
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical processes induced by the irradiation of 4-hydroxybenzophenone in different solvents†
Francesco
Barsotti
a,
Marcello
Brigante
*b,
Mohamed
Sarakha
b,
Valter
Maurino
a,
Claudio
Minero
a and
Davide
Vione
*ac
aDipartimento di Chimica, Università di Torino, Via P. Giuria 5, 10125 Torino, Italy
bClermont Université, Université Blaise Pascal, Institut de Chimie de Clermont-Ferrand, BP 10448, F-63000 Clermont-Ferrand, France
cCentro NatRisk, Via L. Da Vinci 44, 10095 Grugliasco (TO), Italy. E-mail: marcello.brigante@univ-bpclermont.fr; davide.vione@unito.it
First published on 25th September 2015
Abstract
The singlet and triplet excited states of 4-hydroxybenzophenone (4BPOH) undergo deprotonation in the presence of water to produce the anionic ground-state, causing fluorescence quenching and photoactivity inhibition. The same process does not take place in an aprotic solvent such as acetonitrile. In acetonitrile, 4BPOH is fluorescent (interestingly, one of its fluorescence peaks overlaps with peak C of humic substances), it yields singlet oxygen upon irradiation and induces the triplet-sensitised transformation of phenol (with a rate constant of (6.6 ± 0.3) × 107 M−1 s−1 (μ ± σ) between phenol itself and a triplet 4BPOH). The 4BPOH shows an intermediate behaviour in a partially protic solvent such as 2-propanol, where some deprotonation of the excited states is observed. In acetonitrile/2-propanol mixtures (at least up to 50% of 2-propanol) there is also some evidence of alcohol oxidation by the 4BPOH triplet state, while the experimental data are silent concerning such a possibility in pure 2-propanol. Considering that benzophenones are important components of chromophoric dissolved organic matter (CDOM) in surface waters, the present findings could have significance for the photoactivity of the hydrophilic surface layers vs. the hydrophobic cores of CDOM.
Introduction
Benzophenones are important chromophores and photosensitisers present in chromophoric dissolved organic matter (CDOM) in natural waters, and particularly in humic substances.1,2 Recent evidence has provided additional arguments in favour of an important role played by benzophenones in the optical and photochemical properties of CDOM and humic materials.3,4A major feature of benzophenones is the fact that the energy levels of their first excited singlet (S1) and triplet (T1) states are very near. This issue, combined with the fact that their S1 and T1 are strongly coupled by spin–orbit interactions, favours the inter-system crossing (ISC) from S1 to T1 that has elevated (often near-unity) quantum yields. Therefore, light absorption exciting the electrons from S0 to S1 is followed to a large degree by relaxation of the excited benzophenones into T1, which triggers important triplet-sensitised processes.5 In the case of unsubstituted benzophenone (BP), both S1 and T1 have n–π* configuration, independently of the polarity of the solvent. The n–π* configuration favours both the ISC and the reactivity of T1, thereby enhancing the triplet-sensitised processes. In the case of substituted benzophenones, the n–π* and π–π* configurations in both the singlet and triplet manifolds are relatively near in energy to each other. Moreover their relative positions depend on the polarity of the solvent, and the prevailing configuration may be different in solvents of different polarities. Interestingly, the π–π* configurations of both S1 and T1 are associated with longer excited-state lifetimes, lower ISC quantum yields and reduced photochemical reactivity.5–9
In the case of 4-hydroxybenzophenone (4BPOH) the S1 state has n–π* configuration, independently of the polarity of the solvent. In contrast, the n–π* and π–π* configurations of T1 are relatively near in energy. Apolar solvents favour n–π*, while polar solvents give higher stability to π–π*. In some cases one can even observe that T1 has a mixed n–π*/π–π* character, because of very similar energy levels of the two configurations, which may produce a double-exponential decay of the phosphorescence signal.5 In acetonitrile, an equilibrium between the n–π* and π–π* triplet states of 4BPOH has been reported.6
The described effect of polarity on the excited states of 4BPOH is important in aprotic solvents, while the picture may change dramatically in protic solvents such as water. Indeed, 4BPOH-S0 is a weak acid (pKa ∼ 8.5), while 4BPOH-S1 and 4BPOH-T1 are strong acids that undergo fast deprotonation/deactivation in an aqueous environment.10 This issue strongly limits the photoinduced reactivity of 4BPOH in protic solvents compared to aprotic (polar or apolar) solvents.11
The fluorescence spectrum of 4BPOH is also interesting, because it shows a band in a region that overlaps with peak C of humic substances (excitation at 300–320 nm, emission at 450–550 nm).12–14 This fact makes 4BPOH a potentially interesting model molecule for humic substances, and it accounts for the importance of studying and correlating its photophysics and photochemistry in different solvents.
Humic substances are a complex mixture of compounds that originate from a wide variety of sources.15,16 Their components are assembled in a micelle-like fashion where hydrophilic moieties are in contact with water, while the hydrophobic moieties form waterless inner cores.17,18 Humic compounds are also important sunlight absorbers,19 with key consequences for the penetration of biologically harmful UV radiation in the water column.20,21 Sunlight absorption produces a variety of reactive transient species (triplet states, singlet oxygen 1O2 and hydroxyl radical ˙OH)22–25 that are involved in the transformation of dissolved pollutants.26–28 The photoactivity of humic substances is linked to their molecular weight,29–31 because the larger fractions undergo important inter-molecular interactions that favour internal conversion and quench both photoactivity and fluorescence.4,32–35 However, the hydrophobic cores of the larger moieties show elevated concentration values of singlet oxygen.36–38
Because 4BPOH shares some features in common with humic compounds, which make it a potentially interesting model molecule, the present work investigates the photochemical processes induced by 4BPOH in different solvents, namely an aprotic one (acetonitrile), a partially protic one (2-propanol), as well as water. The results may apply to photoinduced reactions taking place in aqueous solution or in hydrophobic environments, such as those occurring in different CDOM compartments.17,18
Experimental
Reagents and materials
4-Hydroxybenzophenone (4BPOH, 98%), benzophenone (BP, 99%), furfuryl alcohol (FFA, 98%), 2-propanol (99.8%), 2-nitrobenzaldehyde (98%), acetonitrile (gradient grade), HClO4 (70% in water), NaOH (98%) and methanol (gradient grade) were purchased from Sigma-Aldrich, and nitrogen (5.5, 99.9995%), argon (5.5, 99.9995%) and oxygen (6.0, 99.9999%) from SAPIO. Water used was of Milli-Q quality.Fluorescence measurements
The excitation–emission matrix (EEM) fluorescence spectra were taken with a VARIAN Cary Eclipse fluorescence spectrophotometer, with an excitation range from 200 to 400 nm at 10 nm steps, and an emission range from 200 to 600 nm with a scan rate of 1200 nm min−1. Excitation and emission slits were both set at 10 nm. Spectra were taken in a fluorescence quartz cuvette (Hellma) with 1 cm optical path length. The Raman signal of water was taken as a reference for the lamp intensity and signal stability within different measurements.Laser flash photolysis experiments
The laser apparatus has been previously described.39 The laser excitation wavelength was fixed at 355 nm with an energy of ∼94 mJ per pulse. An appropriate volume of stock solutions was mixed before each experiment to obtain the desired concentrations. The pH was acidified using HClO4, and deoxygenated and oxygen-saturated solutions were used (when necessary) after 20 min of bubbling with argon or pure oxygen, respectively.The second-order rate constant between 4BPOH-T1 (hereafter, 34BPOH*) and quenchers was calculated from the regression line of the absorbance logarithm decay against the quencher concentration. The error bars were derived at the 3σ level from the scattering of the experimental data.
