
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Safe generation and use of bromine azide under continuous flow conditions – selective 1,2-bromoazidation of olefins†
David
Cantillo
,
Bernhard
Gutmann
and
C.
Oliver Kappe
*
Institute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at
First published on 2nd December 2015
Abstract
Bromine azide (BrN3), a useful but extremely toxic and explosive reagent for the preparation of vicinal 1,2-bromine azide compounds, was safely generated and reacted in situ with alkenes in a continuous flow photoreactor. BrN3 was generated by a novel procedure from NaBr and NaN3 in water, and efficiently extracted into an organic phase containing the alkene thus avoiding decomposition. The resulting addition products have been used for the preparation of several useful building blocks.
Halogen azides XN3 (X = F, Cl, Br, I) are, together with hydrazoic acid (X = H), the simplest possible azide compounds and among the few covalent inorganic azides that have been studied in the last decades. They have been investigated in great depth experimentally and theoretically to clarify their structure and bonding situation.1–3 However, the extraordinary tendency toward explosion and the considerable toxicity presents a significant challenge to the experimental chemist. These materials are thus commonly regarded as high energy species rather than as genuinely valuable synthetic reagents. For instance, bromine azide (BrN3) is extremely explosive and only little is known about its physical and chemical properties. In fact, until recently, iodine azide was the only halogen azide, for which a crystal structure was available. The other halogen azides are extremely sensitive to even smallest variations in temperature and pressure, with explosions occurring at pressure variations of Δp ≥ 67 μbar.1 The reaction energy for the disintegration of BrN3 to Br2 and N2 is very large (404 kJ mol−1), and the decomposition is accompanied by the formation of large amounts of toxic and corrosive gases.1 The Br–N3 bond in this molecule is particularly weak, with a thermodynamic strength near zero.2 Only very recently, Schulz and coworkers succeeded in obtaining BrN3 crystals using a miniature zone-melting procedure and the crystal structure of bromine azide could be elucidated.3 The chemistry of the halogen azides was systematically explored by the group of Hassner.4 Hassner and co-workers for the first time demonstrated the direct addition of halogen azides to olefins in the 1960s. This reaction has finally provided broad access to synthetically highly valuable vicinally functionalized azides and vinyl azides. The addition of IN3 to alkenes usually proceeds by an ionic mechanism in a Markovnikov fashion and with anti-selectivity, whereas ClN3 predominantly reacts by a radical pathway. This reflects the increasing tendency for homolytic cleavage in the order IN3 < BrN3 < ClN3. Accordingly, for BrN3 both radical and ionic reactions have been reported depending on the reaction conditions (Fig. 1).4,5
