
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Changes in air quality and tropospheric composition due to depletion of stratospheric ozone and interactions with changing climate: implications for human and environmental health
S.
Madronich
*a,
M.
Shao
b,
S. R.
Wilson
c,
K. R.
Solomon
d,
J. D.
Longstreth
e and
X. Y.
Tang
b
aAtmospheric Chemistry Division, National Center for Atmospheric Research, Boulder, Colorado 80307, USA. E-mail: sasha@ucar.edu
bPeking University, College of Environmental Science and Engineering, Beijing 100871, China
cSchool of Chemistry, University of Wollongong, NSW, 2522, Australia
dCentre for Toxicology and School of Environmental Sciences, University of Guelph, ON, N1G 2W1, Canada
eThe Institute for Global Risk Research, LLC, Bethesda, Maryland 20817, USA
First published on 7th November 2014
Abstract
UV radiation is an essential driver for the formation of photochemical smog, which includes ground-level ozone and particulate matter (PM). Recent analyses support earlier work showing that poor outdoor air quality is a major environmental hazard as well as quantifying health effects on regional and global scales more accurately. Greater exposure to these pollutants has been linked to increased risks of cardiovascular and respiratory diseases in humans and is associated globally with several million premature deaths per year. Ozone also has adverse effects on yields of crops, leading to loss of billions of US dollars each year. These detrimental effects also may alter biological diversity and affect the function of natural ecosystems. Future air quality will depend mostly on changes in emission of pollutants and their precursors, but changes in UV radiation and climate will contribute as well. Significant reductions in emissions, mainly from the energy and transportation sectors, have already led to improved air quality in many locations. Air quality will continue to improve in those cities/states that can afford controls, and worsen where the regulatory infrastructure is not available. Future changes in UV radiation and climate will alter the rates of formation of ground-level ozone and photochemically-generated particulate matter and must be considered in predictions of air quality. The decrease in UV radiation associated with recovery of stratospheric ozone will, according to recent global atmospheric model simulations, lead to increases in ground-level ozone at most locations. If correct, this will add significantly to future ground-level ozone trends. However, the spatial resolution of these global models is insufficient to inform policy at this time, especially for urban areas. UV radiation affects the atmospheric concentration of hydroxyl radicals, ˙OH, which are responsible for the self-cleaning of the atmosphere. Recent measurements confirm that, on a local scale, ˙OH radicals respond rapidly to changes in UV radiation. However, on large (global) scales, models differ in their predictions by nearly a factor of two, with consequent uncertainties for estimating the atmospheric lifetime and concentrations of key greenhouse gases and air pollutants. Projections of future climate need to consider these uncertainties. No new negative environmental effects of substitutes for ozone depleting substances or their breakdown-products have been identified. However, some substitutes for the ozone depleting substances will continue to contribute to global climate change if concentrations rise above current levels.
Introduction
The degradation of air quality is one of the major environmental hazards facing modern society. Human activities result in the emission of many chemicals to the atmosphere, which are either toxic themselves, or produce noxious compounds when exposed to ambient ultraviolet (UV) radiation. UV radiation is an essential driver for the generation of ground-level ozone (O3) and some particulate matter (PM, frequently called aerosol) including sulfate, nitrate, and organic aerosols. These pollutants have major health implications for humans and the environment. Future changes in tropospheric UV radiation, whether from stratospheric ozone changes or other factors such as clouds, are likely to contribute to trends in air quality and associated health effects.UV radiation makes hydroxyl (˙OH) radicals, the so-called cleaning agents of the troposphere. These radicals limit the atmospheric lifetime of many gases that are important to both tropospheric and stratospheric chemistry as well as climate change, including methane (CH4), hydrogen-containing halocarbons (e.g. hydrofluorocarbons, hydrochlorofluorocarbons and hydrobromocarbons), and the oxides of sulfur and nitrogen (SO2 and NOx).
This paper provides an assessment of how UV radiation affects air quality, particularly ground-level O3 and PM, in the context of the growing body of knowledge on their health impacts, geographic distributions, and long-term trends. It should be recognised that many factors drive these trends, including changes in both natural and anthropogenic emissions, as well as climate variability through changes in temperature, moisture, and atmospheric circulation patterns. Tropospheric UV radiation is one of these factors, and its effects can be approximately superimposed onto the effects of the other factors, but complex non-linear interactions must be considered to obtain reliable estimates.
Since the previous assessment of the interactions between ozone depletion, climate change, and air-quality in the troposphere,10,11 significant advances are noted in: (i) understanding and quantifying the important consequences of poor air quality for human health, separating the effects of O3 and PM; (ii) understanding changes in air pollution on urban and regional scales, in terms of changes in anthropogenic emissions (increases or decreases, depending on location) as well as long-range transport; (iii) understanding long-term changes of key tropospheric oxidants (O3 and ˙OH) on continental and global scales, as anthropogenic emissions continue in an environment where both stratospheric ozone and climate are also changing.
An equally important advance is a better understanding of uncertainties inherent in numerical models used to predict the future chemical composition of the atmosphere. These computer models endeavor to represent and integrate the many chemical, physical, and biological processes that control air quality as well as climate. Recent inter-comparisons among the models (see below) highlight important differences that cast some doubt on the reliability of future projections, while also indicating a path to model improvements.
Ozone-depleting substances (ODS) could also affect air quality. While the long-lived chlorofluorocarbons (CFCs) break down almost exclusively in the stratosphere, the halogenated replacements break down in the troposphere. The cycling of the halogenated species in the troposphere needs to be assessed to ensure that there are no other significant short- and long-term effects that will result from the replacements for the halocarbons. Health effects could result from exposure to these substances, and are therefore included in this assessment (previously this was included in the health assessment, which now focuses on UV-mediated effects).
This paper provides summaries of the state of knowledge on ground-level O3, PM, and ˙OH radicals, and addresses some of the more complex but still largely unquantified interactions between air quality, climate change, and human activities. An important additional interaction between air quality and stratospheric ozone depletion is the introduction of substitute compounds for ODSs pursuant to international agreements. The last part of this paper provides an update on selected substitutes whose potential environmental and health impacts should be considered.
In summary, our assessment updates and reinforces several key conclusions. Air pollution is increasingly recognised as a major environmental hazard and a risk to human health, globally leading to several million premature deaths per year. Air pollution also damages vegetation and reduces agricultural yields, with associated economic losses estimated as $10–20 billion annually. UV radiation is an essential ingredient for the formation of ground-level O3 and some PM, and of ˙OH radicals that control the global self-cleaning capacity of the troposphere. Future trends in UV radiation will modulate future trends in air quality. Air quality is sensitive to other changes in the environment including atmospheric circulation, hydrological cycles, and temperatures, all of which are likely to change due to the combined effects of changing stratospheric ozone and climate. No new negative environmental effects of the substitutes for the ODSs have been identified.
Ground-level ozone
Ozone-related morbidities manifested as acute and chronic bronchitis, asthma and/or atopic dermatitis,22 appendicitis,23 venous thromboembolic disease, and pulmonary embolisms24 have been reported. Ozone may also interact synergistically with viral infections. A study in Hong Kong reported interactive effects between viral infections and concentration of O3 that resulted in increased risks for hospitalisation for respiratory disease.25 The mechanism for this apparent synergism was not reported.
A number of studies have extrapolated the effects of exposure to O3 into the future. Based on the OECD19 study, premature deaths from ground-level ozone will increase to about 0.75 million per year worldwide by 2050. The greatest increases are predicted in India (130 premature deaths per million per decade by 2050) but those in OECD countries will be almost as large (95 premature deaths per million per decade by 2050), mostly as a result of greater sensitivity in an ageing population.19 Modelling of the interactions between concentrations of tropospheric ozone, precursors of ozone in the atmosphere, and climate change suggests that, by 2050, concentrations of O3 will increase in developing countries and decrease in developed countries,26 due to regional differences in future emissions.
In a review of studies on the effects of O3 on plants, it was concluded that reductions in the yields of 18 to 27% result from exposure to O3 at concentrations of 70 to 100 ppb, at the upper end of typical regional concentrations.30 Not all crops are equally sensitive to ozone. In a sensitive crop, such as soybean, yields in several cultivars were shown to decrease by as much as a factor of 2 with long-term exposure to 20–30 ppb (24 hour mean) of added ozone.31 Reductions in photosynthesis upon exposure to O3 were estimated in three different forest types and found to be statistically significant for an orange orchard, and observable for ponderosa pine.32 The potential for damage from O3 in crop plants was assessed in relation to the IPCC (pessimistic) A2 scenario33 for 2100.1 Changes in gross plant productivity resulting from changes in tropospheric O3 were projected to range from −40 to +15%, depending on location (Fig. 1). In another example, the ranges of global crop losses for wheat and soybean in 2030 as estimated from the IPCC A2 scenario were 5.4–26% and 15–19%, respectively.34 In this same study, yield reductions in the B1 (optimistic) scenario were 4.0–17% for wheat and 9.5–15% for soybean, with monetised annual losses estimated to range from $12–21 billion (year 2000 dollar equivalents).
 | ||
| Fig. 1 Global assessment of the projected percentage changes in gross primary productivity (GPP) due to O3 under the Intergovernmental Panel on Climate Change A2 scenario in 2100 within the World Wildlife Foundation Global 200 priority conservation areas. From Ainsworth et al.1 Reproduced with permission of the Royal Society. | ||
Although most of the research on the effects of O3 in ecosystems has been directed toward plants, effects on soil microorganisms have also been reported.35 The abundance and diversity of methanogenic bacteria in soils of rice paddies were found to be reduced after exposure to elevated concentrations of O3 (60 ppb) in ground-level air.
Overall, productivity and yields of crops will likely be reduced as a result of increases in concentrations of tropospheric O3. There is some hope that genetic selection of plants tolerant to O3 will mitigate these adverse effects on production of food and fibre but other plants in the ecosystem are likely to suffer greater adverse effects. These are expected to impact diversity and functions of natural ecosystems. Other effects of climate change, e.g., mediated by temperature and precipitation, will also affect yields of crops.
| Stratospheric ozone formation: | Reaction |
|---|---|
| O2 + hν (λ < 240 nm) → O + O | (1) |
| O + O2 → O3 | (2) |
| Tropospheric ozone and ˙OH formation: | |
|---|---|
| O3 + hν (λ < 330 nm) → O* + O2 | (3) |
| O* + H2O → ˙OH + ˙OH | (4) |
| ˙OH + VOC + O2 → HO2 (or organic analog RO2) + other products | (5) |
| HO2 + NO → NO2 + ˙OH | (6) |
| NO2 + hν (λ < 420 nm) → NO + O | (7) |
| O + O2 → O3 | (2) |
| Secondary tropospheric radical sources: | |
|---|---|
| CH2O + hν (λ < 340 nm) + 2 O2 → HO2 + HO2 + CO | (8) |
| H2O2 + hν (λ < 350 nm) → ˙OH + ˙OH | (9) |
| HONO + hν (λ < 395 nm) → ˙OH + NO | (10) |
Formation of O3 in polluted urban atmospheres has been recognised since the 1950s,36 and occurs when mixtures of volatile organic compounds (VOCs) and nitrogen oxides (NOx = NO + NO2) are exposed to the UV radiation available in the troposphere. The chemistry leading to tropospheric O3 formation is generally complex because it (i) requires the absorption of multiple photons, (ii) is augmented by several catalytic cycles, and (iii) can be fuelled by many different VOCs, including those of anthropogenic and biogenic origin. Table 2 provides a highly simplified schematic of the major chemical pathways. The production of tropospheric O3 is autocatalytic because photolysis of an initial amount of O3 (Table 2, reactions 3 and 4) results in two ˙OH radicals, each of which can then continue through the reaction sequence to regenerate more O3. Note that two photons are required for this process (Table 2, reactions 3 and 7) with their combined minimum energy being more than sufficient to break O2 directly.
Reaction 3 in Table 2, the photolysis of ozone to yield excited oxygen atoms O*, is the primary source of radicals (˙OH, HO2, and RO2) involved in production of O3. It is also very sensitive to the overhead ozone column (see Table 1 of McKenzie et al.37). Other sources of radicals include the photolysis of formaldehyde (CH2O), hydrogen peroxide (H2O2), and nitrous acid (HONO). These latter compounds are typically the products of previous chemical reactions initiated by O3 and ˙OH and so they are sensitive to the production of primary radicals (Table 2, reaction 3) and therefore to UV-B radiation as affected by changes in stratospheric ozone.
Urban ozone trends differ in different cities. Considerable progress has been made in reducing urban O3 in Europe, the United States, and some other locations.2,38–40Fig. 2 shows the reductions in ground level ozone achieved in Los Angeles and Mexico City over the past several decades. In Beijing, from 2005 to 2011, ground-based measurements give daytime average O3 increasing at 2.6 ppb (5%) per year.41 In comparison, increases of 3% per year over 2002–2010 have been reported for the tropospheric O3 column above Beijing.42 Other Asian cities showing increases include Hong Kong (0.55 ppb per year over 1994–2007), Seoul (about 5 ppb over 1991–2007), and Tokyo with a doubling of days with O3 exceedances (incidences where air quality standards are exceeded).40
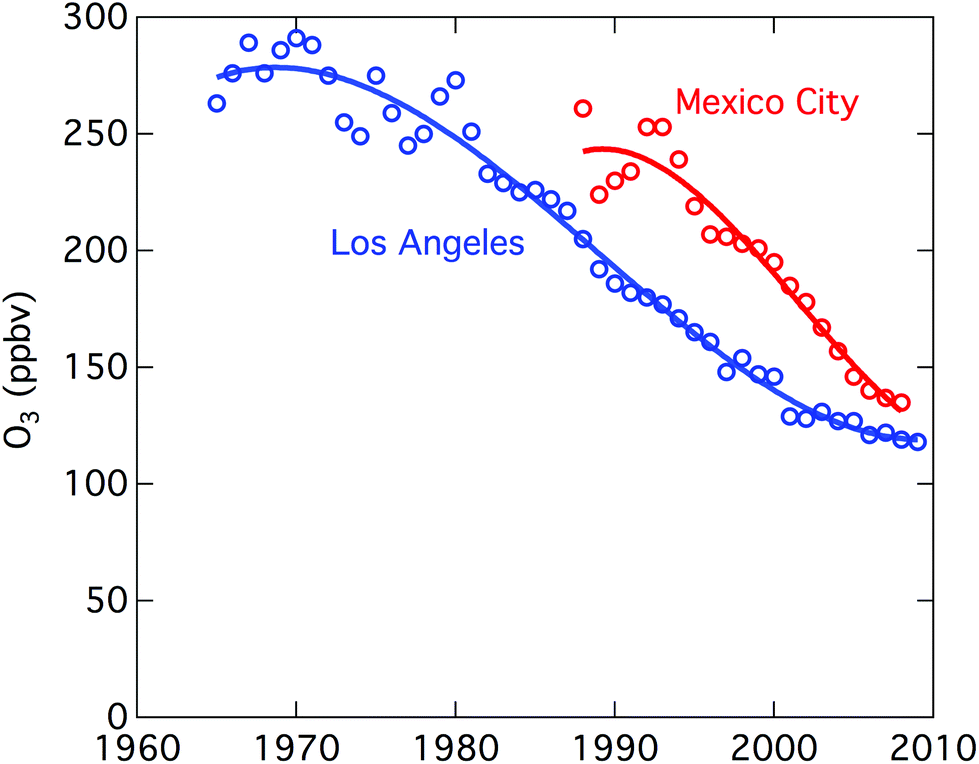 | ||
| Fig. 2 Improvements in air quality in Los Angeles and Mexico City. Plotted is the 3-year average of the 4th highest maximum ozone 8-hour average. (From Parrish et al.2). | ||
Regional ozone (ozone averaged over large areas extending well beyond cities) is increasing at some locations, particularly in densely populated areas, for example 6–7% per decade in the Indo-Gangetic Plains.43 Regional production of ozone was also shown to be a factor limiting air quality improvements from local emission reductions during the Beijing Olympics.44 Background ozone continues to increase at many locations, such as central and northwestern Europe, but is decreasing in eastern and southwestern Europe.38,39 Increases of 0.25 ppb per year have been reported for ground-level ozone at Mace Head, Ireland.45 In the western U.S., mid-tropospheric (3–10 km) ozone has increased by 0.6 ppb per year in the springtime over 1995–2008, and may be indicative of long range transport.46 Changes in the seasonal cycle observed at various mid-latitude locations in the Northern Hemisphere are consistent with increasing emissions of precursors.47 On the other hand, a review of measurements made with balloon-borne instruments (ozone sondes) and surface observations at remote locations showed that most of the increases in both hemispheres occurred in the early part of the 20–40 year record, with more recent changes characterised by little or no increase.48
Changes in atmospheric circulation are also likely to have affected trends of tropospheric ozone. Some increases may have been due to changes in the rate of stratosphere–troposphere air exchange.49 Trends at Mauna Loa, Hawaii, were influenced by changes in circulation due to El Niño, transporting air masses from different regions of Asia to Hawaii.50
Prediction of tropospheric concentrations of O3via numerical models remains problematic, with large differences at regional38 and global scales.6,51 This is illustrated in Fig. 3, where several global models, represented by different lines, are seen to differ by as much as 25% for current conditions, and future predictions show a wider range depending on the choice of future scenario as well as model. However, it should be noted that future photochemical ozone formation will in any case depend to a large extent on details of future emissions, particularly those associated with different fuel choices (e.g. diesel, gasoline, or biofuels).
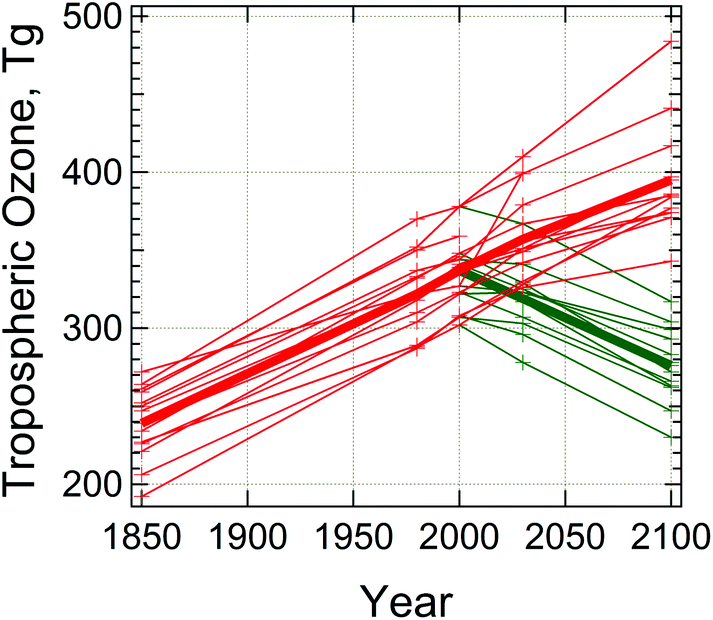 | ||
| Fig. 3 Global tropospheric ozone burden simulated by different models. Drawn from data in Table 1 of Young et al.6 Thin lines are for individual model results, thick lines are multi-model averages, for two scenarios of future emissions, RCP2.6 (green) and RCP8.5 (red), as defined by the Intergovernmental Panel on Climate Change.9 | ||
The specific response of tropospheric O3 to future changes in stratospheric O3 was modeled by Zeng et al.52 and Zhang et al.53 with both studies showing large-scale increases in tropospheric O3, as a net result of slowing both production and loss in response to declining UV levels. However the low resolution of their models (several degrees latitude × longitude) is insufficient to discern urban effects where higher levels of NOx are expected to maintain an opposite (positive) relationship between UV radiation and ground-level ozone54,55 that would indicate improvement in air quality in response to recovery of stratospheric ozone. Global models also do not agree well with measurements of the background atmosphere (e.g.ref. 56), indicating that there is still significant work to be done in understanding this chemistry. These uncertainties make it difficult to identify precisely which geographic regions will experience decreases in tropospheric ozone as stratospheric ozone recovers, and which ones will suffer increases. Nevertheless, all models agree that over large regions tropospheric ozone will increase.
Particulate matter
Particulate matter (PM) in the atmosphere consists of small solid or liquid particles suspended in air, also called aerosols. The size of PM is recognised as important for health effects, with PM smaller than 2.5 μm (termed PM2.5) being inhaled deeper into lungs than larger particles, typically measured as all particles below 10 μm (PM10).Where control of emissions into the troposphere has resulted in decreases in PM2.5, fewer health effects have been observed. In a study of life expectancy in 545 U.S. counties, reductions in PM2.5 of 10 μg m−3 from 2000 to 2007 were associated with an increase in mean life expectancy of 0.35 years (SD = 0.16 years).60
Overall, the global relevance of particulates to human health is very large, and substantial changes are expected to occur in response to changes in climate.61 Future predictions are uncertain due to limitations of atmospheric models and their assumptions62 and, specifically for human health effects, the difficulty to clearly separate effects of O3 and PM2.5.63 Future changes in aerosols are uncertain but may be substantial regionally. A multi-model analysis of past and future trends in aerosol, described in Fig. 8 of Bais et al.,64 indicates large changes in industrialised regions, particularly in China.
The formation of sulfate and nitrate aerosols is well understood in terms of the ˙OH oxidation of SO2 and NO2 giving sulfuric and nitric acids, respectively. While the majority of this production occurs in the gas phase, the sulfate and nitrate condense rapidly to form particles, particularly if ammonia is present. Chemical reactions in cloud and rain water can also contribute.65
Considerable progress has been made recently in understanding secondary organic aerosols (SOA), which previous observations had shown to be seriously underestimated by models. While many details remain poorly understood, numerous studies support the basic conceptual model that hydrocarbons are oxidised (by ˙OH and NO3 radicals, and O3) into a myriad of heavier, more functionalised molecules as well as smaller fragments.66 Molecules with multiple functional groups (e.g., alcohols, ketones, aldehydes, organic acids, nitrates and peroxides) typically have lower vapour pressures and therefore are likely to condense onto particles. However, quantification remains a problem due to the large number of chemical species contributing to particle mass. Significant advances in modelling have been made by classifying these multifunctional compounds according to relevant properties, such as vapour pressure,67,68 solubility,69 oxidation state,70 atomic ratios (O, C, H, etc.),71,72 and carbon number and polarity.73 In practice, for ambient aerosols many of these properties are not known and therefore cannot be used to constrain predictions. However, these modelling frameworks now allow exploratory sensitivity analyses to help identify the most important processes for more accurate parameterisation.
Removal of aerosols from the atmosphere is poorly understood. Ultimately removal from the atmosphere occurs by wet or dry deposition. Incorporation of aerosol particles into raindrops (wet deposition) leads to lifetimes estimated to range from 0.5 to 2 weeks.74–76 Dry deposition of particles is generally slower.65,77
 | ||
| Fig. 5 Annual average aerosol optical depth at 550 nm from the MODIS and MISR satellite instruments (top and bottom, for the years 2004–2006) and models (middle, for the year 2000). From Schindell et al.4 | ||
Heavily populated urban locations are of special interest, and some recently reported measurements in megacities are summarised in Table 3. World Health Organization (WHO) guidelines are frequently exceeded by all cities listed. Reductions in PM concentrations are occurring in many cities, in some cases well-documented by long-term urban monitoring networks, and evidently related to emissions-lowering technologies for both fixed and mobile sources. However, measurements at many polluted locations are still sparse, making an assessment of trend difficult.
| City | PM10 (μg m−3) | PM2.5 (μg m−3) | Measurement period |
|---|---|---|---|
| a Positive (+) and negative (−) trends are indicated. Values shown are ranges observed over the measurement period. Compiled from data reported by Zhu et al.;40 see original for details of the sampling intervals. | |||
| WHO guidelines | 20 | 10 | Annual mean |
| 50 | 25 | 24-hour mean | |
| Cairo | 90–260 | 30–220 | 1999–2002 |
| Dakar | 30–60 | 2008–2009 | |
| Bangkok | 40–90 (−) | 1995–2008 | |
| Beijing | 150–180 (−) | 95–155 | 1999–2008 |
| Delhi | 50–300 | 50–250 | 2004–2009 |
| Dhaka | >100 (+) | >30 (+) | 2002–2006 |
| Hong Kong | 40–50 (−) | 20–40 | 1998–2008 |
| Jakarta | 60–100 (−) | 2001–2007 | |
| Manila | 40–50 | 20–30 (−) | 2001–2008 |
| Seoul | 60–80 (−) | 1995–2007 | |
| Shanghai | 90–110 (−) | 2002–2007 | |
| Tokyo | 15–30 (−) | 2001–2008 | |
| Tehran | 65–370 | 2003 | |
| Santiago | 50–100 (−) | 20–30 (−) | 2000–2008 |
| Sao Paulo | 40–70 (−) | 1996–2006 | |
| Los Angeles | 40–80 (−) | 1998–2008 | |
| Houston | 30–40 (−) | 1998–2008 | |
| New York City | 30–70 (−) | 1998–2008 | |
| Mexico City | 50–180 (−) | 20–25 (−) | 1990–2010 |
| London | 20–35 | 1994–2004 | |
| Moscow | 35–50 (+) | 2006–2008 | |
| Milan | 35–60 (−) | 2000–2009 | |
| Istanbul | 45 | 20 | 2002–2003 |
Future concentrations of aerosols are subject to similar scenario assumptions as other pollutants.4,79,80 Globally averaged sulfate concentrations have already decreased over the last two decades and are expected to continue decreasing. Organic and black carbon are expected to continue to increase over the next few decades globally, but then decrease, with timing and magnitude depending on the specific scenario.
On smaller geographic scales, aerosol concentrations are sensitive to local and regional emissions, and may improve or worsen depending on regulatory strategies. For example, a multi-model analysis of past and future trends in aerosols, described in Fig. 8 of Bais et al.64 shows strong regional reductions of aerosol concentrations by 2090, particularly in China.
Hydroxyl radicals
Understanding ˙OH is fundamental to understanding the chemistry of ozone and secondary aerosols as well. Cycling between ˙OH and HO2 (Reactions 5 and 6, Table 2) is essential for tropospheric ozone formation. Notably, ˙OH itself has a lifetime of only seconds, but it affects O3 on the time scale of hours to days, CO over months, and methane over a decade. For this reason, direct detection of ˙OH has focused on local short-term measurements, while longer-term impacts, for example, on methane lifetimes, have been estimated from global models.
Several techniques have been developed over the past few decades to detect and measure concentrations of ˙OH. Measurements have been reviewed by Heard and Pilling81 and more recently by Stone et al.82 Most of the recent measurements are consistent with model calculations within a factor of approximately two, (e.g., urban locations including New York City,83 Tokyo,84 Mexico City,85,86 and Houston87). Studies in forested locations, specifically West Africa88 and northern Michigan89 also show reasonable agreement with models.
Measured ˙OH is much greater, by as much as an order of magnitude, than predicted by models in environments containing high concentrations of biogenic hydrocarbons (e.g., isoprene, methyl butenol, and terpenes) and low concentrations of NOx, including over the tropical forest of Suriname,90–92 Borneo,93,94 the Pearl River Delta (PRD),95,96 and suburban Beijing during low-NOx episodes.97 This apparent underestimation of ˙OH by models has led to a re-examination of the chemistry of isoprene at low NOx, and to the suggestion that at least part of the ˙OH initially lost by reaction with isoprene is later regenerated by secondary reactions.98,99 Simulations using an environmental smog chamber also indicate the need for some recycling of ˙OH by isoprene chemistry under very low NOx conditions, although not to an extent that would explain the large discrepancies between observations and models found over tropical Suriname (Fuchs et al., 2013).100
Several inter-comparisons between different ˙OH instruments show good agreement in some circumstances but also disagreement in others.82,101–103 The largest discrepancies appear to occur in environments dominated by biogenic hydrocarbons. For example, Mao et al.104 found a factor of two difference between laser-induced fluorescence and a chemical analysis methods at Blodget Forest, California.
Instruments have been developed recently to measure the total ˙OH reactivity, i.e., the rate at which ˙OH molecules are removed by reaction with the many constituents of sampled air (e.g., CO, VOCs, and NOx). The reactivity of ˙OH provides an important constraint on the budget of ˙OH since it must essentially balance the rate of production. Measurements show that this reactivity is larger than predicted from the simple sum of typically known constituents, indicating the presence of substantial but unmeasured amounts of other reactive compounds, by values ranging from 25–35% in Tokyo,105 about 30% in a terpene-rich mid-latitude forest,106 a factor of 2 in the Pearl River Delta107 and 60–90% in a boreal forest.108,109 The missing compounds are presumed to be a multitude of partly oxygenated organic compounds (aldehydes, ketones, etc.) formed during the ˙OH-initiated photo-degradation of VOCs.
Despite unresolved differences among various ˙OH instruments and models, some fundamental aspects of the photo-chemistry have been clearly demonstrated. Important in the present context is the theoretical expectation that, for relatively clean conditions, concentrations of ˙OH should scale more or less linearly with the photolysis of O3 to generate O* (Reaction 3, Table 2), j(O3), which in turn is dependent on the amount of UV radiation. This linear correlation has now been re-confirmed by direct measurements of ˙OH and j(O3) over a year at Mace Head, Ireland110 and is in agreement with earlier observations in the tropical Atlantic111 and in the European Alps.112 Because tropospheric j(O3) values are sensitive to the overhead ozone column, with a ∼1.5% increase in j(O3) for each 1% decrease in the O3 column (see Table 1 of McKenzie et al.,37), these studies reaffirm the importance of stratospheric ozone to tropospheric ˙OH and to the photochemistry of the lower atmosphere.
Estimates over longer time scales are largely based on models, which differ significantly. This was demonstrated by the recent intercomparison of 16 global chemistry-transport models for predictions of the methane lifetime, which is limited by reaction with ˙OH.5,7 The modelled mean lifetime of methane was 8.6 ± 1.2 years (range 6.4–11.6 years) for the year 2000. Pre-industrial (1850) to present day (2000) changes in ˙OH were either positive or negative (see Fig. 6), depending largely on how each model specified relative changes in emissions of CO and NOx. Pike and Young115 showed that global concentrations of ˙OH (and therefore the lifetime of CH4) were sensitive to how models represent ˙OH recycling by isoprene, which remains uncertain, as discussed above. If such buffering of ˙OH by biogenic VOCs is pervasive, it casts doubt on the strong sensitivities to anthropogenic emissions of CO and NOx shown in Fig. 6 for current models.
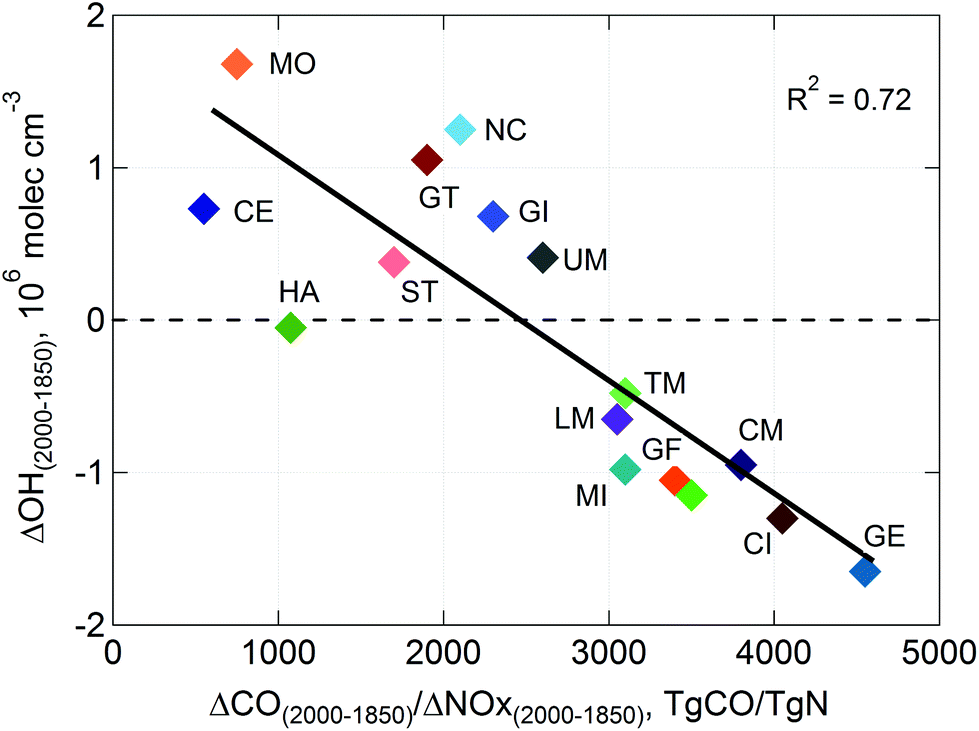 | ||
| Fig. 6 Changes in globally averaged hydroxyl radicals (˙OH) between pre-industrial times (1850) and present day (2000) calculated by 16 different models (model-mean for the year 2000 ˙OH ∼1.1x106 molec cm−3) for relative changes in emissions of carbon monoxide (ΔCO) and nitrogen oxides (ΔNOx) specified within each model (From Naik et al.5). | ||
An alternate approach to estimating changes in ˙OH based on sulfate isotopic studies suggested a 10% decrease in global ˙OH since pre-industrial times,116 broadly in line with some of the models reported by Naik et al.5 However, the accuracy of this method remains untested.
The multi-model mean changes in predicted ˙OH concentrations at the surface for the year 2100 and two different emission scenarios, RCP2.6 and RCP8.5, have been calculated7 and are shown in Fig. 7. Substantial reductions in ˙OH are expected throughout much of the southern hemisphere due to large increases in methane (CH4) in the RCP 8.5 scenario, while regional increases and decreases occur in both scenarios due to changes in shorter-lived precursors NOx and CO. Another model study focused on the recovery of stratospheric O3 to 1980 levels (holding all other factors constant) predicted that global concentrations of ˙OH will decrease by 1.7% due to the lower tropospheric UV radiation levels.53
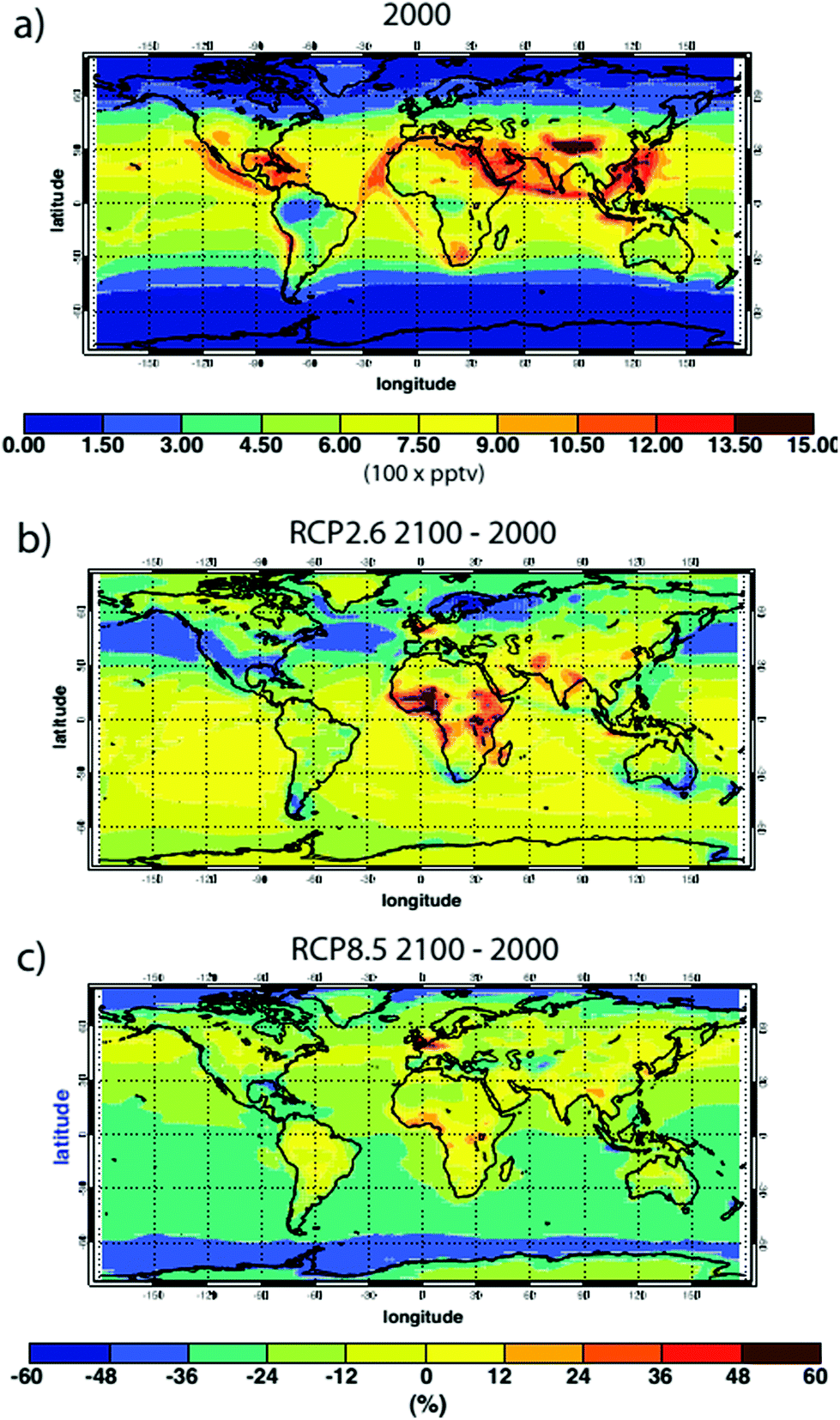 | ||
| Fig. 7 (a) Annual average surface ˙OH concentration, mean of 14 models, for the year 2000. (b) Model-mean % change in surface ˙OH concentrations in 2100 relative to 2000 for the IPCC RCP 2.6 emission scenario; (c) same for RCP 8.5 scenarios (From Voulgarakis et al.7). | ||
Global models are also sensitive to climate change, including changes in temperature, humidity, stratospheric ozone, and uncertain NOx emissions from natural sources such as biomass burning and lightning.117–119 Thus, multi-model averages such as those shown in Fig. 7 do not truly reflect the model variability or actual uncertainties. However, in clean marine atmospheres, concentrations of ˙OH are well predicted by models,120,121 but other unidentified oxidants appear to be important.120 These other oxidants could be halogens.
Climate-mediated changes in air quality
Air pollution is a complex, multifaceted problem that can only be correctly considered when integrated within the whole Earth system. Direct emissions from human activities are well recognized, but emissions that would otherwise be considered natural can also change due to, for example, deforestation, biomass burning, and even feedback between air quality, climate change (especially the hydrological cycle), and ecosystem health. Atmospheric transport of pollutants and their precursors is subject to circulation patterns that are likely to change under a changing climate. In particular, changes in the frequency of stagnation episodes that limit the dispersion of pollutants may have large impacts on air quality in affected areas. Chemical transformations, e.g., those making ground-level O3 from the photo-oxidation of hydrocarbons and nitrogen oxides, are sensitive to climate variables including temperature and moisture, as well as UV radiation. Removal of pollutants occurs mainly via contact with the Earth's surfaces (dry deposition) or scavenging by precipitation (wet deposition). Both could change significantly in the future, e.g., changes in land-use altering rates of dry deposition, and changes in precipitation patterns modifying wet deposition rates.Several potentially major interactions are discussed here. Many other feedback processes are possible and may be plausible but are not fully understood and cannot yet be quantified reliably. These are, by and large, outside the scope of the present assessment, except to the extent that we recognise their existence and therefore provide a cautionary note that the general aspects of our assessment need to be evaluated carefully for any given location with full consideration of these additional factors.
Background ozone concentrations can be influenced by long-range air transport, and this is likely to be an important factor in the future, transporting air between continents.122 The background ozone concentrations can also be altered by changes in stratosphere/troposphere exchange and by the changes in global atmospheric composition, notably some greenhouse gases.119 By analysing the natural variability of stratospheric transport, it has been estimated that changes in stratospheric circulation due to climate change will lead to around a 2% increase in tropospheric ozone in the northern mid-latitudes by the end of this century.123 Although small, the significant geographic extent implies that this might be an additional factor affecting air quality.
Increased temperatures at ground level are expected to increase biogenic emissions of reactive organics (e.g., ref. 124 and 125). However, emissions may well depend on other factors as well, such as water stress on the plants.32 Indeed, changes in the climate, coupled to decreases in air quality, can substantially alter biogenic activity in ways that are difficult to predict.
Models suggest that other critical processes are also likely to be altered by climate change. Variations in cloudiness can alter the rate of photochemical production of ozone. Increased surface heating can result in changes in atmospheric movement (wind speed, both horizontal and vertical). Cloudiness can also be altered by human activity, with evidence that controls on air pollution have increased solar radiation at some locations.126 Finally, changes in rainfall patterns and cloudiness can alter the rate of removal of both reactive precursors and ozone itself.26 Estimates predict increasing ozone concentrations at ground level throughout the 21st century, driven by all of these meteorological factors, which are regionally dependent. This can be offset by changes in emissions from human activity, which may either augment or reverse the overall trend, depending on the levels of controls implemented.26,127 Significant regional air quality changes may result, even if only episodically.128
Change in climate also may affect human health indirectly. A study on allergic respiratory diseases and bronchial asthma showed that while exacerbation is related to air-pollutants, amounts of allergen in the air are also important.137 The presence of allergenic pollens in the atmosphere might be prolonged by climate change and increase frequency and severity of these diseases.
Halogenated organic and other substitutes in the troposphere
Brominated substances. Natural bromo-carbons bromoform and dibromomethane are emitted from the oceans, and their emission strengths and role in the atmosphere are becoming better understood. These compounds release bromine upon oxidation in the atmosphere that is generally observed as bromine monoxide (BrO). The presence of bromine can lead to depletion of ground level ozone.138 However, observations in other marine locations do not find such events occurring.139 In the tropics, where sources are believed to be the biggest and vertical motion is enhanced, it has been estimated that these compounds have a lifetime in the atmosphere of 1 to 3 weeks. However, even with this relatively short lifetime, over 90% of the bromine that is transported to the stratosphere comes from these species.140
Bromine monoxide (BrO) is also calculated to be a significant oxidant for dimethylsulfide (20%) in the clean marine environment of the southern hemisphere.141 This oxidation is a significant source of aerosol in this environment (nearly 20% of the total) and so has direct impacts upon cloud formation and light scattering, and hence climate. A large fraction of this BrO is derived from sea salt, and thus is an important part of the natural bromine background to which anthropogenic brominated organics are added. The overall significance of changes due to climate, as noted above, is not yet known.
n-Propyl bromide (C3H7Br), or 1-bromopropane, was introduced in the early to mid-1990s as an intermediate in a variety of closed commercial manufacturing processes for products such as pesticides, pharmaceuticals, and quaternary ammonium compounds. From the mid-1990s, it began to be used as a less toxic substitute for methylene chloride in open air uses such as vapour and immersion degreasing and cleaning of electronics and metals.142 In 2003, it was first proposed by the SNAP program as an acceptable alternative for CFC-113 and methyl chloroform in a limited number of specific applications where emissions could be tightly controlled for both environmental and exposure concerns. Specifically, these included use as a solvent in industrial equipment for metals, electronics, and precision cleaning and in aerosol solvents and adhesive end-uses. However, the final rule issued in 2007143 allowed use only as a solvent for industrial equipment; other uses such as aerosol solvents and adhesives are listed as unacceptable by the Agency.
The SNAP program decisions were based on health data related to reproductive and neurological end-points for which they considered a work place standard of an 8-hour time-weight acceptable exposure limit (AEL) of 25 ppm to be acceptable.144 Subsequent to that information, reports have indicated that additional adverse effects have been added to the toxicological dossier for n-propyl bromide including immunotoxicity (significant decreases in a specific antibody) in rodents following 10 week inhalation exposures at levels of 125–500 ppm (mice) or 1000 ppm (rat)145 and multi-site carcinogenicity following two-year chronic inhalation exposures at 250 or 500 ppm in rats and mice.142
Chlorinated substances. Chloroform (CHCl3) has a number of poorly known natural sources as well as anthropogenic sources.146 Peat bogs may be a large unrecognized natural source of chloroform (10% of the total).147,148 Sources such as these are likely to be sensitive to both climate and land use change, and so represent an uncertainty in future predictions.
Production and consumption of carbon tetrachloride (CCl4) are regulated under the Montreal Protocol. Observations show that atmospheric CCl4 mixing ratios are decreasing at a rate slower than expected from assumed phase-out schedules149,150 The sources and sinks of CCl4, and their uncertainties, are beyond the scope of this paper, but it is probable that the slower decline is due to yet unidentified emissions. If such sources involve personal exposure, e.g., in solvent use, potential health effects may be anticipated.151
Chlorofluorinated substances. Historically the first compounds developed as replacements for refrigerants, the largest sector where CFCs were used, were saturated hydrochlorofluorocarbons (HCFCs) and then saturated hydrofluorocarbons (HFCs), both classes of compounds which because of their hydrogen content, were susceptible to attack from hydroxyl radicals in the atmosphere, resulting in shorter atmospheric lifetimes than CFCs. Readers are referred to earlier reports for a review about the human and environmental risks for many of these replacements.
There is one compound in this category, trans-1-chloro,3,3,3-trifluoropropene (HCFO 1233zd(E)), which is just now being developed as a foam blowing agent, refrigerant, and solvent. A recent toxicology study152 reported that HCFO 1233zd(E) was not acutely toxic and was not associated with any genetic toxicity in a battery of tests. The compound had an acute 4-hour 50% lethal concentration value (LC50) of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm in rats, and a no observed effect level (NOEL) in canine cardiac sensitization studies of 25000 ppm. The heart was identified as the apparent target organ on the basis of histopathological observations from a 2 week range finding study in male and female rats exposed levels of 0, 2000, 5700 and 20000 ppm 6 hours per day for 5 days per week, Males at the mid and high doses and females at the high dose developed multifocal mononuclear infiltrates of cardiac tissue. In a 12-week study at 4000, 10000 and 15000 ppm, 6 hours per day for 5 days per week, a NOEL/lowest observed adverse effect level for multifocal mononuclear infiltrates of the heart was 4000 ppm. In a full inhalation developmental toxicity study at concentrations of 0, 4000, 10000 and 150000 ppm, HCFO 1233zd(E) was not associated with any effects on uterine, placental or fetal weights, nor were there any fetal abnormalities observed so that the study resulted in a NOEL for developmental toxicity of 15000 ppm.
000 ppm in rats, and a no observed effect level (NOEL) in canine cardiac sensitization studies of 25000 ppm. The heart was identified as the apparent target organ on the basis of histopathological observations from a 2 week range finding study in male and female rats exposed levels of 0, 2000, 5700 and 20000 ppm 6 hours per day for 5 days per week, Males at the mid and high doses and females at the high dose developed multifocal mononuclear infiltrates of cardiac tissue. In a 12-week study at 4000, 10000 and 15000 ppm, 6 hours per day for 5 days per week, a NOEL/lowest observed adverse effect level for multifocal mononuclear infiltrates of the heart was 4000 ppm. In a full inhalation developmental toxicity study at concentrations of 0, 4000, 10000 and 150000 ppm, HCFO 1233zd(E) was not associated with any effects on uterine, placental or fetal weights, nor were there any fetal abnormalities observed so that the study resulted in a NOEL for developmental toxicity of 15000 ppm.
Fluorinated substances. Sulfuryl fluoride (SO2F2) is a substitute for the ozone-depleting fumigant methyl bromide used on crops and soils. Several reports on its use in agriculture have been published recently153–156 and it will likely become more widely used in the future. As previously stated,11 its atmospheric oxidation lifetime is estimated to be large (>300 years). However, SO2F2 is relatively soluble in water and is expected to partition into cloud water and ultimately rain out of the atmosphere with a half-life of about 2 weeks. The major sink is the oceans where the ultimate breakdown products are inorganic sulfate and fluoride. These breakdown products are not of concern for environment or health. However, SO2F2 has a large global warming potential (GWP) and its use is likely to increase in the future, so that monitoring of concentrations in the atmosphere should continue.
Hydrochlorofluorocarbons and hydrofluorocarbons (HCFCs and HFCs): As has been discussed previously,11,157 several of the hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs) used as substitutes for ozone-depleting CFCs and a new fluorinated olefin (HFO) can break down into trifluoroacetic acid (TFA). TFA is stable in the environment but is water soluble and accumulates in playas, land-locked lakes, and the oceans where it combines with cations such as sodium, potassium, calcium, and magnesium (Na+, K+, Ca2+, and Mg2+). More than 95% of the salts of TFA found in the oceans are naturally produced. These salts are inert and not of toxicological or environmental concern in the small concentrations (≈200 ng L−1) that are present in the oceans.
Perfluoro-n-butyl iodide (PFBI) is a compound being investigated as a substitute for CFCs. Its use is being explored as a replacement in aircraft maintenance operations as an alternative cleaner for liquid and gaseous oxygen aerospace systems and as a wipe cleaner solvent. A recent publication158 summarised earlier preliminary toxicity testing results in which an acute inhalation of no observed adverse effect level (NOAEL) of 3900 ppm for cardiac sensitisation was established. Based on the results of a preliminary 4-week inhalation toxicity test, a 13-week inhalation study was conducted in which 15 male and 10 female rats per group were exposed by inhalation via nose-only exposure for 6 hours per day and 5 days per week to nominal concentrations of 500, 1500, and 5000 ppm PFBI. The 13-week inhalation exposure at 1500 ppm was associated with increases in concentrations of thyroid hormones and thyroid stimulating hormone in serum of male and female rats. These concentrations returned to control values following the 4-week recovery period without exposure in males (the only animals given a recovery period). The authors noted that, based on published information, they assumed that the return to normal following the recovery period indicated that the hormonal changes were only transient and not adverse and that the changes in female rats would also return to normal. Thus the NOAEL was selected to be 1500 ppm. The 3900 ppm from the earlier work served as an occupational acute exposure limit for PFBI, while the 1500 ppm NOAEL was converted to an occupational exposure limit (OEL) by adjusting it to an 8-hour/day time-weighted average exposure and applying uncertainty factors for animal to human, inter-human variability and subchronic to chronic extrapolation to arrive at a final value of 40 ppm.
Modelling of susceptibility of the analogous compound (perfluoro-n-propyl iodide) to photolysis suggested a half-life of a few hours in sunlight. Based on rapid degradation, PFBI has zero GWP and is not an ODS. Its atmospheric degradation is likely to produce TFA, analogously to many other fluorinated organics, e.g. some HCFCs and HFCs (see above).
Hydrofluoroolefins (HFOs). Among the other new compounds being used as substitutes are those belonging to the class known as hydrofluoroolefins (HFOs). Two examples currently being developed for use in the refrigeration, foam-blowing and/or aerosol sectors, 1,3,3,3 tetrafluoropropene (HFO 1234ze) and 2,3,3,3 tetrafluoropropene (HFO-1234yf) have recently been characterised toxicologically.159–161 In the case of HFO 1234ze, the compound was not acutely toxic at levels as high as 207
000 ppm following 4 hours of exposure and showed no activity in a battery of genetic toxicity tests. In addition, the compound did not induce cardiac sensitization at levels as high as 120000. Following a 2 week range finding study at 5000, 20000 and 50000 ppm 6 hours per day, 5 days per week, the liver and heart were identified as the target organs; however, in a subsequent 4 week study at 1000, 5000, 10000, and 15000, and a 90 day study at 1500, 5000, and 15000, only the heart was identified as the target organ. The associated finding was multifocal mononuclear cell infiltrates with no evidence of fibrosis and no evidence of increased severity with increased duration of exposure.
No new papers on the relevance of TFA to human health and the environment have been published in the literature since the date of the previous assessment (2010). Thus, projected future increased loadings of TFA to playas, land-locked lakes, and the oceans due to continued use of HCFCs, HFCs, and replacement products are still judged to present negligible risks for aquatic organisms and humans.
Gaps in knowledge
A key air quality constituent is ozone, and any future changes in ozone have significant outcomes for both human and environmental health. However, the direction and the magnitude of change in tropospheric ozone due to recovery of stratospheric ozone are still under debate, may depend on location, and need to be better quantified. Modulation by UV radiation will modify the impacts of climate change and regulation of local emissions. This represents a significant gap in our understanding. Computer modelling now has the potential to address this issue, but still requires the development and refinement of parameterisations for physical and chemical processes, as well as future scenarios under different socio-economic assumptions.Particulate matter (aerosol) plays a significant role in climate change and also in air quality. While the role of UV radiation in PM formation is known, the sensitivity of PM properties to changes in UV radiation has not been sufficiently quantified.
The oxidation capacity of the atmosphere remains poorly characterised in a number of environmentally sensitive regions, with an order of magnitude difference between measurements and models. Both measurements and our understanding of the key chemical processes have large uncertainties. One example of this lack of understanding is the uncertainty in future methane concentrations, with models predicting ˙OH driven lifetimes that differ by a factor of 2.
Acknowledgements
M. Shao is supported by the Natural Science Foundation of CHINA for Outstanding Young Scholars (Grant No. 41125018). The National Center for Atmospheric Research is sponsored by the National Science Foundation. S. Madronich was supported by the U.S. Global Change Research Program. The participation of Associate Professor S. Wilson was supported through funding from the Australian Government's Ozone Science Strategy.References
- E. A. Ainsworth, C. R. Yendrek, S. Sitch, W. J. Collins and L. D. Emberson, The effects of tropospheric ozone on net primary productivity and implications for climate change, Annu. Rev. Plant Biol., 2012, 63, 637–661 CrossRef CAS PubMed.
- D. D. Parrish, H. B. Singh, L. Molina and S. Madronich, Air quality progress in North American megacities: A review, Atmos. Environ., 2011, 45, 7015–7025 CrossRef CAS.
- R. V. Martin, C. E. Sioris, K. Chance, T. B. Ryerson, T. H. Bertram, P. J. Wooldridge, R. C. Cohen, J. A. Neuman, A. Swanson and F. M. Flocke, Evaluation of space-based constraints on global nitrogen oxide emissions with regional aircraft measurements over and downwind of eastern North America, J. Geophys. Res., [Atmos.], 2006, 111, D15308 CrossRef.
- D. T. Shindell, J. F. Lamarque, M. Schulz, M. Flanner, C. Jiao, M. Chin, P. J. Young, Y. H. Lee, L. Rotstayn, N. Mahowald, G. Milly, G. Faluvegi, Y. Balkanski, W. J. Collins, A. J. Conley, S. Dalsoren, R. Easter, S. Ghan, L. Horowitz, X. Liu, G. Myhre, T. Nagashima, V. Naik, S. T. Rumbold, R. Skeie, K. Sudo, S. Szopa, T. Takemura, A. Voulgarakis, J. H. Yoon and F. Lo, Radiative forcing in the ACCMIP historical and future climate simulations, Atmos. Chem. Phys., 2013, 13, 2939–2974 CrossRef CAS.
- V. Naik, A. Voulgarakis, A. M. Fiore, L. W. Horowitz, J. F. Lamarque, M. Lin, M. J. Prather, P. J. Young, D. Bergmann, P. J. Cameron-Smith, I. Cionni, W. J. Collins, S. B. Dalsøren, R. Doherty, V. Eyring, G. Faluvegi, G. A. Folberth, B. Josse, Y. H. Lee, I. A. MacKenzie, T. Nagashima, T. P. C. van Noije, D. A. Plummer, M. Righi, S. T. Rumbold, R. Skeie, D. T. Shindell, D. S. Stevenson, S. Strode, K. Sudo, S. Szopa and G. Zeng, Preindustrial to present day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 2013, 13, 5277–5298 Search PubMed.
- P. J. Young, A. T. Archibald, K. W. Bowman, J. F. Lamarque, V. Naik, D. S. Stevenson, S. Tilmes, A. Voulgarakis, O. Wild, D. Bergmann, P. Cameron-Smith, I. Cionni, W. J. Collins, S. B. Dalsøren, R. M. Doherty, V. Eyring, G. Faluvegi, L. W. Horowitz, B. Josse, Y. H. Lee, I. A. MacKenzie, T. Nagashima, D. A. Plummer, M. Righi, S. T. Rumbold, R. B. Skeie, D. T. Shindell, S. A. Strode, K. Sudo, S. Szopa and G. Zeng, Pre-industrial to end 21st century projections of tropospheric ozone from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 2013, 13, 2063–2090 CAS.
- A. Voulgarakis, V. Naik, J. F. Lamarque, D. T. Shindell, P. J. Young, M. J. Prather, O. Wild, R. D. Field, D. Bergmann, P. Cameron-Smith, I. Cionni, W. J. Collins, S. B. Dalsøren, R. M. Doherty, V. Eyring, G. Faluvegi, G. A. Folberth, L. W. Horowitz, B. Josse, I. A. MacKenzie, T. Nagashima, D. A. Plummer, M. Righi, S. T. Rumbold, D. S. Stevenson, S. A. Strode, K. Sudo, S. Szopa and G. Zeng, Analysis of present day and future OH and methane lifetime in the ACCMIP simulations, Atmos. Chem. Phys., 2013, 13, 2563–2587 Search PubMed.
- A. Hilboll, A. Richter and J. P. Burrows, Long-term changes of tropospheric NO2 over megacities derived from multiple satellite instruments, Atmos. Chem. Phys., 2013, 13, 4145–4169 CAS.
- IPCC, Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report, Intergovernmental Panel on Climate Change Report No., Geneva, 2014.
- UNEP, Environmental effects of ozone depletion and its interactions with climate change: Progress Report 2009, Photochem. Photobiol. Sci., 2010, 9, 275–294 Search PubMed.
- X. Tang, S. R. Wilson, K. R. Solomon, M. Shao and S. Madronich, Changes in tropospheric composition and air quality due to stratospheric ozone depletion and interactions with changes in climate, Photochem. Photobiol. Sci., 2011, 10, 280–291 CAS.
- S. C. Anenberg, J. Schwartz, D. Shindell, M. Amann, G. Faluvegi, Z. Klimont, G. Janssens-Maenhout, L. Pozzoli, R. Van Dingenen, E. Vignati, L. Emberson, N. Z. Muller, J. J. West, M. Williams, V. Demkine, W. K. Hicks, J. Kuylenstierna, F. Raes and V. Ramanathan, Global air quality and health co-benefits of mitigating near-term climate change through methane and black carbon emission controls, Environ. Health Perspect., 2012, 120, 831–839 CrossRef PubMed.
- S. C. Anenberg, L. W. Horowitz, D. Q. Tong and J. J. West, An estimate of the global burden of anthropogenic ozone and fine particulate matter on premature human mortality using atmospheric modeling, Environ. Health Perspect., 2010, 118, 1189–1195 CrossRef CAS PubMed.
- S. C. Anenberg, J. J. West, L. W. Horowitz and D. Q. Tong, The global burden of air pollution on mortality: Anenberg et al. respond, Environ. Health Perspect., 2011, 119, A158–A159 CrossRef.
- S. P. de Almeida, E. Casimiro and J. Calheiros, Short-term association between exposure to ozone and mortality in Oporto, Portugal, Environ. Res., 2011, 111, 406–410 CrossRef PubMed.
- Y. L. Hsieh, Y. H. Yang, T. N. Wu and C. Y. Yang, Air pollution and hospital admissions for myocardial infarction in a subtropical city: Taipei, Taiwan, J. Toxicol. Environ. Health, Part A, 2010, 73, 757–765 CrossRef CAS PubMed.
- I. Hůnová, M. Malý, J. Řezáčová and M. Braniš, Association between ambient ozone and health outcomes in Prague, Int. Arch. Occup. Environ. Hlth., 2012, 86, 89–97 CrossRef PubMed.
- S. S. Lim, T. Vos, A. D. Flaxman, G. Danaei, K. Shibuya, H. Adair-Rohani, M. A. AlMazroa, M. Amann, H. R. Anderson, K. G. Andrews, M. Aryee, C. Atkinson, L. J. Bacchus, A. N. Bahalim, K. Balakrishnan, J. Balmes, S. Barker-Collo, A. Baxter, M. L. Bell, J. D. Blore, F. Blyth, C. Bonner, G. Borges, R. Bourne, M. Boussinesq, M. Brauer, P. Brooks, N. G. Bruce, B. Brunekreef, C. Bryan-Hancock, C. Bucello, R. Buchbinder, F. Bull, R. T. Burnett, T. E. Byers, B. Calabria, J. Carapetis, E. Carnahan, Z. Chafe, F. Charlson, H. Chen, J. S. Chen, A. T.-A. Cheng, J. C. Child, A. Cohen, K. E. Colson, B. C. Cowie, S. Darby, S. Darling, A. Davis, L. Degenhardt, F. Dentener, D. C. Des Jarlais, K. Devries, M. Dherani, E. L. Ding, E. R. Dorsey, T. Driscoll, K. Edmond, S. E. Ali, R. E. Engell, P. J. Erwin, S. Fahimi, G. Falder, F. Farzadfar, A. Ferrari, M. M. Finucane, S. Flaxman, F. G. R. Fowkes, G. Freedman, M. K. Freeman, E. Gakidou, S. Ghosh, E. Giovannucci, G. Gmel, K. Graham, R. Grainger, B. Grant, D. Gunnell, H. R. Gutierrez, W. Hall, H. W. Hoek, A. Hogan, H. D. Hosgood Iii, D. Hoy, H. Hu, B. J. Hubbell, S. J. Hutchings, S. E. Ibeanusi, G. L. Jacklyn, R. Jasrasaria, J. B. Jonas, H. Kan, J. A. Kanis, N. Kassebaum, N. Kawakami, Y.-H. Khang, S. Khatibzadeh, J.-P. Khoo and C. Kok, et al., A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010, Lancet, 2012, 380, 2224–2260 CrossRef.
- OECD, OECD Environmental Outlook to 2050: The Consequences of Inaction: Key Findings on Health and Environment, Organization for Economic Cooperation and Development Report No., Paris, 2012. http://www.oecd.org/environment/outlookto2050.
- Y. Fang, V. Naik, L. W. Horowitz and D. L. Mauzerall, Air pollution and associated human mortality: the role of air pollutant emissions, climate change and methane concentration increases from the preindustrial period to present, Atmos. Chem. Phys., 2013, 13, 1377–1394 CAS.
- N. Fann, A. D. Lamson, S. C. Anenberg, K. Wesson, D. Risley and B. J. Hubbell, Estimating the national public health burden associated with exposure to ambient PM2.5 and ozone, Risk Anal., 2012, 32, 81–95 CrossRef PubMed.
- J. T. Lee, Y. S. Cho and J. Y. Son, Relationship between ambient ozone concentrations and daily hospital admissions for childhood asthma/atopic dermatitis in two cities of Korea during 2004-2005, Int. J. Environ. Health Res., 2010, 20, 1–11 CrossRef CAS PubMed.
- G. G. Kaplan, D. Tanyingoh, E. Dixon, M. Johnson, A. J. Wheeler, R. P. Myers, S. Bertazzon, V. Saini, K. Madsen, S. Ghosh and P. J. Villeneuve, Ambient ozone concentrations and the risk of perforated and nonperforated appendicitis: a multicity case-crossover study, Environ. Health Perspect., 2013, 121, 939–943 Search PubMed.
- R. E. Dales, S. Cakmak and C. B. Vidal, Air pollution and hospitalization for venous thromboembolic disease in Chile, J. Thromb. Haemost., 2010, 8, 669–674 CrossRef CAS PubMed.
- C. M. Wong, T. Q. Thach, P. Y. Chau, E. K. Chan, R. Y. Chung, C. Q. Ou, L. Yang, J. S. Peiris, G. N. Thomas, T. H. Lam, T. W. Wong and A. J. Hedley, Part 4. Interaction between air pollution and respiratory viruses: time-series study of daily mortality and hospital admissions in Hong Kong, Research Report, 2010, 283–362 Search PubMed.
- H. Lei, D. J. Wuebbles and X.-Z. Liang, Projected risk of high ozone episodes in 2050, Atmos. Environ., 2012, 59, 567–577 CrossRef CAS.
- Royal Society, Ground-level ozone in the 21st century:future trends, impacts and policy implications, The Royal Society Report No. RS Policy document 15/08, London, UK, October 2008 2008, p. 148. http://royalsociety.org/policy/publications/2008/ground-level-ozone/.
- S. Wilkinson, G. Mills, R. Illidge and W. J. Davies, How is ozone pollution reducing our food supply?, J. Exp. Bot., 2012, 63, 527–536 CrossRef CAS PubMed.
- K. W. Jaggard, A. Qi and E. S. Ober, Possible changes to arable crop yields by 2050, Philos. Trans. R. Soc. London, Ser. B, 2010, 365, 2835–2851 CrossRef PubMed.
- C. P. Leisner and E. A. Ainsworth, Quantifying the effects of ozone on plant reproductive growth and development, Glob. Change Biol., 2012, 18, 606–616 CrossRef.
- A. M. Betzelberger, C. R. Yendrek, J. Sun, C. P. Leisner, R. L. Nelson, D. R. Ort and E. A. Ainsworth, Ozone exposure response for U.S. soybean cultivars: linear reductions in photosynthetic potential, biomass, and yield, Plant Physiol., 2012, 160, 1827–1839 CrossRef CAS PubMed.
- S. Fares, R. Vargas, M. Detto, A. H. Goldstein, J. Karlik, E. Paoletti and M. Vitale, Tropospheric ozone reduces carbon assimilation in trees: estimates from analysis of continuous flux measurements, Glob. Change Biol., 2013, 19, 2427–2443 CrossRef PubMed.
- IPCC, Special Report on Emissions Scenarios: A Special Report of Working Group III of the Intergovernmental Panel on Climate Change, Intergovernmental Panel on Climate Change Report No., Geneva, 2000.
- S. Avnery, D. L. Mauzerall, J. Liu and L. W. Horowitz, Global crop yield reductions due to surface ozone exposure: 2. Year 2030 potential crop production losses and economic damage under two scenarios of O3 pollution, Atmos. Environ., 2011, 45, 2297–2309 CrossRef CAS.
- Y. Feng, X. Lin, Y. Yu, H. Zhang, H. Chu and J. Zhu, Elevated ground-level O3 negatively influences paddy methanogenic archaeal community, Sci. Rep., 2013, 3, 3193 Search PubMed.
- A. J. Haagen-Smit, C. E. Bradley and M. M. Fox, Ozone formation in photochemical oxidation of organic substances, Ind. Eng. Chem. Res., 1953, 45, 2086–2089 CrossRef CAS.
- R. L. McKenzie, P. J. Aucamp, A. F. Bais, L. O. Björn, M. Ilyas and S. Madronich, Ozone depletion and climate change: Impacts on UV radiation, Photochem. Photobiol. Sci., 2011, 10, 182–198 CAS.
- A. Colette, C. Granier, Ø. Hodnebrog, H. Jakobs, A. Maurizi, A. Nyiri, B. Bessagnet, A. D'Angiola, M. D'Isidoro, M. Gauss, F. Meleux, M. Memmesheimer, A. Mieville, L. Rouïl, F. Russo, S. Solberg, F. Stordal and F. Tampieri, Air quality trends in Europe over the past decade: a first multi-model assessment, Atmos. Chem. Phys., 2011, 11, 11657–11678 CrossRef CAS.
- R. C. Wilson, Z. L. Fleming, P. S. Monks, G. Clain, S. Henne, I. B. Konovalov, S. Szopa and L. Menut, Have primary emission reduction measures reduced ozone across Europe? An analysis of European rural background ozone trends 1996–2005, Atmos. Chem. Phys., 2012, 12, 437–454 CAS.
- T. Zhu, M. Melamed, D. Parrish, M. Gauss, L. Gallardo, M. Lawrence, A. Knare and C. Liousse, WMO/IGAC Impacts of Megacities on Air Pollution and Climate, World Meteorological Organization Report No. World Meteorological Organization (WMO) Global Atmosphere Watch (GAW) Report No. 205, Geneva, January 2013, http://www.wmo.int/pages/prog/arep/gaw/gaw-reports.html.
- Q. Zhang, B. Yuan, M. Shao, X. Wang, S. Lu, K. Lu, M. Wang, L. Chen, C. C. Chang and S. C. Liu, Variations of ground-level O3 and its precursors in Beijing in summertime between 2005 and 2011, Atmos. Chem. Phys., 2014, 14, 6089–6101 CAS.
- Y. Wang, P. Konopka, Y. Liu, H. Chen, R. Müller, F. Plöger, M. Riese, Z. Cai and D. Lü, Tropospheric ozone trend over Beijing from 2002–2010: ozonesonde measurements and modeling analysis, Atmos. Chem. Phys., 2012, 12, 8389–8399 CrossRef CAS.
- D. M. Lal, S. D. Ghude, S. D. Patil, S. H. Kulkarni, C. Jena, S. Tiwari and M. K. Srivastava, Tropospheric ozone and aerosol long-term trends over the Indo-Gangetic Plain (IGP), India, Atmos. Res., 2012, 116, 82–92 CrossRef CAS.
- T. Wang, W. Nie, J. Gao, L. K. Xue, X. M. Gao, X. F. Wang, J. Qiu, C. N. Poon, S. Meinardi, D. Blake, S. L. Wang, A. J. Ding, F. H. Chai, Q. Z. Zhang and W. X. Wang, Air quality during the 2008 Beijing Olympics: secondary pollutants and regional impact, Atmos. Chem. Phys., 2010, 10, 7603–7615 CrossRef CAS.
- R. G. Derwent, A. J. Manning, P. G. Simmonds, T. G. Spain and S. O'Doherty, Analysis and interpretation of 25 years of ozone observations at the Mace Head Atmospheric Research Station on the Atlantic Ocean coast of Ireland from 1987 to 2012, Atmos. Environ., 2013, 80, 361–368 CrossRef CAS.
- O. R. Cooper, D. D. Parrish, A. Stohl, M. Trainer, P. Nedelec, V. Thouret, J. P. Cammas, S. J. Oltmans, B. J. Johnson, D. Tarasick, T. Leblanc, I. S. McDermid, D. Jaffe, R. Gao, J. Stith, T. Ryerson, K. Aikin, T. Campos, A. Weinheimer and M. A. Avery, Increasing springtime ozone mixing ratios in the free troposphere over western North America, Nature, 2010, 463, 344–348 CrossRef CAS PubMed.
- D. D. Parrish, K. S. Law, J. Staehelin, R. Derwent, O. R. Cooper, H. Tanimoto, A. Volz-Thomas, S. Gilge, H. E. Scheel, M. Steinbacher and E. Chan, Lower tropospheric ozone at northern midlatitudes: Changing seasonal cycle, Geophys. Res. Lett., 2013, 40, 1631–1636 CrossRef CAS.
- S. J. Oltmans, A. S. Lefohn, D. Shadwick, J. M. Harris, H. E. Scheel, I. Galbally, D. W. Tarasick, B. J. Johnson, E. G. Brunke, H. Claude, G. Zeng, S. Nichol, F. Schmidlin, J. Davies, E. Cuevas, A. Redondas, H. Naoe, T. Nakano and T. Kawasato, Recent tropospheric ozone changes – A pattern dominated by slow or no growth, Atmos. Environ., 2013, 67, 331–351 CrossRef CAS.
- P. G. Hess and R. Zbinden, Stratospheric impact on tropospheric ozone variability and trends: 1990-2009, Atmos. Chem. Phys., 2013, 13, 649–674 CAS.
- M. Lin, L. W. Horowitz, S. J. Oltmans, A. M. Fiore and S. Fan, Tropospheric ozone trends at Mauna Loa Observatory tied to decadal climate variability, Nat. Geosci., 2014, 7, 136–143 CrossRef CAS.
- D. S. Stevenson, P. J. Young, V. Naik, J. F. Lamarque, D. T. Shindell, A. Voulgarakis, R. B. Skeie, S. B. Dalsoren, G. Myhre, T. K. Berntsen, G. A. Folberth, S. T. Rumbold, W. J. Collins, I. A. MacKenzie, R. M. Doherty, G. Zeng, T. P. C. van Noije, A. Strunk, D. Bergmann, P. Cameron-Smith, D. A. Plummer, S. A. Strode, L. Horowitz, Y. H. Lee, S. Szopa, K. Sudo, T. Nagashima, B. Josse, I. Cionni, M. Righi, V. Eyring, A. Conley, K. W. Bowman, O. Wild and A. Archibald, Tropospheric ozone changes, radiative forcing and attribution to emissions in the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 2013, 13, 3063–3085 CAS.
- G. Zeng, O. Morgenstern, P. Braesicke and J. A. Pyle, Impact of stratospheric ozone recovery on tropospheric ozone and its budget, Geophys. Res. Lett., 2010, 37 Search PubMed.
- H. Zhang, S. Wu, Y. Huang and Y. Wang, Effects of stratospheric ozone recovery on photochemistry and ozone air quality in the troposphere, Atmos. Chem. Phys., 2014, 14, 4079–4089 CrossRef.
- S. C. Liu and M. Trainer, Responses of the tropospheric ozone and odd hydrogen radicals to column ozone change, J. Atmos. Chem., 1988, 6, 221–233 CrossRef CAS.
- A. M. Thompson, R. W. Stewart, M. A. Owens and J. A. Herwehe, Sensitivity of tropospheric oxidants to global chemical and climate change, Atmos. Environ., 1989, 23, 519–532 CrossRef CAS.
- D. D. Parrish, J. F. Lamarque, V. Naik, L. Horowitz, D. T. Shindell, J. Staehelin, R. Derwent, O. R. Cooper, H. Tanimoto, A. Volz-Thomas, S. Gilge, H. E. Scheel, M. Steinbacher and M. Fröhlich, Long-term changes in lower tropospheric baseline ozone concentrations: Comparing chemistry-climate models and observations at northern mid-latitudes, J. Geophys. Res., [Atmos.], 2014, 2013JD021435 Search PubMed.
- USEPA, Integrated Science Assessment for Particulate Matter, U.S. Environmental Protection Agency, Office of Research and Development Report No. EPA/600/R-08/139F, Research Triangle Park, NC, 2010, p. 2228.
- Y. Chen, A. Ebenstein, M. Greenstone and H. Li, Evidence on the impact of sustained exposure to air pollution on life expectancy from China's Huai River policy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12936–12941 CrossRef CAS PubMed.
- A. van Donkelaar, R. Martin, R. C. Levy, A. M. da Silva, M. Krzyzanowski, N. E. Chubarova, E. Semutnikova and A. J. Cohen, Satellite-based estimates of ground-level fine particulate matter during extreme events: A case study of the Moscow fires, Atmos. Environ., 2011, 45, 6225–6232 CrossRef CAS.
- A. W. Correia, C. A. Pope, 3rd, D. W. Dockery, Y. Wang, M. Ezzati and F. Dominici, Effect of air pollution control on life expectancy in the United States: an analysis of 545 U.S. counties for the period from 2000 to 2007, Epidemiol., 2013, 24, 23–31 CrossRef PubMed.
- J. T. Spickett, H. L. Brown and K. Rumchev, Climate change and air quality: the potential impact on health, Asia. Pac. J. Public. Health., 2011, 23, 37S–345 CrossRef PubMed.
- E. S. Post, A. Grambsch, C. Weaver, P. Morefield, J. Huang, L. Y. Leung, C. G. Nolte, P. Adams, X. Z. Liang, J. H. Zhu and H. Mahoney, Variation in estimated ozone-related health Impacts of climate change due to modeling choices and assumptions, Environ. Health Perspect., 2012, 120, 1559–1564 CrossRef PubMed.
- R. L. Prueitt and R. L. Goodman, The global burden of ozone on respiratory mortality: No clear evidence for association, Environ. Health Perspect., 2011, 119, A158 CrossRef PubMed.
- A. F. Bais, R. L. McKenzie, P. J. Aucamp, M. Ilyas, S. Madronich, G. Bernhard and K. Tourpali, Ozone depletion and climate change: Impacts on UV radiation, Photochem. Photobiol. Sci., 2014, 14 10.1039/c1034pp90032d.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Hoboken, NJ, USA, 2006 Search PubMed.
- X. Bi, B. R. Simoneit, G. Sheng, S. Ma and J. Fu, Composition and major sources of organic compounds in urban aerosols, Atmos. Res., 2008, 88, 256–265 CrossRef CAS.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. E. Cubison, J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of organic aerosols in the atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- N. M. Donahue, K. M. Henry, T. F. Mentel, A. Kiendler-Scharr, C. Spindler, B. Bohn, T. Brauers, H. P. Dorn, H. Fuchs, R. Tillmann, A. Wahner, H. Saathoff, K.-H. Naumann, O. Möhler, T. Leisner, L. Müller, M.-C. Reinnig, T. Hoffmann, K. Salo, M. Hallquist, M. Frosch, M. Bilde, T. Tritscher, P. Barmet, A. P. Praplan, P. F. DeCarlo, J. Dommen, A. S. H. Prévôt and U. Baltensperger, Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13503–13508 CrossRef CAS PubMed.
- A. Hodzic, S. Madronich, B. Aumont, J. Lee-Taylor, T. Karl, M. Camredon and C. Mouchel-Vallon, Limited influence of dry deposition of semivolatile organic vapors on secondary organic aerosol formation in the urban plume, Geophys. Res. Lett., 2013, 40, 3302–3307 CrossRef CAS.
- J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. H. Kessler, M. R. Canagaratna, K. R. Wilson, K. E. Altieri, L. R. Mazzoleni, A. S. Wozniak, H. Bluhm, E. R. Mysak, J. D. Smith, C. E. Kolb and D. R. Worsnop, Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 2011, 3, 133–139 CrossRef CAS PubMed.
- C. L. Heald, J. H. Kroll, J. L. Jimenez, K. S. Docherty, P. F. DeCarlo, A. C. Aiken, Q. Chen, S. T. Martin, D. K. Farmer and P. Artaxo, A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 2010, 37, L08803 CrossRef.
- C. D. Cappa and K. R. Wilson, Multi-generation gas-phase oxidation, equilibrium partitioning, and the formation and evolution of secondary organic aerosol, Atmos. Chem. Phys., 2012, 12, 9505–9528 CAS.
- J. F. Pankow and K. C. Barsanti, The carbon number-polarity grid: A means to manage the complexity of the mix of organic compounds when modeling atmospheric organic particulate matter, Atmos. Environ., 2009, 43, 2829–2835 CrossRef CAS.
- B. Croft, J. R. Pierce, R. V. Martin, C. Hoose and U. Lohmann, Uncertainty associated with convective wet removal of entrained aerosols in a global climate model, Atmos. Chem. Phys., 2012, 12, 10725–10748 CrossRef CAS.
- B. Croft, J. R. Pierce and R. V. Martin, Interpreting aerosol lifetimes using the GEOS-Chem model and constraints from radionuclide measurements, Atmos. Chem. Phys., 2014, 14, 4313–4325 CrossRef.
- N. I. Kristiansen, A. Stohl and G. Wotawa, Atmospheric removal times of the aerosol-bound radionuclides 137Cs and 131I measured after the Fukushima Dai-ichi nuclear accident - a constraint for air quality and climate models, Atmos. Chem. Phys., 2012, 12, 10759–10769 CrossRef CAS.
- R. Zalakeviciute, M. L. Alexander, E. Allwine, J. L. Jimenez, B. T. Jobson, L. T. Molina, E. Nemitz, S. N. Pressley, T. M. VanReken, I. M. Ulbrich, E. Velasco and B. K. Lamb, Chemically-resolved aerosol eddy covariance flux measurements in urban Mexico City during MILAGRO 2006, Atmos. Chem. Phys., 2012, 12, 7809–7823 CAS.
- J. L. Hand, B. A. Schichtel, M. Pitchford, W. C. Malm and N. H. Frank, Seasonal composition of remote and urban fine particulate matter in the United States, J. Geophys. Res., [Atmos.], 2012, 117, D05209 Search PubMed.
- M. Amann, Z. Klimont and F. Wagner, Regional and Global Emissions of Air Pollutants: Recent Trends and Future Scenarios, Annu. Rev. Environ. Resources, 2013, 38, 31–55 CrossRef.
- S. J. Smith and T. C. Bond, Two hundred fifty years of aerosols and climate: the end of the age of aerosols, Atmos. Chem. Phys., 2014, 14, 537–549 CAS.
- D. E. Heard and M. J. Pilling, Measurement of OH and HO2 in the troposphere, Chem. Rev., 2003, 103, 5163–5198 CrossRef CAS PubMed.
- D. Stone, L. K. Whalley and D. E. Heard, Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 2012, 41, 6348–6404 RSC.
- X. Ren, W. H. Brune, J. Mao, M. J. Mitchell, R. L. Lesher, J. B. Simpas, A. R. Metcalf, J. J. Schwab, C. Cai and Y. Li, Behavior of OH and HO2 in the winter atmosphere in New York City, Atmos. Environ., 2006, 40, 252–263 CrossRef.
- Y. Kanaya, R. Cao, H. Akimoto, M. Fukuda, Y. Komazaki, Y. Yokouchi, M. Koike, H. Tanimoto, N. Takegawa and Y. Kondo, Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004, J. Geophys. Res., [Atmos.], 2007, 112, DOI:10.1029/2007JD008670.
- S. Dusanter, D. Vimal, P. S. Stevens, R. Volkamer, L. T. Molina, A. Baker, S. Meinardi, D. Blake, P. Sheehy, A. Merten, R. Zhang, J. Zheng, E. C. Fortner, W. Junkermann, M. Dubey, T. Rahn, B. Eichinger, P. Lewandowski, J. Prueger and H. Holder, Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign – Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 2009, 9, 6655–6675 CAS.
- G. Li, W. Lei, M. Zavala, R. Volkamer, S. Dusanter, P. Stevens and L. T. Molina, Impacts of HONO sources on the photochemistry in Mexico City during the MCMA-2006/MILAGO Campaign, Atmos. Chem. Phys., 2010, 10, 6551–6567 CrossRef CAS.
- S. Chen, X. Ren, J. Mao, Z. Chen, W. H. Brune, B. Lefer, B. Rappenglück, J. Flynn, J. Olson and J. H. Crawford, A comparison of chemical mechanisms based on TRAMP-2006 field data, Atmos. Environ., 2010, 44, 4116–4125 CrossRef CAS.
- D. Stone, M. J. Evans, R. Commane, T. Ingham, C. F. A. Floquet, J. B. McQuaid, D. M. Brookes, P. S. Monks, R. Purvis, J. F. Hamilton, J. Hopkins, J. Lee, A. C. Lewis, D. Stewart, J. G. Murphy, G. Mills, D. Oram, C. E. Reeves and D. E. Heard, HOx observations over West Africa during AMMA: Impact of isoprene and NOx, Atmos. Chem. Phys., 2010, 10, 9415–9429 CrossRef CAS.
- S. M. Griffith, R. F. Hansen, S. Dusanter, P. S. Stevens, M. Alaghmand, S. B. Bertman, M. A. Carroll, M. Erickson, M. Galloway, N. Grossberg, J. Hottle, J. Hou, B. T. Jobson, A. Kammrath, F. N. Keutsch, B. L. Lefer, L. H. Mielke, A. O'Brien, P. B. Shepson, M. Thurlow, W. Wallace, N. Zhang and X. L. Zhou, OH and HO2 radical chemistry during PROPHET 2008 and CABINEX 2009 – Part 1: Measurements and model comparison, Atmos. Chem. Phys., 2013, 13, 5403–5423 Search PubMed.
- D. Kubistin, H. Harder, M. Martinez, M. Rudolf, R. Sander, H. Bozem, G. Eerdekens, H. Fischer, C. Gurk, T. Klüpfel, R. Königstedt, U. Parchatka, C. L. Schiller, A. Stickler, D. Taraborrelli, J. Williams and J. Lelieveld, Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 2010, 10, 9705–9728 CAS.
- J. Lelieveld, T. M. Butler, J. N. Crowley, T. J. Dillon, H. Fischer, L. Ganzeveld, H. Harder, M. G. Lawrence, M. Martinez, D. Taraborrelli and J. Williams, Atmospheric oxidation capacity sustained by a tropical forest, Nature, 2008, 452, 737–740 CrossRef CAS PubMed.
- M. Martinez, H. Harder, D. Kubistin, M. Rudolf, H. Bozem, G. Eerdekens, H. Fischer, T. Klüpfel, C. Gurk, R. Königstedt, U. Parchatka, C. L. Schiller, A. Stickler, J. Williams and J. Lelieveld, Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: airborne measurements, Atmos. Chem. Phys., 2010, 10, 3759–3773 CrossRef CAS.
- T. A. M. Pugh, A. R. MacKenzie, C. N. Hewitt, B. Langford, P. M. Edwards, K. L. Furneaux, D. E. Heard, J. R. Hopkins, C. E. Jones, A. Karunaharan, J. Lee, G. Mills, P. Misztal, S. Moller, P. S. Monks and L. K. Whalley, Simulating atmospheric composition over a South-East Asian tropical rainforest: performance of a chemistry box model, Atmos. Chem. Phys., 2010, 10, 279–298 CrossRef CAS.
- L. K. Whalley, P. M. Edwards, K. L. Furneaux, A. Goddard, T. Ingham, M. J. Evans, D. Stone, J. R. Hopkins, C. E. Jones, A. Karunaharan, J. D. Lee, A. C. Lewis, P. S. Monks, S. J. Moller and D. E. Heard, Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 2011, 11, 7223–7233 CrossRef CAS.
- A. Hofzumahaus, F. Rohrer, K. Lu, B. Bohn, T. Brauers, C. C. Chang, H. Fuchs, F. Holland, K. Kita, Y. Kondo, X. Li, S. Lou, M. Shao, L. Zeng, A. Wahner and Y. Zhang, Amplified trace gas removal in the troposphere, Science, 2009, 324, 1702–1704 CrossRef CAS PubMed.
- K. D. Lu, F. Rohrer, F. Holland, H. Fuchs, B. Bohn, T. Brauers, C. C. Chang, R. Häseler, M. Hu, K. Kita, Y. Kondo, X. Li, S. R. Lou, S. Nehr, M. Shao, L. M. Zeng, A. Wahner, Y. H. Zhang and A. Hofzumahaus, Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 2012, 12, 1541–1569 CAS.
- K. D. Lu, A. Hofzumahaus, F. Holland, B. Bohn, T. Brauers, H. Fuchs, M. Hu, R. Häseler, K. Kita, Y. Kondo, X. Li, S. R. Lou, A. Oebel, M. Shao, L. M. Zeng, A. Wahner, T. Zhu, Y. H. Zhang and F. Rohrer, Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006, Atmos. Chem. Phys., 2013, 13, 1057–1080 CAS.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, J. H. Kroll, J. H. Seinfeld and P. O. Wennberg, Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 2009, 9, 1479–1501 CrossRef CAS.
- J. Peeters and J. F. Műller, HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: experimental evidence and global impact, Phys. Chem. Chem. Phys., 2010, 12, 14227–14235 RSC.
- H. Fuchs, A. Hofzumahaus, F. Rohrer, B. Bohn, T. Brauers, H. P. Dorn, R. Haseler, F. Holland, M. Kaminski, X. Li, K. Lu, S. Nehr, R. Tillmann, R. Wegener and A. Wahner, Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 2013, 6, 1023–1026 CrossRef CAS.
- H. Fuchs, H. P. Dorn, M. Bachner, B. Bohn, T. Brauers, S. Gomm, A. Hofzumahaus, F. Holland, S. Nehr, F. Rohrer, R. Tillmann and A. Wahner, Comparison of OH concentration measurements by DOAS and LIF during SAPHIR chamber experiments at high OH reactivity and low NO concentration, Atmos. Measure. Technol., 2012, 5, 1611–1626 CrossRef CAS.
- X. Ren, J. Mao, W. H. Brune, C. A. Cantrell, R. L. Mauldin Iii, R. S. Hornbrook, E. Kosciuch, J. R. Olson, J. H. Crawford, G. Chen and H. B. Singh, Airborne intercomparison of HOx measurements using laser-induced fluorescence and chemical ionization mass spectrometry during ARCTAS, Atmos. Measure. Technol., 2012, 5, 2025–2037 CAS.
- E. Schlosser, T. Brauers, H. P. Dorn, H. Fuchs, R. Häseler, A. Hofzumahaus, F. Holland, A. Wahner, Y. Kanaya, Y. Kajii, K. Miyamoto, S. Nishida, K. Watanabe, A. Yoshino, D. Kubistin, M. Martinez, M. Rudolf, H. Harder, H. Berresheim, T. Elste, C. Plass-Dülmer, G. Stange and U. Schurath, Technical Note: Formal blind intercomparison of OH measurements: results from the international campaign HOxComp, Atmos. Chem. Phys., 2009, 9, 7923–7948 CrossRef CAS.
- J. Mao, X. Ren, L. Zhang, D. M. Van Duin, R. C. Cohen, J. H. Park, A. H. Goldstein, F. Paulot, M. R. Beaver, J. D. Crounse, P. O. Wennberg, J. P. DiGangi, S. B. Henry, F. N. Keutsch, C. Park, G. W. Schade, G. M. Wolfe, J. A. Thornton and W. H. Brune, Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 2012, 12, 8009–8020 CAS.
- A. Yoshino, Y. Nakashima, K. Miyazaki, S. Kato, J. Suthawaree, N. Shimo, S. Matsunaga, S. Chatani, E. Apel, J. Greenberg, A. Guenther, H. Ueno, H. Sasaki, J.-y. Hoshi, H. Yokota, K. Ishii and Y. Kajii, Air quality diagnosis from comprehensive observations of total OH reactivity and reactive trace species in urban central Tokyo, Atmos. Environ., 2012, 49, 51–59 CrossRef CAS.
- Y. Nakashima, S. Kato, J. Greenberg, P. Harley, T. Karl, A. Turnipseed, E. Apel, A. Guenther, J. Smith and Y. Kajii, Total OH reactivity measurements in ambient air in a southern Rocky mountain ponderosa pine forest during BEACHON-SRM08 summer campaign, Atmos. Environ., 2014, 85, 1–8 CrossRef CAS.
- S. Lou, F. Holland, F. Rohrer, K. Lu, B. Bohn, T. Brauers, C. C. Chang, H. Fuchs, R. Häseler, K. Kita, Y. Kondo, X. Li, M. Shao, L. Zeng, A. Wahner, Y. Zhang, W. Wang and A. Hofzumahaus, Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 2010, 10, 11243–11260 CrossRef CAS.
- A. C. Nölscher, J. Williams, V. Sinha, T. Custer, W. Song, A. M. Johnson, R. Axinte, H. Bozem, H. Fischer, N. Pouvesle, G. Phillips, J. N. Crowley, P. Rantala, J. Rinne, M. Kulmala, D. Gonzales, J. Valverde-Canossa, A. Vogel, T. Hoffmann, H. G. Ouwersloot, J. Vilà-Guerau de Arellano and J. Lelieveld, Summertime total OH reactivity measurements from boreal forest during HUMPPA-COPEC 2010, Atmos. Chem. Phys., 2012, 12, 8257–8270 Search PubMed.
- V. Sinha, J. Williams, J. Lelieveld, T. M. Ruuskanen, M. K. Kajos, J. Patokoski, H. Hellen, H. Hakola, D. Mogensen, M. Boy, J. Rinne and M. Kulmala, OH reactivity neasurements within a boreal forest: Evidence for unknown reactive emissions, Environ. Sci. Technol., 2010, 44, 6614–6620 CrossRef CAS PubMed.
- H. Berresheim, J. McGrath, M. Adam, R. L. Mauldin, B. Bohn and F. Rohrer, Seasonal measurements of OH: NOx, and J(O1D) at Mace Head, Ireland, Geophys. Res. Lett., 2013, 40, 1659–1663 CrossRef CAS.
- T. Brauers, M. Hausmann, A. Bister, A. Kraus and H.-P. Dorn, OH radicals in the boundary layer of the Atlantic Ocean: 1. Measurements by long-path laser absorption spectroscopy, J. Geophys. Res., [Atmos.], 2001, 106, 7399 CrossRef CAS.
- F. Rohrer and H. Berresheim, Strong correlation between levels of tropospheric hydroxyl radicals and solar ultraviolet radiation, Nature, 2006, 442, 184–187 CrossRef CAS PubMed.
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Non-CO2 greenhouse gases and climate change, Nature, 2011, 476, 43–50 CrossRef CAS PubMed.
- G. Monteil, S. Houweling, E. J. Dlugockenky, G. Maenhout, B. H. Vaughn, J. W. C. White and T. Rockmann, Interpreting methane variations in the past two decades using measurements of CH4 mixing ratio and isotopic composition, Atmos. Chem. Phys., 2011, 11, 9141–9153 CrossRef CAS.
- R. C. Pike and P. J. Young, How plants can influence tropospheric chemistry: the role of isoprene emissions from the biosphere, Weather, 2009, 64(12), 332–336 CrossRef.
- E. D. Sofen, B. Alexander and S. A. Kunasek, The impact of anthropogenic emissions on atmospheric sulfate production pathways, oxidants, and ice core Δ17O(SO42−), Atmos. Chem. Phys., 2011, 11, 3565–3578 CrossRef CAS.
- C. D. Holmes, M. J. Prather, O. A. Søvde and G. Myhre, Future methane, hydroxyl, and their uncertainties: key climate and emission parameters for future predictions, Atmos. Chem. Phys., 2013, 13, 285–302 Search PubMed.
- J. G. John, A. M. Fiore, V. Naik, L. W. Horowitz and J. P. Dunne, Climate versus emission drivers of methane lifetime against loss by tropospheric OH from 1860–2100, Atmos. Chem. Phys., 2012, 12, 12021–12036 CrossRef CAS.