Steady irradiation experiments
Solutions (5 mL volume) containing 0.1 mM 4BPOH and other reagents (when necessary, such as 0.1 mM phenol or 0.1 mM FFA) were placed in cylindrical Pyrex glass cells (4.0 cm diameter, 2.3 cm height, with a lateral neck tightly closed with a screw cap). The solvent was either water, acetonitrile or 2-propanol. Irradiation of the cells took place under a Philips TL 01 (20 W) lamp with an emission maximum at 313 nm. The solutions in the cells were irradiated from the top and the optical path length was 0.4 cm. The lamp irradiance over the solutions was 2.6 ± 0.1 W m−2, measured with a CO.FO.ME.GRA (Milan, Italy) power meter equipped with a UV-sensitive probe (290–400 nm). The lamp emission spectra (spectral photon flux density p°(λ), given in Einstein L−1 s−1 nm−1) were obtained by combining spectrophotometric measures (CCD spectrophotometer Ocean Optics USB 2000, calibrated with a DH-2000-CAL radiation source) with chemical actinometry using 2-nitrobenzaldehyde. The detailed procedure to determine p°(λ) is described in detail elsewhere.25Fig. 1 shows the spectral photon flux density of the lamp, together with the absorption spectra of 4BPOH (molar absorption coefficients) in both acetonitrile and 2-propanol. The latter were taken with a Varian Cary 100 Scan double-beam UV-vis spectrophotometer, using quartz cuvettes (Hellma, optical path length 1 cm). The main reason for the higher absorption coefficient of 4BPOH in 2-propanol above 300 nm is that the absorption maximum in this solvent, compared to acetonitrile, is shifted from 285 to 295 nm (bathochromic shift). In contrast, the absorbance at the maximum is similar in both the cases.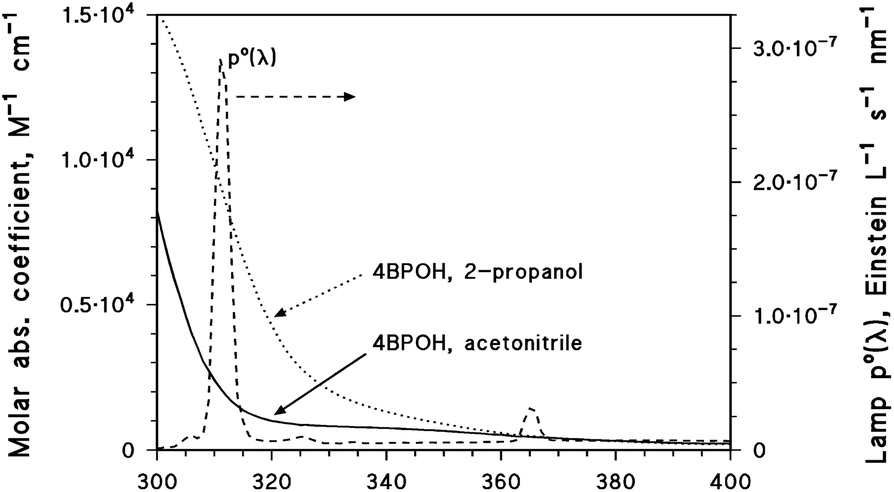 | ||
| Fig. 1 Absorption spectra (molar absorption coefficients) of 4BPOH in acetonitrile and 2-propanol. Emission spectrum (spectral photon flux density) of the lamp used for the steady irradiation experiments. | ||
After the scheduled irradiation times, the cells were withdrawn from the lamp. Measured aliquots of the irradiated solutions (2 mL) were diluted with an equal volume of water and analysed by high-performance liquid chromatography with diode-array detection (HPLC-DAD). A VWR-Hitachi Elite instrument, equipped with a L-2300 autosampler (25 μL injection volume), a L-2130 quaternary pump for low-pressure gradients, a Duratec DDG-75 online degasser, a L-2300 column oven (operated at 40 °C), a L-2445 DAD detector and a column RP-C18 LiChroCART packed with a LiChrospher 100 RP-18 material (VWR, 4 mm × 125 mm × 5 μm) was used.
The eluent was a mixture of 3.5 mM H3PO4 in water (A) and of methanol (B). Gradient elution (1 mL min−1 flow rate) was as follows: from 5% B to 70% B in 15 min, maintained at 70% B for 7 min, back to 5% B in 5 min and maintained at 5% B for an additional 5 min (post-run equilibration). The detection wavelengths were 290 nm for 4BPOH, 210 nm for phenol, and 230 nm for FFA. The retention times were 15.5 min for 4BPOH, 9.0 min for phenol and 5.3 min for FFA. The column dead time was 1.4 min.
Additional runs were carried out to measure the formation of 4-phenoxyphenol (4PP) from 1 mM phenol + 1 mM 4BPOH in acetonitrile and 2-propanol. Isocratic elution (1 mL min−1 flow rate) used 50% A and 50% B, with 229 nm detection wavelength and 10.2 min retention time for 4PP.
The time evolution data of 4BPOH, phenol and FFA were fitted with pseudo-first order kinetic functions of the form Ct = C0e−kt, where Ct is the concentration of the substrate at the time t, C0 its initial concentration and k the pseudo-first order degradation rate constant. The initial transformation rate was calculated as R0 = kC0. The uncertainty in the rates is reported as ±σ and it mainly depends on the uncertainty in k, which represents the goodness of the fit of the exponential functions to the experimental data. The reproducibility of replicated experiments was around 15%.
The calculation of the quantum yields of 4BPOH and phenol degradation was based on the photon flux absorbed by the photosensitiser, 4BPOH (P4BPOHa). The latter was calculated as follows:40
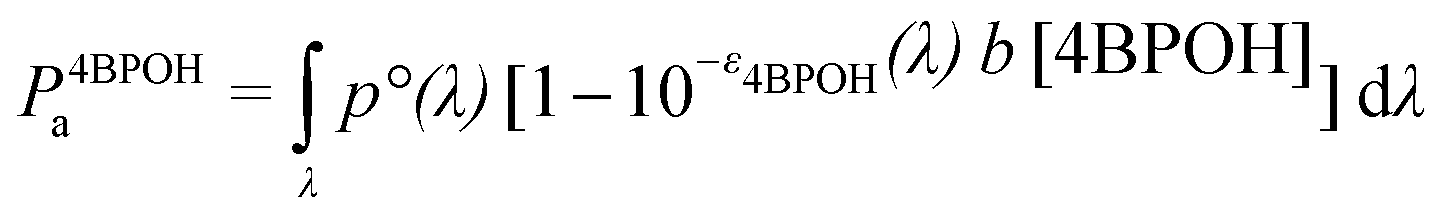 | (1) |
Results and discussion
Fluorescence measurements
Fig. 2 shows the fluorescence EEM spectra of 0.1 mM 4BPOH in different solvents (acetonitrile, 2-propanol and water, the latter at the natural pH ∼ 6). Considering first the fluorescence spectra in the organic solvents, one can notice a band at Ex/Em ∼ 225/325 nm that is included in the region of phenolic compounds (which is consistent with the OH group on the aromatic ring of 4BPOH).12 | ||
