (Fig. S35†): δ = 3.5 9 (b, –CH2O, 23 × 1H), 3.40 (s, CH3O–, 3H), 2.20 (b, –CF2CH2CH2–, 1H), 1.89 (b, –CF2CH2CH2–, 1H); 19F NMR (376.41 MHz, C6D6, 25 °C) (Fig. S36†): δ = −80 to −84 (CF(CF3)CF2O–), −84.04 (CF3CF2CF2O–, 3F), −82.37 (CF3CF2CF2O–, 2F), −112 to −118 (b, –CF(CF3)CF2CH2CH2–, 2F), −131.56 (s, CF3CF2CF2–, 2F), −146.05 (m, –CF(CF3)CF2–, 9.9 × 1F); 13C NMR (101 MHz, C6D6, 25 °C) (Fig. S37†): δ = 118.0 (qd, 1JCF = 290.9, 2JCF = 28.2 Hz, –OCF(CF3)CF2–), 117.6 (qt, 1JCF = 286.15 Hz, 2JCF = 32.93 Hz, CF3CF2CF2O–, 1C), 117.5 (qd, 1JCF = 286.15, 2JCF = 34.40 Hz, CF3CF2CF2O, 1C), 114.7 (td, 1JCF = 285.74, 2JCF = 31.26 Hz, –OCF(CF3)CF2–, 8.9 × 1C), 105.2 (tsext, 1JCF= 267.03 Hz, 2JCF = 36.68 Hz, CF3CF2CF2O–, 1C), 101.8 (dsext, 1JCF = 270.7, 2JCF = 36.7 Hz, –OCF(CF3)CF2–), 69.42 (bs, –CH2O–, 21 × 1C), 57.59 (s, CH3, 1C), 27.80 (m, –CF2CH2CH2CH2O–, 1C), 20.55 (s, –CH2CH2CH2O–, 1C).
4.3.8.1. Model reactions of 1-iodoperfluorohexane with radical initiators. General procedure: In a round-bottomed flask (10 mL), C6F13I (0.5002 g, 1.12 mmol) and the appropriate initiator (TBPPi, AIBN, or BPO) were added. Oxygen was removed by purging the mixture with nitrogen gas for at least 10 minutes. The reaction mixture was heated at the required temperature.
RXN #21: TBPPi (0.6702 g, 2.42 mmol) was added to C6F13I and heated at 75 °C for 3 h. 19F-NMR yield = 92% C6F13CH2CI(CH3)2.
RXN #22: AIBN in chloroform (0.2002 g, 1.21 mmol; 1.002 g, 8.37 mmol) was added to C6F13I and heated to 90 °C for 3 h. No reaction.
RXN #23: AIBN in chloroform (0.3678 g, 2.24 mmol; 1.4712 g, 12.3 mmol), Na2S2O4 (0.0436 g, 0.266 g), and H2O (0.108) were added to C6F13I and heated to 90 °C for 3 h.
RXN #24: BPO (0.3315 g, 1.36 mmol), copper(II) acetate (0.0102 g, 0.056 mmol), and acetic acid (1 mL) were added to C6F13I heated to 90 °C for 3 h. 19F-NMR yield = >99% C6F13-Aromatic compounds (see Fig. S14†).
4.3.8.2. Model reactions of F[CF(CF3)CF2O]8.9CF(CF3)CF2I with radical initiators. General procedure: In a round-bottomed flask (10 mL) equipped with a condenser, oligo(HFPO)-CF(CF3)CF2I (M = 1773 g mol−1, 1.003 g, 0.564 mmol) and the appropriate initiator (TBPPi, AIBN, or BPO) were added. Oxygen was removed by purging the mixture with nitrogen gas for at least 5 minutes. The reaction mixture was heated at the required temperature.
RXN #25: TBPPi (0.329 g, 1.154 mmol) was added to oligo(HFPO)-CF(CF3)CF2I and heated to 75 °C for 4 h. 19F-NMR Yield = 95.5% of F[CF(CF3)CF2O]8.9CF(CF3)CF2CH2CI(CH3)2 and 4.5% F[CF(CF3)CF2O]8.9CF(CF3)CF2H
RXN #26: AIBN (0.1895 g, 1.154 mmol) was added to oligo(HFPO)-CF(CF3)CF2I and heated to 90 °C for 4 h. No reaction.
