
Photoswitchable anticancer activity via trans–cis isomerization of a combretastatin A-4 analog†
Jonathon E.
Sheldon
ad,
M. Michael
Dcona
bd,
Charles E.
Lyons
d,
John C.
Hackett
cd and
Matthew C. T.
Hartman
*ad
aDepartment of Chemistry, Virginia Commonwealth University, 1001 W. Main St., Richmond, VA 23284-2006, USA. E-mail: mchartman@vcu.edu
bDepartment of Internal Medicine, Virginia Commonwealth University, 1101 E. Marshall St., Richmond, VA 23298-0663, USA
cDepartment of Physiology and Biophysics, Virginia Commonwealth University, 1101 E. Marshall St., Richmond, VA 23298-0551, USA
dMassey Cancer Center, Virginia Commonwealth University, 401 College St., Richmond, VA 23292-0037, USA
First published on 27th October 2015
Abstract
Combretastatin A-4 (CA4) is highly potent anticancer drug that acts as an inhibitor of tubulin polymerization. The core of the CA4 structure contains a cis-stilbene, and it is known that the trans isomer is significantly less potent. We prepared an azobenzene analog of CA4 (Azo-CA4) that shows 13–35 fold enhancement in potency upon illumination. EC50 values in the light were in the mid nM range. Due to its ability to thermally revert to less toxic trans form, Azo-CA4 also has the ability to automatically turn its activity off with time. Azo-CA4 is less potent than CA-4 because it degrades in the presence of glutathione as evidenced by UV-Vis spectroscopy and ESI-MS. Nevertheless, Azo-CA4 represents a promising strategy for switchable potency for treatment of cancer.
Introduction
Off-target toxicity persists as a challenge in the development and improvement of cancer chemotherapeutics. The use of light is one means to target therapy at the tumor site. In photodynamic therapy (PDT), a photosensitizer is employed to create cytotoxic reactive oxygen species (ROS) at a point of irradiation.1,2 One of the clinical challenges involving PDT is that hypoxic tumor microenvironments can limit the ability to create sufficient ROS for therapy.3,4 As an alternative, researchers have used light to trigger drug activation or drug delivery. Examples include the release of anti-cancer drugs from a negatively charged, cell-impermeable small molecule along with release from cancer-targeting dendrimers, nanoparticles and liposomes.5–13 Although these methods for targeted drug delivery are promising, in all cases the drug activation is irreversible upon illumination. Very recently, Presa and coworkers have developed Pt(II) complexes with ligands that can be switched between more toxic and less toxic forms depending on the wavelength of light used.14 One potential problem for the clinical application of each of these light-activated drug systems is that the activated drug can diffuse away from the illuminated site of the tumor, causing unwanted damage to the tissues it encounters. An ideal light-activated drug would automatically revert to a less potent form over time in order to limit this off-target toxicity. Photoswitchable drugs also offer other unique opportunities in photopharmacology.15CA4 (Fig. 1) is a tubulin polymerization inhibitor and anticancer drug that exhibits selective cytotoxicity toward tumor vasculature.16,17 A variety of cancer cells are highly sensitive to CA4 treatment. EC50 values are often under 10 nM.18,19 A prodrug of CA4 (CA4-phosphate) is currently being tested in clinical trials in combination with existing chemotherapy agents.20,21 In addition, a photocaged version of CA4 has been developed to monitor microtubule dynamics in cells.22 CA4 contains a cis-stilbene structure (Fig. 1a), which is known to be 60-fold more potent than the trans isomer.23 Light could be used to switch between the two isomers, but this process requires <300 nm UV light which is toxic to cells.24 Azobenzenes contain a molecular backbone that resembles the stilbenoid backbone of CA4, with the olefinic carbon atoms replaced with nitrogens (Fig. 1a). Yet, unlike stilbenes, azobenzenes are able to be switched from the trans to cis form with low intensity light that is compatible with cells.25,26 Moreover, when irradiation is terminated, the cis form of azobenzenes relaxes back to the thermodynamically stable trans form with a half-life that ranges from milliseconds to hours.27–29 In principle, therefore, an azobenzene analog of CA4 (Azo-CA4, Fig. 1a) will exhibit the dual properties we desired: enhancement in potency with light and automatic turn-off over time.
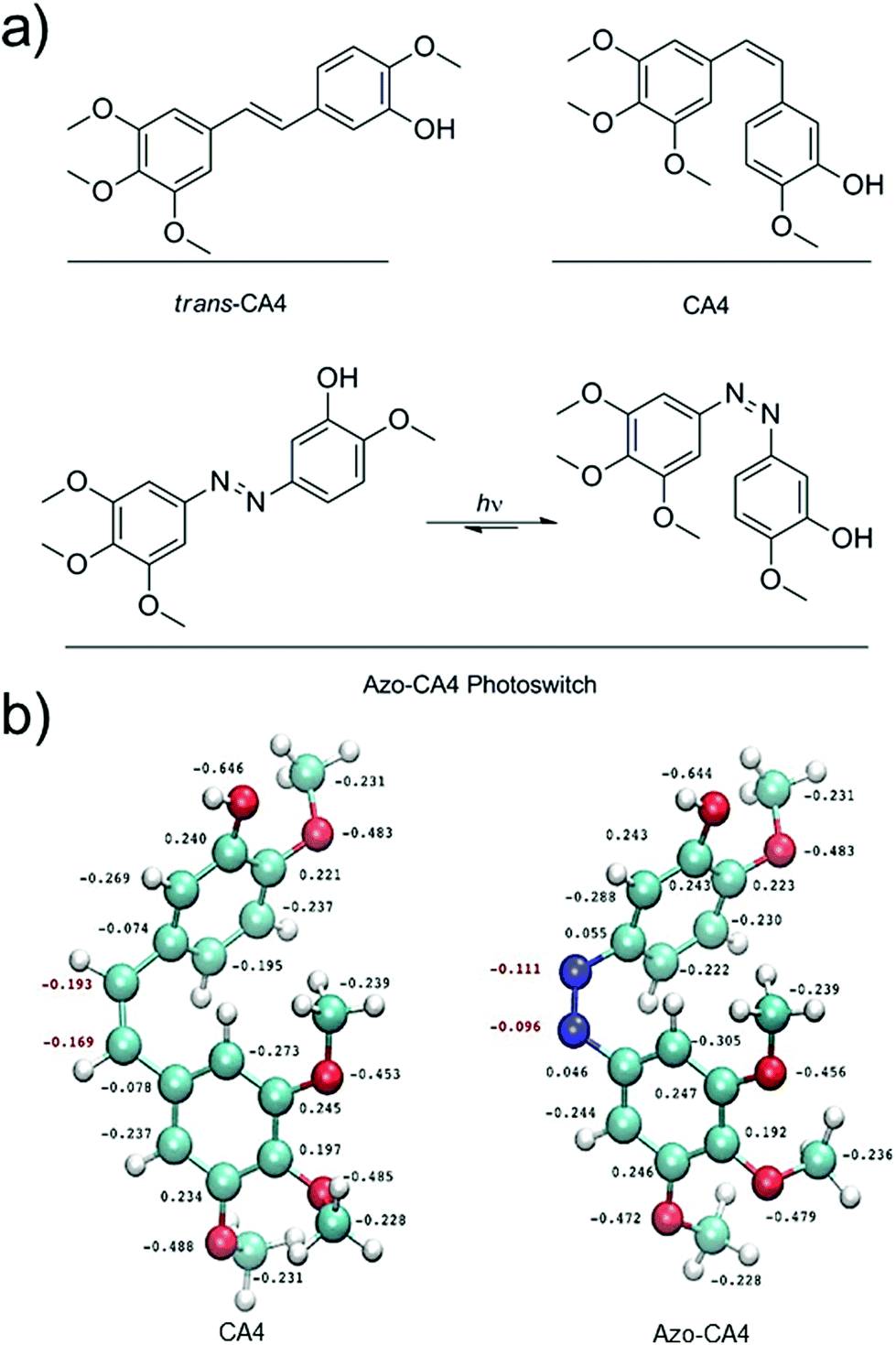 | ||
| Fig. 1 (a) Design of Azo-CA4, a photoswitchable analog of CA4. (b) RI-MP2/def2-TZVP derived electronic structure of CA4 (left) and Azo-CA4 (right). Labeled charges for non-hydrogen atoms were computed using the natural population analysis method. | ||
Very recently, Borowiak et al.30 and Engdahl et al.31 reported the synthesis of azobenzene analogs of CA-4 and have extensively investigated their use as tools for controlling tubulin polymerization in cells. They have also reported EC50 values using various light regimes. Herein we add to the knowledge of these exciting compounds, focusing on the effectiveness of their ability to automatically turn off their activity over time, their effects on endothelial cells, and potential limitations for in vivo use.
Experimental
General methods
All reagents and solvents were purchased from Sigma-Aldrich, VWR, or Fisher Scientific. Reagents for cellular assays were purchased from ATCC, Invitrogen-Gibco, or VWR. The tubulin polymerization assay kit was purchased from Cytoskeleton Inc. 1H NMR spectra and 13C spectra were recorded at room temperature on a Bruker 400 MHz Ultrashield Plus. Proton and Carbon chemical shifts are expressed in parts per million (ppm, δ scale) and are referenced to the solvent peak. UV-Vis experiments were carried out on either NanoDrop ND-1000 or an Agilent 8453 UV/Vis. Cell Titer Blue (CTB) fluorescence was measured on BioTek Synergy H1 Hybrid plate reader. The UV lamp used in all studies consisted of a simple aquarium light fixture containing two Philips PL-S 9W/2P BLB bulbs. The light was placed at distance of 5 cm from the top of plate. The intensity was measured to be 4.4 mW cm−2.Compound synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeOH and EtOH (24 mL) in the presence of 10% Pd/C (400 mg, 0.38 mmol) under Ar. The reaction vessel was purged for 20 minutes with H2 gas, sealed, and stirred for 3 h. The reaction was filtered through Celite and evaporated to yield 1.69 g (94.5%) of a dark brown oil. 1H NMR (400 MHz, CDCl3) δ 6.68 (d, J = 8.4 Hz, 1H), 6.28 (d, J = 2.8 Hz, 1H), 6.25 (dd, J = 8.4, 2.7 Hz, 1H), 3.72 (s, 3H), 3.30 (s, 2H), 0.99 (s, 9H), 0.15 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 146.12, 144.32, 140.65, 114.43, 109.49, 108.20, 56.62, 25.76, 18.43, −4.36, −4.64. ESI-MS (M + H)+ C13H24NO2Si+ calc. 254.1571. obsd 254.1566.
:1 mixture of MeOH and EtOH (24 mL) in the presence of 10% Pd/C (400 mg, 0.38 mmol) under Ar. The reaction vessel was purged for 20 minutes with H2 gas, sealed, and stirred for 3 h. The reaction was filtered through Celite and evaporated to yield 1.69 g (94.5%) of a dark brown oil. 1H NMR (400 MHz, CDCl3) δ 6.68 (d, J = 8.4 Hz, 1H), 6.28 (d, J = 2.8 Hz, 1H), 6.25 (dd, J = 8.4, 2.7 Hz, 1H), 3.72 (s, 3H), 3.30 (s, 2H), 0.99 (s, 9H), 0.15 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 146.12, 144.32, 140.65, 114.43, 109.49, 108.20, 56.62, 25.76, 18.43, −4.36, −4.64. ESI-MS (M + H)+ C13H24NO2Si+ calc. 254.1571. obsd 254.1566.
Molar extinction coefficient determination
A 3.1 mM stock solution of Azo-CA4 was prepared in 23% DMSO in water. The solution was stirred vigorously and kept in the dark. 400 μL of the solution was placed in the each of three tubes and diluted to 1 mL with water to give a final concentration of 1.26 mM Azo-CA4. 2-fold serial dilutions were made and the absorption of each solution was measured on a NanoDrop ND-1000 spectrophotometer. The molar absorptivity, based on three independent trials, was found to be 19150 L mol−1 cm−1.
Half-life of thermal relaxation determination
A solution of 300 μM Azo-CA4 (100 μL) in either 10% CH3CN in PBS, 20% CH3CN, 0.25% DMSO in PBS, 0.9% DMSO in PBS, or 0.25% DMSO in H2O were placed in a 96 well plate and were irradiated with 380 nm light (4.4 mW cm−2) for 1 minute. Absorbance readings were taken from triplicate wells every 1 min for 3 h at 37 °C and subtracted from the fully relaxed absorbance reading. Data was fitted to a 3-parameter exponential curve (SigmaPlot software).Azo-CA4 photoswitching integrity assay
A 64 μM solution of Azo-CA4 in DMSO was placed in a cuvette in an Agilent 8453 UV/VIS at 37 °C for 23 hours. The sample was illuminated for 1 min every 3 hours and the absorbance was measured every 5 minutes.pH sensitivity experiments
A 183 μM solution of Azo-CA4 in 3.2% DMSO in 50 mM phosphate buffer (pH 8.0) was prepared. The absorption spectrum and pH were measured after multiple additions of small volumes of concentrated HCl.Glutathione reduction assay
Azo-CA4 (300 μM in 5.7% DMSO/PBS pH 7.2) in the presence of fully reduced glutathione (10 mM in PBS pH 7.2) and TCEP (5 mM in PBS pH 7.2) was placed in 3 separate wells (100 μL per well) of a 96 well plate at 37 °C. Experiments were performed under UV (380 nm) irradiation (1 min every 2 hours, 4.4 mW cm−2) or protected from light exposure. For experiments in the light, absorbance readings were collected immediately prior to the subsequent light exposure. Absorbance readings were measured on a BioTek Synergy H1 Hybrid plate reader.Characterization of glutathione reduction products by ESI-MS
Samples were loaded on a self-packed fused silica (Polymicro Technologies) trap column (360 micron o.d. × 100 micron i.d.) with a Kasil frit packed with 5–15 micron irregular phenyl C-18 YMC packing. The trap column was connected to an analytical column (360 micron o.d. × 50 micron i.d.) with a fritted tip at 5 micron or less (New Objective) packed with 5 μm phenyl C-18 YMC packing. Peptides were trapped and then eluted into a Thermo Finnigan LCQ deca XP max mass spectrometer with an acetonitrile gradient from 0% to 80% over one hour at a flow rate of between 50–150 nl per minute. The mass spectrometer was operated in data dependent mode. First a MS scan from mass 200 or 300–2000 m/z was collected to determine the mass of molecules eluting at that time, the top two most abundant masses were fragmented into MS/MS scans and placed on an exclusion list. This sequence MS followed by 2 MS/MS scans with exclusion was repeated throughout the 15 minute gradient. Ion chromatograms were plotted of the masses of interest.In vitro tubulin polymerization
Fluorescence in vitro tubulin polymerization assays using porcine tubulin were conducted using a fluorescence based polymerization assay kit (Cat. #BK011P; Cytoskeleton, Denver, CO) and following the manufacturers protocol. Each assay was exposed to varying concentrations of Azo-CA4 (80 μM–800 nM). Samples were either exposed to light (380 nm, irradiated for 1 minute) or kept in the dark. Irradiated samples of Azo-CA4 were added to tubulin containing wells directly after exposure to light. Azo-CA4 samples kept in the dark were added at the same time and the kinetic assay was started immediately and proceeded for 1 hour at 37 °C (Ex. 350 nm, Em. 435 nm). Samples were normalized to assays lacking Azo-CA4. IC50 values were calculated by fitting the points to the four parameter logistic equation using SigmaPlot software.Cell lines and culture conditions
The HUVEC line was cultured in the recommended Basal media supplemented with VEGF growth factors, 5% Fetal Bovine Serum (FBS, Life Technologies), 2% L-glutamine and 1% of a 10000 units per mL penicillin and 10000 μg mL−1 streptomycin solution. The MDA-MB-231 line was cultured in DMEM media with supplemented with 10% FBS and 1% of a 10000 units per mL penicillin and 10000 μg mL−1 streptomycin solution. The T24 line was cultured in McCoy's 5a Medium (ATCC) and supplemented with 10% FBS and 1% of a 10000 units per mL penicillin and 10000 μg mL−1 streptomycin solution.
Cell viability assays
Cells (18750 cells per mL for HUVEC, 125000 cells per mL for MDA-MB-231 and T24 125000 cells per mL) were seeded in dark, flat bottom 96 well plates with clear lids for >2 hours in 80 μL media. After the growing period, Azo-CA4 and CA4 were added at varying concentrations in 20 μL 0.25% DMSO in media. Various concentrations of Azo-CA4 (30 μM–58.6 nM for experiments in the light and 100 μM–195 nM for experiments in the dark) and CA4 (30 nM–0.059 nM) were replicated 5 times on each plate. Cells were incubated with drug for 48 hours with one 96 well plate undergoing irradiation at 380 nm for 1 minute every hour or every 2 hours for the duration of the experiment and the other kept in the absence of light. 5 wells in each experiment were devoted to media with no cells or cells with vehicle. At the end of the 48 hour time period cells were incubated for 2 hours with 20 μL Cell Titer Blue reagent (CTB) (Promega) at which time fluorescence was measured on a BioTek Synergy H1 Hybrid plate reader (Ex. 560 nm, Em. 590 nm). Cell viability was expressed as a percentage of maximum (cells with vehicle) and corrected for background fluorescence of the media. Data is shown as an average ± S.D. of three independent experiments. GI50 values were calculated using SigmaPlot software using a four parameter logistic equation.
Results
Prior to synthesis of Azo-CA4, we performed a computational study (Fig. 1b) to compare its structure to CA4. Both molecules were fully optimized at the RI-MP2/def2-TZVP level of theory and natural population analyses were performed.32 Azo-CA4 was, indeed, highly similar in structure to CA4 (Fig. 1b). The optimized geometries and natural charges calculated for non-hydrogen atoms are almost identical with the exception of the nitrogens on the azo bridge. Because the aromatic groups and their methoxy substituents are the major contributors to tubulin binding in CA4,33 based on structure alone, we would expect that Azo-CA4 would be a potent tubulin inhibitor and act similarly to CA4.To synthesize Azo-CA4 (Scheme 1), we first protected the phenolic oxygen of nitroaniline 1 with a TBDMS group followed by reduction of the nitro group to give 2. Compound 2 was coupled to 3,4,5-trimethoxyaniline with diacetoxyiodobenzene34 to yield azobenzene 3 which was separable from homo-aniline coupling side products. Finally, 3 was deprotected with fluoride ion to yield Azo-CA4, 4.
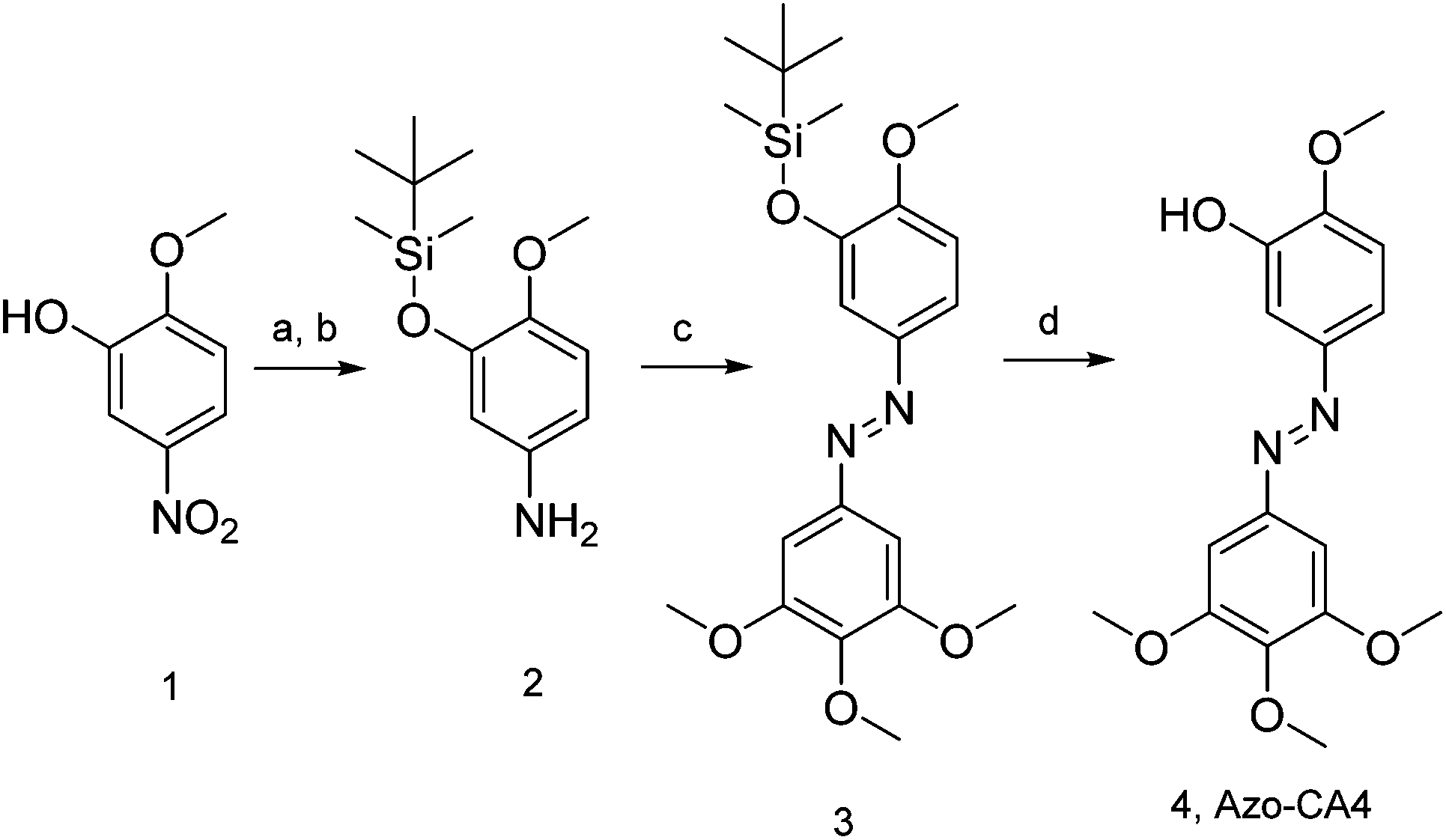 | ||
| Scheme 1 Synthesis of Azo-CA4. Reaction conditions: (a) TBDMS-Cl, DMF 92%. (b) 10% Pd/C, H2, MeOH/EtOH, 95%. (c) 3,4,5-Trimethoxyaniline, (diacetoxyiodo)benzene, CH2Cl2, 6%. (d) KF, AcOH, 95%. | ||
We analyzed the photoswitching properties of Azo-CA4 (Fig. 2). The absorbance spectrum of trans azo-CA4 showed a λmax of 379 nm and an extinction coefficient consistent with other reports (Fig. 2a and ESI Fig. S1†).30,35 After 30 seconds of irradiation with 380 nm light, the compound's absorption pattern had dramatically shifted toward an absorption profile that is characteristic for cis azobenzenes36,37 (Fig. 2a). Over time Azo-CA4 reverted back to the trans form with a half-life of 88 min at 37 °C (Fig. 2b, c and ESI Fig. S2 and Table S1†). Multiple cycles of switching and relaxation could be performed over 24 h without degradation (Fig. 2d). Surprisingly, the half-life for thermal reversion measured under our conditions was considerably longer than that reported previously.30 To check if the presence of DMSO or buffers caused this change, we measured the photoswitching kinetics under a variety of buffer conditions and light intensities, including those identical to that reported by Borowiak et al.,30 (Fig. S2 and S3†). Our half-lives for reversion (75–100 min) are consistently much longer than the reported value (6.2 min) even when measured under identical conditions (20% CH3CN in PBS) (Table S1†).
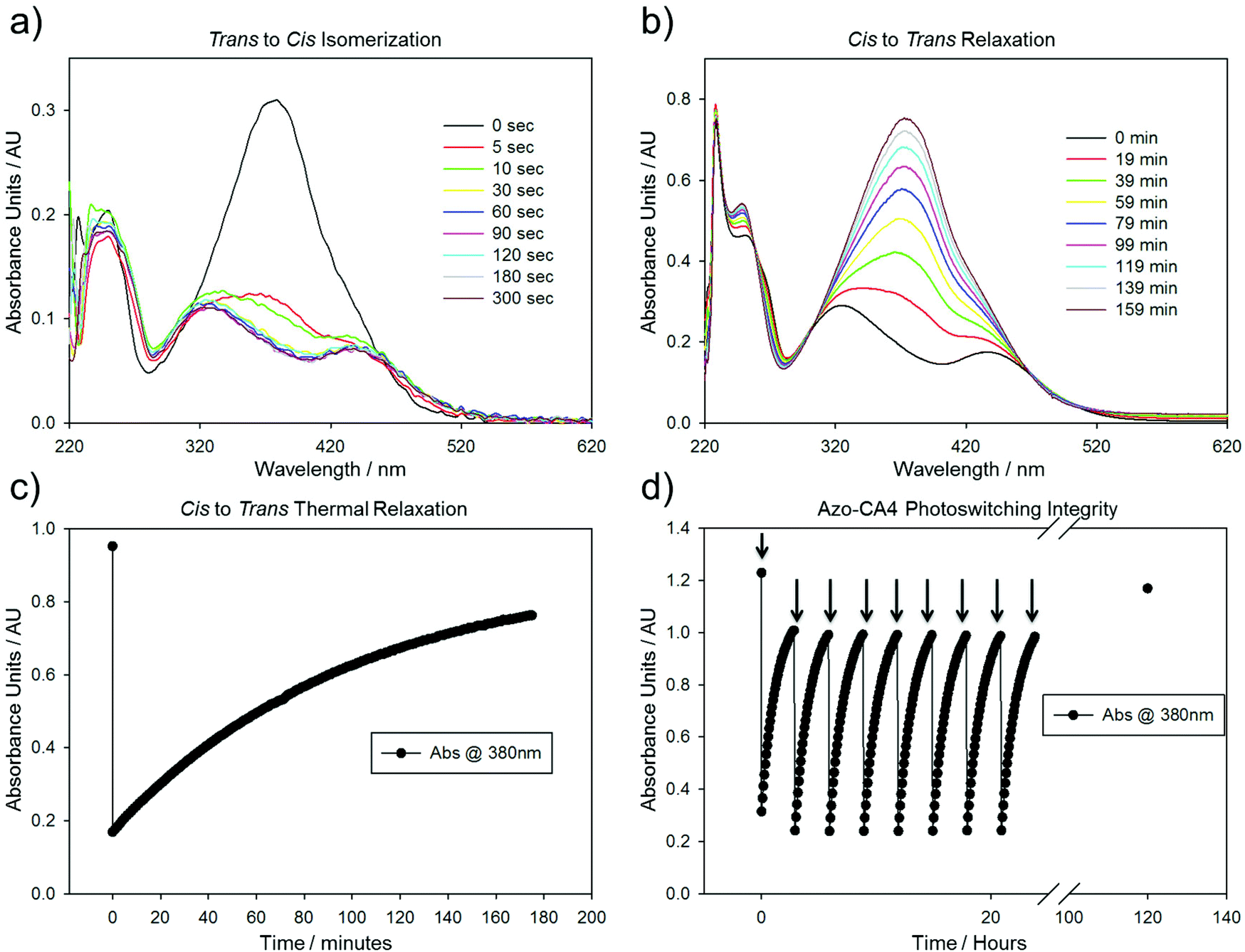 | ||
| Fig. 2 Absorption experiments performed on Azo-CA4. (a) Absorption spectra of Azo-CA4 (157 μM) after exposure to 380 nm light (4.4 mW cm−2) for various amounts of time. (b) Absorption spectra demonstrating thermal relaxation over time of Azo-CA4 in the dark at 37 °C. Prior to placing in the dark, Azo-CA4 was treated for 1 minute with 380 nm light (4.4 mW cm−2). (c) Time course of Azo-CA4 relaxation in DMSO monitored by absorbance at 380 nm. (d) Time course of 380 nm absorbance of Azo-CA4 over time in DMSO. The sample was irradiated with 380 nm light (4.4 mW cm−2) for 1 minute every 3 hours at 37 °C (arrows). The final point indicates a fully dark-adapted Azo-CA4 left in the dark for 100 hours after the last irradiation. | ||
We then demonstrated that Azo-CA4 inhibits tubulin polymerization in a similar manner to CA4 (ESI Fig. S4†). In vitro experiments were carried out with Azo-CA4 pre-activated with light or in the dark. As expected, in the light Azo-CA4 was a more potent inhibitor of tubulin polymerization than in the dark; light activated Azo-CA4 had an IC50 of 5.1 μM (ESI Fig. S4†), similar to the IC50 of CA4 (1.9 μM)38 and was 2.8-fold more potent than the dark adapted compound.
Borowiak et al.30 and Engdahl et al.31 investigated the toxicity of Azo-CA4 on several cancer cell lines. CA4 is a known vasculature-disrupting agent, and we therefore tested the effects of Azo-CA4 on human umbilical vein endothelial cells (HUVECs).39,40 For comparison, we also evaluated the effects of Azo-CA4 on a breast adenocarcinoma epithelial cell line (MDA-MB-231) that has shown sensitivity to CA4.41 Assays with CA4 itself in these cell lines showed EC50s in the low nM range, consistent with other reports (ESI Fig. S5†). With Azo-CA4, we observed significant enhancement of potency in the presence of light vs. untreated controls for both cell lines (Fig. 3a and b). Based on the half-lives we determined for cis Azo-CA4, we designed several pulse patterns to check for light-enabled toxicity ranging from a single 1 minute pulse at the beginning of the experiment, to a 1 minute pulse every 60 minutes (Fig. 3c). As expected with the turn-off mechanism, we saw decreasing EC50 values as the duration between pulses decreased (Table 1). For the 1 h pulse regime, Azo-CA4 was 22-fold more toxic to HUVEC cells than dark controls. Light alone at the same doses was not toxic for either cell line (Fig. 3d).
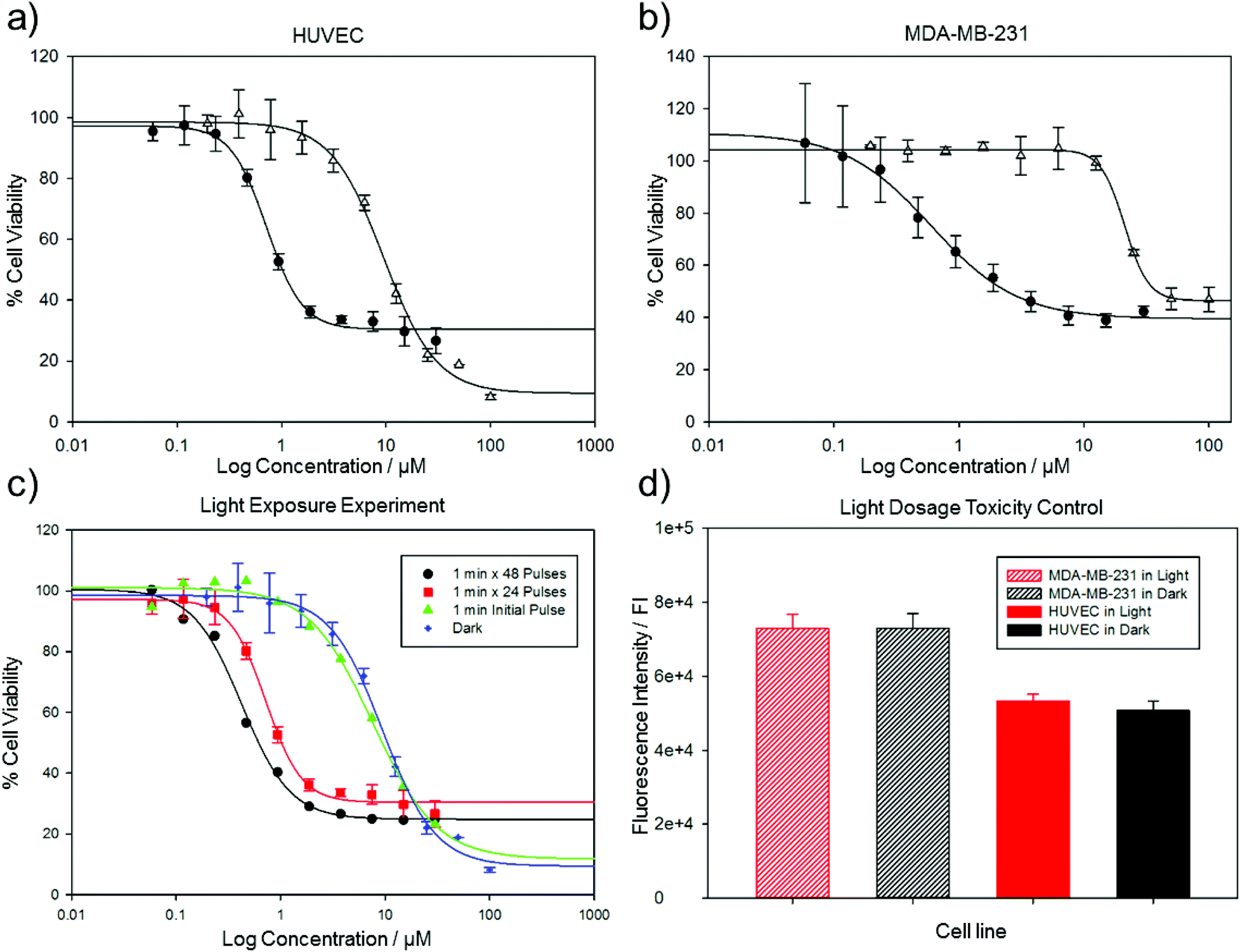 | ||
| Fig. 3 Analysis of the inhibition of Azo-CA4 in cell culture. Cells were treated with varying concentrations of Azo-CA4 for 48 hours and were either exposed to 380 nm light (4.4 mW cm−2) (closed circles) or kept in the dark (open triangles). In (a) HUVEC cells were treated with light for 1 min every 2 h. In (b) MDA-MD-231 cells were treated with light for 1 min every 1 h. In (c) HUVEC cells were treated with 1 minute light pulses at varying intervals: no light (black circles), 1 × 1 minute at the beginning of the experiment (green triangles), 24 × 1 minute (every 2 h), and 48 × 1 minute (every 1 h). Cell viability assays reported as a percentage of vehicle controls. The value in parentheses is the EC50. In (d), the cells were treated with 380 nm light (4.4 mW cm−2, 1 minute every hour) or kept in the dark for 48 h followed by analysis of cell density with Cell Titer Blue. | ||
| MDA-MB-231a | HUVECb | HUVECc | HUVECd | |
|---|---|---|---|---|
| a Cell viability assay with light pulse for 1 min every 1 hour. Error represents triplicate experiments. b Cell viability assay with light pulse for 1 min every 1 hour. Error represents 5 wells. c Cell viability assay with light pulse for 1 min every 2 hours. Error represents triplicate experiments. d Cell viability assay with light pulse for 1 min at beginning of experiment Error represents 5 wells. | ||||
| Azo-CA4 light | 0.6 ± 0.07 μm | 0.4 ± 0.03 μm | 0.7 ± 0.04 μm | 7.7 ± 1.5 μm |
| Azo-CA4 dark | 21.1 ± 0.6 μm | 9.5 ± 0.8 μm | — | — |
| CA4 | 0.0037 ± 0.2 μm | 0.0021 ± 0.2 μm | — | — |
The significant gap between the cytotoxicity of CA4 and Azo-CA4 in these cell lines suggested that Azo-CA4 may be unstable in cells. Since azobenzenes are known to react with glutathione (GSH),42,43 we evaluated the extent to which GSH could inactivate Azo-CA4 (Fig. 4 and ESI Fig. S6†). Both forms of Azo-CA4 were degraded by GSH over the course of several hours. Interestingly, the light-activated cis-Azo-CA4 compounds were degraded more rapidly than trans-Azo-CA4, and this effect could limit the potency at higher light doses when more cis-Azo-CA4 is present.
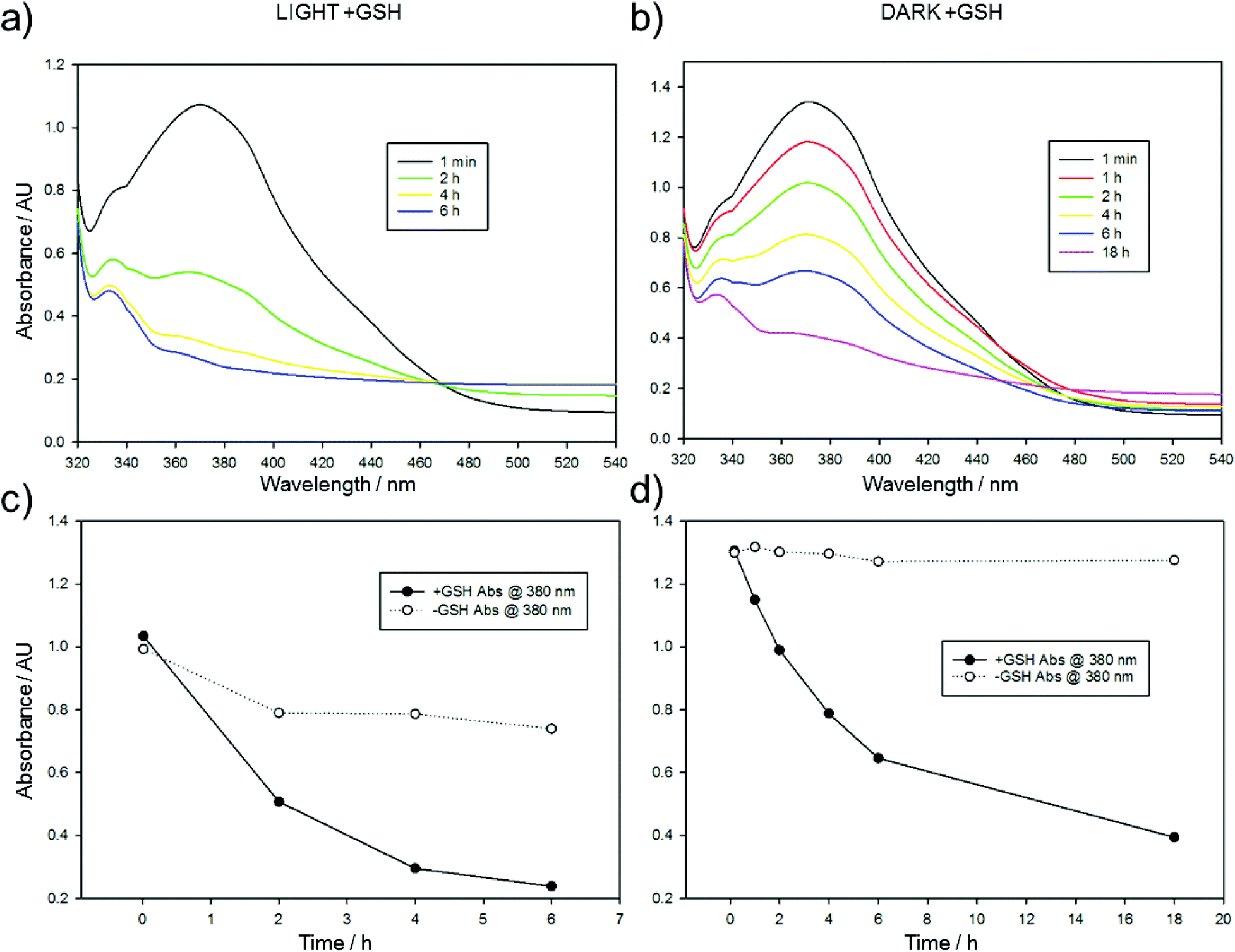 | ||
| Fig. 4 Glutathione exposure in light. Azo-CA4 (300 μM in 5.7% DMSO/PBS pH 7.2) in the presence of fully reduced glutathione (10 mM in PBS pH 7.2) and TCEP (5 mM in PBS pH 7.2) was placed in 3 separate wells (100 μL per well) of a 96 well plate at 37 °C and (a) exposed to light (380 nm, 4.4 mW cm−2) for 1 min every 2 h. Absorbance spectra were taken at different time points directly preceding exposure to light and are expressed as an average of triplicate readings. (b) Is identical to (a) except the reaction was kept in the dark, and the Azo-CA4 concentration was 400 μM. (c) and (d) plot the change absorbance of 380 nm over time of the data in (a) and (b) compared to identical reactions lacking glutathione. The decrease in absorbance of the minus GSH control in (c) is due to incomplete relaxation back to the trans state. | ||
To investigate this further, we treated our compound with light and GSH and followed the course of the reaction by LC-MS/MS (Fig. 5 and ESI Fig. S7–S9†). As expected, the peak corresponding to the cis-Azo-CA4 decreased over time (Fig. 5a and ESI Fig. S7†). Surprisingly, a new peak of the same mass was formed during the reaction. This peak did not co-elute with trans-Azo-CA4 (ESI Fig. S8†), and therefore must be due to a rearrangement product or products. (Note also that the samples were irradiated with light immediately prior to injection which would have eliminated the majority of the trans isomer even if was present beforehand). Additionally, transient formation of a peak of 624 Da was observed during the reaction. The MS-MS fragmentation of this peak showed neutral loss peaks of 307, 275, and 129, all of which are characteristic for GSH adducts (ESI Fig. S7†).44 This peak was not present in a control reaction lacking Azo-CA4 (ESI Fig. S9†), lending credence to the fact that this species is an adduct of glutathione and Azo-CA4.
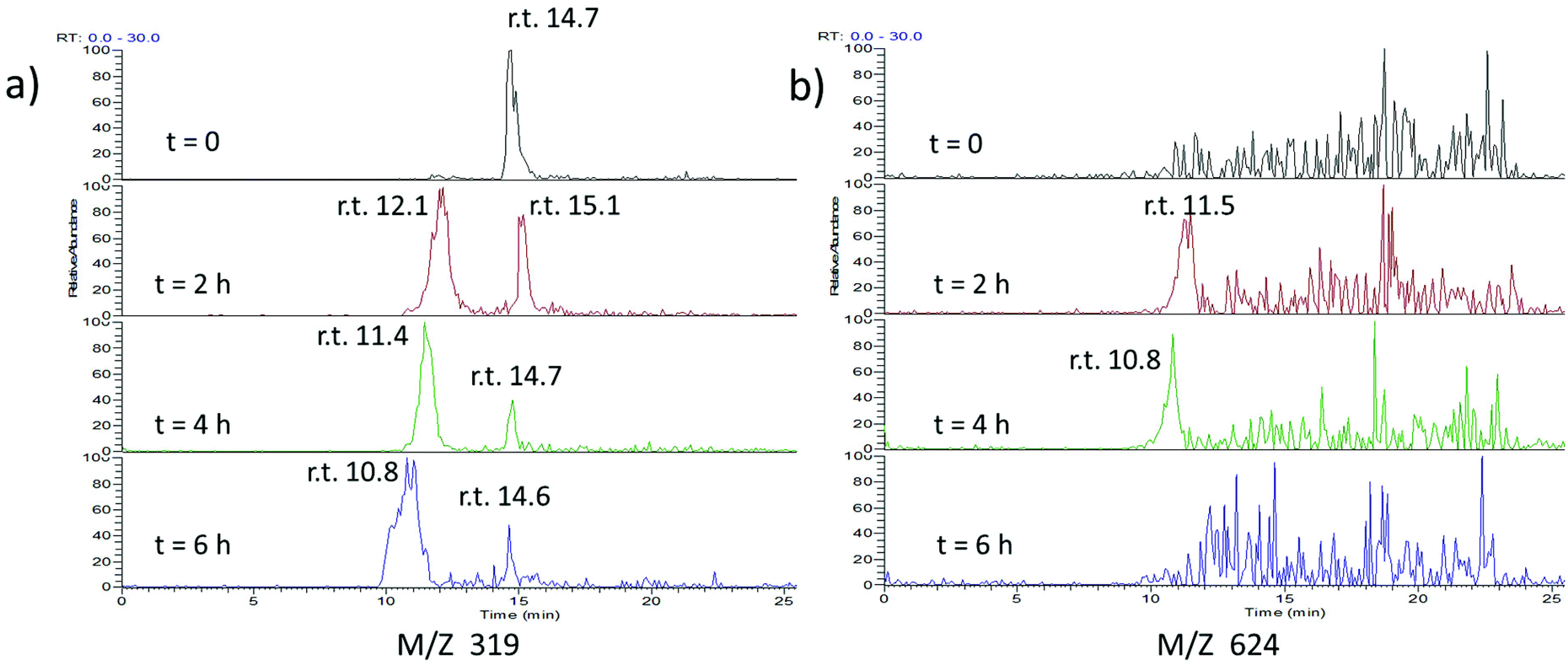 | ||
| Fig. 5 ESI-MS analysis of the reaction of glutathione and Azo-CA4. Azo-CA4 (300 μM in 5.7% DMSO/PBS pH 7.2) in the presence of fully reduced glutathione (10 mM in PBS pH 7.2) and TCEP (5 mM in PBS pH 7.2) was placed in 3 separate wells (100 μL per well) of a 96 well plate at 37 °C and exposed to light (380 nm, 4.4 mW cm−2) for 1 min every 2 h immediately preceding analysis. At the times given, a sample was removed and injected onto an analytical column containing 5 μm phenyl C-18 YMC and analyzed by ESI-MS. (a) Traces for ions of M + H identical to Azo-CA4 (319 Da). (b) Traces corresponding to m/z of 624 Da. Slight retention time drifts are expected due to manual loading of the capillary containing sample onto the instrument (see Fig. S7† for the LC-MS spectra of co-injected standards). | ||
Discussion
The half-life of thermal reversion of Azo-CA4 and its future analogs is a critical piece of their potential use as automatic turn-off drugs and tools.15 It is therefore significant that our values for the half-lives of Azo-CA4 vary significantly from those determined by Borowiak et al. under the same conditions; our value is about 14-fold longer (85 min vs. 6.2 min).30 Although in cells the half-life of Azo-CA4 can be altered by thiols, the cellular environment,15 and by tubulin binding itself, all of our cell culture data supports the longer half-life value. For example, our measured EC50 values for Azo-CA4 with MDA-231 cells are very similar to those determined by Borowiak et al. on this cell line,30 yet we pulsed with light once per hour, and they pulsed every 15 seconds. If the half-life was 6.2 minutes as reported,30 we should have observed significantly weaker potency with our pulse regimen.Glutathione reactions with azobenzenes have been studied on a number of systems. For most azobenzenes, nucleophilic attack of GS− anion on the diazo nitrogen gives the intermediate sulfenylhydrazide45 (Scheme 2). The sulfenyl hydrazide is then either attacked by a second glutathione anion to displace the reduced hydrazide (Scheme 2) or is hydrolyzed by water to give back the azobenzene plus a sulfenic acid. The hydrazide is easily oxidized back to the azobenzene, and therefore, the net result of each of these reactions is re-formation of the original trans-azobenzene. By a similar mechanism, glutathione catalyzes the relaxation of cis-azobenzenes back into their trans form.15,46 With Azo-CA4; however, our data shows that reaction with glutathione leads to the destruction of the azobenzene structure. This has been observed before by Woolley with other electron rich methoxy-substituted azobenzenes.41,42,47 Woolley proposed that the enhanced basicity of the electron rich azo nitrogens leads to protonation which primes the compound for nucleophilic attack by GSH. However, this proposed mechanism leads to the production of the same sulfenyl hydrazide (Scheme 2) that would be expected to result in reversion back to the azobenzene. We measured the pKa of Azo-CA4, and it is below 4.6 (ESI Fig. S10†), so it will not be protonated at physiological pH. Moreover, we did not see any ions corresponding either to the mass of the phenylhydrazide or the sulfenylhydrazide during the course of the reaction. Instead, our LC-MS data shows that a transient adduct of Azo-CA4 and GSH with a mass of 624 is formed during the GSH reaction. This mass is two less than would be expected for the sulfenyl hydrazide. The formation of this 624 Da species could potentially be explained by nucleophilic attack of the azo nitrogen onto GSSG to form the sulfenyl diazo cation. To test this, we incubated Azo-CA4 with GSSG, but no reaction was observed over 18 h (data not shown) which rules out this potential mechanism. One could envision other possibilities for formation of the sulfenyl diazo cation, or loss of the two hydrogens via a mechanism involving electrocyclization of the two rings. Since the light-activated Azo-CA4 degrades more rapidly than that in the dark (Fig. 4c), photochemical pathways for degradation that also involve GSH could also be envisioned. More work needs to be done to distinguish between these and other possibilities. Importantly, to our knowledge, this type of degradation has only been observed with electron-rich azobenzenes, which suggests that addition of electron withdrawing substituents could eliminate or slow the degradation pathway.
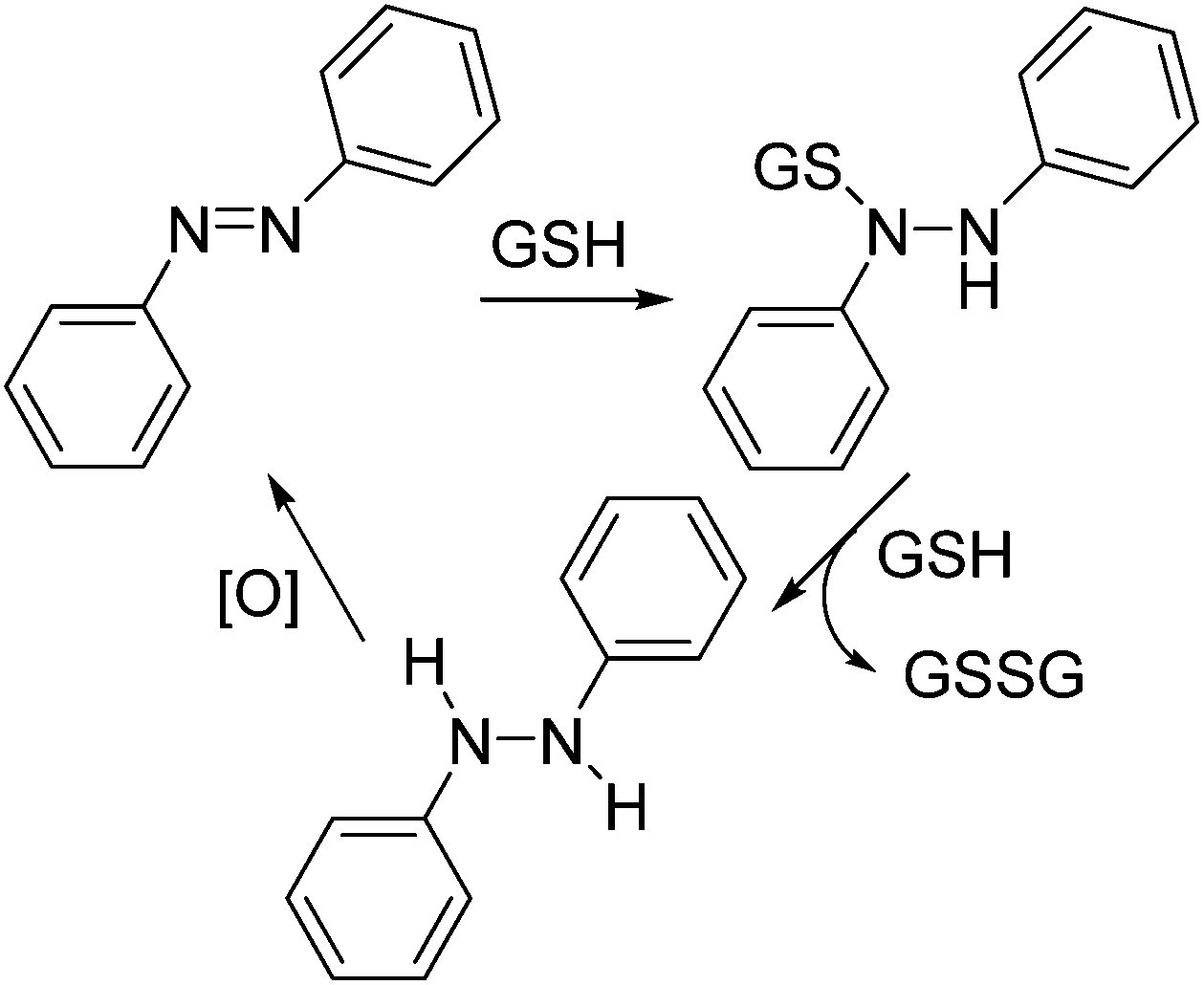 | ||
| Scheme 2 Reactions of azobenzenes with glutathione. Reported reaction cycle of azobenzenes with glutathione. | ||
In summary, Azo-CA4 exhibits remarkable enhancements in potency against HUVEC cells that are consistent with its ability to switch into the more toxic cis form in the presence of light. Moreover, over time, Azo-CA4 automatically converts back to its less toxic form. Provided the glutathione reactivity issue can be solved, Azo-CA4 and future, similar compounds offer the potential for automatic turn-off of drug activity in sites distal from the site of activation and thus present a novel means for tightly controlling drug potency and antitumor activity.
Acknowledgements
The authors are grateful to the Ohio Supercomputer Center for a generous allocation of computational resources. This work was supported by grants awarded to M.C.T.H. (R15CA167582) and J.C.H. (R01GM092827) from the National Institutes of Health. We thank Dr. Richard Moran, Dr. Massimo Bertino, David Hacker, Erica Peterson, Samantha Katner, and Dr. Stacie Richardson for experimental assistance and scientific discussion.References
- D. E. Dolmans, D. Fukumura and R. K. Jain, Photodynamic therapy for cancer, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- C. Robertson, D. H. Evans and H. Abrahamse, Photodynamic therapy (PDT): a short review on cellular mechanisms and cancer research applications for PDT, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- B. W. Henderson and T. J. Dougherty, How Does Photodynamic Therapy work?, Photochem. Photobiol., 1992, 55, 145–157 CrossRef CAS PubMed.
- B. C. Wilson and M. S. Patterson, The physics, biophysics and technology of photodynamic therapy, Phys. Med. Biol., 2008, 53, R61 CrossRef CAS PubMed.
- M. Michael Dcona, Q. Yu, J. A. Capobianco and M. C. T. Hartman, Near infrared light mediated release of doxorubicin using upconversion nanoparticles, Chem. Commun., 2015, 51, 8477–8479 RSC.
- M. M. Dcona, D. Mitra, R. W. Goehe, D. A. Gewirtz, D. A. Lebman and M. C. T. Hartman, Photocaged permeability: a new strategy for controlled drug release, Chem. Commun., 2012, 48, 4755–4757 RSC.
- S. Ki Choi, T. Thomas, M. Li, A. Kotlyar, A. Desai, J. Baker and R. James, Light-controlled release of caged doxorubicin from folate receptor-targeting PAMAM dendrimer nanoconjugate, Chem. Commun., 2010, 46, 2632–2634 RSC.
- J. You, G. Zhang and C. Li, Exceptionally High Payload of Doxorubicin in Hollow Gold Nanospheres for Near-Infrared Light-Triggered Drug Release, ACS Nano, 2010, 4, 1033–1041 CrossRef CAS PubMed.
- J. You, P. Zhang, F. Hu, Y. Du, H. Yuan, J. Zhu, Z. Wang, J. Zhou and C. Li, Near-Infrared Light-Sensitive Liposomes for the Enhanced Photothermal Tumor Treatment by the Combination with Chemo, Pharm. Res., 2014, 31, 554–565 CrossRef CAS PubMed.
- S. S. Agasti, A. Chompoosor, C. You, P. Ghosh, C. K. Kim and V. M. Rotello, Photoregulated Release of Caged Anticancer Drugs from Gold Nanoparticles, J. Am. Chem. Soc., 2009, 131, 5728–5729 CrossRef CAS PubMed.
- L. L. Fedoryshin, A. J. Tavares, E. Petryayeva, S. Doughan and U. J. Krull, Near-Infrared-Triggered Anticancer Drug Release from Upconverting Nanoparticles, ACS Appl. Mater. Interfaces, 2014, 6, 13600–13606 CAS.
- X. Hu, J. Tian, T. Liu, G. Zhang and S. Liu, Photo-Triggered Release of Caged Camptothecin Prodrugs from Dually Responsive Shell Cross-Linked Micelles, Macromolecules, 2013, 46, 6243–6256 CrossRef CAS.
- Q. Xing, N. Li, Y. Jiao, D. Chen, J. Xu, Q. Xu and J. Lu, Near-infrared light-controlled drug release and cancer therapy with polymer-caged upconversion nanoparticles, RSC Adv., 2015, 5, 5269–5276 RSC.
- A. Presa, R. F. Brissos, A. B. Caballero, I. Borilovic, L. Korrodi-Gregório, R. Pérez-Tomás, O. Roubeau and P. Gamez, Photoswitching the Cytotoxic Properties of Platinum(II) Compounds, Angew. Chem., Int. Ed., 2015, 54, 4561–4565 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, Photopharmacology: beyond proof of principle, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- G. R. Pettit, S. B. Singh, M. R. Boyd, E. Hamel, R. K. Pettit, J. M. Schmidt and F. Hogan, Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6(1a), J. Med. Chem., 1995, 10, 1666–1672 CrossRef.
- G. R. Pettit, G. M. Cragg, D. L. Herald, J. M. Schmidt and P. Lohavanijaya, Isolation and structure of combretastatin, Can. J. Chem., 1982, 60, 1374–1376 CrossRef CAS.
- V. M. Sharma, K. V. Adi Seshu, C. Vamsee Krishna, P. Prasanna, V. Chandra Sekhar, A. Venkateswarlu, S. Rajagopal, R. Ajaykumar, D. S. Deevi, N. V. S. Rao Mamidi and R. Rajagopalan, Novel 6,7-diphenyl-2,3,8,8a-tetrahydro-1H-indolizin-5-one analogues as cytotoxic agents, Bioorg. Med. Chem. Lett., 2003, 13, 1679–1682 CrossRef CAS.
- R. Dorr, K. Dvorakova, K. Snead, D. Alberts, S. Salmon and G. R. Pettit, Antitumor activity of combretastatin-A4 phosphate, a natural product tubulin inhibitor, Invest. New Drugs, 1996, 14, 131–137 CrossRef CAS.
- P. Nathan, M. Zweifel, A. R. Padhani, D. M. Koh, M. Ng, D. J. Collins, A. Harris, C. Carden, J. Smythe, N. Fisher, N. J. Taylor, J. J. Stirling, S. P. Lu, M. O. Leach, G. J. Rustin and I. Judson, Phase I trial of combretastatin A4 phosphate (CA4P) in combination with bevacizumab in patients with advanced cancer, Clin. Cancer Res., 2012, 18, 3428–3439 CrossRef CAS PubMed.
- G. J. S. Rustin, S. M. Galbraith, H. Anderson, M. Stratford, L. K. Folkes, L. Sena, L. Gumbrell and P. M. Price, Phase I Clinical Trial of Weekly Combretastatin A4 Phosphate: Clinical and Pharmacokinetic Results, J. Clin. Oncol., 2003, 21, 2815–2822 CrossRef CAS PubMed.
- M. Wühr, E. S. Tan, S. K. Parker, H. W. Detrich and T. J. Mitchison, A model for cleavage plane determination in early amphibian and fish embryos, Curr. Biol., 2010, 20, 2040–2045 CrossRef PubMed.
- N. J. Lawrence, D. Rennison, M. Woo, A. T. McGown and J. A. Hadfield, Antimitotic and cell growth inhibitory properties of combretastatin A-4-like ethers, Bioorg. Med. Chem. Lett., 2001, 11, 51–54 CrossRef CAS.
- T. Figueiras, M. Neves-Petersen and S. Petersen, Activation Energy of Light Induced Isomerization of Resveratrol, J. Fluoresc., 2011, 21, 1897–1906 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Azobenzene photoswitches for biomolecules, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- I. K. Lednev, T. Ye, P. Matousek, M. Towrie, P. Foggi, F. V. R. Neuwahl, S. Umapathy, R. E. Hester and J. N. Moore, Femtosecond time-resolved UV-visible absorption spectroscopy of trans-azobenzene: dependence on excitation wavelength, Chem. Phys. Lett., 1998, 290, 68–74 CrossRef CAS.
- J. Broichhagen, M. Schönberger, S. C. Cork, J. A. Frank, P. Marchetti, M. Bugliani, A. M. J. Shapiro, S. Trapp, G. A. Rutter, D. J. Hodson and D. Trauner, Optical control of insulin release using a photoswitchable sulfonylurea, Nat. Commun., 2014, 5 Search PubMed.
- S. Pittolo, X. Gómez-Santacana, K. Eckelt, X. Rovira, J. Dalton, C. Goudet, J. Pin, A. Llobet, J. Giraldo, A. Llebaria and P. Gorostiza, An allosteric modulator to control endogenous G protein-coupled receptors with light, Nat. Chem. Biol., 2014, 10, 813–815 CrossRef CAS PubMed.
- J. R. Kumita, O. S. Smart and G. A. Woolley, Photo-Control of Helix Content in a Short Peptide, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS.
- M. Borowiak, W. Nahaboo, M. Reynders, K. Nekolla, P. Jalinot, J. Hasserodt, M. Rehberg, M. Delattre, S. Zahler and A. Vollmar, Photoswitchable inhibitors of microtubule dynamics optically control mitosis and cell death, Cell, 2015, 162, 403–411 CrossRef CAS PubMed.
- A. J. Engdahl, E. A. Torres, S. E. Lock, T. B. Engdahl, P. S. Mertz and C. N. Streu, Synthesis, Characterization, and Bioactivity of the Photoisomerizable Tubulin Polymerization Inhibitor azo-Combretastatin A4, Org. Lett., 2015, 17, 4546–4549 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS PubMed.
- Y. Jin, P. Qi, Z. Wang, Q. Shen, J. Wang, W. Zhang and H. Song, 3D-QSAR Study of Combretastatin A-4 Analogs Based on Molecular Docking, Molecules, 2011, 16, 6684–6700 CrossRef CAS PubMed.
- K. Monir, M. Ghosh, S. Mishra, A. Majee and A. Hajra, Phenyliodine(III) Diacetate (PIDA) Mediated Synthesis of Aromatic Azo Compounds through Oxidative Dehydrogenative Coupling of Anilines: Scope and Mechanism, Eur. J. Org. Chem., 2014, 1096–1102 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- P. P. Birnbaum, J. H. Linford and D. W. G. Style, The absorption spectra of azobenzene and some derivatives, Trans. Faraday Soc., 1953, 49, 735–744 RSC.
- D. Liu, J. Karanicolas, C. Yu, Z. Zhang and G. A. Woolley, Site-specific incorporation of photoisomerizable azobenzene groups into ribonuclease S, Bioorg. Med. Chem. Lett., 1997, 7, 2677–2680 CrossRef CAS.
- M. Cushman, D. Nagarathnam, D. Gopal, A. K. Chakraborti, C. M. Lin and E. Hamel, Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization, J. Med. Chem., 1991, 34, 2579–2588 CrossRef CAS.
- L. Vincent, P. Kermani, L. M. Young, J. Cheng, F. Zhang, K. Shido, G. Lam, H. Bompais-Vincent, Z. Zhu, D. J. Hicklin, P. Bohlen, D. J. Chaplin, C. May and S. Rafii, Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling, J. Clin. Invest., 2005, 115, 2992–3006 CAS.
- G. G. Dark, S. A. Hill, V. E. Prise, G. M. Tozer, G. R. Pettit and D. J. Chaplin, Combretastatin A-4, an Agent That Displays Potent and Selective Toxicity toward Tumor Vasculature, Cancer Res., 1997, 57, 1829–1834 CAS.
- S. Parihar, A. Kumar, A. K. Chaturvedi, N. K. Sachan, S. Luqman, B. Changkija, M. Manohar, O. Prakash, D. Chanda, F. Khan, C. S. Chanotiya, K. Shanker, A. Dwivedi, R. Konwar and A. S. Negi, Synthesis of combretastatin A4 analogues on steroidal framework and their anti-breast cancer activity, J. Steroid Biochem. Mol. Biol., 2013, 137, 332–344 CrossRef CAS PubMed.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, Photoswitching Azo Compounds in Vivo with Red Light, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos and G. A. Woolley, Robust visible light photoswitching with ortho-thiol substituted azobenzenes, Chem. Commun., 2013, 49, 10314–10316 RSC.
- C. M. Murphy, C. Fenselau and P. L. Gutierrez, Fragmentation characteristic of glutathione conjugates activated by high-energy collisions, J. Am. Soc. Mass Spectrom., 1992, 3, 815–822 CrossRef CAS.
- C. Boulègue, M. Löweneck, C. Renner and L. Moroder, Redox potential of azobenzene as an amino acid residue in peptides, ChemBioChem, 2007, 8, 591–594 CrossRef PubMed.
- R. F. Standaert and S. B. Park, Abc amino acids: design, synthesis, and properties of new photoelastic amino acids, J. Org. Chem., 2006, 71, 7952–7966 CrossRef CAS PubMed.
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos and G. A. Woolley, Robust visible light photoswitching with ortho-thiol substituted azobenzenes, Chem. Commun., 2013, 49, 10314–10316 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02005k |
| This journal is © The Royal Society of Chemistry 2016 |