| Fig. 2 EEM fluorescence spectra of 0.1 mM 4BPOH in acetonitrile (a), 2-propanol (b) and water (c). | ||
More interestingly, the band with Ex/Em ∼ 325/475 nm overlaps with peak C of humic substances.12 Because benzophenones are known chromophores/photosensitisers occurring in CDOM, the fact that a compound of this class shows fluorescence in the humic region deserves certain attention. In contrast to the results obtained in organic solvents, the fluorescence spectrum of 4BPOH in water shows little or no fluorescence emission. This issue is consistent with literature reports that the excited states of 4BPOH (including the excited singlet state(s)) undergo rapid (radiationless) deactivation by deprotonation in aqueous solution.41,42
Laser flash photolysis experiments
The laser excitation of 4BPOH in acetonitrile produces a transient spectrum with absorption maxima at 330 and 520 nm, with the characteristic features of 34BPOH* (see Fig. 3).43 The transient spectra taken at relatively long times (about 0.30 μs) show the occurrence of a small peak between 550 and 600 nm, which might be consistent with the ketyl radical of 4BPOH (4BP(H˙)OH). The latter could arise upon reaction between 34BPOH* and the ground-state 4BPOH (34BPOH* + 4BPOH → 4BP(H˙)OH + 4BPO˙), while the reaction between 34BPOH* and acetonitrile is rather unlikely.6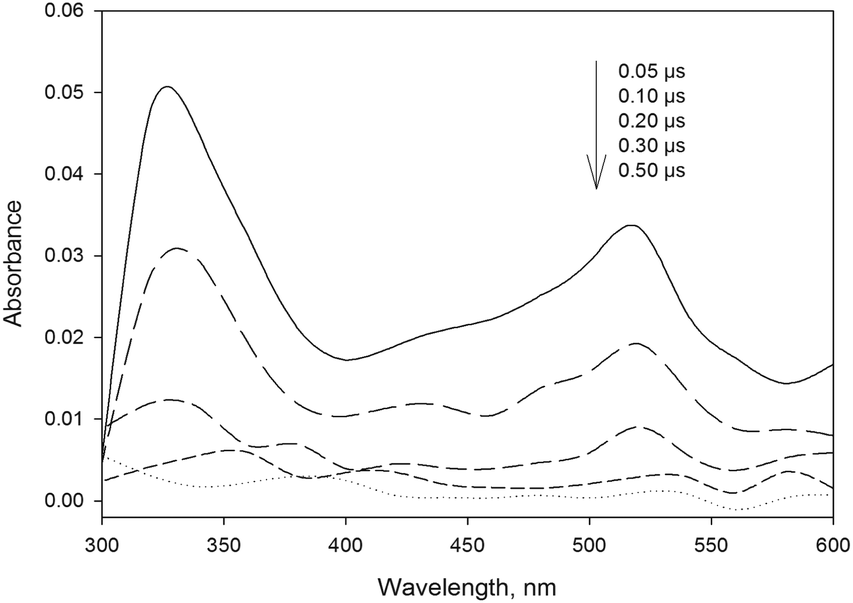 | ||
| Fig. 3 Time trend of the transient absorption spectrum obtained upon LFP (355 nm, 94 mJ) excitation of 4BPOH (0.1 mM) in aerated CH3CN solution at ambient temperature (295 ± 2 K). | ||
The first-order decay constant of the signal measured at 520 nm (k520 nm) is in the range of 107 s−1 in aerated solutions. Coherently with the triplet state assignment, k520 nm depends on the content of dissolved oxygen. In fact, k520 nm increased when passing from an Ar-bubbled system to an aerated and an O2-bubbled one. The concentration of dissolved oxygen was assessed based on the published data of its solubility in acetonitrile.44 From the plotted data (see Fig. ESI1†) one obtains a second-order reaction rate constant of (3.6 ± 0.1) × 109 M−1 s−1 between 34BPOH* and O2, with the probable formation of 1O2 that is typical of triplet-state reactivity.45
Fig. 4a shows the decay traces of the transient absorbance at 520 nm in water/acetonitrile mixtures, as a function of the volume percentage of water (up to 6%). One can observe that the trace absorbance just after the laser pulse is lower in the presence of higher percentages of water, and that the trace decay becomes faster when increasing the water content. This result can be accounted for by the deprotonation of both the singlet and the triplet states of 4BPOH,41,42 which would both be favoured in the presence of higher percentages of water. The deprotonation of the singlet state would compete with the formation of the triplet, thereby decreasing the triplet absorbance just after the laser pulse.
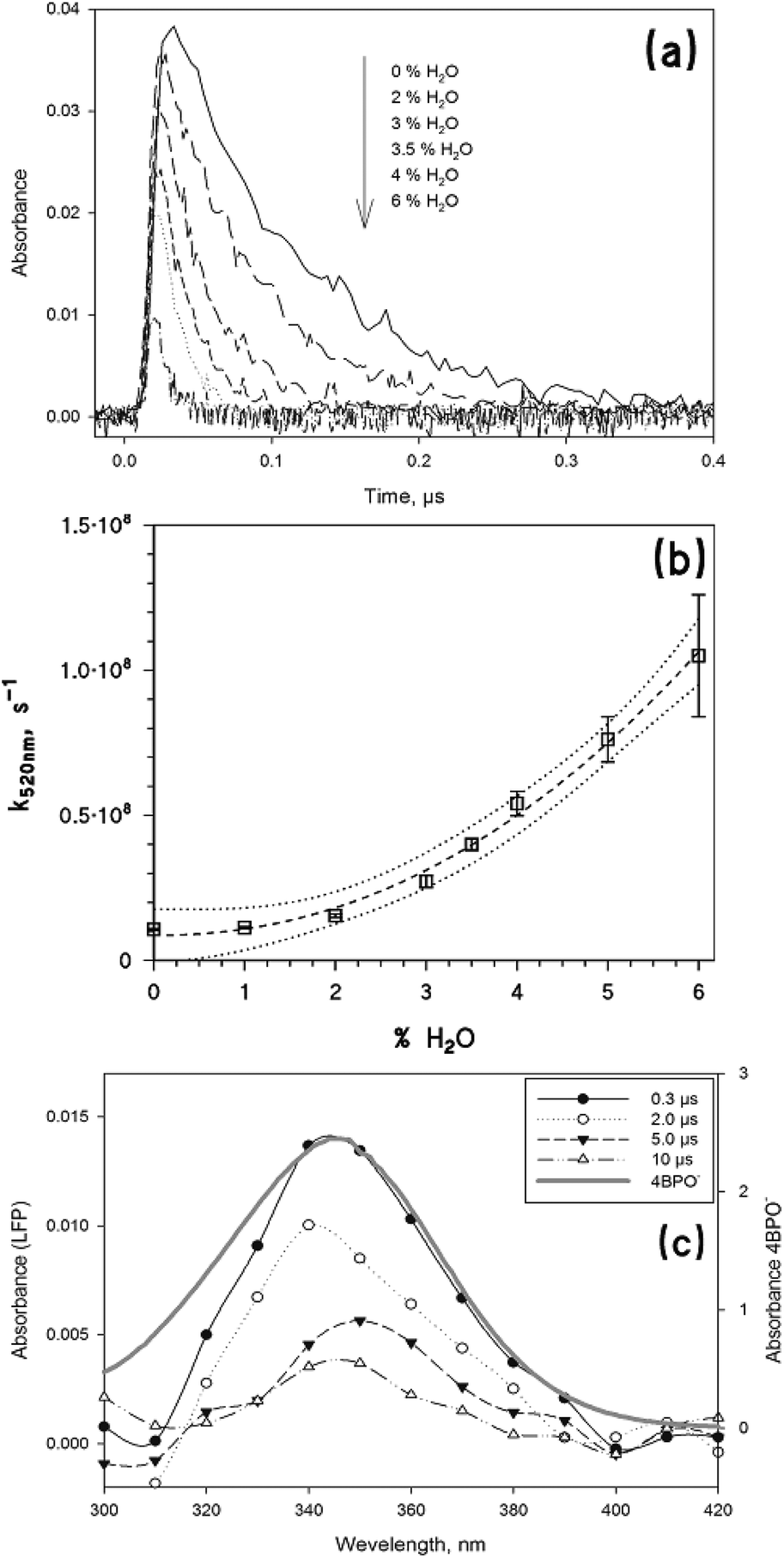 | ||
| Fig. 4 (a) Trace decay at 520 nm as a function of the percentage of water in CH3CN. The transients were obtained upon LFP irradiation (355 nm, 94 mJ) of 0.1 mM 4BPOH in aerated solutions. (b) Trend of the first-order decay constant of the absorbance at 520 nm, as a function of the percentage of water in binary mixtures H2O/CH3CN. 4BPOH 0.1 mM, laser excitation at 355 nm, 94 mJ per pulse. The fit curve (dashed) is a polynomial, the dotted ones are the 95% confidence bands of the fit. (c) Time trend of the transient absorption spectrum obtained upon LFP (355 nm, 94 mJ) excitation of 4BPOH (0.1 mM) in an aerated H2O solution at ambient temperature (295 ± 2 K). The grey solid line shows the UV-vis spectrum of anionic 4BPOH (4BPO−, 0.1 mM) in water at pH 10.5. | ||
The deprotonation of the triplet state would enhance the relevant trace decay, as reported in Fig. 4b which shows that k520 nm increases with the volume percentage of water. In the presence of pure water as the solvent, the signal corresponding to the triplet state of 4BPOH could no longer be observed. Most likely, 34BPOH* is not formed in water because the deprotonation of the excited singlet state is much faster than the ISC. Moreover, even if formed to some extent in water, or in water–acetonitrile mixtures with a rather elevated water content, 34BPOH* is expected (i) to undergo very fast deprotonation, and (ii) to be of π–π* configuration, which would be strongly favoured by the polar solvent.5,6 Both issues would produce negligible photochemical reactivity of 34BPOH* under the relevant conditions.
The excited-state deprotonation is known to yield an anionic species with an absorption maximum around 350 nm.11,42 In both water and water–acetonitrile mixtures we observed the formation of a transient with the maximum absorbance at 350 nm, which was longer-lived compared to the triplet state. The transient absorption spectrum is very similar to that of the anionic form of 4BPOH (hereafter, 4BPO−), as shown in Fig. 4c, which justifies the identification of the observed species with the ground-state 4BPO−.
Interestingly, k350 nm (the pseudo-first order decay constant of 4BPO−) increases linearly when increasing the volume percentage of water, which may suggest an acid–base process involving 4BPO− (Fig. ESI2†). Considering that the decay of the 350 nm signal brings the absorbance back to its initial value (namely the value observed before the laser pulse, in the presence of 4BPOH alone), the most likely process is the protonation of 4BPO− back to 4BPOH. Such a deprotonation–protonation sequence would be accounted for by the fact that the triplet state of 4BPOH (pKa < −2) is a much stronger acid than the corresponding ground-state (pKa ∼ 8.5).42 The above hypothesis is further confirmed by the fact that k350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm depends on pH (see Fig. ESI3†). The pH trend would be consistent with a reaction between 4BPO− and H3O+ at pH < 5, while at pH > 5 the prevailing reaction would take place with water.
nm depends on pH (see Fig. ESI3†). The pH trend would be consistent with a reaction between 4BPO− and H3O+ at pH < 5, while at pH > 5 the prevailing reaction would take place with water.
The triplet states of benzophenones often behave as effective photosensitisers for the transformation of phenolic compounds.46,47 The reactivity between 34BPOH* and phenol was assessed by studying the effect of phenol concentration on the value of k520nm in acetonitrile. The value of k520nm increases linearly with increasing phenol (see Fig. ESI4†), from which trend a second-order rate constant k34BPOH*,phenol = (6.6 ± 0.3) × 107 M−1 s−1 can be obtained between 34BPOH* and phenol. The reaction yields a species with an absorption maximum around 380 nm, which is longer-lived compared to 34BPOH*. It could be assigned to the phenoxy radical,48 which suggests that 34BPOH* would react with phenol by hydrogen abstraction (or by electron transfer followed by deprotonation, which does however look unlikely in aprotic acetonitrile). The formation of phenoxy radicals has indeed been reported upon reaction between carbonyl triplets and phenols.7
Additional experiments of laser irradiation showed that negligible reaction would take place between 34BPOH* and furfuryl alcohol (FFA). Considering that the latter is a 1O2 probe,24,25,38 it would be possible to use FFA to assess the formation of 1O2 from irradiated 4BPOH (vide infra), without the potential bias of a reaction between FFA and 34BPOH*.
As an additional solvent, 2-propanol was chosen because it has intermediate proticity between aprotic acetonitrile and water.8,49,50 The initial absorbance of the triplet state after the laser pulse decreased linearly with the increasing percentage of 2-propanol (see Fig. ESI5†), which suggests that a lesser amount of 34BPOH* would be formed in the presence of the alcohol. This result is consistent with the deprotonation of the excited singlet state in the presence of 2-propanol.
Moreover, the first-order rate constant of 34BPOH* decay (k520nm) increased linearly with increasing 2-propanol (see Fig. ESI6†), coherently with a reaction between 34BPOH* and the alcohol. The corresponding second-order rate constant, derived as the slope of the plot k520nmvs. 2-propanol, is k34BPOH*,2-propanol = (3.4 ± 0.3) × 106 M−1 s−1. The relevant process could be an acid–base and/or a redox reaction, and the literature shows an important disagreement over this issue. While some authors assume that 34BPOH* is able to abstract hydrogen from alcohols including 2-propanol,42 others explicitly exclude this possibility and only consider an acid–base process where the alcohol acts as a H+ acceptor for the deprotonation of 34BPOH*.51 For instance, no hydrogen abstraction is reported to take place between 34BPOH* and ethanol in ethanol solution, where the chemical reactivity of 34BPOH* should be decreased by its π–π* configuration.8
To gain insight into the process details, the reaction with 2-propanol was also studied by laser irradiation of benzophenone (BP), which differs from 4BPOH due to the absence of the OH group on the aromatic ring. In this case, it is well known that the BP triplet state abstracts a H atom from the alcohol to form the ketyl radical of BP,52 which has a comparable lifetime as the BP triplet state and partially overlaps with its absorption spectrum.53 The laser irradiation of BP was carried out in a system containing 50% acetonitrile and 50% 2-propanol. Fig. 5a shows, as a function of wavelength, the first-order decay constant of the flash photolysis traces (kdecay, upper graph), as well as the maximum absorbance value reached by each trace, soon after the laser pulse (lower graph). The maximum trace absorbance gives insight into the absorption spectrum of the transient(s) formed by laser irradiation. The absorbance peaks around 320 and 520 nm can be assigned to the triplet state of BP;43,53 interestingly, these peaks correspond to the values of kdecay ∼ 4 × 106 s−1. However, the decay constant shows different values in other wavelength intervals, and in particular it is kdecay ∼ 1 × 107 s−1 at around 450 and 600 nm. The variations of kdecay suggest that the reported maximum trace absorbance is the result of the contribution of more than one species. Literature data indicate that the transient absorption upon laser irradiation of BP and 2-propanol results from both the triplet state and the ketyl radical of BP.53 Because the BP triplet state absorbs radiation around 320 and 520 nm,43 one can identify the species with kdecay ∼ 4 × 106 s−1 as the triplet state of BP and that with kdecay ∼ 1 × 107 s−1 as the ketyl radical. The latter would thus absorb radiation at around 450 and 600 nm. Interestingly, the ketyl radical is not formed upon laser irradiation of BP in pure acetonitrile.
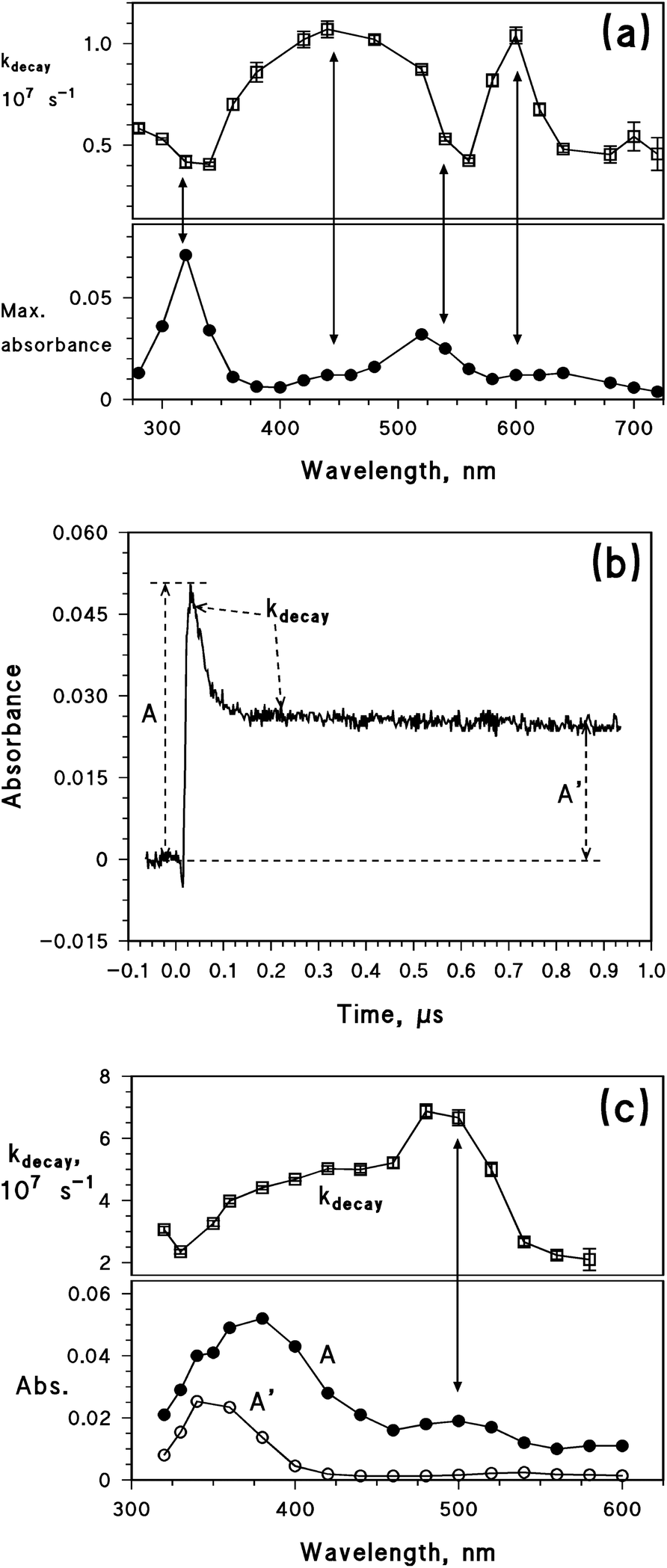 | ||
| Fig. 5 (a) Upper plot: first-order decay constant (kdecay) of the laser trace as a function of wavelength, upon irradiation (355 nm, 94 mJ per pulse) of 0.1 mM benzophenone in 1:1 2-propanol:acetonitrile. Lower plot: maximum absorbance of the laser trace under the same conditions. (b) Laser trace (350 nm) upon irradiation of 0.1 mM 4BPOH in 1:1 2-propanol:acetonitrile (355 nm, 94 mJ per pulse). The absorbance values A and A′ and the time interval used to determine kdecay are also highlighted. (c) Upper plot: first-order decay constant (kdecay) of the laser trace as a function of wavelength, upon irradiation (355 nm, 94 mJ per pulse) of 0.1 mM 4BPOH in 1:1 2-propanol:acetonitrile. Lower plot: wavelength trends of A and A′ under the same conditions. | ||
The above-discussed laser irradiation of BP in 50% acetonitrile + 50% 2-propanol yielded transient traces that, after an exponential decay, went back to the absorbance value observed before the laser pulse. The situation changed considerably upon laser irradiation of 4BPOH in the same solvent mixture. In this case, the trace reached a maximum (A) and then decayed exponentially down to a constant value (A′) that was higher than the initial absorbance (see Fig. 5b). This issue means that a longer-lived species was formed in the presence of 4BPOH, while the exponential decay itself is silent as to the occurrence of one or more short-lived transients.