RXN #27: AIBN in chloroform (0.1895 g, 1.154 mmol; 0.9475 g, 7.94 mmol) was added to oligo(HFPO)-CF(CF3)CF2I and heated to 90 °C for 4 h. No reaction.
RXN #28: AIBN in chloroform (0.1895 g, 1.128 mmol; 0.742 g, 6.22 mmol), Na2S2O4 (0.0236 g, 0.133 g), H2O (0.055 ml) was added to oligo(HFPO)-CF(CF3)CF2I and heated to 90 °C for 4 h. No reaction.
RXN #29: BPO (0.279 g, 1.154 mmol), copper(II) acetate (0.071 g, 0.389 mmol), and acetic acid (0.3 ml) were added to oligo(HFPO)-CF(CF3)CF2I and heated to 90 °C for 8 h. 19F-NMR yield = 7.5% F[CF(CF3)CF2O]8.9CF(CF3)CF2-Aromatics and 0.5% F[CF(CF3)CF2O]8.9CF(CF3)CF2H.
Acknowledgements
The authors thank the E.I. du Pont de Nemours and Co., Inc. (especially Dr Jon Howell), the Daikin Industries, and the Elf Atochem for generous gifts of oligo(HFPO)-CF(CF3)CF2I, ammonium perfluorooctanoate, and C6F13I, respectively, the Fundation Balard-Chaire Total for supporting Professor Chadron Mark Friesen who spent one academic year at Ingénierie et Architectures Macromoléculaires, Institut Charles Gerhardt, and the European Commission for funding Jiří Lapčík with an Erasmus grant.References
- E. Kissa, Fluorinated Surfactants and Repellents, Marcel Dekker, Inc, New York, 2nd edn, 2001 Search PubMed.
- (a) C. Taylor, Annual Surfactants Review, 1999 Search PubMed; (b) K. Skrabania, A. Laschewsky, H. von Berlepsch and C. Böttcher, Langmuir, 1999, 25, 7594–7606 CrossRef PubMed; (c) A. Laschewsky, Adv. Polym. Sci., 1995, 124, 1–39 CrossRef CAS.
- (a) M. P. Krafft and J. G. Riess, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1185–1198 CrossRef CAS; (b) M. P. Krafft and J. G. Riess, Chem. Rev., 2007, 109, 1714–1785 CrossRef PubMed; (c) J. G. Riess, Curr. Opin. Colloid Interface Sci., 2009, 14, 294–304 CrossRef CAS PubMed.
- (a) J. G. Riess, Chem. Rev., 2001, 101, 2797–2920 CrossRef CAS PubMed; (b) J. G. Riess, Curr. Opin. Colloid Interface Sci., 2001, 14, 294–304 CrossRef PubMed.
- G. Kostov, F. Boschet and B. Ameduri, J. Fluorine Chem., 2009, 130, 1192–1199 CrossRef CAS PubMed.
- (a) R. Renner, Environ. Sci. Technol., 2001, 35, 154A–160A CrossRef CAS; (b) H. J. Lehmler, Chemosphere, 2001, 58, 1471–1496 CrossRef PubMed.
- E. I. du Pont de Nemours and Company . DuPont Global Strategy, Comprehensive Source Reduction, Presentation to EPA, January 31, 2005, U. S. EPA Administrative Record AR226-1914, 2005.
- J. Kovarova and Z. Svobodova, Neuroendocrinol. Lett., 2008, 29, 599–608 CAS.
- (a) R. Kaplánek, O. Paleta, I. Ferjentsiková and M. Kodíce, J. Fluorine Chem., 2009, 130, 308–316 CrossRef PubMed; (b) L. Caillier, E. Taffin de Givenchy, R. Levy, Y. Vandenberghe, S. Geribaldi and F. Guittard, J. Colloid Interface Sci., 2009, 332, 201–207 CrossRef CAS PubMed.
- L. Ahrens, J. Environ. Monit., 2011, 13, 20–31 RSC.
- O. A. De Silva, C. Spencer, B. F. Scott, S. Backus and D. C. G. Muir, Environ. Sci. Technol., 2011, 45, 8060–8066 CrossRef PubMed.
- A. A. Rand and S. A. Mabury, Environ. Sci. Technol., 2011, 45, 8053–8059 CrossRef CAS PubMed.
- (a) J. H. Simons, J. Electrochem. Soc., 1949, 95, 47–59 CrossRef CAS PubMed; (b) J. Burdon, I. W. Parson and J. C. Tatlow, Tetrahedron, 1949, 28, 43–51 CrossRef; (c) F. G. Drakesmith and D. A. Hughes, J. Appl. Electrochem., 1949, 6, 23–38 CrossRef; (d) J. S. Clarke and A. T. Juhn, J. Electroanal. Chem., 1949, 85, 299–306 CrossRef.
- B. Ameduri and B. Boutevin, Well-Architectured Fluoropolymers: Synthesis, Properties and Applications, Elsevier, Amsterdam, 2004 Search PubMed.
- A. B. Lindstrom, M. J. Strynar and E. L. Libelo, Environ. Sci. Technol., 2011, 45, 7954–7961 CrossRef CAS PubMed.
- J. C. D'Eon and S. A. Mabury, Environ. Sci. Technol., 2011, 45, 7974–7984 CrossRef PubMed.
- United Nations Environment Programme Report of the Conference of the Parties of the Stockholm Convention on Persistent Organic Pollutants on the work of its fourth meeting. http://chm.pops.int/Portals/0/Repository/COP4/UNEP-POPS-COP. 4–38 (accessed November 10, 2002).
- Y. Zushi, J. Hogarh and S. Masunaga, Clean Technol. Environ. Policy, 2011, 1–12 Search PubMed.
- O. Midasch, H. Drexler, N. Hart, M. W. Beckman and F. Angerer, J. Int. Arch Occup. Environ. Health, 2007, 80, 643–652 CrossRef CAS PubMed.
- E. Brede, M. Wilhelm, T. Göen, J. Müller, K. Rauchfuss and M. Kraft, et al. , Int. J. Hyg. Environ. Health, 2011, 213, 217–223 CrossRef PubMed.
- K. Steenland, T. Fletcher and D. A. Savitz, Environ. Health Perspect., 2010, 118, 1100–1108 CrossRef CAS PubMed.
- J. R. Parsons, M. Saez, J. Dolfing and P. de Voogt, Reviews of Environmental Contamination Toxicology, Springer, New York, 2008, vol. 196, pp. 53–71 Search PubMed.
- G. W. Olsen, J. M. Burris, D. J. Ehresman, J. W. Froehlich, A. M. Seacat and J. L. Butenhoff, et al. , Environ. Health Perspect., 2007, 115, 1298–1305 CrossRef CAS PubMed.
- H. Hori, E. Hayakaiva, H. Einaga, S. Kutsuna, K. Koike and T. Ibusuki, et al. , Environ. Sci. Technol., 2004, 38, 6118–6124 CrossRef CAS.
- M. Houde, A. O. De Silva, D. C. G. Muir and R. J. Letcher, Environ. Sci. Technol., 2011, 45, 7962–7973 CrossRef CAS PubMed.
- G. Olsen, S. Chang, P. Noker, G. Gorman, D. Ehresman, P. Lieder and J. Butenhohh, Toxicology, 2009, 256, 65–74 CrossRef CAS PubMed.
- (a) H. Fromme, O. Midasch, D. Twardella, J. Angerer, S. Boehmer and B. Liebl, Int. Arch. Occup. Environ. Health, 2007, 80, 313–319 CrossRef CAS PubMed; (b) T. Fromel and T. P. Knepper, Chemosphere, 2010, 80, 1387–1392 CrossRef PubMed.
