
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total syntheses of disulphated glycosphingolipid SB1a and the related monosulphated SM1a†
Haruka
Hirose
a,
Hideki
Tamai
a,
Chao
Gao
b,
Akihiro
Imamura
*a,
Hiromune
Ando
ac,
Hideharu
Ishida
a,
Ten
Feizi
b and
Makoto
Kiso
*ac
aDepartment of Applied Bioorganic Chemistry, Faculty of Applied Biological Sciences, Gifu University, 1-1 Yanagido, Gifu-shi, Gifu 501-1193, Japan. E-mail: aimamura@gifu-u.ac.jp; kiso@gifu-u.ac.jp; Tel: +81-58-293-3453
bGlycosciences Laboratory, Department of Medicine, Imperial College London, Hammersmith Campus, Du Cane Road, London W12 0NN, UK
cInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida Ushinomiya-cho, Sakyo-ku, Kyoto 606-8501, Japan
First published on 24th September 2015
Abstract
Total syntheses of two natural sulphoglycolipids, disulphated glycosphingolipid SB1a and the structurally related monosulphated SM1a, are described. They have common glycan sequences and ceramide moieties and are associated with human epithelial carcinomas. The syntheses featured efficient glycan assembly and the glucosyl ceramide cassette as a versatile building block. The binding of the synthetic sulphoglycolipids by the carcinoma-specific monoclonal antibody AE3 was investigated using carbohydrate microarray technology.
Introduction
Sulphoglycolipids are acidic glycosphingolipids that contain one or two sulphate (HSO3−) groups, and are components of cell membranes.1 To date, several sulphoglycolipids have been isolated and characterised.1,2 Interestingly, many of them have the same glycan sequence as ganglio-series3 or globo-series gangliosides4 but they contain sulphate groups instead of sialic acid residues. These sulphated glycolipids with a ganglio-series core framework are found in mammalian nervous tissue and in the kidney, where they may play a role in osmotic adaptation.5 In addition, many of these sulphated glycolipids are known to be associated with various carcinomas.6 SB1a, for instance, a di-sulphated analogue of the sialoglycolipid GD1a, has been found to be abundantly expressed on the surface of the human hepatocellular carcinoma cell line PLC/PRF/15 but not in glycolipid extracts from a cirrhotic liver or normal liver.6a Although association has been suggested between sulphoglycolipid accumulation and cancerous behaviours of epithelial cancer cells,6b–d,7 the precise roles of these sulphoglycolipids in carcinomas and other biological systems has not been clarified. To elucidate these roles, structurally homogeneous forms of sulphoglycolipids are in great demand. In the course of searching for carbohydrate sequences recognized by the epithelial cancer-specific monoclonal antibody (mAb) AE3, microarray analyses were performed with approximately 500 sequence-defined glycan probes. These revealed an unpredicted binding of the antibody to SM1a,8 the monosulphated analogue of the sialoglycolipid GM1. The mAb AE3 was bound relatively weakly to the three structurally-related gangliosides asialo-GM1, GM1 and GM1 (Gc). There was no binding to the di-sulphated analogue SB1a. As the SM1a and SB1a included in the screening analyses were from natural sources, it was important to rule out the presence of any minor contaminants which could not be detected by mass spectrometry. The structures of SM1a (1) and SB1a (2), which were first isolated from rat kidney,9 were assigned as shown in Fig. 1.9,2a Characteristically, their ceramide compositions were mainly C24 non-hydroxylated fatty acids (tetracosanoic acid) and C18 4-hydroxysphinganine (D-ribo-phytosphingosine). Very recently, Li and co-workers reported the first total synthesis of SM1a; however, its ceramide part consisted of unnatural unsaturated sphingosine.10 Previously, our group accomplished the syntheses of a large number of sialoglycans, including gangliosides11 and their sulphated analogues,12 using a chemical approach. Here we report the first total syntheses of the disulphated glycolipid SB1a and the monosulphated SM1a, each with a ceramide tail as found in natural products. The binding of the synthesized sulphoglycolipids by mAb AE3 was investigated using carbohydrate microarray technology.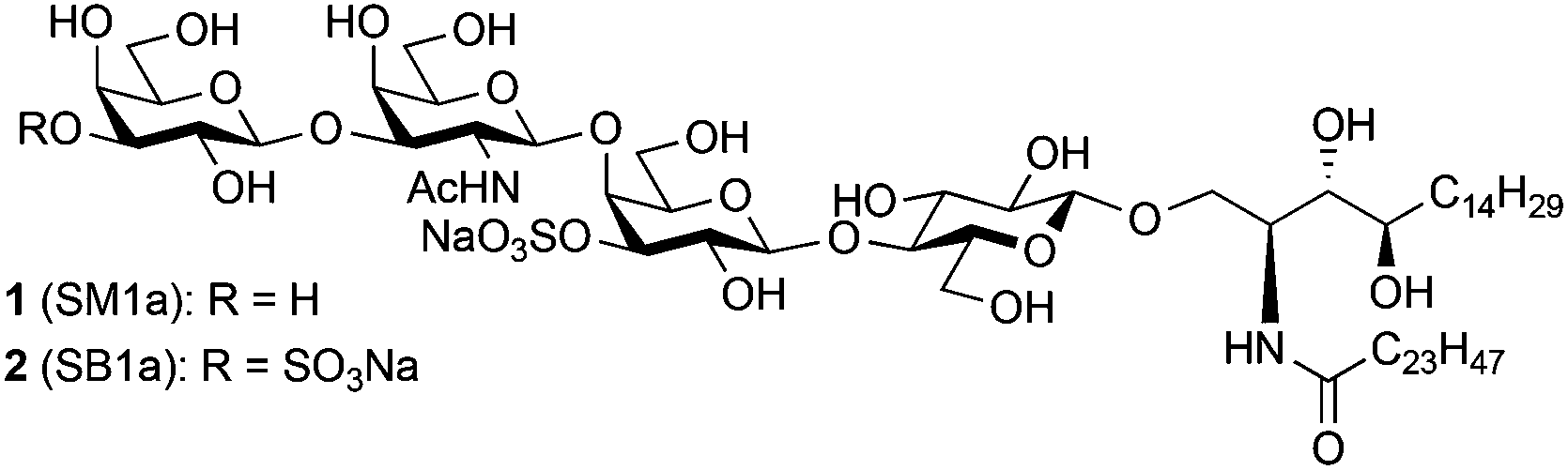 | ||
| Fig. 1 Structures of SM1a and SB1a. | ||
Results and discussion
Fig. 2 presents the retrosynthetic analyses of the target compounds. Obviously, the systematic synthesis of SB1a and SM1a is possible because they have a gangliotetraose skeleton and a ceramide part. The key disconnection toward total synthesis is performed between the inner galactose and the reducing end glucose residues to produce key fragments, i.e. the non-reducing end trisaccharide unit and the glucosyl ceramide (GlcCer) cassette. Each C3 position of the galactose residues should be protected by the different selectively removable protecting groups because they are potential sites to introduce the sulphate group. To date, we have succeeded in the efficient total synthesis of natural gangliosides such as GQ1b,13 GalNAc-GD1a,14 GalNAc-GM1b,15 X2,16 LLG-3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 and GAA-7,18 using the GlcCer cassette approach.19 This approach has been proven to be able to overcome a long-standing issue in glycolipid synthesis, namely that coupling of the flexible, poor reactive ceramide acceptor and the oligosaccharyl donor results in low yield and loss of the valuable oligosaccharide unit. Encouraged by previous successes, we decided to apply the GlcCer cassette approach to the present total syntheses. The present GlcCer cassette 4 was designed as shown in Fig. 2 and could be divided into the known glucose 7 and ceramide 8 derivatives. The common trisaccharyl donor 3 could be assembled from the known monosaccharide derivatives 5 and 6.
17 and GAA-7,18 using the GlcCer cassette approach.19 This approach has been proven to be able to overcome a long-standing issue in glycolipid synthesis, namely that coupling of the flexible, poor reactive ceramide acceptor and the oligosaccharyl donor results in low yield and loss of the valuable oligosaccharide unit. Encouraged by previous successes, we decided to apply the GlcCer cassette approach to the present total syntheses. The present GlcCer cassette 4 was designed as shown in Fig. 2 and could be divided into the known glucose 7 and ceramide 8 derivatives. The common trisaccharyl donor 3 could be assembled from the known monosaccharide derivatives 5 and 6.
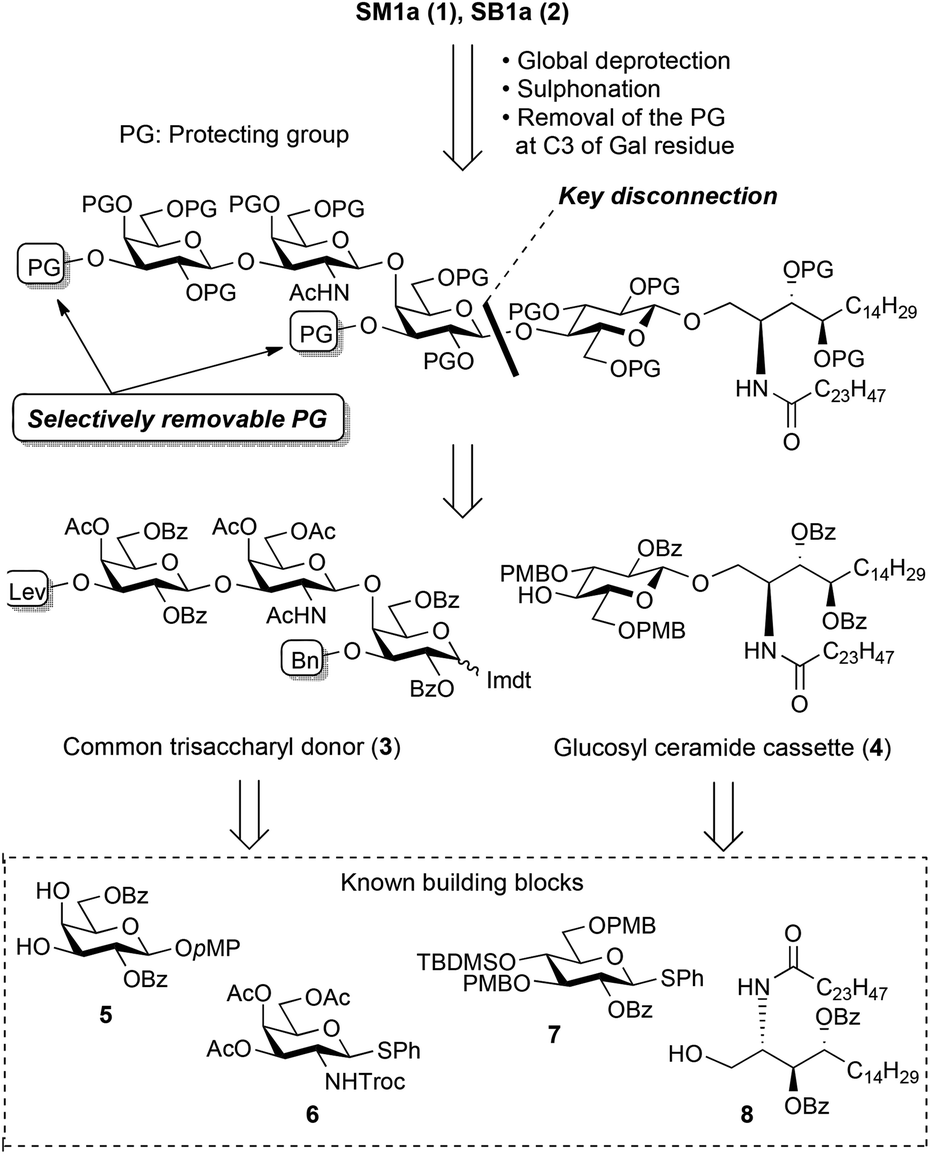 | ||
| Fig. 2 Retrosynthetic analysis of the target compounds, SB1a and SM1a. | ||
First, following the above synthetic strategy, the common trisaccharyl donor 3 was prepared from the known galactose derivative 520 and N-Troc-protected galactosamine derivative 6.21 As shown in Scheme 1, selective introduction of the benzyl group at the C3 position of 5 was performed in a one-pot fashion with two steps, tin acetal formation and subsequent benzylation, giving the 4-OH galactosyl acceptor 9 in 93% yield. The benzyl group was extremely stable under both acidic and basic reaction conditions, and it could be chemoselectively removed by hydrogenolysis prior to sulphonation.
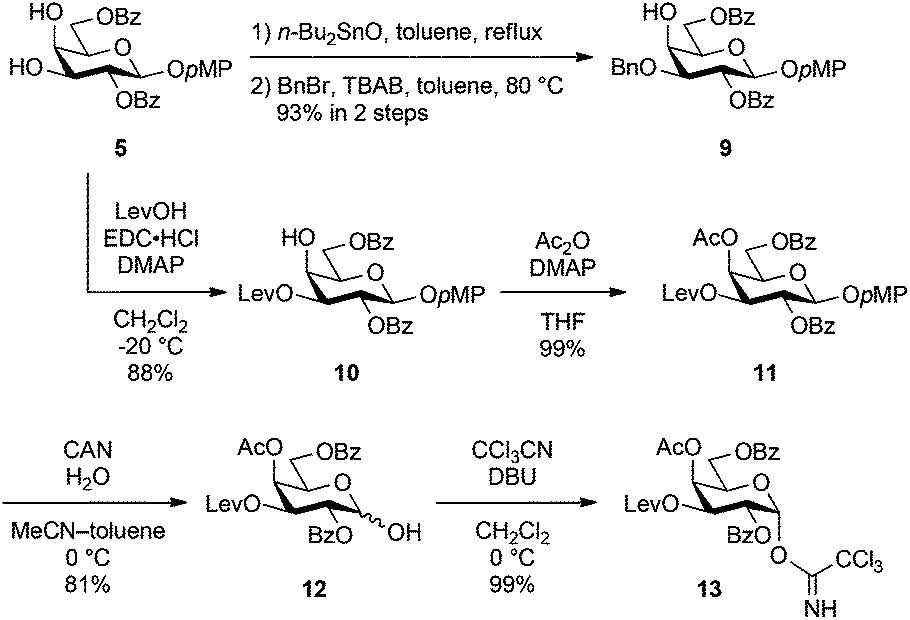 | ||
| Scheme 1 Synthesis of galactose derivatives 9 and 13. | ||
Thereafter, the non-reducing end galactose unit was also prepared from 5. The 3-OH of 5 was selectively protected as a levulinoyl ester by treatment with levulinic acid, EDC·HCl and DMAP in CH2Cl2 at −20 °C, affording 10 in 88% yield. Regioselectivity was produced on the basis of the difference in reactivity between equatorial 3-OH and axial 4-OH. The selectively removable Lev group in the presence of other acyl protecting groups was installed for SB1a. After acetylation of 4-OH (giving 11), the p-methoxyphenyl (pMP) group at the anomeric position was removed with CAN and H2O in an optimised mixed solvent system22 to give hemiacetal 12 in 81% yield. Finally, the introduction of the trichloroacetimidoyl group23 yielded the non-reducing end galactosyl donor 13 in a nearly quantitative yield.
Scheme 2 illustrates the synthesis of the inner disaccharide GalNβ(1 → 4)Gal acceptor 16. The glycosylation of 9 with the known galactosaminyl donor 6 equipped with the 2,2,2-trichloroethoxycarbonyl (Troc) group at C2 as a β-stereo-directing element was performed in the presence of NIS and TfOH24 in CH2Cl2 at −40 °C, giving disaccharide 14 as a single β-isomer in 76% yield. The reductive removal of the Troc group by treatment with zinc and acetic acid in MeCN gave the amino derivative 15 in good yield. MeCN was selected as a solvent to obtain the pure amino derivative before the following acetyl migration step because our previous study on the synthesis of a ganglioside found that MeCN effectively suppressed random acetyl group migration.17 To rapidly convert 15 into the acceptor form 16, acetyl migration from 3-O to 2-N was exerted. Although this reaction was attempted several times under different conditions, the undesired formation of 17, which was the over-migrated product from 4-O to 3-O, could not be avoided. The results are summarised in Table 1. As shown in entry 1, the reaction was performed in 1,4-dioxane under acidic conditions at 60 °C. The migration of the acetyl group was very sluggish and prolonged reaction time increased the generation of the over-migrated side-product 17. Moreover, 19% of 15 were recovered. In entry 2, to facilitate acetyl migration, the reaction temperature was increased up to 90 °C. However, the reaction was an abysmal failure, providing a complex mixture after 16 h. Changing the solvent to DMF did not affect the outcome significantly (entry 3). However, in AcOH/DMF 1:4 at 90 °C (entry 4), the reaction time decreased and the best result (56% of 16) was obtained. Although basic conditions using triethylamine were also examined, the yield of 16 was not improved (entry 5).
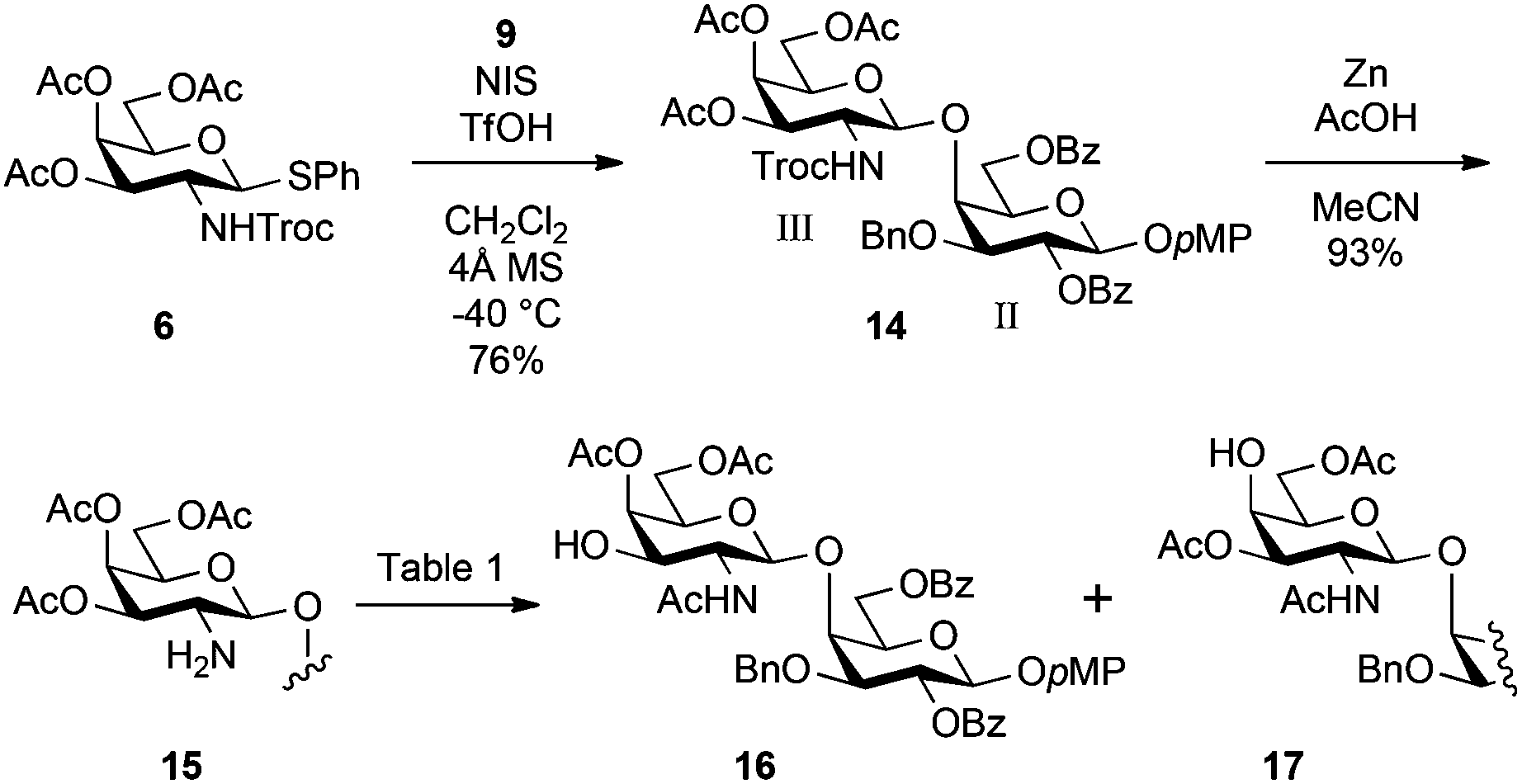 | ||
| Scheme 2 Synthesis of disaccharide acceptor 16. | ||
| Entry | Additive/solvent | Temp. (°C) | Time (h) | Yield of 16 (%) | Yield of 17 (%) | Recovered 15 (%) |
|---|---|---|---|---|---|---|
| 1 | AcOH/1,4-dioxane (1:4) |
60 | 78 | 51 | 19 | 30 |
| 2 | AcOH/1,4-dioxane (1:4) |
90 | 16 | Complex mixture | — | |
| 3 | AcOH/DMF (1:4) |
60 | 48 | 49 | 19 | 32 |
| 4 | AcOH/DMF (1:4) |
90 | 4 | 56 | 18 | 26 |
| 5 | Et3N/DMF (1:4) |
60 | 72 | 49 | 11 | 40 |
As shown in Scheme 3, access to the trisaccharyl donor 3 as a pivotal common unit began with glycosylation of 16 with 13 in the presence of TMSOTf in CH2Cl2 at 0 °C, giving trisaccharide 18 in 77% yield. The selective removal of the p-methoxyphenyl group with CAN and H2O followed by the introduction of the trichloroacetimidoyl group afforded the common trisaccharyl donor 3 in 80% yield over two steps.
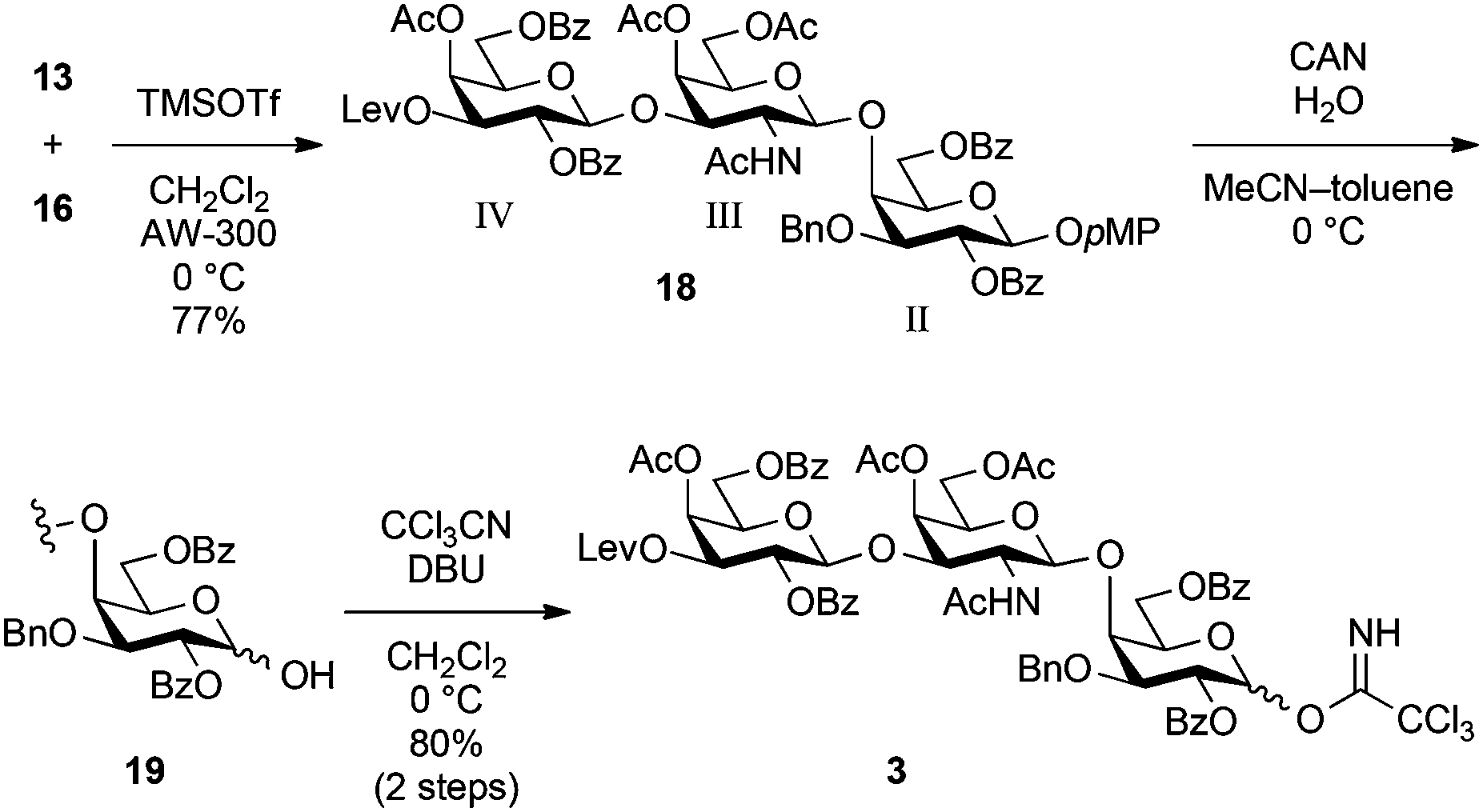 | ||
| Scheme 3 Synthesis of common trisaccharide donor 3. | ||
Scheme 4 shows the assembly of the targeted tetrasaccharyl ceramide framework. First, the GlcCer cassette 4 was constructed by the coupling of the known glucosyl donor 716 and ceramide acceptor 825 mediated by a NIS-TfOH promoter system in CH2Cl2 at 0 °C to afford 20 in 71% yield. A previous study on the GlcCer cassette approach has shown the importance of the protection of hydroxyl groups in the Glc residue to increase the reactivity of the 4-OH of the GlcCer cassette. The best combination for protection was the presence of electron-donating groups on both O4 and O6 and an electron-withdrawing group on O2.13–15 Furthermore, the TBDMS protection of the C4 hydroxy group on a Glc donor is also important for efficient coupling of glucose and ceramide.17 Thus, we used the Glc donor 7 in this study. The removal of the TBDMS group in 20 by exposure to TBAF gave an 87% yield of the GlcCer cassette 4, which was the coupling partner for the trisaccharyl donor 3 in the final glycosylation.
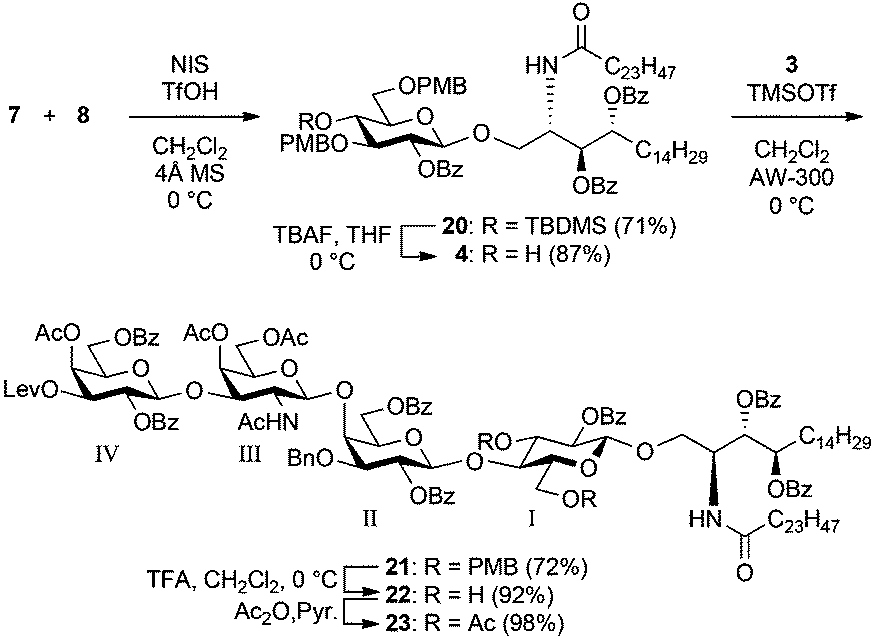 | ||
| Scheme 4 Synthesis of GlcCer cassette 4 followed by glycosylation with 3. | ||
The key glycosylation reaction performed between the equimolar amounts of trisaccharyl donor 3 and GlcCer cassette 4 was allowed to proceed in the presence of a catalytic amount of TMSOTf in CH2Cl2 at 0 °C, providing the tetrasaccharyl ceramide framework 21 in 72% yield. Following this, the p-methoxybenzyl groups in 21 were removed under acidic conditions (giving 22). Subsequent acetylation afforded 23, which was used for the following reaction sequences as the core framework of both target compounds.
The selective removal of the benzyl group in 23 by hydrogenation was examined. The results are summarised in Table 2. The Pearlman's catalyst (Pd(OH)2–C) was chosen as a Pd catalyst and the reaction was performed in EtOAc under ambient pressure of H2 gas. Consequently, only a poor yield (5%) of the desired product 24 was obtained and most of the starting material 23 was recovered (entry 1). Although increasing the Pd catalyst amount gave a marginally better result (9%), the effect was very limited (entry 2). As entry 3 shows, the reaction solvent was changed to EtOH, which has been often used as a solvent for hydrogenolysis. After stirring for 72 h under ambient pressure, the reaction was subsequently subjected to a work-up process, providing 24 in 50% yield. Further prolonged stirring produced unwanted by-products, one of which was identified as a product in which the benzene ring of the benzoyl groups was reduced. The separation of the crude mixture of 24 and by-products by silica gel column chromatography was a laborious task. We speculated that steric hindrance surrounding the benzyl group made access of the benzyl group onto the surface of palladium carbon difficult.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. of Pd(OH)2-C | Solvent | Pressure (MPa) | Time (h) | % Yield of 24 |
| 1 | 0.1 | EtOAc | — | 24 | 5 |
| 2 | 1.8 | EtOAc | — | 11 | 9 |
| 3 | 3.6 | EtOH | — | 72 | 50 |
| 4 | 3.6 | EtOH | 0.3 | 23 | 77 |
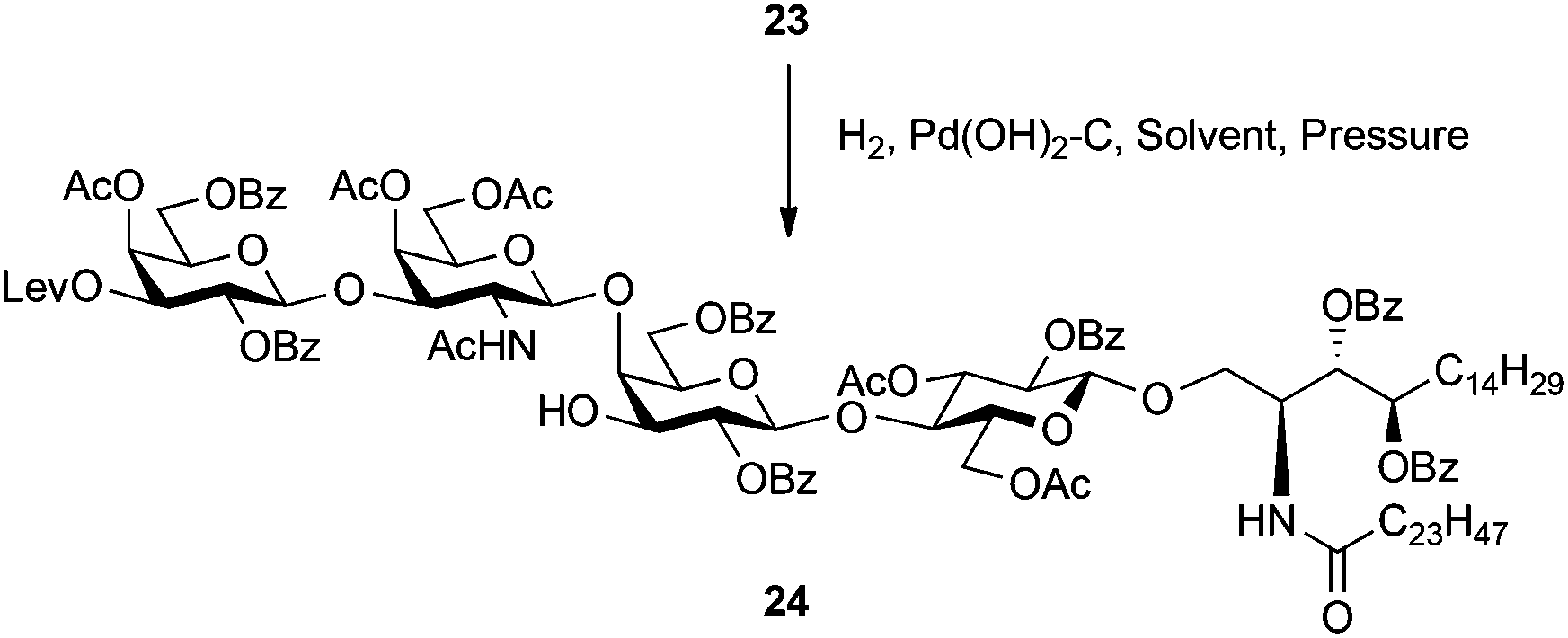
Toward solving the problem, the reaction was carried out under a H2 atmosphere at higher pressure (0.3 MPa) (entry 4). As anticipated, the reaction rate became faster and the yield of 24 was increased up to 77%. Although reduction of the benzene ring of the benzoyl group was observed, the limited reaction time (<24 h) minimised the formation of the by-product. The hydroxyl group of the obtained 24 was available for sulphonation toward SM1a.
Scheme 5 shows the final steps to mono-sulphated glycosphingolipid SM1a. For sulphonation of the hydroxyl group at the C3 position of the inner galactose residue, 24 was reacted with SO3·Pyr in pyridine at 80 °C, yielding 25 in 91% yield. Finally, saponification of whole acyl protecting groups using 0.5 M aq. NaOH in MeOH was performed to furnish one of the target compounds SM1a (1) as an Na form in good yield.
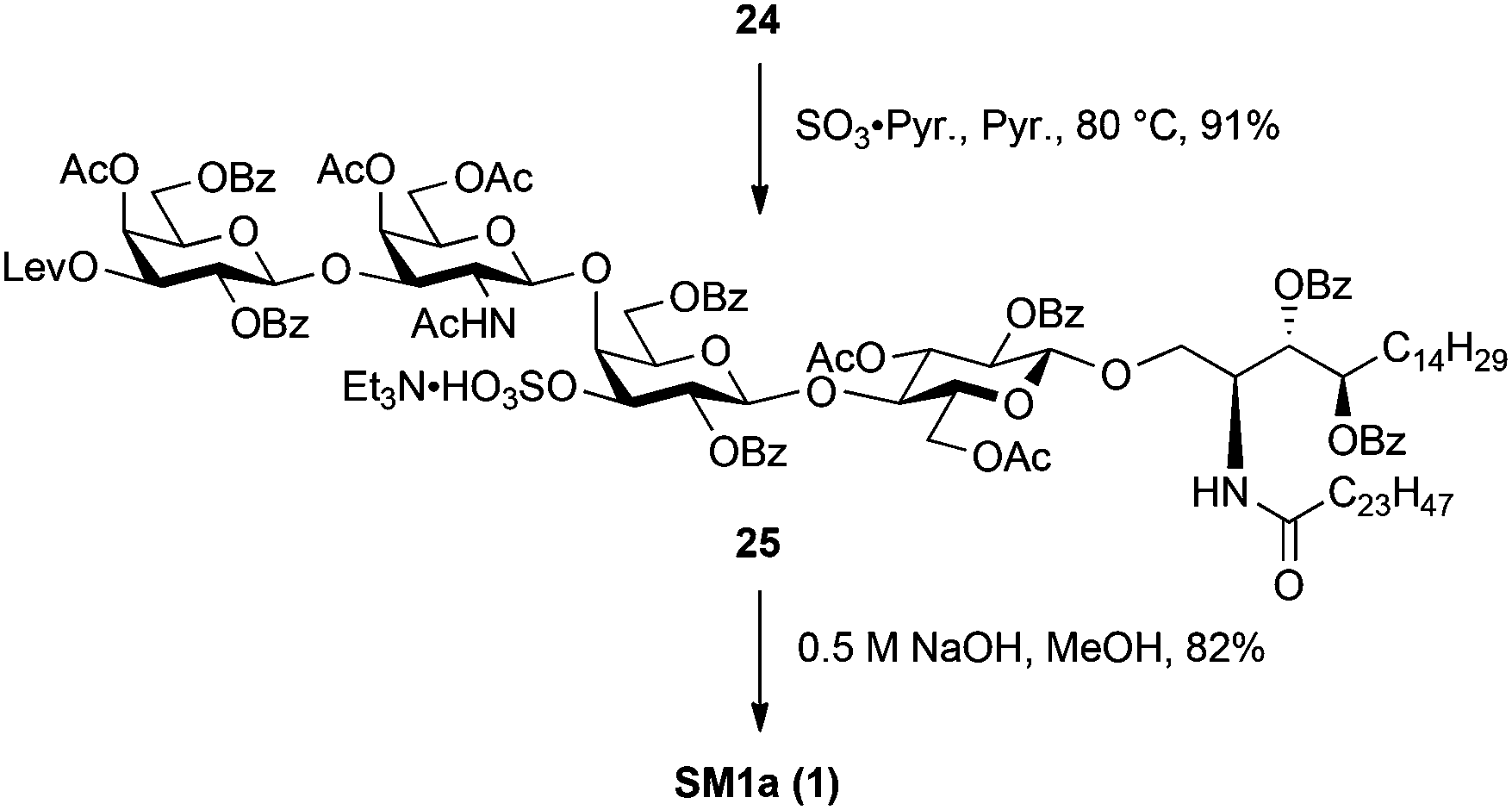 | ||
| Scheme 5 Sulphonation followed by global deprotection toward SM1a. | ||
SB1a was also derived from 24via three steps (Scheme 6). First, the levulinoyl group was removed by treatment with NH2NH2·AcOH in THF, giving diol 26 in excellent yield. Sulphonation of two hydroxyl groups was performed using the same procedure as that of SM1a, affording the fully protected SB1a 27 in 96% yield. Finally, cleavage of acyl groups under saponification conditions successfully delivered the targeted SB1a (2) in excellent yield.
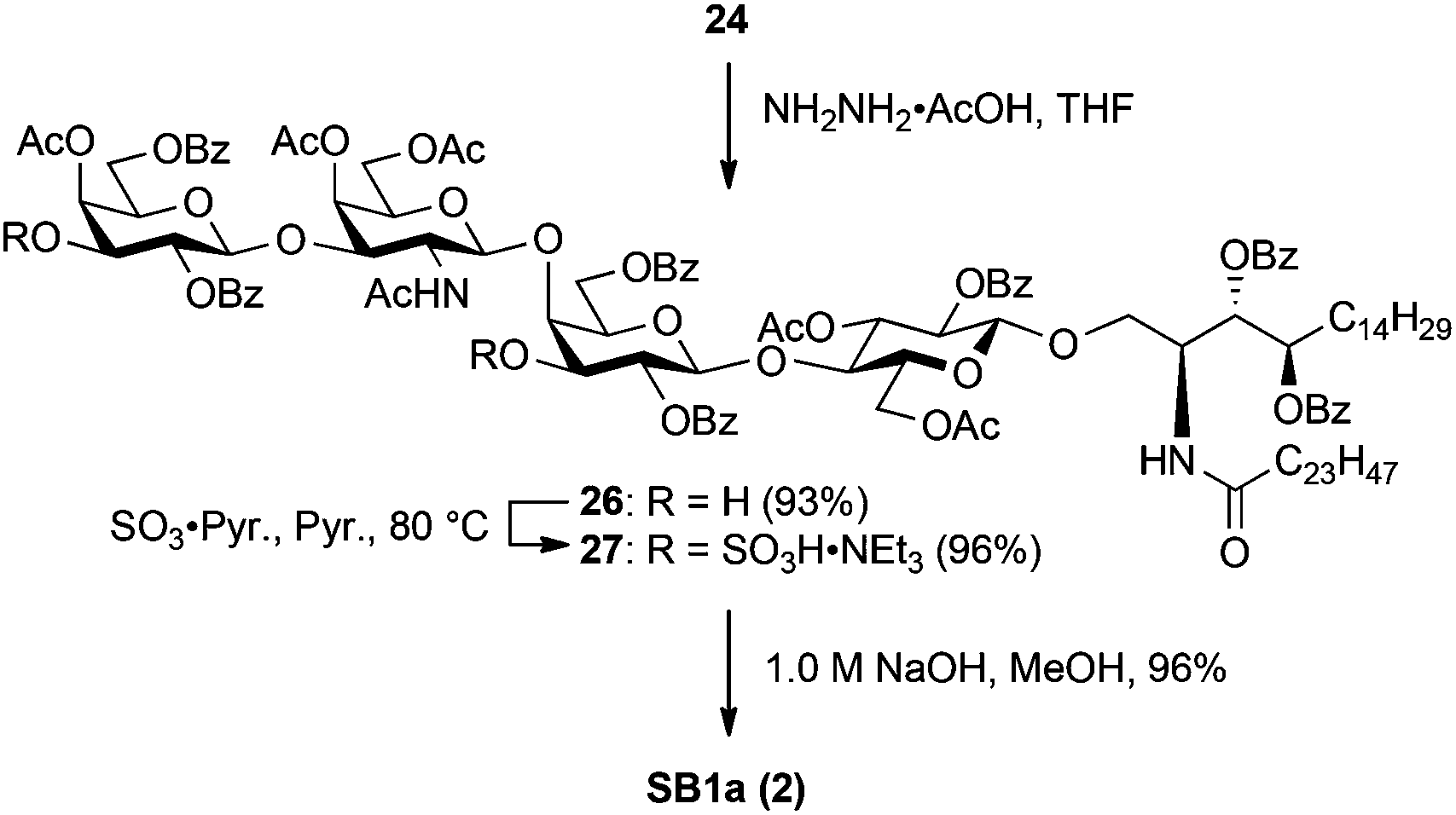 | ||
| Scheme 6 Conversion of 24 into SB1a through sulphonation followed by deprotection. | ||
The synthetic sulphoglycolipids SM1a and SB1a were printed in a small focused microarray together with the naturally occurring SM1a. The array was then probed with mAb AE3 to compare the binding signals elicited (Fig. 3). The synthetic and the naturally occurring SM1a were equally well bound by mAb AE3, whereas there were no binding signals with the synthetic disulphated glycolipid SB1a. This result corroborated the previous finding in the screening array analysis that AE3 binding was restricted to the monosulphated glycolipid SM1a with an unsubstituted terminal galactose residue. Sulphation of this galactose, as in SB1a, obviated the AE3 binding.
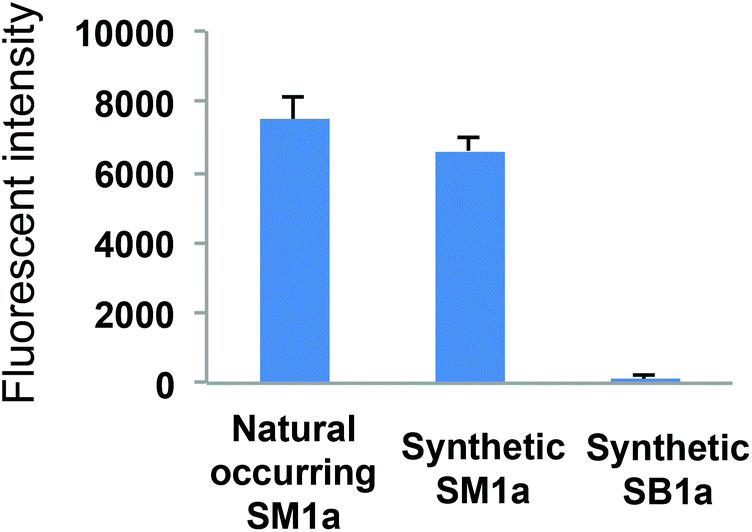 | ||
| Fig. 3 Microarray analyses of mAb AE3 with the naturally occurring and chemically synthesized SM1a and synthetic SB1a. The results shown are binding intensities with duplicated spots of the three glycolipids printed at 5 fmol in the arrays with error bars representing half of the difference between the two values. | ||
Conclusions
The total synthesis of disulphated glycosphingolipid SB1a was accomplished for the first time, as well as synthesis of the related monosulphated SM1a with a natural ceramide moiety. These glycolipids were systematically synthesised by efficient glycan assembly using common building blocks and the GlcCer cassette approach. The success of the total synthesis of sulphoglycolipids demonstrates the broad utility of the GlcCer cassette approach for glycolipid synthesis. Furthermore, binding analysis using the two synthetic sulphoglycolipids corroborated the previous observations on the recognition of the SM1a glycolipid by mAb AE3, although the antibody was originally raised against a cancer-associated mucin-type glycoprotein. These findings raise the possibility that sulphated O-glycan analogs of SM1a occur on cancer-associated mucins.Experimental section
General methods
All reactions were carried out under a positive pressure of argon, unless otherwise noted. All chemicals were purchased from commercial suppliers and used without further purification, unless otherwise noted. Molecular sieves were purchased from Wako Chemicals Inc. (Miyazaki, Japan) and dried at 300 °C for 2 h in a muffle furnace prior to use. Solvents as reaction media were dried over molecular sieves and used without purification. TLC analysis was performed on Merck TLC plates (silica gel 60F254 on glass plates). Compound detection was either by exposure to UV light (2536 Å) or by soaking in a solution of 10% H2SO4 in ethanol followed by heating. Silica gel (80 mesh and 300 mesh) manufactured by Fuji Silysia Co. was used for flash column chromatography. The quantity of silica gel was usually estimated as 100 to 200-fold weight of the sample to be charged. Solvent systems in chromatography were specified in v/v. Evaporation and concentration were carried out in vacuo. 1H-NMR and 13C-NMR spectra were recorded with Bruker AVANCE III 500/800 spectrometers. Chemical shifts in 1H-NMR spectra are expressed in ppm (δ) relative to the signal of Me4Si, adjusted to δ 0.00 ppm. Data are presented as follows: Chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, dd = double of doublet, td = triple doublet, m = multiplet and/or multiple resonances), integration, coupling constant in hertz (Hz), position of the corresponding proton. COSY methods were used to confirm the NMR signal assignments. The sugar units were numbered with Roman numerals I, II, III, IV beginning at the reducing (“upstream”) end of the molecule. The individual resonances were numbered with Roman numeral superscripts as, for example, the following: H-3I, H-3II, H-3III, H-3IV. High-resolution mass (ESI-TOF MS) spectra were run on the Bruker micrOTOF. Optical rotations were measured with a “Horiba SEPA-300” high-sensitive polarimeter.Synthetic procedures
:1), the reaction mixture was quenched with a solution of cesium fluoride (5 wt% in H2O). The mixture was extracted with CHCl3, and then the organic layer was washed with brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 80:1) to give 9 (223 mg, 93%) as an amorphous compound; [α]D +38.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.07–7.15 (m, 15H, Ph), 6.89–6.57 (2 d, 4H, Ar), 5.72 (dd, 1H, J1,2 8.1 Hz, J2,3 9.7 Hz, H-2), 4.91 (d, 1H, H-1), 4.76–4.64 (m, 3H, H-6a, H-6b, PhCH2), 4.57 (d, 1H, Jgem 12.3 Hz, PhCH2), 4.16 (s, 1H, H-4), 3.95 (m, 1H, H-5), 3.75 (dd, 1H, J3,4 3.4 Hz, H-3), 3.67 (s, 3H, OCH3), 2.76 (s, 1H, OH); 13C NMR (125 MHz, CDCl3) δ 166.3, 165.3, 155.4, 151.3, 136.9, 133.2, 133.1, 129.9, 129.8, 128.5, 128.4, 128.4, 128.1, 127.9, 118.8, 114.3, 100.9, 78.2, 72.6, 71.8, 71.0, 66.2, 63.5, 55.5; HRMS (ESI) m/z: found [M + Na]+ 607.1936, C34H32O9 calcd for [M + Na]+ 607.1939.
:1), the reaction mixture was diluted with CHCl3, and then successively washed with water and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by crystallization (n-hexane/EtOAc) to give 10 (207 mg, 88%) as a white crystal; M.p. 94–96 °C (from n-hexane/EtOAc); [α]D +32.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.07–7.42 (m, 10H, Ph), 6.93–6.61 (2 d, 4H, Ar), 5.83 (dd, 1H, J1,2 8.1 Hz, J2,3 10.3 Hz, H-2), 5.13 (dd, 1H, J3,4 3.2 Hz, H-3), 5.04 (d, 1H, H-1), 4.72–4.65 (m, 2H, H-6a, H-6b), 4.32 (dd, 1H, J4,OH 7.3 Hz, H-4), 4.09 (dd, 1H, J5,6a 6.1 Hz, J5,6b 6.5 Hz, H-5), 3.69 (s, 3H, OCH3), 3.22 (d, 1H, OH), 2.74–2.44 (m, 4H, CH2CH2), 2.13 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 208.0, 171.9, 166.3, 155.5, 151.2, 133.3, 133.2, 129.8, 129.7, 129.5, 128.4, 128.4, 118.9, 114.3, 101.0, 74.1, 72.6, 69.4, 66.6, 63.2, 55.5, 38.2, 29.6, 28.3; HRMS (ESI) m/z: found [M + Na]+ 615.1837, C32H32O11 calcd for [M + Na]+ 615.1837.
:1), the reaction mixture was quenched with MeOH and diluted with EtOAc. The organic layer was then washed with satd aq. NaHCO3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 100:1) to give 11 (384 mg, 99%) as an amorphous compound; [α]D +14.4° (c 3.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.04–7.42 (m, 10H, Ph), 6.92–6.63 (2 d, 4H, Ar), 5.77 (dd, 1H, J1,2 8.1 Hz, J2,3 10.4 Hz, H-2), 5.62 (d, 1H, J3,4 3.3 Hz, H-4), 5.33 (dd, 1H, H-3), 5.10 (d, 1H, H-1), 4.56 (dd, 1H, J5,6a 7.6 Hz, Jgem 11.3 Hz, H-6a), 4.44 (dd, 1H, J5,6b 5.6 Hz, H-6b), 5.62 (t, 1H, H-5), 3.68 (s, 3H, OCH3), 2.65–2.39 (m, 4H, CH2CH2), 2.23 (s, 3H, CH3), 2.01 (s, 3H, Ac); 13C NMR (125 MHz, CDCl3) δ 205.6, 171.6, 170.1, 165.8, 165.1, 155.5, 150.9, 133.3, 133.2, 129.7, 129.6, 129.33, 129.2, 128.3, 118.7, 114.3, 100.9, 71.1, 71.1, 69.2, 67.2, 61.2, 55.4, 37.5, 29.3, 27.7, 20.1; HRMS (ESI) m/z: found [M + Na]+ 657.1946, C34H34O12 calcd for [M + Na]+ 657.1942.
:1), the reaction mixture was diluted with EtOAc. The organic layer was successively washed with satd aq. NaHCO3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 150:1 → 100:1) to give 12 (455 mg, α/β 5:1, 81%) as a white solid; 1H NMR (500 MHz, CDCl3) δ 8.05–7.42 (m, 12.2H, Ph), 5.70–5.68 (m, 2H, H-1α, H-2α), 5.62 (dd, 1H, J3,4 3.3 Hz, J4,5 1.0 Hz, H-4α), 5.56 (d, 0.2H, J3,4 1.8 Hz, H-4β), 5.39 (dd, 1H, J2,3 10.0 Hz, H-3α), 5.35–5.33 (m, 0.4H, H-2β, H-3β), 4.87 (t, 0.2H, J1,2 8.0 Hz, J1,OH 8.2 Hz, H-1β), 4.65 (t, 1H, J5,6a, J5,6b 6.7 Hz, H-5α), 4.53 (dd, 0.2H, J5,6a 6.6 Hz, Jgem 11.3 Hz, H-6aβ), 4.47 (dd, 1H, Jgem 11.3 Hz, H-6aα), 4.36 (dd, 0.2H, J5,6b 6.9 Hz, H-6bβ), 4.29 (dd, 1H, J5,6b 6.9 Hz, H-6bα), 4.15 (t, 0.2H, H-5β), 4.10 (d, 0.2H, OHβ), 3.43 (d, 1H, OHα), 2.73–2.37 (m, 5H, CH2CH2α,β), 2.19 (s, 3.6H, CH3α,β), 2.04 (s, 3.7H, Acα,β); 13C NMR (125 MHz, CDCl3) δ 206.0, 171.8, 170.3, 166.1, 166.0, 133.7, 133.5, 133.3, 130.0, 129.7, 129.4, 129.2, 128.5, 128.4, 96.2, 90.9, 71.9, 71.1, 70.5, 69.0, 68.4, 67.5, 67.4, 66.4, 62.1, 61.8, 37.7, 37.7, 29.6, 29.5, 27.8, 27.7, 20.7, 20.6; HRMS (ESI) m/z: found [M + Na]+ 551.1524, C27H28O11 calcd for [M + Na]+ 551.1524.
:1), the reaction mixture was concentrated and then purified by silica gel column chromatography (CHCl3/MeOH 100:1) to give 13 (572 mg, 99%) as an amorphous compound; [α]D +77.7° (c 0.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.59 (s, 1H, NH), 8.01–7.41 (m, 10H, Ph), 6.76 (dd, 1H, J1,2 3.0 Hz, H-1), 5.73–5.66 (m, 3H, H-2, H-3, H-4), 4.65 (t, 1H, J5,6a, J5,6b 6.6 Hz, H-5), 4.48 (dd, 1H, Jgem 11.4 Hz, H-6a), 4.36 (dd, 1H, H-6b), 2.72–2.42 (m, 4H, CH2CH2), 2.22 (s, 3H, CH3), 2.06 (s, 3H, Ac); 13C NMR (125 MHz, CDCl3) δ 205.8, 171.7, 170.1, 165.8, 165.5, 160.5, 133.6, 133.3, 129.9, 129.7, 129.3, 128.7, 128.4, 128.4, 93.6, 90.7, 69.3, 67.8, 67.7, 67.4, 61.9, 37.7, 29.5, 27.7, 20.6; HRMS (ESI) m/z: found [M + Na]+ 694.0615, C29H28Cl3NO11 calcd for [M + Na]+ 694.0620.
:1). The reaction mixture was quenched with satd aq. NaHCO3 and then filtered through a pad of Celite, and then washed with CHCl3. The combined filtrate and washings were washed with satd aq. Na2S2O3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/EtOAc 25:1 → 15:1 → 5:1) and the product obtained was then subjected to crystallization to give 14 (696 mg, 76%) as a white crystal; M.p. 187 °C (from n-hexane/EtOAc); [α]D +1.5° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.07–7.22 (m, 15H, Ph), 6.89–6.56 (2 d, 4H, Ar), 5.71 (dd, 1H, J1,2 8.2 Hz, J2,3 9.9 Hz, H-2II), 5.38 (br d, 1H, J2,NH 7.4 Hz, NHIII), 5.32 (d, 1H, J3,4 2.9 Hz, H-4III), 4.98 (br d, 1H, J2,3 10.3 Hz, H-3III), 4.90 (d, 1H, H-1II), 4.86 (d, 1H, Jgem 12.1 Hz, Cl3CCH2), 4.80 (d, 1H, J1,2 8.5 Hz, H-1III), 4.76–4.71 (m, 2H, H-6aII, Cl3CCH2), 4.65 (d, 1H, Jgem 11.6 Hz, PhCH2), 4.60–4.57 (m, 2H, H-6bII, PhCH2), 4.20 (d, 1H, J3,4 1.7 Hz, H-4II), 4.12–4.05 (m, 2H, H-5III, H-6aIII), 3.98–3.91 (m, 2H, H-5II, H-2III), 3.83–3.68 (m, 2H, H-6bIII, H-3II), 3.67 (s, 3H, OCH3), 2.15, 2.03, 1.97 (3 s, 9H, 3Ac); 13C NMR (125 MHz, CDCl3) δ 170.4, 170.2, 170.2, 166.3, 164.9, 155.4, 154.4, 151.4, 136.8, 133.2, 133.2, 129.9, 129.8, 129.7, 129.7, 128.8, 128.5, 128.5, 128.1, 118.8, 114.3, 101.6, 101.1, 96.0, 79.4, 74.4, 74.1, 73.5, 72.3, 71.4, 70.7, 70.4, 66.6, 64.0, 61.3, 55.5, 52.7, 20.6, 20.6, 20.5; HRMS (ESI) m/z: found [M + Na]+ 1068.1985, C49H50Cl3NO18 calcd for [M + Na]+ 1068.1986.
:1, 8.4 mL) was added zinc powder (2.1 g). After stirring for 20 min at ambient temperature as the reaction was monitored by TLC (CHCl3/MeOH 15:1), the reaction mixture was filtered through a pad of Celite, and then washed with EtOAc. The filtrate and washings were combined and extracted with EtOAc. The organic layer was successively washed with satd aq. NaHCO3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 100:1) to give 15 (342 mg, 93%) as a white solid; [α]D +38.0° (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.07–7.18 (m, 15H, Ph), 6.89–6.55 (2 d, 4H, Ar), 5.80 (dd, 1H, J1,2 8.1 Hz, J2,3 10.0 Hz, H-2II), 5.32 (d, 1H, J3,4 2.6 Hz, H-4III), 4.92 (d, 1H, H-1II), 4.74–4.70 (m, 2H, H-3III, H-6aIII), 4.67–4.57 (m, 3H, H-6bIII, PhCH2), 4.52 (d, 1H, J1,2 7.9 Hz, H-1III), 4.21 (d, 1H, J3,4 2.7 Hz, H-4II), 4.14–4.07 (m, 2H, H-6aIII, H-6bIII), 3.94 (dd, 1H, J5,6a 4.1 Hz, J5,6b 8.1 Hz, H-5II), 3.82 (dd, 1H, J5,6a 6.6 Hz, J5,6b 7.3 Hz, H-5III), 3.76 (dd, 1H, J2,3 10.5 Hz, H-3II), 3.67 (s, 3H, OCH3), 3.29 (dd, 1H, J2,3 10.8 Hz, H-2III), 2.13, 2.05, 2.02 (3 s, 9H, 3Ac), 1.65 (s, 2H, NH2); 13C NMR (125 MHz, CDCl3) δ 170.4, 170.4, 170.3, 166.3, 165.2, 155.4, 151.4, 137.0, 133.2, 133.2, 129.9, 129.8, 129.7, 128.5, 128.5, 128.4, 128.2, 128.0, 118.6, 114.3, 105.3, 100.9, 78.7, 74.2, 73.9, 72.6, 72.5, 71.2, 70.7, 66.3, 64.1, 61.7, 55.5, 51.8, 20.8, 20.7, 20.6; HRMS (ESI) m/z: found [M + Na]+ 894.2944, C46H49NO16 calcd for [M + Na]+ 894.2944.
:4, 11.5 mL) was stirred for 4 h at 90 °C as the reaction was monitored by TLC (CHCl3/MeOH 20:1). The reaction mixture was neutralized with ice-cooled satd aq. NaHCO3 and extracted with EtOAc. The organic layer was then washed with H2O and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 80:1) to give 16 (56 mg, 56%) as an amorphous compound; [α]D +14.0° (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.08–7.14 (m, 15H, Ph), 6.89–6.57 (m, 5H, Ar, NH), 6.08 (d, 1H, J1,OH 2.0 Hz, OH), 5.75 (dd, 1H, J1,2 8.1 Hz, J2,3 9.9 Hz, H-2II), 5.27 (d, 1H, J3,4 3.2 Hz, H-4III), 4.92 (d, 1H, J1,2 8.1 Hz, H-1II), 4.77 (dd, 1H, J5,6a 3.7 Hz, Jgem 12.1 Hz, H-6aII), 4.66 and 4.62 (2 d, 2H, Jgem 10.7 Hz, PhCH2), 4.28 (dd, 1H, J5,6b 8.1 Hz, Jgem 12.1 Hz, H-6bII), 4.47 (d, 1H, J1,2 8.6 Hz, H-1III), 4.19 (d, 1H, J3,4 2.9 Hz, H-4II), 4.12 (d, 2H, H-6aIII, H-6bIII), 4.00 (dd, 1H, J5,6a 3.6 Hz, J5,6b 8.1 Hz, H-5II), 3.92–3.88 (m, 2H, H-2III, H-3II), 3.77 (t, 1H, J5,6a, J5,6b 6.4 Hz, H-5III), 3.68 (s, 3H, OCH3), 3.58 (m, 1H, H-3III), 2.16, 2.05, 1.90 (3 s, 9H, 3Ac); 13C NMR (125 MHz, CDCl3) δ 175.2, 170.6, 170.4, 166.4, 165.1, 155.5, 151.2, 135.8, 133.5, 133.3, 129.8, 129.7, 129.5, 129.1, 128.9, 128.6, 128.6, 128.5, 118.8, 114.3, 102.5, 100.7, 80.0, 75.8, 74.5, 74.0, 72.3, 71.4, 71.3, 68.0, 64.0, 62.1, 55.8, 55.5, 22.6, 20.8, 20.7; HRMS (ESI) m/z: found [M + Na]+ 894.2944, C46H49NO16 calcd for [M + Na]+ 894.2944.
:1). The reaction mixture was quenched with satd aq. NaHCO3 and filtered through a pad of Celite, and then washed with CHCl3. The combined filtrate and washings were washed with satd aq. Na2S2O3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/EtOAc 25:1 → 15:1 → 5:1) and the product was then subjected to crystallization to give 18 (574 mg, 77%) as a white crystal; M.p. 184 °C (from n-hexane/EtOAc); [α]D +160.0° (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.06–7.07 (m, 25H, Ph), 6.81–6.50 (2 d, 4H, Ar), 5.62–5.58 (m, 2H, NH, H-2II), 5.52 (d, 1H, J3,4 3.0 Hz, H-4IV), 5.46–5.42 (m, 2H, H-4III, H-2IV), 5.22 (dd, 1H, J2,3 10.5 Hz, J3,4 3.4 Hz, H-3IV), 5.18 (d, 1H, J1,2 8.3 Hz, H-1III), 5.05 (dd, 1H, J2,3 10.9 Hz, J3,4 3.4 Hz, H-3III), 4.81 (d, 1H, J1,2 8.1 Hz, H-1IV), 4.78 (d, 1H, J1,2 7.9 Hz, H-1II), 4.63 (dd, 1H, J5,6b 3.7 Hz, Jgem 12.2 Hz, H-6aII), 4.58–4.48 (m, 3H, H-6aIV, H-6bII, PhCH2), 4.36 (d, 1H, Jgem 12.8 Hz, PhCH2), 4.31 (dd, 1H, J5,6b 7.1 Hz, Jgem 11.2 Hz, H-6bIV), 4.10–4.02 (m, 2H, H-5IV, H-6aIII), 3.99 (d, 1H, J3,4 2.5 Hz, H-4II), 3.94 (dd, 1H, J5,6b 6.8 Hz, Jgem 11.5 Hz, H-6bIII), 3.84 (dd, 1H, J5,6b 8.3 Hz, H-5II), 3.79 (t, 1H, H-5III), 3.64 (s, 3H, OCH3), 3.56 (dd, 1H, J2,3 10.0 Hz, H-3II), 3.24–3.19 (m, 1H, H-2III), 2.63–2.36 (m, 4H, CH2CH2), 2.21, 2.15, 2.01, 1.72 (4 s, 15H, 3Ac, CH3); 13C NMR (125 MHz, CDCl3) δ 205.7, 172.6, 171.7, 170.5, 170.3, 170.0, 166.2, 165.8, 165.1, 165.0, 155.3, 151.3, 137.1, 133.6, 133.2, 129.9, 129.8, 129.8, 129.7, 129.7, 129.7, 129.5, 129.4, 128.7, 128.4, 128.4, 128.3, 128.0, 118.6, 114.2, 101.7, 100.8, 99.1, 78.0, 74.4, 72.9, 72.4, 71.9, 71.0, 70.7, 69.4, 67.1, 64.2, 62.5, 61.5, 55.5, 37.7, 29.6, 29.4, 27.8, 23.2, 20.8, 20.7, 20.7; HRMS (ESI) m/z: found [M + Na]+ 1404.4469, C73H75NO26 calcd for [M + Na]+ 1404.4470.
:5:3, 12 mL) was added ammonium cerium(IV) nitrate (1.33 g, 2.40 mmol) at 0 °C. After stirring for 40 min as the reaction was monitored by TLC (CHCl3/MeOH 20:1), the reaction mixture was extracted with EtOAc. The organic layer was successively washed with satd aq. NaHCO3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 150:1 → 100:1) to give 19. The obtained residue was then dissolved in CH2Cl2 (12 mL). To the solution were added trichloroacetonitrile (685 μL, 4.84 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (283 μL, 2.66 mmol) at 0 °C. After stirring for 1 h as the reaction was monitored by TLC (CHCl3/MeOH 20:1), the reaction mixture was concentrated and then purified by silica gel column chromatography (CHCl3/MeOH 80:1) to give 3 (275 mg, 80% over 2 steps) as an amorphous compound; 3α: [α]D +42.9° (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.39 (s, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH), 8.05–7.14 (m, 25H, Ph), 6.51 (d, 1H, J1,2 3.6 Hz, H-1II), 5.64 (dd, 1H, J2,3 10.4 Hz, H-2II), 5.51–5.49 (m, 2H, NHII, H-4III), 5.21 (dd, 1H, J2,3 10.5 Hz, J3,4 3.5 Hz, H-3IV), 5.17 (d, 1H, J1,2 8.3 Hz, H-1III), 4.98 (dd, 1H, J2,3 10.8 Hz, J3,4 3.5 Hz, H-3III), 4.77 (d, 1H, J1,2 7.9 Hz, H-1IV), 4.63–4.58 (m, 2H, H-6aIV, PhCH2), 4.52 (dd, 1H, J5,6b 6.4 Hz, Jgem 11.3 Hz, H-6aII), 4.43 (d, 1H, Jgem 12.5 Hz, PhCH2), 4.38–4.29 (m, 3H, H-5IV, H-6bIV, H-6bII), 4.15 (d, 1H, J3,4 2.0 Hz, H-4II), 4.09–4.00 (m, 3H, H-3II, H-5II, H-6aIII), 3.91 (dd, 1H, J5,6b 7.2 Hz, Jgem 11.6 Hz, H-6bIII), 3.78 (t, 1H, H-5III), 3.24–3.19 (m, 1H, H-2III), 2.64–2.34 (m, 4H, CH2CH2), 2.20–1.36 (5 s, 15H, 4Ac, CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 172.1, 171.7, 170.4, 170.2, 169.9, 166.2, 165.8, 165.3, 164.9, 160.2, 137.1, 133.5, 133.3, 133.1, 133.0, 129.8, 129.7, 129.6, 129.5, 129.5, 129.4, 129.2, 128.6, 128.4, 128.3, 128.3, 127.9, 101.7, 99.2, 94.1, 90.9, 74.7, 74.0, 73.9, 71.9, 71.6, 71.0, 70.9, 70.7, 69.4, 69.2, 68.5, 67.0, 64.5, 62.6, 61.4, 60.3, 55.3, 37.6, 29.6, 29.4, 27.7, 23.0, 20.8, 20.7, 20.6; HRMS (ESI) m/z: found [M + Na]+ 1441.3147, C68H69Cl3N2O25 calcd for [M + Na]+ 1441.3147.
:1). The reaction mixture was quenched with satd aq. NaHCO3 and filtered through a pad of Celite, and then washed with CHCl3. The combined filtrate and washings were washed with satd aq. Na2S2O3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (toluene/EtOAc 30:1) to give 20 (150 mg, 71%) as a white solid; [α]D +35.0° (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.01–7.35 (m, 15H, Ph), 7.27–6.60 (4 d, 8H, Ar), 6.16 (d, 1H, JNH,2 = 9.0 Hz, NH), 5.58 (dd, 1H, J2,3 7.8 Hz, J3,4 3.5 Hz, H-3Cer), 5.38–5.35 (m, 1H, H-4Cer), 5.11 (near t, 1H, J1,2, J2,3 8.5 Hz, H-2I), 4.56–4.47 (m, 4H, ArCH2, H-1I, H-2Cer), 4.30–4.15 (2 d, 4H, Jgem 11.7 Hz, ArCH2), 4.03 (dd, 1H, J1,2 3.6 Hz, Jgem 10.1 Hz, H-1aCer), 3.78 and 3.68 (2 s, 6H, OCH3), 3.64 (dd, 1H, H-1bCer), 3.59–3.55 (m, 3H, H-3I, H-4I, H-6aI), 3.41 (t, 1H, J5,6a, J5,6b 6.4 Hz, H-5I), 3.32 (dd, 1H, Jgem 10.6 Hz, H-6bI), 1.88–1.70 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.60–1.09 (m, 66H, 33CH2), 0.89–0.83 (m, 6H, 2CH3), 0.81 (s, 9H, tBu), −0.03, −0.05 (s, 6H, 2SiCH3); 13C NMR (125 MHz, CDCl3) δ 174.0, 166.0, 165.2, 165.1, 159.1, 158.8, 133.1, 132.9, 132.8, 130.3, 130.2, 130.1, 130.0, 129.8, 129.8, 129.7, 129.6, 129.1, 129.0, 128.3, 128.3, 113.7, 113.4, 100.3, 82.7, 74.4, 74.2, 73.7, 73.2, 70.9, 68.9, 66.4, 55.2, 55.0, 47.9, 36.3, 31.9, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.2, 29.1, 25.9, 25.5, 25.4, 22.7, 17.9, 14.1, −3.8, −4.8; HRMS (ESI) m/z: found [M + Na]+ 1518.9706, C73H75NO26 calcd for [M + Na]+ 1518.9701.
:1), the reaction mixture was concentrated. The residue was purified by silica gel column chromatography (n-hexane/EtOAc 2:1) to give 4 (73 mg, 87%) as a white solid; [α]D +19.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.00–7.26 (m, 15H, Ph), 7.16–6.65 (4 d, 8H, Ar), 6.09 (d, 1H, JNH,2 9.1 Hz, NH), 5.54 (dd, 1H, J2,3 7.8 Hz, J3,4 3.7 Hz, H-3Cer), 5.37–5.34 (m, 1H, H-4Cer), 5.06 (dd, 1H, J1,2 7.9 Hz, J2,3 9.3 Hz, H-2I), 4.63–4.52 (m, 3H, ArCH2, H-2Cer), 4.41 (d, 1H, H-1I), 4.34–4.26 (2 d, 4H, Jgem 11.5 Hz, ArCH2), 3.99 (dd, 1H, J1,2 3.4 Hz, Jgem 10.1 Hz, H-1aCer), 3.79, 3.70 (2 s, 6H, 2OCH3), 3.68–3.56 (m, 3H, H-3I, H-4I, H-1bCer), 3.47–3.37 (m, 3H, H-5I, H-6aI, H-6bI), 2.87 (s, 1H, OH), 1.91–1.73 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.46–1.09 (m, 66H, 33CH2), 0.94–0.86 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 172.9, 166.0, 165.2, 165.0, 159.3, 159.1, 133.2, 133.0, 129.8, 129.8, 129.7, 129.6, 129.6, 129.5, 129.3, 128.4, 128.4, 128.3, 113.8, 113.7, 100.6, 81.1, 73.9, 73.7, 73.5, 73.3, 73.1, 72.8, 70.3, 66.8, 55.2, 55.1, 47.8, 36.3, 31.9, 29.7, 29.7, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.2, 25.5, 25.4, 22.7, 14.1; HRMS (ESI) m/z: found [M + Na]+ 1404.8831, C85H123NO14 calcd for [M + Na]+ 1404.8836.
:1). The reaction mixture was quenched with satd aq. NaHCO3 and filtered through a pad of Celite, and then washed with CHCl3. The combined filtrate and washings were washed with satd aq. Na2S2O3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by gel filtration (Sephadex LH-20, CHCl3/MeOH 1:1) and silica gel column chromatography (n-hexane/EtOAc 4:3 → 1:1) to give 21 (122 mg, 72%) as a white solid; [α]D +28.0° (c 2.4, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.03–6.60 (m, 50H, Ar), 6.02 (d, 1H, JNH,2 = 8.9 Hz, NHCer), 5.53–5.49 (m, 3H, H-4IV, NHIII, H-3Cer), 5.45 (d, 1H, J3,4 3.4 Hz, H-4III), 5.40 (dd, 1H, J1,2 7.9 Hz, J2,3 10.5 Hz, H-2IV), 5.34–5.30 (m, 2H, H-2II, H-4Cer), 5.17 (dd, 1H, J3,4 3.4 Hz, H-3IV), 5.09 (d, 1H, J1,2 8.3 Hz, H-1III), 5.05–4.97 (m, 2H, H-2I, H-3III), 4.76 (d, 1H, H-1IV), 4.73 (d, 1H, Jgem 10.5 Hz, ArCH2), 4.59 (dd, 1H, J5,6a 5.1 Hz, Jgem 11.8 Hz, H-6aII), 4.55–4.46 (m, 5H, H-2Cer, H-1II, H-6aIV, ArCH2), 4.31–4.27 (m, 4H, ArCH2, H-1I, H-6bIV), 4.11–4.05 (m, 2H, H-5IV, H-6bIII), 4.02–3.88 (m, 5H, H-4II, ArCH2, H-4I, H-1aCer, H-6aIII), 3.84 (dd, 1H, J1b,2 6.9 Hz, Jgem 11.5 Hz, H-1bCer), 3.74 (t, 1H, J5,6a, J5,6b 6.1 Hz, H-5III), 3.69–3.64 (m, 7H, 2OCH3, H-3I), 3.55 (dd, 1H, Jgem 10.3 Hz, H-6bIII), 3.46 (t, 1H, J5,6a, J5,6b 6.0 Hz, H-5II), 3.42 (dd, 1H, J5,6a 3.8 Hz, Jgem 11.4 Hz, H-6aI), 3.35–3.30 (m, 2H, H-3II, H-6bI), 3.19–3.13 (m, 2H, H-5I, H-2III), 2.62–2.31 (m, 4H, CH3COCH2CH2COO), 2.18–1.93 (4 s, 12H, 3Ac, CH3COCH2CH2COO), 1.83–1.69 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.60–1.00 (m, 66H, 33CH2), 0.99–0.81 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 172.9, 172.3, 171.7, 170.4, 170.2, 170.0, 166.2, 165.9, 165.8, 165.1, 165.0, 164.9, 164.9, 159.1, 158.9, 137.3, 133.4, 133.1, 132.9, 132.8, 130.3, 130.2, 130.0, 129.9, 129.7, 129.7, 129.6, 129.5, 129.5, 129.4, 129.2, 128.6, 128.4, 128.4, 128.3, 128.3, 128.1, 127.9, 113.6, 113.4, 101.7, 100.6, 100.2, 99.3, 80.0, 76.2, 75.0, 74.8, 73.9, 73.6, 73.3, 73.1, 72.9, 72.3, 71.9, 71.4, 71.0, 70.9, 70.6, 69.4, 69.2, 67.5, 67.0, 66.7, 63.7, 62.5, 61.4, 55.4, 55.1, 55.0, 47.7, 37.6, 36.2, 31.9, 29.7, 29.7, 29.6, 29.5, 29.5, 29.4, 29.4, 29.3, 29.2, 27.7, 27.7, 25.4, 25.3, 23.0, 22.6, 21.4, 20.7, 20.7, 20.5, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2662.2887, C151H190N2O38 calcd for [M + Na]+ 2662.2889.
:1), the reaction mixture was diluted with CHCl3. The organic layer was successively washed with ice-cooled satd aq. NaHCO3 and brine. The organic solution was dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (n-hexane/EtOAc 4:3) to give 22 (100 mg, 92%) as a white solid; [α]D +40.0° (c 0.9, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.05–7.06 (m, 40H, Ph), 6.31 (d, 1H, JNH,2 9.7 Hz, NHCer), 5.60 (dd, 1H, J2,3 9.3 Hz, J3,4 2.8 Hz, H-3Cer), 5.52–5.50 (m, 2H, H-4IV, NHIII), 5.45–5.32 (m, 4H, H-4III, H-2IV, H-2II, H-4Cer), 5.19 (dd, 1H, J2,3 10.5 Hz, J3,4 3.5 Hz, H-3IV), 5.08 (d, 1H, J1,2 8.3 Hz, H-1III), 5.05 (dd, 1H, J1,2 8.1 Hz, J2,3 9.6 Hz, H-2I), 4.97 (dd, 1H, J2,3 10.9 Hz, J3,4 3.4 Hz, H-3III), 4.76 (d, 1H, J1,2 7.9 Hz, H-1IV), 4.73 (dd, 1H, J5,6a 2.6 Hz, Jgem 12.6 Hz, H-6aII), 4.53–4.49 (m, 5H, H-1II, H-2Cer, H-6aIV, ArCH2, OH), 4.35 (d, 1H, J1,2 7.9 Hz, H-1I), 4.32–4.21 (m, 3H, ArCH2, H-6bIV, H-6bII), 4.07 (t, 1H, J5,6a, J5,6b 6.9 Hz, H-5IV), 4.02 (dd, 1H, J5,6a 5.1 Hz, Jgem 11.6 Hz, H-6aIII), 3.92–3.88 (m, 2H, H-4II, H-6bIII), 3.83 (t, 1H, J2,3, J3,4 9.3 Hz, H-3I), 3.78–3.75 (m, 3H, H-1aCer, H-5II, H-5III), 3.64 (t, 1H, J3,4, J4,5 9.2 Hz, H-4I), 3.50–3.45 (m, 2H, H-1bCer, H-3II), 3.17–3.10 (m, 2H, H-2III, H-6aI), 3.02 (br d, 1H, J5,6a 9.5 Hz, H-5I), 2.90 (br d, 1H, Jgem 11.7 Hz, H-6bI), 2.67 (dd, 1H, JOH,6a 3.9 Hz, JOH,6b 10.4 Hz, OH), 2.62–2.21 (m, 4H, CH3COCH2CH2CO), 2.20–2.00 (4 s, 12H, 3Ac, CH3COCH2CH2CO), 1.97–1.40 (m, 7H, H-5aCer, H-5bCer, NHCOCH2, Ac), 1.38–1.17 (m, 66 H, 33CH2), 0.91–0.85 (m, 6 H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.7, 173.0, 171.7, 170.5, 170.2, 169.8, 166.5, 166.1, 165.8, 165.6, 165.0, 164.9, 136.8, 133.2, 132.8, 130.0, 129.9, 129.8, 129.7, 129.6, 129.5, 129.4, 129.3, 129.3, 128.6, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 101.7, 99.5, 99.2, 80.4, 77.6, 73.9, 73.7, 73.1, 72.2, 71.9, 71.0, 70.7, 69.4, 69.3, 64.1, 62.6, 61.5, 55.3, 48.0, 37.7, 36.5, 31.9, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.3, 29.2, 28.4, 27.8, 25.5, 23.0, 22.6, 20.8, 20.7, 20.7, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2422.1740, C135H174N2O36 calcd for [M + Na]+ 2422.1739.
:1) to give 23 (63 mg, 98%) as a white solid; [α]D +19.0° (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.05–7.06 (m, 40H, Ph), 6.04–6.02 (m, 2H, NHIII, NHCer), 5.53–5.50 (m, 2H, H-3Cer, H-4IV), 5.45–5.42 (m, 2H, H-4III, H-2IV), 5.32–5.23 (m, 3H, H-4Cer, H-3I, H-2II), 5.20 (dd, 1H, J2,3 10.5 Hz, J3,4 3.4 Hz, H-3IV), 5.14 (d, 1H, J1,2 8.3 Hz, H-1III), 5.05 (dd, 1H, J2,3 10.9 Hz, J3,4 3.4 Hz, H-3III), 4.98 (dd, 1H, J1,2 7.8 Hz, J2,3 9.7 Hz, H-2I), 4.79 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.58 (dd, 1H, J5,6a 4.5 Hz, Jgem 12.0 Hz, H-6aII), 4.53–4.48 (m, 3H, H-1II, H-2Cer, H-6aIV), 4.34–4.24 (m, 5H, H-1I, ArCH2, H-6bIV, H-6bII), 4.07 (t, 1H, J5,6a, J5,6b 6.8 Hz, H-5IV), 3.98–3.85 (m, 5H, H-6aIII, H-6aI, H-4II, H-1aCer, H-6bIII), 3.80–3.73 (m, 2H, H-6bI, H-5III), 3.66 (dd, 1H, J5,6a 4.7 Hz, J5,6b 8.0 Hz, H-5II), 3.60 (t, 1H, J3,4, J4,5 9.5 Hz, H-4I), 3.49–3.43 (m, 2H, H-1bCer, H-3II), 3.37–3.34 (m, 1H, H-5I), 3.13–3.09 (m, 1H, H-2III), 2.62–2.33 (m, 4H, CH3COCH2CH2COO), 2.19–1.89 (5 s, 15H, 4Ac, CH3COCH2CH2COO), 1.88–1.75 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.62 (s, 3H, Ac), 1.46–1.13 (m, 69H, 33CH2, Ac), 0.91–0.86 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 172.8, 172.6, 171.6, 170.4, 170.2, 170.2, 169.8, 166.2, 166.0, 165.8, 165.1, 165.0, 164.8, 164.6, 137.0, 133.4, 133.3, 133.1, 133.0, 132.8, 130.0, 129.8, 129.7, 129.7, 129.6, 129.5, 129.4, 129.3, 129.1, 128.6, 128.6, 128.4, 128.3, 128.2, 128.1, 127.9, 101.6, 100.3, 100.0, 98.8, 78.1, 75.0, 74.4, 73.6, 72.7, 72.6, 72.1, 71.9, 71.8, 71.7, 71.1, 71.0, 70.9, 70.6, 69.3, 67.1, 66.8, 63.7, 62.5, 61.5, 55.4, 47.7, 37.6, 36.3, 31.9, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.4, 29.3, 29.2, 28.8, 27.7, 25.4, 25.4, 23.0, 22.6, 20.7, 20.7, 20.7, 20.6, 20.3, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2506.1951, C139H178N2O38 calcd for [M + Na]+ 2506.1950.
:1), the reaction mixture was filtered through a pad of Celite and the precipitate was washed with EtOH. The filtrate was concentrated and the residue obtained was purified by silica gel column chromatography (CHCl3/MeOH 80:1) to give 24 (31 mg, 77%) as a white solid; [α]D +20.5° (c 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.05–7.33 (m, 35 H, Ar), 6.08 (d, 1 H, JNH,2 9.3 Hz, NHCer), 6.01 (d, 1H, JNH,2 6.5 Hz, NHIII), 5.54–5.49 (m, 3H, H-3Cer, H-1III, H-4IV), 5.44–5.41 (m, 2H, H-4III, H-2IV), 5.32–5.20 (m, 3H, H-4IV, H-3I, H-3IV), 5.01–4.94 (m, 2H, H-2II, H-2I), 4.75 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.59 (dd, 1H, J5,6a 4.6 Hz, Jgem 11.9 Hz, H-6aII), 4.52–4.45 (m, 3H, H-2Cer, H-3III, H-6aIV), 4.39 (d, 1H, J1,2 7.7 Hz, H-1II), 4.33–4.26 (m, 3H, H-1I, H-6bIV, H-6bII), 4.11–4.07 (m, 2H, H-5IV, H-4II), 4.01–3.94 (m, 2H, H-6aIII, H-6bIII), 3.90–3.83 (m, 3H, H-6aI, H-5III, H-1aCer), 3.77 (dd, 1H, J5,6a 4.8 Hz, J5,6b 7.2 Hz, H-5II), 3.73–3.64 (m, 3H, H-4I, H-1bCer, H-3II), 3.38 (dd, 1H, J5,6a 3.3 Hz, J5,6b 9.9 Hz, H-5I), 3.25 (d, 1H, JOH,3 10.1 Hz, OH), 3.14 (m, 1H, H-2III), 2.62–2.32 (m, 4H, CH3COCH2CH2COO), 2.19, 2.15, 1.99, 1.94, 1.89 (5 s, 15H, 4Ac, CH3COCH2CH2COO), 1.84–1.76 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.71, 1.59 (2 s, 6H, 2Ac), 1.52–1.13 (m, 66H, 33CH2), 0.94–0.85 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 173.2, 172.8, 171.7, 170.4, 170.3, 170.2, 169.7, 166.2, 166.1, 165.8, 165.1, 165.0, 164.8, 133.7, 133.5, 133.2, 133.1, 132.9, 130.0, 129.9, 129.8, 129.7, 129.7, 129.6, 129.5, 129.5, 129.2, 129.1, 129.1, 128.7, 128.7, 128.6, 128.5, 128.4, 128.3, 101.8, 100.1, 98.9, 75.5, 75.0, 74.6, 73.7, 73.1, 72.7, 72.5, 72.3, 72.0, 71.8, 71.7, 70.9, 69.6, 69.5, 67.0, 67.0, 63.6, 62.4, 62.1, 61.5, 55.9, 47.7, 37.6, 36.3, 31.9, 30.0, 29.7, 29.7, 29.6, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.2, 28.7, 27.7, 25.5, 25.4, 22.8, 22.7, 20.8, 20.8, 20.7, 20.6, 20.4, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2416.1478, C132H172N2O38 calcd for [M + Na]+ 2416.1480.
:1), the reaction was quenched with triethylamine, and pyridine was co-evaporated with toluene. The residue was purified by silica gel column chromatography (CHCl3/MeOH/Et3N 120:1:1.2) to give 25 (9.8 mg, 91%) as a white solid; [α]D +9.8° (c 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 10.3 (br s, 1H, Et3NH), 8.04–7.33 (m, 35H, Ar), 7.12 (d, 1H, JNH,2 7.3 Hz, NHIII), 6.01 (d, 1H, JNH,2 9.2 Hz, NHCer), 5.50–5.48 (m, 2H, H-3Cer, H-4IV), 5.40–5.35 (m, 2H, H-4III, H-2IV), 5.30–5.27 (m, 1H, H-4Cer), 5.22 (t, 1H, J2,3, J3,4 9.5 Hz, H-3I), 5.15 (dd, 1H, J2,3 10.5 Hz, J3,4 3.4 Hz, H-3IV), 5.00 (dd, 1H, J1,2 8.4 Hz, J2,3 9.8 Hz, H-2II), 4.96–4.92 (m, 2H, H-1III, H-2I), 4.79 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.60–4.58 (m, 3H, H-6aII, H-3II, H-4II), 4.50–4.46 (m, 2H, H-2Cer, H-6aIV), 4.42 (d, 1H, J1,2 8.1 Hz, H-1II), 4.33–4.26 (m, 3H, H-1I, H-6bIV, H-6bII), 4.08 (t, 1H, J5,6a, J5,6b 6.8 Hz, H-5IV), 4.04–4.00 (m, 2H, H-6aI, H-3III), 3.87–3.74 (m, 6H, H-6aIII, H-6bIII, H-2III, H-5II, H-1aCer, H-6aI), 3.68–3.62 (m, 2H, H-5III, H-4I), 3.47 (dd, 1H, J1b,2 3.7 Hz, Jgem 10.1 Hz, H-1bCer), 3.34–3.31 (m, 1H, H-5I), 3.07–3.03 (m, 6H, 3NCH2CH3), 2.61–2.30 (m, 4H, CH3COCH2CH2CO), 2.18–1.68 (6 s, 18H, 5Ac, CH3COCH2CH2CO), 1.76–1.71 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.49–1.17 (m, 69H, 33CH2, Ac), 0.89–0.83 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.7, 172.8, 171.6, 170.6, 170.1, 170.0, 169.4, 166.0, 165.9, 165.0, 165.0, 164.9, 133.4, 133.3, 133.1, 132.9, 130.0, 129.7, 129.6, 129.6, 129.4, 129.1, 128.6, 128.6, 128.5, 128.5, 128.4, 128.3, 101.7, 99.8, 77.6, 76.0, 73.5, 72.8, 72.3, 71.3, 71.1, 70.9, 69.8, 69.4, 67.2, 62.6, 61.7, 47.7, 46.0, 37.6, 36.3, 31.9, 30.0, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.5, 29.4, 29.3, 29.2, 28.8, 27.7, 25.4, 25.4, 22.8, 22.7, 20.7, 20.6, 20.6, 20.4; HRMS (ESI) m/z: found [M − H]− 2472.1078, C138H186N2O41S calcd for [M − H]− 2472.1083.
:9:2), the reaction mixture was concentrated. The residue was purified by gel filtration (Sephadex LH-20, CHCl3/MeOH/H2O 5:4:1) to give 1 (4.5 mg, 82%); [α]D +15.0° (c 0.5, CHCl3/MeOH/H2O 5:4:1); 1H NMR (500 MHz, CD3OD) δ 4.56 (d, 1H, J1,2 8.6 Hz, H-1III), 4.33 (d, 1H, J1,2 7.8 Hz, H-1), 4.27 (d, 1H, J1,2 7.6 Hz, H-1), 4.21 (d, 1H, J1,2 7.8 Hz, H-1), 4.13 (dd, 1H, J2,3 10.6 Hz, H-2III), 3.94 (d, 1H, J3,4 2.4 Hz, H-4III), 2.11 (t, 2H, NHCOCH2CH2), 1.95 (s, 3H, Ac), 1.53–1.15 (m, 68H, 34CH2), 0.80 (2 t, 6H, 2CH3); 13C NMR (200 MHz, DMSO-d6) δ 171.9, 171.0, 104.6, 103.4, 103.3, 81.0, 80.7, 78.0, 75.9, 75.3, 75.2, 74.8, 73.8, 73.4, 73.2, 73.0, 72.3, 70.7, 70.6, 69.8, 69.6, 69.1, 68.2, 67.2, 65.2, 62.8, 60.8, 60.5, 60.2, 59.4, 39.9, 31.4, 31.3, 30.7, 29.3, 29.28, 29.21, 29.20, 29.16, 29.1, 29.0, 28.8, 28.73, 28.70, 25.5, 25.4, 23.3, 22.2, 22.1, 14.0; HRMS (ESI) m/z: found [M − H]− 1435.8351, C138H186N2O41S calcd for [M − H]− 1435.8352.
:1), the reaction mixture was diluted with EtOAc and successively washed with satd aq. NH4Cl and brine. The organic layer was then dried over Na2SO4 and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 60:1) to give 26 (42 mg, 93%) as a colorless syrup; [α]D +19.8° (c 0.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.07–7.33 (m, 35H, Ph), 6.34 (d, 1H, J2,NH 6.3 Hz, NHIII), 6.22 (d, 1H, J2,NH 9.2 Hz, NHCer), 5.55 (dd, 1H, J2,3 8.6 Hz, J3,4 3.3 Hz, H-3Cer), 5.48 (d, 1H, J1,2 7.9 Hz, H-1III), 5.43 (2 d, 2H, J3,4 3.5 Hz, H-4IV, H-4III), 5.29 (m, 1H, H-4Cer), 5.24, 5.23 (2 t, 2H, H-3I, H-2IV), 4.99 and 4.98 (2 t, J1,2 7.8 Hz, 2H, H-2III, H-2II), 4.71 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.60 (dd, 1H, Jgem 11.8 Hz, J5,6a 4.6 Hz, H-6aII), 4.52–4.45 (m, 3H, H-2II, H-3III, H-6aIV), 4.37 (d, 1H, H-1II), 4.32 (d, 1H, J1,2 7.7 Hz, H-1I), 4.31–4.28 (m, 3H, H-6bIV, H-6bII), 4.09 (d, 1H, J3,4 2.4 Hz, H-4II), 4.01–3.93 (m, 4H, H-3IV, H-5IV, H-6aIII, H-5III), 3.86–3.77 (m, 4H, OH, H-1aCer, H-6bIII, H-5II), 3.71–3.67 (m, 2H, H-3II, H-4I), 3.63 (t, 1H, Jgem 9.5 Hz, J5,6a 9.5 Hz, H-6aI), 3.47 (dd, 1H, Jgem 9.7 Hz, J1b,2 3.5 Hz, H-1bCer), 3.37–3.32 (m, 2H, H-6bI, H-5I), 3.14 (m, 1H, H-2III), 2.66 (d, 1H, J3,OH 5.5 Hz, OH), 2.19–1.74 (m, 16H, 4Ac, 2CH2), 1.68 and 1.64 (2 s, 6H, 2Ac), 1.47–1.12 (m, 66H, 33CH2), 0.88, 0.87 (2 t, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 173.3, 172.9, 171.1, 170.5, 170.4, 170.3, 169.7, 166.3, 166.2, 166.1, 165.9, 165.8, 165.2, 164.8, 133.7, 133.2, 133.1, 132.9, 130.1, 129.9, 129.85, 129.80, 129.76, 129.71, 129.70, 129.6, 129.5, 129.4, 129.3, 129.1, 128.7, 128.6, 128.5, 128.44, 128.40, 128.3, 101.5, 100.2, 99.1, 77.6, 75.3, 74.9, 74.7, 73.8, 73.3, 73.1, 73.0, 72.7, 72.4, 72.3, 72.0, 71.8, 71.7, 71.3, 71.0, 69.7, 69.6, 67.0, 63.7, 62.5, 62.1, 62.0, 55.9, 47.8, 36.3, 31.9, 29.74, 29.65, 29.60, 29.56, 29.55, 29.49, 29.35, 29.2, 28.6, 25.5, 25.4, 23.0, 22.7, 20.8, 20.7, 20.6, 20.4, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2318.1113, C127H166N2O36 calcd for [M + Na]+ 2318.1113.
:1), the reaction was quenched with triethylamine. Pyridine was then co-evaporated with toluene. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH/Et3N 30:1:0.2) and then gel filtration (Sephadex LH-20, CHCl3/MeOH 1:1) was performed to give 27 (37 mg, 96%) as a white solid; [α]D +9.8° (c 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 9.50 (br s, 2H, Et3NH), 7.99–7.27 (m, 35H, Ph), 6.97 (d, 1H, J2, NH 9.0 Hz, NHIII), 5.98 (d, 1H, J2, NH 9.2 Hz, NHCer), 5.70 (d, 1H, J3,4 3.3 Hz, H-4III), 5.42 (dd, 1H, H-3Cer), 5.31 (d, 1H, J3,4 3.1 Hz, H-4III), 5.25 (dd, 1H, J1,2 7.8 Hz, J2,3 10.1 Hz, H-2IV), 5.22 (m, 1H, H-4Cer), 5.15 (t, 1H, J2,3, J3,4 9.5 Hz, H-3I), 4.92 (dd, 1H, J1,2 7.8 Hz, J3,4 9.7 Hz, H-2I), 4.76 (d, 1H, J1,2 7.4 Hz, H-1III), 4.70 (d, 1H, H-1IV), 4.61 (dd, 1H, H-3IV), 4.53 (dd, 1H, Jgem 12.3 Hz, J5,6a 3.3 Hz, H-6aIII), 4.49–4.45 (m, 2H, H-3II, H-4II), 4.41 (m, 1H, H-2Cer), 4.33 (d, 1H, H-1II), 4.31–4.19 (m, 4H, H-1I, H-3II, H-6bIII, H-6aII), 3.96 (t, 1H, J5,6 6.5 Hz, H-5III), 3.93 (near d, 1H, Jgem 10.7 Hz, H-6aIV), 3.86–3.70 (m, 5H, H-2III, H-1aCer, H-6aI, H-6bIV, H-5II), 3.57–3.52 (m, 3H, H-4I, H-6bI, H-5IV), 3.40 (dd, 1H, Jgem 13.0 Hz, J1b,2 4.7 Hz, H-1bCer), 3.27 (m, 1H, H-5I), 3.01 (2 q, 12H, 2NCH2CH3), 2.19–1.60 (m, 19H, 5Ac, 2CH2), 1.39–1.04 (m, 89H, Ac, 34CH2, 6NCH2CH3), 0.80 (2 t, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 173.0, 172.9, 170.6, 170.2, 170.0, 169.9, 169.6, 167.7, 166.2, 166.1, 166.0, 165.3, 165.2, 165.1, 164.9, 133.5, 133.4, 133.1, 133.0, 132.9, 132.4, 130.9, 130.1, 130.0, 129.9, 129.8, 129.7, 129.66, 129.64, 129.55, 129.53, 129.4, 129.0, 128.7, 128.6, 128.5, 128.48, 128.40, 128.36, 128.30, 128.25, 103.0, 102.0, 101.2, 99.8, 78.1, 77.7, 77.3, 76.4, 76.2, 74.9, 73.7, 72.8, 72.7, 72.3, 71.5, 71.2, 71.1, 70.2, 69.8, 69.5, 68.6, 68.1, 66.9, 64.3, 62.8, 62.5, 61.8, 51.5, 47.7, 46.9, 46.8, 46.5, 38.7, 36.2, 31.9, 30.3, 30.0, 29.7, 29.6, 29.5, 29.49, 29.47, 29.4, 29.3, 29.2, 29.1, 28.9, 28.4, 25.4, 25.3, 23.7, 22.9, 22.6, 22.3, 20.8, 20.7, 20.6, 202, 14.1, 14.0, 10.9, 9.0, 8.8, 8.6; HRMS (ESI) m/z: found [M − H + Na]− 2476.0103, C127H164N2O42S2 calcd for [M − H + Na]− 2476.0103.
:45:10), the reaction mixture was concentrated. The resulting residue was purified by silica gel column chromatography (Iatrobeads, CHCl3/MeOH/H2O 6:4:0 → 6:4:0.5) to give 2 (28 mg, 96%); [α]D +13.0° (c 0.5, CHCl3/MeOH/H2O 5:4:1); 1H NMR (500 MHz, CD3OD) δ 4.47 (d, 1H, J1,2 7.1 Hz, H-1), 4.43 (d, 1H, J1,2 7.6 Hz, H-1), 4.30 (2 d, 2H, J1,2 7.9 Hz, 2 H-1), 2.21 (t, 2H, Jgem, J1,2 7.2 Hz, H-1aCer, H-1bCer), 2.07 (br s, 3H, Ac), 1.63–1.16 (m, 68H, 34CH2), 0.89 (2 t, 6H, 2CH3); 13C NMR (200 MHz, DMSO-d6) δ 171.9, 170.4, 104.3, 103.6, 103.5, 103.4, 80.8, 80.2, 79.1, 77.9, 75.7, 75.1, 74.9, 74.8, 74.7, 74.3, 73.9, 73.5, 73.3, 70.6, 69.8, 69.5, 69.04, 69.02, 67.3, 66.2, 60.8, 60.24, 60.18, 59.4, 50.5, 50.2, 50.1, 39.9, 35.6, 35.5, 31.34, 31.32, 30.7, 29.32, 29.27, 29.20, 29.19, 19.15, 29.09, 29.04, 29.03, 29.01, 29.00, 28.8, 28.7, 28.6, 25.5, 25.4, 23.34, 23.30, 22.13, 22.12, 14.0; HRMS (ESI) m/z: found [M − H + Na]− 1537.7741, C68H126N2O30S2 calcd for [M − H + Na]− 1537.7740.
NH), 8.05–7.14 (m, 25H, Ph), 6.51 (d, 1H, J1,2 3.6 Hz, H-1II), 5.64 (dd, 1H, J2,3 10.4 Hz, H-2II), 5.51–5.49 (m, 2H, NHII, H-4III), 5.21 (dd, 1H, J2,3 10.5 Hz, J3,4 3.5 Hz, H-3IV), 5.17 (d, 1H, J1,2 8.3 Hz, H-1III), 4.98 (dd, 1H, J2,3 10.8 Hz, J3,4 3.5 Hz, H-3III), 4.77 (d, 1H, J1,2 7.9 Hz, H-1IV), 4.63–4.58 (m, 2H, H-6aIV, PhCH2), 4.52 (dd, 1H, J5,6b 6.4 Hz, Jgem 11.3 Hz, H-6aII), 4.43 (d, 1H, Jgem 12.5 Hz, PhCH2), 4.38–4.29 (m, 3H, H-5IV, H-6bIV, H-6bII), 4.15 (d, 1H, J3,4 2.0 Hz, H-4II), 4.09–4.00 (m, 3H, H-3II, H-5II, H-6aIII), 3.91 (dd, 1H, J5,6b 7.2 Hz, Jgem 11.6 Hz, H-6bIII), 3.78 (t, 1H, H-5III), 3.24–3.19 (m, 1H, H-2III), 2.64–2.34 (m, 4H, CH2CH2), 2.20–1.36 (5 s, 15H, 4Ac, CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 172.1, 171.7, 170.4, 170.2, 169.9, 166.2, 165.8, 165.3, 164.9, 160.2, 137.1, 133.5, 133.3, 133.1, 133.0, 129.8, 129.7, 129.6, 129.5, 129.5, 129.4, 129.2, 128.6, 128.4, 128.3, 128.3, 127.9, 101.7, 99.2, 94.1, 90.9, 74.7, 74.0, 73.9, 71.9, 71.6, 71.0, 70.9, 70.7, 69.4, 69.2, 68.5, 67.0, 64.5, 62.6, 61.4, 60.3, 55.3, 37.6, 29.6, 29.4, 27.7, 23.0, 20.8, 20.7, 20.6; HRMS (ESI) m/z: found [M + Na]+ 1441.3147, C68H69Cl3N2O25 calcd for [M + Na]+ 1441.3147.
:1). The reaction mixture was quenched with satd aq. NaHCO3 and filtered through a pad of Celite, and then washed with CHCl3. The combined filtrate and washings were washed with satd aq. Na2S2O3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (toluene/EtOAc 30:1) to give 20 (150 mg, 71%) as a white solid; [α]D +35.0° (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.01–7.35 (m, 15H, Ph), 7.27–6.60 (4 d, 8H, Ar), 6.16 (d, 1H, JNH,2 = 9.0 Hz, NH), 5.58 (dd, 1H, J2,3 7.8 Hz, J3,4 3.5 Hz, H-3Cer), 5.38–5.35 (m, 1H, H-4Cer), 5.11 (near t, 1H, J1,2, J2,3 8.5 Hz, H-2I), 4.56–4.47 (m, 4H, ArCH2, H-1I, H-2Cer), 4.30–4.15 (2 d, 4H, Jgem 11.7 Hz, ArCH2), 4.03 (dd, 1H, J1,2 3.6 Hz, Jgem 10.1 Hz, H-1aCer), 3.78 and 3.68 (2 s, 6H, OCH3), 3.64 (dd, 1H, H-1bCer), 3.59–3.55 (m, 3H, H-3I, H-4I, H-6aI), 3.41 (t, 1H, J5,6a, J5,6b 6.4 Hz, H-5I), 3.32 (dd, 1H, Jgem 10.6 Hz, H-6bI), 1.88–1.70 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.60–1.09 (m, 66H, 33CH2), 0.89–0.83 (m, 6H, 2CH3), 0.81 (s, 9H, tBu), −0.03, −0.05 (s, 6H, 2SiCH3); 13C NMR (125 MHz, CDCl3) δ 174.0, 166.0, 165.2, 165.1, 159.1, 158.8, 133.1, 132.9, 132.8, 130.3, 130.2, 130.1, 130.0, 129.8, 129.8, 129.7, 129.6, 129.1, 129.0, 128.3, 128.3, 113.7, 113.4, 100.3, 82.7, 74.4, 74.2, 73.7, 73.2, 70.9, 68.9, 66.4, 55.2, 55.0, 47.9, 36.3, 31.9, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.2, 29.1, 25.9, 25.5, 25.4, 22.7, 17.9, 14.1, −3.8, −4.8; HRMS (ESI) m/z: found [M + Na]+ 1518.9706, C73H75NO26 calcd for [M + Na]+ 1518.9701.
:1), the reaction mixture was concentrated. The residue was purified by silica gel column chromatography (n-hexane/EtOAc 2:1) to give 4 (73 mg, 87%) as a white solid; [α]D +19.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.00–7.26 (m, 15H, Ph), 7.16–6.65 (4 d, 8H, Ar), 6.09 (d, 1H, JNH,2 9.1 Hz, NH), 5.54 (dd, 1H, J2,3 7.8 Hz, J3,4 3.7 Hz, H-3Cer), 5.37–5.34 (m, 1H, H-4Cer), 5.06 (dd, 1H, J1,2 7.9 Hz, J2,3 9.3 Hz, H-2I), 4.63–4.52 (m, 3H, ArCH2, H-2Cer), 4.41 (d, 1H, H-1I), 4.34–4.26 (2 d, 4H, Jgem 11.5 Hz, ArCH2), 3.99 (dd, 1H, J1,2 3.4 Hz, Jgem 10.1 Hz, H-1aCer), 3.79, 3.70 (2 s, 6H, 2OCH3), 3.68–3.56 (m, 3H, H-3I, H-4I, H-1bCer), 3.47–3.37 (m, 3H, H-5I, H-6aI, H-6bI), 2.87 (s, 1H, OH), 1.91–1.73 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.46–1.09 (m, 66H, 33CH2), 0.94–0.86 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 172.9, 166.0, 165.2, 165.0, 159.3, 159.1, 133.2, 133.0, 129.8, 129.8, 129.7, 129.6, 129.6, 129.5, 129.3, 128.4, 128.4, 128.3, 113.8, 113.7, 100.6, 81.1, 73.9, 73.7, 73.5, 73.3, 73.1, 72.8, 70.3, 66.8, 55.2, 55.1, 47.8, 36.3, 31.9, 29.7, 29.7, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.2, 25.5, 25.4, 22.7, 14.1; HRMS (ESI) m/z: found [M + Na]+ 1404.8831, C85H123NO14 calcd for [M + Na]+ 1404.8836.
:1). The reaction mixture was quenched with satd aq. NaHCO3 and filtered through a pad of Celite, and then washed with CHCl3. The combined filtrate and washings were washed with satd aq. Na2S2O3 and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by gel filtration (Sephadex LH-20, CHCl3/MeOH 1:1) and silica gel column chromatography (n-hexane/EtOAc 4:3 → 1:1) to give 21 (122 mg, 72%) as a white solid; [α]D +28.0° (c 2.4, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.03–6.60 (m, 50H, Ar), 6.02 (d, 1H, JNH,2 = 8.9 Hz, NHCer), 5.53–5.49 (m, 3H, H-4IV, NHIII, H-3Cer), 5.45 (d, 1H, J3,4 3.4 Hz, H-4III), 5.40 (dd, 1H, J1,2 7.9 Hz, J2,3 10.5 Hz, H-2IV), 5.34–5.30 (m, 2H, H-2II, H-4Cer), 5.17 (dd, 1H, J3,4 3.4 Hz, H-3IV), 5.09 (d, 1H, J1,2 8.3 Hz, H-1III), 5.05–4.97 (m, 2H, H-2I, H-3III), 4.76 (d, 1H, H-1IV), 4.73 (d, 1H, Jgem 10.5 Hz, ArCH2), 4.59 (dd, 1H, J5,6a 5.1 Hz, Jgem 11.8 Hz, H-6aII), 4.55–4.46 (m, 5H, H-2Cer, H-1II, H-6aIV, ArCH2), 4.31–4.27 (m, 4H, ArCH2, H-1I, H-6bIV), 4.11–4.05 (m, 2H, H-5IV, H-6bIII), 4.02–3.88 (m, 5H, H-4II, ArCH2, H-4I, H-1aCer, H-6aIII), 3.84 (dd, 1H, J1b,2 6.9 Hz, Jgem 11.5 Hz, H-1bCer), 3.74 (t, 1H, J5,6a, J5,6b 6.1 Hz, H-5III), 3.69–3.64 (m, 7H, 2OCH3, H-3I), 3.55 (dd, 1H, Jgem 10.3 Hz, H-6bIII), 3.46 (t, 1H, J5,6a, J5,6b 6.0 Hz, H-5II), 3.42 (dd, 1H, J5,6a 3.8 Hz, Jgem 11.4 Hz, H-6aI), 3.35–3.30 (m, 2H, H-3II, H-6bI), 3.19–3.13 (m, 2H, H-5I, H-2III), 2.62–2.31 (m, 4H, CH3COCH2CH2COO), 2.18–1.93 (4 s, 12H, 3Ac, CH3COCH2CH2COO), 1.83–1.69 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.60–1.00 (m, 66H, 33CH2), 0.99–0.81 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 172.9, 172.3, 171.7, 170.4, 170.2, 170.0, 166.2, 165.9, 165.8, 165.1, 165.0, 164.9, 164.9, 159.1, 158.9, 137.3, 133.4, 133.1, 132.9, 132.8, 130.3, 130.2, 130.0, 129.9, 129.7, 129.7, 129.6, 129.5, 129.5, 129.4, 129.2, 128.6, 128.4, 128.4, 128.3, 128.3, 128.1, 127.9, 113.6, 113.4, 101.7, 100.6, 100.2, 99.3, 80.0, 76.2, 75.0, 74.8, 73.9, 73.6, 73.3, 73.1, 72.9, 72.3, 71.9, 71.4, 71.0, 70.9, 70.6, 69.4, 69.2, 67.5, 67.0, 66.7, 63.7, 62.5, 61.4, 55.4, 55.1, 55.0, 47.7, 37.6, 36.2, 31.9, 29.7, 29.7, 29.6, 29.5, 29.5, 29.4, 29.4, 29.3, 29.2, 27.7, 27.7, 25.4, 25.3, 23.0, 22.6, 21.4, 20.7, 20.7, 20.5, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2662.2887, C151H190N2O38 calcd for [M + Na]+ 2662.2889.
:1), the reaction mixture was diluted with CHCl3. The organic layer was successively washed with ice-cooled satd aq. NaHCO3 and brine. The organic solution was dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (n-hexane/EtOAc 4:3) to give 22 (100 mg, 92%) as a white solid; [α]D +40.0° (c 0.9, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.05–7.06 (m, 40H, Ph), 6.31 (d, 1H, JNH,2 9.7 Hz, NHCer), 5.60 (dd, 1H, J2,3 9.3 Hz, J3,4 2.8 Hz, H-3Cer), 5.52–5.50 (m, 2H, H-4IV, NHIII), 5.45–5.32 (m, 4H, H-4III, H-2IV, H-2II, H-4Cer), 5.19 (dd, 1H, J2,3 10.5 Hz, J3,4 3.5 Hz, H-3IV), 5.08 (d, 1H, J1,2 8.3 Hz, H-1III), 5.05 (dd, 1H, J1,2 8.1 Hz, J2,3 9.6 Hz, H-2I), 4.97 (dd, 1H, J2,3 10.9 Hz, J3,4 3.4 Hz, H-3III), 4.76 (d, 1H, J1,2 7.9 Hz, H-1IV), 4.73 (dd, 1H, J5,6a 2.6 Hz, Jgem 12.6 Hz, H-6aII), 4.53–4.49 (m, 5H, H-1II, H-2Cer, H-6aIV, ArCH2, OH), 4.35 (d, 1H, J1,2 7.9 Hz, H-1I), 4.32–4.21 (m, 3H, ArCH2, H-6bIV, H-6bII), 4.07 (t, 1H, J5,6a, J5,6b 6.9 Hz, H-5IV), 4.02 (dd, 1H, J5,6a 5.1 Hz, Jgem 11.6 Hz, H-6aIII), 3.92–3.88 (m, 2H, H-4II, H-6bIII), 3.83 (t, 1H, J2,3, J3,4 9.3 Hz, H-3I), 3.78–3.75 (m, 3H, H-1aCer, H-5II, H-5III), 3.64 (t, 1H, J3,4, J4,5 9.2 Hz, H-4I), 3.50–3.45 (m, 2H, H-1bCer, H-3II), 3.17–3.10 (m, 2H, H-2III, H-6aI), 3.02 (br d, 1H, J5,6a 9.5 Hz, H-5I), 2.90 (br d, 1H, Jgem 11.7 Hz, H-6bI), 2.67 (dd, 1H, JOH,6a 3.9 Hz, JOH,6b 10.4 Hz, OH), 2.62–2.21 (m, 4H, CH3COCH2CH2CO), 2.20–2.00 (4 s, 12H, 3Ac, CH3COCH2CH2CO), 1.97–1.40 (m, 7H, H-5aCer, H-5bCer, NHCOCH2, Ac), 1.38–1.17 (m, 66 H, 33CH2), 0.91–0.85 (m, 6 H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.7, 173.0, 171.7, 170.5, 170.2, 169.8, 166.5, 166.1, 165.8, 165.6, 165.0, 164.9, 136.8, 133.2, 132.8, 130.0, 129.9, 129.8, 129.7, 129.6, 129.5, 129.4, 129.3, 129.3, 128.6, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 101.7, 99.5, 99.2, 80.4, 77.6, 73.9, 73.7, 73.1, 72.2, 71.9, 71.0, 70.7, 69.4, 69.3, 64.1, 62.6, 61.5, 55.3, 48.0, 37.7, 36.5, 31.9, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.3, 29.2, 28.4, 27.8, 25.5, 23.0, 22.6, 20.8, 20.7, 20.7, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2422.1740, C135H174N2O36 calcd for [M + Na]+ 2422.1739.
:1) to give 23 (63 mg, 98%) as a white solid; [α]D +19.0° (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.05–7.06 (m, 40H, Ph), 6.04–6.02 (m, 2H, NHIII, NHCer), 5.53–5.50 (m, 2H, H-3Cer, H-4IV), 5.45–5.42 (m, 2H, H-4III, H-2IV), 5.32–5.23 (m, 3H, H-4Cer, H-3I, H-2II), 5.20 (dd, 1H, J2,3 10.5 Hz, J3,4 3.4 Hz, H-3IV), 5.14 (d, 1H, J1,2 8.3 Hz, H-1III), 5.05 (dd, 1H, J2,3 10.9 Hz, J3,4 3.4 Hz, H-3III), 4.98 (dd, 1H, J1,2 7.8 Hz, J2,3 9.7 Hz, H-2I), 4.79 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.58 (dd, 1H, J5,6a 4.5 Hz, Jgem 12.0 Hz, H-6aII), 4.53–4.48 (m, 3H, H-1II, H-2Cer, H-6aIV), 4.34–4.24 (m, 5H, H-1I, ArCH2, H-6bIV, H-6bII), 4.07 (t, 1H, J5,6a, J5,6b 6.8 Hz, H-5IV), 3.98–3.85 (m, 5H, H-6aIII, H-6aI, H-4II, H-1aCer, H-6bIII), 3.80–3.73 (m, 2H, H-6bI, H-5III), 3.66 (dd, 1H, J5,6a 4.7 Hz, J5,6b 8.0 Hz, H-5II), 3.60 (t, 1H, J3,4, J4,5 9.5 Hz, H-4I), 3.49–3.43 (m, 2H, H-1bCer, H-3II), 3.37–3.34 (m, 1H, H-5I), 3.13–3.09 (m, 1H, H-2III), 2.62–2.33 (m, 4H, CH3COCH2CH2COO), 2.19–1.89 (5 s, 15H, 4Ac, CH3COCH2CH2COO), 1.88–1.75 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.62 (s, 3H, Ac), 1.46–1.13 (m, 69H, 33CH2, Ac), 0.91–0.86 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 172.8, 172.6, 171.6, 170.4, 170.2, 170.2, 169.8, 166.2, 166.0, 165.8, 165.1, 165.0, 164.8, 164.6, 137.0, 133.4, 133.3, 133.1, 133.0, 132.8, 130.0, 129.8, 129.7, 129.7, 129.6, 129.5, 129.4, 129.3, 129.1, 128.6, 128.6, 128.4, 128.3, 128.2, 128.1, 127.9, 101.6, 100.3, 100.0, 98.8, 78.1, 75.0, 74.4, 73.6, 72.7, 72.6, 72.1, 71.9, 71.8, 71.7, 71.1, 71.0, 70.9, 70.6, 69.3, 67.1, 66.8, 63.7, 62.5, 61.5, 55.4, 47.7, 37.6, 36.3, 31.9, 29.7, 29.6, 29.6, 29.5, 29.5, 29.4, 29.4, 29.3, 29.2, 28.8, 27.7, 25.4, 25.4, 23.0, 22.6, 20.7, 20.7, 20.7, 20.6, 20.3, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2506.1951, C139H178N2O38 calcd for [M + Na]+ 2506.1950.
:1), the reaction mixture was filtered through a pad of Celite and the precipitate was washed with EtOH. The filtrate was concentrated and the residue obtained was purified by silica gel column chromatography (CHCl3/MeOH 80:1) to give 24 (31 mg, 77%) as a white solid; [α]D +20.5° (c 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.05–7.33 (m, 35 H, Ar), 6.08 (d, 1 H, JNH,2 9.3 Hz, NHCer), 6.01 (d, 1H, JNH,2 6.5 Hz, NHIII), 5.54–5.49 (m, 3H, H-3Cer, H-1III, H-4IV), 5.44–5.41 (m, 2H, H-4III, H-2IV), 5.32–5.20 (m, 3H, H-4IV, H-3I, H-3IV), 5.01–4.94 (m, 2H, H-2II, H-2I), 4.75 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.59 (dd, 1H, J5,6a 4.6 Hz, Jgem 11.9 Hz, H-6aII), 4.52–4.45 (m, 3H, H-2Cer, H-3III, H-6aIV), 4.39 (d, 1H, J1,2 7.7 Hz, H-1II), 4.33–4.26 (m, 3H, H-1I, H-6bIV, H-6bII), 4.11–4.07 (m, 2H, H-5IV, H-4II), 4.01–3.94 (m, 2H, H-6aIII, H-6bIII), 3.90–3.83 (m, 3H, H-6aI, H-5III, H-1aCer), 3.77 (dd, 1H, J5,6a 4.8 Hz, J5,6b 7.2 Hz, H-5II), 3.73–3.64 (m, 3H, H-4I, H-1bCer, H-3II), 3.38 (dd, 1H, J5,6a 3.3 Hz, J5,6b 9.9 Hz, H-5I), 3.25 (d, 1H, JOH,3 10.1 Hz, OH), 3.14 (m, 1H, H-2III), 2.62–2.32 (m, 4H, CH3COCH2CH2COO), 2.19, 2.15, 1.99, 1.94, 1.89 (5 s, 15H, 4Ac, CH3COCH2CH2COO), 1.84–1.76 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.71, 1.59 (2 s, 6H, 2Ac), 1.52–1.13 (m, 66H, 33CH2), 0.94–0.85 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.6, 173.2, 172.8, 171.7, 170.4, 170.3, 170.2, 169.7, 166.2, 166.1, 165.8, 165.1, 165.0, 164.8, 133.7, 133.5, 133.2, 133.1, 132.9, 130.0, 129.9, 129.8, 129.7, 129.7, 129.6, 129.5, 129.5, 129.2, 129.1, 129.1, 128.7, 128.7, 128.6, 128.5, 128.4, 128.3, 101.8, 100.1, 98.9, 75.5, 75.0, 74.6, 73.7, 73.1, 72.7, 72.5, 72.3, 72.0, 71.8, 71.7, 70.9, 69.6, 69.5, 67.0, 67.0, 63.6, 62.4, 62.1, 61.5, 55.9, 47.7, 37.6, 36.3, 31.9, 30.0, 29.7, 29.7, 29.6, 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.2, 28.7, 27.7, 25.5, 25.4, 22.8, 22.7, 20.8, 20.8, 20.7, 20.6, 20.4, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2416.1478, C132H172N2O38 calcd for [M + Na]+ 2416.1480.
:1), the reaction was quenched with triethylamine, and pyridine was co-evaporated with toluene. The residue was purified by silica gel column chromatography (CHCl3/MeOH/Et3N 120:1:1.2) to give 25 (9.8 mg, 91%) as a white solid; [α]D +9.8° (c 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 10.3 (br s, 1H, Et3NH), 8.04–7.33 (m, 35H, Ar), 7.12 (d, 1H, JNH,2 7.3 Hz, NHIII), 6.01 (d, 1H, JNH,2 9.2 Hz, NHCer), 5.50–5.48 (m, 2H, H-3Cer, H-4IV), 5.40–5.35 (m, 2H, H-4III, H-2IV), 5.30–5.27 (m, 1H, H-4Cer), 5.22 (t, 1H, J2,3, J3,4 9.5 Hz, H-3I), 5.15 (dd, 1H, J2,3 10.5 Hz, J3,4 3.4 Hz, H-3IV), 5.00 (dd, 1H, J1,2 8.4 Hz, J2,3 9.8 Hz, H-2II), 4.96–4.92 (m, 2H, H-1III, H-2I), 4.79 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.60–4.58 (m, 3H, H-6aII, H-3II, H-4II), 4.50–4.46 (m, 2H, H-2Cer, H-6aIV), 4.42 (d, 1H, J1,2 8.1 Hz, H-1II), 4.33–4.26 (m, 3H, H-1I, H-6bIV, H-6bII), 4.08 (t, 1H, J5,6a, J5,6b 6.8 Hz, H-5IV), 4.04–4.00 (m, 2H, H-6aI, H-3III), 3.87–3.74 (m, 6H, H-6aIII, H-6bIII, H-2III, H-5II, H-1aCer, H-6aI), 3.68–3.62 (m, 2H, H-5III, H-4I), 3.47 (dd, 1H, J1b,2 3.7 Hz, Jgem 10.1 Hz, H-1bCer), 3.34–3.31 (m, 1H, H-5I), 3.07–3.03 (m, 6H, 3NCH2CH3), 2.61–2.30 (m, 4H, CH3COCH2CH2CO), 2.18–1.68 (6 s, 18H, 5Ac, CH3COCH2CH2CO), 1.76–1.71 (m, 4H, H-5aCer, H-5bCer, NHCOCH2), 1.49–1.17 (m, 69H, 33CH2, Ac), 0.89–0.83 (m, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 205.7, 172.8, 171.6, 170.6, 170.1, 170.0, 169.4, 166.0, 165.9, 165.0, 165.0, 164.9, 133.4, 133.3, 133.1, 132.9, 130.0, 129.7, 129.6, 129.6, 129.4, 129.1, 128.6, 128.6, 128.5, 128.5, 128.4, 128.3, 101.7, 99.8, 77.6, 76.0, 73.5, 72.8, 72.3, 71.3, 71.1, 70.9, 69.8, 69.4, 67.2, 62.6, 61.7, 47.7, 46.0, 37.6, 36.3, 31.9, 30.0, 29.7, 29.7, 29.6, 29.6, 29.5, 29.5, 29.5, 29.4, 29.3, 29.2, 28.8, 27.7, 25.4, 25.4, 22.8, 22.7, 20.7, 20.6, 20.6, 20.4; HRMS (ESI) m/z: found [M − H]− 2472.1078, C138H186N2O41S calcd for [M − H]− 2472.1083.
:9:2), the reaction mixture was concentrated. The residue was purified by gel filtration (Sephadex LH-20, CHCl3/MeOH/H2O 5:4:1) to give 1 (4.5 mg, 82%); [α]D +15.0° (c 0.5, CHCl3/MeOH/H2O 5:4:1); 1H NMR (500 MHz, CD3OD) δ 4.56 (d, 1H, J1,2 8.6 Hz, H-1III), 4.33 (d, 1H, J1,2 7.8 Hz, H-1), 4.27 (d, 1H, J1,2 7.6 Hz, H-1), 4.21 (d, 1H, J1,2 7.8 Hz, H-1), 4.13 (dd, 1H, J2,3 10.6 Hz, H-2III), 3.94 (d, 1H, J3,4 2.4 Hz, H-4III), 2.11 (t, 2H, NHCOCH2CH2), 1.95 (s, 3H, Ac), 1.53–1.15 (m, 68H, 34CH2), 0.80 (2 t, 6H, 2CH3); 13C NMR (200 MHz, DMSO-d6) δ 171.9, 171.0, 104.6, 103.4, 103.3, 81.0, 80.7, 78.0, 75.9, 75.3, 75.2, 74.8, 73.8, 73.4, 73.2, 73.0, 72.3, 70.7, 70.6, 69.8, 69.6, 69.1, 68.2, 67.2, 65.2, 62.8, 60.8, 60.5, 60.2, 59.4, 39.9, 31.4, 31.3, 30.7, 29.3, 29.28, 29.21, 29.20, 29.16, 29.1, 29.0, 28.8, 28.73, 28.70, 25.5, 25.4, 23.3, 22.2, 22.1, 14.0; HRMS (ESI) m/z: found [M − H]− 1435.8351, C138H186N2O41S calcd for [M − H]− 1435.8352.
:1), the reaction mixture was diluted with EtOAc and successively washed with satd aq. NH4Cl and brine. The organic layer was then dried over Na2SO4 and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH 60:1) to give 26 (42 mg, 93%) as a colorless syrup; [α]D +19.8° (c 0.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.07–7.33 (m, 35H, Ph), 6.34 (d, 1H, J2,NH 6.3 Hz, NHIII), 6.22 (d, 1H, J2,NH 9.2 Hz, NHCer), 5.55 (dd, 1H, J2,3 8.6 Hz, J3,4 3.3 Hz, H-3Cer), 5.48 (d, 1H, J1,2 7.9 Hz, H-1III), 5.43 (2 d, 2H, J3,4 3.5 Hz, H-4IV, H-4III), 5.29 (m, 1H, H-4Cer), 5.24, 5.23 (2 t, 2H, H-3I, H-2IV), 4.99 and 4.98 (2 t, J1,2 7.8 Hz, 2H, H-2III, H-2II), 4.71 (d, 1H, J1,2 7.8 Hz, H-1IV), 4.60 (dd, 1H, Jgem 11.8 Hz, J5,6a 4.6 Hz, H-6aII), 4.52–4.45 (m, 3H, H-2II, H-3III, H-6aIV), 4.37 (d, 1H, H-1II), 4.32 (d, 1H, J1,2 7.7 Hz, H-1I), 4.31–4.28 (m, 3H, H-6bIV, H-6bII), 4.09 (d, 1H, J3,4 2.4 Hz, H-4II), 4.01–3.93 (m, 4H, H-3IV, H-5IV, H-6aIII, H-5III), 3.86–3.77 (m, 4H, OH, H-1aCer, H-6bIII, H-5II), 3.71–3.67 (m, 2H, H-3II, H-4I), 3.63 (t, 1H, Jgem 9.5 Hz, J5,6a 9.5 Hz, H-6aI), 3.47 (dd, 1H, Jgem 9.7 Hz, J1b,2 3.5 Hz, H-1bCer), 3.37–3.32 (m, 2H, H-6bI, H-5I), 3.14 (m, 1H, H-2III), 2.66 (d, 1H, J3,OH 5.5 Hz, OH), 2.19–1.74 (m, 16H, 4Ac, 2CH2), 1.68 and 1.64 (2 s, 6H, 2Ac), 1.47–1.12 (m, 66H, 33CH2), 0.88, 0.87 (2 t, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 173.3, 172.9, 171.1, 170.5, 170.4, 170.3, 169.7, 166.3, 166.2, 166.1, 165.9, 165.8, 165.2, 164.8, 133.7, 133.2, 133.1, 132.9, 130.1, 129.9, 129.85, 129.80, 129.76, 129.71, 129.70, 129.6, 129.5, 129.4, 129.3, 129.1, 128.7, 128.6, 128.5, 128.44, 128.40, 128.3, 101.5, 100.2, 99.1, 77.6, 75.3, 74.9, 74.7, 73.8, 73.3, 73.1, 73.0, 72.7, 72.4, 72.3, 72.0, 71.8, 71.7, 71.3, 71.0, 69.7, 69.6, 67.0, 63.7, 62.5, 62.1, 62.0, 55.9, 47.8, 36.3, 31.9, 29.74, 29.65, 29.60, 29.56, 29.55, 29.49, 29.35, 29.2, 28.6, 25.5, 25.4, 23.0, 22.7, 20.8, 20.7, 20.6, 20.4, 14.1; HRMS (ESI) m/z: found [M + Na]+ 2318.1113, C127H166N2O36 calcd for [M + Na]+ 2318.1113.
:1), the reaction was quenched with triethylamine. Pyridine was then co-evaporated with toluene. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH/Et3N 30:1:0.2) and then gel filtration (Sephadex LH-20, CHCl3/MeOH 1:1) was performed to give 27 (37 mg, 96%) as a white solid; [α]D +9.8° (c 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 9.50 (br s, 2H, Et3NH), 7.99–7.27 (m, 35H, Ph), 6.97 (d, 1H, J2, NH 9.0 Hz, NHIII), 5.98 (d, 1H, J2, NH 9.2 Hz, NHCer), 5.70 (d, 1H, J3,4 3.3 Hz, H-4III), 5.42 (dd, 1H, H-3Cer), 5.31 (d, 1H, J3,4 3.1 Hz, H-4III), 5.25 (dd, 1H, J1,2 7.8 Hz, J2,3 10.1 Hz, H-2IV), 5.22 (m, 1H, H-4Cer), 5.15 (t, 1H, J2,3, J3,4 9.5 Hz, H-3I), 4.92 (dd, 1H, J1,2 7.8 Hz, J3,4 9.7 Hz, H-2I), 4.76 (d, 1H, J1,2 7.4 Hz, H-1III), 4.70 (d, 1H, H-1IV), 4.61 (dd, 1H, H-3IV), 4.53 (dd, 1H, Jgem 12.3 Hz, J5,6a 3.3 Hz, H-6aIII), 4.49–4.45 (m, 2H, H-3II, H-4II), 4.41 (m, 1H, H-2Cer), 4.33 (d, 1H, H-1II), 4.31–4.19 (m, 4H, H-1I, H-3II, H-6bIII, H-6aII), 3.96 (t, 1H, J5,6 6.5 Hz, H-5III), 3.93 (near d, 1H, Jgem 10.7 Hz, H-6aIV), 3.86–3.70 (m, 5H, H-2III, H-1aCer, H-6aI, H-6bIV, H-5II), 3.57–3.52 (m, 3H, H-4I, H-6bI, H-5IV), 3.40 (dd, 1H, Jgem 13.0 Hz, J1b,2 4.7 Hz, H-1bCer), 3.27 (m, 1H, H-5I), 3.01 (2 q, 12H, 2NCH2CH3), 2.19–1.60 (m, 19H, 5Ac, 2CH2), 1.39–1.04 (m, 89H, Ac, 34CH2, 6NCH2CH3), 0.80 (2 t, 6H, 2CH3); 13C NMR (125 MHz, CDCl3) δ 173.0, 172.9, 170.6, 170.2, 170.0, 169.9, 169.6, 167.7, 166.2, 166.1, 166.0, 165.3, 165.2, 165.1, 164.9, 133.5, 133.4, 133.1, 133.0, 132.9, 132.4, 130.9, 130.1, 130.0, 129.9, 129.8, 129.7, 129.66, 129.64, 129.55, 129.53, 129.4, 129.0, 128.7, 128.6, 128.5, 128.48, 128.40, 128.36, 128.30, 128.25, 103.0, 102.0, 101.2, 99.8, 78.1, 77.7, 77.3, 76.4, 76.2, 74.9, 73.7, 72.8, 72.7, 72.3, 71.5, 71.2, 71.1, 70.2, 69.8, 69.5, 68.6, 68.1, 66.9, 64.3, 62.8, 62.5, 61.8, 51.5, 47.7, 46.9, 46.8, 46.5, 38.7, 36.2, 31.9, 30.3, 30.0, 29.7, 29.6, 29.5, 29.49, 29.47, 29.4, 29.3, 29.2, 29.1, 28.9, 28.4, 25.4, 25.3, 23.7, 22.9, 22.6, 22.3, 20.8, 20.7, 20.6, 202, 14.1, 14.0, 10.9, 9.0, 8.8, 8.6; HRMS (ESI) m/z: found [M − H + Na]− 2476.0103, C127H164N2O42S2 calcd for [M − H + Na]− 2476.0103.
:45:10), the reaction mixture was concentrated. The resulting residue was purified by silica gel column chromatography (Iatrobeads, CHCl3/MeOH/H2O 6:4:0 → 6:4:0.5) to give 2 (28 mg, 96%); [α]D +13.0° (c 0.5, CHCl3/MeOH/H2O 5:4:1); 1H NMR (500 MHz, CD3OD) δ 4.47 (d, 1H, J1,2 7.1 Hz, H-1), 4.43 (d, 1H, J1,2 7.6 Hz, H-1), 4.30 (2 d, 2H, J1,2 7.9 Hz, 2 H-1), 2.21 (t, 2H, Jgem, J1,2 7.2 Hz, H-1aCer, H-1bCer), 2.07 (br s, 3H, Ac), 1.63–1.16 (m, 68H, 34CH2), 0.89 (2 t, 6H, 2CH3); 13C NMR (200 MHz, DMSO-d6) δ 171.9, 170.4, 104.3, 103.6, 103.5, 103.4, 80.8, 80.2, 79.1, 77.9, 75.7, 75.1, 74.9, 74.8, 74.7, 74.3, 73.9, 73.5, 73.3, 70.6, 69.8, 69.5, 69.04, 69.02, 67.3, 66.2, 60.8, 60.24, 60.18, 59.4, 50.5, 50.2, 50.1, 39.9, 35.6, 35.5, 31.34, 31.32, 30.7, 29.32, 29.27, 29.20, 29.19, 19.15, 29.09, 29.04, 29.03, 29.01, 29.00, 28.8, 28.7, 28.6, 25.5, 25.4, 23.34, 23.30, 22.13, 22.12, 14.0; HRMS (ESI) m/z: found [M − H + Na]− 1537.7741, C68H126N2O30S2 calcd for [M − H + Na]− 1537.7740.
Microarray construction and analysis
The naturally occurring SM1a and the synthetic SM1a and SB1a were robotically arrayed in duplicate at 2 and 5 fmol per spot on nitrocellulose-coated glass slides using a non-contact instrument as described.26 Antibody binding analyses were performed essentially as described.27 In brief, after blocking with 3% w/v bovine serum albumin (Sigma) in HEPES-buffered saline (5 mM HEPES pH 7.4, 150 mM NaCl, 5 mM CaCl2), the microarray was probed with the undiluted mAb AE3-containing culture supernatant. Binding was detected using biotinylated anti-mouse IgM (Sigma, μ-chain specific) at a 1:200 dilution, followed by Alexa Fluor-647-labelled streptavidin (1 μg ml−1; Molecular Probes). Imaging and data analysis were as described.7,28 The results shown are at 5 fmol per spot.
Acknowledgements
The iCeMS is supported by World Premier International Research Center Initiative (WPI), MEXT, Japan. This work was financially supported in part by MEXT of Japan (a Grant-in-Aid for Scientific Research (B) No. 22380067 to M.K.; a Grant-in-Aid for Young Scientists (A) No. 23688014 and Grant-in-Aid on Innovative Areas No. 26110704, Deciphering sugar chain-based signals regulating integrative neuronal functions, (to H.A); the Alliance of Glycobiologists grant U01 CA168925-01 and The Wellcome Trust grant WT099197MA (to T.F) and a fellowship from China Scholarship (to CG). We thank Kiyoko Ito (Gifu University) for providing technical assistance.References
- I. Ishizuka, Prog. Lipid Res., 1997, 36, 245–319 CrossRef CAS.
- (a) K. Tadano-Aritomi, H. Kubo, P. Ireland, M. Okuda, T. Kasama, S. Handa and I. Ishizuka, Carbohydr. Res., 1995, 273, 41–52 CrossRef CAS; (b) I. Ishizuka and Y. Niimura, Glycobiology, 2006, 16, 729–735 CrossRef PubMed.
- (a) K. Tadano and I. Ishizuka, J. Biol. Chem., 1982, 257, 1482–1490 CAS; K. Tadano and I. Ishizuka, J. Biol. Chem., 1982, 257, 9294–9299 Search PubMed; (b) K. Tadano, I. Ishizuka, M. Matsuo and S. Matsumoto, J. Biol. Chem., 1982, 257, 13413–13420 CAS; (c) N. Iida, T. Toida, Y. Kushi, S. Handa, P. Fredman, L. Svennerholm and I. Ishizuka, J. Biol. Chem., 1989, 264, 5974–5980 CAS.
- (a) K. Tadano-Aritomi, T. Kasama, S. Handa and I. Ishizuka, Eur. J. Biochem., 1992, 209, 305–313 CrossRef CAS PubMed; (b) K. Tadano-Aritomi, M. Okuda, I. Ishizuka, H. Kubo and P. Ireland, Carbohydr. Res., 1994, 265, 49–59 CrossRef CAS.
- (a) K. Nagai, I. Ishizuka and S. Oda, J. Biochem., 1984, 95, 1501–1511 CAS; (b) Y. Niimura and I. Ishizuka, Biochim. Biophys. Acta, 1990, 1052, 248–254 CrossRef CAS; (c) Y. Niimura and I. Ishizuka, Comp. Biochem. Physiol., 1991, 100B, 535–541 CAS.
- (a) N. Hiraiwa, N. Iida, I. Ishizuka, S. Itai, K. Shigeta, R. Kannagi, Y. Fukuda and H. Imura, Cancer Res., 1988, 48, 6769–6774 CAS; (b) N. Hiraiwa, Y. Fukuda, H. Imura, K. Tadano-Aritomi, K. Nagai, I. Ishizuka and R. Kannagi, Cancer Res., 1990, 50, 2917–2928 CAS; (c) T. Kobayashi, K. Honke, K. Kamio, N. Sakakibara, S. Gasa, N. Miyao, T. Tsukamoto, I. Ishizuka, T. Miyazaki and A. Makita, Br. J. Cancer, 1993, 67, 76–80 CrossRef CAS PubMed; (d) K. Shida, Y. Misonou, H. Korekane, Y. Seki, S. Noura, M. Ohue, K. Honke and Y. Miyamoto, Glycobiology, 2009, 19, 1018–1033 CrossRef CAS PubMed.
- A. S. Palma, Y. Liu, R. A. Childs, C. Herbert, D. Wang, W. Chai and T. Feizi, Biochem. Biophys. Res. Commun., 2011, 408, 548–552 CrossRef CAS PubMed.
- (a) Y. Niimura and I. Ishizuka, Glycobiology, 2006, 16, 729–735 CrossRef CAS PubMed; (b) K. Honke, M. Tsuda, Y. Hirahara, N. Miyao, T. Tsukamoto, M. Satoh and Y. Wada, Cancer Res., 1998, 58, 3800–3805 CAS; (c) Y. Misonou, K. Shida, H. Korekane, Y. Seki, S. Noura, M. Ohue and Y. Miyamoto, J. Proteome Res., 2009, 8, 2990–3005 CrossRef CAS PubMed; (d) N. Hiraiwa, N. Iida, I. Ishizuka, S. Itai, K. Shigeta, R. Kannagi, Y. Fukuda and H. Imura, Cancer Res., 1988, 48, 6769–6774 CAS.
- K. Tanano and I. Ishizuka, J. Biol. Chem., 1982, 257, 13413–13420 Search PubMed.
- P. Zhang, K. Wang, J. Zhang, C. Li and H. Guan, Eur. J. Org. Chem., 2015, 570–583 CrossRef CAS PubMed.
- M. Kiso, H. Ishida, H. Ando and A. Imamura, Ganglioside Synthesis, in Glycoscience: Biology and Medicine, ed. N. Taniguchi, T. Endo, G. W. Hart, P. H. Seeberger and C.-H. Wong, Springer, Tokyo, 2015, vol. 1, pp. 331–338 Search PubMed.
- (a) A. Hasegawa, K. Ito and M. Kiso, J. Carbohydr. Chem., 1995, 14, 353–368 CrossRef CAS PubMed; (b) S. Komba, H. Ishida, M. Kiso and A. Hasegawa, Carbohydr. Res., 1996, 285, C1–C8 CrossRef CAS; (c) S. Komba, H. Ishida, M. Kiso and A. Hasegawa, Bioorg. Med. Chem., 1996, 4, 1833–1847 CrossRef CAS; (d) S. Komba, C. Galustian, H. Ishida, T. Feizi, R. Kannagi and M. Kiso, Angew. Chem., Int. Ed., 1999, 38, 1131–1133 CrossRef CAS; (e) T. Komori, S. Kondo, H. Ando, H. Ishida and M. Kiso, Carbohydr. Res., 2002, 337, 1679–1686 CrossRef CAS.
- A. Imamura, H. Ando, H. Ishida and M. Kiso, J. Org. Chem., 2009, 74, 3009–3023 CrossRef CAS PubMed.
- K. Fujikawa, S. Nakashima, M. Konishi, T. Fuse, N. Komura, T. Ando, H. Ando, N. Yuki, H. Ishida and M. Kiso, Chem. – Eur. J., 2011, 17, 5641–5651 CrossRef CAS PubMed.
- M. Konishi, A. Imamura, K. Fujikawa, H. Ando, H. Ishida and M. Kiso, Molecules, 2013, 18, 15153–15181 CrossRef CAS PubMed.
- S. Nakashima, H. Ando, R. Saito, H. Tamai, H. Ishida and M. Kiso, Chem. Asian J., 2012, 7, 1041–1051 CrossRef CAS PubMed.
- H. Tamai, H. Ando, H.-N. Tanaka, R. Hosoda-Yabe, T. Yabe, H. Ishida and M. Kiso, Angew. Chem., Int. Ed., 2011, 50, 2330–2333 CrossRef CAS PubMed.
- H. Tamai, A. Imamura, J. Ogawa, H. Ando, H. Ishida and M. Kiso, Eur. J. Org. Chem., 2015, 5199–5211 CrossRef CAS PubMed.
- H. Sakamoto, S. Nakamura, T. Tsuda and S. Hashimoto, Tetrahedron Lett., 2009, 41, 7691–7695 CrossRef.
- G. Gu, Y. Du and J. Pan, Carbohydr. Res., 2002, 337, 1313–1317 CrossRef CAS.
- T. Fuse, H. Ando, A. Imamura, N. Sawada, H. Ishida, M. Kiso, T. Ando, S.-C. Li and Y.-T. Li, Glycoconjugate J., 2006, 23, 329–343 CrossRef CAS PubMed.
- Y. Matsuzaki, Y. Ito, Y. Nakahara and T. Ogawa, Tetrahedron Lett., 1993, 34, 1061–1064 CrossRef CAS.
- R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef PubMed.
- (a) P. Konradsson, D. R. Mootoo, R. E. McDevitt and B. Fraser-Reid, J. Chem. Soc., Chem. Commun., 1990, 270–272 RSC; (b) G. H. Veeneman, S. H. van Leeuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331–1334 CrossRef CAS.
- K. Koike, Y. Nakahara and T. Ogawa, Agric. Biol. Chem., 1990, 54, 663–667 CrossRef CAS.
- (a) Y. Liu, R. Childs, A. Palma, M. Campanero-Rhodes, M. Stoll, W. Chai and T. Feizi, Neoglycolipid-Based Oligosaccharide Microarray System: Preparation of NGLs and Their Noncovalent Immobilization on Nitrocellulose-Coated Glass Slides for Microarray Analyses, in Carbohydrate Microarrays: Methods and Protocols, ed. Y. Chevolot, Humana Press, 2012, vol. 808, pp. 117–136. Available from http://link.springer.com/protocol/10.1007%2F978-1-61779-373-8_8 Search PubMed; (b) A. Palma, T. Feizi, Y. Zhang, M. Stoll, A. Lawson, E. Díaz-Rodríguez, M. Campanero-Rhodes, J. Costa, S. Gordon, G. Brown and W. Chai, J. Biol. Chem., 2006, 281, 5771–5779 CrossRef CAS PubMed.
- C. Gao, Y. Liu, H. Zhang, Y. Zhang, M. N. Fukuda, A. S. Palma, R. P. Kozak, R. A. Childs, M. Nonaka, Z. Li, D. L. Siegel, P. Hanfland, D. M. Peehl, W. Chai, M. I. Greene and T. Feizi, J. Biol. Chem., 2014, 289, 16462–16477 CrossRef CAS PubMed.
- M. S. Stoll and T. Feizi, Proceeding of the Beilstein Symposium on Glyco-Bioinformatics, Potsdam, Germany, 2009. Available from http://www.beilstein-institut.de/glycobioinf2009/ProceedingsWeb/Stoll/Stoll.html Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c5ob01744k |
| This journal is © The Royal Society of Chemistry 2015 |