
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel set of symmetric methylene blue derivatives exhibits effective bacteria photokilling – a structure–response study†
Anita
Gollmer
a,
Ariane
Felgenträger
a,
Wolfgang
Bäumler
a,
Tim
Maisch
a and
Andreas
Späth
*b
aDepartment of Dermatology, University Medical Center Regensburg, Germany
bDepartment of Organic Chemistry, University of Regensburg, Germany. E-mail: andreas.spaeth@chemie.uni-regensburg.de; Tel: +49-941-943-4087
First published on 21st October 2014
Abstract
This study focuses on the structure–response relationship of symmetrically substituted phenothiazinium dyes. Four hydrophilic derivatives with the ability of additional hydrogen bonding (5, 6) or additional electrostatic interaction (3, 4) were synthesized, photophysically characterized and compared to the parent compound methylene blue (MB, 1) and a lipophilic derivative (2) without additional coordination sites. Derivative 5 was most effective against Gram-positive Staphylococcus aureus and Gram-negative Escherichia coli reaching a maximum photodynamic efficacy of >5log10 steps (≥99.999%) of bacteria killing in 10 minutes (5 μM, 30 J cm−2) without inherent dark toxicity after one single treatment with the incoherent light source PDT1200 (λmax = 660 nm, 50 mW cm−2). Interestingly, one derivative with two additional primary positive charges (3) showed selective killing of Escherichia coli (5 μM, 30 J cm−2, 4log10 steps inactivation (≥99.99%)) and no antimicrobial effect on Staphylococcus aureus. This might allow the development of a new generation of photosensitizers with higher antimicrobial efficacy and selectivity for future applications.
Introduction
Multiple drug resistance is one of the upcoming threats of our century.1 This means, the disease-causing microorganism is able to resist different antimicrobials, especially antibiotics, but also antifungal or antiviral drugs.2 Thus many conventional antibacterial strategies fail and there exists an increasing spread of multi-resistant bacteria.3,4 Besides the development of novel antibiotics, other methods for controlling the spread of pathogenic bacteria have been extensively studied.5–8Especially useful are disinfection methods, which minimize selection-pressure, unlike antibiotics. One of the most promising methods is the photodynamic inactivation of bacteria (PIB).4 The bacteria are incubated with per se non-toxic dyes (photosensitizers, PS). After a short time of incubation, the PS can be excited by visible light. This leads to the production of highly reactive oxygen species (ROS) directly at the bacteria during illumination, which oxidatively damage cellular structures such as membranes or DNA and as a result the bacteria are killed.9 Among the ROS generated, it is well acknowledged that singlet oxygen (1O2) plays the major role in these photodynamic reactions.10
Phenothiazinium dyes belong to the most prominent class of such PS due to their absorption in the red region of the visible spectrum (ε > 5 × 104 L mol−1 cm−1, λmax ∼ 660 nm), their low dark toxicity and their attachment and penetration abilities.10,11 MB 1 and other phenothiazinium derivatives have shown promising antimicrobial photodynamic efficacy towards bacteria such as Staphylococcus aureus (S. aureus),12Escherichia coli (E. coli)13,14 and methicillin resistant S. aureus.15 These PS are also effective against funguses such as Candida species,16,17 tropical pathogens18 and viruses,19 and are therefore applied in the antimicrobial field. MB 1 and its known derivatives have shown to achieve a log10-reduction of >5log10 steps (>99.999%) of bacteria at light doses up to 30 J cm−2, using intensities of 125 mW cm−2 in a concentration range of 2 to 10 μM in suspension.20
Phenothiazinium compounds are frequently used for PIB in oral treatments, especially in endodontics. Incubation times and total illumination times differ, but are usually in the minute time scale.21–25 In all these cited studies an antimicrobial efficacy of 5 to 6log10 reduction was achieved. In nearly all the studies covering phenothiazinium compounds their dark toxicity became apparent. The authors observed at least one log10 step of reduction of the number of bacteria without illumination. For clinical application dark toxicity is a critical factor. It is a key factor of each photosensitizer that the photodynamic process can be controlled by light.
The synthesis of MB 1 and other phenothiazinium derivatives was summarized by Wainwright et al.26 Mostly simple substituents like alkyl, alkylaryl or hydroxyalkyl residues were introduced at the auxochromic sites.27–29 Examples of phenothiazine dyes with highly polar substituents or additional coordination sites are rare.30 Only one example carrying multiple positive charges was presented, but no antimicrobial efficacy or selectivity was published.31 Structure–response relationships between these substitution patterns also comparing lipophilic examples are missing.
Gram-negative bacteria like E. coli are more difficult to inactivate by PIB. This is a result of their altered cell wall structure and cellular architecture in comparison to Gram-positive bacteria.32 As there is no need of the PS penetrating the cell membrane for good PIB efficacy,33 a hydrophilic character of the dye is not a disadvantage, as long as it is ensured that most of the PS cannot be washed away of the target organism. Additional electrostatic binding sites or the ability to establish additional hydrogen bonds support stronger and faster attachment to the cell wall of bacteria. In addition, these structural features may also lead to desirable selectively and/or increased efficacy against Gram-negative bacteria. In contrast to also effective but dark toxic lipophilic dye amphiphiles, which penetrate the cell wall and localize intracellular,34 hydrophilic dyes should not penetrate the bacterial wall. Disorders in the membrane or interaction with DNA or RNA are avoided and therefore dark toxicity is on a lower level.
Recently, we reported on phenothiazinium derivatives with one altered substituent on the auxochromic sites (Fig. 2).35 With the ability of additional hydrogen bonding and/or additional cationic charges the derivatives have shown to be highly effective upon illumination against S. aureus and E. coli with up to 7log10 steps without inherent dark toxicities. The additional positive charges in the substituents also proved to be advantageous suppressing aggregation and therefore enabled to expand the therapeutic concentration window. Six membered ring substituents enhanced the photostability of the compounds. In general a singlet oxygen quantum yield (ΦΔ) of 30–40% was determined for compound 7–12.
The scope of published studies only covers lipophilic to moderately hydrophilic phenothiazinium derivatives. Until now, hydrophilic or very hydrophilic derivatives were not covered systematically.
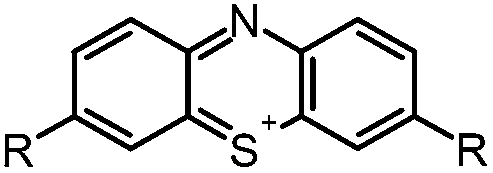
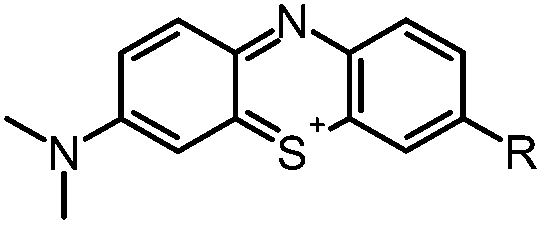
Aim of the present study is to investigate the antibacterial effect of additional positive charges or hydrogen bond acceptors on symmetrically substituted, now hydrophilic phenothiazine derivatives (Fig. 1). More polarity of the molecules might cause the PS to remain outside the cell, suppressing dark toxicity. Positive charges might though lead to better attachment to the exterior of the cell and fast antimicrobial action.33 In addition, improved and selective killing of Gram-negative bacteria may occur. Two substituents in the lead structure MB 1 were changed in order to examine the influence of these substituents on photophysical properties such as ΦΔ, stability and aggregation. Furthermore the new set of symmetrically 3,7-substituted phenothiazinium derivatives is compared with the asymmetrically one-fold substituted derivatives published earlier by our group to establish a structure-relationship of these novel set of compounds.35 Few of the compounds are known as an iodide or bromide salt, but not as a chloride. As iodides can react with singlet oxygen, we consequently investigated the chloride salts of all compounds for the first time.
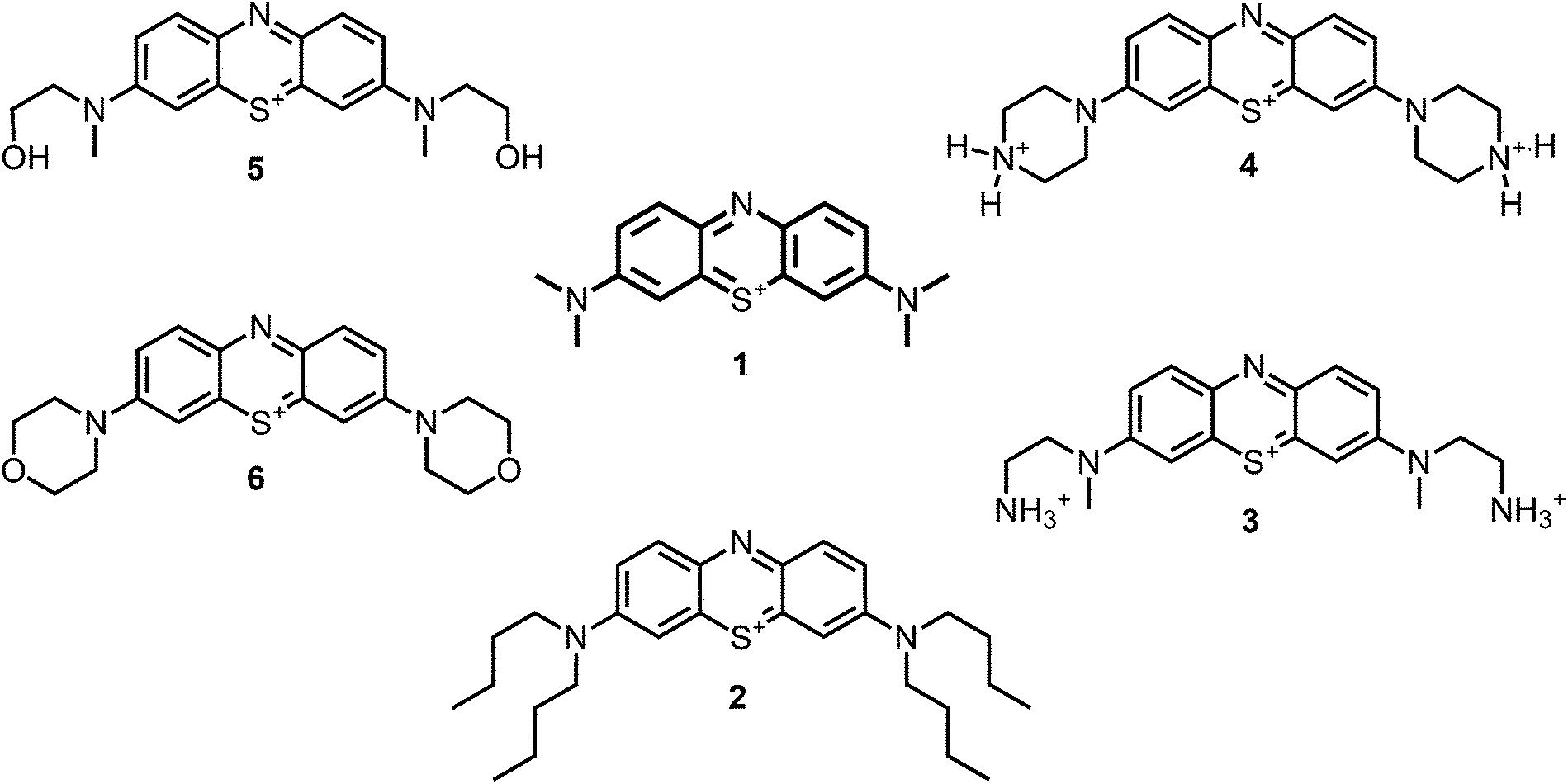 | ||
| Fig. 1 Chemical structures of the compounds investigated (2–6) in comparison to the lead compound MB 1; counterions are chloride in all cases and were avoided for clarity. | ||
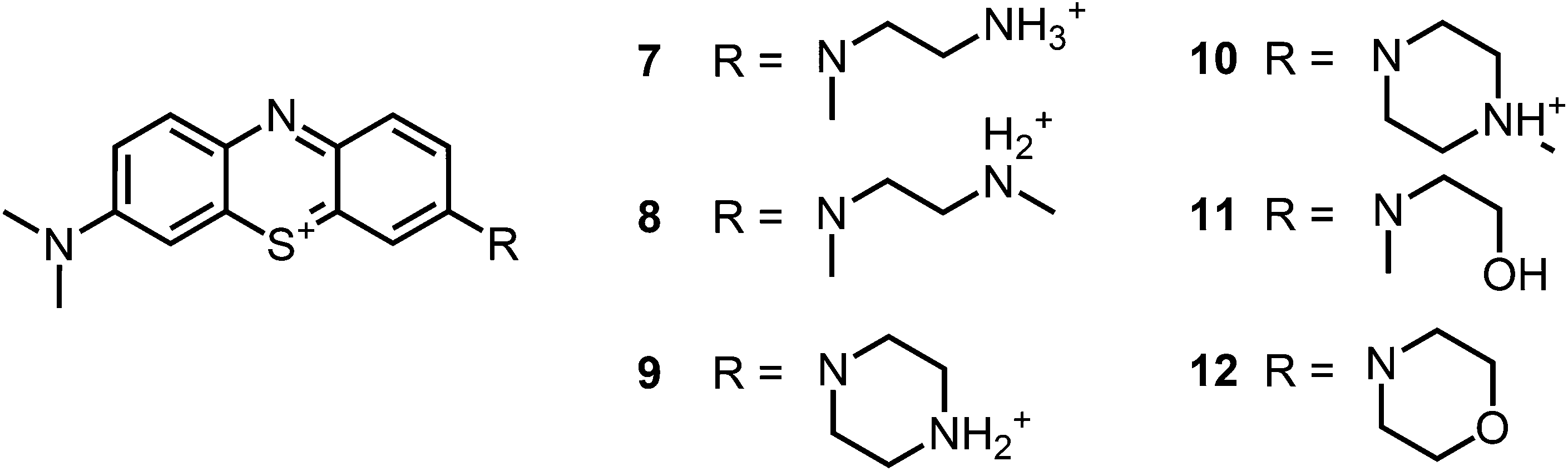 | ||
| Fig. 2 Chemical structures of the analogue compounds (7–12) previously published by our group; counterions are chloride in all cases and were avoided for clarity.35 | ||
Experimental section
General materials and methods
Commercial reagents and starting materials were purchased from Acros Organics, TCI Europe, Fluka, Merck or Sigma-Aldrich and used without further purification. The dry solvents acetone, dichloromethane, dimethylsulfoxide and dimethylformamide were purchased from Roth (RotiDry Sept) or Sigma-Aldrich (puriss., absolute), stored over molecular sieves under nitrogen and were used as received.Thin layer chromatography (TLC) analyses were performed on silica gel 60 F-254 with 0.2 mm layer thickness and detection via UV light at 254 nm/366 nm or through staining with ninhydrin in ethanol. Flash column chromatography was performed on Merck silica gel (Si 60 40–63 μm) either manually or on a Biotage® solera™ flash purification system. Column chromatography was performed on silica gel (70–230 mesh) from Merck. Melting points were measured on a SRS melting point apparatus (MPA100 Opti Melt) and are uncorrected.