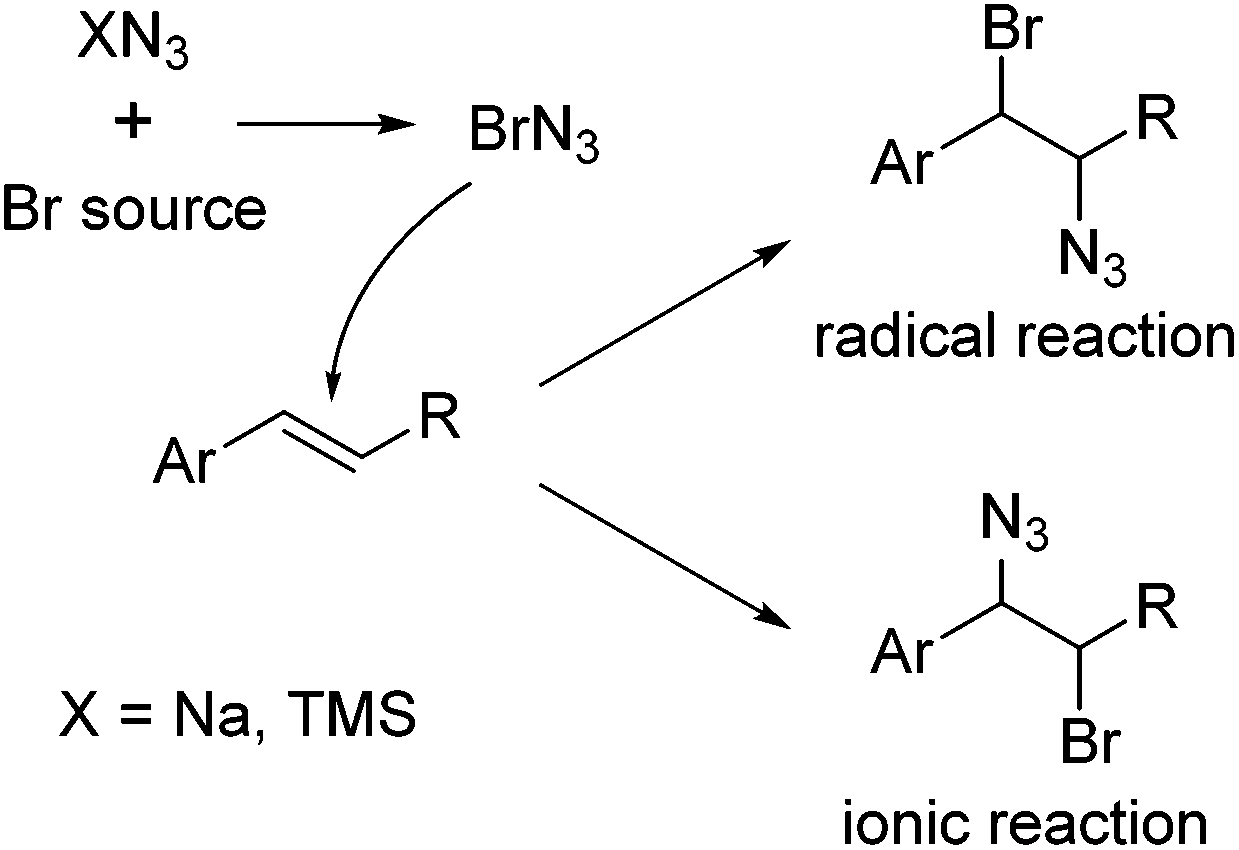 | ||
| Fig. 1 Generation of BrN3 and addition to an alkene. Homolytic or heterolytic cleavage of the Br–N3 bond leads to the radical reaction or the ionic reaction products, respectively. | ||
Early syntheses of bromine azide started from Br2 gas and solid NaN3.6 Combinations with N-bromosuccinimide and NaN3 or TMSN3 allow the generation of BrN3in situ and have become popular for the preparation of vicinal bromoazides from olefins.7 Very recently the group of Finn described an elegant procedure for the in situ generation and trapping of chlorine azide in a biphasic system in batch.8 Chlorine azide was generated from NaN3, NaOCl, and AcOH in aqueous solution and immediately reacted with olefinic compounds in the organic phase. This methodology significantly increases the safety during the laboratory scale generation and use of ClN3 (the reactions were carried out in a 0.6 mmol scale). On the other hand, operation with larger reaction volumes clearly would become problematic.
Recently, continuous flow processing has emerged as an important tool which enables to safely perform chemistries typically “forbidden” in batch.9,10 In a microreactor, highly reactive and/or toxic intermediates can be generated in situ within a closed system and converted directly to nonhazardous intermediates or products by combining multiple reagent streams.11 Moreover, the outstanding heat and mass transfer characteristics of microreactors, together with the fact that the reaction is resolved along the length of the reaction channel, enables a precise control of the residence time of intermediates or products, sometimes down to the microsecond level, by a thermal or chemical quench of the solution. Since the actual reaction volumes in a microreactor/flow device are very small, safety concerns associated with hazardous reagents are further minimized.11 An example of the generation of a halogen azide generated in a microreactor was recently described by the group of Wirth. IN3 was generated from iodine monochloride (ICl) and tetrabutylammonium azide and was reacted with aldehydes yielding carbamoyl azides.12
To overcome the problems associated with BrN3 we envisaged a continuous flow protocol for its safe generation and utilization. Herein we report the generation of BrN3 by a novel route starting from inexpensive NaBr and NaN3 as halogen and azide sources, and Oxone as oxidizing agent. Photolysis in a subsequent continuous flow photoreactor in the presence of an alkene afforded vicinal bromo azides with good yields and essentially complete anti-Markovinkov selectivity.
The continuous flow setup consisted of three feeds (Fig. 2) containing the olefin in an organic solvent (dichloromethane or ethyl acetate) (Feed A), an aqueous solution of NaBr and NaN3 (Feed B), and an excess of Oxone in water (Feed C). The three feeds were introduced in the reactor using sample loops (2 mmol substrate scale, see ESI† for details), and mixed using a PEEK cross-assembly (0.5 mm i.d.). BrN3 is rapidly formed upon mixing of NaN3/NaBr and oxone in the water phase. A reddish color develops in the aqueous phase upon mixing of the feeds, indicating the formation of BrN3, and the colored compound is immediately extracted into the organic phase. BrN3 is highly water sensitive and reacts with water to form toxic and explosive hydrazoic acid (HN3) and HOBr. HN3 and HOBr then further react to ultimately form dinitrogen gas, hydrogen bromide and water. Therefore a rapid extraction of BrN3 into the organic phase is essential to prevent excessive decomposition in the aqueous phase. This efficient extraction was achieved by the segmented flow regime obtained within the tubing. In the organic phase the bromine azides reacts with the olefin (Fig. 2). A 20% excess of NaBr/NaN3 was used to ensure complete conversion of the olefin and thus facilitate the isolation of the 1,2-bromine azide product. Addition of the halogen azide to electron rich alkenes is a fast process with selectivity (ionic vs. radical addition) dependent on the solvent polarity.4a However, when electron withdrawing groups are attached to the olefin, the addition becomes slower, and partial decomposition of the halogen azide can lead to poor yields for the desired addition products. Thus, the continuous flow setup for the generation and extraction of BrN3 was coupled with a flow photochemical reactor to enhance the radical addition for electron-poor substrates. The photochemical reactor13 simply consisted of a transparent FEP tubing (i.d. 0.8 mm, 18 mL volume) wrapped around a compact fluorescent lamp (max. 365 nm) (see ESI† for details). The system was pressurized using a 7 bar back-pressure regulator to properly control the residence time (nitrogen gas is formed due to partial decomposition of BrN3). The reaction mixture was immediately quenched after the photoreactor with an aqueous solution containing an excess of Na2S2O3 after a residence time of approximately 10 min. Using this continuous flow setup large amounts of material can be processed by simply running the flow setup for longer periods of time. Yet, even in case that no addition to the olefin would occur a maximum quantity of 3 mmol of BrN3 would be present in the reactor at any time.
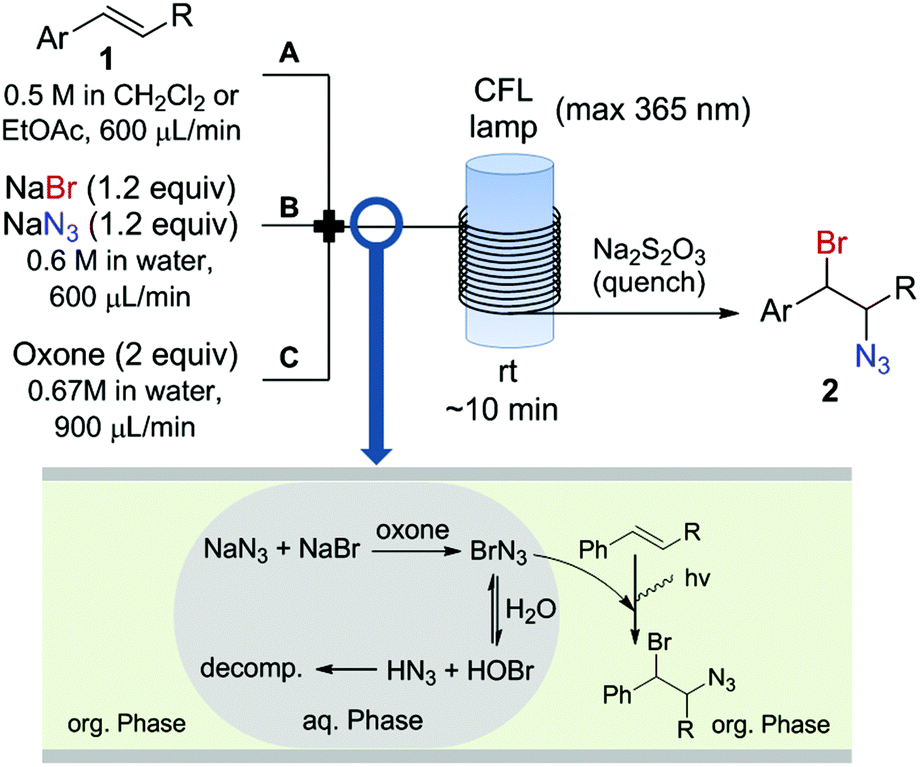 | ||
| Fig. 2 Continuous flow setup utilized for the generation and use of BrN3. The halogen azide is formed in the aqueous phase and rapidly extracted to the organic solvent reacting with the alkene. R = H, Aryl, COOEt. | ||
An initial set of experiments using styrene (1a) and ethyl cinnamate (1b) as model substrates was carried out with the continuous flow setup (Table 1). A 0.5 M solution of substrate in the organic solvent, 1.2 equivalents of NaBr and NaN3, and 2 equivalents of Oxone were introduced in the flow reactor (Fig. 2). The fast addition of BrN3 to styrene did not require irradiation, and full conversion of the substrate was achieved independently of the use of the lamp. Yet, improved selectivity towards the radical process was attained under irradiation (entries 1–4). As expected the solvent had an important influence on the selectivity, with the product of the radical pathway favored when the less polar DCM was used (entries 3–4). In the case of ethyl cinnamate (1b) poor conversions were observed in the absence of light (entries 6 and 8). Prolonged reaction times (>1 h) did not significantly improve the results due to decomposition of BrN3. Gratifyingly, the addition reaction was considerably enhanced under light irradiation and full conversion was obtained both in DCM and EtOAc as solvents (entries 5 and 7). Product resulting from the ionic addition pathway was not observed for this substrate, probably inhibited by the presence of the carboxylate group, and the main side product observed was the 1,2-dibromide (ca. 4%). DCM as solvent and light irradiation were chosen as general conditions for the continuous flow 1,2-bromoazidation of alkenes. GC-MS and HPLC analyses of the crude reaction mixtures demonstrated extremely clean reaction profiles (see Fig. S2 in the ESI†). Thus, the pure 1,2-bromine azide adducts could be isolated in very good yields by simple separation of the organic phase and evaporation of the solvent after washing with brine.
| Entry | Substrate | Solvent | Light irradiation | Conv. (selectivity)b (%) |
|---|---|---|---|---|
| a Conditions: reactions were carried out using the conditions depicted in Fig. 2 on a 2 mmol scale. b Conversions and selectivity were determined by GC-MS for 1a, and HPLC (215 nm) for 1b. Selectivity refers to the proportion of the desired 1,2-bromine azide addition product resulting from the radical pathway (cf.Fig. 1). | ||||
| 1 |
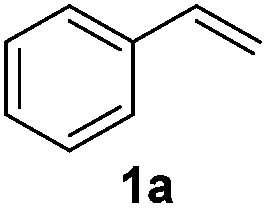
|
EtOAc | On | 99(43) |
| 2 | EtOAc | Off | 99(10) | |
| 3 | DCM | On | 99(82) | |
| 4 | DCM | Off | 99(74) | |
| 5 |
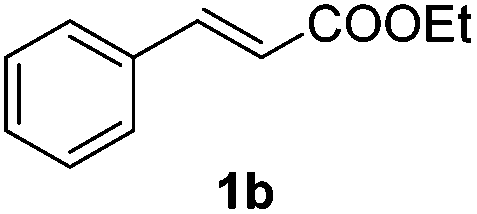
|
EtOAc | On | 99(94) |
| 6 | EtOAc | Off | 43(83) | |
| 7 | DCM | On | 98(96) | |
| 8 | DCM | Off | 41(96) | |
A series of aromatic alkenes were converted into the corresponding bromine azide adducts using the continuous flow setup (Scheme 1). Styrenes (2a, 2c–e) and cinnamates (2b, 2f) decorated with electron withdrawing and electron donating groups gave the corresponding 1,2-bromine azide adducts with very good to excellent yields. Stilbenes (2g–i) were also reacted under the same conditions.‡ It should be noted that the radical addition is not an anti-selective process and resulted in mixtures of products that could not be separated. In the case of the cinnamates 2b, 2f and stilbenes 2g–i mixtures of two pairs of diastereomers showing separated sets of NMR signals were isolated.
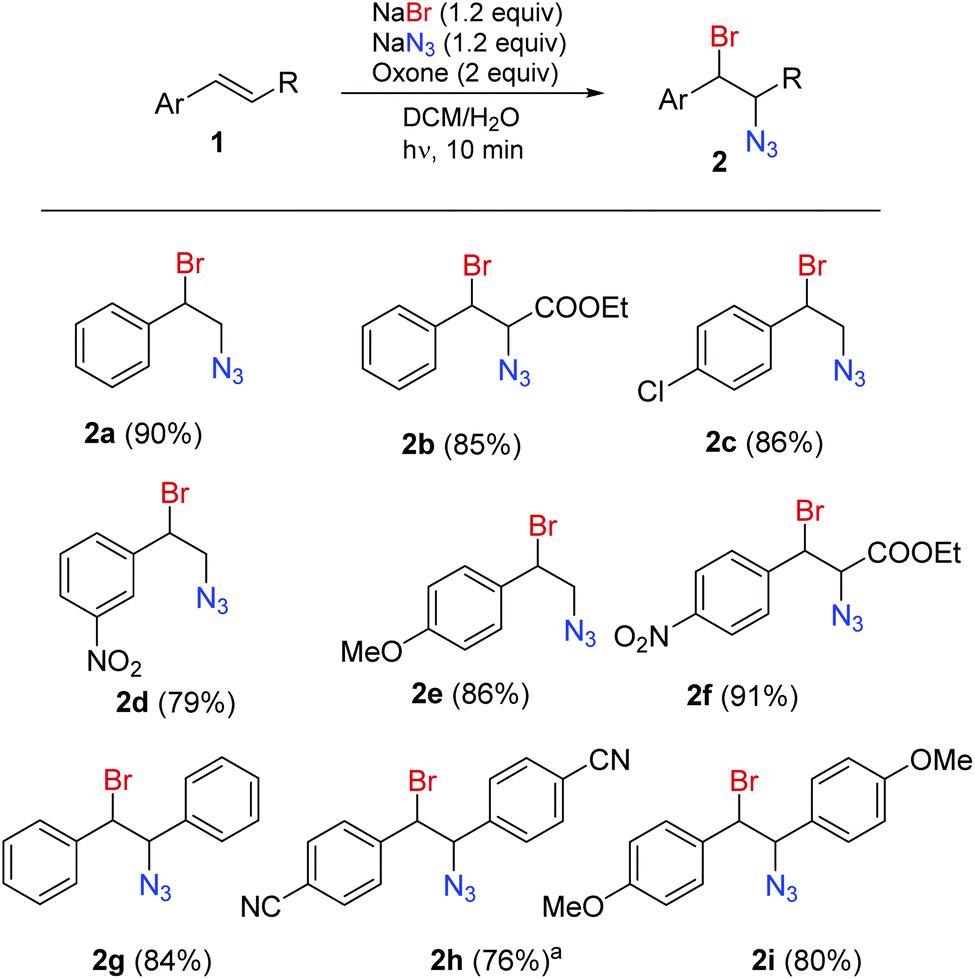 | ||
| Scheme 1 Continuous-flow light-enhanced 1,2-bromoazidation of alkenes (isolated yields). For Experimental details, see the ESI.†a Compound 2h was prepared in batch. | ||
The high purity profiles with which the 1,2-bromine azide reaction mixtures were obtained allowed us to directly use the crude products without purification for exploring several follow-up reactions (Scheme 2). Compound 2a could be transformed to 2-phenyl aziridine 3 by simply adding LiAlH4 in dry diethyl ether solution.14 Aziridines are typically prepared from haloamines and aminoalcohols15 or oximes.16 Aziridination of alkenes is possible via nitrene addition.17 The current method provides an efficient two-step procedure for the aziridination of alkenes. Compound 3 was obtained in a overall yield of 62% (with respect to the starting styrene) (Scheme 2a).
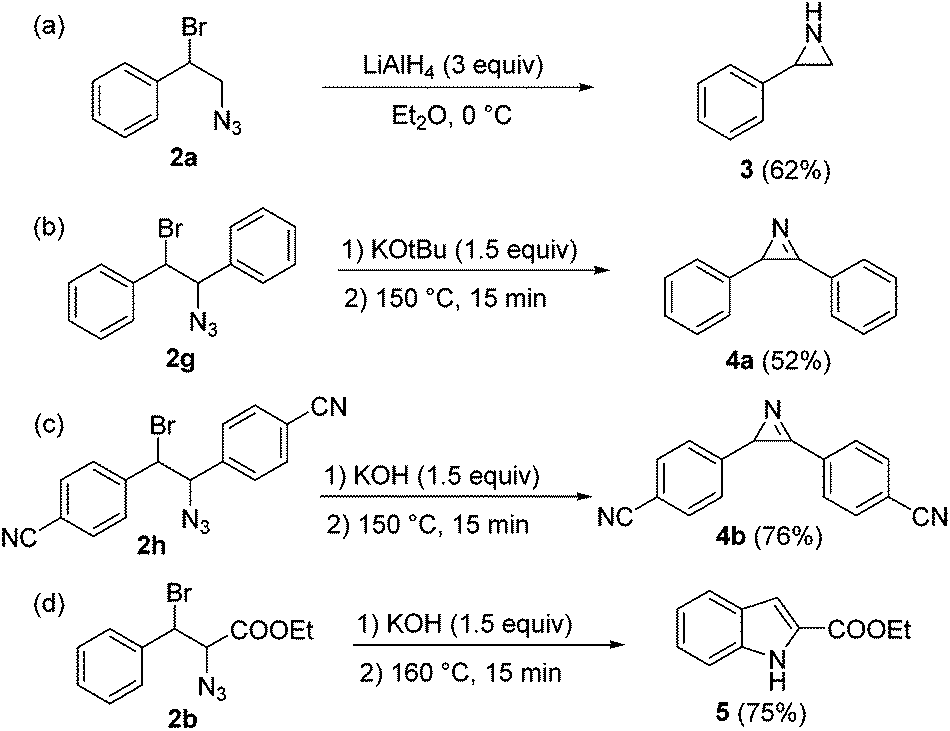 | ||
| Scheme 2 Transformations of 1,2-bromine azides. | ||
One of the most useful applications of 1,2-bromine azides is their transformation into vinyl azides, followed by thermolysis to lead azirines.18 Azirines are found in some natural products, and can be used for the generation of nitrile ylides under UV irradiation.19 Treatment of compounds 2g and 2h with base and themolysis in toluene at 150 °C§ (Scheme 2b and c) resulted in the corresponding pure azirines. Interestingly, different bases were required for the elimination of HBr from 2g and 2h. While KOtBu was needed for the elimination in the case of 2g, KOH sufficed for substrates bearing electron withdrawing groups (2h). Notably, treatment of 2h with KOtBu provoked partial, undesired elimination of hydrazoic acid from the substrate.
Azirines can be transformed into indoles using rhodium catalysts under suitable conditions.20 A special case is the Hemetsberger–Knittel indole synthesis, which enables the preparation of indole-2-carboxylic esters from the corresponding 3-aryl-2-azido-propenoic esters.21 The continuous flow protocol presented herein when applied to substituted alkyl cinnamate substrates thus results in a facile and convenient route for the preparation of indole-2-carboxylic esters. 1,2-Bromine azide 2b was treated with KOH (1.5 equiv.), washed with water, and heated in toluene at 160 °C for 15 min. Notably, the HPLC analysis of the crude reaction mixture revealed that indole 5 had been formed in very high purity. Evaporation of the solvent and crystallization from cyclohexane yielded the pure ethyl indole-2-carboxylate 5 with 75% yield.
The mechanism of the Hemetsberger–Knittel indole synthesis is not very well understood, but stable azirine intermediates have been proposed as well as transient nitrene species (Scheme 3).22,23 In order to shed light on the mechanism, heating of the vinyl azide formed after addition of the base to 2b was repeated at lower temperature (120 °C) and monitored by HPLC analysis. The results revealed very rapid formation of the azirine intermediate via thermal extrusion of N2 and a subsequent, slow transformation of the azirine into the final indole product. Indeed, the azirine intermediate could be detected by HPLC analysis (see ESI† for details). Preliminary DFT calculations on the azide thermolysis and indole formation suggest concerted transition states instead of nitrene intermediates as previously suggested (see ESI† for details).
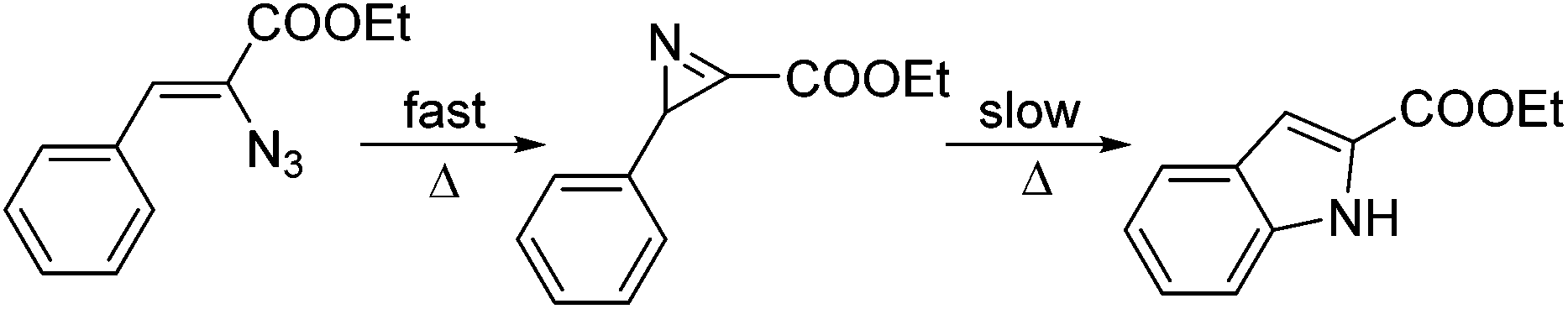 | ||
| Scheme 3 Mechanism of the Hemetsberger–Knittel indole synthesis. | ||
In summary, we have developed a continuous flow protocol for the safe generation and use of BrN3. The procedure is based on a biphasic system. The hazardous reagent is generated in aqueous solution using a novel protocol from NaBr and NaN3, and Oxone as oxidizing agent. Upon formation, BrN3 is rapidly extracted to an organic solvent containing the alkene substrate using a segmented flow regime. The 1,2-bromoazidation can be significantly enhanced by light irradiation. Therefore, a continuous flow photochemical reactor has been integrated in our setup. Addition of BrN3 to electron-poor alkenes takes place efficiently avoiding undesired decomposition of the reagent. The 1,2-bromine azide adducts are obtained in high purity, and can be directly used for the generation of several building blocks such as aziridines, azirines, and indoles using facile, literature known procedures.
Notes and references
- I. C. Tornieporth-Oetting and T. M. Klapötke, Angew. Chem., Int. Ed., 1995, 34, 511 CrossRef CAS.
- T. L. Henshaw, S. J. David, M. A. MacDonald, J. V. Gilbert, D. H. Stedman and R. D. Coombe, J. Phys. Chem., 1987, 91, 2281 Search PubMed.
- B. Lyhs, D. Bläser, C. Wölper, S. Schulz and G. Jansen, Angew. Chem., Int. Ed., 2012, 51, 1970 CrossRef CAS PubMed.
- (a) A. Hassner and F. Boerwinkle, J. Am. Chem. Soc., 1968, 90, 216 CrossRef CAS; (b) A. Hassner and F. W. Fowler, J. Org. Chem., 1968, 33, 2686 CrossRef CAS; (c) A. Hassner, F. Boerwinkle and A. B. Lavy, J. Am. Chem. Soc., 1970, 92, 4879 CrossRef CAS.
- K. Dehnicke, Angew. Chem., Int. Ed., 1967, 6, 240 CrossRef CAS.
- D. A. Spencer, J. Chem. Soc., Trans., 1925, 127, 216 RSC.
- (a) D. Van Ende and A. Krief, Angew. Chem., Int. Ed., 1974, 13, 279 Search PubMed; (b) I. Saikia and P. Phukan, C. R. Chim., 2012, 15, 688 CrossRef CAS.
- R. A. Valiulin, S. Mamidyala and M. G. Finn, J. Org. Chem., 2015, 80, 2740 CrossRef CAS PubMed.
- (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef CAS PubMed; (b) B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298 CrossRef CAS PubMed; (c) S. T. R. Muller and T. Wirth, ChemSusChem, 2015, 8, 245 CrossRef PubMed; (d) S. B. Otvos and F. Fülop, Catal. Sci. Technol., 2015, 5, 4926 RSC.
- For examples of azide preparation in continuous flow, see: (a) C. J. Smith, N. Nikbin, S. V. Ley, H. Lange and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1938 RSC; (b) M. M. E. Delville, P. J. Nieuwland, P. Janssen, K. Koch, J. C. M. van Hest and F. P. J. T. Rutjes, Chem. Eng. J., 2011, 167, 556 CrossRef CAS; (c) C. E. M. Salvador, B. Pieber, P. M. Neu, A. Torvisco, C. K. Z. Andrade and C. O. Kappe, J. Org. Chem., 2015, 80, 4590 CrossRef CAS PubMed.
- (a) Flow Chemistry, ed. F. Darvas, V. Hessel and G. Dorman, De Gruyter, Berlin, 2014 Search PubMed; (b) Microreactors in Preparative Chemistry, ed. W. Reschetilowski, Wiley-VCH, Weinheim, 2013 Search PubMed; (c) Microreactors in Organic Synthesis and Catalysis, ed. T. Wirth, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed; (d) Handbook of Micro Reactors, ed. V. Hessel, J. C. Schouten, A. Renken, Y. Wang and J.-i. Yoshida, Wiley-VCH, Weinheim, 2009 Search PubMed; (e) Chemical Reactions and Processes under Flow Conditions, ed. S. V. Luis and E. Garcia-Verdugo, RSC Green Chemistry, 2010 Search PubMed.
- J. C. Brandt and T. Wirth, Beilstein J. Org. Chem., 2009, 5 CrossRef CAS PubMed , No. 30.
- D. Cantillo, O. de Frutos, J. A. Rincon, C. Mateos and C. O. Kappe, J. Org. Chem., 2014, 79, 223 CrossRef CAS PubMed.
- A. Hassner, J. Matthews and F. W. Fowler, J. Am. Chem. Soc., 1969, 91, 5046 CrossRef CAS PubMed.
- H. Wenker, J. Am. Chem. Soc., 1935, 57, 2328 CrossRef CAS.
- K. N. Campbell and J. F. Mckenna, J. Org. Chem., 1939, 4, 198 CrossRef CAS.
- M. A. Loreto, L. Pellacani, P. A. Tardella and E. Toniato, Tetrahedron Lett., 1984, 25, 4271 CrossRef CAS.
- A. Hassner and J. Keogh, Tetrahedron Lett., 1975, 19, 1575 CrossRef.
- A. F. Khlebnikov and M. S. Novikov, Tetrahedron, 2013, 69, 3363 CrossRef CAS.
- B. J. Stokes, H. Dong, B. E. Leslie, A. L. Pumphrey and T. G. Driver, J. Am. Chem. Soc., 2007, 129, 7500 CrossRef CAS PubMed.
- H. Hemetsberger and D. Knittel, Monatsh. Chem., 1972, 103, 194 CrossRef CAS.
- D. Knittel, Synthesis, 1985, 186 CrossRef CAS.
- W. L. Heaner IV, C. S. Gelbaum, L. Gelbaum, P. Pollet, K. W. Richman, W. DuBay, J. D. Butler, G. Wells and C. L. Liotta, RSC Adv., 2013, 3, 13232–13242 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and copies of NMR spectra of all prepared compounds. See DOI: 10.1039/c5ob02425k |
| ‡ 4,4′-Dicyanostilbene was poorly soluble in organic solvents and the reaction could not be performed in continuous flow. In this case, the 1,2-bromoazidation was carefully performed in batch adding the solution of Oxone to a biphasic mixture of the alkyne in DCM and NaBr/NaN3 in water. |
| § Thermolysis in DCM and ethyl acetate also gave the desired compounds. However, higher purity was obtained in toluene as solvent. |
| This journal is © The Royal Society of Chemistry 2016 |