- H. Fromme, S. Tittlemier, W. Volkel, M. Wilhelm and D. Twardella, Int. J. Hyg. Environ. Health, 2009, 212, 239–270 CrossRef CAS PubMed.
- M. Lorber and P. P. Egeghy, Environ. Sci. Technol., 2011, 45, 8006–8014 CrossRef CAS PubMed.
- D. Trudel, L. Horowitz, M. Wormuth, M. Scheringer, I. T. Cousins and K. Hungerbuhler, Risk Anal., 2008, 28, 807–807 CrossRef PubMed.
- L. S. Haug, C. Thomsen, A. L. Brantsaeter, H. E. Kvalem, M. Haugen and G. Becher, et al. , Environ. Int., 2010, 36, 772–778 CrossRef CAS PubMed.
- (a) U.S. Environmental Protection Agency. PFOA stewardship Program. http://www.epa.gov/oppt/pfoa/pubs/pfoarisk.htm (accessed April 5th, 2006); (b) J. M. Barroso, Off. J. Eur. Union Law, 2010, 223, 29–36 Search PubMed.
- V. Barry, A. Winquist and K. Steenland, Environ. Health Perspect., 2013, 121, 1313–1318 Search PubMed.
- C. Zhang, H. Yan, F. Li, X. Hu and Q. Zhou, J. Hazard. Mater., 2013, 260, 689–699 CrossRef CAS PubMed.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. J. van Leeuwen, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
- T. Schuster, S. Schellenberger, R. Friedrich, M. Klapper and K. Müllen, J. Fluorine Chem., 2013, 154, 30–36 CrossRef CAS PubMed.
- R. Renner, Environ. Sci. Technol., 2006, 40, 12–13 CrossRef.
- R. J. Dams, M. S. Terrazas, M. J. Sierakowski and G. G. I. Moore, US Patent US, 0148671 A1, 2006 (assigned to 3M) Search PubMed.
- S. K. Ritter, Chem. Eng. News, 2010, 88, 12–17 Search PubMed.
- A. Zaggia and B. Ameduri, Curr. Opin. Colloid Interface Surf., 2012, 17, 188–195 CrossRef CAS PubMed.
- M. Peschka, N. Fichtner, W. Hierse, P. Kirsch, E. Montenegro and M. Seidel, et al. , Chemosphere, 2008, 72, 1534–1540 CrossRef CAS PubMed.
- (a) M. Klapper, S. Nenov, R. Haschick and K. Müllen, Acc. Chem. Res., 2008, 41, 1190–1201 CrossRef CAS PubMed; (b) F. E. Golling, T. Schuster, C. Geidel, L. Mammen, D. Vollmer, M. Klapper and K. Müllen, Advances in Fluorine containing polymers, Amer. Chem. Soc., Washington D.C., 2012 Search PubMed; (c) T. Schuster, J. W. Krumpfer, S. Schellenberger, R. Friedrich, M. Klapper and K. Müllen, J. Colloid Interface Sci., 2014, 428, 276–285 CrossRef CAS PubMed.
- J. Eastoe, S. E. Rogers, L. J. Martin, A. Paul, F. Guittard, E. Guittard, R. K. Heenan and J. R. P. Webster, Langmuir, 2006, 22, 2034–2038 CrossRef CAS PubMed.
- A. Dramé, E. Taffin de Givenchy, S. Y. Dieng, S. Amigoni, M. Oumar, A. Diouf, T. Darmanin and F. Guittard, Langmuir, 2013, 29, 14815–14822 CrossRef PubMed.
- P. Kappler and M. J. Lina, WO, 121060 A1, 2005 (assigned to Arkema) Search PubMed.
- G. Boutevin, D. Tiffes, C. Loubat, B. Boutevin and B. Ameduri, J. Fluorine Chem., 2012, 134, 77–84 CrossRef PubMed.
- F. Boschet, G. K. Kostov, B. Boutevin, A. Jackson and B. Ameduri, Polym. Chem., 2012, 3, 217–223 RSC.
- G. K. Kostov, F. Boschet, J. Buller, L. Badache, S. M. Brandstadter and B. Ameduri, Macromolecules, 2011, 44, 1841–1855 CrossRef CAS.
- (a) J. L. Howell and E. W. Pérez, US, 0073588 A1, 2003, assigned to Du Pont de Nemours Search PubMed; (b) L. Han, Y. Zhang, H. Li and L. Li, Colloids Surf., A, 2009, 34, 176–180 CrossRef PubMed; (c) S. V. Kostjuk, E. Ortega, F. Ganachaud, B. Ameduri and B. Boutevin, Macromolecules, 2009, 42, 612–619 CrossRef CAS; (d) N. Durand, D. Mariot, B. Ameduri, B. Boutevin and F. Ganachaud, Langmuir, 2009, 27, 4057–4067 CrossRef PubMed.
- United States Environmental Protection Agency, Computational Toxicology Research, http://actor.epa.gov/actor/GenericChemical?casrn=60164-51-4 (accessed June 20, 2014).
- H. Li, H.-Q. Chen, S. Qing and Y.-M. Zhang, J. Polym. Res., 2011, 18, 645–650 CrossRef CAS.
- (a) C. M. Friesen, The Synthesis and Characterization of Lubricant Derivatives for New Industrial Applications and Improvements in Thermal Oxidative Environments, Ph.D. Dissertation, University of Alabama, Tuscaloosa, 2000 Search PubMed; (b) G. A. Mountain, B. J. Jelier, C. Bagia, C. M. Friesen and J. M. Janjic, J. Fluorine Chem., 2014, 162, 38–44 CrossRef CAS PubMed.
- A. Bravo, H.-R. Bjørsvik, F. Fontana, L. Liguori, A. Mele and F. Minisci, J. Org. Chem., 1997, 62, 7128–7136 CrossRef CAS PubMed.
- M. P. Gelin and B. Ameduri, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 160–171 CrossRef CAS.
- (a) G. G. Furin, Russ. Chem. Rev., 2000, 69(6), 491–522 CrossRef CAS PubMed; (b) P. M. Murphy, C. S. Baldwin and R. C. Buck, J. Fluorine Chem., 2012, 138, 3–23 CrossRef CAS PubMed.
- D. Lahiouhel, B. Ameduri and B. Boutevin, J. Fluorine Chem., 2001, 107(1), 81–88 CrossRef CAS.
- G. Gambaretto, L. Conte, G. Fornasieri, C. Zarantonello, D. Tonei, A. Sassi and R. Bertani, J. Fluorine Chem., 2003, 121(1), 57–63 CrossRef CAS.
- T. Nakamura, S. H. Thang, W. K. Busfield, S. Suyama, I. D. Jenkins and E. Rizzardo, J. Am. Chem. Soc., 1996, 118, 10824–10828 CrossRef CAS.
- C. M. Friesen, C. D. Montgomery and S. A. J. U. Temple, J. Fluorine Chem., 2012, 144, 24–32 CrossRef CAS PubMed.
- M. J. P. Smith, N. R. Cameron and J. A. Mosely, Analyst, 2012, 137(19), 4524–4453 RSC.
- B. Ameduri and B. Boutevin, Macromolecules, 1990, 23, 2433–2439 CrossRef CAS.
- F. Ceretta, A. Zaggia, L. Conte and B. Ameduri, J. Fluorine Chem., 2012, 135, 220–224 CrossRef CAS PubMed.
- J. Guiot, B. Ameduri and B. Boutevin, Macromolecules, 2002, 35, 8694–8707 CrossRef CAS.
- G. Kostov, M. Holan, B. Ameduri and M. H. Hung, Macromolecules, 2012, 45, 7375–7387 CrossRef CAS.
- C. M. Friesen, K. N. Hay, J. L. Howell and D. A. Nyvall, US, 287559A1, 2006 (assigned to DuPont) Search PubMed.
- Q. Yu, R. Zhang, S. Deng, J. Huang and G. Yu, Water Res., 2009, 43, 1150–1158 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py00965g |
| This journal is © The Royal Society of Chemistry 2015 |