The plot shown in Fig. 5c was obtained under similar conditions as that of Fig. 5a, the only difference being that it is referred to 4BPOH instead of BP. In this case as well, the solvent was a 1:1 mixture of acetonitrile and 2-propanol. Also in this case there is evidence of the occurrence of more than one species. First of all, the transient absorption spectrum (A) shows two maxima (around 380 and 500 nm) that, differently from the case of 34BPOH* alone (see Fig. 3), have quite different absorbances. Secondly, kdecay varies from ∼2 × 107 to ∼7 × 107 s−1. The plateau absorbance A′ has a maximum around 350 nm, which agrees with the absorption spectrum of 4BPO−. The latter species is reported to be formed in alcoholic solvents such as ethanol.8 Further evidence in favour of the identification of the longer-lived species with 4BPO− is its decay kinetics, which is very slow in 2-propanol and is accelerated upon addition of water traces (see Fig. ESI7†). It is reasonable that 4BPO−, once formed, undergoes protonation that would be much faster in water compared to 2-propanol.
Fig. 5c suggests that in addition to 4BPO− which is relatively long-lived, two transient species with shorter and similar lifetimes occur in the system. The triplet state 34BPOH* is known to absorb at ∼330 and ∼500–520 nm,43 thus it should be the species with kdecay ∼ 7 × 107 s−1. The other species would account for the observed kdecay ∼ 2 × 107 s−1 at around 575 nm, and possibly also around 320 nm. In analogy with the results obtained with BP, the second species might be the ketyl radical of 4BPOH (4BP(H˙)OH). This radical is reported to have an absorption maximum at 560 nm,6 which would be reasonably consistent with the observed wavelength trend of kdecay. If the identification of the second species with 4BP(H˙)OH is correct, there would be support for the possibility of an electron-transfer reaction between 34BPOH* and 2-propanol. However, one should also consider that the experiments reported in Fig. 5c were carried out in 1:1 2-propanol:acetonitrile. In pure acetonitrile there is an equilibrium between the n–π* and π–π* configurations of 34BPOH*,6 which allows for the occurrence of the photochemically reactive n–π*. The equilibrium could be maintained in the mixtures of acetonitrile and 2-propanol, while in pure 2-propanol one cannot exclude the occurrence of π–π* alone. Unfortunately, flash photolysis experiments in pure 2-propanol are inconclusive because the lack of 34BPOH* detection could be due either to a very short lifetime of π–π*, or (more likely) to a very fast deprotonation of 34BPOH* (in either configuration) to give 4BPO−.
Scheme 1 shows the hypothesised processes involving irradiated 4BPOH. Excited-state deprotonation and 4BPO− protonation would be faster in water than in 2-propanol.
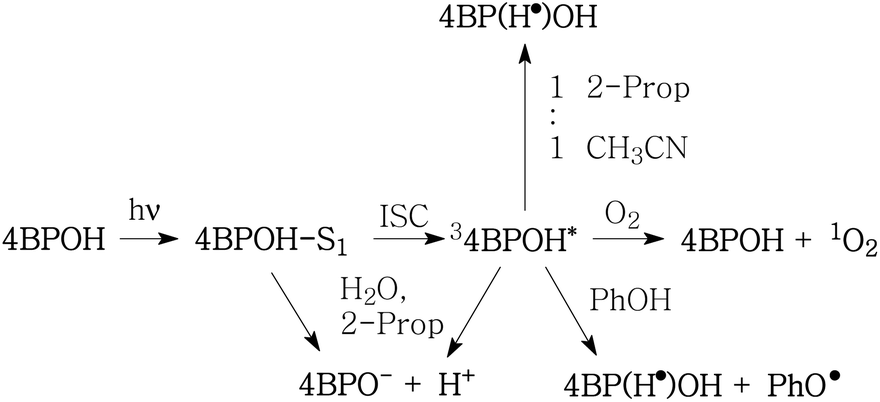 | ||
| Scheme 1 Hypothesised processes involving the transient species formed upon irradiation of 4BPOH. 2-Prop: 2-propanol; PhOH: phenol; PhO˙: phenoxyl; ISC: inter-system crossing. | ||
Steady irradiation experiments
The ability of 4BPOH to photosensitise the degradation of phenol was tested in water, acetonitrile and 2-propanol. The transformation rates RS of the two substrates (S = 4BPOH or phenol, both at 0.1 mM initial concentration) under different conditions are shown in Fig. 6a. Fig. 6b shows the relevant quantum yields of transformation (calculated as ΦS = RS(P4BPOHa)−1).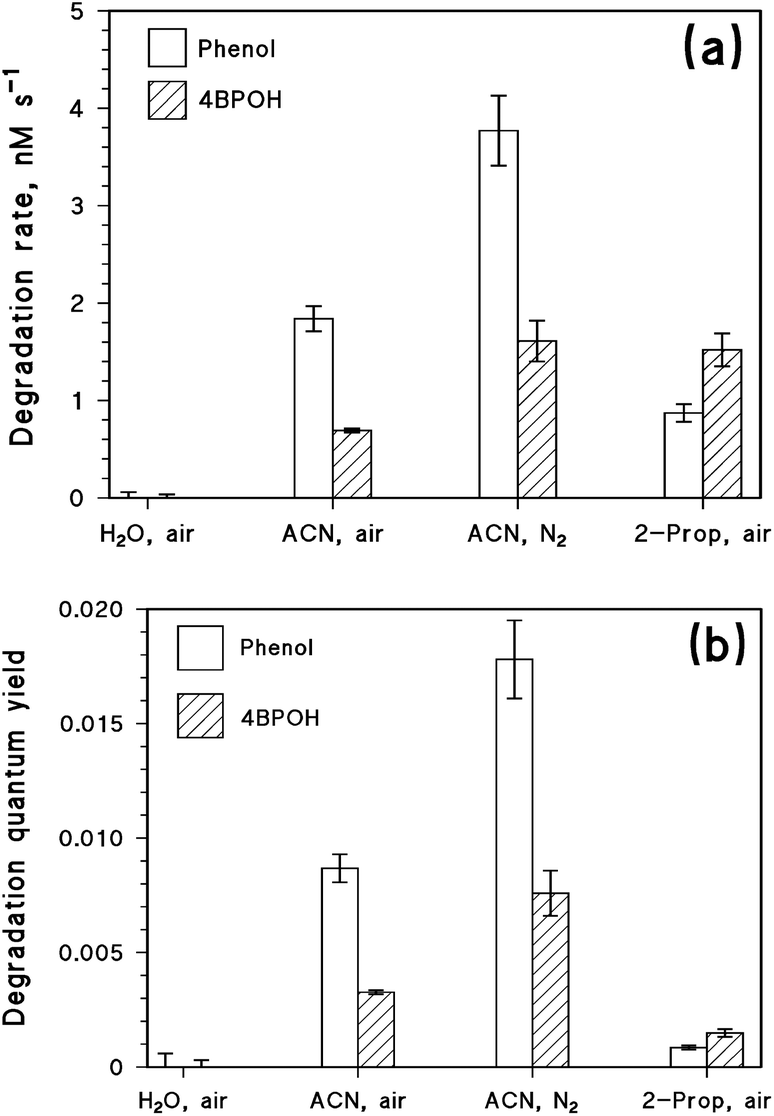 | ||
| Fig. 6 Degradation rates (a) and degradation quantum yields (b) of phenol and 4BPOH in mixtures (initial concentration 0.1 mM for both compounds), upon UVB irradiation in different solvents (water, acetonitrile, and 2-propanol) and different conditions (air or nitrogen atmosphere). The error bars represent ±σ. The rates and quantum yields in water were negligible, and only the related uncertainty (detection limit) is reported. ACN = acetonitrile. | ||
No degradation of either 4BPOH or phenol was observed in water, while both compounds were photodegraded in the organic solvents. Focusing first on the degradation rate of phenol, in aerated solutions it followed the order acetonitrile > 2-propanol > water. An analogous trend was observed for the quantum yields, just with larger differences between acetonitrile and 2-propanol compared to the rates: the reason is that 4BPOH absorbs lamp radiation to a higher extent in 2-propanol compared to acetonitrile ( ). Moreover, phenol degradation was enhanced with acetonitrile under a nitrogen atmosphere. These results can be explained easily in the framework of the photochemical processes depicted in Scheme 1, considering that 34BPOH* would oxidise phenol to the phenoxy radical and, therefore, trigger phenol transformation. Additional evidence for the formation of the phenoxy radical is the occurrence of the compound 4PP (formed upon dimerisation of phenoxyl54) upon irradiation of 1 mM phenol + 1 mM 4BPOH in acetonitrile (15–20 μM 4PP was produced for irradiation times >24 h) and 2-propanol (1–1.5 μM 4PP was formed at the same irradiation time scale).
). Moreover, phenol degradation was enhanced with acetonitrile under a nitrogen atmosphere. These results can be explained easily in the framework of the photochemical processes depicted in Scheme 1, considering that 34BPOH* would oxidise phenol to the phenoxy radical and, therefore, trigger phenol transformation. Additional evidence for the formation of the phenoxy radical is the occurrence of the compound 4PP (formed upon dimerisation of phenoxyl54) upon irradiation of 1 mM phenol + 1 mM 4BPOH in acetonitrile (15–20 μM 4PP was produced for irradiation times >24 h) and 2-propanol (1–1.5 μM 4PP was formed at the same irradiation time scale).
The lack of phenol degradation in water would be accounted for by the fast deprotonation/quenching of the 4BPOH excited states in this solvent. Indeed, no signal of 34BPOH* could be detected upon laser irradiation of 4BPOH in aqueous solution. In the case of 2-propanol as the solvent, the reaction between 34BPOH* and phenol would be in competition with triplet deprotonation and possibly with the oxidation of the solvent (although some evidence of the oxidation of 2-propanol by 34BPOH* is only available for mixtures with acetonitrile, and not in pure alcohol). These reactions would reduce the availability of 34BPOH* for the degradation of phenol in 2-propanol, but they would not be as fast as the decay of excited 4BPOH in water. Finally, because 34BPOH* does not react with acetonitrile, phenol photosensitised degradation was fastest in this solvent.
The faster degradation of phenol in the absence of oxygen (acetonitrile as the solvent, Fig. 6), due to the reaction with 34BPOH*, is accounted for by the quenching of 34BPOH* by oxygen to yield 1O2. Coherently, Fig. ESI1† suggests that the decay constant of 34BPOH* in acetonitrile becomes almost double when passing from a deoxygenated to an aerated system. This is fully consistent with the observed degradation of phenol, which was about two times faster under a nitrogen atmosphere compared to air (Fig. 6). In the aerated system, 0.1 mM phenol would scavenge approximately 0.06% of 34BPOH*, a percentage that would become approximately double in the absence of oxygen. However, in an aerated solution there would be formation of 1O2 that could also contribute to phenol degradation. The first-order decay constant of 1O2 in acetonitrile is about 2.5 × 104 s−1,55 while the second-order reaction rate constant between phenol and 1O2 seems to be strongly solvent-dependent. Indeed, while the rate constants of 106–107 M−1 s−1 are reported for water,56 in organic solvents one has the rate constant values of about 104 M−1 s−1.57,58 The difference could at least in part be explained by the higher reactivity of the phenolate anion toward 1O2, coherently with the fact that the rate constant in water increases with increasing pH.54 If the reaction rate constants between phenol and 1O2 in acetonitrile were in the range of 104 M−1 s−1, only ∼0.004% of 1O2 would react with 0.1 mM phenol and the reaction with singlet oxygen would play a secondary role in phenol degradation (∼7% of the total). Triplet 4BPOH would thus induce more effective degradation of phenol under a N2 atmosphere, where triplet quenching by O2 would not be operational.
Focusing now on the degradation of 4BPOH in different solvents, one observes first of all the absence of transformation in water, which can be explained as per the above discussion. The 4BPOH degradation in acetonitrile was faster in the absence of oxygen, which quenches 34BPOH* to give back the ground-state molecule in a null cycle of excitation–deactivation:
| 34BPOH* + O2 → 4BPOH + 1O2 | (2) |
The degradation rate of 4BPOH was higher in 2-propanol than in acetonitrile (Fig. 6a). One should consider that 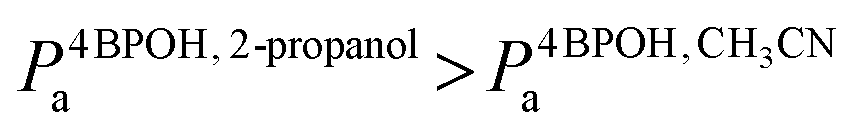 (by a factor of ∼4.5) and, as far as the transformation quantum yields are concerned, it was
(by a factor of ∼4.5) and, as far as the transformation quantum yields are concerned, it was 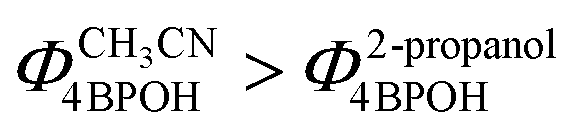 (Fig. 6b). Therefore, while the difference in the rates can be accounted for by the higher radiation absorption of 4BPOH in 2-propanol, the lower quantum yield in the alcoholic solvent is consistent with the quenching of 34BPOH* by an acid–base and possibly also a redox process.
(Fig. 6b). Therefore, while the difference in the rates can be accounted for by the higher radiation absorption of 4BPOH in 2-propanol, the lower quantum yield in the alcoholic solvent is consistent with the quenching of 34BPOH* by an acid–base and possibly also a redox process.
Finally, the formation of 1O2 by irradiated 4BPOH (0.1 mM initial concentration) was assessed in water, acetonitrile and 2-propanol by addition of 0.1 mM FFA. The initial rate of FFA transformation (RFFA) was negligible in water, and it was RFFA = (8.66 ± 0.68) × 10−10 M s−1 in acetonitrile and RFFA = (1.11 ± 0.14) × 10−10 M s−1 in 2-propanol. Because 34BPOH* shows negligible reactivity with FFA, as suggested by LFP experiments, the observed degradation of FFA would be accounted for by the production of 1O2 in the irradiated systems.
Conclusions
The singlet and triplet excited states of 4BPOH undergo deprotonation in the presence of water to produce the ground-state 4BPO−, which quickly adds H+ to yield back the ground-state 4BPOH. This process quenches the fluorescence and the photosensitising activity of 4BPOH in aqueous solution. In aprotic acetonitrile, 4BPOH shows significant fluorescence emission with a fluorescence band (Ex/Em ∼ 325/475 nm) that overlaps with peak C of humic substances. In acetonitrile, 34BPOH* mainly reacts with O2 to produce 1O2 and it is also involved in the degradation of phenol, with a second-order reaction rate constant k34BPOH*,phenol = (6.6 ± 0.3) × 107 M−1 s−1. The reaction would produce the phenoxy radical, and presumably also the ketyl radical of 4BPOH.With the solvent 2-propanol, which shows some protic properties and can be oxidised, light-excited 4BPOH undergoes partial deprotonation to 4BPO− (but to a considerably lesser extent than in water), and very slow reprotonation. There is also some evidence that 34BPOH* could abstract an H atom from 2-propanol in alcohol:acetonitrile mixtures. In pure 2-propanol, it is possible that 34BPOH* is in a π–π* configuration that is non-reactive toward the oxidation of alcohols. However, in pure 2-propanol one does not observe 34BPOH* signals at least because of the fast deprotonation, thus this work is silent about the ability of 34BPOH* to oxidise 2-propanol in pure alcoholic solvents. The partial 34BPOH* quenching in 2-propanol, whatever the actual pathway(s), decreases the photosensitising ability of 4BPOH in 2-propanol compared to acetonitrile. However, the ability of irradiated 4BPOH to produce 1O2 and degrade phenol is still significant in the alcoholic solvent.
Considering that benzophenones (possibly including 4BPOH) are important constituents of CDOM and particularly of humic substances, the present findings may be significant for the photoactivity of the hydrophobic and hydrophilic humic moieties. Because a compound such as 4BPOH would not be fluorescent and would not produce 1O2 in the presence of water, but would do so in its absence, it might contribute to the occurrence of elevated 1O2 levels in the hydrophobic cores of humic materials.
Acknowledgements
DV acknowledges financial support by Università di Torino – EU Accelerating Grants, project TO_Call2_2012_0047 (DOMNAMICS). The authors also thank the University Blaise Pascal for supporting a one-month stay of DV at ICCF – Clermont Ferrand.Notes and references
- S. Canonica, B. Hellrung, P. Mueller and J. Wirz, Aqueous oxidation of phenylurea herbicides by triplet aromatic ketones, Environ. Sci. Technol., 2006, 40, 6636–6641 CrossRef CAS.
- J. Wenk, S. N. Eustis, K. McNeill and S. Canonica, Quenching of excited triplet states by dissolved natural organic matter, Environ. Sci. Technol., 2013, 47, 12802–12810 CrossRef CAS PubMed.
- K. S. Golanoski, S. Fang, R. Del Vecchio and N. V. Blough, Investigating the mechanism of phenol photooxidation by humic substances, Environ. Sci. Technol., 2012, 46, 3912–3920 CrossRef CAS PubMed.
- C. M. Sharpless and N. V. Blough, The importance of charge-transfer interactions in determining chromophoric dissolved organic matter (CDOM) optical and photochemical properties, Environ. Sci.: Processes Impacts, 2014, 16, 654–671 CAS.
- D. K. Palit, Photophysics and excited state relaxation dynamics of p-hydroxy and p-amino-substituted benzophenones: a review, Res. Chem. Intermed., 2005, 31, 205–225 CrossRef CAS PubMed.
- A. C. Bhasikuttan, A. K. Singh, D. K. Palit, A. V. Sapre and J. P. Mittal, Laser flash-photolysis studies on the monohydroxy derivatives of benzophenone, J. Phys. Chem. A, 1998, 102, 3470–3480 CrossRef CAS.
- P. K. Das, M. V. Encinas and J. C. Scaiano, Laser flash photolysis study of the reactions of carbonyl triplets with phenols and photochemistry of p-hydroxypropiophenone, J. Am. Chem. Soc., 1981, 103, 4154–4162 CrossRef CAS.
- M. Hoshino and M. Koizumi, Order of quencher participation in photochemistry. I. Proton transfer from the excited p-hydroxybenzophenone in mixed solvents of cyclohexane and alcohols, Bull. Chem. Soc. Jpn., 1972, 45, 2731–2736 CrossRef CAS.
- D. K. Palit, H. Pal, T. Mukherjee and J. P. Mittal, Photodynamics of the S1 state of some hydroxy- and amino-substituted naphthoquinones and anthraquinones, J. Chem. Soc., Faraday Trans., 1990, 86, 3861 RSC.
- M. I. Sancho, A. H. Jubert, S. E. Blanco, F. H. Ferretti and E. A. Castro, Determination of dissociation constants of p-hydroxybenzophenone in aqueous organic mixtures – Solvent effects, Can. J. Chem., 2008, 86, 462–469 CrossRef CAS.
- Y. Du, J. D. Xue, C. S. Ma, W. M. Kwok and D. L. Phillips, Time-resolved resonance Raman and density functional theory study of the deprotonation reaction of the triplet state of para-hydroxybenzophenone in mixed acetonitrile/water solutions, J. Raman Spectrosc., 2008, 39, 1518–1525 CrossRef CAS PubMed.
- P. G. Coble, Characterization of marine and terrestrial DOM in seawater using excitation-emission spectroscopy, Mar. Chem., 1996, 51, 325–346 CrossRef CAS.
- M. M. D. Sierra, M. Giovanela, E. Parlanti and E. J. Soriano-Sierra, Fluorescence fingerprint of fulvic and humic acids from varied origins as viewed by single-scan and excitation/emission matrix techniques, Chemosphere, 2005, 58, 715–733 CrossRef CAS PubMed.
- F. Barsotti, Theoretical and experimental investigations on the fluorescence properties of humic substances, M.Sc. Thesis, University of Torino, Italy, 2014 Search PubMed.
- N. Mladenov, R. Sommaruga, R. Morales-Baquero, I. Laurion, L. Camarero, M. C. Dieguez, A. Camacho, A. Delgado, O. Torres, Z. Chen, M. Felip and I. Reche, Dust inputs and bacteria influence dissolved organic matter in clear alpine lakes, Nat. Commun., 2011, 2, 405 CrossRef CAS PubMed.
- S. Halbedel, O. Buettner and M. Weitere, Linkage between the temporal and spatial variability of dissolved organic matter and whole-stream metabolism, Biogeosciences, 2013, 10, 5555–5569 CrossRef.
- D. Smejkalova and A. Piccolo, Aggregation and disaggregation of humic supramolecular assemblies by NMR diffusion ordered Spectroscopy (DOSY-NMR), Environ. Sci. Technol., 2008, 42, 699–706 CrossRef CAS.
- E. Appiani and K. McNeill, Photochemical production of singlet oxygen from particulate organic matter, Environ. Sci. Technol., 2015, 49, 3514–3522 CrossRef CAS PubMed.
- L. Bracchini, A. M. Dattilo, M. Falcucci, S. A. Loiselle, V. Hull, C. Arena and C. Rossi, Spatial and temporal variations of the inherent and apparent optical properties in a shallow coastal lake, J. Photochem. Photobiol., B, 2005, 80, 161–177 CrossRef CAS PubMed.
- I. Laurion, M. Ventura, J. Catalan, R. Psenner and R. Sommaruga, Attenuation of ultraviolet radiation in mountain lakes: Factors controlling the among- and within-lake variability, Limnol. Oceanogr., 2000, 45, 1274–1288 CrossRef.
- C. Ruiz-Gonzalez, R. Simo, R. Sommaruga and J. M. Gasol, Away from darkness: a review on the effects of solar radiation on heterotrophic bacterioplankton activity, Front. Microbiol., 2013, 4, 131 Search PubMed.
- C. Tai, Y. B. Li, Y. G. Yin, Y. Cai and G. B. Jiang, Free radical photochemistry of dissolved organic matter in natural water, Prog. Chem., 2012, 24, 1388–1397 CAS.
- E. De Laurentiis, M. Minella, V. Maurino, C. Minero, M. Brigante, G. Mailhot and D. Vione, Photochemical production of organic matter triplet states in water samples from mountain lakes, located below or above the tree line, Chemosphere, 2012, 88, 1208–1213 CrossRef CAS PubMed.