- O. Morgenstern, G. Zeng, N. Luke Abraham, P. J. Telford, P. Braesicke, J. A. Pyle, S. C. Hardiman, F. M. O'Connor and C. E. Johnson, Impacts of climate change, ozone recovery, and increasing methane on surface ozone and the tropospheric oxidizing capacity, J. Geophys. Res., [Atmos.], 2013, 118, 1028–1041 CAS.
- Z. H. Beygi, H. Fischer, H. D. Harder, M. Martinez, R. Sander, J. Williams, D. M. Brookes, P. S. Monks, J. Lelieveld and V. P. N. Ca, Oxidation photochemistry in the Southern Atlantic boundary layer: unexpected deviations of photochemical steady state, Atmos. Chem. Phys. Disc., 2011, 11, 8497–8513 CAS.
- R. Sommariva, A. L. Haggerstone, L. J. Carpenter, N. Carslaw, D. J. Creasey, D. E. Heard, J. D. Lee, A. C. Lewis, M. J. Pilling and J. Zádor, OH and HO2 chemistry in clean marine air during SOAPEX-2, Atmos. Chem. Phys., 2004, 4, 839–856 CrossRef CAS.
- H. Lei, D. J. Wuebbles, X.-Z. Liang and S. Olsen, Domestic versus international contributions on 2050 ozone air quality: How much is convertible by regional control?, Atmos. Environ., 2013, 68, 315–325 CrossRef CAS.
- J. L. Neu, T. Flury, G. L. Manney, M. L. Santee, N. J. Livesey and J. Worden, Tropospheric ozone variations governed by changes in stratospheric circulation, Nat. Geosci., 2014, 7, 340–344 CrossRef CAS.
- P. Jimenez-Guerrero, J. J. Gomez-Navarro, R. Baro, R. Lorente, N. Ratola and J. P. Montavez, Is there a common pattern of future gas-phase air pollution in Europe under diverse climate change scenarios?, Clim. Change, 2013, 121, 661–671 CrossRef CAS.
- E. Katragkou, P. Zanis, I. Kioutsioukis, I. Tegoulias, D. Melas, B. C. Krüger and E. Coppola, Future climate change impacts on summer surface ozone from regional climate-air quality simulations over Europe, J. Geophys. Res., [Atmos.], 2011, 116 Search PubMed.
- C. M. Gan, J. Pleim, R. Mathur, C. Hogrefe, C. N. Long, J. Xing, S. Roselle and C. Wei, Assessment of the effect of air pollution controls on trends in shortwave radiation over the United States from 1995 through 2010 from multiple observation networks, Atmos. Chem. Phys., 2014, 14, 1701–1715 CAS.
- A. Colette, B. Bessagnet, R. Vautard, S. Szopa, S. Rao, S. Schucht, Z. Klimont, L. Menut, G. Clain, F. Meleux, G. Curci and L. Rouil, European atmosphere in 2050, a regional air quality and climate perspective under CMIP5 scenarios, Atmos. Chem. Phys., 2013, 13, 7451–7471 CAS.
- M. Demuzere and N. P. M. van Lipzig, A new method to estimate air-quality levels using a synoptic-regression approach. Part II: Future O3 concentrations, Atmos. Environ., 2010, 44, 1356–1366 CrossRef CAS.
- D. J. Jacob and D. A. Winner, Effect of climate change on air quality, Atmos. Environ., 2009, 43, 51–63 CrossRef CAS.
- P. Paasonen, A. Asmi, T. Petäjä, M. K. Kajos, M. Äijälä, H. Junninen, T. Holst, J. P. D. Abbatt, A. Arneth, W. Birmili, H. D. van der Gon, A. Hamed, A. Hoffer, L. Laakso, A. Laaksonen, W. Richard Leaitch, C. Plass-Dülmer, S. C. Pryor, P. Räisänen, E. Swietlicki, A. Wiedensohler, D. R. Worsnop, V.-M. Kerminen and M. Kulmala, Warming-induced increase in aerosol number concentration likely to moderate climate change, Nat. Geosci., 2013, 6, 438–442 CrossRef CAS.
- K. S. Carslaw, O. Boucher, D. V. Spracklen, G. W. Mann, J. G. L. Rae, S. Woodward and M. Kulmala, A review of natural aerosol interactions and feedbacks within the Earth system, Atmos. Chem. Phys., 2010, 10, 1701–1737 CrossRef CAS.
- R. Abrutzky, Health effects of climate and air pollution in Buenos Aires: A first time series analysis, J. Environ. Protect., 2012, 03, 262–271 CrossRef CAS.
- S. Pattenden, B. Armstrong, A. Milojevic, M. R. Heal, Z. Chalabi, R. Doherty, B. Barratt, R. S. Kovats and P. Wilkinson, Ozone, heat and mortality: acute effects in 15 British conurbations, Occ. Environ. Med., 2010, 67, 699–707 CrossRef CAS PubMed.
- C. Ren, M. S. O'Neill, S. K. Park, D. Sparrow, P. Vokonas and J. Schwartz, Ambient temperature, air pollution, and heart rate variability in an aging population, Am. J. Epid., 2011, 173, 1013–1021 CrossRef PubMed.
- P. H. Fischer, B. Brunekreef and E. Lebret, Air pollution related deaths during the 2003 heat wave in the Netherlands, Atmos. Environ., 2004, 38, 1083–1085 CrossRef CAS.
- J. R. Stedman, The predicted number of air pollution related deaths in the UK during the August 2003 heatwave, Atmos. Environ., 2004, 38, 1087–1090 CrossRef CAS.
- G. D'Amato, L. Cecchi, M. D'Amato and G. Liccardi, Urban air pollution and climate change as environmental risk factors of respiratory allergy: an update, J. Investig. Allergol. Clin. Immunol., 2010, 20, 95–102 Search PubMed.
- K. A. Read, A. S. Mahajan, L. J. Carpenter, M. J. Evans, B. V. E. Faria, D. E. Heard, J. R. Hopkins, J. D. Lee, S. J. Moller, A. C. Lewis, L. Mendes, J. B. McQuaid, H. Oetjen, A. Saiz-Lopez, M. J. Pilling and J. M. C. Plane, Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 2008, 453, 1232–1235 CrossRef CAS PubMed.
- J. C. G. Martin, A. S. Mahajan, T. D. Hay, C. Prados-Roman, C. Ordonez, S. M. MacDonald, J. M. C. Plane, M. Sorribas, M. Gil, J. F. P. Mora, M. V. A. Reyes, D. E. Oram, E. Leedham and A. Saiz-Lopez, Iodine chemistry in the eastern Pacific marine boundary layer, J. Geophys. Res., [Atmos.], 2013, 118, 887–904 Search PubMed.
- R. Hossaini, R. Hossaini, M. P. Chipperfield, B. M. Monge-Sanz, N. A. D. Richards, E. Atlas and D. R. Blake, Bromoform and dibromomethane in the tropics: a 3-D model study of chemistry and transport, Atmos. Chem. Phys., 2010, 10, 719–735 CrossRef CAS.
- T. J. Breider, M. P. Chipperfield, N. A. D. Richards, K. S. Carslaw, G. W. Mann and D. V. Spracklen, Impact of BrO on dimethylsulfide in the remote marine boundary layer, Geophys. Res. Lett., 2010, 37 Search PubMed.
- NTP, Toxicology and Carcinogenesis Studies of 1-Bromopropane (CAS No. 106-94-5) in F344/N Rats and B6C3F1 Mice (Inhalation Studies), National Toxicology Program Report No. NTP TR 564, Research Triangle Park, NC, August 2011 2011, p. 195.
- USEPA, Protection of stratospheric ozone: Listing of substitutes for ozone-depleting substances-n-propyl bromide in solvent cleaning, Fed. Regist., 2007, 72, 30142–30167 Search PubMed.
- USEPA, Q and A: 2007 Final and Proposed Regulations for n-Propyl Bromide (nPB), United States Environmental Protection Agency http://www.epa.gov/ozone/snap/solvents/2007nPBRegsQA.html, accessed March 03, 2014.
- S. E. Anderson, A. E. Munson, L. F. Butterworth, D. Germolec, D. L. Morgan, J. A. Roycroft, J. Dill and B. J. Meade, Whole-body inhalation exposure to 1-bromopropane suppresses the IgM response to sheep red blood cells in female B6C3F1 mice and Fisher 344/N rats, Inhal. Toxicol., 2010, 22, 125–132 CrossRef CAS PubMed.
- D. R. Worton, W. T. Sturges, J. Schwander, R. Mulvaney, J. M. Barnola and J. Chappellaz, 20th century trends and budget implications of chloroform and related tri-and dihalomethanes inferred from firn air, Atmos. Chem. Phys., 2006, 6, 2847–2863 CrossRef CAS.
- P. G. Simmonds, R. G. Derwent, A. J. Manning, S. O'Doherty and G. Spain, Natural chloroform emissions from the blanket peat bogs in the vicinity of Mace Head, Ireland over a 14-year period, Atmos. Environ., 2010, 44, 1284–1291 CrossRef CAS.
- M. A. H. Khan, M. E. Whelan and R. C. Rhew, Effects of temperature and soil moisture on methyl halide and chloroform fluxes from drained peatland pasture soils, J. Environ. Monit., 2012, 14, 241–249 RSC.
- X. Xiao, R. G. Prinn, P. J. Fraser, R. F. Weiss, P. G. Simmonds, S. O'Doherty, B. R. Miller, P. K. Salameh, C. M. Harth, P. B. Krummel, A. Golombek, L. W. Porter, J. H. Butler, J. W. Elkins, G. S. Dutton, B. D. Hall, L. P. Steele, R. H. J. Wang and D. M. Cunnold, Atmospheric three-dimensional inverse modeling of regional industrial emissions and global oceanic uptake of carbon tetrachloride, Atmos. Chem. Phys., 2010, 10, 10421–10434 CrossRef CAS.
- Q. Liang, P. A. Newman, J. S. Daniel, S. Reimann, B. D. Hall, G. Dutton and L. J. M. Kuijpers, Constraining the carbon tetrachloride (CCl4) budget using its global trend and inter-hemispheric gradient, Geophys. Res. Lett., 2014, 41, 2014GL060754 Search PubMed.
- G. Malaguarnera, E. Cataudella, M. Giordano, G. Nunnari, G. Chisari and M. Malaguarnera, Toxic hepatitis in occupational exposure to solvents, World J. Gastroenterol., 2012, 18, 2756–2766 CrossRef CAS PubMed.
- A. Tveit, G. M. Rusch, H. Muijser, M. J. van den Hoven and G. M. Hoffman, The acute, genetic, developmental and inhalation toxicology of trans-1-chloro,3,3,3-trifluoropropene (HCFO-1233zd(E)), Drug Chem. Toxicol., 2014, 37, 83–92 CrossRef CAS PubMed.
- L. F. Bonifacio, E. Sousa, P. Naves, M. L. Inacio, J. Henriques, M. Mota, P. Barbosa, M. J. Drinkall and S. Buckley, Efficacy of sulfuryl fluoride against the pinewood nematode, Bursaphelenchus xylophilus (Nematoda: Aphelenchidae), in Pinus pinaster boards, Pest Manag. Sci., 2014, 70, 6–13 CAS.
- K. A. Buckman, J. F. Campbell and B. Subramanyam, Tribolium castaneum (Coleoptera: Tenebrionidae) associated with tice mills: Fumigation efficacy and population rebound, J. Econ. Entomol., 2013, 106, 499–512 CrossRef PubMed.
- A. Cao, M. Guo, D. Yan, L. Mao, Q. Wang, Y. Li, X. Duan and P. Wang, Evaluation of sulfuryl fluoride as a soil fumigant in China, Pest Manag. Sci., 2014, 70, 219–227 CrossRef CAS PubMed.
- J. Susaya, K. H. Kim, Z. H. Shon and R. J. Brown, Demonstration of long-term increases in tropospheric O3 levels: causes and potential impacts, Chemosphere, 2013, 92, 1520–1528 CrossRef CAS PubMed.
- S. R. Wilson, K. R. Solomon and X. Tang, Changes in tropospheric composition and air quality due to stratospheric ozone depletion and climate change, Photochem. Photobiol. Sci., 2007, 6, 301–310 CAS.
- D. R. Mattie, G. M. Hoffman, L. Narayanan, T. R. Sterner, M. J. Wagner and D. E. Dodd, A 13-week nose-only inhalation toxicity study for perfluoro-n-butyl iodide (PFBI) in rats with recommended occupational exposure levels, Inhal. Toxicol., 2010, 22, 847–860 CrossRef CAS PubMed.
- G. M. Rusch, A. Tveit, H. Muijser, M. M. Tegelenbosch-Schouten and G. M. Hoffman, The acute, genetic, developmental and inhalation toxicology of trans-1,3,3,3-tetrafluoropropene (HFO-1234ze), Drug Chem. Toxicol., 2013, 36, 170–180 CrossRef CAS PubMed.
- T. Schmidt, R. Bertermann, G. M. Rusch, G. M. Hoffman and W. Dekant, Biotransformation of 2,3,3,3-tetrafluoropropene (HFO-1234yf) in male, pregnant and non-pregnant female rabbits after single high dose inhalation exposure, Toxicol. Appl. Pharmacol., 2012, 263, 32–38 CrossRef CAS PubMed.
- A. Tveit, G. M. Rusch, H. Muijser and M. M. Tegelenbosch-Schouten, The acute, developmental, genetic and inhalation toxicology of 2,3,3,3-tetrafluoropropene (HFO-1234yf), Drug Chem. Toxicol., 2013, 36, 412–420 CrossRef CAS PubMed.
Footnote |
| † A ppm of X, parts per million, is a mixing ratio of 1 molecule of X per million molecules of air. Similarly, ppb is parts per billion. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |