
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The rapid synthesis and dynamic behaviour of an isophthalamide [2]catenane†
Calum N.
Marrs
and
Nicholas H.
Evans
*
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.h.evans@lancaster.ac.uk
First published on 15th September 2015
Abstract
A serendipitous [2]catenane has been prepared in three steps from commercially available starting materials. The interlocked topology of the catenane has been confirmed by single crystal X-ray structural determination. The rings of the catenane may rotate relative to one another – a process that may be controlled by varying solvent or temperature.
Introduction
Catenanes,1 along with their interlocked counterparts rotaxanes,2 are not only aesthetically pleasing molecules, but are increasingly being used in nanotechnological applications3 many of which exploit the relative motion of their interlocked components.4 Interlocked molecules have become accessible to the synthetic chemist by the development of various template synthesis methodologies that overcome the entropically unfavourable association of multiple molecular components that is required for their preparation. Templates and templating interactions that have been used to prepare catenanes and rotaxanes include: metal cations,5 anions,6 π–π stacking,7 hydrogen bonding8 and (very recently) halogen bonding.9 While a large number of synthetic strategies have been reported to date, many involve lengthy, multi-step procedures to obtain the precursors required for the final reaction step that leads to covalent capture of the interlocked molecule. This represents a considerable hurdle for a chemist wishing to investigate the properties and possible application of catenanes and rotaxanes, and so the discovery of new expedient synthetic routes to these molecules has the potential to aid future studies.Herein we report a serendipitously discovered [2]catenane that was prepared via a short synthetic route (Fig. 1). By reacting a bis-amine (prepared in two steps) with commercially available isophthaloyl chloride, an isophthalamide [2]catenane was produced – in just three reaction steps. In addition to being characterised by NMR and IR spectroscopies and mass spectrometry, the structure of the catenane has been unequivocally confirmed by solid state structural determination. The X-ray structure reveals inter-ring hydrogen bonds which it is believed template formation of the interlocked catenane. NMR studies reveal the rings rotate relative to one another in a process that, at room temperature, is fast on the NMR timescale in d6-DMSO, while it is appreciably slower in chlorinated solvents.
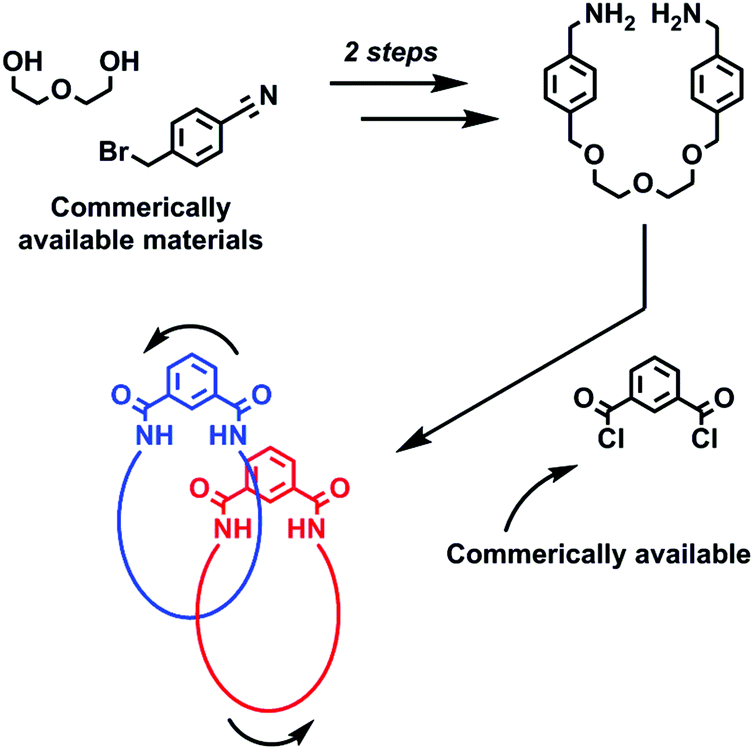 | ||
| Fig. 1 Schematic representation of the expedient three step synthesis of a dynamic isophthalamide [2]catenane. | ||
Results and discussion
When attempting to prepare isophthalamide macrocycle 1 (Scheme 1) by reacting equimolar amounts of a previously reported dimethanamine10 and isophthaloyl chloride in dichloromethane using semi-high dilution macrocyclisation conditions (≈3 mmol L−1), TLC analysis of the crude reaction mixture revealed two closely eluting compounds. Careful silica gel column chromatography allowed for the separate isolation of these compounds. The characterisation (NMR spectroscopy and mass spectrometry, see ESI†) of the first eluted (i.e. less polar) species was consistent with the structure of the anticipated macrocycle 1 (in an isolated yield of 12%). The growth of a single crystal allowed for solid state structure determination, which provided conclusive proof of macrocycle formation (see ESI†). | ||
| Scheme 1 Synthesis of macrocycle 1 and catenane 2. | ||
The more polar compound 2 possessed a very broad 1H NMR spectrum in CDCl3 at room temperature, implying dynamic processes that fall between the fast and slow NMR timescales, and hinting at possible catenane formation (also isolated with a yield of 12%). 1H NMR spectra of 1 and 2 were then recorded in d6-DMSO, which consisted of sharp, well-resolved resonances for both compounds (Fig. 2). Comparing the spectrum of 2 with that of 1, there are significant upfield shifts in aromatic protons f and g, as well as the alkyl protons e, h, i and j, which would be consistent with a freely-rotating catenane structure, where the protons of one ring are able to reside between the aromatic rings of the other macrocycle. Amide proton d is also significantly upfield for 2 compared to 1. At first sight this might appear inconsistent with a catenane structure for 2, as it is expected that in an interlocked structure a carbonyl oxygen on one ring would hydrogen bond to the isophthamide cleft of the other ring. However, we rationalise the appearance of the 1H NMR spectra, by suggesting that a DMSO solvent molecule would be able to efficiently hydrogen bond to the isophthalamide cleft of macrocycle 1 through its highly polarised S+–O− bond, but it would not be able to for the more sterically congested proposed catenane structure of 2.
 | ||
| Fig. 2 1H NMR spectra of (a) macrocycle 1 and (b) catenane 2 (d6-DMSO, 400 MHz, 298 K). See Scheme 1 for atom labels. | ||
Unequivocal evidence for the interlocked topology of catenane 2 was provided by single crystal X-ray structure determination (Fig. 3). A single crystal was grown by slow evaporation of a chloroform solution of the catenane, with the solved structure revealing inter-ring hydrogen bonding involving each isophthalamide cleft. As depicted in Fig. 3, the isophthalamide N–Hs of the left-hand ring are hydrogen bonding to one of the carbonyl oxygen atoms of the right-hand ring (N–H⋯O distances: 2.196 Å and 2.465 Å). In addition, an N–H and the internal isophthalamide C–H of the right-hand ring are hydrogen bonding to the central oxygen in the polyether chain of the left-hand ring.
 | ||
| Fig. 3 X-ray structure of catenane 2. Hydrogen atoms (except N–Hs and isophthalamide internal C–Hs) and disorder in one of the rings are omitted for clarity. | ||
For catenane 2 to form implies that a templating interaction is in operation during the reaction. The presence of inter-ring hydrogen bonding in the X-ray structure of 2 provides evidence to support the hypothesis that the formation of the interlocked molecule is driven by hydrogen bond templation. The proposed mechanism for catenane formation is that a partially cyclized ring threads through macrocycle 1, templated by formation of the inter-component hydrogen bonds to be found in catenane 2, before reaction of the remaining acid chloride and amine to close the second ring of the catenane. At this point, it should be emphasised that other examples of hydrogen bond templated catenanes have been discovered serendipitously; first by Hunter8a and Vögtle8b and second by Leigh.8c In contrast to these previously reported systems, catenane 2 possesses only one isophthalamide group per ring, rather than two, and hence represents to the best of our knowledge the first example of a new class of isophthalamide containing catenane produced by hydrogen bond templation.
We have undertaken further investigations into the dynamic behaviour of catenane 2 in solution by use of 1H NMR spectroscopy. Upon varying the solvent composition from pure CDCl3 to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CDCl3:d6-DMSO at 298 K the 1H NMR spectrum of catenane 2 notably sharpens (Fig. 4). Increasing the proportion of the hydrogen bond accepting dimethyl sulfoxide creates a solvent mixture that disrupts the inter-ring hydrogen bonds so that ring rotation becomes fast on the NMR timescale.
:1 CDCl3:d6-DMSO at 298 K the 1H NMR spectrum of catenane 2 notably sharpens (Fig. 4). Increasing the proportion of the hydrogen bond accepting dimethyl sulfoxide creates a solvent mixture that disrupts the inter-ring hydrogen bonds so that ring rotation becomes fast on the NMR timescale.
 | ||
| Fig. 4
1H NMR spectra of catenane 2 recorded in CDCl3:d6-DMSO solvent mixtures (2.5 mM, 400 MHz, 298 K). See Scheme 1 for atom labels. | ||
We also looked at the effect of temperature upon the dynamic behaviour of catenane 2 by use of VT 1H NMR spectroscopy in CD2Cl4 (see Fig. 5). At 298 K, the spectrum is very broad, except for the triplet attributed to proton a. Tetrachloroethane, like chloroform, is a poor hydrogen bond acceptor compared to dimethyl sulfoxide, and so ring rotation is much slower than in d6-DMSO. However, heating the sample to 318 K, results in the number of peaks reducing, and upon continued heating to 378 K, all the peaks become sharp and well-resolved, as the ring rotation once again becomes fast on the NMR timescale.
 | ||
| Fig. 5 1H NMR spectra of catenane 2 recorded at T = 298 K to 378 K in CD2Cl4 (400 MHz). See Scheme 1 for atom labels. | ||
Conclusions
In summary, a [2]catenane, where each ring possesses one isophthalamide unit, has been synthesised in just three steps from commercially available starting materials. The rings of the catenane are able to rotate relative to one another; the rate of this process may be modulated by solvent or temperature. Further investigations into this new class of catenanes, including the preparation of examples where the motion of the rings may be controlled by chemical stimuli, are being undertaken in our laboratories.Experimental
General information
Commercially available solvents and chemicals were used without further purification. Deionised water was used in all cases. Analytical TLC was carried out on aluminium backed silica gel sheets with fluorescent indicator (254 nm). Column chromatography was carried out on silica gel with a 60 Å particle size.IR spectra were recorded on an Agilent Technologies Cary 630 FTIR spectrometer. NMR spectra were recorded on a Bruker 400 MHz Ultra Shield Plus, with the NMR data for macrocycle 1 and catenane 2 reported below being assigned according to the atom labels to be found in Fig. 6. Mass spectra were recorded on a Thermofisher LTQ Orbitrap XL at the EPSRC UK National Mass Spectrometry Facility at Swansea University. Melting points were recorded on a Gallenkamp capillary melting point apparatus and are uncorrected.
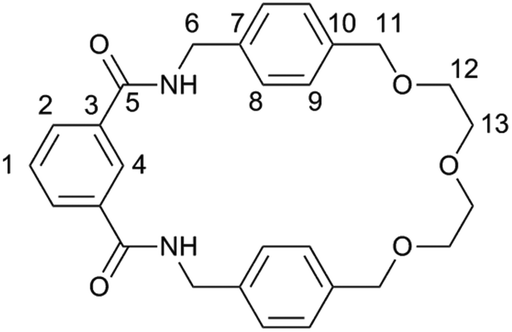 | ||
| Fig. 6 Carbon atom labels used in the assignment of the NMR data. | ||
Experimental procedures
:10 EtOAc/CH2Cl2) to yield macrocycle 1 (Rf = 0.55, 68 mg, 12%) and catenane 2 (Rf = 0.50, 70 mg, 12%) as white solids.
Macrocycle 1. Mp 198–200 °C. νmax/cm−1 (neat) 3320 (N–H), 2860 (C–H), 1660 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1630 (CO), 1530 (N–H), 1080 (C–O). δH(400 MHz; CDCl3) 7.91 (2H, dd, 3J = 7.7 Hz 4J = 1.7 Hz, C2H), 7.80 (1H, s, C4H), 7.45 (1H, t, 3J = 7.7 Hz, C1H), 7.21–7.27 (8H, m, C8H & C9H), 6.85 (2H, t, 3J = 5.4 Hz, NH), 4.46–4.48 (8H, m, C6H & C11H), 3.58–3.68 (8H, m, C12H & C13H). δC(100 MHz; CDCl3) 167.0 (C5), 137.6, 137.1, 134.6, 130.9, 129.5, 128.5, 128.1, 123.7 (8 Ar C environments), 72.8 (C11), 70.6, 69.4 (C12 & C13), 43.9 (C6). m/z (ES) 492.2480 ([M + NH4]+, C28H34N3O5 requires 492.2493.
O), 1630 (CO), 1530 (N–H), 1080 (C–O). δH(400 MHz; CDCl3) 7.91 (2H, dd, 3J = 7.7 Hz 4J = 1.7 Hz, C2H), 7.80 (1H, s, C4H), 7.45 (1H, t, 3J = 7.7 Hz, C1H), 7.21–7.27 (8H, m, C8H & C9H), 6.85 (2H, t, 3J = 5.4 Hz, NH), 4.46–4.48 (8H, m, C6H & C11H), 3.58–3.68 (8H, m, C12H & C13H). δC(100 MHz; CDCl3) 167.0 (C5), 137.6, 137.1, 134.6, 130.9, 129.5, 128.5, 128.1, 123.7 (8 Ar C environments), 72.8 (C11), 70.6, 69.4 (C12 & C13), 43.9 (C6). m/z (ES) 492.2480 ([M + NH4]+, C28H34N3O5 requires 492.2493.
Catenane 2. Mp 224–226 °C. νmax/cm−1 (neat) 3350 (N–H), 2860 (C–H), 1650 (C
O), 1620 (CO), 1510 (N–H), 1070 (C–O). δH(400 MHz; d6-DMSO) 8.17 (2H, s, C4H), 8.04 (4H, br s, NH), 7.97 (4H, dd, 3J = 7.7 Hz 4J = 1.6 Hz, C2H), 7.59 (2H, t, 3J = 7.7 Hz, C1H), 6.92–7.04 (16H, m, C8H & C9H), 4.15–4.18 (16H, m, C6H & C11H), 3.10 (8H, br s, OCH2CH2O), 2.93 (8H, br s, OCH2CH2O). δC(100 MHz; d6-DMSO) 165.6 (C5), 137.3, 136.2, 134.2, 130.7, 128.6, 128.5, 124.9 (8 Ar C environments), 72.2 (C11), 69.3, 68.1 (C12 & C13), 43.5 (C6). m/z (ES) 949.4374 ([M + H]+, C56H61N4O10 requires 949.4382.
Acknowledgements
N. H. E. wishes to thank Lancaster University for financial support. We thank Drs Fraser White, Daniel Baker and Marcus Winter (Agilent Technologies, Yarnton, UK) and Dr Mike Coogan (Lancaster University) for assistance with the collection, solution and interpretation of the X-ray crystallographic data of macrocycle 1 and catenane 2, and the EPSRC UK National Mass Spectrometry facility at Swansea University, UK.Notes and references
- (a) N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658–4683 RSC; (b) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- (a) C.-O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS; (b) V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed; (c) S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symers and J. Wu, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS PubMed; (d) S.-T. Tung, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S. H. Chiu, Angew. Chem., Int. Ed., 2013, 52, 13269–13272 CrossRef CAS PubMed; (e) C. Lincheneau, B. Jean-Denis and T. Gunnlaugsson, Chem. Commun., 2014, 50, 2857–2860 RSC; (f) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- (a) G. M. Hübner, J. Gläser, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 383–386 CrossRef; (b) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS PubMed; (c) M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364–15365 CrossRef CAS PubMed; (d) E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS PubMed; (e) B. Q. Huang, S. M. Santos, V. Felix and P. D. Beer, Chem. Commun., 2008, 4610–4612 RSC; (f) J.-J. Lee, J. M. Baumes, R. D. Connell, A. G. Oliver and B. D. Smith, Chem. Commun., 2011, 47, 7188–7190 RSC; (g) M. J. Langton and P. D. Beer, Chem. Commun., 2014, 50, 8124–8127 RSC; (h) G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed.
- (a) P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396–1399 CrossRef; (b) P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS PubMed; (c) D. G. Hamilton, J. K. M. Sanders, J. E. Davies, W. Clegg and S. J. Teat, Chem. Commun., 1997, 897–898 RSC; (d) G. Barin, A. Coskun, M. M. G. Fouda and J. F. Stoddart, ChemPlusChem, 2012, 77, 159–185 CrossRef CAS.
- (a) C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303–5311 CrossRef CAS; (b) F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619–1622 CrossRef; (c) A. G. Johnston, D. A. Leigh, R. J. Pritchard and M. D. Deegan, Angew. Chem., Int. Ed. Engl., 1995, 34, 1209–1212 CrossRef CAS; (d) F. G. Gatti, D. A. Leigh, S. A. Nepogodiev, A. M. Z. Slawin, S. J. Teat and J. K. Y. Wong, J. Am. Chem. Soc., 2001, 123, 5983–5989 CrossRef CAS PubMed; (e) N. S. Simpkins, D. F. Weske, L. Male, S. J. Coles and M. B. Pitak, Chem. Commun., 2013, 49, 5010–5012 RSC; (f) A. Tron, P. J. Thornton, M. Rocher, H.-P. J. de Rouville, J.-P. Desvergne, B. Kauffmann, T. Buffeteau, D. Cavagnat, J. H. R. Tucker and N. D. McClenaghan, Org. Lett., 2014, 16, 1358–1361 CrossRef CAS PubMed.
- L. C. Gilday, T. Lang, A. Caballero, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2013, 52, 4356–4360 CrossRef CAS PubMed.
- A. Vidonne and D. Philp, Tetrahedron, 2008, 64, 8464–8475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional notes on experimental procedures; characterisation spectra of macrocycle 1 and catenane 2; crystallographic data for macrocycle 1 and catenane 2. CCDC 1416976 and 1416977. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01770j |
| This journal is © The Royal Society of Chemistry 2015 |