
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrienamine catalyzed asymmetric synthesis and biological investigation of a cytochalasin B-inspired compound collection†
Magnus
Sellstedt
a,
Melanie
Schwalfenberg
a,
Slava
Ziegler
a,
Andrey P.
Antonchick
a and
Herbert
Waldmann
*ab
aMax-Planck-Institute für Molekulare Physiologie, Otto-Hahn-Strasse 11, 44227 Dortmund, Germany. E-mail: herbert.waldmann@mpi-dortmund.mpg.de
bTechnische Universität Dortmund, Otto-Hahn-Strasse 6, 44221 Dortmund, Germany
First published on 18th November 2015
Abstract
Due to their enhanced metabolic needs many cancers need a sufficient supply of glucose, and novel inhibitors of glucose import are in high demand. Cytochalasin B (CB) is a potent natural glucose import inhibitor which also impairs the actin cytoskeleton leading to undesired toxicity. With a view to identifying selective glucose import inhibitors we have developed an enantioselective trienamine catalyzed synthesis of a CB-inspired compound collection. Biological analysis revealed that indeed actin impairment can be distinguished from glucose import inhibition and led to the identification of the first selective glucose import inhibitor based on the basic structural architecture of cytochalasin B.
Introduction
Cancer cells adapt to their often hypoxic environment and satisfy their increased need for fast nutrient utilization and metabolic building block generation by shifting their metabolism from oxidative phosphorylation (OXPHOS) to glycolysis even when sufficient oxygen is available. This reprogramming of energy metabolism, termed the Warburg effect, is considered a hallmark of cancer.1 To compensate for the lower efficiency of ATP generation in glycolysis as compared to OXPHOS cancer cells upregulate glucose uptake through dysregulated expression of glucose transporters to facilitate import of glucose. In particular, overexpression of glucose transporter GLUT1 has been reported in many types of human cancers.2 Small molecule inhibition of glucose uptake via GLUT1 is a promising strategy for the development of novel anti-cancer drug candidates and inhibitors of GLUT1 and related members of the GLUT family are in high demand.3 The isoindolinone natural product cytochalasin B (CB; Fig. 1) is a potent GLUT inhibitor4 widely used as a biological tool compound. However, CB also inhibits actin polymerization which prevents its use as a drug5 and impairs its application as a tool compound in biology. Clearly, the discovery of CB analogs that inhibit GLUT activity but do not impair the actin cytoskeleton would be of major interest. However, to date only sparse data are available that correlate CB activity and structure4 and a CB derivative that preferentially inhibits glucose uptake has not been identified. We have now synthesized a CB-inspired compound collection employing enantioselective trienamine catalysis as a key transformation. Biological evaluation of the library revealed the first CB-analogue that inhibits glucose uptake in cancer cells but does not impair actin polymerization.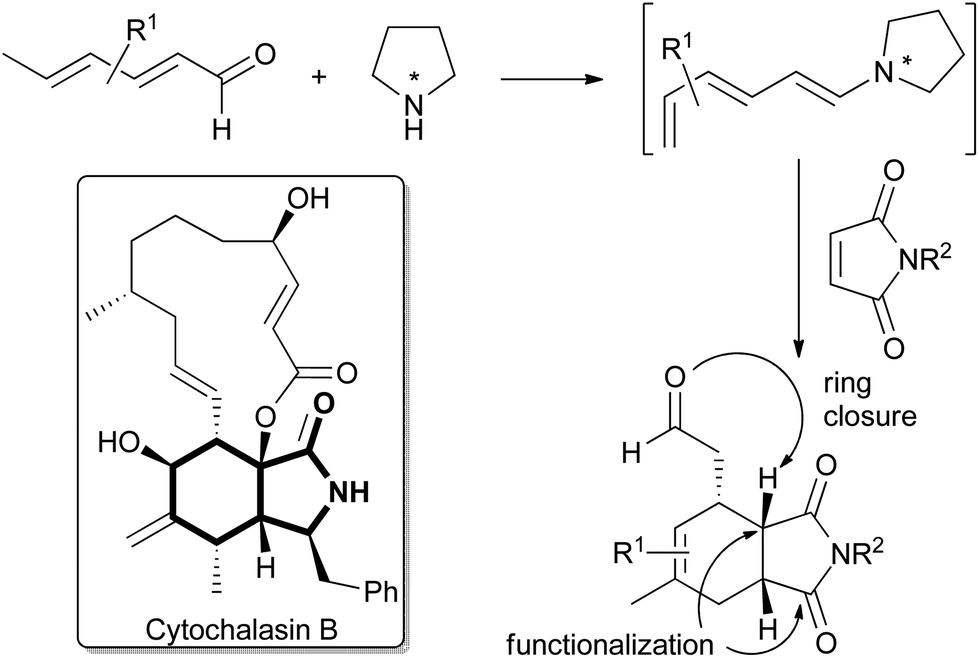 | ||
| Fig. 1 Structure of cytochalasin B (CB) with the semi-saturated isoindolinone motif highlighted and the outline of the synthesis strategy employing enantioselective trienamine organocatalysis. | ||
For the synthesis of cytochalasin B and other cytochalasans an inter- or intramolecular Diels–Alder reaction between a diene- or a triene part and an α,β-unsaturated amide has been employed as a key step.6 Recently, trienamine catalysis has emerged as a powerful method to steer the steric course of asymmetric Diels–Alder reactions,7 and we decided to employ this method in the preparation of a cytochalasin inspired compound library. Chen and Jorgensen and co-workers have described that linear dienals, which in the presence of proline-derived catalysts form asymmetric trienamines, react with highly activated dienophiles such as cyanoacrylates to form Diels–Alder adducts in high enantiomeric excesses.8
However, weaker dienophiles, such as maleimides, did not yield the expected products. Subsequently, maleimides have successfully been reacted with branched aryl-9 and methyl-10 dienones by using cinchinoa alkaloid derived catalysts. However, the trienamine catalysed reaction between dienals and maleimides which would yield the semi-saturated isoindolinone core of the cytochalasans (Fig. 1) has to the best of our knowledge not yet been reported.
Results and discussion
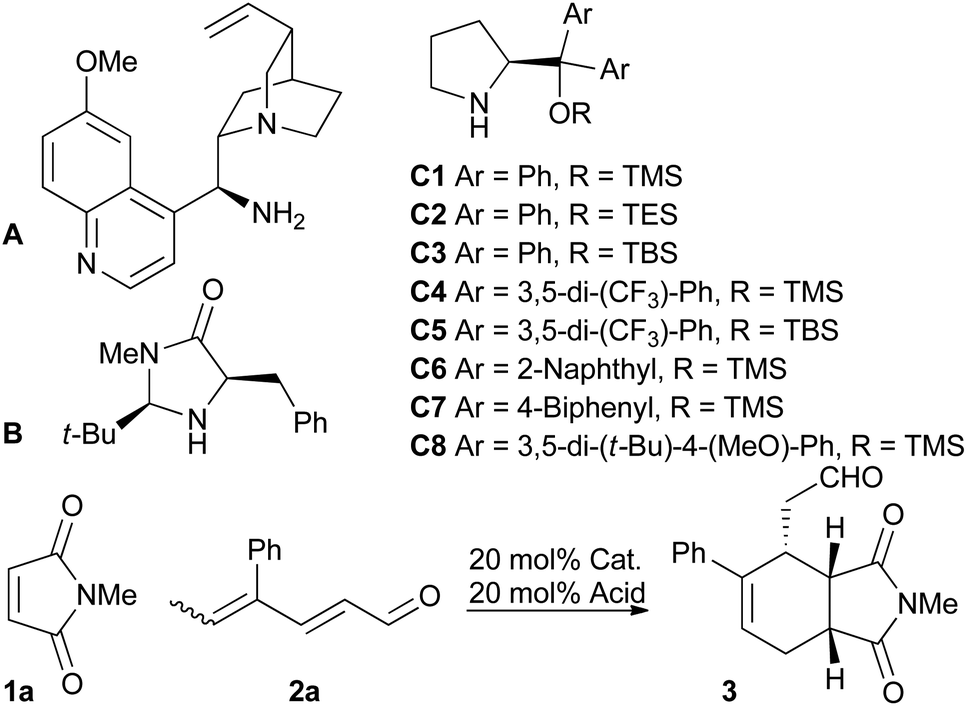
We screened various catalysts for the trienamine reaction between dienal 2a,11 and maleimide 1a (Table 1). While we were unable to isolate any product using the cinchona-based catalyst A previously used with aryldienones,9 the use of the Macmillan catalyst12B afforded a low yield (21%) of ent-3 after 90 hours in 28% ee. Fortunately, the use of diaryl prolinol silyl ethers C proved more successful. The Jorgensen catalysts13C4 and C5 gave much better ee's of 78% and 81%, respectively. These catalysts, however, significantly decreased the reaction rate compared to the diphenyl analogs C1 and C3. The bulky, more electron rich catalyst C8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 gave the highest ee (82%) with a reaction rate similar to that of the diphenyl substituted catalysts. The choice of solvent had a substantial impact on the ee, and chloroform was the best among the tested solvents. The acid had only small effects on the ee, but the yield increased by using benzoic acid instead of 2-fluorobenzoic acid (85% and 65%, respectively). Decreasing the temperature to −10 °C gave slightly higher ee and also increased the yield. However, as expected, this prolonged the reaction time significantly. The absolute stereochemical outcome of the asymmetric trienamine reaction was confirmed by the preparation of and comparison with a known compound15 (see the ESI†).
14 gave the highest ee (82%) with a reaction rate similar to that of the diphenyl substituted catalysts. The choice of solvent had a substantial impact on the ee, and chloroform was the best among the tested solvents. The acid had only small effects on the ee, but the yield increased by using benzoic acid instead of 2-fluorobenzoic acid (85% and 65%, respectively). Decreasing the temperature to −10 °C gave slightly higher ee and also increased the yield. However, as expected, this prolonged the reaction time significantly. The absolute stereochemical outcome of the asymmetric trienamine reaction was confirmed by the preparation of and comparison with a known compound15 (see the ESI†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Acid | Temp. [°C] | Time [h] | Yield [%] | eeb [%] |
| a Reaction conditions: 1a (0.15 mmol), 2a (0.165 mmol), catalyst (0.03 mmol), acid (0.03 mmol), solvent (1.5 mL). b The ee was determined by chiral HPLC using a Chiralpak IA column. c 40 mol% acid. d n.r. = no reaction. OFBA = 2-fluorobenzoic acid, BA = benzoic acid, TMS = trimethylsilyl, TES = triethylsilyl, TBS = tert-butyldimethylsilyl, TFA = trifluoroacetic acid. | |||||||
| 1 | A | CHCl3 | TFAc | 40 | 24 | n.r.d | — |
| 2 | B | CHCl3 | OFBA | 40 | 90 | 21 | −28 |
| 3 | C1 | CHCl3 | OFBA | 40 | 18 | 78 | 34 |
| 4 | C2 | CHCl3 | OFBA | 40 | 17 | 85 | 44 |
| 5 | C3 | CHCl3 | OFBA | 40 | 24 | 74 | 46 |
| 6 | C4 | CHCl3 | OFBA | 40 | 40 | 41 | 78 |
| 7 | C5 | CHCl3 | OFBA | 40 | 64 | 68 | 81 |
| 8 | C6 | CHCl3 | OFBA | 40 | 20 | 62 | 24 |
| 9 | C7 | CHCl3 | OFBA | 40 | 21 | 68 | 6 |
| 10 | C8 | CHCl3 | OFBA | 40 | 22 | 65 | 82 |
| 11 | C8 | PhMe | OFBA | 40 | 19 | 61 | 58 |
| 12 | C8 | EtOH | OFBA | 40 | 41 | 50 | 76 |
| 13 | C8 | DME | OFBA | 40 | 20 | 45 | 45 |
| 14 | C8 | EtOAc | OFBA | 40 | 16 | 59 | 49 |
| 15 | C8 | CHCl3 | BA | 40 | 20 | 85 | 82 |
| 16 | C8 | CHCl3 | AcOH | 40 | 23 | 62 | 80 |
| 17 | C8 | CHCl3 | OFBA | −10 | 120 | 79 | 83 |
To explore the scope of this reaction, various dienals and maleimides were subjected to trienamine catalysis with catalyst C8 (Table 2). The alkyl substituted dienals 2b–d11 were more unstable than the phenyl substituted dienal 2a, and a slightly higher excess (1.5 equiv.) of these dienals was used. The reaction was run at room temperature and the resulting aldehydes were directly treated with a Wittig-reagent to avoid isolation of the somewhat sensitive products. The resulting α,β-unsaturated esters 4–7 were formed with 72–92% ee (Table 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Prod. | R1, R2 | Time 1st step (h) | Yield (%) | ee (%) |
Endo:exo |
E:Z |
| 1 | 4a | H, H | 22 | 37 | 78 | 18:1 |
9:1 |
| 2 | 4b | Bn, H | 22 | 71 | 72 | 19:1 |
19:1 |
| 3 | 4c | Ph, H | 40 | 75 | 92 | 11:1 |
>20:1 |
| 4 | 5a | Me, H | 24 | 34 | 81 | 9:1 |
>20:1 |
| 5 | 5b | Bn, H | 45 | 25 | 73 | >20:1 |
>20:1 |
| 6 | 5c | Ph, H | 45 | 46 | 73 | >20:1 |
>20:1 |
| 7 | 6a | Bn, H | 70 | 58 | 85 | >20:1 |
14:1 |
| 8 | 6b | Ph, H | 47 | 78 | 85 | >20:1 |
15:1 |
| 9 | 7a | Me, H | 21 | 71 | 76 | >20:1 |
>20:1 |
| 10 | 7b | Bn, H | 21 | 58 | 81 | >20:1 |
>20:1 |
| 11 | 7c | Ph, H | 21 | 57 | 79 | >20:1 |
>20:1 |
| 12 | 7d | Ph, Me | 120 | 25 | 82 | >20:1 |
>20:1 |
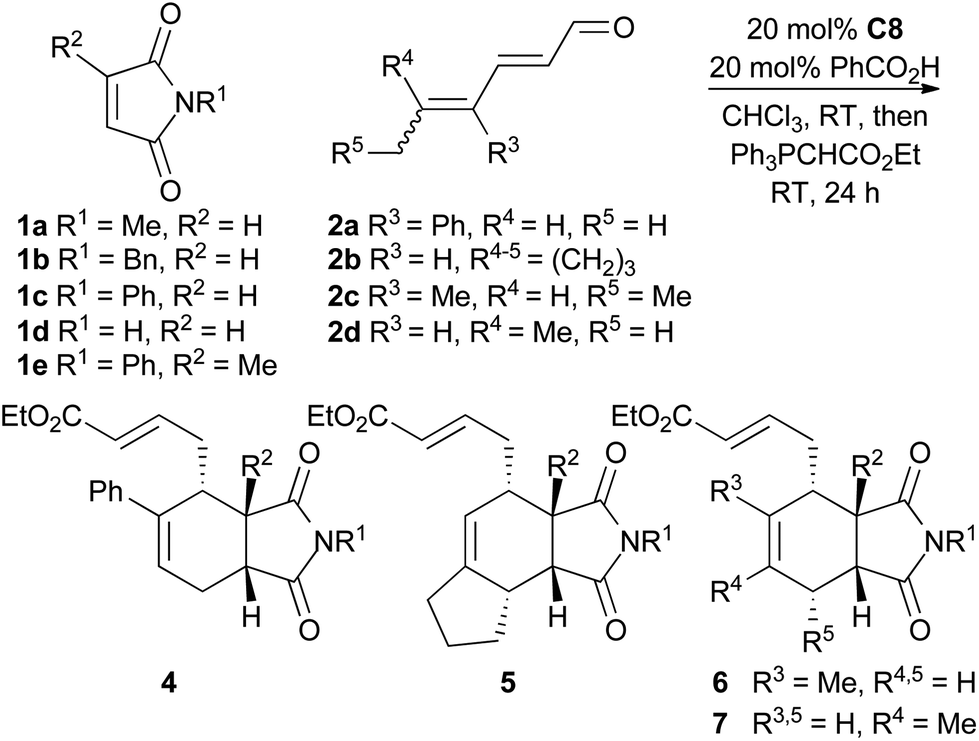
The reaction tolerated a variety of maleimides and dienals. However, linear dienals such as hexadienal failed to give any notable conversion under these conditions, and when a high concentration of hexadienal was used, the maleimide instead underwent a Diels–Alder reaction with the dienal rather than the trienamine. This is in agreement with the same observation for aryl dienones.9 Generally the products were formed in good yields, but compounds 5, which resulted from the least stable dienal, 2b, were obtained in lower yields. Also the 2-methyl substituted maleimide gave a slower reaction and lower yield. The endo-selectivity was good to excellent in all cases.
The prepared α,β-unsaturated esters are themselves interesting as reactants to construct new, more complex natural product-like heterocycles, as exemplified by the synthesis of compound 8 by means of a subsequent highly diastereoselective intramolecular Michael addition (Scheme 1). The configuration of the newly formed stereocenter was determined by NOE measurements. Compound 8 has structural similarities to cytotoxic natural products such as quadrone16 and suberosenol A.17
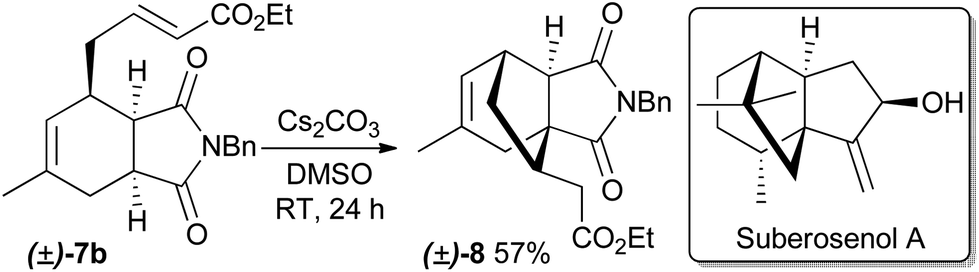 | ||
| Scheme 1 Intramolecular Michael addition of 7b. | ||
For the synthesis of a CB-inspired compound collection we envisioned reduction of the intermediate aldehyde formed in the cycloaddition reaction to avoid potential reactivity problems. For establishment of a suitable synthesis sequence initially the cycloadducts were synthesized as racemates in a thermal non-enantioselective Diels–Alder reaction as shown in Scheme 2. To this end, the dienes were equipped either with a methyl ether (9a) or an allyl ether (9b) which would enable subsequent ring closure by means of ring closing metathesis. The obtained imides 10 were then selectively reduced with DIBAL,18 activated as sulfones, and then coupled with Grignard-reagents using a zinc-mediated reaction19 to produce 11a–g with an additional substituent in the pyrrolidine ring by analogy to the structure of CB. Imides with unprotected N-H did not provide the expected products, but the corresponding TBS-protected imides conveniently gave the substituted N-H amides 11f–g without a separate deprotection step. Boc-protection of these two amides was necessary for further reactions. To further approximate the CB structure, α-hydroxylation of the amide is necessary. For α-hydroxylation of related compounds the Davis oxaziridine has previously been employed,20 and 12b–c were synthesized using similar conditions. To prepare compounds 12a and 12d, however, the reaction with dioxygen18 was more effective. The tertiary alcohols 12 proved unreactive to coupling with acids using coupling reagents such as carbodiimides,20 PyBOP, or HBTU. The alcohols were also unreactive to acid chlorides and 4-DMAP and required deprotonation with sodium hydride to react with acid chlorides. Finally metathesis of 13d–e successfully afforded the macrocyclic compounds 14a–b21 with the same ring-size as CB. Several of the synthesized compounds were also prepared asymmetrically using trienamine catalysis, followed by reduction with sodium borohydride, and then either methylation with TMS-diazomethane, or allylation with tert-butyl-allyl carbonate22 (Scheme 3).
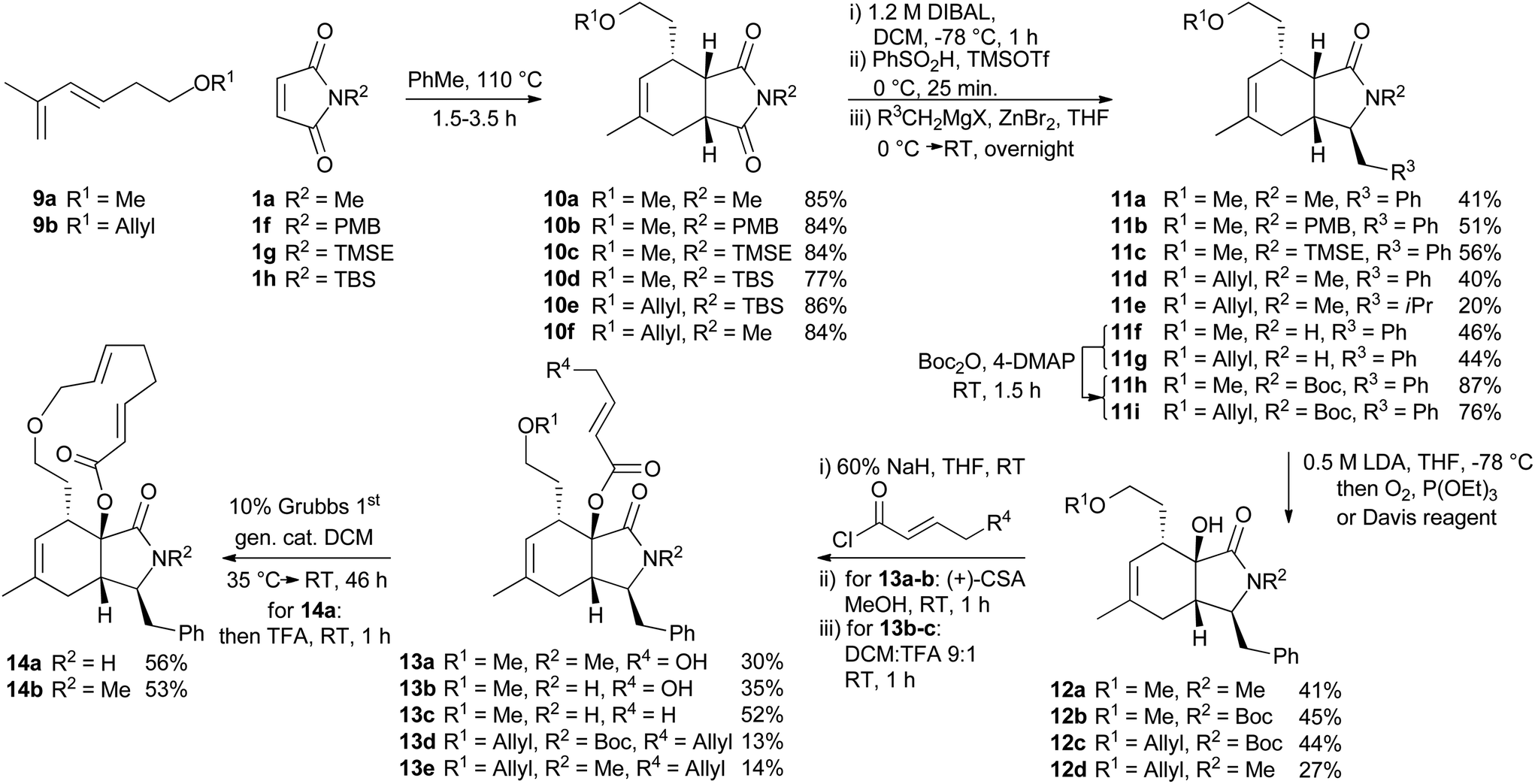 | ||
| Scheme 2 Synthesis of a CB inspired compound collection. PMB = para-methoxybenzyl, TMSE = 2-trimethylsilylethyl, Boc = tert-butoxycarbonyl. | ||
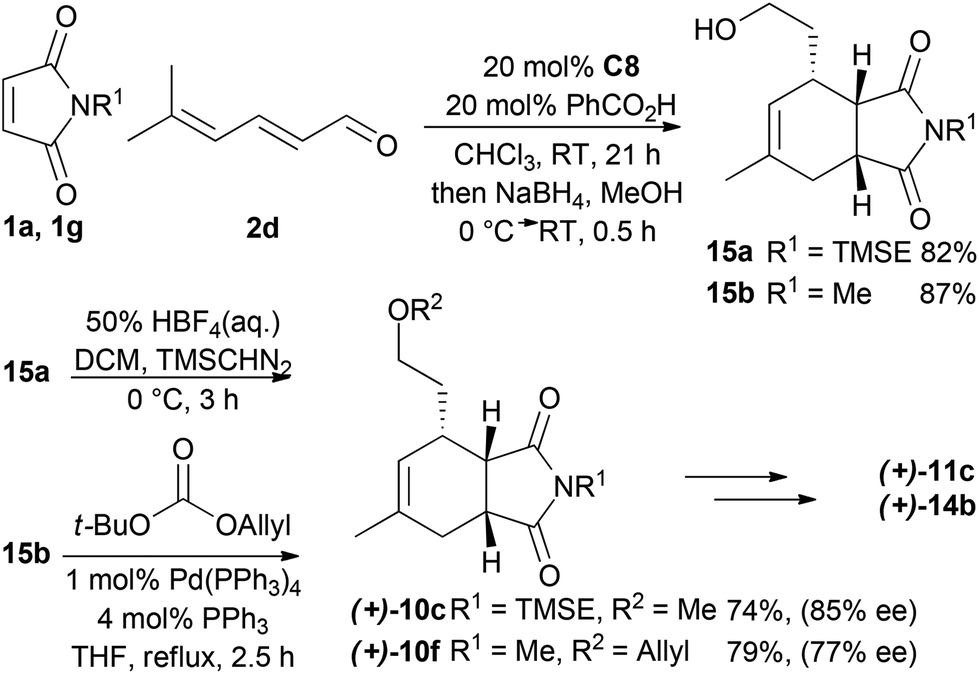 | ||
| Scheme 3 Asymmetric synthesis of selected compounds. | ||
The compounds 4–8 and 10–15 were then investigated for inhibition of glucose import in the human colon cancer cell line HCT116 using a 2-deoxyglucose-based assay.23 Compounds which showed significant inhibition at the initially employed concentration of 180 μM were also analysed at 90 μM and 45 μM. The results revealed that compounds which embody the partially saturated isoindolinone core of CB but lack the medium-sized lactone ring can inhibit glucose import as was in particular observed for 11c and 12c (Fig. 2A). The absolute configuration of these cycloadducts is only of minor importance. These compounds carry only a substituent in the cyclohexane ring and a benzyl group in the pyrrolidine. Analogous bicyclic compounds with an additional substituent α to the carbonyl group (13) were not active. Notably, compound 14a which contains the macrocyclic ring was active in the glucose import assay. Bicyclic compound 12c and tricyclic CB analog 14a were finally investigated for possible influence on the actin cytoskeleton (Fig. 2B). Very gratifyingly, at 180 μM concentration both compounds did not impair the actin cytoskeleton in cells.
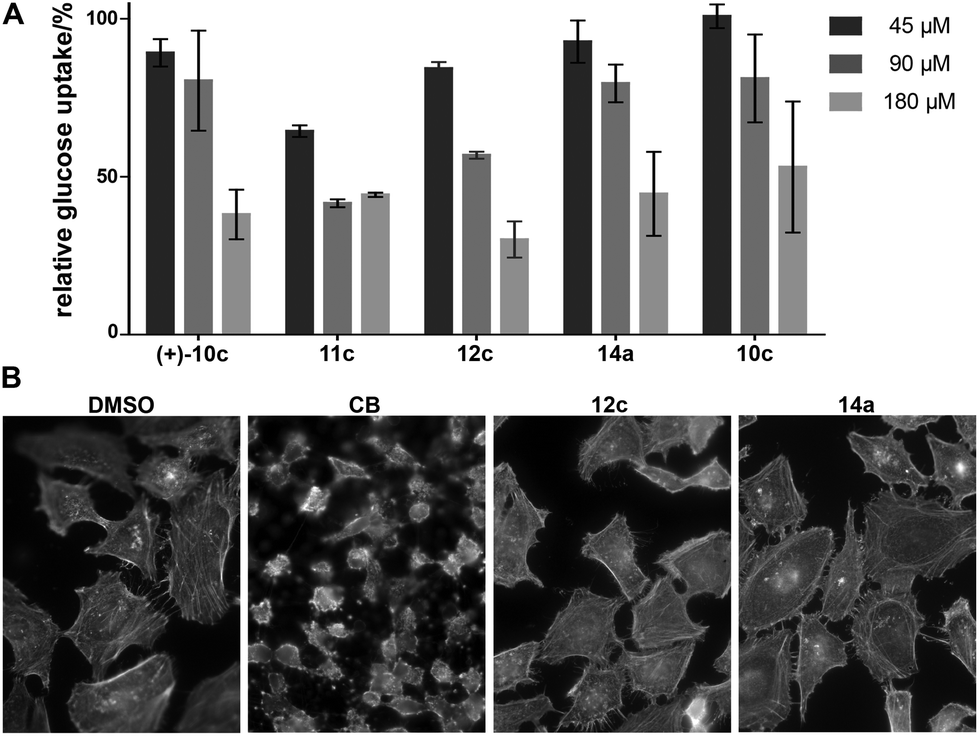 | ||
| Fig. 2 Influence of selected compounds on glucose uptake and the actin cytoskeleton. (A) Glucose uptake was determined in HCT116 cells upon treatment with 2-deoxy-D-glucose (2DG) and with the compounds or DMSO as a control for 30 min followed by detection of 2DG uptake by means of an enzyme coupled reaction using glucose-6-phospate dehydrogenase, NADP+ and diaphorase to reduce weakly fluorescent resazurin to highly fluorescent resorufin. Data are shown as mean values (n = 3) ± s.d. and were normalized to DMSO. (B) The influence of 12c and 14a on the actin cytoskeleton was investigated in HeLa cells. Cells were treated with the compound or DMSO and CB as a control for 1 h prior to fixation and staining of actin using phalloidin labelled with TRITC. Nuclei were stained with DAPI. | ||
Conclusions
Our results demonstrate that it is indeed possible to differentiate the glucose import inhibiting activity of cytochalasin B from its influence on actin polymerization. Structurally significantly simplified CB analogs 12c and 14a are the first compounds that on the one hand resemble the characteristic structural architecture of the natural product at the scaffold level, yet are glucose import inhibitors only. Clearly, these compounds do not reach the potency of the natural product itself, and our data in accord with previous findings4,5 indicate that structural fine-tuning of the macrocycle and the partially saturated isoindolinone scaffold is necessary for full activity. However, they also suggest that by means of appropriate structure–activity correlation it may be possible to develop potent glucose import inhibitors based on the structure of cytochalasin B which are devoid of undesired impact on the actin cytoskeleton.Experimental
Detailed experimental procedures and analytical data are described in the ESI.†Acknowledgements
We gratefully acknowledge the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013) (grant no. 268309) (H. Waldmann) and the Swedish Research Council (grant no. 350-2012-6611) (M. Sellstedt) for financial support.Notes and references
- (a) D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed; (b) G. Kroemer and J. Pouyssegur, Cancer Cell, 2008, 13, 472–482 CrossRef CAS PubMed; (c) M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029–1033 CrossRef CAS PubMed.
- (a) R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS PubMed; (b) N. El Mjiyad, A. Caro-Maldonado, S. Ramirez-Peinado and C. Munoz-Pinedo, Oncogene, 2011, 30, 253–264 CrossRef CAS PubMed.
- (a) J. B. Park, J. Nat. Prod., 2001, 64, 381–384 CrossRef CAS PubMed; (b) Y. Patel and A. Harmon, FASEB J., 2004, 18, A897–A897 Search PubMed; (c) X. Cao, L. Fang, S. Gibbs, Y. Huang, Z. Dai, P. Wen, X. Zheng, W. Sadee and D. Sun, Cancer Chemother. Pharmacol., 2007, 59, 495–505 CrossRef CAS PubMed; (d) T. E. Wood, S. Dalili, C. D. Simpson, R. Hurren, X. Mao, F. S. Saiz, M. Gronda, Y. Eberhard, M. D. Minden, P. J. Bilan, A. Klip, R. A. Batey and A. D. Schimmer, Mol. Cancer Ther., 2008, 7, 3546–3555 CrossRef CAS PubMed; (e) M. Shanmugam, S. K. McBrayer, J. Qian, K. Raikoff, M. J. Avram, S. Singhal, V. Gandhi, P. T. Schumacker, N. L. Krett and S. T. Rosen, J. Biol. Chem., 2009, 284, 26816–26830 CrossRef CAS PubMed; (f) D. A. Chan, P. D. Sutphin, P. Nguyen, S. Turcotte, E. W. Lai, A. Banh, G. E. Reynolds, J.-T. Chi, J. Wu, D. E. Solow-Cordero, M. Bonnet, J. U. Flanagan, D. M. Bouley, E. E. Graves, W. A. Denny, M. P. Hay and A. J. Giaccia, Sci. Transl. Med., 2011, 3 CAS; (g) I. García-Álvarez, L. Garrido and A. Fernández-Mayoralas, ChemMedChem, 2007, 2, 496–504 CrossRef PubMed; (h) O. A. Ulanovskaya, J. Cui, S. J. Kron and S. A. Kozmin, Chem. Biol., 2011, 18, 222–230 CrossRef CAS PubMed; (i) D. Wang, P.-C. Chu, C.-N. Yang, R. Yan, Y.-C. Chuang, S. K. Kulp and C.-S. Chen, J. Med. Chem., 2012, 55, 3827–3836 CrossRef CAS PubMed; (j) Y. Liu, Y. Cao, W. Zhang, S. Bergmeier, Y. Qian, H. Akbar, R. Colvin, J. Ding, L. Tong, S. Wu, J. Hines and X. Chen, Mol. Cancer Ther., 2012, 11, 1672–1682 CrossRef CAS PubMed; (k) C. Granchi and F. Minutolo, ChemMedChem, 2012, 7, 1318–1350 CrossRef CAS PubMed; (l) I. Heisler, T. Müller, S. Golz, J. Telser, H. Rehwinkel, H. Siebeneicher, B. Buchmann, L. Zorn, K. Eis and M. Koppitz, Patent, WO2013182612, 2013 Search PubMed.
- (a) A. L. Rampal, H. B. Pinkofsky and C. Y. Jung, Biochem., 1980, 19, 679–683 CrossRef CAS; (b) J. F. Griffin, A. L. Rampal and C. Y. Jung, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 3759–3763 CrossRef CAS PubMed.
- (a) T. Hirose, Y. Izawa, K. Koyama, S. Natori, K. Iida, I. Yahara, S. Shimaoka and K. Maruyama, Chem. Pharm. Bull., 1990, 38, 971–974 CrossRef CAS PubMed; (b) I. Yahara, F. Harada, S. Sekita, K. Yoshihira and S. Natori, J. Cell Biol., 1982, 92, 69–78 CrossRef CAS PubMed.
- (a) K. Scherlach, D. Boettger, N. Remme and C. Hertweck, Nat. Prod. Rep., 2010, 27, 869–886 RSC; (b) G. Stork, Y. Nakahara, Y. Nakahara and W. J. Greenlee, J. Am. Chem. Soc., 1978, 100, 7775–7777 CrossRef CAS; (c) A. Craven, D. J. Tapolczay, E. J. Thomas and J. W. F. Whitehead, J. Chem. Soc., Chem. Commun., 1985, 145–147 RSC.
- (a) H. Jiang, L. Albrecht and K. A. Jorgensen, Chem. Sci., 2013, 4, 2287–2300 RSC; (b) J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491–1500 CrossRef CAS PubMed.
- Z.-J. Jia, H. Jiang, J.-L. Li, B. Gschwend, Q.-Z. Li, X. Yin, J. Grouleff, Y.-C. Chen and K. A. Jorgensen, J. Am. Chem. Soc., 2011, 133, 5053–5061 CrossRef CAS PubMed.
- X.-F. Xiong, Q. Zhou, J. Gu, L. Dong, T.-Y. Liu and Y.-C. Chen, Angew. Chem., Int. Ed., 2012, 51, 4401–4404 CrossRef CAS PubMed.
- P.-Q. Chen, Y.-C. Xiao, C.-Z. Yue and Y.-C. Chen, Org. Chem. Front., 2014, 1, 490–493 RSC.
- Z.-J. Jia, Q. Zhou, Q.-Q. Zhou, P.-Q. Chen and Y.-C. Chen, Angew. Chem., Int. Ed., 2011, 50, 8638–8641 CrossRef CAS PubMed.
- A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458–2460 CrossRef CAS PubMed.
- J. Franzen, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjaersgaard and K. A. Jorgensen, J. Am. Chem. Soc., 2005, 127, 18296–18304 CrossRef CAS PubMed.
- Y.-k. Liu, C. Ma, K. Jiang, T.-Y. Liu and Y.-C. Chen, Org. Lett., 2009, 11, 2848–2851 CrossRef CAS PubMed.
- CCDC 864374, available from http://www.ccdc.cym.uk/data_request/cif.
- R. L. Ranieri and G. J. Calton, Tetrahedron Lett., 1978, 499–502 CrossRef CAS.
- J. H. Sheu, K. C. Hung, G. H. Wang and C. Y. Duh, J. Nat. Prod., 2000, 63, 1603–1607 CrossRef CAS.
- M. Y. Kim, J. E. Starrett and S. M. Weinreb, J. Org. Chem., 1981, 46, 5383–5389 CrossRef CAS.
- Y. Arai, M. Matsui, A. Fujii, T. Kontani, T. Ohno, T. Koizumi and M. Shiro, J. Chem. Soc., Perkin Trans. 1, 1994, 25–39 RSC.
- A. M. Haidle and A. G. Myers, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12048–12053 CrossRef CAS PubMed.
- The E: Z-selectivity in the macrocyclizations were about 1.5–2:1. The Z-isomers obtained were not fully analytically pure, but appeared to be significantly less active than their E-counterparts as glucose uptake inhibitors.
- J. M. Kraus, H. C. Gits and R. B. Silverman, Tetrahedron Lett., 2012, 53, 1319–1322 CrossRef CAS PubMed.
- N. Yamamoto, M. Ueda, T. Sato, K. Kawasaki, K. Sawada, K. Kawabata and H. Ashida, Curr. Protoc. Pharmacol., 2011, 55, 12.14.51–12.14.22 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02272j |
| This journal is © The Royal Society of Chemistry 2016 |