NMR spectra were recorded on Bruker Avance 300 (1H 300.13 MHz, 13C 75.47 MHz, T = 300 K), Bruker Avance 400 (1H 400.13 MHz, 13C 100.61 MHz, T = 300 K), Bruker Avance 600 (1H 600.13 MHz, 13C 150.92 MHz, T = 300 K) and Bruker Avance III 600 Kryo (1H 600.25 MHz, 13C 150.95 MHz, T = 300 K) instruments. The chemical shifts are reported in δ[ppm] relative to external standards (solvent residual peak). The spectra were analyzed by first order, the coupling constants J are given in hertz [Hz]. Characterization of the signals: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet, psq = pseudo quintet, dd = double doublet, dt = doublet of triplets, ddd = double double doublet. Integration is determined as the relative number of atoms. Assignment of signals in 13C-spectra was determined with 2D-spectroscopy (COSY, HSQC and HMBC) or DEPT technique (pulse angle: 135°) and given as (+) for CH3 or CH, (−) for CH2 and (Cq) for quaternary Cq. Error of reported values: chemical shift 0.01 ppm (1H NMR) and 0.1 ppm (13C NMR), coupling constant J 0.1 Hz. The solvents used for the measurements are reported for each spectrum.
IR spectra were recorded with a Bio-Rad FT-IR-FTS 155 spectrometer. Fluorescence spectra were recorded on a ‘Cary Eclipse’ fluorescence spectrophotometer and absorption spectra on a “Cary BIO 50” UV/VIS/NIR spectrometer from Varian. All measurements were performed in 1 cm quartz cuvettes (Hellma) and UV-grade solvents (Baker or Merck) at 25 °C.
Mass spectra were recorded on Varian CH-5 (EI), Finnigan MAT95 (EI-, CI- and FAB-MS), Agilent Q-TOF 6540 UHD (ESI-MS, APCI-MS), Finnigan MAT SSQ 710 A (EI-MS, CI-MS) or Thermo Quest Finnigan TSQ 7000 (ES-MS, APCI-MS) spectrometer. Xenon serves as the ionization gas for FAB.
Synthesis and purification of the compounds
MB chloride (1) was purchased from Sigma Aldrich (Germany) and was used without further purification.The purity of all synthesized compounds was determined by NMR spectroscopic methods (Bruker Avance 300, DMSO-d6) and HPLC-MS confirming a purity of >95%. All derivatives for the antimicrobial investigations were supplied in their chloride form. The phenothiazinium derivatives and 5,10,15,20-Tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluenesulfonate) (TMPyP) were dissolved and diluted in Millipore H2O to obtain a stock solution of 1 mM for each molecule and kept in the dark at 4 °C until use.
Phenothiazin-5-ium Tetraiodide Hydrate (8) was synthesized in accordance with known literature protocol.36 2-(N-butyloxycarbonyl-2-aminoethyl)-1-(methyl)amine (9) was prepared as described earlier.35 Derivative (2-I) was prepared according to the literature protocol.34
Synthesis of hydroxyl- or ether-bis-dialkylaminophenothiazinium compounds
A solution of 2-N-methylaminoethanol (225 mg, 3 mmol) in methanol (50 mL) was added dropwise at room temperature to a stirred solution of phenothiazin-5-ium tetraiodide hydrate (8) (720 mg, 1 mmol) in methanol (100 mL) over a period of 1 h. The mixture was over night at room temperature. The organic solution was concentrated by evaporation to about 10 mL and left to cool. The deposited solid was collected by filtration, washed with diethyl ether and dried.
The crude salt was dissolved in dichloromethane (100 mL) and another portion of 2-N-methylaminoethanol (450 mg, 6 mmol) in dichloromethane (20 mL) was added dropwise over a period of 1 h. The solution was stirred over night at room temperature and was then washed with water (50 mL). The organic layer was dried over MgSO4 and the solvent was concentrated at reduced pressure not exciding a water bath temperature of 40 °C. Diethylether (60 mL) was added to precipitate the product. The solid was collected by filtration, washed with diethyl ether and dried. The crude material was purified by column chromatography over aluminium oxide (Brockmann I, std. Grade, 150 mesh, Aldrich, basic, activated) with dichloromethane → dichloromethane–ethanol 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → dichloromethane–methanol 5:1 → methanol as the eluent.
:1 → dichloromethane–methanol 5:1 → methanol as the eluent.
Dark blue, purple-metallic shining glass (26%, 0.26 mmol). 1H-NMR (400 MHz, DMSO-d6): δ[ppm] = 7.89 (m, 2H), 7.52 (m, 4H), 3.84 (m, 4H), 3.67 (m, 4 H), 3.31 (s, 6 H); – 13C-NMR (100 MHz, DMSO-d6): δ[ppm] = 154.1 (quat), 138.0 (+), 135.1 (quat), 119.4 (+), 107.0 (+), 58.6 (−), 55.1 (−), 40.1 (+); – IR (neat): ν[cm−1] = MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 344.1 (100, M+); – MW = 344.46 + 126.90 g mol−1; MF = C18H22N3O2SI.
Solid phenothiazinium tetraiodide (2.16 g, 3 mmol) was suspended in methanol (100 mL). A solution of morpholine (1.03 g, 1 mL, 12 mmol) in methanol (100 mL) was added dropwise over a period of 90 min. The resulting solution was allowed to stir at room temperature for 5 h. The reaction progress was monitored by thin layer chromatography (silica gel, using dichloromethane–ethanol 8
 :1 cont. 2% acetic acid). If necessary, stirring was continued over night. The solution was extracted three times with 5% w/v hydroiodic acid (50 mL) and three times with water (50 mL). Post-extraction, the organic layer was dried over magnesium sulfate, concentrated to a low volume and precipitated with dry diethyl ether. Reprecipitation was carried out until spectrophotometric analysis gave a peak ratio (λmax = 660:λmax = 290) of >2.2. The product was further purified by flash chromatography on silica gel using gradient elution in dichloromethane–methanol 19:1 → 5:1 and dichloromethane–methanol 5:1, finally re-crystallisation from dichloromethane.
:1 cont. 2% acetic acid). If necessary, stirring was continued over night. The solution was extracted three times with 5% w/v hydroiodic acid (50 mL) and three times with water (50 mL). Post-extraction, the organic layer was dried over magnesium sulfate, concentrated to a low volume and precipitated with dry diethyl ether. Reprecipitation was carried out until spectrophotometric analysis gave a peak ratio (λmax = 660:λmax = 290) of >2.2. The product was further purified by flash chromatography on silica gel using gradient elution in dichloromethane–methanol 19:1 → 5:1 and dichloromethane–methanol 5:1, finally re-crystallisation from dichloromethane.
Reddish shining, blue-purple lustrous crystals (52%, 0.52 mmol). 1H-NMR (300 MHz, DMSO-d6): δ[ppm] = 7.93 (d, 8.8 Hz, 2H), 7.77 (s, 2H), 7.68 (d, 8.8 Hz, 2 H), 3.87 (m, 4H), 3.78 (m, 4 H); – 13C-NMR (75 MHz, DMSO-d6): δ[ppm] = 153.1 (quat), 138.0 (+), 134.1 (quat), 119.2 (+), 107.1 (+), 65.9 (−), 47.7 (−); – IR (neat): ν[cm−1] = MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): e/z (%) = 368.1 (100, M+); – MW = 368.48 + 126.90 g mol−1; MF = C20H22N3O2SI.
Synthesis of boc-protected methyleneblue derivatives
The boc-protected amine (2 mmol) was added dropwise to a well-stirring solution of phenothiazinium tetraiodide monohydrate (730 mg, 1 mmol) in methanol (100 mL) at room temperature under nitrogen. The reaction mixture was stirred for 6 h and evaporated under reduced pressure to leave a dark residue, which was immediately used for the second step without further purification. To a solution of this salt in dichloromethane (200 mL) was added dropwise a solution of triethylamine (0.3 g, 0.4 mL, 3 mmol) in dichloromethane (50 mL). After stirring for 5 minutes the second portion of the boc-protected amine (3 mmol) in dichloromethane (100 mL) was added over a period of 2 h. The solution was stirred over night at room temperature and was then washed with water (3 × 250 mL). The organic layer was dried over MgSO4 and the solvent was evaporated at reduced pressure not exciding a water bath temperature of 40 °C. The crude material was purified by repeated flash chromatography with silica gel using dichloromethane–ethanol 10:1 as the eluent.
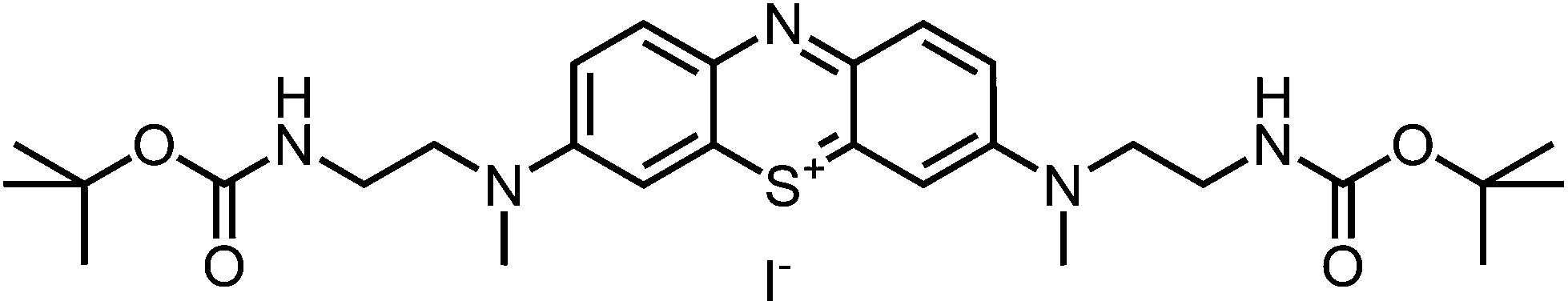
Dark blue, purple-metallic shining glass (59%, 0.59 mmol). 1H-NMR (300 MHz, CDCl3): δ[ppm] = 7.97 (d, 8.6 Hz, 2H), 7.38 (m, 4 H), 3.84 (m, 4H), 3.46 (m, 4 H), 3.33 (s, 6 H), 1.35 & 1.25 (s, 18 H); – 13C-NMR (75 MHz, CDCl3): δ[ppm] = 156.4 (quat), 154.2 (quat), 138.4 (+), 135.9 & 134.7 (quat), 119.0 (+), 107.0 (+), 79.5 (quat), 53.3 (−), 40.4 (+), 38.0 (−), 28.4 (+); – MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 542.3 (100, M+); – MW = 542.73 + 126.90 g mol−1; MF = C28H40N5SO4I.
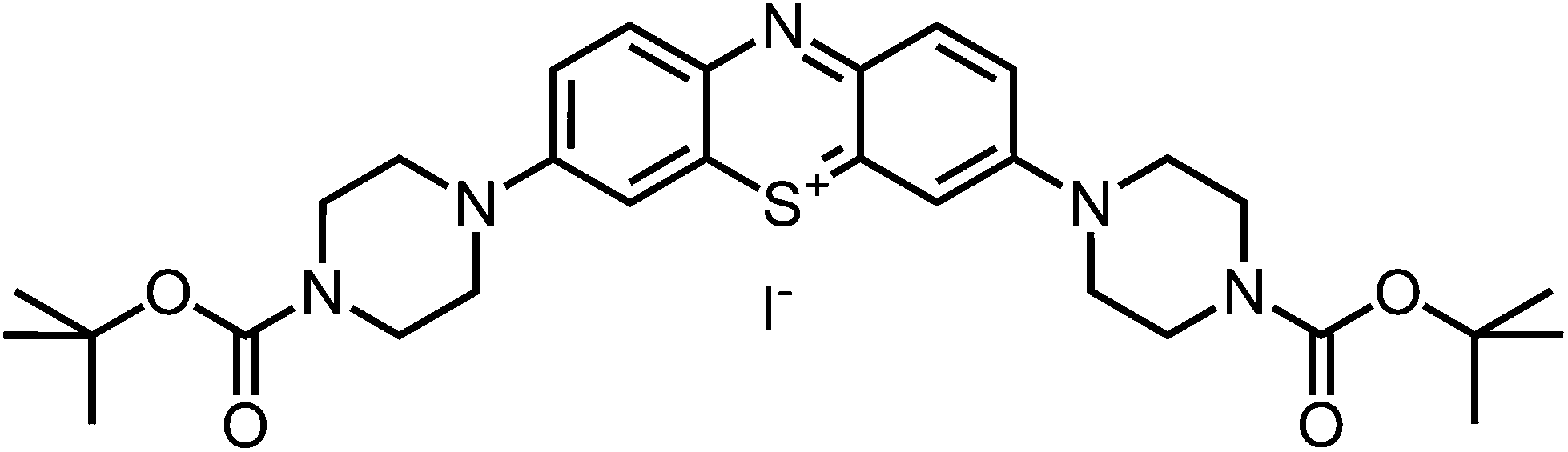
Dark blue, purple-metallic shining solid (64%, 0.64 mmol). 1H-NMR (400 MHz, CDCl3): δ[ppm] = 7.93 (d, 8.6 Hz, 2H), 7.84 (m, 2 H), 7.42 (dd, 8.8 Hz, 1.3 Hz, 2 H), 3.91 (m, 8 H), 3.40–3.70 (m, 8 H), 1.42–1.48 (m, 18 H); – 13C-NMR (100 MHz, CDCl3): δ[ppm] = 154.6 (quat), 153.9 (quat), 138.8 (+), 136.6 & 135.7 (quat), 118.9 (+), 107.9 (+), 80.8 (quat), 47.7 (−), 42.0–44.0 (−), 28.3 (+); – MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 566.3 (100, M+); – MW = 566.75 + 126.90 g mol−1; MF = C30H40N5SO4I.
Deprotection of boc-protected methyleneblue derivatives
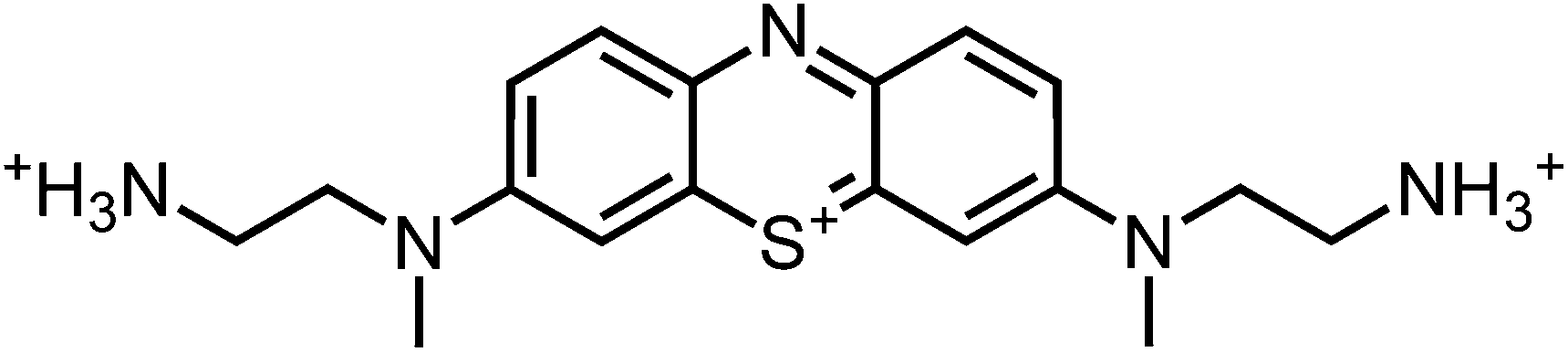
The boc-protected methylenblue derivative (0.5 mmol) was dissolved in dichloromethane (4 mL). Trifluoro acetic acid (TFA) (285 mg, 0.2 mL, 2.5 mmol) in dichloromethane (2 mL, 10% TFA) was added dropwise and the reaction mixture was stirred for 5 h at room temperature. The solution was transferred to four blue caps, the product was precipitated by addition of diethylether (13.5 mL per tube) and centrifuged. The solution was decanted off the precipitate, it was resuspended in diethylether (15 mL per tube) and settled by centrifugation again. The solvent was decanted off and the residue was dried at reduced pressure without heating.MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 342.2 (100, M+), 171.6 (31, (M+ + H)2+), 163.1 (48, (M+ + H)2+ –NH3); – MW = 344.51 + 3 × 114.02 g mol−1; MF = C24H29N5F3O6S.
MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 366.2 (65, M+), 183.6 (100, (M+ + H)2+), 122.7 (12, (M+ + H)3+); – MW = 368.53 + 3 × 114.02 g mol−1; MF = C26H29N5F3O6S.

Ion exchange protocol for methyleneblue derivatives
The column was packed with ion exchanger (Amberlite IRA-958). The resin was rinsed with acidic sodium chloride solution (10% aqueous NaCl cont. 0.1% HCl, 100 mL) and conditioned with dilute hydrochloric acid (0.1%)–acetonitrile–methanol (3:1:1). The MB derivative (0.5 mmol) was dissolved in hydrochloric acid (1 M, 10 mL) and lyophilized. A solution of this mixed salt was dissolved in hydrochloric acid (0.1%)–acetonitrile–methanol (3:1:1) (6 mL) and was slowly passed through a column (height 10 cm, diameter 1 cm; transferred with 4 mL of the solvent mixture to the conditioned anion exchanger (Amberlite IRA-958) eluting with 20 mL of the solvent mixutre. The solvents methanol and acetonitrile were evaporated at reduced pressure not exciding a water bath temperature of 40 °C. The aqueous solution was lyophilized to give the product as dark blue solid.
MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 284.1 (100, M+); – MW = 284.41 + 35.45 g mol−1; MF = C16H18N3SCl.
1H-NMR (400 MHz, CDCl3): δ[ppm] = 7.87 (d, 8.6 Hz, 2H), 7.78 (s, 2H), 7.14 (d, 8.8 Hz, 2 H), 3.66 (m, 4H), 1.73 (m, 4 H), 1.49 (m, 4 H), 1.03 (t, 7.6 Hz, 6 H); – 13C-NMR (100 MHz, CDCl3): δ[ppm] = 152.7 (quat), 138.4 (+), 135.8 (quat), 118.0 (+), 107.3 (+), 52.4 (−), 29.9 (−), 20.2 (−), 13.9 (+); – IR (neat): ν[cm−1] = 3362, 2952, 2930, 2868, 2707, 2073, 1585, 1481, 1449, 1391, 1331, 1286, 1250, 1215, 1135, 1107, 1038, 929, 878, 806, 745, 672, 617; – MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): e/z (%) = 452.3 (100, M+); – UV (H2O, 10 μM): λ/[nm] (ε/[M−1 cm−1]) = 679 (63
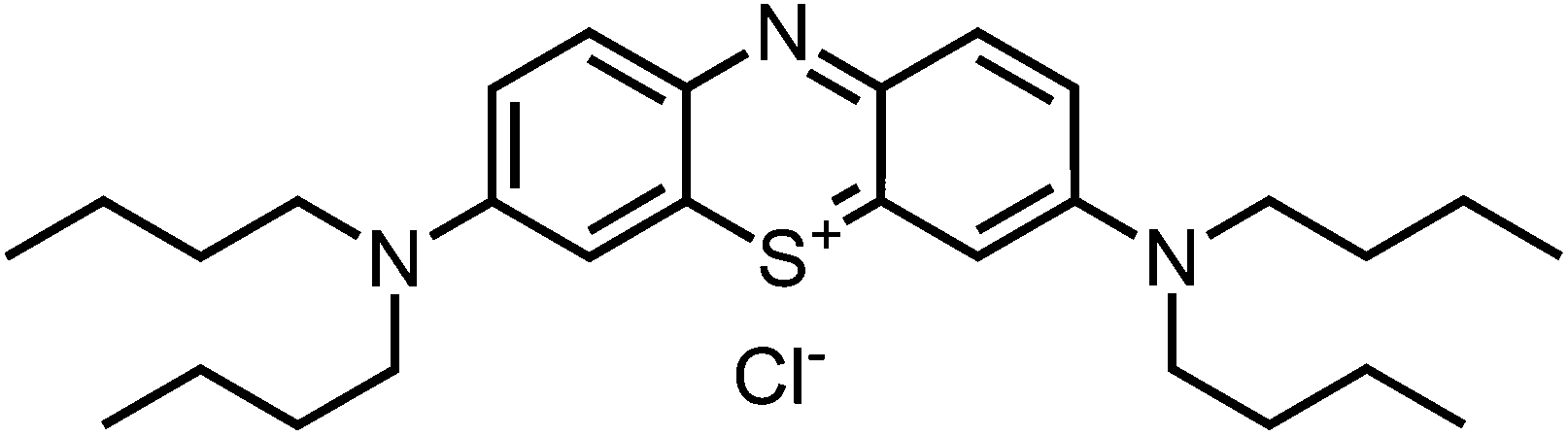 200), 298 (37300); – MW = 452.73 + 35.45 g mol−1; MF = C28H42N3SCl.
200), 298 (37300); – MW = 452.73 + 35.45 g mol−1; MF = C28H42N3SCl.
1H-NMR (300 MHz, DMSO-d6): δ[ppm] = 8.43 (bs, 6 H), 8.02 (d, 8.6 Hz, 2H), 7.82 (m, 2 H), 7.68 (m, 2 H), 4.06 (m, 4H), 3.16 (m, 4 H); – 13C-NMR (75 MHz, DMSO-d6): δ[ppm] = 154.0 (quat), 138.1 (+), 135.7 (quat), 118.7 (+), 106.8 (+), 53.1 (−), 40.6 (+), 38.9 (−); – IR (neat): ν[cm−1] = 3351, 3218, 2965, 2872, 2741, 2170, 2056, 1585, 1516, 1481, 1437, 1386, 1364, 1321, 1278, 1241, 1174, 1131, 1087, 1035, 976, 947, 880, 830, 805, 666; – MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 342.2 (100, M+), 171.6 (20, (M+ + H)2+), 163.1 (56, (M+ + H)2+ –NH3); – UV (H2O, 10 μM): λ/[nm] (ε/[M−1 cm−1]) = 617 (48
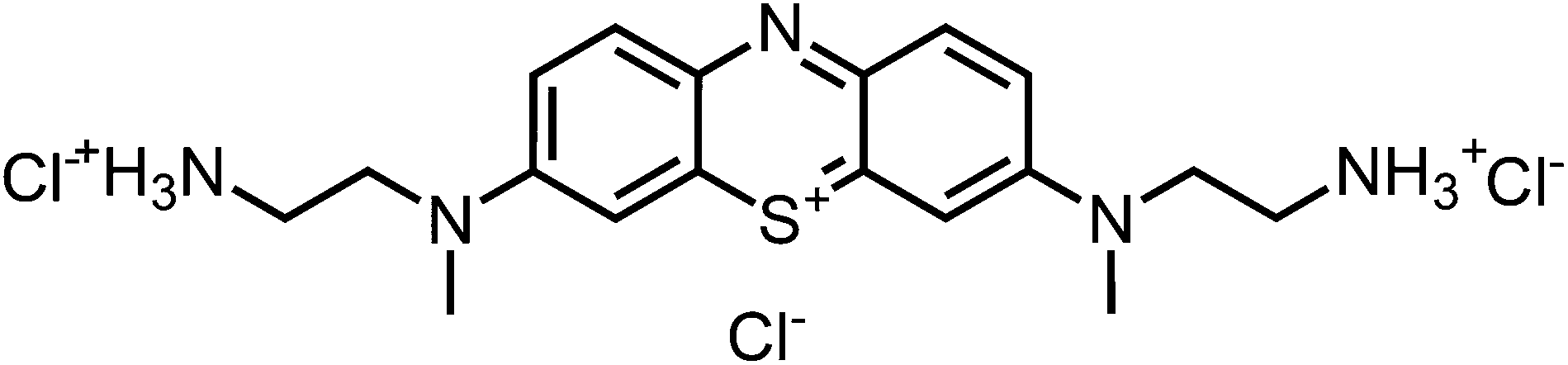 700), 291 (33500); – MW = 344.51 + 3 × 35.45 g mol−1; MF = C18H26N5SCl3.
700), 291 (33500); – MW = 344.51 + 3 × 35.45 g mol−1; MF = C18H26N5SCl3.
1H-NMR (300 MHz, DMSO-d6): δ[ppm] = 9.60–9.80 (m, 4 H), 8.08 (d, 8.6 Hz, 2H), 7.90 (s, 2 H), 7.76 (m, 2 H), 4.15 (m, 8 H), 3.48 (m, 8 H); – 13C-NMR (75 MHz, DMSO-d6): δ[ppm] = IR (neat): ν[cm−1] = 3375, 2920, 2707, 2454, 1651, 1587, 1489, 1439, 1392, 1358, 1229, 1132, 1082, 1023, 930, 878, 822, 572; – MS (ESI-MS, CH2Cl2–MeOH + 10 mmol NH4OAc): e/z (%) = 366.2 (76, M+), 183.6 (100, (M+ + H)2+), 122.7 (17, (M+ + H)3+); – UV (H2O, 10 μM): λ/[nm] (ε/[M−1 cm−1]) = 642 (45
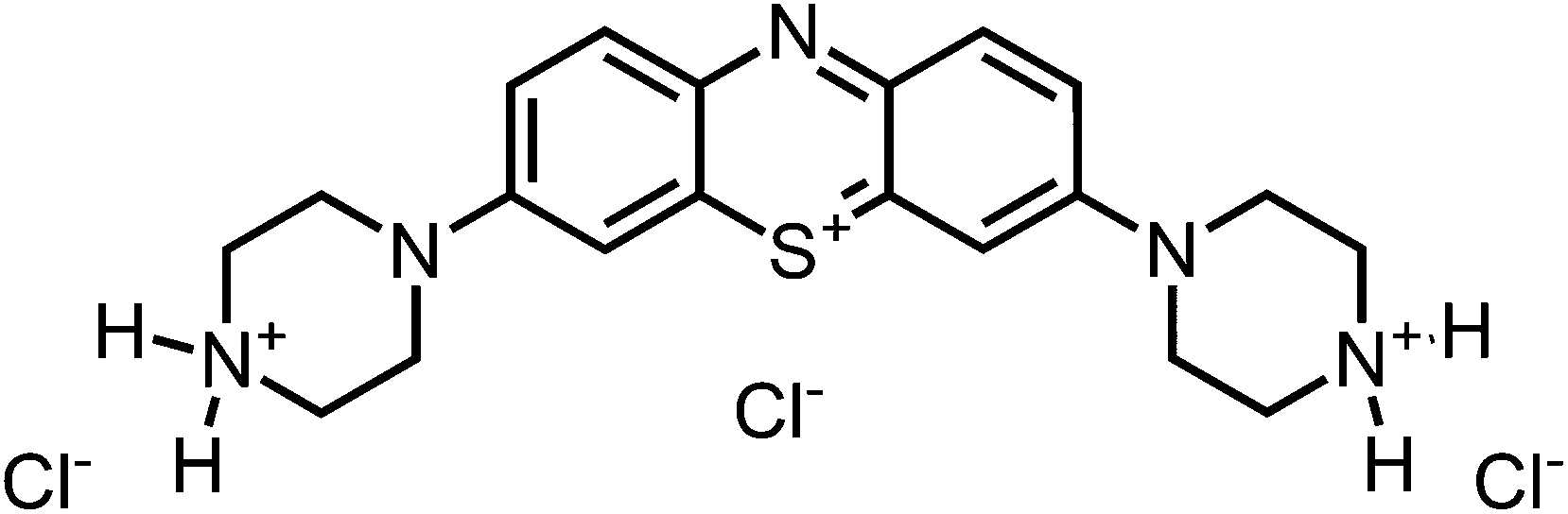 900), 295 (31200); – MW = 368.53 + 3 × 35.45 g mol−1; MF = C20H26N5SCl3.
900), 295 (31200); – MW = 368.53 + 3 × 35.45 g mol−1; MF = C20H26N5SCl3.
1H-NMR (300 MHz, MeOD): δ[ppm] = 7.86 (d, 9.1 Hz, 2H), 7.50 (m, 4H), 3.83 (m, 4H), 3.70 (m, 4 H), 3.35 (s, 6 H); – 13C-NMR (75 MHz, MeOD): δ[ppm] = 154.0 (quat), 137.6 (+), 135.0 (quat), 119.3 (+), 106.9 (+), 58.6 (−), 55.0 (−), 39.9 (+); – IR (neat): ν[cm−1] = 3211, 2916, 2872, 2706, 1587, 1523, 1483, 1435, 1387, 1318, 1221, 1189, 1134, 1035, 968, 875, 823, 768, 660; – UV (H2O, 10 μM): λ/[nm] (ε/[M−1 cm−1]) = 663 (58
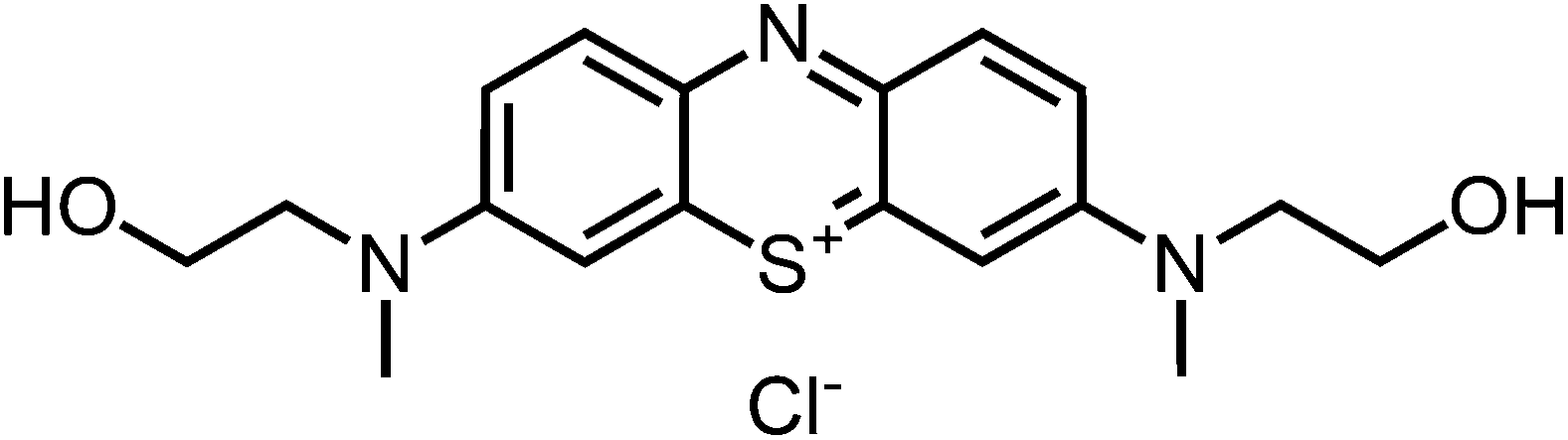 100), 296 (35600); – MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): e/z (%) = 344.1 (100, M+); – MW = 344.46 + 35.45 g mol−1; MF = C18H22N3O2SCl.
100), 296 (35600); – MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): e/z (%) = 344.1 (100, M+); – MW = 344.46 + 35.45 g mol−1; MF = C18H22N3O2SCl.
1H-NMR (300 MHz, MeOD): δ[ppm] = 7.94 (d, 8.6 Hz, 2H), 7.76 (m, 2H), 7.69 (dd, 8.8 Hz, 1,2 Hz, 2 H), 3.87 (m, 4H), 3.80 (m, 4 H); – 13C-NMR (75 MHz, MeOD): δ[ppm] = 154.1 (quat), 140.0 (+), 134.6 (quat), 120.4 (+), 107.9 (+), 67.6 (−), 49.1 (−); – IR (neat): ν[cm−1] = 3450, 2965, 2902, 2864, 2690, 1590, 1518, 1486, 1450, 1394, 1358, 1310, 1228, 1141, 1111, 1044, 1032, 944, 881, 835, 768, 747, 665, 619; – MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): e/z (%) = 368.1 (100, M+); – UV (H2O, 10 μM): λ/[nm] (ε/[M−1 cm−1]) = 664 (60
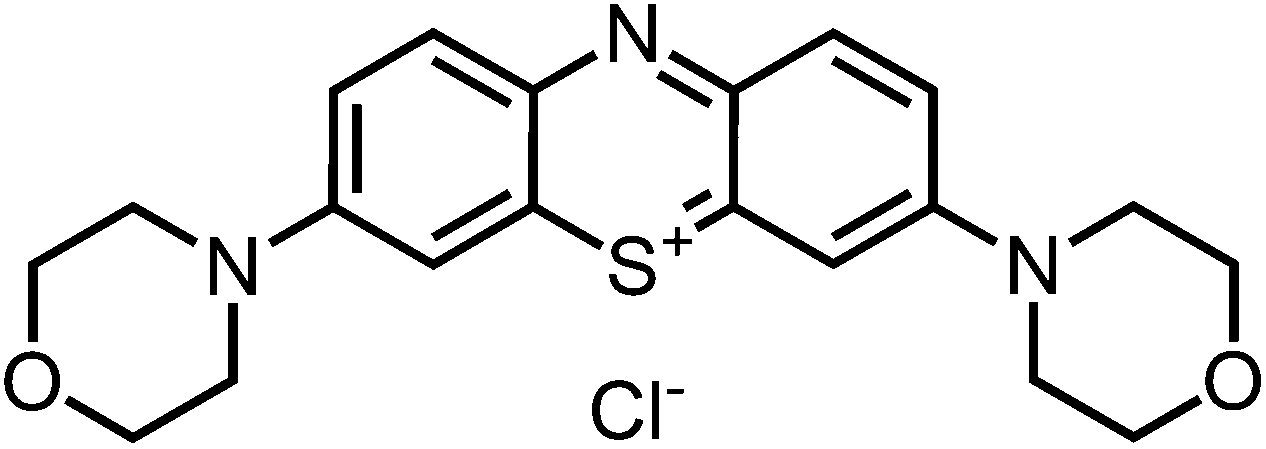 400), 297 (39100); – MW = 368.48 + 35.45 g mol−1; MF = C20H22N3O2SCl.
400), 297 (39100); – MW = 368.48 + 35.45 g mol−1; MF = C20H22N3O2SCl.
Absorption spectroscopy
Steady state absorption spectra were recorded at room temperature with a DU640 spectrophotometer (Beckman Instruments GmbH, Germany) in a concentration range of 10 μM to 200 μM of the respective dyes. In order to examine the aggregation of MB 1 and its symmetrically substituted derivatives, the absorption cross-section σ [cm2] was calculated from the measured transmission using the following equation: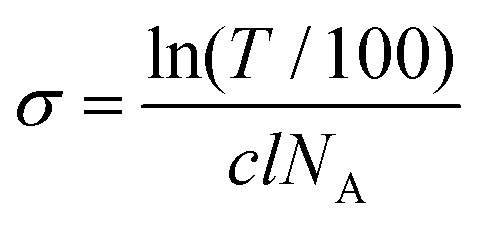 | (1) |
For the photostability experiments, solutions with the respective PS (PS concentration was initially 5 μM) were filled in a quartz cuvette with a path length of 1 cm (QS-101, Hellma Optik, Jena, Germany), irradiated for 250 s at 600 nm during magnetic stirring with a total energy of 16.25 J (power was 65 mW). For irradiation a tunable OPO laser (EKSPLA, Lithuania) was used, which has a frequency of f = 1 kHz. Upon irradiation, the transmission T was recorded. The absorption was calculated by the following equation:
| A = 100 − T | (2) |
These settings are comparable with the settings that were used by Felgentraeger et al. to examine asymmetrically substituted phenothiazinium derivatives which are also compared in this study.35
For the uptake/attachment experiments of (3), the bacteria (OD = 0.6) were incubated with 10 μM of (3) for 60 min (500 μL bacterial suspension + 500 μL of (3) in H2O, in Eppendorf tubes 1.5 mL) to get a final concentration of 5 μM. Upon incubation, the bacteria were centrifuged (13000 rpm, 5 min) and the absorption of the supernatant was determined. Subsequently a washing step was performed, the bacterial pellet was resuspended and again centrifuged and the absorption of the second supernatant was recorded.
The polarity of the PS was estimated by measuring the octanol-water coefficient. Distribution of 1 × 10−4 mol of each phenothiazinium salt between both phases was measured by UV/Vis spectroscopy after 10 minutes of stirring at room temperature.
Direct measurement of singlet oxygen
In order to produce singlet oxygen, the respective PS were filled in a quartz cuvette and excited with the tunable laser system. Direct detection of singlet oxygen was performed by time-resolved measurements of the singlet oxygen luminescence at 1270 nm (10 nm FWHM filter) in near-backward direction with respect to the excitation beam using an infrared-sensitive photomultiplier (R5509-42, Hamamatsu Photonics Deutschland GmbH, Herrsching, Germany).37Quantum yield of singlet oxygen formation (ΦΔ)
Φ Δ of the novel symmetrically substituted derivatives of MB 1 were determined by the direct detection method and compared to the ΦΔ of TMPyP. The ΦΔ of TMPyP in H2O is known to be 0.77 ± 0.04 from literature and own control measurements.38 A concentration of 2.5 μM of each of the newly synthesized derivatives was used and irradiated at λ = 600 nm. Since the irradiation wavelength was 600 nm, a concentration of 10 μM for TMPyP was chosen for a sufficient absorption at this wavelength. The samples were air-saturated at 25 °C and illuminated at different powers ranging from 9 to 80 mW. The detected singlet oxygen photons were summed up and plotted against the absorbed energy.TMPyP as standard was purchased from Sigma Aldrich (Germany) and was used without further purification.
Phototoxicity experiments
The bacterial strains S. aureus (ATCC 25923) and E. coli (ATCC 25922) were grown overnight aerobically at 37 °C in Mueller-Hinton broth (Gibco Life Technologies GmbH, Germany) on an orbital shaker. When the bacterial cultures reached the stationary phase of growth, the cells were harvested by centrifugation (3000 rpm, 10 min), washed with Millipore H2O and were resuspended in Millipore H2O yielding an optical density of 0.6 at 600 nm corresponding to ∼108–109 cells mL−1 for the use in the phototoxicity experiments. The bacteria were transferred into a 96-well microtitre plate (100 μL per well) and incubated either for 10 min or for 60 min in the dark with 100 μL per well of different concentrations of the PS ranging from 0 μM to 100 μM. At the end of the incubation period the bacterial suspensions were illuminated with an incoherent light source PDT1200 provided by Herbert Waldmann GmbH & Co.KG (Germany). The emission of the light source can be convolved with the absorption of MB (1) and its derivatives and their ΦΔ in order to calculate the theoretical effective phototoxicity caused by singlet oxygen (vide infra).35 The samples were irradiated with an intensity of 50 mW cm−2 for 10 min yielding an overall light dose of 30 J cm−2. Controls were neither sensitized with a PS nor exposed to light or were incubated only with the PS at the highest concentration. Upon irradiation, the survival of the bacteria was determined by the colony forming units (CFU) assay using the Miles, Misra & Irwin technique.39 Therefore, serially 10-fold diluted aliquots of treated and control cells were plated on Mueller-Hinton agar and the numbers of CFU mL−1 were counted after 24 h of incubation at 37 °C.For the uptake/attachment experiments of (3), the bacteria were centrifuged (13000 rpm, 5 min) after incubation and a washing step was performed afterwards. This procedure was performed twice. Subsequently, the bacteria were irradiated and treated as mentioned above.
Data analysis
Each individual experiment was performed in triplicate and all data are shown as means with standard deviation of the mean. A reduction of at least three orders of magnitude of log10 steps was considered biologically relevant with regard to the guidelines for hand hygiene.40Results and discussion
For this study, a set of symmetric phenothiazinium derivatives modified on both auxochromic positions, 3 and 7, was synthesized. Four of these compounds are hydrophilic (3, 4, 5 and 6) and one derivative is lipophilic (2).The impact of additional hydrogen bonds (5 and 6) or additional cationic charges (3 and 4) on both sides of the chromophore on the photophysical properties and antimicrobial photodynamic efficacy was investigated. We have chosen cyclic (4 and 6) and acyclic moieties (2, 3 and 5) as well as secondary (4) and primary ammonium groups (3) that have been introduced at the auxochromic sites of the phenothiazinium structure, to cover a larger scope of modifications.
In this study we used chloride (Cl−) as the respective counterion for all positively charged derivatives, because iodide (I−) as the counterion can react with singlet oxygen to triiodide.41 Therefore iodide as the counterion may consume singlet oxygen and influences the photodynamic action of the PS. No influence of chloride as a counterion on ROS generation is known.
Synthesis
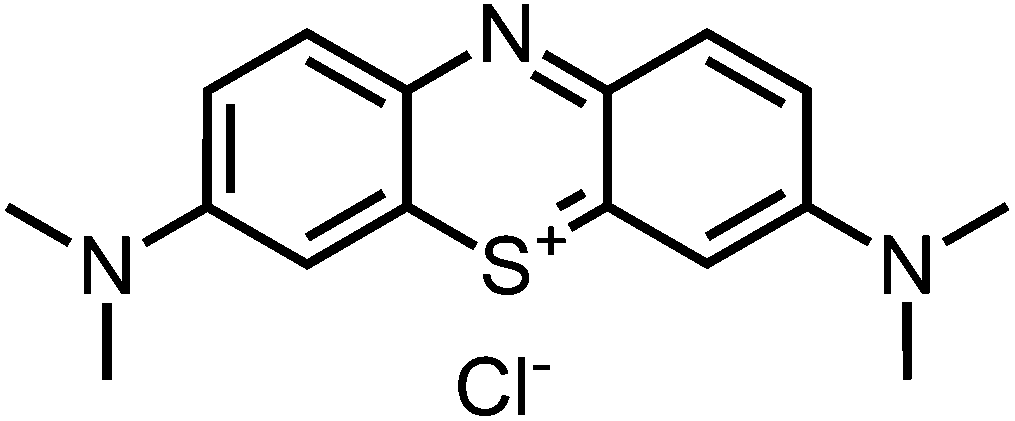
Compound 2-I34 and compound 6-I42 are literature known as iodide salt (see Scheme 3).34,43 We decided to investigate 2-I as the chloride salt (2).Phenothiazinium tetraiodide (14),36 freshly prepared from phenothiazine (13), was converted to MB (1) (Scheme 1) and a variety of bis-fold substituted derivatives (Schemes 2, 3) by reaction with secondary amines in dichloromethane.
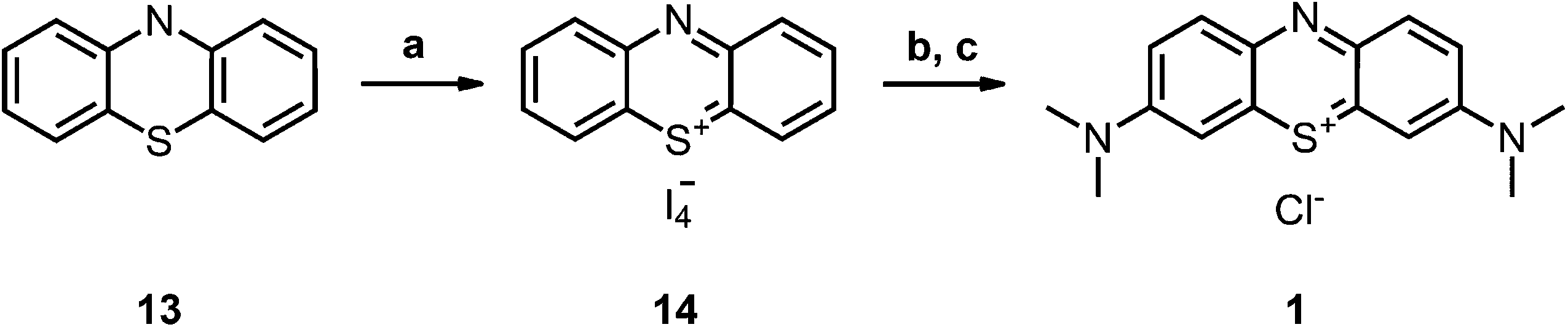 | ||
| Scheme 1 Synthesis of the precursors and MB 1; conditions: (a) DCM, I2, RT, 2 h, quant.; (b) HNMe2, MeOH, DCM, RT, 6 h, 74%; (c) ion exchanger Amberlite IRA958, water. | ||
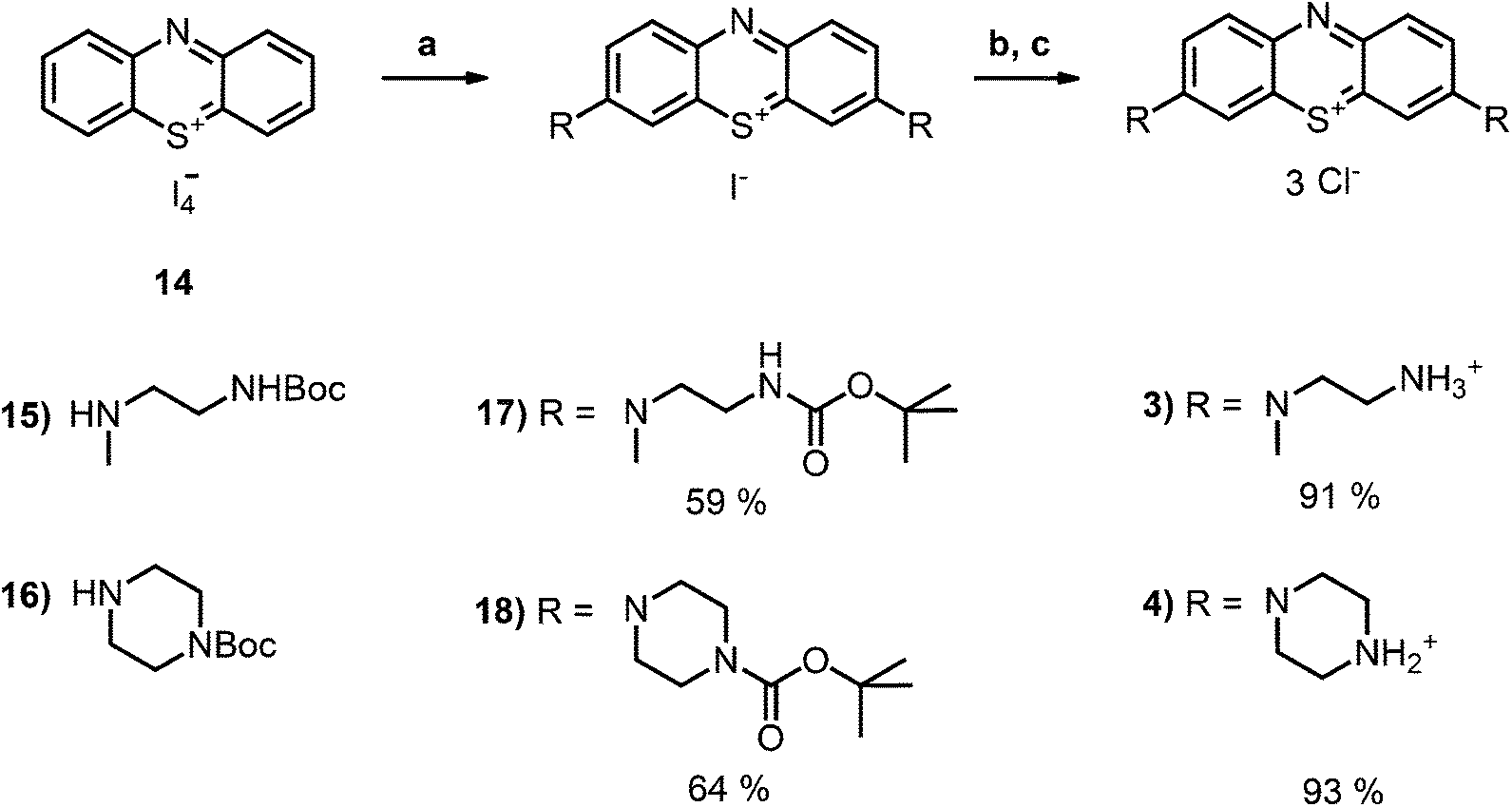 | ||
| Scheme 2 Synthesis of the boc-protected derivatives 17 and 18 and their transformation to the deprotected chromophores as chloride salts (3 and 4); conditions: (a) DCM, boc-protected amine 15 or 16, NEt3, RT, 5 h; (b) DCM, TFA, RT, 4 h; (c) ion exchanger Amberlite IRA958, water. | ||
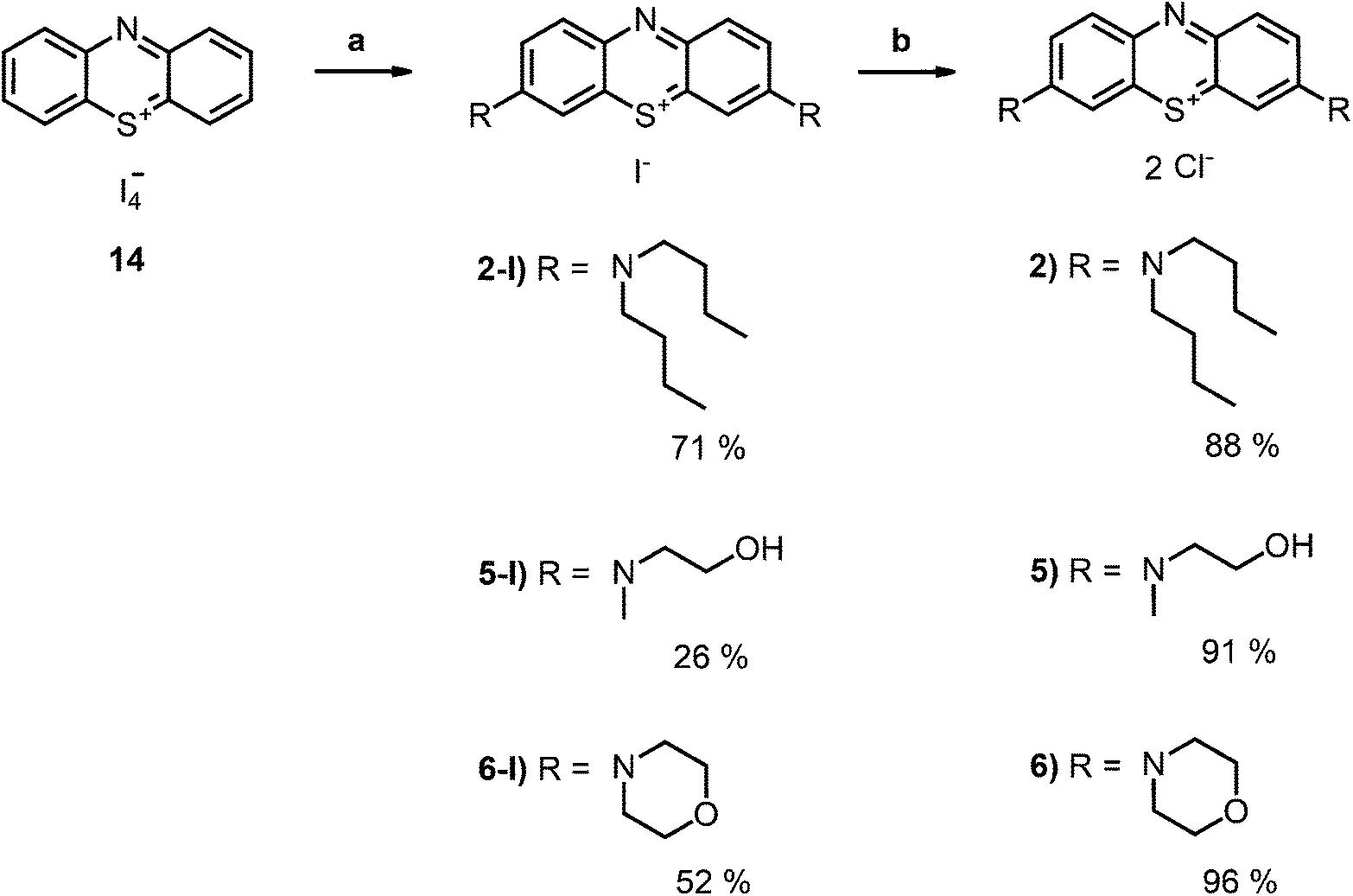 | ||
| Scheme 3 Synthesis of the second set of MB derivatives as chloride salts (2, 5, 6); conditions: (a) DCM, secondary amine: 2-(N-methylamino)ethanol, morpholine or N,N-dibutylamine, RT, 5 h; (b) ion exchanger Amberlite IRA958, water. | ||
The desired boc-protected derivatives 17 and 18 were prepared starting from (14) in good yields using an excess of the appropriate boc-protected amine (15 or 16) in presence of triethylamine in dichloromethane. After deprotection with TFA using standard conditions, the counterion was exchanged versus chloride using amberlite IRA958. Both steps resulted in excellent to nearly quantitative yields (Scheme 2).
Phenothiazinium compounds 2, 4 and 5 were prepared using similar conditions and reacting N,N-dibutylamine to give 2-I, 2-(N-methylamino)ethanol to yield 5-I or 4-morpholine to give 6-I, respectively, with moderate to good yields. After purification by flash chromatography and crystallisation, the counterion was exchanged with chloride in quantitative yield following the same protocol as before (Scheme 3).
We present here a straight forward synthesis and purification protocol for the preparation of a variety of polar, symmetrically substituted phenothiazinium compounds without need of HPLC purification. The purity of all compounds was checked by NMR and HPLC-MS and was ≥98%.
Photophysics: singlet oxygen generation ability and aggregation
Here, we investigated the aggregation of the novel derivatives within a concentration range from 10 μM to 200 μM (Table 1, Fig. 3). In Table 1 the absorption maximum of each derivative is specified. The peak of (3) at 617 nm does not match the peak of (7), the corresponding asymmetric derivative. The other molecules have the same absorption maxima as the asymmetric analogues. The peak of (5) and (6) match the absorption peak of (1) within experimental accuracy, as do the corresponding asymmetric derivatives 11 and 12, respectively, which were analyzed by Felgentraeger et al.35 Compared to the asymmetric derivative 12, (6) does not show any dimerization within experimental accuracy. Further, (4) does not show any dimerization effect such as the asymmetric analogue (9). However, (3) shows a weak red shift with increasing concentration (Fig. 3 left). Long term experiments have shown that the aggregation state of (3) is stable, because the absorption maxima did not shift over time (6 months), but remained at the same position when diluting the original stock solutions. The aggregation state of the stock solutions of compound 3 was neither influenced by temperature (40 °C and 100 °C, 30 min, respectively) nor by ultrasonic treatment (30 min). The lipophilic derivative 2 shows the formation of an absorption peak at 627 nm and the main peak, which is assigned to the monomer at 679 nm, is diminished with increasing PS concentration. This effect is known as hypochromicity. Both effects are known for (1) (vide supra). In line with this, (5) shows similar spectroscopic behavior as (2), but has a much weaker aggregation tendency than the latter. The hypochromic effect is observed at higher concentration, than for (2). Consequently, the new symmetric and hydrophilic derivatives 3–6 show not such a pronounced (3, 5) or no aggregation behavior (4, 6) such as MB 1, which might influence the phototoxic efficacy of the respective dyes in a positive manner. The suppressed tendency to aggregate is also beneficial, because a larger concentration span can be used without negative influence on the photophysics of the compounds.
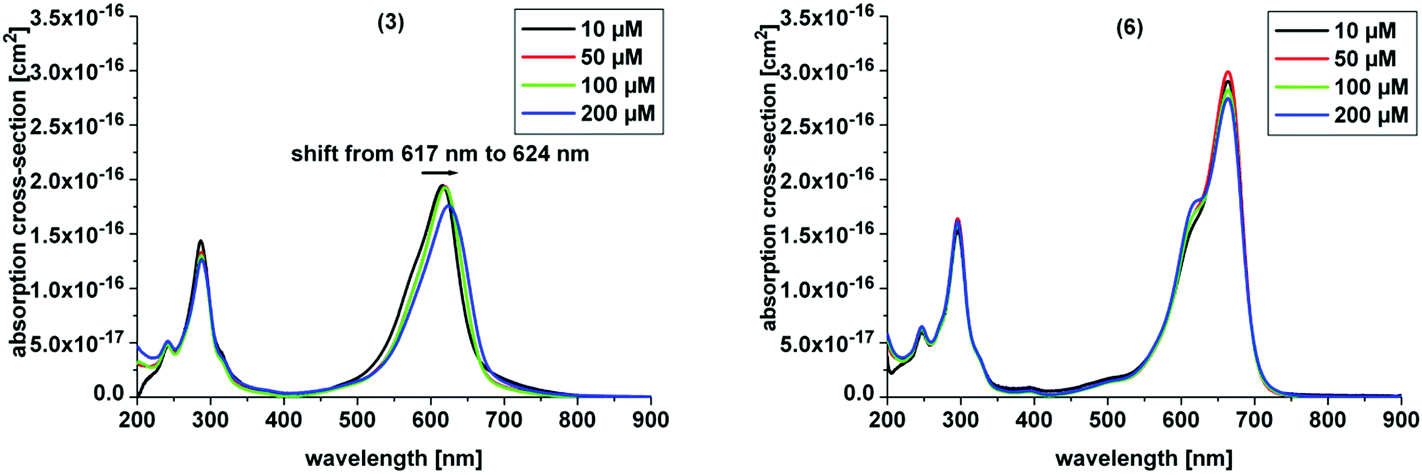 | ||
| Fig. 3 Selected absorption spectra of the discussed phenothiazinium derivatives with increasing concentration ranging from 10 μM to 200 μM in H2O. (3) (left) Shows a red shift in the spectra with increasing concentration, (6) (right) shows no aggregation with increasing concentration. | ||
000 laser pulses at 600 nm which equals a total energy of 16.25 J (power was 65 mW for 250 s)
|
|
MW cation [g mol−1] | λ abs, max [nm] ε [L mol−1 cm−1] | Dimerization | Photo-stability [%] | |
|---|---|---|---|---|---|
| 1 |
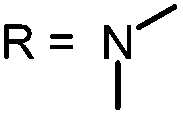
|
373.9 | 665 | Yes | 97 |
| 2 |
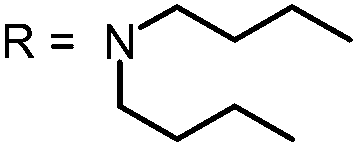
|
452.73 | 679 | Yes | 89 |
| 3 |
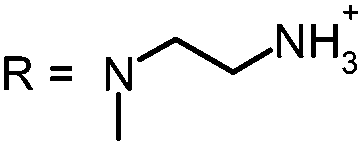
|
344.51 | 617 | Yes | 62 |
| 4 |
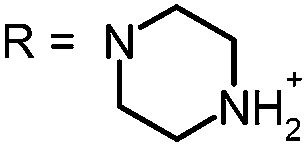
|
368.53 | 642 | No | 92 |
| 5 |
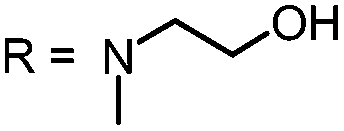
|
344.51 | 663 | Yes | 96 |
| 6 |
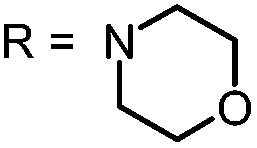
|
368.48 | 664 | No | 99 |
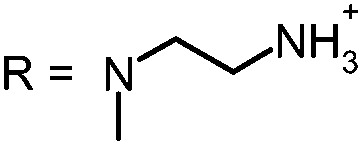
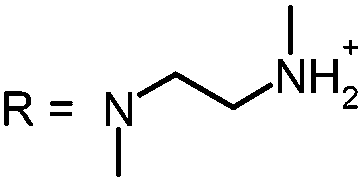
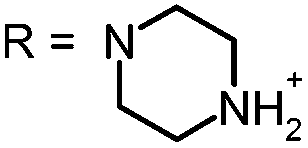
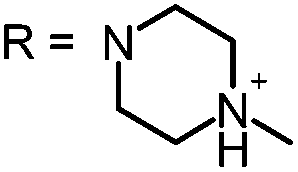
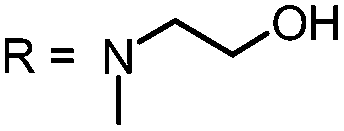
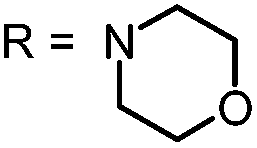
In our study the phenothiazinium derivatives were diluted to a final concentration of 5 μM and irradiated at 600 nm for 250 s with a power of 65 mW yielding an energy of 16.25 J. Hereby, the same amount of light energy per time unit is absorbed by each derivative. Photostability was recorded by absorption spectroscopy. The value to estimate photostability was given with the ratio of the absorption maxima after and before irradiation (Table 1). It was shown that only (3) is photoinstable for energies up to 16.25 J (Fig. 4).
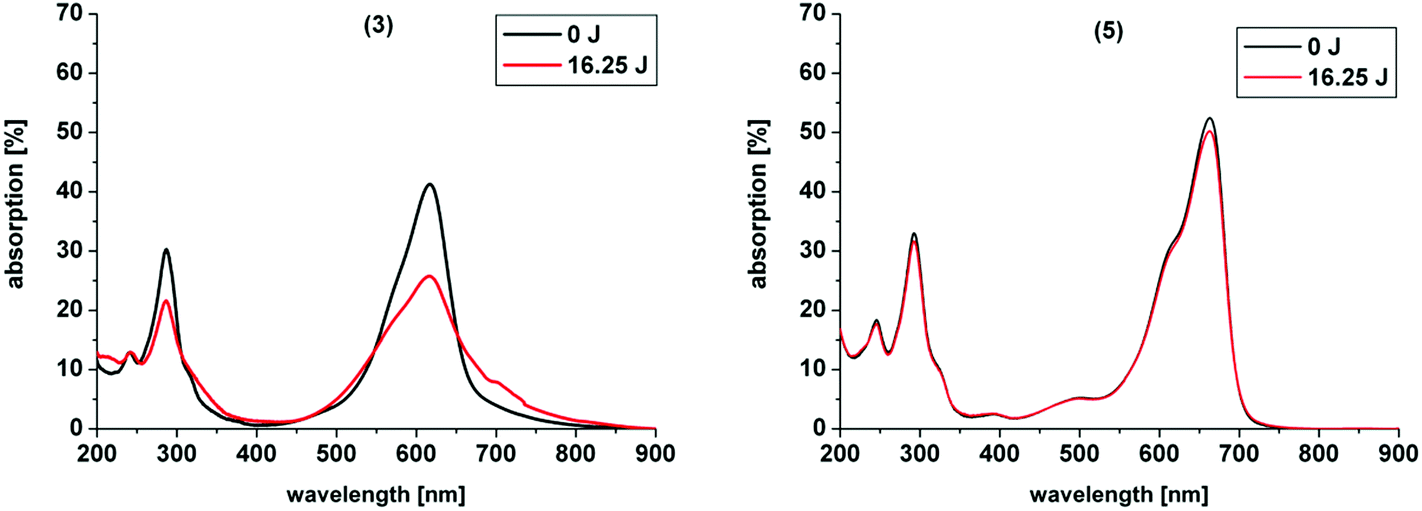 | ||
| Fig. 4 Photostability of compounds 3 and 5 upon irradiation at 600 nm for 250 s and a power of 65 mW yielding a total energy of 16.25 J. (3) showed a pronounced decrease in absorption upon irradiation. | ||
The asymmetric analogue 7 that was analyzed by Felgentraeger et al. showed as well a decrease in the main absorption region and in the UV-range.35 A possible explanation for the degradation of (3) might be that ammonium groups are good leaving groups or can be oxidized after deprotonation in aqueous media by singlet oxygen.49 Therefore, degradation of (3) upon irradiation is eased in comparison to the other examples. After degradation of one of the substituents at the nitrogen in the 3- or 7-position of the chromophore system deprotonation and reduction are enabled, followed by elimination of an amine group and decomposition occurs of the whole ring system, as commonly known for MB 1.26 In contrast, simple alkyl chains as in (2) are less in risk to be oxidized and to degrade, resulting in a far higher stability. Six-membered rings are known to be very stable moieties in organic chemistry. Ring-opening and degradation is even more hampered. Thus, the cyclic substituents in (4) and (6) are more stabilized and less accessible to photodegradation by the common mechanism and show the highest photostabilities of all compounds presented.
The polarity of the PS was estimated by measuring the octanol-water coefficient. Distribution of 1 × 10−4 mol of each phenothiazinium salt between both phases was measured by UV/Vis spectroscopy after 10 minutes of stirring at room temperature. Table 2 summarizes the results and gives data for the photophysical measurements as described above.
Φ Δ of the novel symmetric compounds such as (2), (3), (4), (5) and (6) were compared with TMPyP (10 μM) as reference PS whereas ΦΔ of (1) is displayed as literature value.51 The quantum yields of the PS are shown in the following order (eqn (3)) and are summarized in Table 2:
| ΦΔ (4) > ΦΔ (1) > ΦΔ (5) > ΦΔ (3) > ΦΔ (2) > ΦΔ (6) | (3) |
However, Chen et al. proposed that although singlet oxygen is highly important, the rate of bacteria inactivation is determined by the binding of the dye to the bacteria.47 In our study we evaluated the effective phototoxicity of singlet oxygen also taking the emission of the light source, the absorption of the PS and ΦΔ into account.
In order to estimate the effectiveness of singlet oxygen that is generated by the absorbed light energy of the different derivatives in the phototoxicity experiments, the values of the emission spectrum “Em” of the light source were convolved with the values for the absolute absorption “Abs” of the respective dyes for the spectral region between 500 nm and 800 nm and multiplied with the ΦΔ of the phenothiazinium derivatives.35 Hereby, the absorption of the respective dyes at a concentration of 10 μM was measured. According to the following formula (eqn (4)) an effective toxicity, that might be caused by singlet oxygen, “Eff. Tox. of 1O2” was predicted for each PS:
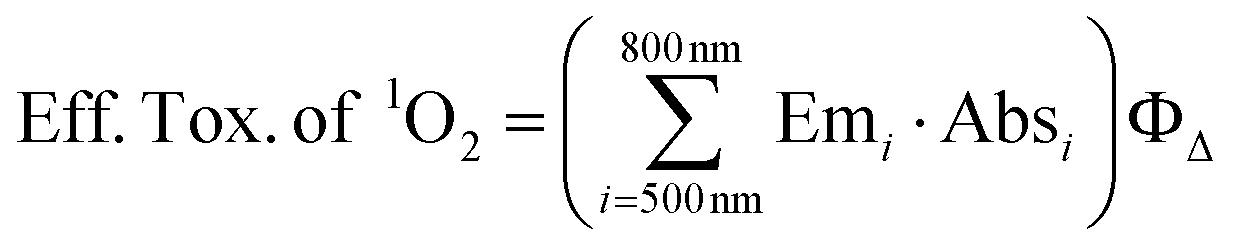 | (4) |
This formula accounts for the effective absorbed energy (i.e. the sum of the product of emission and absorption) of every PS that is used to generate singlet oxygen.
Photobiological studies and photodynamic inactivation of bacteria (PIB)
In our study we investigated the phototoxicity of symmetrically substituted phenothiazinium derivatives against S. aureus and E. coli as representatives for Gram-positive and Gram-negative bacteria.| S. aureus | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | IC | 0 μM + Light | 1 μM + Light | 5 μM + Light | 10 μM + Light | 50 μM + Light | 100 μM + Light | 100 μM DC |
| (1) | 10′ | — | <3 | 5 | 5 | 5 | >3 | — |
| 60′ | — | <3 | 5 | 5 | 5 | 5 | — | |
| (2) | 10′ | — | <3 | 5 | 5 | 5 | 5 | — |
| 60′ | — | <3 | 5 | 5 | 5 | 5 | 5 | |
| (3) | 10′ | — | — | — | — | — | — | — |
| 60′ | — | — | — | — | — | — | — | |
| (4) | 10′ | — | — | <3 | >3 | <3 | <3 | — |
| 60′ | — | — | <3 | >3 | <3 | <3 | — | |
| (5) | 10′ | — | <3 | >5 | >5 | >5 | >5 | — |
| 60′ | — | >3 | >5 | >5 | >5 | >5 | — | |
| (6) | 10′ | — | <3 | >5 | >5 | >5 | >5 | — |
| 60′ | — | <3 | >5 | >5 | >5 | >5 | — | |
| E. coli | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | IC | 0 μM + Light | 1 μM + Light | 5 μM + Light | 10 μM + Light | 50 μM + Light | 100 μM + Light | 100 μM DC |
| (1) | 10′ | — | <3 | 5 | 5 | 5 | 5 | — |
| 60′ | — | — | 5 | 5 | 5 | 5 | — | |
| (2) | 10′ | — | <3 | 5 | 5 | 5 | 5 | 5 |
| 60′ | — | <3 | 5 | 5 | 5 | 5 | 5 | |
| (3) | 10′ | — | — | <3 | <3 | 5 | >3 | — |
| 60′ | — | — | >3 | >3 | 5 | 5 | — | |
| (4) | 10′ | — | — | <3 | >3 | 5 | >3 | — |
| 60′ | — | — | <3 | >3 | >3 | <3 | — | |
| (5) | 10′ | — | <3 | >5 | >5 | >5 | >5 | — |
| 60′ | — | <3 | >5 | >5 | >5 | >5 | — | |
| (6) | 10′ | — | <3 | >5 | >3 | >3 | >5 | — |
| 60′ | — | <3 | >5 | >5 | >5 | >5 | — | |
The lipophilic derivative 2 showed dark toxicity for a concentration of 100 μM whereas the parent compound 1 is not toxic at this concentration under our experimental conditions. The butyl chains in the substituents in the 3- and 7-position of the phenothiazinium chromophore raise the amphiphilic character in comparison to (1). Thus, the ability of compound (2) to penetrate the cell membrane and disturb its structure is higher, leading to increased dark toxicity. This ability is known to be the mode of antimicrobial action in cationic surfactants like benzalkonium chloride or dioctadecyldimethylammonium bromide. These well-known and comparable compounds are carrying long alkyl chains and a positive charge. They are destabilizing bacterial cell membranes by localization in the cell membrane leading to polarization and rupture.53
In general, Gram-positive bacteria are better accessible by PS because their cytoplasmic membrane is surrounded by a relatively porous layer of peptidoglycan and lipoteichoic acid.54 Therefore, they are more easily inactivated by antimicrobial photodynamic therapy than Gram-negative bacteria.32
Fig. 5 shows the inactivation of E. coli by derivative 3. In contrast to this general model, compound 3 shows a higher phototoxic activity against the Gram-negative E. coli, compared to Gram-positive S. aureus. Although (3) has positive charges, nearly no inactivation of S. aureus was observed (Fig. 5). At a concentration of 50 μM a killing efficacy of >5log10 was achieved against E. coli, whereas at this concentration only <1log10 of S. aureus were inactivated.
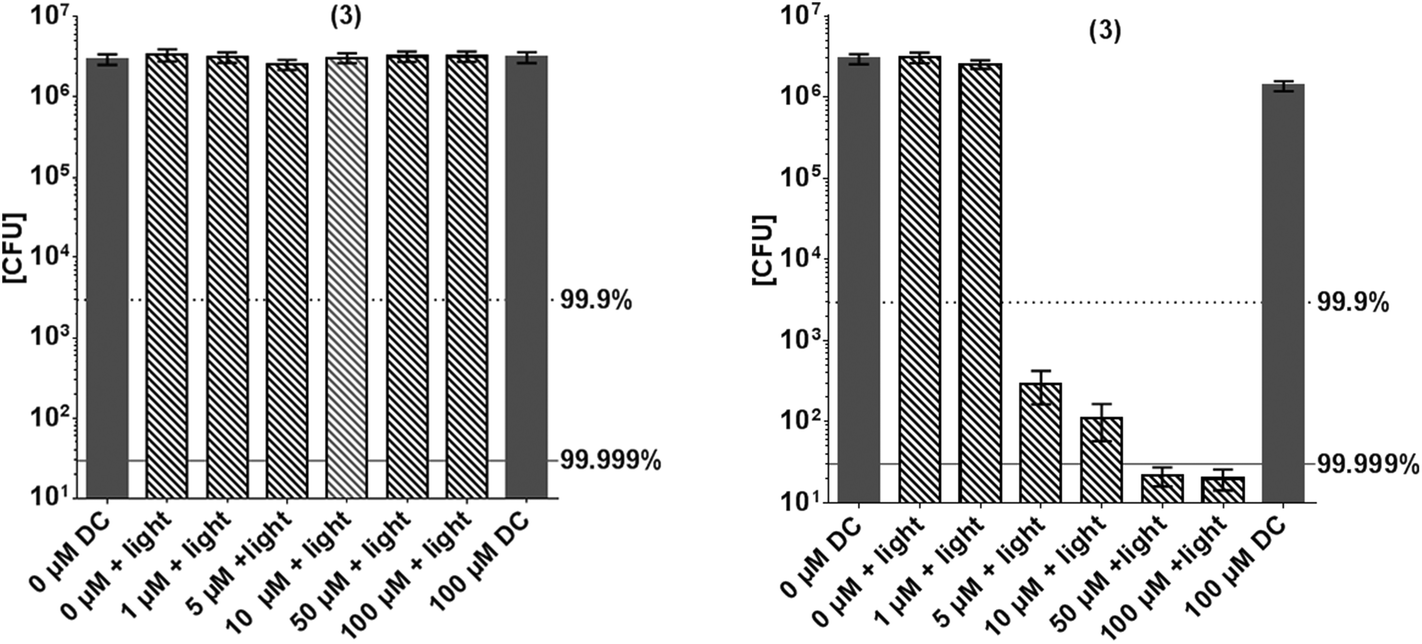 | ||
| Fig. 5 Photodynamic inactivation of S. aureus (left) and E. coli (right) by (3) using the incoherent light source PDT1200 (output-intensity of 50 mW cm−2, 10 min; corresponding to an applied energy of 30 J cm−2), incubation period of (3) for 60 min. Surviving colonies were counted 24 h later. Dark grey column: controls without illumination. Hatched columns: (3) +light activation. Grey dotted line corresponds to a reduction of 3log10 steps (99.9% killing efficacy, black solid line (5log10 steps reduction, 99.999% killing efficacy), black bars related to controls without illumination and without (3) (n = 3 independent experiments: bars represent the mean +95% confidence interval). DC: dark control. | ||
The differences in structure of the bacterial cell wall may be reasoned as explanation for these findings: Gram-positive bacteria possess a thick layer of peptidoglycan on their exterior. This polymer, consisting of sugars and small peptides forms a mesh-like layer having regular cavities. The small peptides linking the sugar chains also contain glutamic acid, whose negatively charged side chain residue protrudes in the cavities formed. Thus, (3) with its unique structure and two additional positive charges may be trapped in the outer layers of the peptidoglycan structure by inclusion and charge interaction. Not reaching the inner cell wall, the ROS generation only takes place in a suitable amount on the surface of S. aureus, not substantially damaging the microorganism. The cell wall of Gram-negative bacteria is far thinner than the one of Gram-positive bacteria. In addition, Gram-negative bacteria are surrounded by an additional outer membrane, which contains a layer of lipopolysaccharides on its exterior. Long sugar chains protrude from the surface. At the end of these sugar chains, near the cell wall, they are phosphorylated on several occasions. The proximity of the sugar surfaces offers the dye a similar environment as for example in cyclodextrines, which can include and bind aromatic chromophores like Fuchsin, Crystal Violet or MB.55 Poly-cyclodextrin structures can saturate with high concentrations of MB 1.56 The binding strength to (1) is strongly enhanced, when the sugar moieties carry negatively charged substituents like carboxylates.57 The stability of these dyes in cyclodextrins is also enhanced, by a protective effect of the water-shielding sugar planes. A similar binding event may take place on the surface of E. coli. The phenothiazinium derivatives diffuse to the area of negative charge accumulation on the lower end of the sugar chains and bind to the lipopolysaccharides.58 This can be explained as follows: (3) is strongly bound by the multiple electrostatic interactions with its positively charged groups and is then “included” by the dense sitting sugar planes protecting the chromophore from destructive influences of the medium. Upon irradiation a sufficient concentration of singlet oxygen is generated close to the thin cell wall of the Gram-negative bacterium causing much more severe damage than to the thick layer in S. aureus. Due to the additional stabilization of the dye by the assumed “inclusion”, the PS is photodynamically active over a longer period of time, when sitting on the surface of the Gram-negative bacteria. If the compound is localized on the exterior, a stronger photobleaching can be assumed.
It is well acknowledged that positive charges at a PS lead structure allow better attachment to the exterior wall of bacteria. Surprisingly, and as mentioned above, S. aureus was not affected by the PIB treatment when (3) is used. Therefore, further experiments were conducted in order to examine the structure–response relationship of this compound in particular. Uptake/attachment experiments show that the compound attaches to both bacterial strains, Gram-positive and Gram-negative bacteria, in a comparable amount (Fig. 6). In the first supernatant it can be seen that slightly more dye is detached from S. aureus, but within experimental accuracy this does not explain the fact that S. aureus is not inactivated at all (Fig. 5). The phototoxicity studies that were performed with additional washing steps show the same effect as the phototoxicity experiments in suspension, E. coli was inactivated whereas S. aureus was not. The above mentioned explanation regarding localization, stabilization and attachment reflects a possible mechanism being responsible for our observations. However, further studies are in progress and shall be presented in a publication in the future, as improving PIB against Gram-negative species is an important issue in current research.59
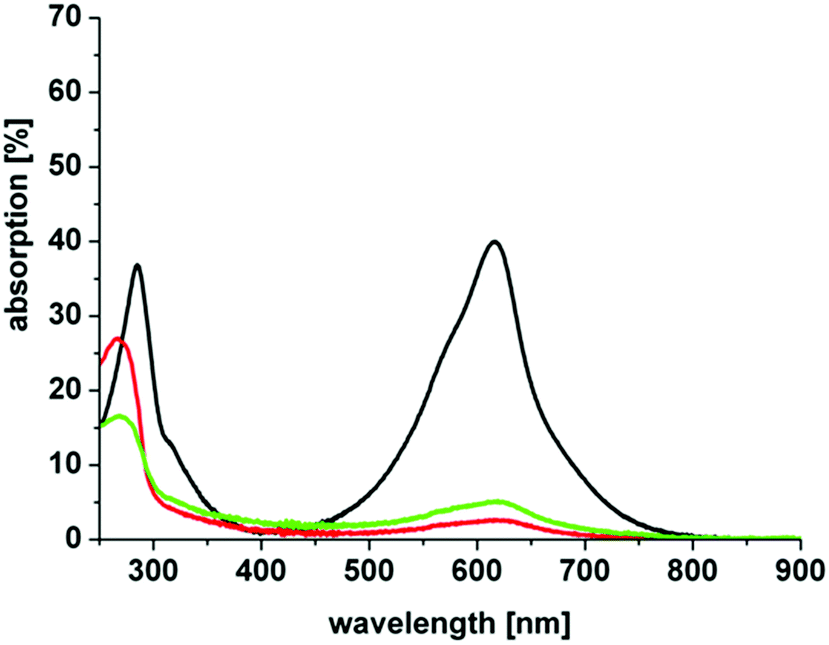 | ||
| Fig. 6 Uptake/attachment of (3) by S. aureus (green line) and E. coli (red line) incubated with 5 μM of (3) in H2O for 60 min. The green and red lines represent the absorption of the first supernatant after incubation, respectively. The black line represents 5 μM of (3) in H2O. The first supernatants show that (3) attaches to both bacterial strains. | ||
Derivative 6, an analogue with cyclic ether substituents, showed better efficacy in comparison to (4), which carries a positively charged nitrogen atom in the six-membered rings located at 3- and 7-position of the phenothiazinium chromophore (Fig. 7).
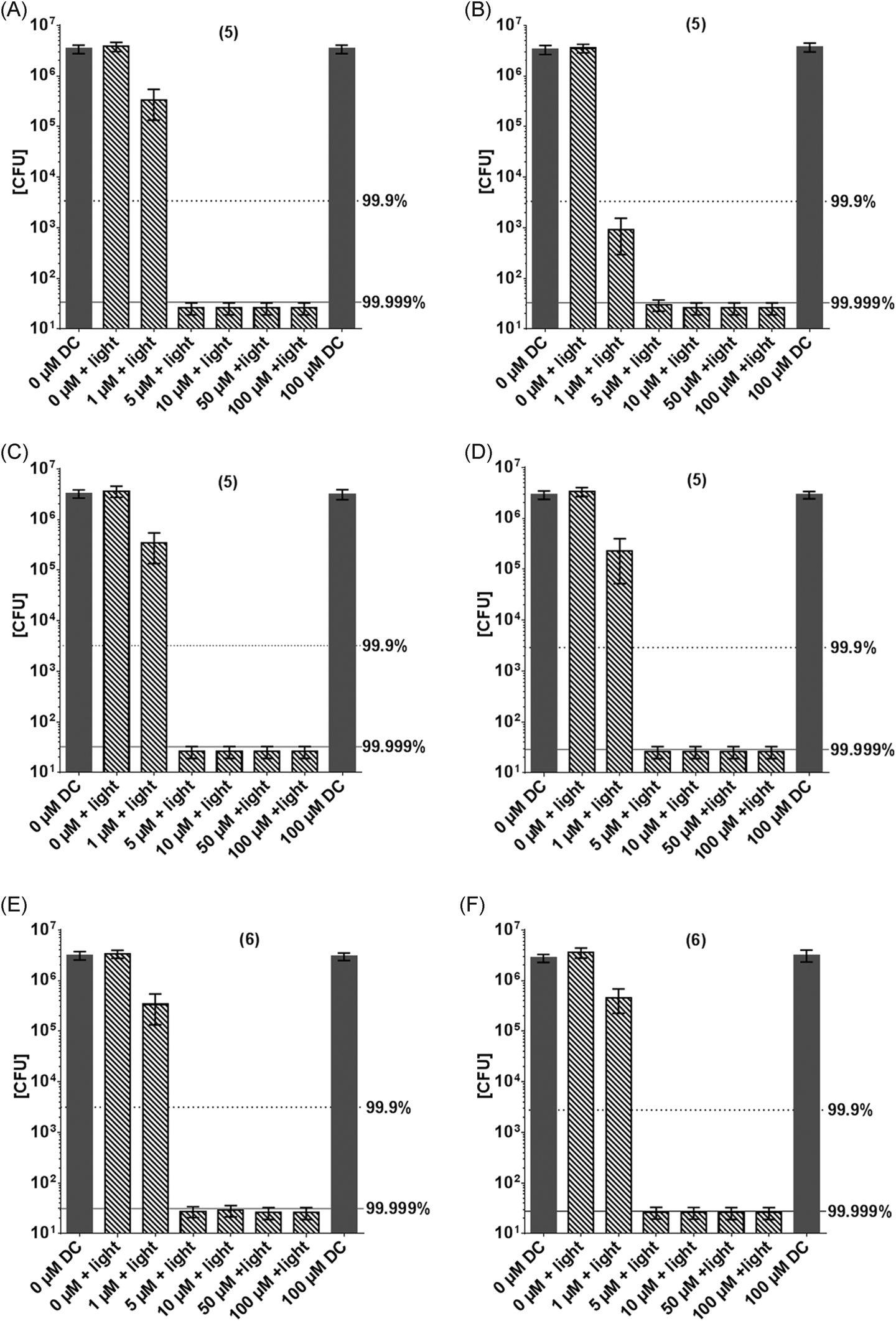 | ||
| Fig. 7 Photodynamic inactivation of S. aureus (A, B & E) and E. coli (C, D & F) by (5) (A–D) and (6) (E & F) using the incoherent light source PDT1200 (output-intensity of 50 mW cm−2, incubation for 10 min (A, C) or 60 min (B, D, E & F), irradiation for 10 min, corresponding to an applied energy of 30 J cm−2. Surviving colonies were counted 24 h later. Dark grey column: controls without illumination or PS only. Hatched columns: PS + light activation. Grey dotted line corresponds to a reduction of 3log10 steps (99.9% killing efficacy, black solid line (5log10 steps reduction, 99.999% killing efficacy), black bars related to controls without illumination and without (5) or (6) (n = 3 independent experiments: bars represent the mean +95% confidence interval). DC: dark control. | ||
The compounds 5 and 6 proved to be the most potent examples investigated in this study, with an antimicrobial efficacy of >7log10 steps at only 5 μM PS concentration at the given irradiation conditions (Fig. 7). They are characterized by a good balance between lipophilicity and hydrophilicity, and have the ability to develop hydrogen bonds in addition to the bond by its positively charged center part. This enables good penetration and adhesion of the dyes in the peptidoglycan structure and also in the lipopolysaccharide layer. Thus, the cell wall of Gram-negative, as well as the one of Gram-positive bacteria is saturated with enough PS to cause quick and severe damage to both species upon illumination.
| Eff. Tox. of 1O2, order of effectivity: (1) ≈ (5) > (6) ≈ (2) > (4) > (3) | (5) |
The toxicity data show the highest effect with (1), (5) and (6), which is reflected by the value for Eff. Tox. of 1O2. Also, (2) shows a high toxicity, corresponding to the predicted effective 1O2 toxicity, but since a substantial dark toxicity adds to this killing effect it will not be taken into further consideration for the correlation. The two lowest values for 1O2 toxicity, determined for (3) and (4), reflect the low photokilling of these two PS. A value for predicting the overall toxicity, however, clearly must take many more features, like PS binding ability, change of the ΦΔ upon binding with bacteria cell structures, site of 1O2 generation, photostability upon irradiation, etc., into account. As reported herein, (3) shows different phototoxic behaviour for S. aureus and E. coli, which cannot simply be explained by the values presented in Table 2. The herein presented “Eff. Tox. for 1O2” should be considered only as a first guideline for PS selection based on the criterion of singlet oxygen phototoxicity.
Conclusions
The four hydrophilic derivatives with the ability of additional hydrogen bonding (5, 6) or additional electrostatic interaction (3, 4) showed fast and effective antimicrobial action against bacteria. With Gram-positive Staphylococcus aureus and Gram-negative Escherichia coli the most effective derivative 5 reached a maximum efficacy of >5log10 steps (≥ 99.999%) of bacteria killing in 10 minutes (5 μM, 30 J cm−2) after one single treatment with the incoherent light source PDT1200 (λmax = 660 nm, 50 mW cm−2). In contrast to the parent compound 1 and the lipophilic derivative 2 they showed no inherent dark toxicity.In this study we confirmed, that phenothiazinium derivatives with cyclic substituents at the auxochromic positions are more stable than acyclic analogs. All hydrophilic derivatives showed good photostability and neglectable aggregation behavior. Additional positive charges are advantageous to suppress aggregation of the compounds. In the concentration range up to 200 μM nearly no aggregation can be observed especially for compound (4) and (6). This might improve the PIB application by extension of the therapeutic concentration window.
In addition we identified one derivative with unique antimicrobial selectivity. Compound 3, comprising two additional primary positive charges, was selective and effective against Gram-negative Escherichia coli (5 μM, 4log10 steps inactivation) over Gram-positive Staphylococcus aureus (100 μM, <1log10 steps inactivation). After correlation to the photophysical and chemical properties of the PS, a reasonable explanation might be that (3) is strongly bound by the multiple electrostatic interactions with its positively charged groups and is then “included” by the dense sitting sugar planes on the surface of the Gram-negative bacterium. Upon irradiation a sufficient concentration of singlet oxygen might be generated close to the thin cell wall of E.coli causing much more severe damage than to the thick layer in S. aureus.
Ongoing experiments aim at more insight of the proposed mode of action, which will be the focus of a subsequent publication.
This new generation of phenothiazinium derivatives might allow the development of more efficient PS with shorter process times and higher antimicrobial activity in comparison to MB 1 and its well-known derivatives.
Abbreviations
| MB | Methylene blue |
| PS | Photosensitizer |
| PIB | Photodynamic inactivation of bacteria |
| CFU | Colony forming unit |
| ROS | Reactive oxygen species |
| TFA | Trifluoro acetic acid |
| TMPyP | 5,10,15,20-Tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluenesulfonate) |
Acknowledgements
We gratefully acknowledge Martin Rappl, Jana Schiller and Ewa Kowalewski for their excellent technical assistance. Anita Gollmer would like to thank the German Research Foundation for funding (DFG-GO-2340/1-1). No conflict of interest is declared.References
- D. Cressey, Nature News Blog, September, 2013 Search PubMed.
- A. P. Magiorakos, A. Srinivasan, R. B. Carey, Y. Carmeli, M. E. Falagas, C. G. Giske, S. Harbarth, J. F. Hindler, G. Kahlmeter, B. Olsson-Liljequist, D. L. Paterson, L. B. Rice, J. Stelling, M. J. Struelens, A. Vatopoulos, J. T. Weber and D. L. Monnet, Clin. Microbiol. Infect., 2012, 18, 268–281 CrossRef CAS PubMed.
- P. J. van Duijn, M. J. Dautzenberg and E. A. Oostdijk, Curr. Opin. Crit. Care, 2011, 17, 658–665 CrossRef PubMed.
- T. Maisch, S. Hackbarth, J. Regensburger, A. Felgentraeger, W. Baeumler, M. Landthaler and B. Roeder, J. Dtsch. Dermatol. Ges., 2011, 9, 360–366 Search PubMed.
- T. Maisch, T. Shimizu, A. Mitra, J. Heinlin, S. Karrer, Y. F. Li, G. Morfill and J. L. Zimmermann, J. Ind. Microbiol. Biotechnol., 2012, 39, 1367–1375 CrossRef CAS PubMed.
- T. G. Klampfl, G. Isbary, T. Shimizu, Y. F. Li, J. L. Zimmermann, W. Stolz, J. Schlegel, G. E. Morfill and H. U. Schmidt, Appl. Environ. Microbiol., 2012, 78, 5077–5082 CrossRef PubMed.
- T. Maisch, T. Shimizu, Y. F. Li, J. Heinlin, S. Karrer, G. Morfill and J. L. Zimmermann, PLoS One, 2012, 7, e34610 CAS.
- S. Matsuzaki, M. Rashel, J. Uchiyama, S. Sakurai, T. Ujihara, M. Kuroda, M. Ikeuchi, T. Tani, M. Fujieda, H. Wakiguchi and S. Imai, J. Infect. Chemother., 2005, 11, 211–219 CrossRef PubMed.
- F. Harris, L. K. Chatfield and D. A. Phoenix, Curr. Drug Targets, 2005, 6, 615–627 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- M. Wainwright, Int. J. Antimicrob. Agents, 2000, 16, 381–394 CrossRef CAS.
- C. M. Cassidy, R. F. Donnelly and M. M. Tunney, J. Photochem. Photobiol., B, 2010, 99, 62–66 CrossRef CAS PubMed.
- S. Menezes, M. A. M. Capella and L. R. Caldas, J. Photochem. Photobiol., B, 1990, 5, 505–517 CrossRef CAS.
- H. Singh and D. D. Ewing, Photochem. Photobiol., 1978, 28, 547–552 CrossRef CAS PubMed.
- P. S. Zolfaghari, S. Packer, M. Singer, S. P. Nair, J. Bennett, C. Street and M. Wilson, BMC Microbiol., 2009, 9 Search PubMed.
- L. M. Giroldo, M. P. Felipe, M. A. de Oliveira, E. Munin, L. P. Alves and M. S. Costa, Lasers Med. Sci., 2009, 24, 109–112 CrossRef PubMed.
- G. G. Carvalho, M. P. Felipe and M. S. Costa, J. Microbiol., 2009, 47, 619–623 CrossRef CAS PubMed.
- M. S. Baptista and M. Wainwright, Braz. J. Med. Biol. Res., 2011, 44, 1–10 CrossRef CAS PubMed.
- S. J. Wagner, A. Skripchenko, D. Robinette, J. W. Foley and L. Cincotta, Photochem. Photobiol., 1998, 67, 343–349 CrossRef CAS PubMed.
- X. Ragas, T. Dai, G. P. Tegos, M. Agut, S. Nonell and M. R. Hamblin, Lasers Surg. Med., 2010, 42, 384–390 CrossRef PubMed.
- C. Komine and Y. Tsujimoto, J. Endod., 2013, 39, 411–414 CrossRef PubMed.
- M. N. Usacheva, M. C. Teichert and M. A. Biel, Lasers Surg. Med., 2001, 29, 165–173 CrossRef CAS PubMed.
- M. B. Fonseca, P. O. Junior, R. C. Pallota, H. F. Filho, O. V. Denardin, A. Rapoport, R. A. Dedivitis, J. F. Veronezi, W. J. Genovese and A. L. Ricardo, Photomed. Laser Surg., 2008, 26, 209–213 CrossRef PubMed.
- P. V. Araujo, K. I. Teixeira, L. D. Lanza, M. E. Cortes and L. T. Poletto, Acta Odontol. Latinoam., 2009, 22, 93–97 Search PubMed.
- J. A. Williams, G. J. Pearson, M. J. Colles and M. Wilson, Caries Res., 2003, 37, 190–193 CrossRef CAS PubMed.
- M. Wainwright and R. M. Giddens, Dyes Pigm., 2003, 57, 245–257 CrossRef CAS.
- S. A. Gorman, A. L. Bell, J. Griffiths, D. Roberts and S. B. Brown, Dyes Pigm., 2006, 71, 153–160 CrossRef CAS PubMed.
- M. Wainwright, K. Meegan, C. Loughran and R. M. Giddens, Dyes Pigm., 2009, 82, 387–391 CrossRef CAS PubMed.
- M. Wainwright, S. D. Brandt, A. Smith, A. Styles, K. Meegan and C. Loughran, J. Photochem. Photobiol., B, 2010, 99, 74–77 CrossRef CAS PubMed.
- D. Creed, W. C. Burton and N. C. Fawcett, J. Chem. Soc., Chem. Commun., 1983, 1521–1523 RSC.
- O. M. New and D. Dolphin, Eur. J. Org. Chem., 2009, 2009, 2675–2686 CrossRef.
- M. Merchat, G. Bertolini, P. Giacomini, A. Villanueva and G. Jori, J. Photochem. Photobiol., B, 1996, 32, 153–157 CrossRef CAS.
- Y. Nitzan, R. Dror, H. Ladan, Z. Malik, S. Kimel and V. Gottfried, Photochem. Photobiol., 1995, 62, 342–347 CrossRef CAS PubMed.
- K. J. Mellish, R. D. Cox, D. I. Vernon, J. Griffiths and S. B. Brown, Photochem. Photobiol., 2002, 75, 392–397 CrossRef CAS.
- A. Felgentraeger, T. Maisch, D. Dobler and A. Spaeth, Biomed Res. Int., 2013, 482167 Search PubMed.
- L. Strekowski, D. F. Hou, R. L. Wydra and R. F. Schinazi, J. Heterocycl. Chem., 1993, 30, 1693–1695 CrossRef CAS.
- J. Baier, T. Fuss, C. Poellmann, C. Wiesmann, K. Pindl, R. Engl, D. Baumer, M. Maier, M. Landthaler and W. Baeumler, J. Photochem. Photobiol., B, 2007, 87, 163–173 CrossRef CAS PubMed.
- P. K. Frederiksen, S. P. McIlroy, C. B. Nielsen, L. Nikolajsen, E. Skovsen, M. Jorgensen, K. V. Mikkelsen and P. R. Ogilby, J. Am. Chem. Soc., 2005, 127, 255–269 CrossRef CAS PubMed.
- A. A. Miles, S. S. Misra and J. O. Irwin, The Journal of hygiene, 1938, 38, 732–749 CrossRef CAS.
- J. M. Boyce and D. Pittet, Infect. Control. Hosp. Epidemiol., 2002, 23, S3–S40 Search PubMed.
- J. Mosinger and B. Mosinger, Experientia, 1995, 51, 106–109 CrossRef CAS.
- S. B. Brown, J. Griffiths, K. J. Mellish, C. C. O'Grady, D. J. H. Roberts, R. G. Tunstall and D. I. Vernon, Biologically active methylene blue derivatives, US Patent, US 20040147508 A1, 2004 Search PubMed.
- S. B. Brown, C. C. O'Grady, J. Griffiths, K. J. Mellish, R. G. Tunstall, D. J. H. Roberts and D. I. Vernon, (Photopharmica Limited, GB), Biologically active methylene blue derivatives, US Patent, 7,732,439, June 8, 2010 Search PubMed.
- M. N. Usacheva, M. C. Teichert and M. A. Biel, J. Photochem. Photobiol., B, 2003, 71, 87–98 CrossRef CAS PubMed.
- S. Sabbahi, Z. Alouini, M. Jemli and A. Boudabbous, Water Sci. Technol., 2008, 58, 1047–1054 CrossRef CAS PubMed.
- J. Chen, T. C. Cesario and P. M. Rentzepis, J. Phys. Chem. A, 2011, 115, 2702–2707 CrossRef CAS PubMed.
- J. Chen, T. C. Cesario and P. M. Rentzepis, Chem. Phys. Lett., 2010, 498, 81–85 CrossRef CAS PubMed.
- P. A. R. Tafulo, R. B. Queiros and G. Gonzalez-Aguilar, Spectrochim. Acta, Part A, 2009, 73, 295–300 CrossRef CAS PubMed.
- A. Spath, C. Leibl, F. Cieplik, K. Lehner, J. Regensburger, K. A. Hiller, W. Baumler, G. Schmalz and T. Maisch, J. Med. Chem., 2014, 57, 5157–5168 CrossRef PubMed.
- T. Kiesslich, A. Gollmer, T. Maisch, M. Berneburg and K. Plaetzer, Biomed Res. Int., 2013, 2013 Search PubMed , 840417.
- Y. Usui and K. Kamogawa, Photochem. Photobiol., 1974, 19, 245–247 CrossRef CAS PubMed.
- I. Walker, S. A. Gorman, R. D. Cox, D. I. Vernon, J. Griffiths and S. B. Brown, Photochem. Photobiol. Sci., 2004, 3, 653–659 CAS.
- D. B. Vieira and A. M. Carmona-Ribeiro, J. Antimicrob. Chemother., 2006, 58, 760–767 CrossRef CAS PubMed.
- M. R. Hamblin and T. Hasan, Photochem. Photobiol. Sci., 2004, 3, 436–450 CAS.
- X. Zhang, L. Shi, G. Xu and C. Chen, J. Inclusion Phenom. Macrocyclic Chem., 2013, 75, 147–153 CrossRef CAS PubMed.
- I. Kacem, T. Laurent, N. Blanchemain, C. Neut, F. Chai, S. Haulon, H. Hildebrand and B. Martel, J. Biomed. Mater. Res., Part A, 2013 DOI:10.1002/jbma.34965.
- G. M. Zhang, S. M. Shuang, C. Dong and J. H. Pan, Spectrochim. Acta, Part A, 2003, 59, 2935–2941 CrossRef.
- M. N. Usacheva, M. C. Teichert, C. E. Sievert and M. A. Biel, Lasers Surg. Med., 2006, 38, 946–954 CrossRef PubMed.
- S. K. Sharma, T. Dai, G. B. Kharkwal, Y. Y. Huang, L. Huang, V. J. De Arce, G. P. Tegos and M. R. Hamblin, Curr. Pharm. Des., 2011, 17, 1303–1319 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: For selected NMR spectra of the compounds, as well as singlet-oxygen measurements, UV-Vis data concerning aggregation and stability. See DOI: 10.1039/c4pp00309h |
| ‡ Literature known compound: Mazur et al., U.S. (1993), US 5220009 A 19930615. |
| § Literature known compound: Brown et al., biologically active MB derivatives, US2004/0147508A1 and WO2005/054217A1; synthesis modified. |
| ¶ Literature known compound: Brokken-Zijp et al., Brit. UK Pat. Appl. (1982), GB 2083488 A 19820324. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |