
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study of the norcaradiene–cycloheptatriene equilibrium in a series of azulenones by NMR spectroscopy; the impact of substitution on the position of equilibrium
Lorraine M.
Bateman
a,
Orla A.
McNamara
a,
N. Rachael
Buckley
a,
Patrick
O'Leary†
a,
Francis
Harrington
a,
Norma
Kelly
a,
Sarah
O'Keeffe
a,
Angela
Stack
a,
Shane
O'Neill
a,
Daniel G.
McCarthy
a and
Anita R.
Maguire
*b
aDepartment of Chemistry, Analytical and Biological Chemistry Research Facility, University College Cork, Ireland
bDepartment of Chemistry and School of Pharmacy, Analytical and Biological Chemistry Research Facility, Synthesis and Solid State Pharmaceutical Centre, University College Cork, Ireland. E-mail: a.maguire@ucc.ie
First published on 8th September 2015
Abstract
A systematic investigation of the influence of substitution at positions C-2 and C-3 on the azulenone skeleton, based on NMR characterisation, is discussed with particular focus on the impact of the steric and electronic characteristics of substituents on the position of the norcaradiene–cycloheptatriene (NCD–CHT) equilibrium. Variable temperature (VT) NMR studies, undertaken to enable the resolution of signals for the equilibrating valence tautomers revealed, in addition, interesting shifts in the equilibrium.
Introduction
The addition of carbenes derived from α-diazocarbonyl compounds to benzenes, resulting in ring expansion to provide cycloheptatrienes, originally developed by Buchner at the end of the 19th century, is among the most synthetically powerful methods of transforming stable aromatic rings to much more reactive systems.1,2 Mechanistically, it is believed that this ring expansion occurs via initial carbene addition to provide a norcaradiene 1N followed by reversible 6π electrocyclic ring-opening to the more stable cycloheptatriene tautomer‡1T with the equilibrium generally favouring the latter (Scheme 1).3 While the earlier studies were conducted either thermally or photochemically, this transformation became practically useful from a synthetic perspective with the advent of rhodium carboxylate catalysts in the 1980s.4–8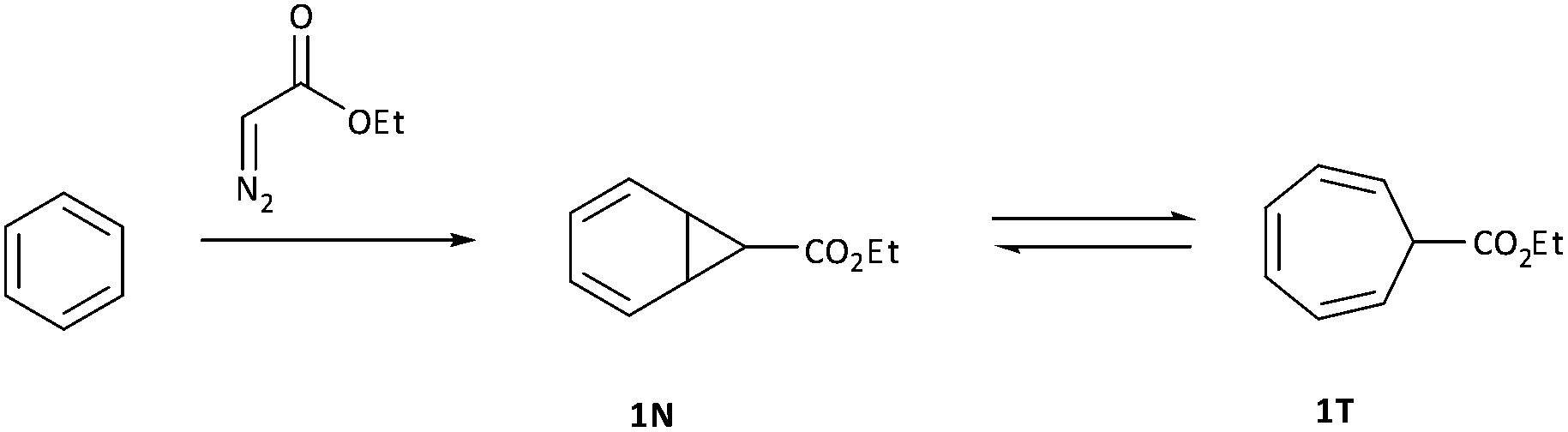 | ||
| Scheme 1 Reaction of ethyl diazoacetate with benzene resulting in cycloheptatriene. | ||
Subsequent to the original work of Buchner, the existence of the norcaradiene tautomer was a topic of much dispute that was ultimately resolved in 1950s.3 The existence of the norcaradiene tautomer has also been irrefutably confirmed through trapping of this species in cycloaddition reactions and structural characterisation of the cycloadducts, for example by Vogel,9 Manitto10 and Doyle.11 With the advent of NMR spectroscopy, a greater understanding of the norcaradiene–cycloheptatriene equilibrium emerged through direct observation of the interconverting species rather than inferring their existence by trapping experiments. In 1955, E. J. Corey first described the use of NMR spectroscopy in an attempt to study 2N and 2T, the enol acetate tautomers of the natural product eucarvone confirming only the presence of the cycloheptatriene structure (Fig. 1).12
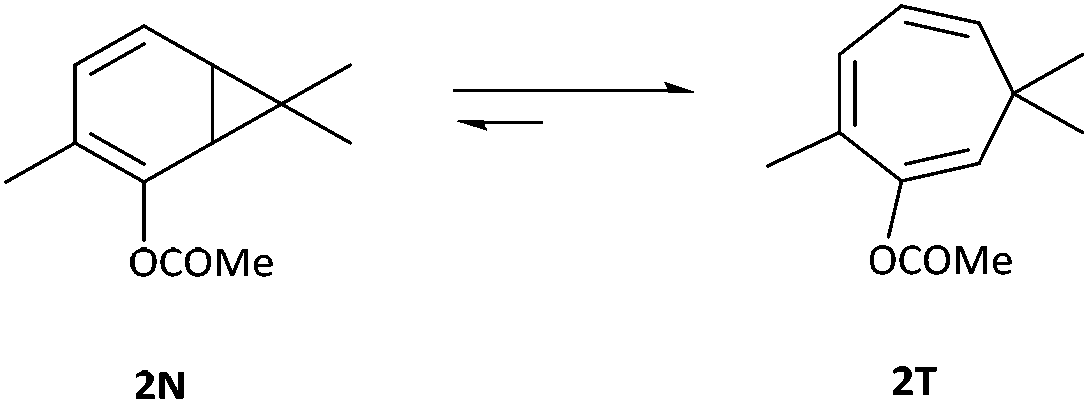 | ||
| Fig. 1 Enol acetate tautomers of eucarvone. | ||
Subsequent reports by Anet,13 Jensen and Smith,14 and Roberts15 further investigated the spectroscopic evidence for the detection of the elusive norcaradiene 3N by VT-1H NMR spectroscopy (Fig. 2). Even at temperatures as low as −150 °C, no signals for the norcaradiene were observed, however Rubin did succeed in directly observing the norcaradiene 3N at low temperature (77 K) using UV, which allowed him to provide an insight into the kinetics of its interconversion to 3T.16 Günther used low temperature NMR spectroscopy to confirm the existence of ‘Buchner's acid’ (Fig. 2) as a fluxional system, with only 3% NCD observed at low temperature (13C NMR at −132.5 °C and 1H NMR at −150 °C).17 Okamoto has described extensive variable-temperature NMR work in the study of the position of the norcaradiene–cycloheptatriene equilibrium of various substituted cycloheptatrienes.18
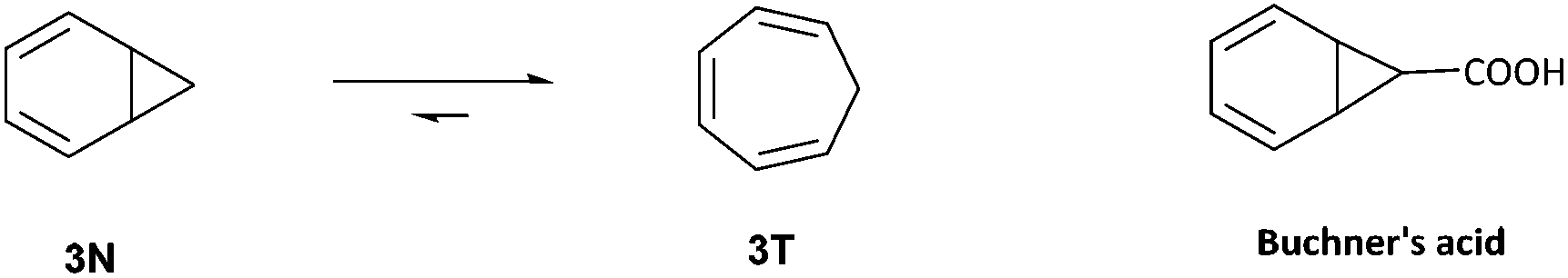 | ||
| Fig. 2 Norcaradiene–cycloheptatriene system for original VT-NMR studies. | ||
Hannemann has reported that the chemical shifts of the 1,6 H and C atoms of pure cycloheptatriene 3N lie in the region δH 5.2–5.3 and δC 120–130 respectively, whereas the corresponding signals in typical norcaradienes 3T resonate at δH 2.8–3.5 and δC 35–45.19 This tautomeric system is influenced by the steric and electronic effects of the substituents on the azulenone framework. In general, the cycloheptatriene (CHT) tautomer is thermodynamically more stable than the norcaradiene (NCD) tautomer, which has a strained cyclopropane ring. Thus, the equilibrium lies on the side of the cycloheptatriene species (Table 1, entry 1). However, introduction of substituents on the norcaradiene framework has a dramatic effect on the relative stability of the two tautomers, through a combination of electronic and conjugative effects. Electron withdrawing substituents such as CHO, COOR, CN at C-7 tend to shift the equilibrium to the norcaradiene side (Table 1, entry 2), while the equilibrium is shifted to the cycloheptatriene side by the presence of electron donating substituents, such as OR, NR2etc. (Table 1, entry 3).20,21 Ciganek proposed that an increase in the NC–C–CN angle due to the dipole–dipole repulsion between the two cyano groups leads to the stabilisation of the norcaradiene form (Table 1, entry 2). Replacing one of the cyano groups with a trifluoromethyl group led to a rapidly equilibrating system that was studied by variable temperature NMR (Table 1, entry 5).22 Analogously, two trifluoromethyl groups on the azulenone framework, push the equilibrium almost entirely to the cycloheptatriene (Table 1, entry 4). Kohmoto's studies have shown that the stability of norcaradienes is largely controlled by the nature of the substituent at the C-7 position.23–25 With a CO2Me substituent, the norcaradiene structure is destabilised however, and cleavage of C1–C6 bond is induced by valence isomerisation (Table 1, entry 6).26
The intramolecular Buchner reaction involving aromatic addition reactions of aryl α-diazoketones, effected by rhodium(II) catalysis, leads to a tricyclic norcaradiene, which exists in dynamic equilibrium with the more stable bicyclic cycloheptatriene tautomer through an electrocyclic ring-opening/ring-closing process (Scheme 2). This approach has attracted attention as a versatile route to azulenone systems, which are a very interesting series of compounds incorporating CHT–NCD valence isomers.7,8,27,28 The synthetic utility of the reaction, particularly in the ability to simultaneously generate multiple stereogenic centres, including a challenging bridgehead centre, in a highly stereoselective fashion, has been demonstrated, most notably in the synthesis of various natural products by McKervey29,30 and Mander31–33 and more recently by Reisman.34,35
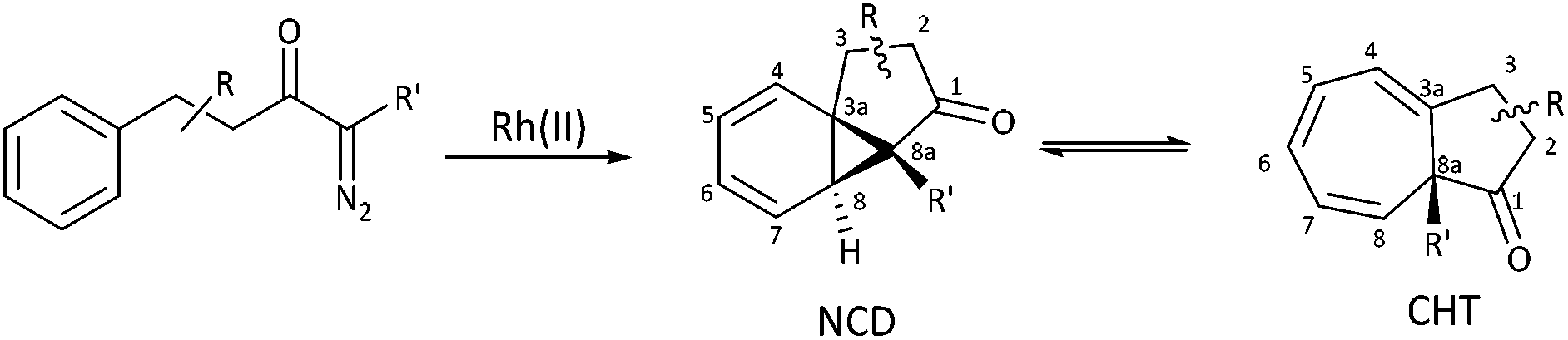 | ||
| Scheme 2 Rhodium(II) catalysed intramolecular Buchner reaction. | ||
Through careful catalyst and substrate selection, high levels of chemoselectivity and regiocontrol can be exercised. Indeed, the enantioselectivity of the intramolecular Buchner reaction of α-diazoketones can even be sensitive to the nature of the counterion present in the catalyst.36
The effect of catalyst and substituent choice on the efficiency, regio- and diastereoselectivity of the reactions of a range of α- and β-substituted α-diazoketones has been recently reported by the Maguire group.42 In addition the dynamic behaviour of methoxyl-substituted azulenones has been described.43
Following Hannemann19 and McKervey's7,8 precedent, NMR spectroscopy is a very useful tool for studying the position of the norcaradiene–cycloheptatriene equilibrium in substituted azulenone systems. The tautomeric equilibrium in the azulenones is, in general, rapid on a NMR timescale, and hence, time-averaged 1H and 13C NMR signals are observed for the system at room temperature. The position of the tautomeric equilibrium can be readily estimated from the C(8)H 1H NMR and C-8 13C NMR chemical shifts since the C-hybridisation exists between a sp2 environment in the cycloheptatriene and a sp3 environment in the norcaradiene.19
Key work by Saba,44 led to the detection of the two tautomeric forms of azulenone 4 using low temperature NMR spectroscopy. Time-averaged signals for the rapid dynamic equilibrium of azulenone 4 were detected at 25 °C (δH C(8)H 3.86 ppm, δC C-8 82.05 ppm) but lowering the temperature to −95 °C led to the observation of two sets of new signals corresponding to the two tautomeric forms of azulenone 4 (Scheme 3). Chemical evidence collected by Saba, also substantiated the existence of the equilibrium by trapping the norcaradiene tautomer in a hetero-Diels Alder reaction with 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) to give the adduct 5 (Scheme 3).44
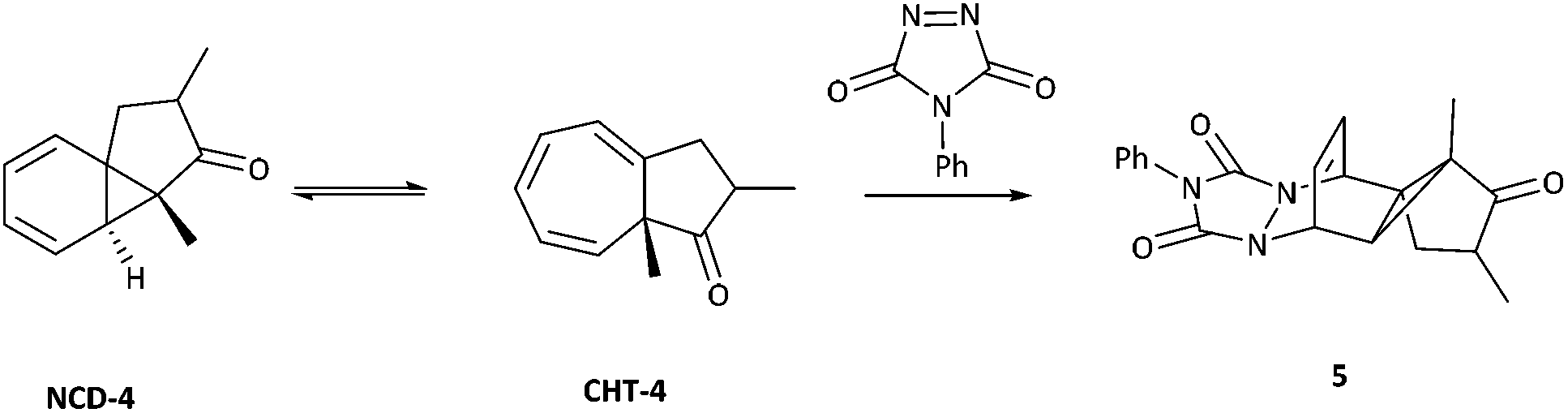 | ||
| Scheme 3 PTAD trapping of the norcaradiene tautomer. | ||
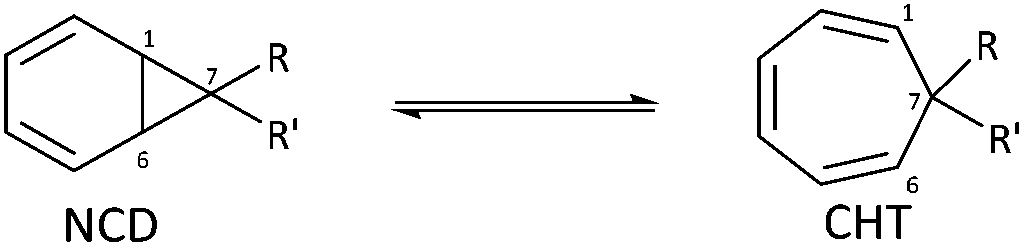
Herein, we discuss the findings of a systematic NMR study on the impact of varying the steric and electronic characteristics of substituents on the position of the norcaradiene–cycloheptatriene equilibrium in a range of azulenones (Scheme 4). These substituents include:
• a hydrogen or a methyl group (R2) at the bridgehead position
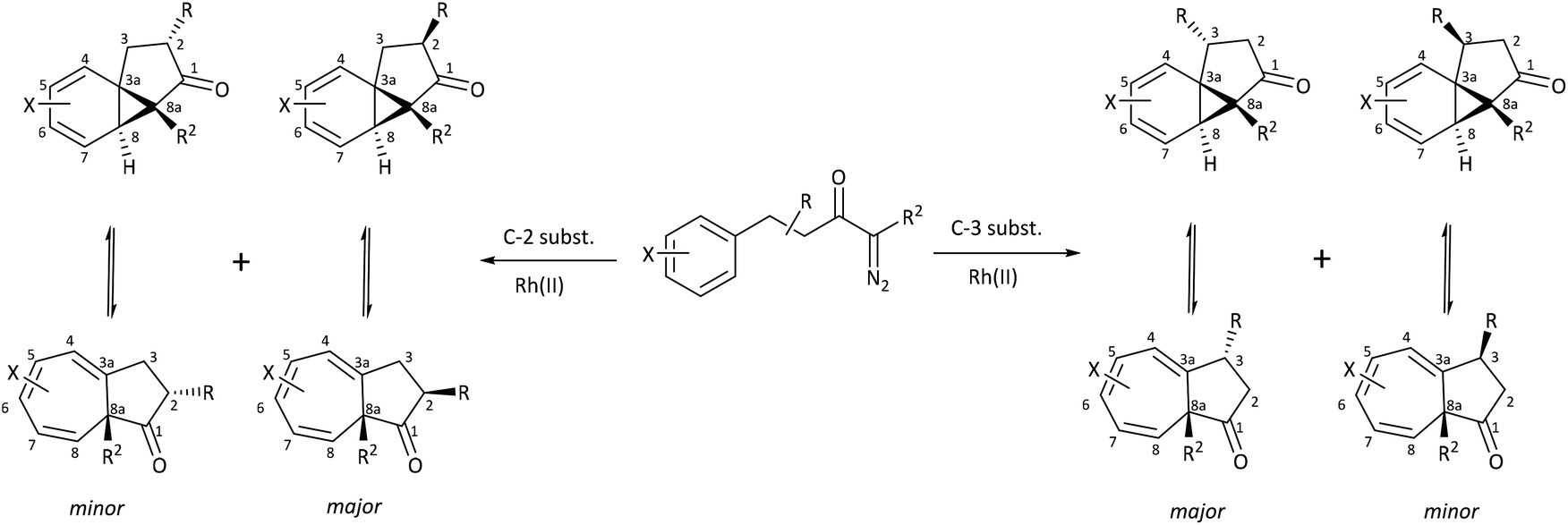 | ||
| Scheme 4 Cyclisation of precursor α-diazoketones leads to an equilibrating norcaradiene–cycloheptatriene product; major and minor diastereomers shown in each case. | ||
• alkyl groups (R) with varying steric demands at positions C-2 and C-3
• a methoxy substituent (X) on the ring system.
A series of room temperature (rt) and variable temperature (VT) 1H and 13C NMR spectroscopy experiments was conducted as described below.
Results and discussion
A series of azulenones with varying substituents R, R2 and X, designed to enable exploration of the influence of substitution on the norcaradiene–cycloheptatriene tautomeric equilibrium, were prepared by efficient rhodium(II) catalysed cyclisation of the precursor α-diazoketones, derived from the appropriate carboxylic acid, following previously reported methodology (Scheme 4).42,43,45,46 With substituents (R) in the linker chain of the diazoketones, the resulting azulenones can be formed in two diastereomeric forms. In practice, the major diastereomer of azulenones substituted at C-2 is the cis-azulenone (C-2 relative to C-8a) while the trans-azulenone (C-3 relative to C-8a) is the major product of the C-3 substituted azulenones due to a conformational preference in the transition states for the aromatic addition.45,47 In many instances, and particularly with sterically demanding groups at position C-3, the azulenones are formed as single diastereomers. The overall diastereoselectivity is sensitive to the nature of the rhodium catalyst.36,42,48The effect of substitution at the bridgehead position, R2 (Scheme 4) has been demonstrated in compounds 6–13. The bridgehead unsubstituted azulenones 6, 8, 10 and 12 derived from the corresponding terminal diazoketones are relatively labile compounds and decompose at room temperature within days. Furthermore, they rearrange on exposure to silica gel to conjugated trienone systems (Scheme 5).
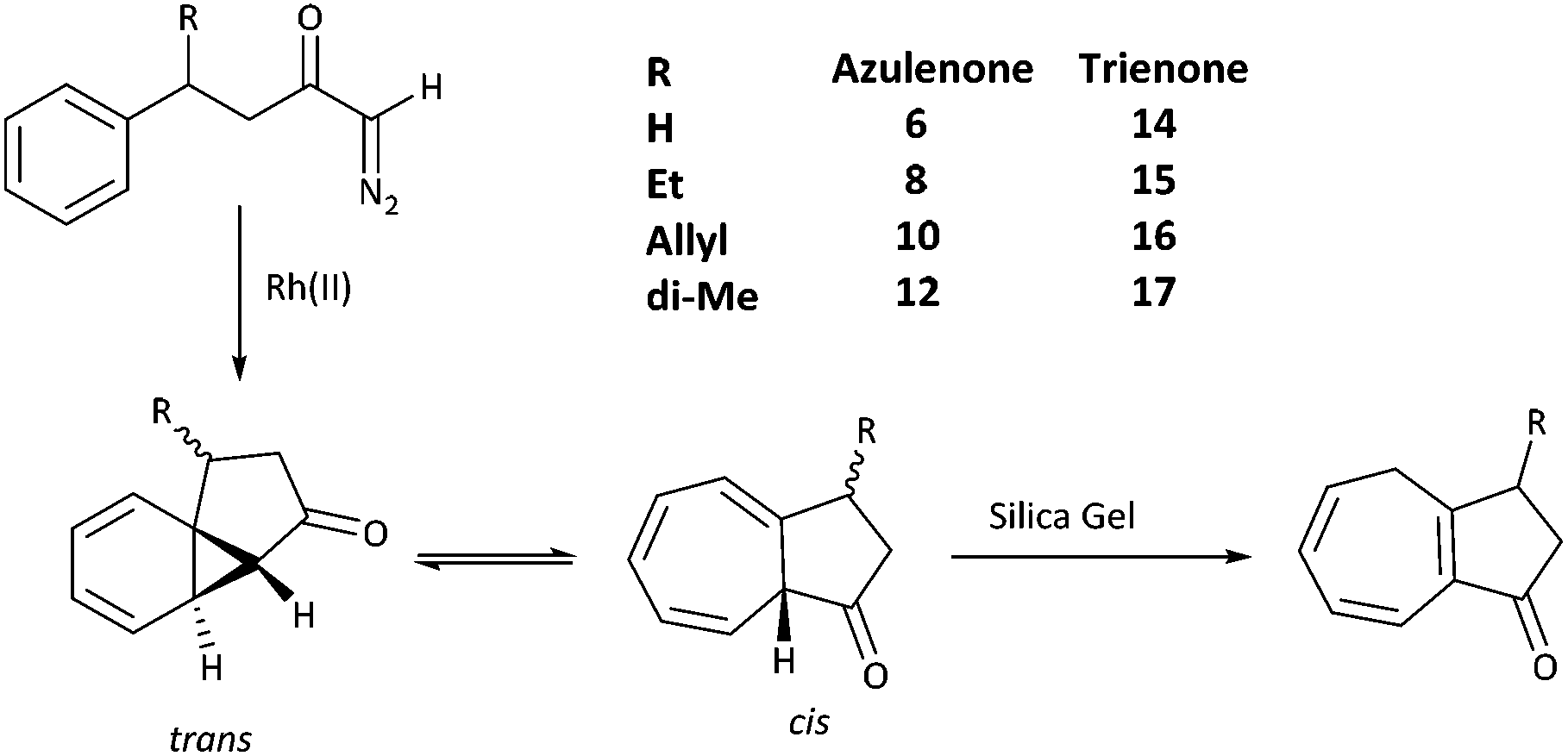 | ||
| Scheme 5 Rearrangement of bridgehead-unsubstituted azulenones to a conjugated trienone. | ||
Treatment with trimethylamine yields the same result. The rearrangement of the unconjugated azulenone on treatment with silica gel, to the more thermodynamically stable isomer, has previously been reported by Scott for the unsubstituted azulenone 6.49
In contrast, the presence of a bridgehead methyl group stabilises the azulenones so that they can be purified by chromatography. Furthermore, increasing the steric demand at C-2 and C-3 (R = Me, Et, n-Pr, i-Pr, n-Bu, t-Bu) results in an increase in stability of the azulenones and indeed the substituted versions are much easier to handle and store than the parent compounds.
Based on Hanneman's analysis, the chemical shift of C(8)H in the cycloheptatriene tautomer is estimated at δH 5.2–5.3 ppm, while the norcaradiene is estimated at δH 2.8–3.5 ppm. In all cases, the observed spectroscopic details are time-averaged when recorded at room temperature, and therefore the position of the tautomeric equilibrium is estimated on Hannemann's basis and included in the tables below. The relative percentages of the cycloheptatriene present are indicative of the trends rather than absolute and should be considered accordingly.
Bridgehead substitution at C-8a
The parent azulenone 6, first described by Scott,28,49 and McKervey,7,8 is a useful starting point in an NMR study of this dynamic equilibrium, existing almost entirely as the cycloheptatriene, with the proton chemical shift of the C(8)H signal occurring at δH 5.06 ppm. Bridgehead substitution of the proton at C-8a with a methyl group results in a shift towards the norcaradiene structure 7 (Table 2, entry 1), which can be rationalised by conformational effects – the bridgehead methyl has the effect of bringing the C-3a and C-8 closer together, resulting in a stabilisation of the norcaradiene (Fig. 3).51 This effect is further demonstrated in the substituted derivatives, particularly the alkyl substituted azulenones (Table 2, entries 2–4).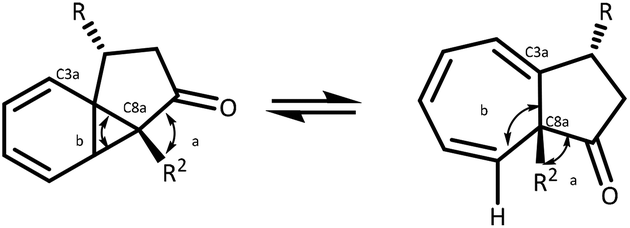 | ||
| Fig. 3 Conformational influence of substituents. | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R2 = H | R2 = Me | ||||||||
| R/R1 | Azulenonea,b | δ H-8 | J 7–8 | % CHT | Azulenonea | δ H-8 | J 7–8 | δ C-8 | % CHT | |
| a Chemical shifts reported in ppm measured at rt.; coupling constants in Hertz; literature references describing the synthesis and characterisation of each of the azulenones included above. b Compounds readily isomerise to the trienone (14–17) and are too unstable to be fully characterised by 13C NMR. c Data for the major trans-diastereomers (C-3 substituent relative to C-8a) only is given in this table (entries 2 & 3). Data for the cis diastereomers of azulenones 9 and 11 is given in Table 3. d Signal masked by signals for the allyl group protons, thus coupling constant could not calculated. | ||||||||||
| 1 | H/H | 6 | 5.06c | 9.4 | 90–95 |
7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 7 |
4.24 | 8.4 | 95.0 | 60–65 |
| 2 | Et/H |
842 |
5.08c | 9.0 | 90–95 |
945 |
3.75 | 7.2 | 77.0 | 40–45 |
| 3 | Allyl/H |
1042 |
5.11 | —d | 95–100 |
1145 |
3.82 | 7.0 | 79.5 | 45–50 |
| 4 | Me/Me |
1250 |
5.12 | 9.3 | 95–100 |
1348 |
4.16 | 8.1 | 89.0 | 55–60 |

As illustrated in Table 2, when the bridgehead substituent R2 = H, variation of the substituent at C-3 does not have a significant influence on the position of equilibrium. However, this is altered when R2 = Me, where C-3 alkyl substitution results in a significant shift towards the norcaradiene. Furthermore, as the steric demand of the C-3 substituent increases, the equilibrium is increasingly shifted towards the norcaradiene form, albeit with modest differences (H to allyl to ethyl). In the case of the geminal-dimethyl substituted derivatives 12 & 13 (Table 2, entry 4), a similar effect is evident with a dramatic increase in the norcaradiene tautomer when the bridgehead methyl substituent is introduced.
Alkyl substitution at C-2 and C-3
Key spectroscopic data for C-2 and C-3 substituted azulenones is summarised in Tables 3 and 4 and our discussion will focus initially on the major diastereomers in each instance, i.e. cis-C-2–C-8a and trans-C-3–C-8a. Substitution at C-3 shifts the position of equilibrium towards the norcaradiene relative to the unsubstituted azulenone 7, as illustrated in Table 3. In contrast, substitution at position C-2 has little impact relative to the unsubstituted analogue and results in a slightly increased preference for the cycloheptatriene structure (compare Tables 3 and 4). Thus, the position of the norcaradiene–cycloheptatriene equilibrium is less sensitive to the substituent in the C-2 substituted azulenones than for the C-3 substituted azulenones (compare Table 3, entries 2–5 with Table 4, entries 2–5). As the C-2 alkyl substituent is more remote from the interconverting norcaradiene–cycloheptatriene ring systems than the C-3 alkyl substituent, this observation is readily rationalised on conformational grounds. Interestingly, the magnitude of the coupling constants (J) of the C(8)H is larger when the signal appears further downfield, consistent with increased sp2 character at C(8)H in the cycloheptatriene tautomer.|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R/R1 | Azulenonea | δ H-8 | J | δ C-8 | % CHT | Azulenonea | δ H-8 | J | δ C-8 | % CHT |
|
a Azulenone isolated as a mixture of diastereomers: C-3 trans or cis w.r.t. C-8a. The trans-azulenone is the major diastereomer in all cases.
b Chemical shifts reported in ppm measured at rt.; coupling constants in Hertz; literature references describing the synthesis and characterisation of each of the azulenones included above.
c Signal not detected.
d
cis-Isomer not identified.
e Azulenones are present in the ratio, a:b = 93:7. The stereochemistry of the major isomer has not been established.
f Signal is broad.
|
|||||||||||
| 1 | H/H | 7 | 4.24 | 8.4 | — | 60–65 | |||||
| 2 | Me/H43 | trans-18 | 3.73 | 7.5 | —c | 40–45 | cis-18 | 4.66 | 8.9 | —c | 75–80 |
| 3 | Et/H45 | trans-9 | 3.75 | 7.2 | 79.18 | 40–45 | cis-9 | 4.85 | 9.2 | —c | 85–90 |
| 4 | n-Pr/H45 | trans-19 | 3.68 | 7.0 | 75.5 | 40–45 | cis-19 | 4.90 | 9.0 | 115.7 | 85–90 |
| 5 | i-Pr/H45 | trans-20 | 3.84 | 7.5 | 87.9 | 45–50 | cis-20 | 4.70 | 9.0 | —c | 80–85 |
| 6 | n-Bu/H45 | trans-21 | 3.70 | 7.0 | 74.7 | 40–45 | cis-21 | 4.90 | 9.0 | 115.5 | 85–90 |
| 7 | t-Bu/H45 | trans-22 | 4.14 | 7.8 | 84.0 | 55–60 | cis-22 | 5.08 | 10.0 | —c | 90–95 |
| 8 | Ph/H45 |
23d |
4.07 | 8.0 | —c | 50–55 | |||||
| 9 | Ph/Me42 |
24ae |
3.45 | 6.8 | 64.6f | 25–30 |
24be |
4.21 | 7.9 | —c | 60–65 |
| 10 | Me/Me48 | 25 | 4.15 | 8.0 | 89.1 | 55–60 | |||||
| 11 | Allyl/H45 | trans-11 | 3.82 | 7.0 | 79.8 | 45–50 | cis-11 | 4.80 | 9.0 | —c | 80–85 |
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | Azulenonea | δ H-8 | J | δ C-8 | % CHT | Azulenonea | δ H-8 | J | δ C-8 | % CHT |
| a Azulenone isolated as a mixture of diastereomers, C-2 trans or cis relative to C-8a. The cis-azulenones are the major diastereomers. While the minor trans-diastereomers are seen with the C-2-alkyl azulenones the phenyl and benzyl derivatives 29 and 30 are formed exclusively as the cis-diastereomer (entries 6&7). b Chemical shifts reported in ppm measured at rt.; coupling constants in Hertz; literature references describing the synthesis and characterisation of each of the azulenones included above. c Signal not detected. | |||||||||||
| 1 | H | 7 | 4.24 | 8.4 | — | 60–65 | |||||
| 2 | Me42 | cis-4 | 4.30 | 8.5 | 97.9 | 65–70 | trans-4 | 5.29 | 9.9 | 124.2 | 95–100 |
| 3 | Et42 | cis-26 | 4.36 | 8.6 | 100.6 | 70–75 | trans-26 | 5.24 | 9.9 | 123.8 | 95–100 |
| 4 | n-Pr42 | cis-27 | 4.37 | 8.6 | 100.4 | 70–75 | trans-27 | 5.24 | 9.9 | —c | 95–100 |
| 5 | i-Pr42 | cis-28 | 4.53 | 9.0 | 106.2 | 75–80 | trans-28 | 5.24 | 10.0 | 124.4 | 95–100 |
| 6 | Ph42 | cis-29 | 4.65 | 8.9 | 105.1 | 75–80 | |||||
| 7 | Bn42,48 | cis-30 | 4.37 | 8.6 | 99.6 | 70–75 | |||||
Interestingly, a distinctive steric effect is observed in the C-3 alkyl substituted azulenone series, which is not observed for the corresponding C-2 substituted analogues; as the alkyl substituent increases in size from methyl to ethyl to n-alkyl, a small but detectable shift towards the norcaradiene form is observed, (cf.Fig. 4 and compare Table 3, entries 1–5 with Table 4, entries 1–5). However, with the more sterically demanding t-butyl and geminal-dimethyl groups at position C-3, a conformational change is evident and the position of equilibrium is essentially frozen by the steric demands of the bulky substituents (Table 3, entries 7 & 10). The position of equilibrium for these azulenones is essentially equivalent to that of the unsubstituted azulenone 7 (Table 3, entry 1) in that they contain almost equal mixtures of the two tautomers. Intermediate effects are seen with an isopropyl substituent (Table 3, entry 5). Where detected, the 13C NMR chemical shifts of the C-8 carbons mirror the observed trends, showing a distinctive shift due to the changing hybridisation of the carbons involved in the equilibration process.
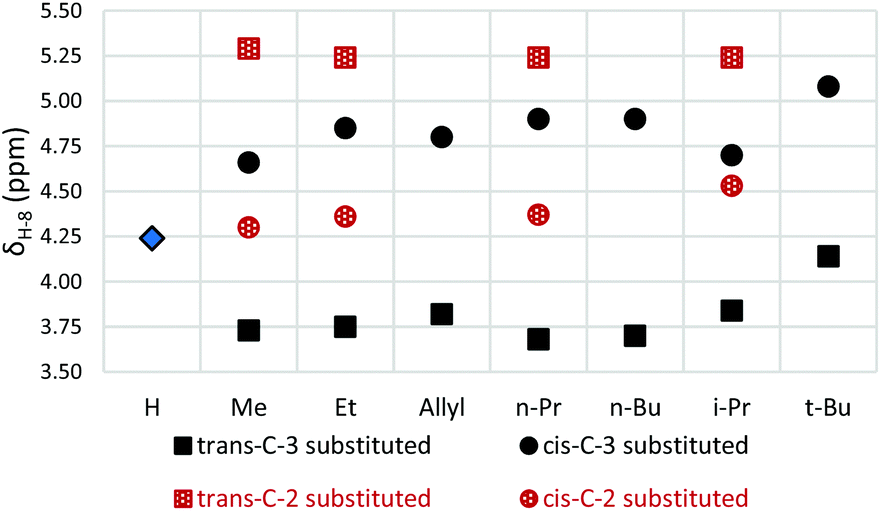 | ||
| Fig. 4 Effect of substitution at C-3 and C-2 on C(8)H 1H NMR chemical shift relative to the unsubstituted azulenone 7. | ||
The stereochemistry at C-2, C-3 and C-8a
As illustrated in Fig. 4, the stereochemistry of the substituents at positions C-2 and C-3 relative to C-8a methyl has a bearing on the position of the norcaradiene–cycloheptatriene equilibrium. In the C-3 substituted azulenones, which are formed predominantly as trans-diastereomers, the equilibrium lies towards the norcaradiene relative to the unsubstituted azulenone 7; while for the minor cis-diastereomers, there is a downfield shift of the C(8)H signal towards the cycloheptatriene. In the C-2 substituted series, for both the cis- and trans-azulenones, the position of equilibrium lies more towards the cycloheptatriene than in the unsubstituted azulenone 7, with the effect being stronger for the minor trans-azulenones, 26–30. Furthermore, the coupling constant (J) for the C(8)H signal is a little larger for the minor diastereomers reflecting the different conformation of the two diastereomers based on the position of equilibrium.For each of the minor diastereomers (cis-C-3 substituted and trans-C-2 substituted), the position of equilibrium is less sensitive to substituent bulk in both cases with little or no shift in equilibrium for C-2 substituted azulenones (Fig. 5) and a small shift towards the cycloheptatriene with increased steric demand at C-3 for the C-3 substituted azulenones (Table 3). Steric strain between the alkyl substituent and the C(4)H in the cis-C-3 norcaradiene destabilise this tautomer, while the presence of the alkyl substituent at position C-3 in the major trans-C-3 azulenones conformationally favours the norcaradiene tautomer by bringing C-3 and C-8 closer together (Fig. 5).
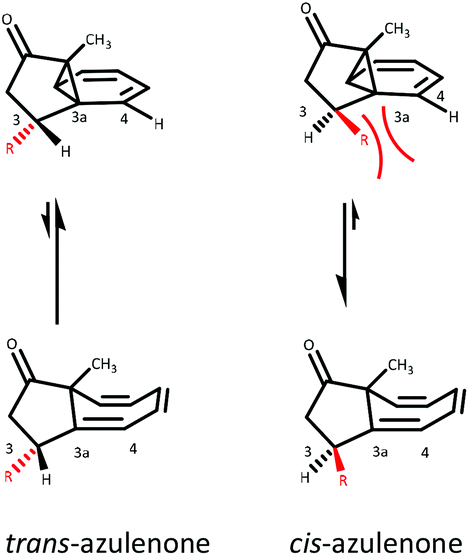 | ||
| Fig. 5 Conformational preference for NCD tautomer in C-3 substituted azulenones. | ||
Variable temperature NMR spectroscopy studies
In an attempt to resolve the signals of the equilibrating structures into those of the individual tautomers, a series of variable temperature 1H and 13C NMR studies were conducted on selected azulenone systems (Table 5). All studies were conducted in deuterated dichloromethane and typically the temperature was lowered in steps of 10 K from 300 K down to as low as 180 K, depending on the specifications of the system used. A reduction in temperature leads to a slowing down of the rate of interchange between the tautomers which is often observed as a broadening of the 13C C-8 [and in some cases the C(8)H] signal. With sufficient cooling, it was anticipated that separate signals would be observed for the individual tautomers, reflecting the population of the norcaradiene and cycloheptatriene forms at different temperatures.|
|
|||
|---|---|---|---|
| Entry | R | Azulenonea | Δ % CHTb |
|
a Resolution of the individual NCD/CHT signals not observed for any of the compounds above as slow exchange limit not reached.
b Position of equilibrium influenced by temperature. Estimation based upon Hannemann's principle;19 values estimate the change in position of equilibrium using δH C(8)H at rt and at low T (180–230 K depending upon experiment).
c Analysed as a mixture (ca. 80% trans-19), then pure trans-isomer analysed separately.
d Analysed as a mixture trans:cis (>98:2).
|
|||
| 1 | H | 7 | 60–65 → 50–55 |
| 2 | 2-Me | cis-4 | 65–70 → 50–55 |
| 3 | 2-i-Pr | cis-28 | 75–80 → 65–70 |
| 4 | 2-Bn | cis-30 | 70–75 → 50–55 |
| 5 | 3-n-Prc | trans-19 | 40–45 → 20–25 |
| cis-19 | 85–90 → 85–90 | ||
| 6 | 3-i-Prd | trans-20 | 45–50 → 0–5 |
| 7 | 3-n-Bu | trans-21 | 40–45 → 15–20 |
| cis-21 | 85–90 → 80–85 | ||
| 8 | 3-t-Bu | trans-22 | 55–60 → 50–55 |
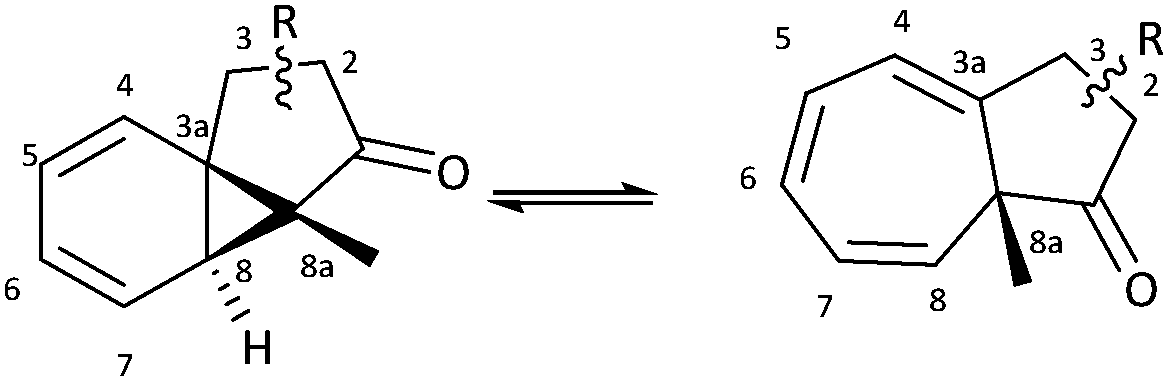
The C-3 alkyl substituted azulenones 19 (80:20 trans:cis, also pure trans), 20 (trans), 21 (cis and trans analysed individually) and 22 (trans), together with the unsubstituted azulenone 7 were initially studied, in an attempt to resolve the respective tautomers (Table 5, entries 1 and 5–8 & Fig. 6). Focussing initially on the major trans diastereomers of azulenones 19, 20 and 21, as illustrated in Fig. 7, upon reduction of the temperature to 193 K, 180 K and 192 K respectively, the key signals of the C(8)H, in each case, broaden as the temperature is reduced, as anticipated. While complete resolution of the individual tautomers was not observed at lowest temperature, since the slow-exchange limit was not reached, the study did nonetheless provide some interesting and unanticipated results, with the spectra of 19, 20 and 21 showing a significant upfield shift of the C(8)H proton signal at lower temperature, indicating an increase in the norcaradiene tautomer. While alteration of chemical shift with variation of temperature is well known, the extent of the variation in this case is much greater than could be rationalised solely on this basis. This effect was greatest in the analysis of trans-20, which exists as an almost equimolar mixture of tautomers at room temperature but showed a dramatic upfield shift of the C(8)H signal to δH 2.70 ppm (est. 0–5% CHT) upon cooling, indicating an increased preference for the norcaradiene tautomer at reduced temperature. Broadening of the C(8)H signal was also observed on cooling until coalescence occurred at 210 K and by 180 K a broad singlet at δH 2.70 ppm, assigned to the C(8)H signal, had appeared. Further decrease in temperature is likely to reach the slow exchange limit and to thus afford tautomeric resolution as the peaks are broad even at low temperature.
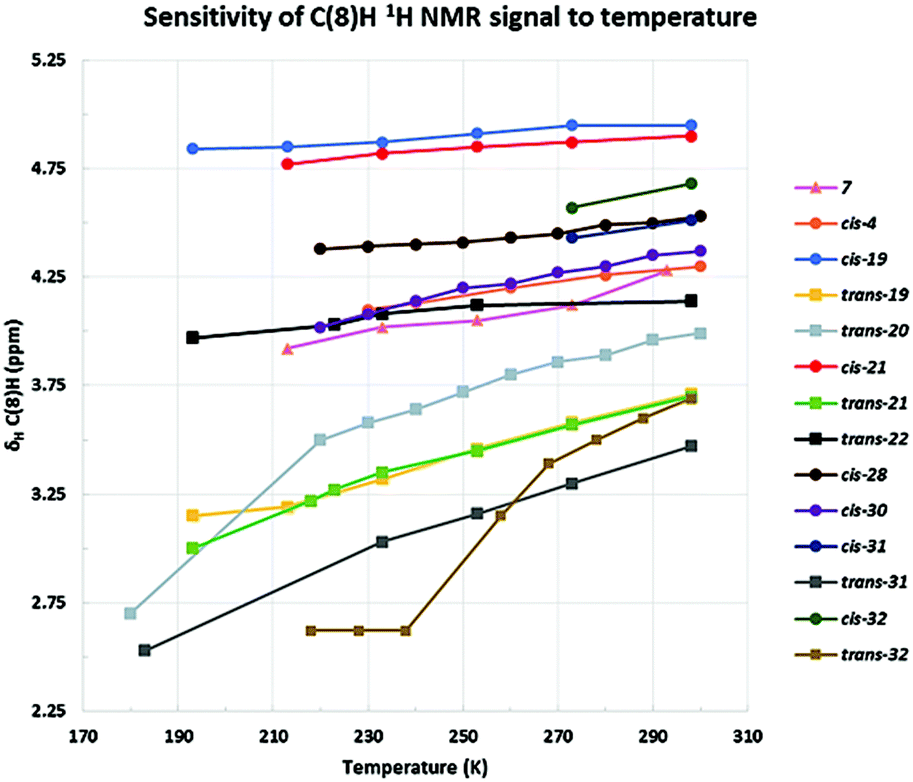 | ||
| Fig. 6 Effect of temperature on the position of NCD–CHT equilibrium as indicated by the C(8)H 1H NMR chemical shift (shift displayed where observed for a given temperature). Signal for cis-31 and cis-32 undetected below 273 K. | ||
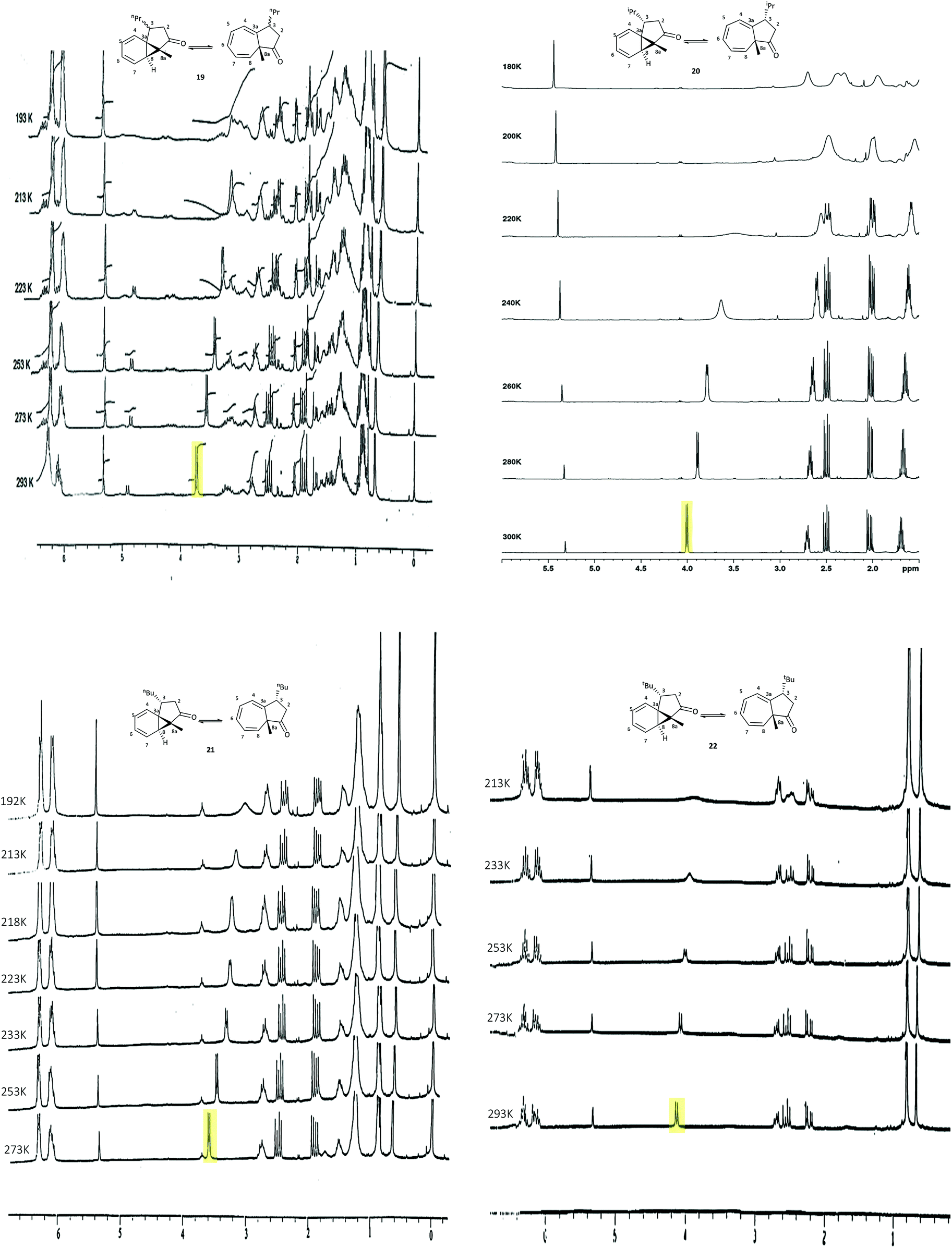 | ||
| Fig. 7 Variable temperature 1H NMR spectra of the azulenones 19 (trans:cis 80:20) (270 MHz), trans-20 (500 MHz), trans-21 (270 MHz), trans-22 (270 MHz) recorded in CD2Cl2; C(8)H highlighted. | ||
Additional evidence to support the assignment of the norcaradiene tautomer was provided by 13C NMR spectroscopy showing the disappearance of the broad C-8 signal (84 ppm at 300 and 240 K) by 180 K and the detection of four distinct, albeit broadened, alkene signals at 122.7, 124.4, 126.5 and 130.0 ppm.
Interestingly, the signals for the minor cis-azulenones 19 and 21 displayed only a minor shift on decreasing the temperature (Table 5, entries 5 and 6); once again resolution of the signals for the interconverting tautomers was not achieved at low temperature. VT-NMR studies on the C-3 t-butyl substituted azulenone 22, behaved very differently. In this case only the trans-isomer is present as the synthesis is highly stereoselective in the presence of the bulky t-butyl substituent. On decreasing the temperature, as illustrated in Fig. 7, the signal for the C(8)H broadened significantly indicating a decreased rate of interconversion, but crucially in this instance there was no detectable shift in the position of equilibrium with temperature, in direct contrast to the outcome with the C-3 n-Pr and n-Bu substituted analogues. It is clear from a combination of the room temperature and VT-NMR studies that the presence of the sterically demanding t-butyl substituent at the C-3 position in 22 has a dominant conformational impact on the structure such that the norcaradiene–cycloheptatriene equilibrium is, in this case, essentially frozen with little sensitivity to temperature or other factors.
Variable temperature studies of the norcaradiene–cycloheptatriene equilibrium usually focus on reaching the slow exchange rate of the interconversion and thus resolving the NMR signals for the two individual tautomers; our observation that the position of equilibrium shifts significantly towards the norcaradiene on lowering the temperature of the azulenone solution, for 19, 20 and 21, is an unusual outcome (Fig. 6).27
The impact of temperature on the position of equilibrium was smaller in the case of the unsubstituted azulenone 7, which showed a shift towards the norcaradiene tautomer of approx. 10% at 213 K. The C(8)H signal was undetectable below this temperature and complete resolution of the tautomers was not observed in our study.
We subsequently explored the C-2 alkyl substituted azulenones 4, 28, 30, derived from the α-substituted diazoketones (Table 5, entries 2–4 & Fig. 6) and once again resolution of the individual tautomers of the azulenones was not observed, since the slow exchange rate of the interconversion was not reached (Fig. 8). In each case only the cis-isomer is present as the synthesis is highly stereoselective (dr >98:2).42 The extent of shift of the C(8)H was less than that observed with the C-3 alkyl series, 19–22 but significant nonetheless at approx. 15%, 10% & 20% respectively. These shifts are analogous to that encountered for the unsubstituted azulenone 7, and show that the position of equilibrium is somewhat less temperature dependant in the C-2 substituted series. This is consistent with the fact that the C-2 alkyl substituent is more remote than the C-3 alkyl substituent from the dynamic norcaradiene–cycloheptatriene ring system resulting in reduced entropic impact on the position of equilibrium.
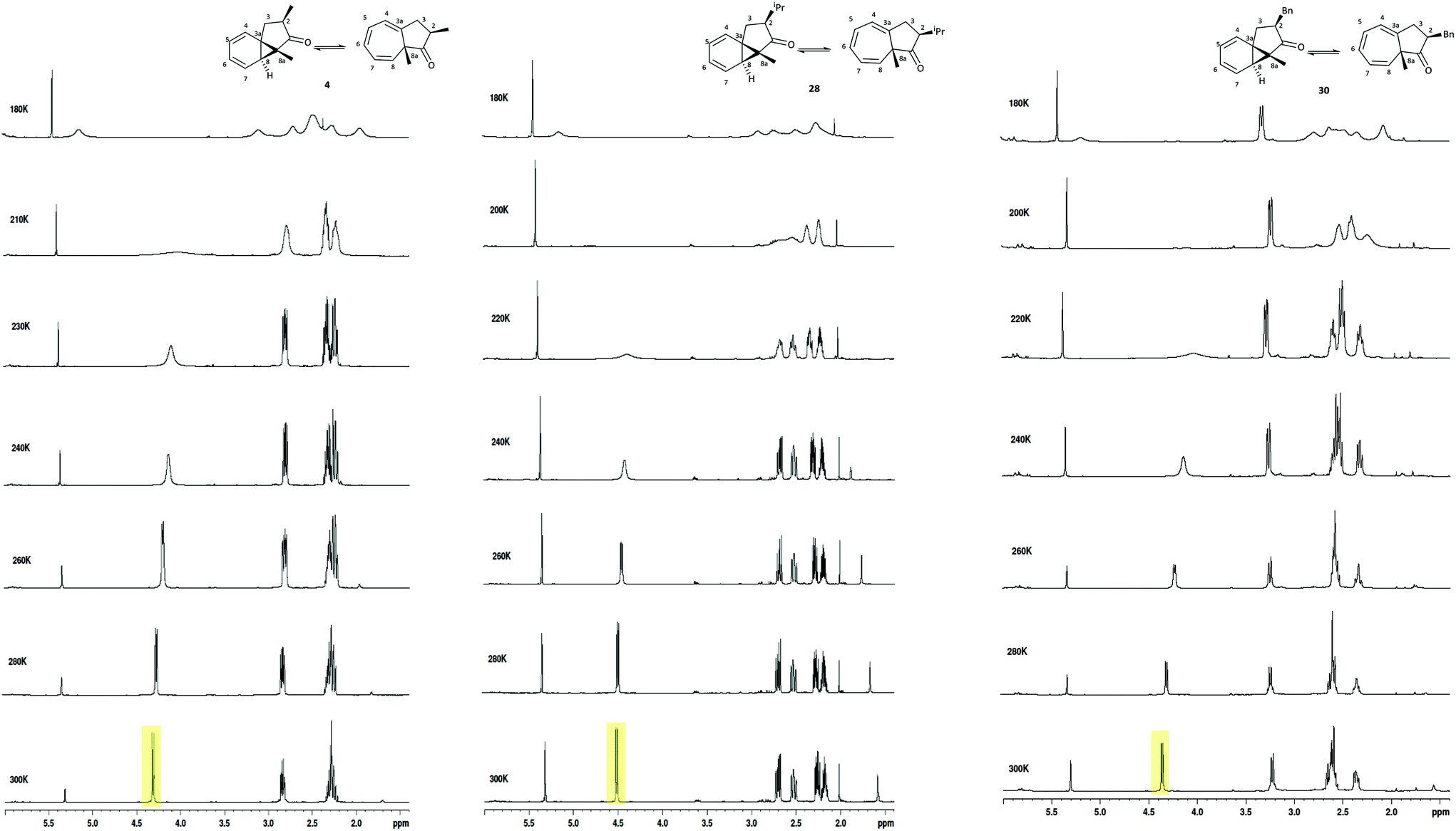 | ||
| Fig. 8 Variable temperature 1H NMR spectra of the azulenone cis-4, cis-28 and cis-30, recorded in CD2Cl2 at 500 MHz. | ||
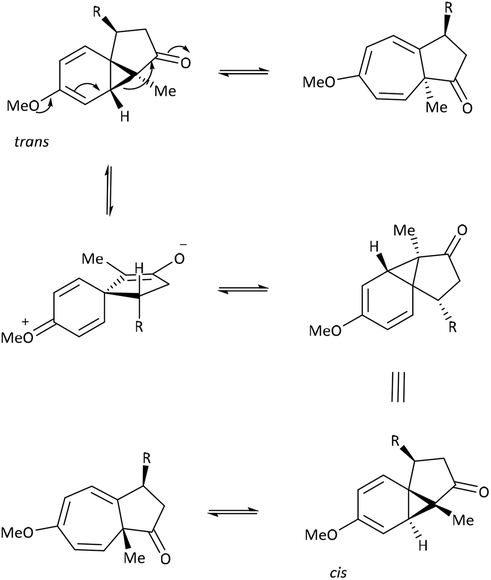
Previous work within our group observed the dynamic equilibria of the C-6 methoxy azulenones cis-31 and cis-32, by variable temperature NMR (Table 6).43 These azulenone systems are complex: in parallel with the equilibrium between the norcaradiene and cycloheptatriene tautomers, interconversion of the trans- and cis-diastereomers occurs simultaneously. Similar to the C-3 substituted systems, resolution of the signals due to the tautomers of each azulenone was not observed since the slow exchange rate of interconversion was not reached, but again some interesting trends were recorded. Decreasing the temperature resulted in an upfield shift in the position of the C(8)H signal of the trans diastereomer, indicating an increased preference for the norcaradiene tautomer at reduced temperatures, in line with the observations noted earlier for azulenones 19 and 21 (Table 5). More significantly, an increase in the ratio of trans:cis azulenone was observed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Azulenone | Resolutiona | Δ % CHTb | drc |
a Observation of the resolution of the individual NCD/CHT signals if slow exchange limit was reached.
b Position of equilibrium influenced by temperature. Estimation based upon Hannemann's principle;19 values estimate change in position of equilibrium using δH C(8)H at rt and at low T (180–218 K depending upon spectrometer). Signals for the cis-isomers were undetectable below 273 K.
c Diastereomeric ratio (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :cis) influenced by temperature.
d Analysed as a mixture (80% trans-31, 80% trans-32, 94% trans-33). :cis) influenced by temperature.
d Analysed as a mixture (80% trans-31, 80% trans-32, 94% trans-33).
|
|||||
| 1 | Me43d |
trans-31 | ✗ | 30–35 → 0–5 | ✓ |
| cis-31 | ✗ | 70–75 → 70–75 | |||
| 2 | 3-i-Pr43d |
trans-32 | ✗ | 40–45 → 0–5 | ✓ |
| cis-32 | ✗ | 75–80 → 75–80 | |||
| 3 | 3-t-Bu43d |
trans-33 | ✓ | Resolution | ✗ |
| cis-33 | ✓ | Resolution | |||
Azulenone 3343 was analysed as a mixture of diastereomers (trans:cis, 94:6) (Table 6, entry 3). At 300 K, the major trans-azulenone C(8)H signal is observed at 3.90 ppm, indicating that the equilibrium contains ∼45% of the cycloheptatriene tautomer. Coalescence is observed for trans-33 at 260 K and resolution of the signals corresponding to both tautomers is achieved by 183 K as the slow exchange limit of the system is reached, where separate doublets are observed at 2.85 ppm and 5.62 ppm for the norcaradiene and cycloheptatriene (Fig. 9). Thus, for the first time in this series, signals for the two interconverting tautomers were successfully resolved by 1H NMR spectroscopy, analogous to Saba's report.44 A more complete analysis of the position of the signals attributed to each component is given in Table 7.
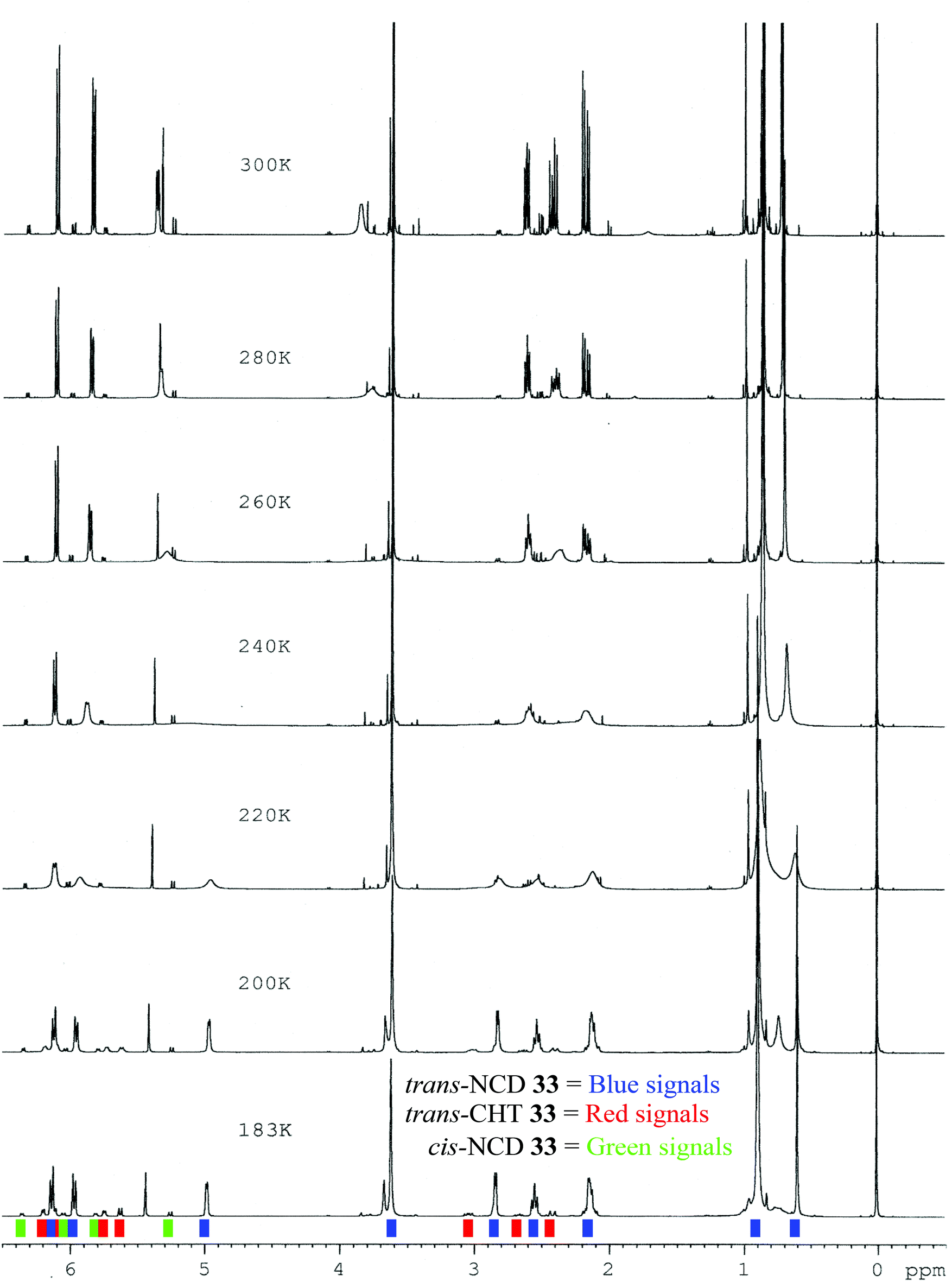 | ||
| Fig. 9 Variable Temperature 1H NMR stacked plot of azulenone 33 (CD2Cl2; 500 MHz); trans-NCD 33 (blue), trans-CHT 33 (red), cis-NCD 33 (green). | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Temp.a (K) | δH H-8 ppm | δH H-7 ppm | δH H-4 & H-5 ppm | |||
| NCD | CHT | NCD | CHT | NCD | CHT | |
| a Spectra recorded in CD2Cl2 at 500 MHz. | ||||||
| 300 | 3.90, br s | 5.45, d | 6.15, d; 5.82, d | |||
| 280 | 3.80, br s | 5.42, br s | 6.15, d; 5.82, d | |||
| 260 | — | 5.38, br s | 6.15, d; 5.82, d | |||
| 240 | — | 5.15 v br s | 6.15, d; 5.82, br d | |||
| 220 | 2.85, br s | — | 4.97, br s | 6.15, d; 5.82, br s | ||
| 200 | 2.85, d | 5.62, br d | 4.98, d | 6.11, d | 6.15, d; 5.97, d | 6.20, d; 5.75, br d |
| 183 | 2.85, d | 5.62, d | 4.98, d | 6.11, d | 6.15, d; 5.97, d | 6.20, d; 5.75, d |
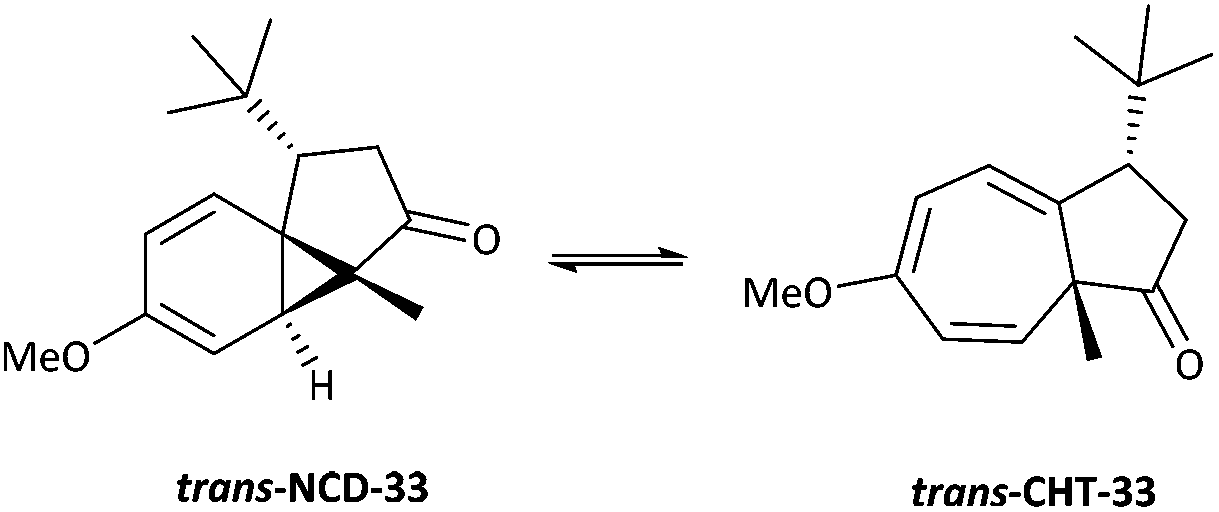
The ability to observe the signals of the two individual tautomers of trans-33, while the slow exchange limit was not reached for the azulenones 4, 20, 28 and 30 by 180 K warrants consideration. This is most likely due to the presence of the sterically demanding t-butyl group, which inhibits the rate of interconversion sufficiently such that at 183 K the slow exchange limit is achieved. As further evidence, in working with the azulenone 33 there was a very significant difference in the rate at which equilibration of the cis and trans isomers took place at room temperature. It took typically 6 days for the mixture to reach 96:4 which appears to be the thermodynamic equilibrium for this compound at room temperature. In contrast the cis–trans interconversion of the methyl substituted derivative 31, is so fast that we have never observed mixtures at anything other than the thermodynamic ratio 80:20. However, with the isopropyl derivative 32 we have managed to capture NMR spectra with cis–trans ratios other than the equilibrium 80:20, which is achieved within 21 h (room temperature, CDCl3) for this compound.43 Accordingly, below room temperature, the rate of cis–trans interconversion of 33 would be slower, such that an alteration in the cis–trans ratio would not be observed in the low temperature NMR studies.
The chemical shift of signals due to C(8)H of the minor cis-azulenone 33 were barely affected on reduction of the temperature, with no broadening observed. This is rationalised on the basis that cis-33 exists almost entirely as its cycloheptatriene tautomer (C(8)H signal observed at 5.38 ppm, estimated 95–100% CHT) and thus there is no apparent interconversion to slow down.
A 13C NMR spectrum of 33 was also recorded at 183 K, along with COSY and C–H correlation experiments, to allow the complete assignment of the 13C NMR spectrum (Fig. 10). This provides further evidence of the presence of the three components in the NMR mixture at 183 K. Three carbonyl resonances are clearly visible at 218.1, 221.4 and 222.9 ppm corresponding to the norcaradiene tautomer of trans-33, the cycloheptatriene tautomer of trans-33 and the cycloheptatriene tautomer of cis-33, respectively. Other significant resonances are the C-8 signals of the norcaradiene tautomer of trans-33 at 36.77 ppm, with the C-8 resonances for the cycloheptatriene tautomer of trans-33 and cis-33 observed at 126.7, which compare well with those reported by Saba.44 Interestingly, the resonances for C-4, C-5 and C-7 of trans-33 in the 13C NMR spectrum of 33 at 300 K are significantly broadened, and no signal due to C-8 of trans-33 is observed, which suggests that at this temperature the rate of norcaradiene to cycloheptatriene interconversion of trans-33 is slow on the 13C-NMR timescale. In contrast, the signals that can be attributed to cis-33 are sharp at this temperature.
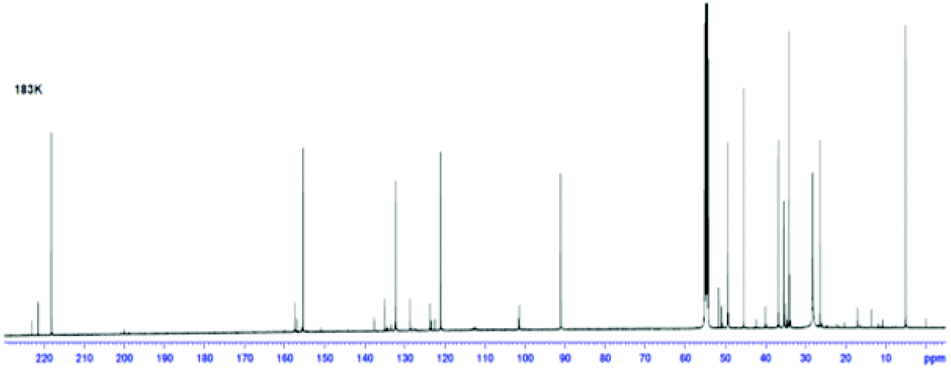 | ||
| Fig. 10 13C NMR spectrum of azulenone 33 at 183 K (CD2Cl2; 500 MHz). | ||
Conclusions
In summary, the norcaradiene–cycloheptatriene tautomeric equilibrium in the series of azulenones studied is influenced by both the position and the steric demands of alkyl substituents on the core molecular framework. NMR spectroscopy has enabled rationalisation of the observed trends on the basis of the position and nature of the substituents. In general substituents at C-3 have a stronger impact on the norcaradiene–cycloheptatriene equilibrium than those at C-2, and, furthermore, when the C-3 substituent is trans- to the bridgehead methyl group the effect is substantially more evident than for the cis-analogues. With the parent unsubstituted azulenone 6, the equilibrium lies very much towards the cycloheptatriene form; introduction of substituents at C(8a) and C-3 (trans) result in a substantial shift towards the norcaradiene tautomer. The minor diastereomers of the azulenones, either cis- or trans-structures depending on whether C-3 or C-2 substituted, lie almost entirely on the side of the cycloheptatriene with little sensitivity to substituent size.Variable temperature NMR studies have shown that the norcaradiene–cycloheptatriene equilibrium in the trans C-3 alkyl substituted azulenones, and to a lesser extent the cis C-2 substituted derivatives, is sensitive to temperature, showing a shift towards the norcaradiene tautomer at lower temperatures. In contrast, with the t-butyl substituent, the equilibrium is unaffected by temperature highlighting the conformational impact of the sterically demanding substituent. Signals for the tautomers of the azulenone 33 were successfully resolved at 183 K allowing full characterisation of each structure by 1H and 13C NMR analysis.
Experimental
1H and 13C NMR spectra were recorded on a Jeol GSX 270 MHz (13C, 67.8 MHz), Bruker AVANCE 300 MHz (13C, 75.5 MHz), Bruker AVANCE 400 MHz (13C, 100.6 MHz) or Bruker AVANCE 500 MHz (13C, 125.8 4 MHz) NMR spectrometers. All spectra recorded at room temperature (∼20 °C) were in deuterated chloroform (CDCl3). 1H and 13C NMR spectra were also recorded in deuterated dichloromethane for VT studies. A minor solvent effect was observed for azulenone 33, δH-8 (CDCl3) 3.81 ppm; δH-8 (CD2Cl2) 3.90 ppm. Chemical shifts (δ) are quoted in parts per million (ppm) downfield from tetramethylsilane and coupling constants (J) in Hertz (Hz). In some cases, 13C NMR spectra were assigned with the aid of DEPT experiments. Synthesis and characterisation of each of the azulenones have been previously described.7,42,43,45,48,50Acknowledgements
The financial support of the following is gratefully acknowledged: SFI Advance Award (14/ADV/RC2751), Pfizer Pharmaceuticals (studentships to P. O. L. and S. O. N.), University College Cork, especially UCC President's Research Fund, Forbairt, Cork Corporation, Cork County Council, the Royal Irish Academy, National University of Ireland (Postdoctoral Fellowship to P. O. L.), Enterprise Ireland, IRCSET (studentships to S. O. K., O. A. M and S. O. N.) PRTLI3 (studentship to A. S.) and Johnson Matthey for a donation of rhodium(II) acetate.Notes and references
- E. Buchner and T. Curtius, Ber., 1885, 18, 2371 CrossRef.
- E. Buchner and T. Curtius, Ber., 1885, 18, 2377 CrossRef.
- W. Doering, G. Laber, R. Vonderwahl, N. F. Chamberlain and R. B. Williams, J. Am. Chem. Soc., 1956, 78, 5448 CrossRef CAS.
- A. J. Anciaux, A. Demonceau, A. J. Hubert, A. F. Noels, N. Petiniot and P. Teyssie, J. Chem. Soc., Chem. Commun., 1980, 765 RSC.
- A. J. Anciaux, A. Demonceau, A. F. Noels, A. J. Hubert, R. Warin and P. Teyssie, J. Org. Chem., 1980, 46, 873 CrossRef.
- A. J. Anciaux, A. Demonceau, A. J. Hubert, A. F. Noels, N. Petiniot and P. Teyssie, J. Org. Chem., 1980, 45, 695 CrossRef CAS.
- M. Kennedy, M. A. McKervey, A. R. Maguire, S. M. Tuladhar and M. F. Twohig, J. Chem. Soc., Perkin Trans. 1, 1990, 1047 RSC.
- M. A. McKervey, S. M. Tuladhar and M. F. Twohig, J. Chem. Soc., Chem. Commun., 1984, 129 RSC.
- E. Vogel and H. Reel, J. Am. Chem. Soc., 1972, 94, 4388 CrossRef CAS.
- P. Manitto, D. Monti and G. Speranza, J. Org. Chem., 1995, 60, 484 CrossRef CAS.
- M. P. Doyle, D. G. Ene, D. C. Forbes and T. H. Pillow, Chem. Commun., 1999, 1691 RSC.
- E. J. Corey, H. J. Burke and W. A. Remers, J. Am. Chem. Soc., 1955, 77, 4941 CrossRef CAS.
- F. A. L. Anet, J. Am. Chem. Soc., 1964, 86, 458 CrossRef CAS.
- F. R. Jensen and L. A. Smith, J. Am. Chem. Soc., 1964, 86, 956 CrossRef CAS.
- J. B. Lambert, L. J. Durham, P. Lepoutere and J. D. Roberts, J. Am. Chem. Soc., 1965, 87, 3896 CrossRef CAS.
- M. B. Rubin, J. Am. Chem. Soc., 1981, 103, 7791 CrossRef CAS.
- R. Wehner and H. Günther, J. Am. Chem. Soc., 1975, 97, 923 CrossRef CAS.
- K. Takeuchi, T. Kitagawa, A. Ueda, Y. Senzaki and K. Okamoto, Tetrahedron, 1985, 41, 5455 CrossRef CAS.
- K. Hannemann, Angew. Chem., Int. Ed. Engl., 1988, 284 CrossRef.
- W. Adam, M. Balci and B. Pietrzak, J. Am. Chem. Soc., 1979, 101, 6285 CrossRef CAS.
- M. Celik and M. Balci, ARKIVOC, 2007, 150 Search PubMed.
- E. Ciganek, J. Am. Chem. Soc., 1965, 87, 652 CrossRef CAS.
- S. Kohmoto, T. Kawatsuji, K. Ogata, M. Yamamoto and K. Yamada, J. Am. Chem. Soc., 1991, 113, 5476 CrossRef CAS.
- S. Kohmoto, T. Funabashi, N. Nakayama, T. Nishio, I. Iida, K. Kishikawa, M. Yamamoto and K. Yamada, J. Org. Chem., 1993, 58, 4764 CrossRef CAS.
- S. Kohmoto, I. Koyano, T. Funabashi, K. Kishikawa, M. Yamamoto and K. Yamada, Tetrahedron Lett., 1995, 36, 553 CrossRef CAS.
- M. Yang, T. R. Webb and P. Livant, J. Org. Chem., 2001, 66, 4945 CrossRef CAS PubMed.
- O. A. McNamara and A. R. Maguire, Tetrahedron, 2011, 67, 9 CrossRef CAS.
- L. T. Scott, J. Chem. Soc., Chem. Commun., 1973, 882 RSC.
- M. Kennedy and M. A. McKervey, J. Chem. Soc., Chem. Commun., 1988, 1028 RSC.
- M. Kennedy and M. A. McKervey, J. Chem. Soc., Perkin Trans. 1, 1991, 2565 RSC.
- J. C. Morris, L. N. Mander and D. C. R. Hockless, Synthesis, 1998, 455 CrossRef CAS.
- B. Frey, L. N. Mander and D. C. R. Hockless, J. Chem. Soc., Perkin Trans. 1, 1998, 1555 RSC.
- H. Zhang, D. C. Appels, D. C. R. Hockless and L. N. Mander, Tetrahedron Lett., 1998, 39, 6577 CrossRef CAS.
- S. Levin, R. R. Nani and S. E. Reisman, Org. Lett., 2010, 12, 780 CrossRef CAS PubMed.
- S. E. Reisman, R. R. Nani and S. Levin, Synlett, 2011, 2437 CrossRef CAS.
- S. O'Neill, S. O'Keeffe, F. Harrington and A. R. Maguire, Synlett, 2009, 2312 Search PubMed.
- H. Günther and H. Hinrichs, Tetrahedron Lett., 1966, 8, 787 CrossRef.
- T. Clark, G. W. Spitznagel, R. Klose and P. v. R. Schleyer, J. Am. Chem. Soc., 1984, 106, 4412 CrossRef CAS.
- D. Gale, W. Middleton and C. Krespan, J. Am. Chem. Soc., 1965, 87, 657 CrossRef CAS.
- D. Gale, W. Middleton and A. Kresge, J. Am. Chem. Soc., 1966, 88, 3617 CrossRef.
- G. Maier, Angew. Chem., Int. Ed. Engl., 1967, 6, 402 CrossRef CAS.
- O. A. McNamara, N. R. Buckley, P. O'Leary, F. Harrington, N. Kelly, S. O'Keeffe, A. Stack, S. O'Neill, S. E. Lawrence, C. N. Slattery and A. R. Maguire, Tetrahedron, 2014, 70, 6870 CrossRef CAS.
- A. R. Maguire, P. O'Leary, F. Harrington, S. E. Lawrence and A. J. Blake, J. Org. Chem., 2001, 66, 7166 CrossRef CAS PubMed.
- A. Saba, Tetrahedron Lett., 1990, 31, 4657 CrossRef CAS.
- A. R. Maguire, N. R. Buckley, P. O'Leary and G. Ferguson, J. Chem. Soc., Perkin Trans. 1, 1998, 4077 RSC.
- P. O'Leary and A. R. Maguire, ARKIVOC, 2009, xi, 130 Search PubMed.
- A. R. Maguire, N. R. Buckley, P. O'Leary and G. Ferguson, Chem. Commun., 1996, 2595 RSC.
- S. O'Keeffe, F. Harrington and A. R. Maguire, Synlett, 2007, 2367 Search PubMed.
- L. T. Scott, M. A. Minton and M. A. Kirms, J. Am. Chem. Soc., 1980, 102, 6311 CrossRef CAS.
- M. Oda, T. Kajioka, K. Ikeshima, R. Miyatake and S. Kuroda, Synth. Commun., 2000, 30, 2335 CrossRef CAS.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Dalton Trans., 1915, 107, 1080 RSC.
Footnotes |
| † Present address: School of Chemistry, NUI Galway, Galway, Ireland. SMAĊT (Synthetic Methods: Asymmetric Catalytic Transformations). |
| ‡ While norcaradiene (NCD) and cycloheptatriene (CHT) are valence isomers, the interconversion is commonly described as NCD–CHT tautomerisation and, accordingly, this terminology is used throughout. |
| This journal is © The Royal Society of Chemistry 2015 |