- E. De Laurentiis, S. Buoso, V. Maurino, C. Minero and D. Vione, Optical and photochemical characterization of chromophoric dissolved organic matter from lakes in Terra Nova Bay, Antarctica. Evidence of considerable photoreactivity in an extreme environment, Environ. Sci. Technol., 2013, 47, 14089–14098 CrossRef CAS PubMed.
- A. Marchisio, M. Minella, V. Maurino, C. Minero and D. Vione, Photogeneration of reactive transient species upon irradiation of natural water samples: Formation quantum yields in different spectral intervals, and implications for the photochemistry of surface waters, Water Res., 2015, 73, 145–156 CrossRef CAS PubMed.
- S. Loiselle, D. Vione, C. Minero, V. Maurino, A. Tognazzi, A. M. Dattilo, C. Rossi and L. Bracchini, Chemical and optical phototransformation of dissolved organic matter, Water Res., 2012, 46, 3197–3207 CrossRef CAS PubMed.
- K. Fenner, S. Canonica, L. P. Wackett and M. Elsner, Evaluating pesticide degradation in the environment: Blind spots and emerging opportunities, Science, 2013, 341, 752–758 CrossRef CAS PubMed.
- D. Vione, M. Minella, V. Maurino and C. Minero, Indirect photochemistry in sunlit surface waters: Photoinduced production of reactive transient species, Chem. – Eur. J., 2014, 20, 10590–10606 CrossRef CAS PubMed.
- M. M. Dong and F. L. Rosario-Ortiz, Photochemical formation of hydroxyl radical from effluent organic matter, Environ. Sci. Technol., 2012, 46, 3788–3794 CrossRef CAS PubMed.
- E. Lee, C. M. Glover and F. L. Rosario-Ortiz, Photochemical formation of hydroxyl radical from effluent organic matter: Role of composition, Environ. Sci. Technol., 2013, 47, 12073–12080 CrossRef CAS PubMed.
- S. Mostafa and F. L. Rosario-Ortiz, Singlet oxygen formation from wastewater organic matter, Environ. Sci. Technol., 2013, 47, 8179–8186 CAS.
- R. Del Vecchio and N. V. Blough, On the origin of the optical properties of humic substances, Environ. Sci. Technol., 2004, 38, 3885–3891 CrossRef CAS.
- O. A. Trubetskoj, O. E. Trubetskaya and C. Richard, Photochemical activity and fluorescence of electrophoretic fractions of aquatic humic matter, Water Resour., 2009, 36, 518–524 CrossRef CAS.
- C. Richard, C. Coelho, G. Guyot, L. Shaloiko, O. Trubetskoj and O. Trubetskaya, Fluorescence properties of the <5 kDa molecular size fractions of a soil humic acid, Geoderma, 2011, 163, 24–29 CrossRef CAS PubMed.
- C. Coelho, G. Guyot, A. ter Halle, L. Cavani, C. Ciavatta and C. Richard, Photoreactivity of humic substances: Relationship between fluorescence and singlet oxygen production, Environ. Chem. Lett., 2011, 9, 447–451 CrossRef CAS.
- D. E. Latch and K. McNeill, Microheterogeneity of singlet oxygen distributions in irradiated humic acid solutions, Science, 2006, 311, 1743–1747 CrossRef CAS PubMed.
- M. Grandbois, D. E. Latch and K. McNeill, Microheterogeneous concentrations of singlet oxygen in natural organic matter isolate solutions, Environ. Sci. Technol., 2008, 42, 9184–9190 CrossRef CAS.
- M. Minella, M. P. Merlo, V. Maurino, C. Minero and D. Vione, Transformation of 2,4,6-trimethylphenol and furfuryl alcohol, photosensitised by Aldrich humic acids subject to different filtration procedures, Chemosphere, 2013, 90, 306–311 CrossRef CAS PubMed.
- M. Brigante, T. Charbouillot, D. Vione and G. Mailhot, Photochemistry of 1-nitronaphthalene: A potential source of singlet oxygen and radical species in atmospheric waters, J. Phys. Chem. A, 2010, 114, 2830–2836 CrossRef CAS PubMed.
- S. E. Braslavsky, Glossary of terms used in photochemistry, 3rd edition, Pure Appl. Chem., 2007, 79, 293–465 CrossRef CAS.
- A. Beckett and G. Porter, Primary photochemical processes in aromatic molecules. Part 10. Photochemistry of substituted benzophenones, Trans. Faraday Soc., 1963, 59, 2051–2057 RSC.
- G. Porter and P. Suppan, Primary photochemical processes in aromatic molecules. Part 12. Excited states of benzophenone derivatives, Trans. Faraday Soc., 1965, 61, 1664–1673 RSC.
- T. S. Godfrey, W. Hilpern and G. Porter, Triplet–triplet absorption spectra of benzophenone and its derivatives, Chem. Phys. Lett., 1967, 490–492 CrossRef CAS.
- C. Franco and J. Olmsted, Photochemical determination of the solubility of oxygen in various media, Talanta, 1990, 37, 905–909 CrossRef CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, Quantum yields for the photosensitized formation of the lowest electronically excited singlet state of molecular oxygen in solution, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS PubMed.
- S. Canonica, U. Jans, K. Stemmler and J. Hoigné, Transformation kinetics of phenols in water. Photosensitization by dissolved natural organic material and aromatic ketones, Environ. Sci. Technol., 1995, 29, 1822–1831 CrossRef CAS PubMed.
- E. De Laurentiis, J. Socorro, D. Vione, E. Quivet, M. Brigante, G. Mailhot, H. Wortham and S. Gligorovski, Phototransformation of 4-phenoxyphenol sensitised by 4-carboxybenzophenone: Evidence of new photochemical pathways in the bulk aqueous phase and on the surface of aerosol deliquescent particles, Atmos. Environ., 2013, 81, 569–578 CrossRef CAS PubMed.
- D. D. Li, R. M. Han, R. Liang, C. H. Chen, W. Z. Lai, J. P. Zhang and L. H. Skibsted, Hydroxyl radical reaction with trans-resveratrol: Initial carbon radical adduct formation followed by rearrangement to phenoxyl radical, J. Phys. Chem. B, 2012, 116, 7154–7161 CrossRef CAS PubMed.
- M. Maus and K. Rurack, Monitoring pH and solvent proticity with donor–acceptor-substituted biphenyls: a new approach towards highly sensitive and powerful fluorescent probes by tuning the molecular structure, New J. Chem., 2000, 24, 677–686 RSC.
- M. Y. Li, C. S. Cline, E. B. Koker, H. H. Carmichael, C. F. Chignell and P. Bilski, Quenching of singlet molecular oxygen by azide anion in solvent mixtures, Photochem. Photobiol., 2001, 74, 760–764 CrossRef CAS.
- M. Hoshino, Low-temperature laser photolysis of 4-hydroxybenzophenone in ethanol solution. Temperature-dependent spectral changes of the excited triplet state, J. Phys. Chem., 1987, 91, 6385–6388 CrossRef CAS.
- P. Natarajan, Quenching of benzophenone triplets by naphthalene, J. Chem. Educ., 1976, 53, 200–201 CrossRef CAS.
- G. Porter and F. Wilkinson, Primary photochemical processes in aromatic molecules. Part 5. Flash photolysis of benzophenone in solution, Trans. Faraday Soc., 1961, 57, 1686–1691 RSC.
- T. N. Das, Oxidation of phenol in aqueous acid: Characterization and reactions of radical cations vis-a-vis the phenoxyl radical, J. Phys. Chem. A, 2005, 109, 3344–3351 CrossRef CAS PubMed.
- F. Wilkinson and J. G. Brummer, Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution, J. Phys. Chem. Ref. Data, 1981, 10, 809–999 CrossRef CAS PubMed.
- D. O. Martire, C. Evans, S. G. Bertolotti, S. E. Braslavsky and N. A. Garcia, Chemosphere, 1993, 26, 1691–1701 CrossRef CAS.
- M. C. Palumbo and N. A. Garcia, Toxicol. Environ. Chem., 1988, 17, 103–116 CrossRef CAS PubMed.
- R. V. Bensasson and M. Rougee, C. R. Acad. Sci., Ser. III, 1988, 307, 807–810 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5pp00214a |
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |