
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Subunit-directed click coupling via doubly cross-linked hemoglobin efficiently produces readily purified functional bis-tetrameric oxygen carriers†
Serena
Singh
,
Ina S.
Dubinsky-Davidchik
,
Ying
Yang
and
Ronald
Kluger
*
Davenport Chemical Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON, Canada M5S 3H6. E-mail: r.kluger@utoronto.ca
First published on 14th September 2015
Abstract
While cross-linked hemoglobin (Hb) tetramers can deliver oxygen as a supplement to red cells, they also cause unacceptable increases in blood pressure, presumably from their penetration of the linings of blood vessels (endothelia) where the internal hemes bind endogenous nitric oxide (NO). This penetration would lower the local concentration of NO that normally induces vasodilation. Enlarging the effective size of the oxygen-carrying protein by coupling two Hbs can prevent their extravasation. Efficient and selective protein–protein coupling to produce those species has been a significant challenge. Introduction of an azide within a protein provides a directionally-oriented reaction site for utilization of the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) in the protein–protein-coupling process based on solubility-directed sequential addition to a bis-alkyne. However, it is known that Hb with an azide-containing cross-link between α-subunits is unreactive in CuAAC. To direct reaction away from the α-subunits of Hb, a specific fumaryl cross-link is installed exclusively between the most reactive sites on those subunits, thereby blocking the α-99 lysyl groups and preventing any further reaction. This modification allows installation of an azide-containing cross-link exclusively between lysine-82 ε-amino groups of the β-subunits of Hb. The multiply interconnected sites establish a geometry that permits initial interfacial interaction of the cross-linked Hb-azide with Cu(I) and a bis-alkyne. After coupling, the protein-linked azide product undergoes CuAAC at the remaining alkyne with a second cross-linked Hb-azide, producing a fully functional cross-linked Hb bis-tetramer whose oxygenation and structural properties include cooperativity and oxygen affinity that should be suitable for testing as an alternative to red cells in transfusions.
Chemical modification of human hemoglobin A (Hb), the oxygen-carrying component of our red cells, has been an important approach to the development of materials that are intended to be safe and effective universal sterile alternatives to red cells in circulation. This class of material is known as a hemoglobin-based oxygen carrier (HBOC).1–3 Since acellular Hb is a tetramer that dissociates into rapidly cleared αβ dimers outside the red cell, stabilization of the intact tetrameric state is essential.4,5 Thus, initial efforts to produce a safe and effective HBOC focused on preventing dissociation of the tetramer, most commonly by chemical cross-linking.6,7 However, clinical trials of cross-linked Hb led to observations of induction of increases in blood pressure to unacceptable levels.8–13 Based on the nature of the observed effects, it is likely that the heme proteins are small enough to extravasate through the endothelia that line blood vessels, consuming endogenous nitric oxide. Since nitric oxide is the essential local signal for relaxation of blood vessels, its resulting reduced concentration leads to vasoconstriction that increases blood pressure.14–17
In an important study, Vandegriff and coworkers18 noted that increasing the size of the circulating Hb species by the addition of chains of polyethylene glycol (PEG) is sufficient to prevent vasoactivity in circulation.19 Presumably, the increased size prevents these species from penetrating the endothelia.20 However, the PEGylation imparts high oxygen affinity along with low cooperativity.21 Those materials have been utilized in specific applications22 and are a potential therapeutic directed to very hypoxic regions of circulation. It has not yet been established in a clinical trial whether cooperativity in oxygen binding and release is necessary for a material to be effective. Calculated curves show that oxygen availability is greatest where there is a steep binding curve within the physiologically significant region. Without cooperativity, the HBOC hyperbolic curves predict materials with low affinity will not be loaded with oxygen in the lungs or high affinity materials with only small amounts of oxygen released in circulation. We reasoned that for producing a more versatile oxygen carrier, the coupling of two Hb tetramers to form a bis-tetramer could yield a product with the necessary size. Although the minimum size needed to avoid extravasation is not known, it is clear from other studies that bis-tetramers are sufficiently large for this purpose.23–26
Specific chemical modification of proteins can introduce bioorthogonal functional groups that lead to formation of derivatives with unique and predictable reactivity.16,27–31 Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC)32,33 has been employed for coupling proteins but the process has been surprisingly inefficient despite the excellent reputation of the process with smaller species.28 In principle, introduction of an azide into a specific site in cross-linked Hb places it on a stabilized location that would allow a reaction to occur with the displayed functional group. Two azide-containing Hb cross-linked tetramers would react with a single bis-alkyne by CuAAC, producing a stable cross-linked bis-tetramer.28,34,35 Furthermore, the reaction of the bis-alkyne with two Hb-azides can be driven by phase transfer catalysis where the increased availability of an initially formed Hb-conjugated alkyne transfers the alkyne into the aqueous phase that contains other Hb-azides, making the process highly efficient in terms of the protein utilization.28,34,35
A significant problem in applying CuAAC to Hb-coupling has been that azide-containing cross-links within the α-subunits of Hb do not participate in CuAAC and can also block reaction in the adjoining β-subunits.28 Therefore, a successful strategy for the formation of a uniformly modified Hb bis-tetramer by CuAAC should ideally be directed specifically to the β-subunits.35 We now report an efficient route to subunit-specific introduction of a bis-amide hydrocarbon cross-link between the α-subunits of Hb. This blocks further reaction in that region of the α-subunits, allowing introduction of a CuAAC-accessible azide on a structured link between the β-subunits. The resulting azide is reactive with bis-alkynes, efficiently producing structurally defined cross-linked bis-tetramers.
Experimental section
Material and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water with 0.1% trifluoroacetic acid. The eluent was monitored at 220 nm. Observed drifts in retention times may be attributed to solvent evaporation over time (solvents were mixed off-line) and possibly variations in column equilibration time, temperature and degassing. The composition of the protein associated with the peaks was investigated by isolation of the fractions and analysis using electrospray ionization (ESI) high resolution mass spectrometry (AIMS Lab, Department of Chemistry, University of Toronto).
000g for 15 min). The supernatant was collected and the final Hb concentration was determined from measuring absorbance at 540 nm.
:water with 0.1% trifluoroacetic acid. The eluent was monitored at 220 nm. Observed drifts in retention times may be attributed to solvent evaporation over time (solvents were mixed off-line) and possibly variations in column equilibration time, temperature and degassing. The composition of the protein associated with the peaks was investigated by isolation of the fractions and analysis using electrospray ionization (ESI) high resolution mass spectrometry (AIMS Lab, Department of Chemistry, University of Toronto).
000g for 15 min). The supernatant was collected and the final Hb concentration was determined from measuring absorbance at 540 nm.
Results
The initial stabilizing link across the α-subunits was achieved using bis(3,5-dibromosalicyl) fumarate in the presence of inositol hexaphosphate.6 To test the efficiency of a subsequent reaction at the β-subunits, the α-α cross-linked product was treated with trimesoyl tris(3,5-dibromosalicylate) (TTDS)36. The product was analyzed by reverse-phase HPLC. The masses of the eluted species were identified by ESI-MS to consist almost entirely of doubly cross-linked Hb (Fig. 1). Based on the pattern of the tryptic digest of the product,37 we deduced the sites at which the cross-link is connected to the protein (α99-fumaryl-α99, β82-trimesyl-β82).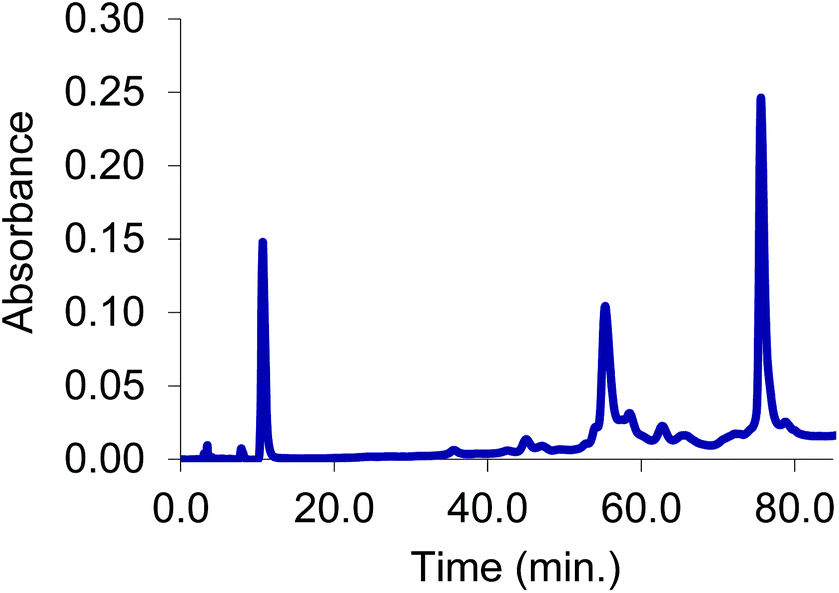 | ||
| Fig. 1 Reverse phase HPLC trace under dissociating conditions of α99-fumaryl-α99, β82-trimesyl-β82 Hb. A minor peak from elution of free heme appears at 11 min, while the major peaks are the β cross-linked subunits occurring at 56 min and α cross-linked subunits at 76 min. | ||
To prepare the azide-containing doubly cross-linked protein for CuAAC coupling, we produced reagents containing azides (1 and 2, Fig. 2) that are analogues of TTDS. Compound 2 was prepared by the multi-step synthesis described in Scheme 1. NBS-bromination of the BOC-protected amine produces the benzylic functionalized product 3. Azide 4 was then obtained from 3 by nucleophilic substitution. Deprotection of 4 gives amine 5, which is combined with an activated diester to produce amide 6. Hydrolysis of the esters in 6 gives diacid 7. EDC-promoted coupling of the diacid and tert-butyl 3,5-dibromosalicylic acid efficiently produces 8. Finally, cleavage of the t-butyl esters with TFA generates 2.
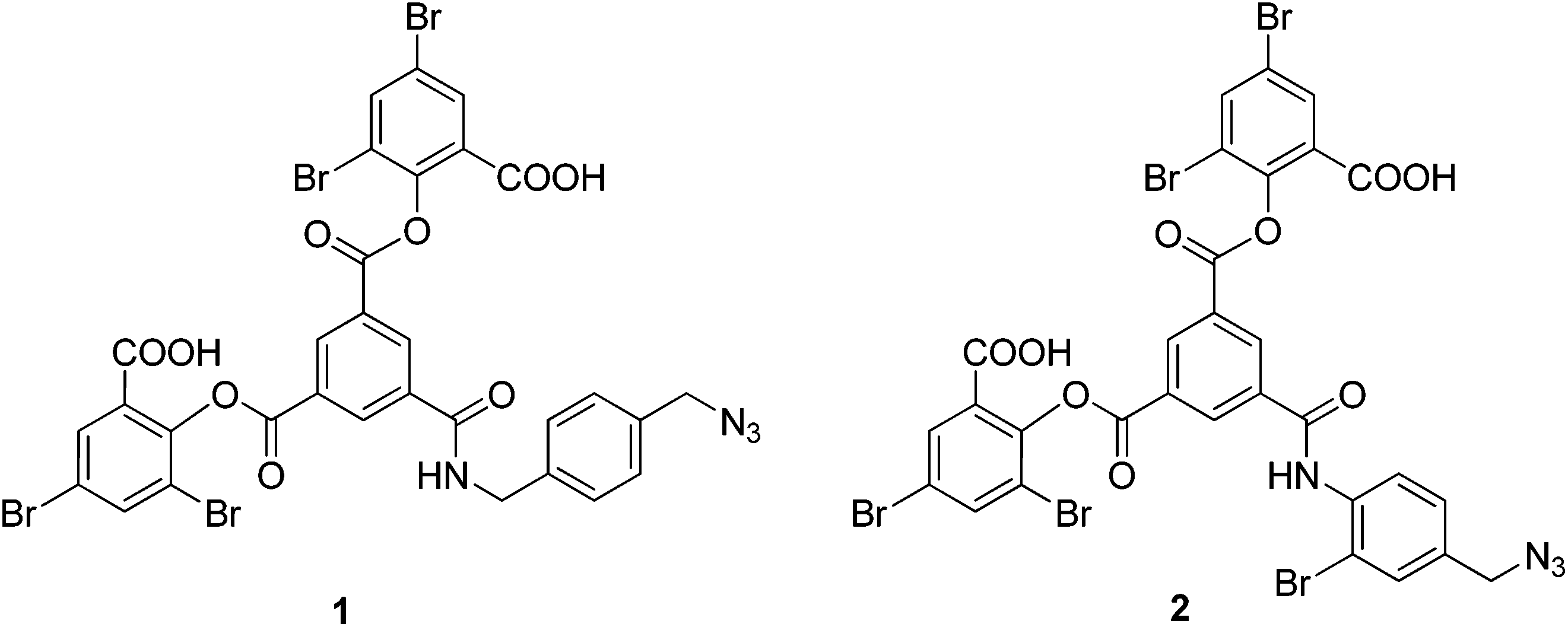 | ||
| Fig. 2 Azido-containing cross-linking reagents for modification of fumaryl cross-linked Hb. | ||
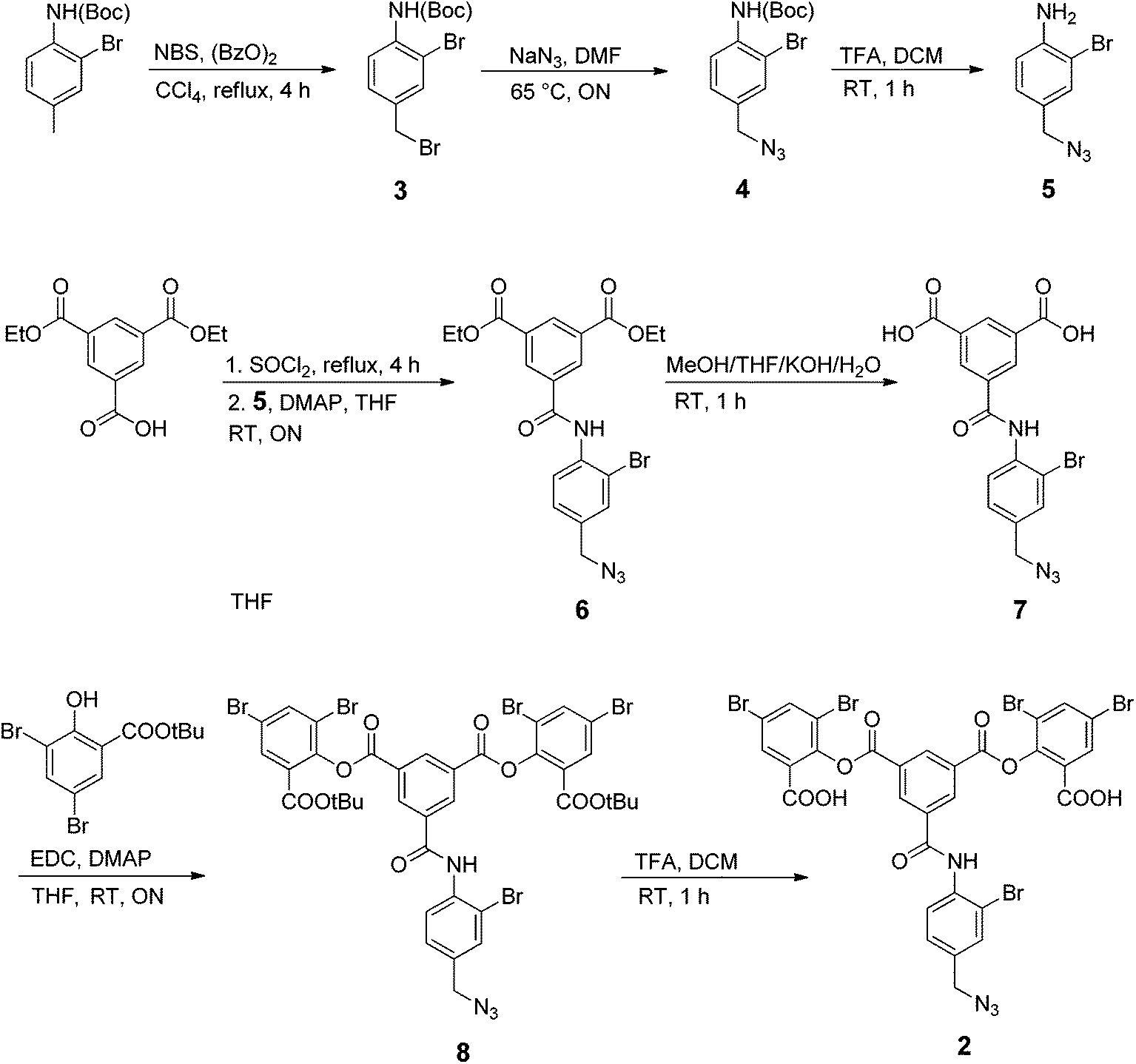 | ||
| Scheme 1 Synthesis of the azide-containing linker 2. | ||
We evaluated the outcome of the reactions of cross-linkers 1 and 2 with α-α-fumaryl cross-linked Hb. HPLC analysis of the products of the reaction of α99-fumaryl-99α, β2 Hb with 5.5 eq. of 1 indicated that approximately 50% of the β subunits are cross-linked by the reaction, with small amounts of by-products resulting from other reactions (see ESI, Fig. 11†). This indicates that reactions of 1 lack the necessary selectivity for an efficient process. The addition of 16 eq. of 1 promoted cross-linking of the majority of the β-subunits but half of this cross-linking occurred at an alternative set of sites within the β-subunits (ESI, Fig. 12†). Those azides did not participate in CuAAC. In contrast, the addition of 15 eq. of azide 2 produces the doubly cross-linked product (α99-fumaryl-α99, β82-azido-β82 Hb, Scheme 2) as the major product with only a minor impurity, most likely an over-modified doubly cross-linked species (Fig. 3). Reaction with 8.5 eq. of 2 produced a defined mixture of 50% of the doubly cross-linked protein and 50% of the unmodified αα-cross-linked species (Fig. 4). Reagent 2, which contains a bromine atom ortho to the central scaffold, replicates the bulk of TTDS, which is a likely source of its regioselectivity. This cross-linker was then utilized to produce the protein required for the CuAAC coupling process via reaction with a bis-alkyne.
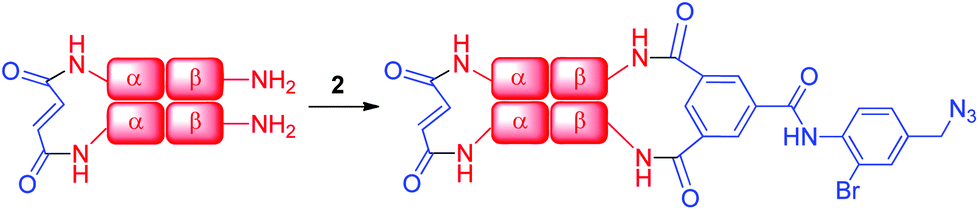 | ||
| Scheme 2 Addition of azide 2 to α99-fumaryl-α99, β2 in sodium borate buffer (0.05 M, pH 9.0) to yield α99-fumaryl-α99, β82-azido-β82 by selective modification of the β-Lys-82 residues. | ||
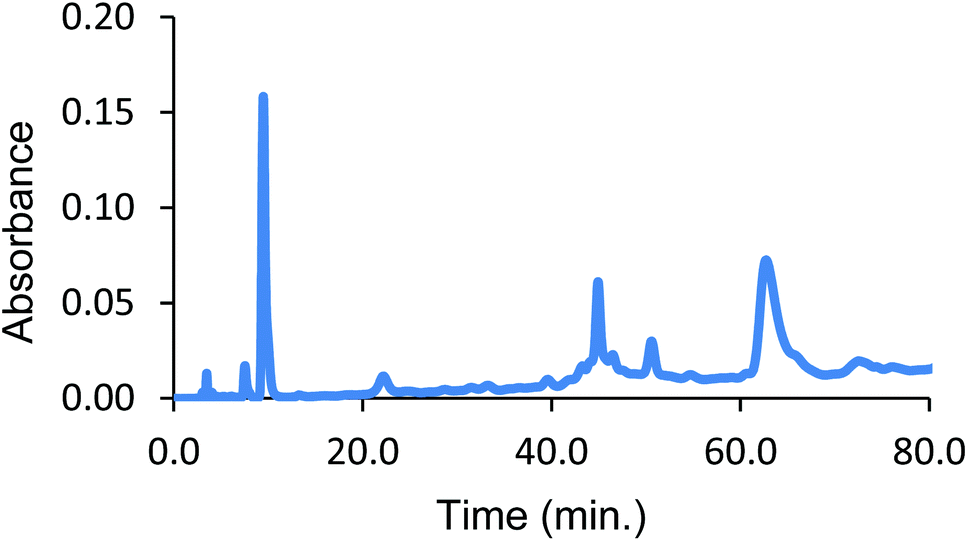 | ||
| Fig. 3 Reverse phase HPLC from cross-linking α99-fumaryl-α99, β2 with 15 eq. of azide 2. Peaks: heme (10 min), ββ-cross-linked subunits (45 min), over-modified ββ-cross-linked subunits (52 min) and αα-cross-linked subunits (62 min). | ||
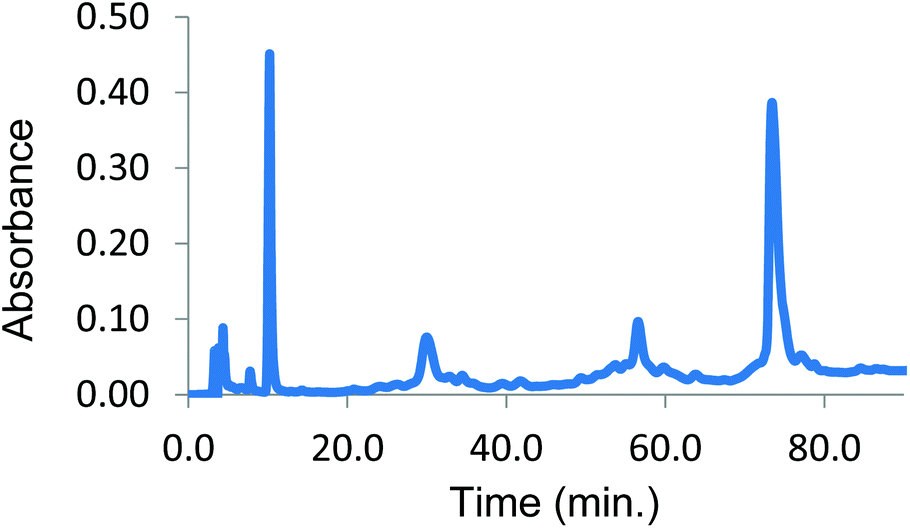 | ||
| Fig. 4 Reverse phase HPLC from cross-linking α99-fumaryl-α99, β2 with 8.5 eq. of azide 2. Peaks: heme (10 min), β subunits (30 min), ββ cross-linked subunits (56 min) and αα cross-linked subunits (73 min). | ||
A basic requirement for successful implementation of CuAAC bis-alkyne protein–protein coupling is that there be only one azide-containing reactant present initially. Therefore, we utilized the material that is 50% β-modified (Fig. 4). We combined the protein-containing solution in phosphate buffer (0.02 M, pH 7.4) with 20 eq. of bis-alkyne 9, 4 eq. of bathophenanthroline ligand 10, 2 eq. of CuSO4 and 40 eq. L-ascorbic acid. This mixture was stirred for 4 h at room temperature in an atmosphere of carbon monoxide according to the previously optimized procedure (Scheme 3).34 Denaturation of α99-fumaryl-α99, β2 is a side-effect of CuAAC (see ESI, Fig. 13†) because Hb species with non-cross-linked β-subunits are particularly vulnerable to denaturation by Cu species.28,40
 | ||
| Scheme 3 Reaction of α99-fumaryl-α99, β82-azido-β82 with bis-alkyne 9 produces a cross-linked bis-tetramer. | ||
Analysis of the product solution by size-exclusion HPLC at high salt concentrations (to induce dissociation of subunits41) reveals that the desired 128 kDa bis-tetramer species is formed from the azido-doubly cross-linked material (Fig. 5). The bis-tetramer in solution was isolated by passing the mixture through a G-100 Sephadex column equilibrated with tris buffer (37.5 mM, pH 7.4) containing 0.5 M magnesium chloride. From SDS-PAGE the 64 kDa band due to the four covalently linked beta-subunits of the bis-tetramer can be identified (see ESI, Fig. 15 and 16†).
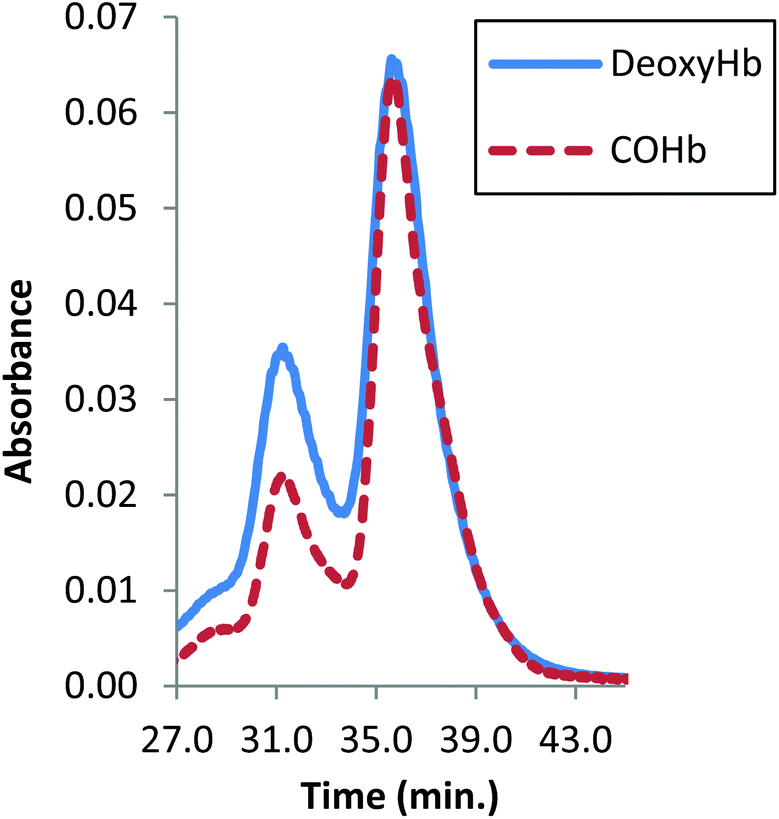 | ||
| Fig. 5 Size-exclusion HPLC from CuAAC as a function of the Hb ligand state. The peak at 32 min is the 128 kDa bis-tetramer and the peak at 37 min is α99-fumaryl-α99, β82-azido-β82 Hb. | ||
The yield of the CuAAC with the azide-containing modified Hb and the bis-alkyne is not improved with further increases in the amounts of the reagents, adjustment of protein concentration, increases in reaction time or increases in temperature (up to 40 °C). We observed no improvements from additional variations of the relative amount of the various reaction components. Utilizing different ligands, copper species and bis-alkynes decreased the relative yield.
In an important and unexpected contrast, the yield from CuAAC increases from 17% to 32% by utilizing deoxyHb (Fig. 5). Addition of inositol hexaphosphate (IHP) or sodium chloride lowers the yield, which is likely to be due to the anionic species blocking the central cationic channel where the required reactions occur. The yield from CuAAC on the modified COHb also improved marginally at pH 6, consistent with a change to a more reactive conformation.42
The purified bis-tetramer has a relatively low affinity for oxygen (P50 = 17.4 torr, Fig. 6, Table 1). Doubly cross-linked Hb (α99-fumaryl-α99, β82-trimesyl-β82) also has a decreased affinity for oxygen compared to acellular Hb. Both species have moderate levels of cooperativity (as indicated by their Hill coefficients, n50). Comparison of the P50 values for native Hb, α99-fumaryl-α99, β2 and α2, β82-trimesyl-β826,43 suggests that the presence of the fumaryl cross-link in the α-subunits contributes significantly to the decrease in oxygen affinity in the resulting bis-tetramer.
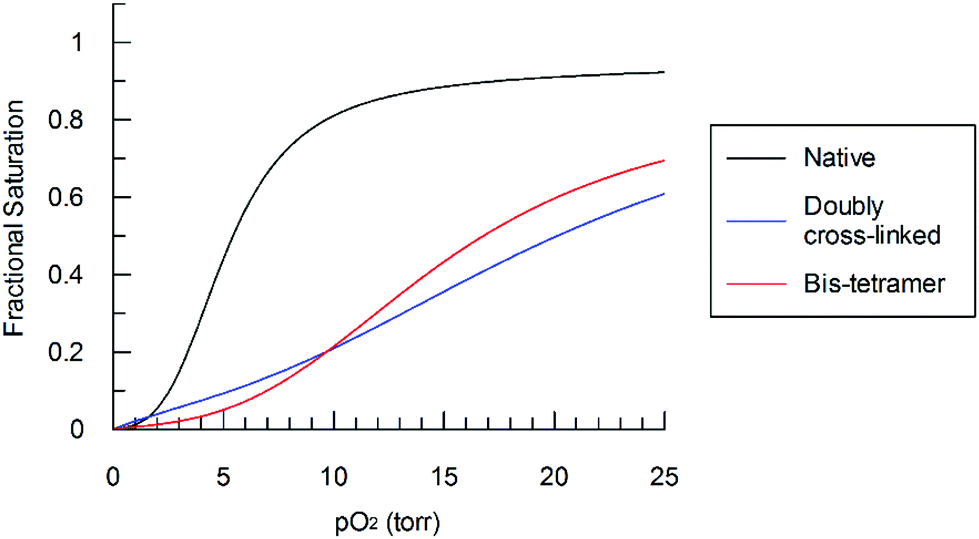 | ||
| Fig. 6 Bis-tetramer oxygen saturation curve compared to those of α99-fumaryl-α99, β82-trimesyl-β82 and native Hb in phosphate buffer (0.01 M, pH 7.4) at 32 °C. | ||
| Hb species | P 50 (torr) | n 50 |
|---|---|---|
| Native | 5.0 ± 0.1 | 3.0 ± 0.1 |
| Native + 1.2 eq. IHP | 28.4 ± 0.2 | 1.8 ± 0.1 |
| Native + 2.3 eq. IHP | 40.0 ± 0.4 | 1.8 ± 0.1 |
| α99-fumaryl-α99, β2 | 13.9 ± 0.3 | 2.6 ± 0.1 |
| α99-fumaryl-α99, β2 + 2.3 eq. IHP | 47.3 ± 0.3 | 1.9 ± 0.1 |
| α2, β82-trimesyl-β82 | 4.8 ± 0.2 | 2.4 ± 0.1 |
| α2, β82-trimesyl-β82 + 1.2 eq. IHP | 9.9 ± 0.2 | 2.2 ± 0.1 |
| α2, β82-trimesyl-β82 + 2.3 eq. IHP | 22.9 ± 0.2 | 2.4 ± 0.1 |
| α99-fumaryl-α99, β82-trimesyl-β82 | 20.6 ± 0.3 | 1.7 ± 0.1 |
| α99-fumaryl-α99, β82-trimesyl-β82 + 2.3 eq. IHP | 23.6 ± 0.4 | 1.8 ± 0.1 |
| Bis-tetramer | 17.4 ± 0.5 | 1.5 ± 0.1 |
IHP dramatically decreases the oxygen affinity of native Hb as it binds to the site that accommodates 2,3-diphosphoglycerate (DPG), an allosteric effector of Hb within red cells.44 Modification of amino groups of Hb in the DPG-binding site decreases the allosteric response to DPG or IHP.45 The decrease of sensitivity also occurs as a consequence of cross-linking between the α-subunits, as observed by Vandegriff and coworkers.46 We observed that cross-linking the β subunits (as in α2, β82-trimesyl-β82) and installing multiple cross-links (as in α99-fumaryl-α99, β82-trimesyl-β82) decreases the allosteric effect of IHP (Table 1). This attenuation also occurs where the concentration of IHP decreases from 0.1 mM (2.3 eq./heme) to 0.05 mM (1.2 eq./heme).
CD spectra of both the bis-tetramer and α99-fumaryl-α99, β82-trimesyl-β82 were acquired in the 200–260 nm and 245–470 nm regions. The spectra of both α99-fumaryl-α99, β82-trimesyl-β82 and the bis-tetramer in the far UV region (Fig. 7) are consistent with the secondary structures of the proteins being well-maintained following chemical modification. A notable feature is the enhanced negative ellipticity at 220 nm relative to that for native Hb. This depression is observed where oxyHb is converted to deoxyHb.47 Variations in the vicinity of 285 nm reflect changes in the environment of aromatic residues at the α1β2 interface.47 For α99-fumaryl-α99, β82-trimesyl-β82 and the bis-tetramer, we observe negative ellipticity in this region and a notable red shift (Fig. 8). These features in the CD spectrum are present in the T-state deoxyHb.47 This presence of the T-state is consistent with the reduced oxygen affinity of the modified protein.
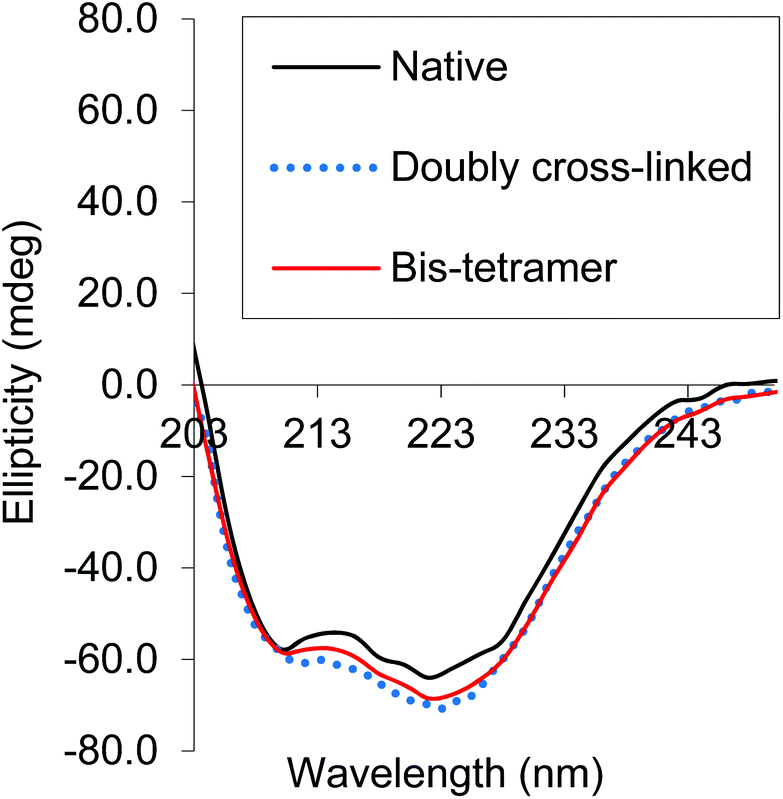 | ||
| Fig. 7 CD spectra of α99-fumaryl-α99, β82-trimesyl-β82 and the bis-tetramer compared to that of native carbonmonoxyHb in the far UV. | ||
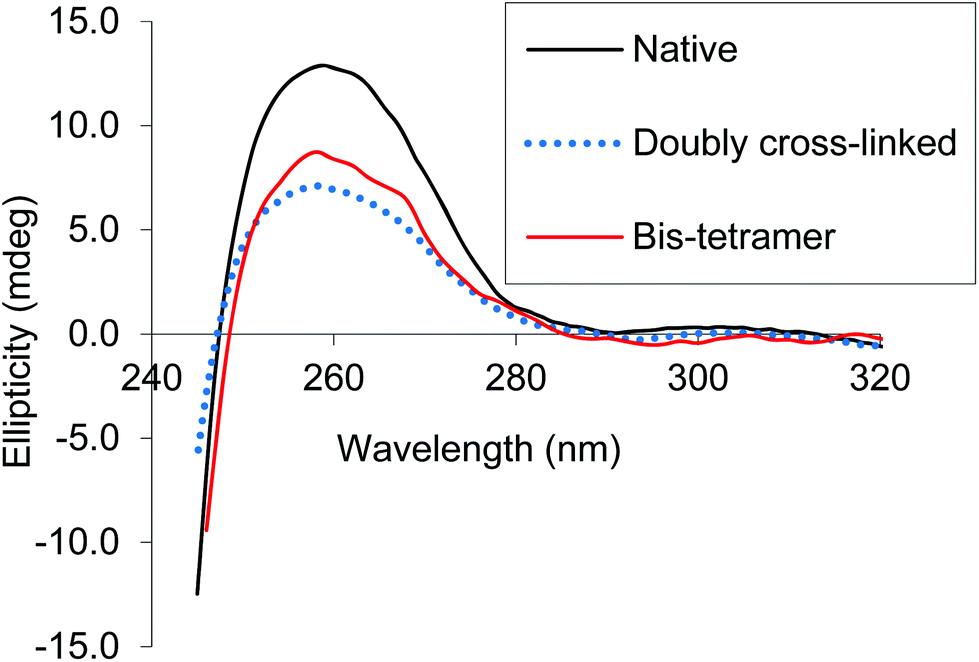 | ||
| Fig. 8 CD spectra of α99-fumaryl-α99, β82-trimesyl-β82 and the bis-tetramer compared to that of native carbonmonoxyHb in the near UV. | ||
Cross-linking of native Hb improves the protein's thermal stability, making it possible to separate cross-linked species from protein lacking cross-links.48 An additive effect from two cross-links could potentially increase thermal stability,49 creating a route for separation from singly cross-linked material. The minimum temperatures at which native and modified COHb species are fully denatured after 30 min are presented in Table 2. The criterion for indicating the complete denaturation of the protein is the absence of absorbance from the heme at 540 nm as the heme is released from denatured protein. Complete denaturation of a solution of native Hb results from heating a solution for 30 min at 79 °C. Denaturing the singly cross-linked species (α2, β82-trimesyl-β82) requires heating at 95 °C. These results indicate that an additional cross-link does not enhance stability. The degree of heat stability is consistent with heterogeneous bulk solution effects.48,50
| CarbonmonoxyHb species | Temperature (°C) |
|---|---|
| Native | 79 |
| α99-fumaryl-α99, β2 | 90 |
| α99-fumaryl-α99, β82-trimesyl-β82 | 94 |
| α2, β82-trimesyl-β82 | 95 |
We also investigated the thermal stability of the doubly cross-linked species in the presence of oxygen, comparing it to α99-fumaryl-α99 cross-linked Hb.51 Turbidity curves were acquired by slowly increasing the temperatures of solutions of the CO-forms of the modified proteins in solutions that were exposed to air while monitoring absorbance at 700 nm (Fig. 9). Hb in solution does not absorb at 700 nm but the formation of aggregates that occur upon unfolding decrease the transparency of the solutions.52 The doubly cross-linked and singly cross-linked species are essentially indistinguishable in terms of their thermal stability whereas the presence of a single cross-link increases overall stability of the protein relative to native Hb, even where auto-oxidation rates are comparable.
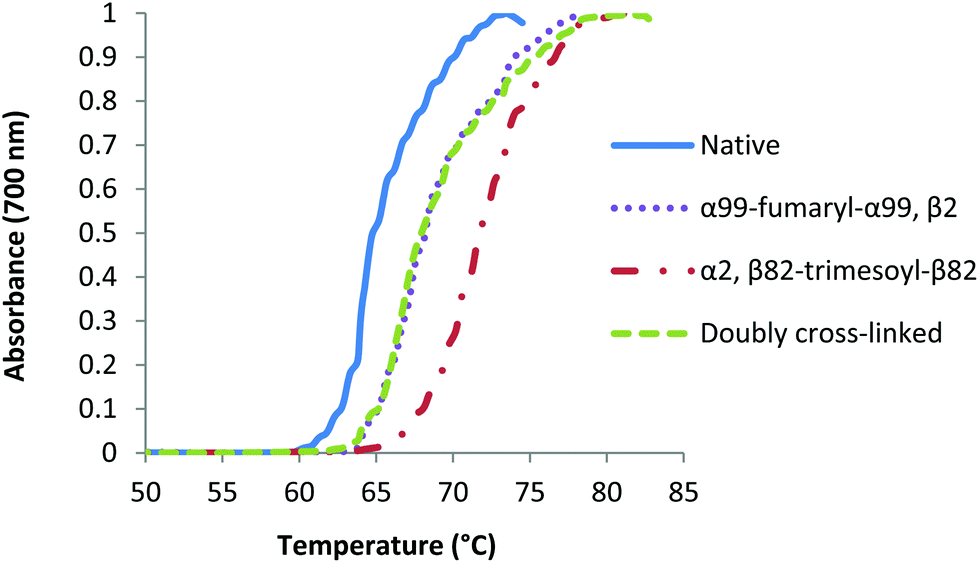 | ||
| Fig. 9 Turbidity curves for native and modified carbonmonoxy Hb species under aerobic conditions in 0.1 M phosphate buffer (0.1 M, pH 7.4). | ||
Discussion
It was recently reported that acetylation of amino groups in the α-subunits of Hb direct cross-linking to give β-subunit modification.35 We reasoned that a fumaryl bridge would serve the same function, with the added benefit of providing a defined product that is readily isolated in high yield.6 The precursor to the bis-tetramer would then be doubly cross-linked and could be correlated with earlier reports.53 It was not known whether this doubly cross-linked hemoglobin could be prepared as efficiently, as the original report of a doubly cross-linked species describes an impure product.53 In that report, a native deoxygenated Hb (deoxyHb) is reacted with two eq. of trimesoyl tris(3,5-dibromosalicylate) (TTDS) followed by treatment with two eq. of bis(3,5-dibromosalicyl) fumarate (DBSF). The cross-linking of the β-subunits with TTDS is quantitative but the second cross-link from reaction with DBSF forms in lower yield at pH 9.0 (Scheme 4, Path B).53 At pH 7.4, the reaction is even less complete.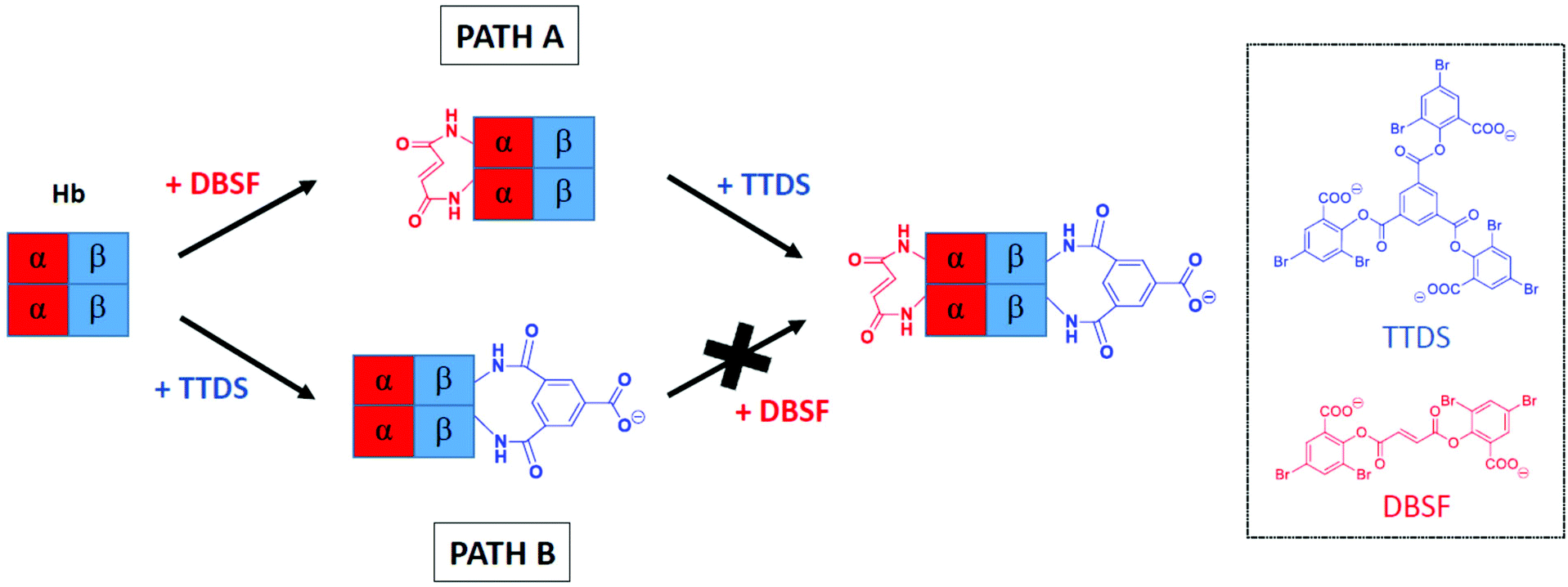 | ||
| Scheme 4 Reversing the order of cross-linking. Path A describes our approach and Path B represents the method established by Jones et al.53 | ||
A logical alternative route to doubly cross-linked Hb reverses the order of reactions, introducing a cross-link between the α-subunits followed by reaction of the β-subunits (Scheme 4, Path A). The state of the protein as R or T can control access to reactive groups54 and thereby control the efficiency of cross-linking. In principle, the materials generated by the two sequences could have different structures and properties as the first cross-link defines the accessible conformational space for the second reaction. In the present study, we find that where the α-subunits are cross-linked ahead of the β-subunits, the doubly cross-linked species is acquired in a higher state of purity than by the original route, with the resulting modified proteins likely to be identical. However, since there are significant differences in the efficiency of the conversion by the two routes, the pathways are not interchangeable. In a formal sense, the results establish that the transition state for the second of the serial cross-linking processes is more closely associated with the reactant than with the product.
The results provide a practical outcome: serial modifications of proteins are not commutative: introduction of the first cross-link specifically limits the accessible conformational space of the modified protein compared to that of the native protein. Introduction of the second link occurs within the subset of conformations defined by the initial cross-link. By reversing the order of reaction, the initial product has access to conformations that would not have been available if the other link were already in place. In other words, the initial link excludes the existence of a subset of conformations and defines a new conformational space by restricting structural movement.55,56 Failure to cross-link the modified protein completely in the second step (Scheme 4, Path B) is an indication that the deoxy conformational space is not coincident with that of native deoxy Hb. This is consistent with the observations of the structures reported from crystallographic analysis.57 Interaction of the protein and the reagent within the α-subunits would be reduced.58 In contrast, Path A (Scheme 4) ensures efficient installation of the second cross-link because the α–α linked fumaryl bis-amide leads to a conformation that is very similar to that of the native protein.59
Our results reveal the utility of cross-linking as a probe for detection of otherwise inaccessible conformations, delineating inter-subunit effects and specific residues in dynamic environments. The use of cross-linkers to probe conformational change is especially useful when X-ray crystallography proves to be inadequate because the conformation of the crystallized protein is different from that in solution.60,61 Introduction of two cross-links between subunits in multi-subunit proteins is potentially a general approach to distinguish specific effects of a cross-link as it defines the accessible space for the second link. These general tactics extend to the specific installation of the azido moiety on the cross-link within the β-subunits (Scheme 2). Azides located within the α-subunits of Hb are unreactive28 and are likely to be within a relatively inaccessible region of the protein. Therefore, the presence of an azide alone is not sufficient to assure its participation as is the case for the reaction of small molecules in solution.33
Although azide 2 is more selective in our comparative analysis, its reaction with cross-linked Hb does not go to completion without over-modification (Fig. 3). In order to obtain the bis-tetramer in a pure state, the reaction was stopped after 50% of the protein was modified (Fig. 4). It was this mixture of singly and doubly cross-linked protein that was submitted to the final coupling stage. The non-quantitative CuAAC yield of 17% (Fig. 5) is unexpected since analogous reactions with modified Hb have proceeded efficiently.34 However, conducting CuAAC on modified Hb in the deoxy state dramatically increases the yield of the coupled product, suggesting that coupling is controlled to some extent by the accessibility of the azide within the doubly cross-linked system.
To deduce whether this deoxyHb effect is specific to CuAAC, we set up a nucleophilic substitution reaction analogous to that of Yang et al.,34 using the non-hydrolyzed form of α99-fumaryl-α99, β82-trimesyl-β82 as the substrate. We observed that even this substitution occurs more readily when the protein is deoxygenated, confirming that the issue was one of accessibility (see ESI, Fig. 19 vs. 20†). This result illustrates the potential utility of CuAAC as a probe of protein conformation. Furthermore, the switchable nature of the doubly cross-linked framework makes it an interesting candidate for drug conjugation. It would be interesting to know if disulfide bond formation40,62 under the CuAAC conditions competes with the protein-coupling process. So far, we can conclude that a well-defined product results from the coupling step with acceptable efficiency in the overall process.
We find that both the pure bis-tetramer and the doubly cross-linked species (α99-fumaryl-α99, β82-trimesyl-β82) possess low oxygen affinity (Fig. 6, Table 1). Circular dichroism (CD) spectra informed us that the source of this low oxygen affinity is associated with enhanced T-state character (Fig. 7 and 8). The oxygen binding properties of the coupled and uncoupled proteins are slightly different because the two Hb units of the bis-tetramer are close enough to communicate.63 Our bis-tetramer is the first with an oxygen affinity close to that of Hb within red cells (P50 = 28.0 torr).64 The bis-tetramer as an HBOC component would not circulate in a DPG-rich environment as occurs within the red cell; therefore, an intrinsically low oxygen affinity is a valuable characteristic for efficient delivery of oxygen to the tissues for a red cell replacement. This holds especially true for doubly cross-linked hemoglobin as the modified protein demonstrates nearly complete insensitivity to allosteric regulation by inositol hexaphosphate (IHP) (Table 1).
Thermal stability data for the modified proteins clearly show that not all cross-links impart equal stability: α99-fumaryl-α99, β2 and α2, β82-trimesyl-β82 responded differently to the conditions. Yang et al.65 showed that α99-fumaryl-α99, β2 metHb and α2, β82-fumaryl-β82 metHb are equally stable. Comparison with those results suggests that stability depends on the structure of the link as well as the sites that it spans. Furthermore, additional cross-links do not necessarily impart cumulative stability, as observed by Ueda et al.49 Despite its dual cross-links, our doubly cross-linked Hb fully denatures at the same temperature as does α2, β82-trimesyl-β82. In principle, with different melting temperatures for fumaryl (singly) cross-linked Hb and doubly cross-linked Hb, separation for the purpose of purification might be possible. However, we were unable to achieve separation of cross-linked species by heat treatment, although this does remove unmodified proteins. We suspect that heterogeneity changes the melting points of the individual components, as observed by Yang and Olsen65 and justified by Despa and coworkers.50
The relative thermal stabilities of the four COHb species are different under aerobic conditions, with α99-fumaryl-α99, β2 and α99-fumaryl-α99, β82-trimesyl-β82 demonstrating equal stability (Fig. 9). Rapid auto-oxidation is a characteristic of fumaryl cross-linked oxyHb species.51 However, in light of the stability that modified COHb possesses under aerobic conditions, we recognize its potential for carbonmonoxyHb-based applications.66
Conclusions
We have shown that functional hemoglobin bis-tetramers are accessible by an efficient three-step procedure: protect, functionalize and connect by a “click” reaction. Double cross-linking, which is a non-commutative process, proceeds with maximum efficiency by directed sequential cross-linking, producing regioselective modification of the β-subunits with an azido moiety poised for solubility-directed coupling by reaction with bis-alkynes. This production of a pure product is a step forward in bis-tetramer design and large scalability, which has been a particular challenge for the alternative of producing protein expression from bacterial sources.23 Finally, the oxygenation properties of the cross-linked hemoglobin bis-tetramer include cooperativity and oxygen affinity that would be suitable for circulation as an alternative to red cells in transfusions, while the extended structure can be expected to prevent extravasation that would produce vasoactivity.Acknowledgements
We thank the Canadian Blood Services for support through an operating grant.References
- H. Sakai, K. Sou, H. Horinouchi, K. Kobayashi and E. Tsuchida, Artif. Organs, 2009, 33, 139–145 CrossRef PubMed.
- R. M. Winslow, J. Intern. Med., 2003, 253, 508–517 CrossRef CAS.
- R. M. Winslow, Biochim. Biophys. Acta, 2008, 1784, 1382–1386 CrossRef CAS PubMed.
- G. Guidotti, W. Konigsberg and L. C. Craig, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 774–782 CrossRef CAS.
- J. R. Hess and R. F. Reiss, Transfus. Med. Rev., 1996, 10, 276–285 CrossRef CAS.
- S. R. Snyder, E. V. Welty, R. Y. Walder, L. A. Williams and J. A. Walder, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7280–7284 CrossRef CAS.
- R. Kluger and F. E. Lui, in Hemoglobin-Based Oxygen Carriers as Red Cell Substitutes and Oxygen Therapeutics, ed. H. W. Kim and A. G. Greenburg, Springer, Berlin, Heidelberg, 2013, DOI:10.1007/978-3-642-40717-8_10, ch. 10, pp. 159–183.
- R. Kluger, Curr. Opin. Chem. Biol., 2010, 14, 538–543 CrossRef CAS PubMed.
- J. Simoni, G. Simoni and J. F. Moeller, Artif. Organs, 2009, 33, 100–109 CrossRef CAS PubMed.
- P. W. Buehler and A. I. Alayash, Transfusion, 2004, 44, 1516–1530 CrossRef CAS PubMed.
- R. J. Rohlfs, E. Bruner, A. Chiu, A. Gonzales, M. L. Gonzales, D. Magde, M. D. Magde, K. D. Vandegriff and R. M. Winslow, J. Biol. Chem., 1998, 273, 12128–12134 CrossRef CAS PubMed.
- D. H. Doherty, M. P. Doyle, S. R. Curry, R. J. Vali, T. J. Fattor, J. S. Olson and D. D. Lemon, Nat. Biotechnol., 1998, 16, 672–676 CrossRef CAS PubMed.
- G. P. Biro, in Hemoglobin-Based Oxygen Carriers as Red Cell Substitutes, ed. H. W. Kim and A. G. Greenburg, Springer, New York, 2013, pp. 543–562 Search PubMed.
- C. Natanson, S. J. Kern, P. Lurie, S. M. Banks and S. M. Wolfe, JAMA, 2008, 299, 2304–2312 CrossRef CAS PubMed.
- J. S. Olson, E. W. Foley, C. Rogge, A.-L. Tsai, M. P. Doyle and D. D. Lemon, Free Radical Biol. Med., 2004, 36, 685–697 CrossRef CAS PubMed.
- R. Kluger, J. S. Foot and A. A. Vandersteen, Chem. Commun., 2010, 46, 1194–1202 RSC.
- K. Sampei, J. A. Ulatowski, Y. Asano, H. Kwansa, E. Bucci and R. C. Koehler, Am. J. Physiol.: Heart Circ. Physiol., 2005, 289, H1191–H1201 CrossRef CAS PubMed.
- K. D. Vandegriff, A. Malavalli, J. Wooldridge, J. Lohman and R. M. Winslow, Transfusion, 2003, 43, 509–516 CrossRef CAS.
- K. D. Vandegriff, M. A. Young, J. Lohman, A. Bellelli, M. Samaja, A. Malavalli and R. M. Winslow, Br. J. Pharmacol., 2008, 154, 1649–1661 CrossRef CAS PubMed.
- B. Belcik and A. Palmer, in Hemoglobin-Based Oxygen Carriers as Red Cell Substitutes and Oxygen Therapeutics, ed. H. W. Kim and A. G. Greenburg, Springer, Berlin, Heidelberg, 2013, DOI:10.1007/978-3-642-40717-8_37, ch. 37, pp. 693–711.
- F. E. Lui and R. Kluger, Biochemistry, 2009, 48, 11912–11919 CrossRef CAS PubMed.
- C. Olofsson, T. Ahl, T. Johansson, S. Larsson, P. Nellgård, S. Ponzer, B. Fagrell, R. Przybelski, P. Keipert, N. Winslow and R. M. Winslow, Anesthesiology, 2006, 105, 1153–1163 CrossRef CAS PubMed.
- C. Fablet, M. C. Marden, B. N. Green, C. Ho, J. Pagnier and V. Baudin-Creuza, Protein Sci., 2003, 12, 690–695 CrossRef CAS PubMed.
- C. Gaucher, É. Domingues-Hamdi, C. Prin-Mathieu, P. Menu and V. Baudin-Creuza, C. R. Biol., 2015, 338, 95–102 CrossRef PubMed.
- F. E. Lui, B. Yu, D. M. Baron, C. Lei, W. M. Zapol and R. Kluger, Transfusion, 2012, 52, 974–982 CrossRef CAS PubMed.
- F. E. Lui and R. Kluger, ChemBioChem, 2010, 11, 1816–1824 CrossRef CAS PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- J. S. Foot, F. E. Lui and R. Kluger, Chem. Commun., 2009, 7315–7317 RSC.
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Brase, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC.
- P. Agarwal, J. van der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed, 2002, 41, 2596–2599 CrossRef CAS.
- Y. Yang and R. Kluger, Chem. Commun., 2010, 46, 7557–7559 RSC.
- A. Wang and R. Kluger, Biochemistry, 2014, 53, 6793–6799 CrossRef CAS PubMed.
- R. Kluger, Y. Song, J. Wodzinska, C. Head, T. S. Fujita and R. T. Jones, J. Am. Chem. Soc., 1992, 114, 9275–9279 CrossRef CAS.
- R. T. Jones, in Methods in Enzymology, ed. J. Everse, K. D. Vandegriff and R. M. Winslow, Academic Press, 1994, vol. 231, pp. 322–343 Search PubMed.
- K. Imai, Biochemistry, 1973, 12, 798–808 CrossRef CAS.
- E. C. DeLand, in Methods in Enzymology, ed. J. Everse, K. D. Vandegriff and R. M. Winslow, Academic Press, 1994, vol. 232, pp. 632–655 Search PubMed.
- C. C. Winterbourn and R. W. Carrell, Biochem. J., 1977, 165, 141–148 CrossRef CAS.
- G. Guidotti, J. Biol. Chem., 1967, 242, 3685–3693 CAS.
- M. K. Safo, J. C. Burnett, F. N. Musayev, S. Nokuri and D. J. Abraham, Acta Crystallogr., 2002, 58, 2031–2037 CrossRef PubMed.
- R. Kluger, J. Wodzinska, R. T. Jones, C. Head, T. S. Fujita and D. T. Shih, Biochemistry, 1992, 31, 7551–7559 CrossRef CAS.
- R. Benesch and R. E. Benesch, Biochem. Biophys. Res. Commun., 1967, 26, 162–167 CrossRef CAS.
- H. Ueno and J. M. Manning, Am. J. Pediatr. Hematol. Oncol., 1988, 10, 348–350 CrossRef CAS.
- K. D. Vandegriff, F. Medina, M. A. Marini and R. M. Winslow, J. Biol. Chem., 1989, 264, 17824–17833 CAS.
- R. Li, Y. Nagai and M. Nagai, J. Inorg. Biochem., 2000, 82, 93–101 CrossRef CAS.
- T. Yang and K. W. Olsen, Arch. Biochem. Biophys., 1988, 261, 283–290 CrossRef CAS.
- T. Ueda, K. Masumoto, R. Ishibashi, T. So and T. Imoto, Protein Eng., 2000, 13, 193–196 CrossRef CAS PubMed.
- F. Despa, D. P. Orgill and R. C. Lee, Ann. Biomed. Eng., 2005, 33, 1125–1131 CrossRef PubMed.
- S. A. Dragan, K. W. Olsen, E. G. Moore and A. Fitch, Bioelectrochemistry, 2008, 73, 55–63 CrossRef CAS PubMed.
- D. M. Reza, M.-M. A. Akbar, N. Parviz, H.-O. Ghourchian and S. Shahrokh, J. Biochem. Mol. Biol., 2002, 35, 364–370 CrossRef CAS.
- R. T. Jones, D. T. Shih, T. S. Fujita, Y. Song, H. Xiao, C. Head and R. Kluger, J. Biol. Chem., 1996, 271, 675–680 CrossRef CAS PubMed.
- D. Caccia, L. Ronda, R. Frassi, M. Perrella, E. Del Favero, S. Bruno, B. Pioselli, S. Abbruzzetti, C. Viappiani and A. Mozzarelli, Bioconjugate Chem., 2009, 20, 1356–1366 CrossRef CAS PubMed.
- M. A. Schumacher, M. M. Dixon, R. Kluger, R. T. Jones and R. G. Brennan, Nature, 1995, 375, 84–87 CrossRef CAS PubMed.
- S. S. Wong, Chemistry of Protein Conjugation and Cross-Linking, Taylor & Francis, 1991 Search PubMed.
- E. J. Fernandez, C. Abad-Zapatero and K. W. Olsen, J. Mol. Biol., 2000, 296, 1245–1256 CrossRef CAS PubMed.
- J. A. Walder, R. H. Zaugg, R. Y. Walder, J. M. Steele and I. M. Klotz, Biochemistry, 1979, 18, 4265–4270 CrossRef CAS.
- R. Chatterjee, E. V. Welty, R. Y. Walder, S. L. Pruitt, P. H. Rogers, A. Arnone and J. A. Walder, J. Biol. Chem., 1986, 261, 9929–9937 CAS.
- M. Laberge and T. Yonetani, Biophys. J., 2008, 94, 2737–2751 CrossRef CAS PubMed.
- S. C. Sahu, V. Simplaceanu, Q. Gong, N. T. Ho, F. Tian, J. H. Prestegard and C. Ho, Biochemistry, 2007, 46, 9973–9980 CrossRef CAS PubMed.
- A. Carr and B. Frei, FASEB J., 1999, 13, 1007–1024 CAS.
- N. Gourianov and R. Kluger, Biochemistry, 2005, 44, 14989–14999 CrossRef CAS PubMed.
- G. Stamatoyannopoulos, A. J. Bellingham, C. Lenfant and C. A. Finch, Annu. Rev. Med., 1971, 22, 221–234 CrossRef CAS PubMed.
- T. Yang and K. W. Olsen, Biochem. Biophys. Res. Commun., 1991, 174, 518–523 CrossRef CAS.
- S. Cao and R. C. Koehler, FASEB J., 2011, 25, 1024.10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01755f |
| This journal is © The Royal Society of Chemistry 2015 |