
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A latent reactive handle for functionalising heparin-like and LMWH deca- and dodecasaccharides†
Gavin J.
Miller
 a,
Karl. R.
Broberg
a,
Claire
Rudd
a,
Madeleine R.
Helliwell
b,
Gordon C.
Jayson
c and
John M.
Gardiner
*a
a,
Karl. R.
Broberg
a,
Claire
Rudd
a,
Madeleine R.
Helliwell
b,
Gordon C.
Jayson
c and
John M.
Gardiner
*a
aManchester Institute of Biotechnology and School of Chemistry, 131 Princess Street, University of Manchester M1 7DN, UK. E-mail: gardiner@manchester.ac.uk; Tel: +44 (0)161 306 4530
bSchool of Chemistry, University of Manchester, Manchester, UK M13 9PL
cInstitute or Cancer Studies and Christie Hospital, University of Manchester, Manchester, UK M20 4BX
First published on 9th September 2015
Abstract
D-Glucosamine derivatives bearing latent O4 functionality provide modified H/HS-type disaccharide donors for a final stage capping approach enabling introduction of conjugation-suitable, non-reducing terminal functionality to biologically important glycosaminoglycan oligosaccharides. Application to the synthesis of the first O4-terminus modified synthetic LMWH decasaccharide and an HS-like dodecasaccharide is reported.
Introduction
The glycosylaminoglycan (GAG) family of carbohydrates constitute the most diverse group of biological carbohydrates. They are pervasive on cell surfaces and within the intra-cellular matrix, playing key roles in mediating regulatory functions relevant to numerous biomedical areas, such as cancer, inflammation, Alzheimer's disease, viral infectivity, and stem cell differentiation.1 Heparin and heparan sulphate (H/HS) are key members of this GAG family of sugars. The complex heterogeneity of the natural oligosaccharides requires syntheses of structurally-defined HS sequences to be able to evaluate the roles of HS substructures, which underpins the potential for ultimate therapeutic applications of synthetic fragments. Such synthetic endeavours have seen a number of important advances in recent years, targeting both anti-coagulant LMWH-replacements,2 but wide interest is directed at the diversity of bio-regulatory roles different synthetic heparin-like oligosaccharides could target.3As part of our program to provide scalable syntheses of structurally-defined H/HS oligosaccharides, encompassing the first reported [GlcN-IdoA]n 12-mer LMWH4 and gram-scale syntheses of heparin-like 12-mers,5 we have also been developing synthetic HS mimetics bearing a site-specifically located functional handle, to provide new GAG tools for chemical biology and biophysical applications of homogenous oligosaccharide conjugates.
Accessibility to new synthetic oligosaccharides bearing functional end groups is highly relevant to the viability of applications to microarray analysis, fluorescent labelling and other biomolecule or nanoparticle conjugations, essential for interrogating biological structure/function effects. Chemical modifications generating new carbohydrate tools are important, not only for the glycosaminoglycan field,6 but for wider potential applicability within the chemical glycobiology community.7
End-modification methods for native linear poly- and oligosaccharides, including GAGs, traditionally employ reductive amination chemistry at the expense of opening the reducing terminal sugar.8 Conjugation-amenable modifications which do not require a free reducing terminus are important. Modifications via the glycosidic terminus have become the default strategy for glycoside conjugation capabilities for GAGs and other oligosaccharides. Most of these approaches introduce glycosidic extensions bearing reactive amine, thiol, alcohol, azide or alkyne functions for further modifications. In the GAG and wider saccharide arena, these have seen valuable applications to arrays and other immobilizations,9 and recent end modifications to assist purification and thereby accelerate synthesis, such as through use of ionic liquid9e or fluorous9f tags. Invariably, the reducing terminus is employed for C1 modification via O-, but also C-, N- or S-glycosides.
Though of high biological and biomedical importance, the challenges presented by chemical synthesis of longer synthetic heparins has limited approaches to end-modifications. A reducing terminal labelling approach via disaccharide iteration (Fig. 1A) requires that the conjugation site or precursor is introduced early and is followed by all the sequence iterations. Use of reactive aldehyde end tags can be envisaged as an alternative to use of a free reducing terminus (aldehyde) by installing the aldehyde on a linker, and though little exploited, has also seen use for amination-based facile dimerization of disaccharides.10 We recently reported introduction of a reducing end latent aldehyde tag (LAT) into a synthetic HS-dodecasaccharide and exploited this for 3H-radiolabelling, enabling the first example of determination of in vivo tissue distribution and metabolism of a pure, biologically-active heparin-like oligosaccharide (c.f. Fig. 1A).11 This reducing terminal modification approach was considerably facilitated by the high efficiency and fidelity of a new 4-mer based iteration 12-mer synthesis to two 2-step cycles, however, in this case still, the latent conjugation site (at the reducing terminus) is included from the beginning of the iterations.
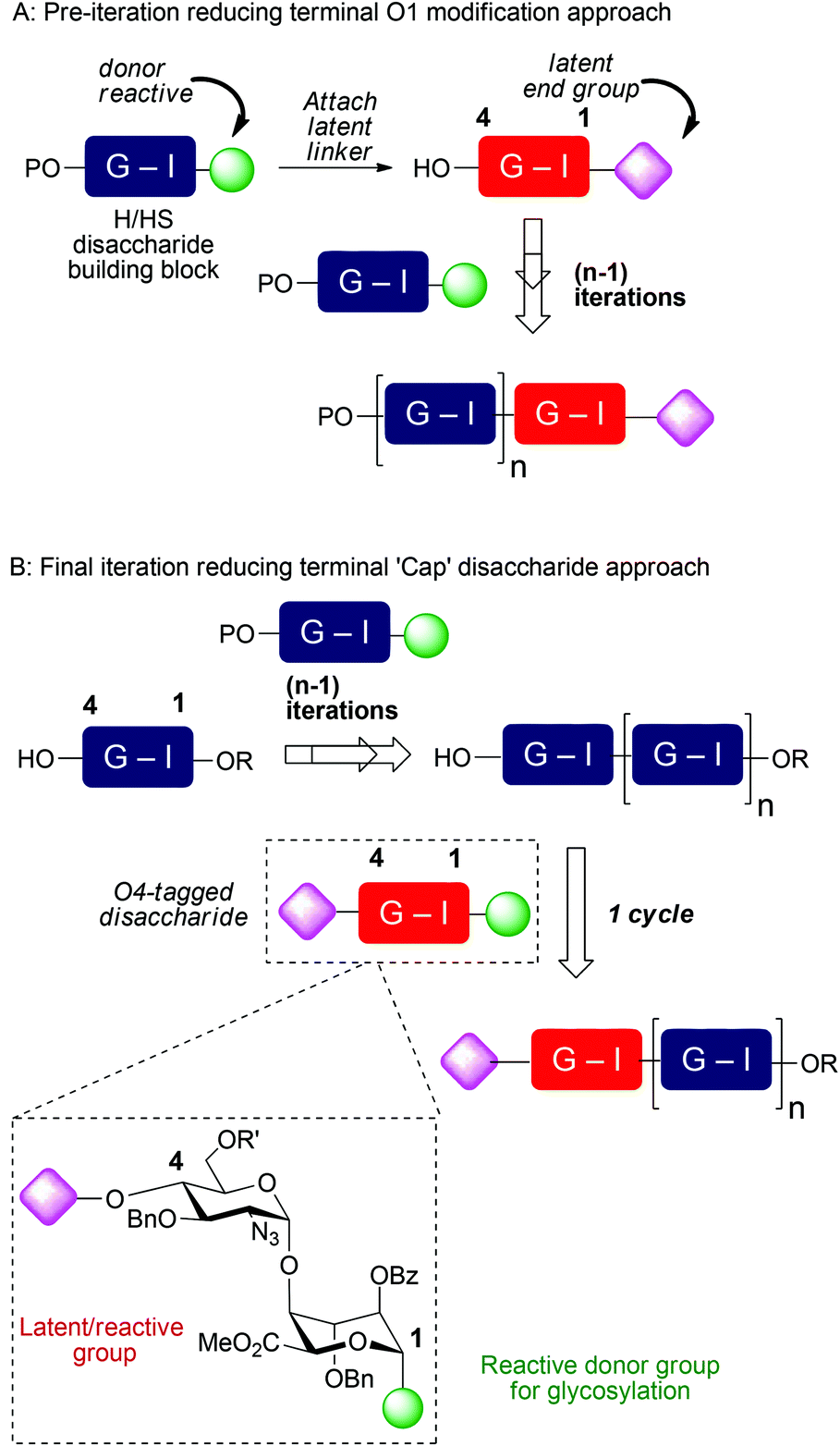 | ||
| Fig. 1 A: Reducing terminal vs. B: final stage capping approaches to introduce functional end groups into HS-like oligosaccharides, R = Me, R′ = Bz or Bn. | ||
Non-reducing terminal conjugation units would obviate the limitation of early-stage introduction of the commonly-employed O1-glycosidic based conjugate-ready modifications for iterative oligosaccharide synthesis and would be anticipated to deliver increasing benefits for longer heparanoids. In such approaches, all but the final iteration step would precede introduction of the latent conjugation site (Fig. 1B). Following our 12-mer HS end-labelling work, we therefore sought to investigate the utility of attaching such a LAT, or similar derivative, to the non-reducing terminus of heparanoid oligosaccharides. This has not been evaluated or exploited for the functional modification of longer, bio-relevant heparin-types or other GAGs. This would be valuable both for H/HS targets, but we envision such end modification would be portable to application in other important GAGs, of different constituent sequences. Given the known biological roles of O6 and O3 D-GlcN functionality in H/HS systems, this would most ideally be located at the O4 terminal position (Fig. 1B). A non-reducing terminal strategy could seek to modify the terminus post-oligomerization, via coupling to a native terminal hydroxyl unit which could be methodologically challenging. Instead, providing new generic end-modified non-reducing ‘capper’ units (e.g. disaccharides) would introduce a terminal, non-reducing end building block (Fig. 1B) bearing the latent conjugation site in the final iterative glycosylation step during oligosaccharide assembly. This convergent strategy would be attractive as it would enable late-stage diversification at the terminus of different acceptor saccharides and accelerate accessibility to HS-sequences of different constituents or containing mixed sulfation patterns, in a parallel manner.
Introducing this to our [GlcN-IdoA]n-based HS oligosaccharide synthetic strategy requires attaching a conjugation-suitable tag containing a terminal reactive group (or latent reactive group) at the D-GlcN-O4-position. Ideally, this would be incorporated into a GlcN-IdoA disaccharide donor unit, two variants of which we have shown are effective iduronate donors, whose coupling rections are routinely high yielding and highly anomerically selective, on even the longest synthetic heparin-like acceptors.4,5,12
Such O4 modification could be approached by attaching the protected handle either directly at disaccharide level or onto the monoglucoside, which could then be coupled with an L-idoA acceptor, providing reagent disaccharides.
Prior examples of D-GlcN derivative O4 alkylations and related modifications include a 4-position formyl moiety (via reduction of a 4-cyano species),13 ethanolamine based linkers for introducing BODIPY fluorophores,14 diethylene glycol derivatives for azide-based linkers,15 thiol terminated alkyl spacer units (via thiol addition to O-allyl)16 and carboxylate terminated linkers.17
Results and discussion
We report here approaches to introduce different oxidation level end groups, namely a carboxylate or an aldehyde surrogate, directly onto a D-GlcN-O4 derivative with an anomeric donor group in place (thiophenyl). Both these functional groups would facilitate further complementary conjugation chemistries. Our strategy was to introduce the O4 unit into suitable D-GlcN monosaccharide derivatives and to then couple to an iduronate acceptor as a direct route to end-modified ‘capper’ disaccharides. We also sought to evaluate such O4 modification via a pre-prepared disaccharide.Alkylation of D-GlcN-O-4 derivatives with carboxylate-bearing tethers
A first strategy considered introducing an O4 tag bearing a carboxylate, envisaging ultimate late-stage applications with suitably reactive labelling nucleophiles. Whilst a suitable ester differentiation for the iduronate might prove necessary, we reasoned that differential reactivity of a spacer-linked carboxylate over the L-IdoA 2-carboxylate could be evaluated or effected by use of a differentially labile terminal ester. Ethylbromoacetate was first employed as a model electrophile for D-GlcN O4-alkylation. Reaction of ethylbromoacetate with the O4 alkoxide salt of glucosamine derivative 118 (Scheme 1) afforded the target ester 2, but in relatively poor yield, owing to degradation of the starting material. However, with novel 2 in hand, saponification to the carboxylate derivative 3 proceeded in 81% yield. A more efficient one step route from 1 to 3 was subsequently effected using alkylation direct with iodoacetic acid in DMF, affording the acid 3 in significantly better overall yield (71%). If advancing this type of linker, a modified L-IdoA ester would thus be the candidate for any subsequent disaccharide formation.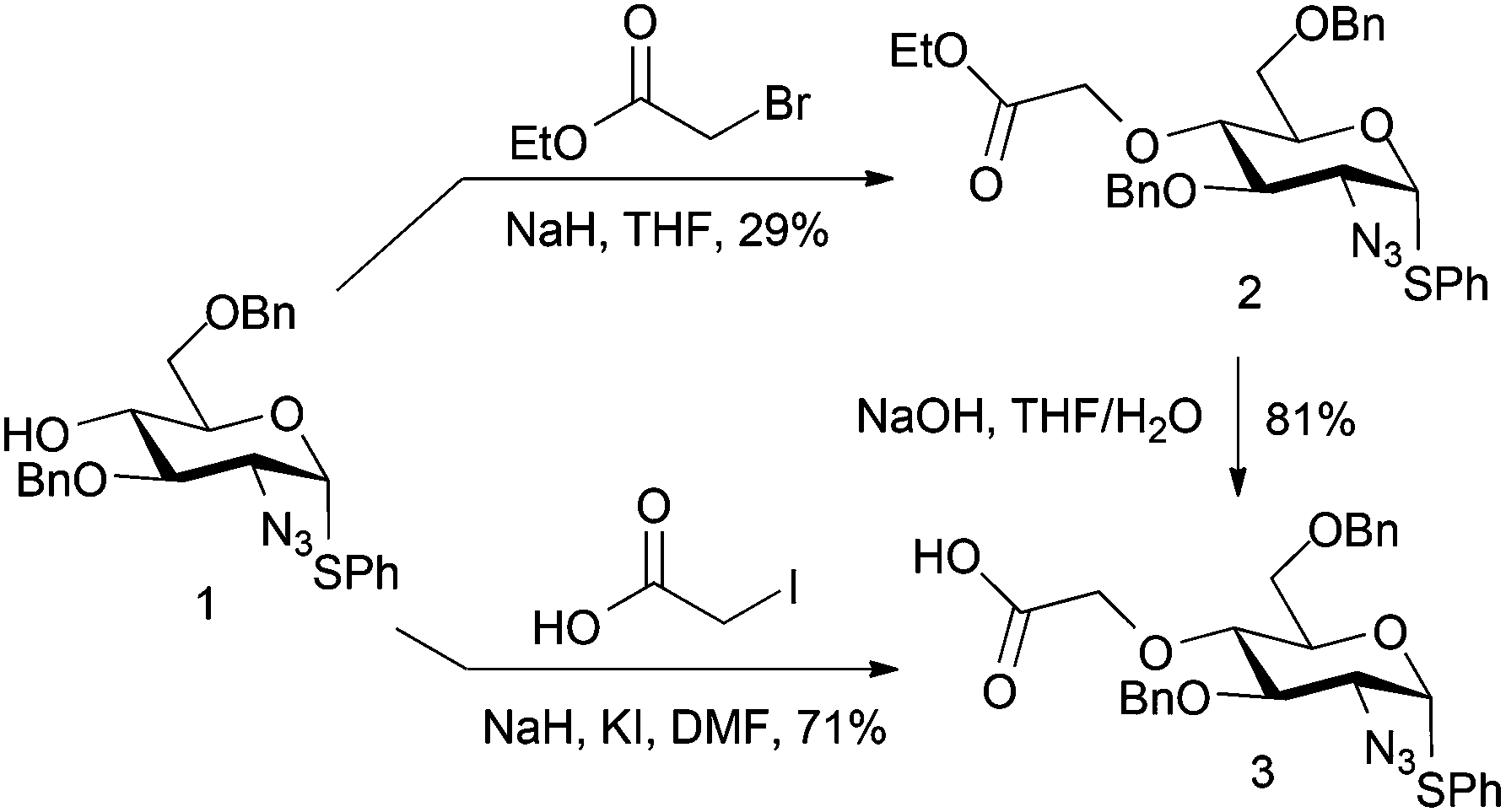 | ||
| Scheme 1 D-GlcN O4 modifications with activated carboxylate/ester electrophiles. | ||
Carboxylate 3 was utilised to evaluate model attachment of two fluorescent units, 419 and 5,20 which contained differing length C2 or C6, diaminoalkyl linker units, offering a divergent access to fluorescently tagged D-GlcN building blocks 6 and 7 (Scheme 2). Coupling of 4 or 5 with 3 using standard amide bond forming reagents afforded the fluoro-tagged D-GlcN derivatives 6 and 7, respectively. This provides evidence for the potential viability of similar derivatization of D-GlcN embedded in longer chains. This approach also envisaged that such novel O4-carboxylate-bearing D-GlcN species could be precursors to extensions with other latent tag units via amide linkages.
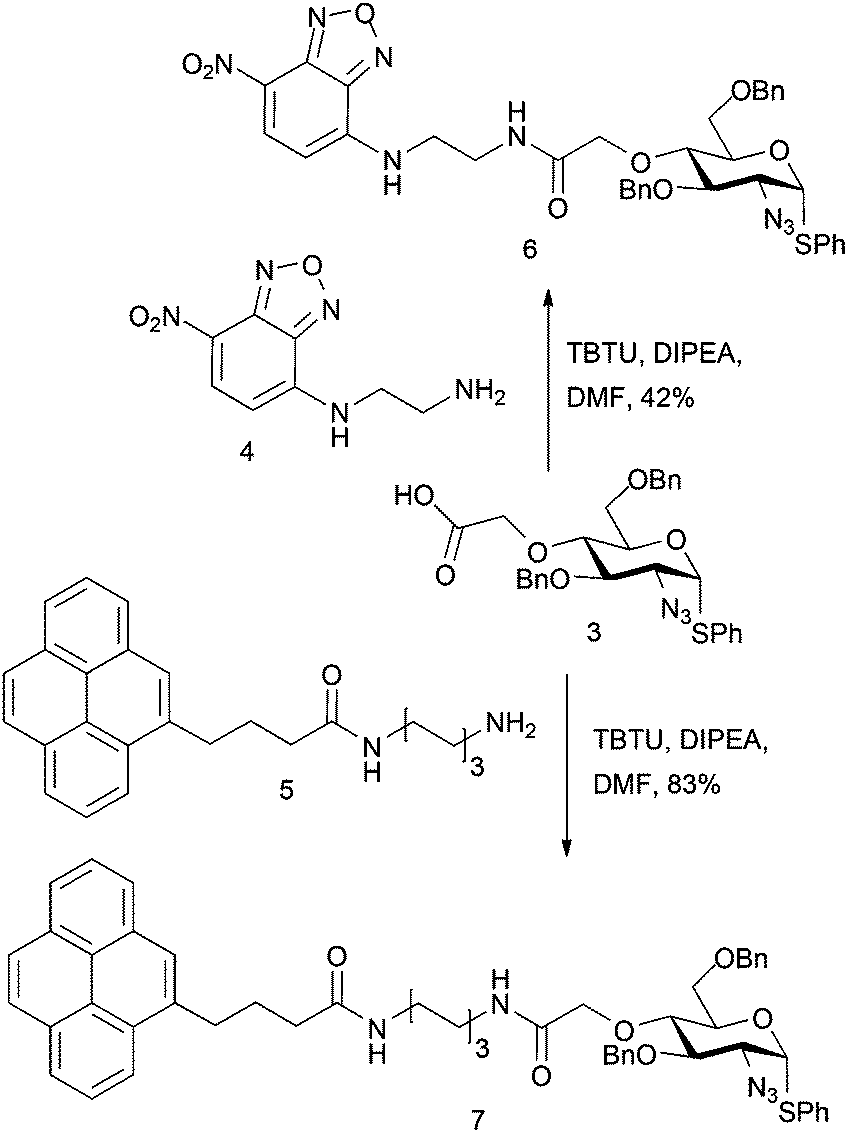 | ||
| Scheme 2 Fluorescently tagged glucosamine derivatives 6 and 7. | ||
We sought to evaluate whether this approach could be applied to the construction of an end-labelled HS-like disaccharide. This would establish the potential to use a pre-labelled HS disaccharide capper unit. Thus, the coupling of NBD-monosaccharide derivative 6 with L-iduronate acceptor 8 was attempted using NIS (1.5 equiv.) and AgOTf (0.1 equiv.).
However, whilst disaccharide coupling occurred to form 9, this was in modest yield, with significant amounts of acceptor recovered. Further inspection of the MS data showed a surprising concomitant iodination of the electron-deficient NBD ring (Scheme 3).
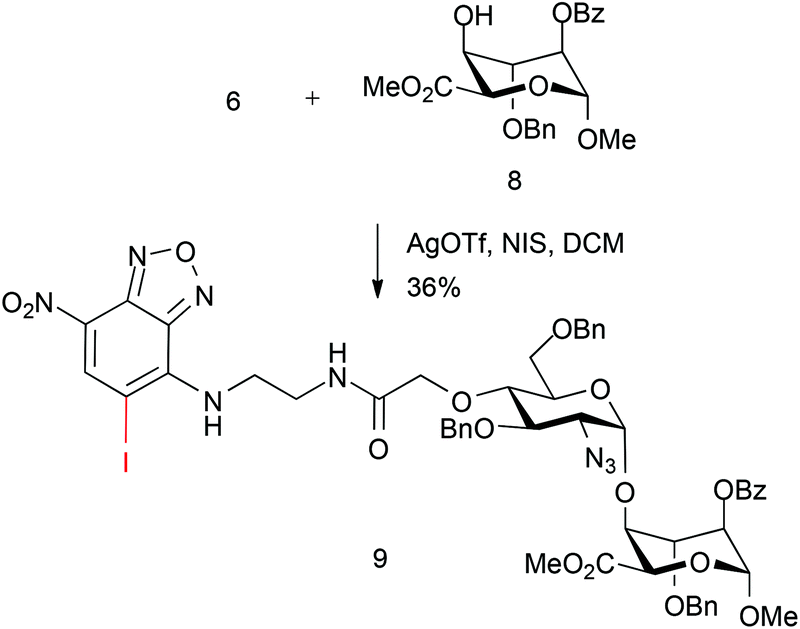 | ||
| Scheme 3 Fluorescently tagged disaccharide synthesis. | ||
Attempts to couple pyrene-labelled glucoazide donor 7 with L-iduronate acceptor 8 returned hydrolysed donor and unreacted acceptor from the reaction mixture, under the same conditions employed for 9. This suggested a poor capability of 7 as a glycosyl donor and, coupled with the unexpected glycosylation reactivity of 6, suggested that a carboxylate-based approach would require further development to be viable.
Our alternative approach was via a D-GlcN O4-aldehyde-level bearing unit, which would remain latent throughout synthetic mono-, di- and oligosaccharide construction. This would parallel our O1-linked LAT approach and enable a final stage aldehyde unmasking on the complete sulphated heparin-type oligosaccharides.21 This would also obviate the need to carry a fluorescently labelled system throughout the remainder of the synthetic transformations to a complex HS target, which might have been necessary, if selective O4-ethanoate coupling were to prove problematic over internal L-IdoA carboxylate reactivity.
Alkylation of D-GlcN derivatives with latent aldehyde O-4 tethers
Alkylation attempts of 1 with aldehyde- or protected aldehyde-containing electrophiles such as 3-bromo-1,1-dimethoxypropane, 4-bromomethylbenzaldehyde, 3-bromomethylbenzaldehyde and 2-(4-(bromomethyl)phenyl)-1,3-dioxolane were unsuccessful, mostly leading to no alkylation product and frequently to degradation of the starting D-GlcN derivative. We thus evaluated use of allylbromide to alkylate the O4 position of 1 with a view to using the derived alkene as an alternative precursor to a 1,2-diol aldehyde surrogate. Allylation of 1 was effected in 97% yield (Scheme 4) and subsequent OsO4-mediated dihydroxylation of 10 furnished diol 11 in 98% yield.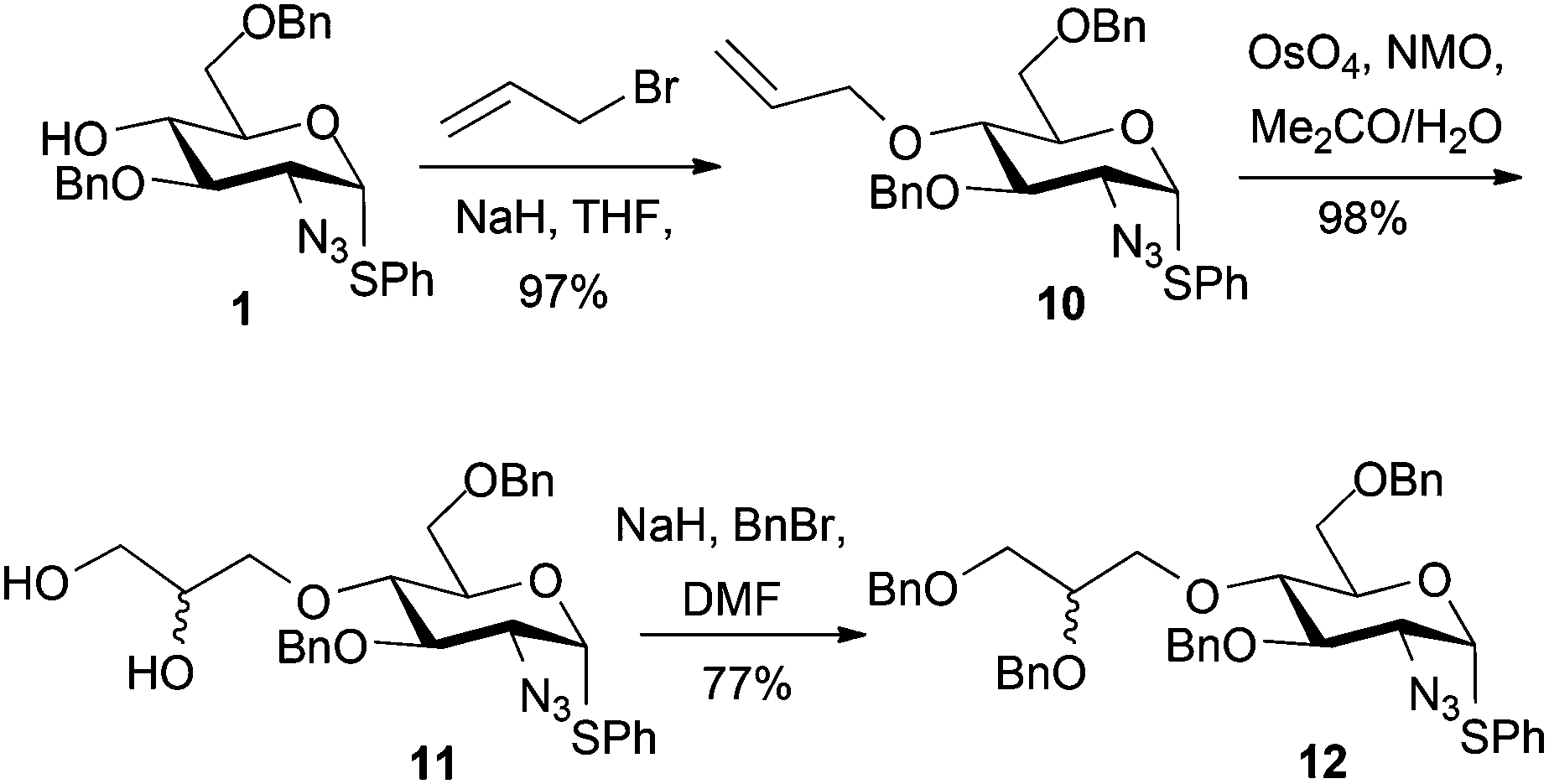 | ||
| Scheme 4 Synthesis of D-GlcN O4-glycidols 12 via allylation–oxidation. | ||
Double benzylation of 11 was completed in high yield to afford 12 with the required masked 1,2-diol unit installed at the O4-position. 1H NMR indicated that the penultimate dihydroxylation reaction to afford diol 11 yielded a mixture of diastereomeric diols (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, R/S). As the diol chiral centre is lost in the anticipated release of the aldehyde at the end of any labelling route, we envisaged this could be pursued using disatereomerically-mixed diol, once late stage analytical methods were in place. The crystal structure of 11 showed a 3:1 diastereomeric ratio (R:S configuration of the diol side chain) in the crystal, with a mixed packing of the two diastereomers (Fig. 2). In addition, the major (side chain R) diastereoisomer adopted two different conformations varying about several torsion angles in the O4 diol-bearing unit, whilst there are no significant differences in any aspects of the sugar ring or derivatives (such as the SPh or azide) on the ring, for either conformer or between diastereomers.
:1, R/S). As the diol chiral centre is lost in the anticipated release of the aldehyde at the end of any labelling route, we envisaged this could be pursued using disatereomerically-mixed diol, once late stage analytical methods were in place. The crystal structure of 11 showed a 3:1 diastereomeric ratio (R:S configuration of the diol side chain) in the crystal, with a mixed packing of the two diastereomers (Fig. 2). In addition, the major (side chain R) diastereoisomer adopted two different conformations varying about several torsion angles in the O4 diol-bearing unit, whilst there are no significant differences in any aspects of the sugar ring or derivatives (such as the SPh or azide) on the ring, for either conformer or between diastereomers.
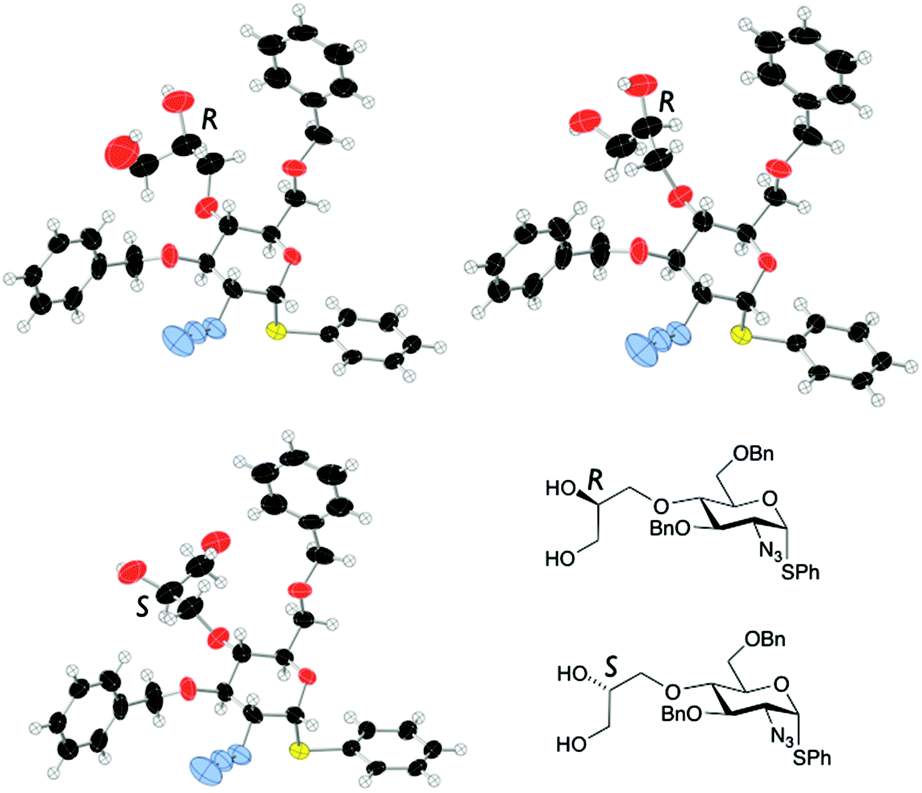 | ||
| Fig. 2 X-Ray crystal structure for diastereoisomers of diol 11 (major: R configuration in diol side chain; minor: S-configuration in side chain). | ||
The monosaccharide building block 12 was suitable for glycosylation with an iduronate acceptor 19 and provided the required, iteration-ready, HS-like disaccharide building block in three steps and 19% overall yield in a manner directly analogous to that for diastereomerically pure 20 and 21 (vide infra).‡ Although it is not critical that the OsO4 mediated dihydroxylation of 10 did not proceed with complete diastereoselectivity (as stereochemistry lost on late-stage aldehyde release), we explored an alternative access to a diastereomerically-pure form of 12 to circumvent any analytical challenges of late stage oligosaccharide synthesis and characterisation using stereoisomeric mixtures, and to afford high purity final targets for analysis and structural proof.
Having successfully employed a dibenzyl glycidol unit as an O1-LAT (via glycosylation)11 we sought to evaluate a similar unit introduced via O4 alkylation. This presented the same potential complications we had encountered in O4-alkylation with a number of unactivated electrophiles (vide supra).
(S)-2,3-Dibenzyloxy propanol was readily converted to its tosylate derivative 1322 and subsequent alkylation of 1 with 13 did proceed, but only in low yield, as the reaction required heating, and as we had seen with other O4 alkylations, resulted in competing starting material degradation (Scheme 5). To circumvent this, employing the corresponding triflate electrophile 1423 enabled efficient alkylation of O4 for two D-GlcN derivatives β-1 and 16 (differentially protected at O6) in good to excellent yields. The slightly lower yield for the formation of 18 was attributed to a competing transesterification reaction, evidenced by formation of a species with very similar Rf to 18 as the reaction progressed. The reaction yield was improved by drop-wise addition of the electrophile at 0 °C.
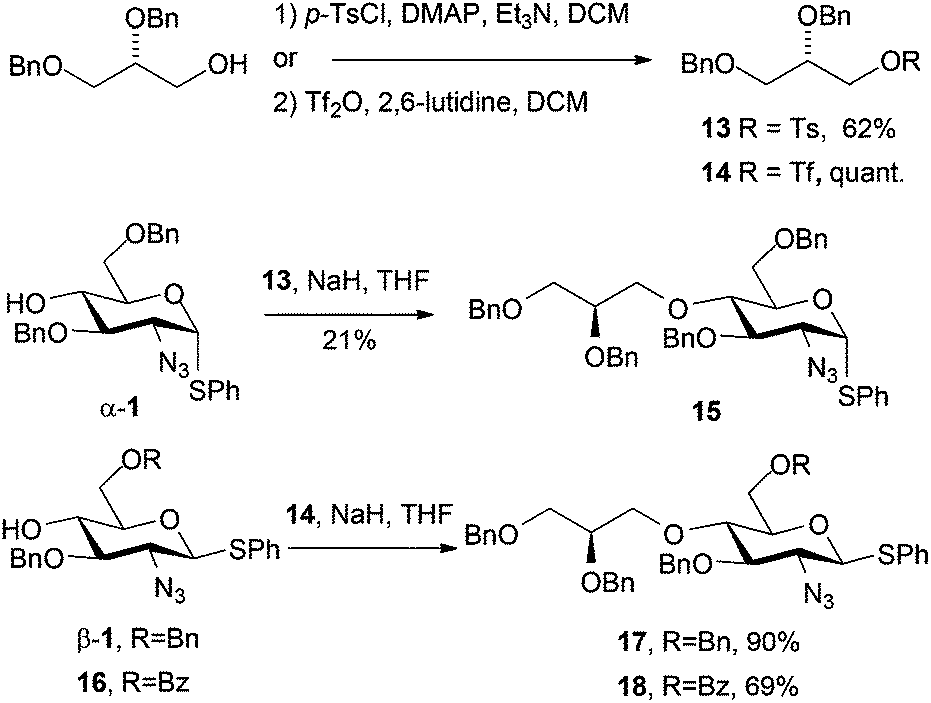 | ||
| Scheme 5 Diastereomerically pure O-glycidol-bearing D-GlcN derivatives 15–18. | ||
Commercially available (S)-2,3-dibenzyloxypropanol is expensive (£100 per g). We thus developed a convenient 5-step route from D-mannitol (3 orders of magnitude cheaper than (S)-2,3-dibenzyloxypropanol), which readily delivered multi-gram quantities of the required alcohol (25% overall yield, 5 steps from D-mannitol, see ESI†).24 This enabled us to access the novel O4-LAT derivatized thioglyosides 17 and 18 in gram quantities and diastereomerically pure form. The inclusion of the differential O6 protection is important as this allows parallel access to backbone O6-sulfation pattern variations in any final HS-oligosaccharide targets.
With these materials in hand we then investigated their utility for synthesis of end-tagged HS-related di- and oligosaccharides.
Synthesis of heparin-like di- and oligosaccharides bearing a functional non-reducing end tether
Thioglycoside derivatives 17 and 18 were readily converted to their respective trichloroacetimidate donors in high yield, following NBS-mediated hydrolysis. Subsequent glycosylations with iduronate acceptor 19 afforded the expected high selectivity for the desired α-1,4 linkage, affording 20 (for H1′ δ: 4.61 ppm, d, J = 3.6 Hz) and 21 (for H1′ δ: 4.72 ppm, d, J = 3.6 Hz). Synthesis of both of these novel O4-modified disaccharides could be readily carried out on multi-hundred mg scale with the synthesis of 20 additionally scaled to gram level (Scheme 6). In each case iduronate accepter 19 was recovered (around 30% for 21 and 35% for 20) following chromatography, suggesting a lower reactivity of the donor thioglycoside in these systems, compared to direct analogues lacking the modified O4-unit.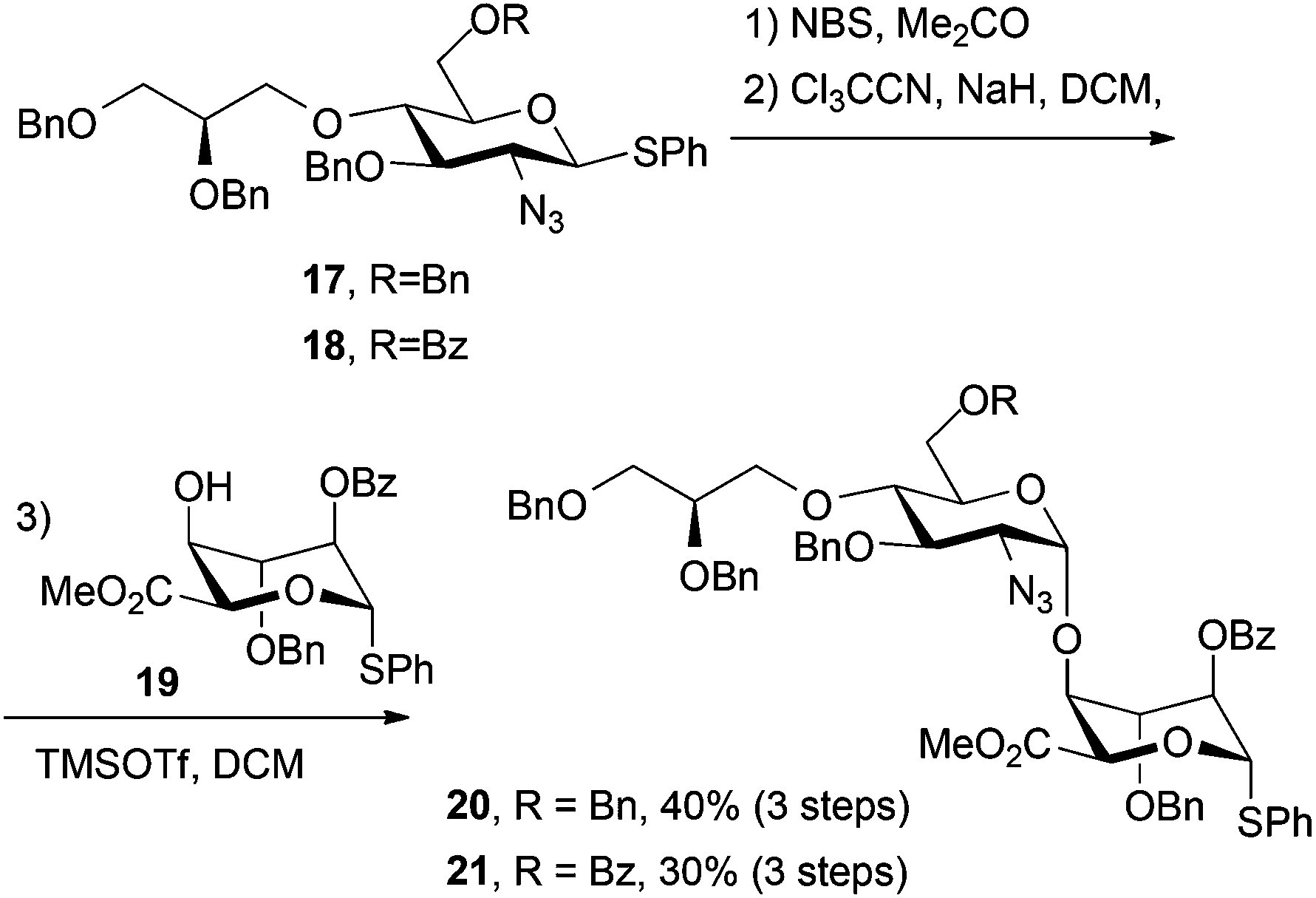 | ||
| Scheme 6 Synthesis of O4-end modified disaccharide donors. | ||
We attempted the direct alkylation of an analogue of disaccharide 21, containing a free 4-OH at the non-reducing end terminus, with triflate 14, but no product was observed. With recovery of iduronate acceptor in disaccharide synthesis and the poor alkylation of a disaccharide, this overall validated the more convergent strategy of including an O4 modification at the disaccharide level, and not by attempting its introduction into longer oligosaccharide sequences.
With disaccharide building blocks 20 and 21 available we sought to demonstrate their application to synthesis of longer O4-tagged HS and LMWH-related oligosaccharides. To illustrate the capacity to access different lengths and backbone sulfation types, the strategy was as outlined in Scheme 7, capping acceptor octa- and decasaccharides with our differentially O6-derivatized O4-LAT-bearing “capper” disaccharides, which, after deprotection and sulfations, thereby aimed to provide O4-terminal diol tag bearing deca- and dodecasaccharides. This would establish this as a route to the longest currently available end-modified synthetic heparin- or LMWH-like oligosaccharides. It would also illustrate applicability to both different O6 sulfation systems, including LMWH. If successful, then release of an aldehyde terminus could be effected analogously to our O1-LAT release using NaIO4.
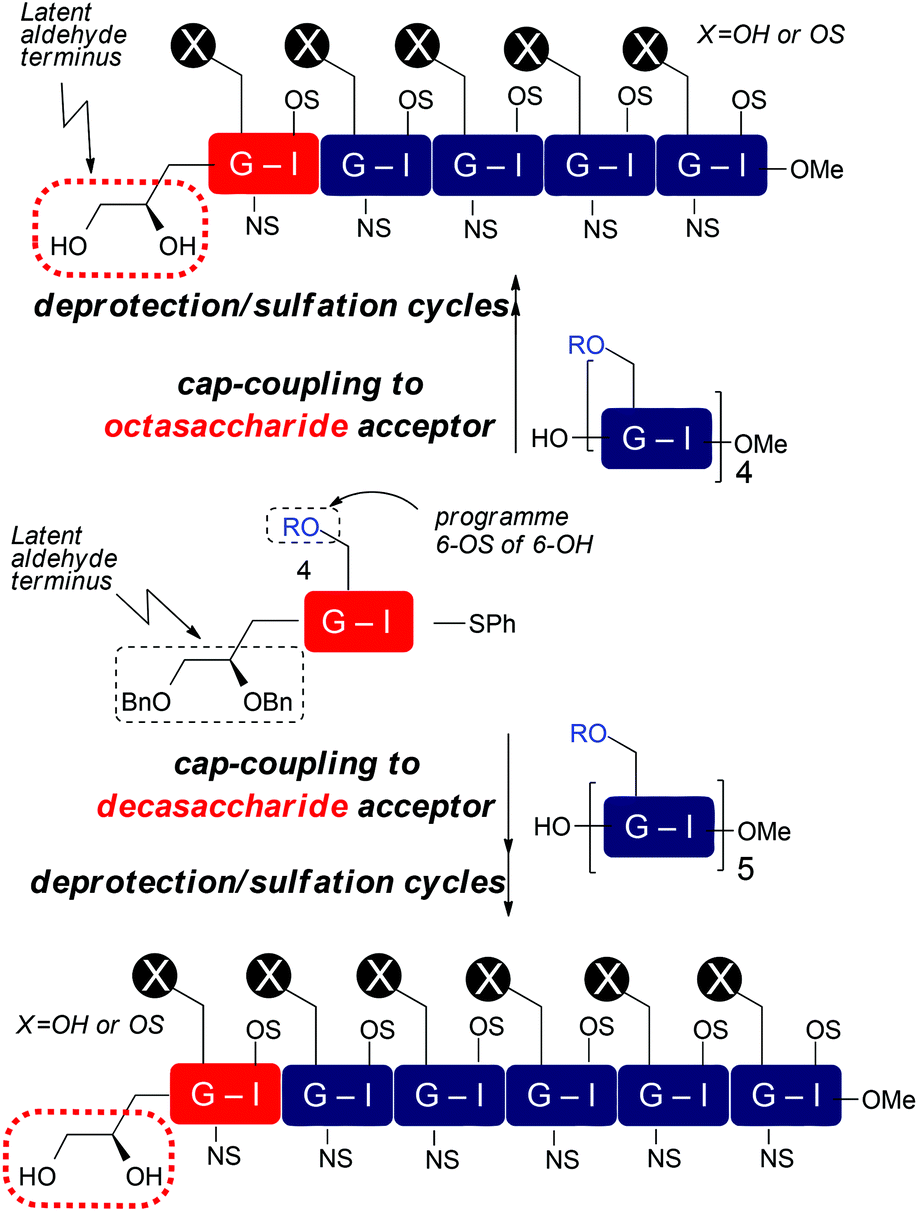 | ||
| Scheme 7 Strategy for end-capping to heparin and LMWHs bearing non-reducing terminal O4 diol tag, R = Bn or Bz. | ||
Accordingly, tagged glycosylation-capping of per-GlcN-6-OBz octasaccharide acceptor 224 with O4-modified disaccharide 21 using NIS and AgOTf at 0 °C, provided the novel end-tagged decasaccharide 25 in good yield and high anomeric purity (Scheme 8). Further, the novel dodecasaccharide 24 was obtained in similarly good yield through tagged glycosylation-capping of decasaccharide acceptor 235a with O4-modified disaccharide 20.
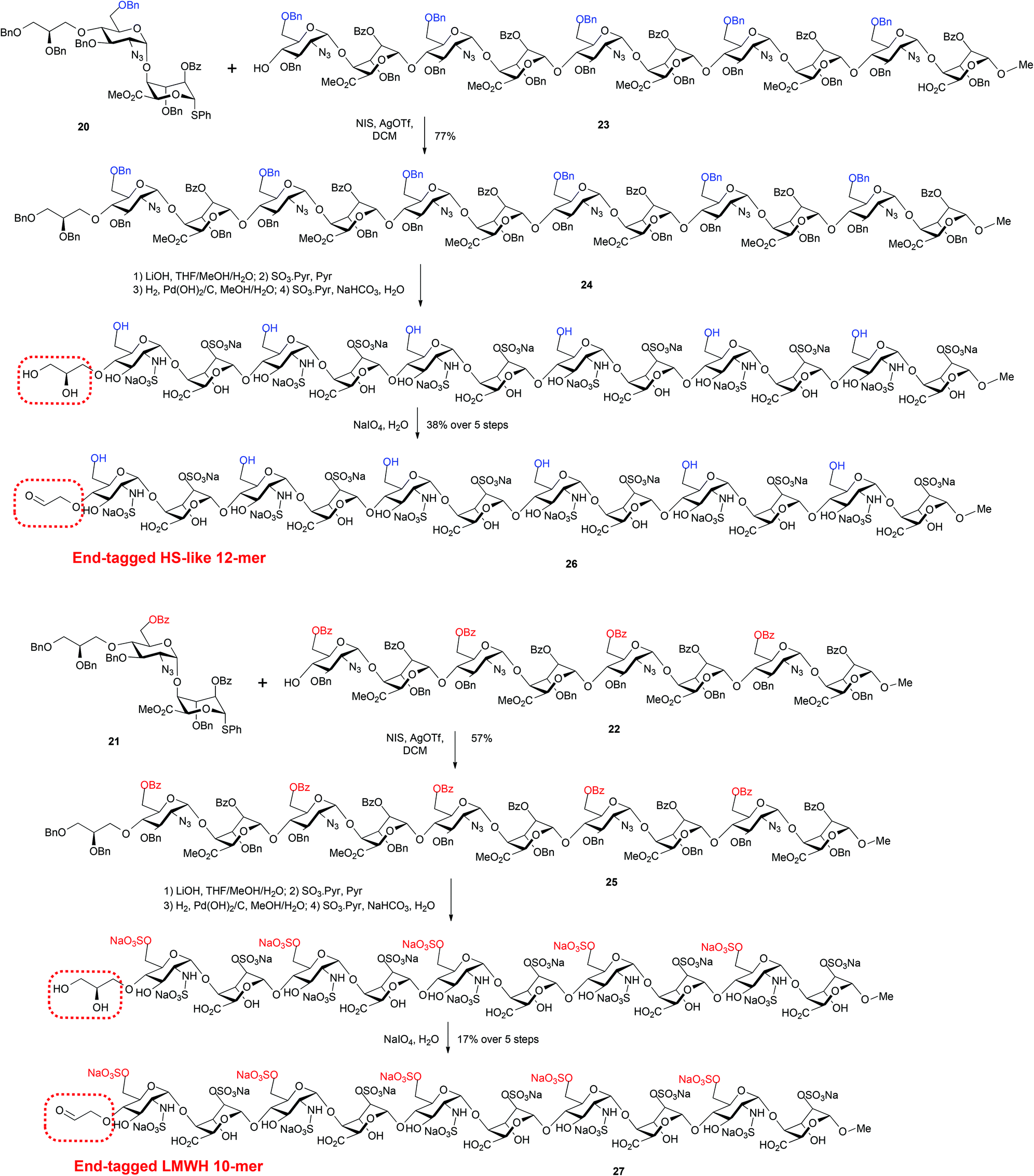 | ||
| Scheme 8 Synthesis of LMWH deca- and HS-like dodecasaccharides with non-reducing end latent aldehyde tethers and conversion to terminal aldehydes. | ||
Finally, the fully protected oligosaccharides 24 and 25 were subjected to standard deprotection and O/N-sulfation steps to afford the native-type species 26 and 27 bearing non-reducing end reactive aldehyde groups (Scheme 8).
Analysis of dodecasaccharide 26 by 800 MHz NMR spectroscopy indicated formation of a hemiacetal between the free aldehyde and the 3-position of the terminal glucosamine unit. This was not unexpected given the potential ease of cyclisation to form a terminal 6-membered ring system. We are however confident that the material was homogenous in terms of the installed sulfation pattern and that the latent motif, whilst preferring to exist in solution predominantly as an acetal, would react as an aldehyde in subsequent conjugation/labelling studies. This was previously demonstrated on our reducing end analogue HS-like dodecasaccharide, which behaved smilarly.11
Conclusions
A reliable and scalable synthesis of two non-reducing end modified GlcN-IdoA disaccharides containing a latent aldehyde group enables inclusion into LMWH- and HS-related deca- and dodecasaccharides. These are the first examples of such O4-modified heparin-like oligosaccharides. This provides a new approach to install an O4 tag onto defined HS oligosaccharide mimetics, which will enable applications in a variety of labelling and conjugation chemistries. The disaccharide units are generic tools that should find utility for final iteration stage coupling to diverse heparin-like oligosaccharides. These reagents also offer scope for exploitation in synthesis of other D-GlcN-containing oligosaccharides.Experimental section
Phenyl 2-azido-3,6-di-O-benzyl-4-O-acetoxyethyl-2-deoxy-1-thio-α-D-glucopyranoside (2)
Glucosamine derivative 118 (2.0 g, 4.2 mmol) was dissolved in dry THF (20 mL) under nitrogen. NaH (185 mg, 4.6 mmol, 60% in mineral oil) was added and the suspension stirred for 30 min at room temperature. Ethylbromoacetate (510 μL, 4.6 mmol) was added and the suspension heated at 55 °C overnight (16 h). TLC analysis (EtOAc/hexane, 3:1) showed starting material remained along with product at a slightly higher Rf. Successive additions of NaH and ethylbromoacetate were made at 16 h and 24 h. After this time a new spot was seen to form on the baseline of the TLC plate and the reaction was stopped (quenching with AcOH). The solution was taken in EtOAc/H2O (20 mL each), the layers separated and the organics washed with saturated aqueous NaHCO3, saturated aqueous NaCl, dried (MgSO4) and solvent removed in vacuo. The crude material was purified by silica gel flash chromatography, eluting with EtOAc/hexane, 8:1, 5:1 to yield 2 (666 mg, 1.1 mmol, 29%) as a clear gum. Rf 0.59 (EtOAc/hexane 1:3); [α]D +120.5 (c = 2.1, DCM); 1H NMR (400 MHz; CDCl3): δ 7.42 (ddt, J = 4.4, 2.8, 1.4 Hz, 2H, ArH), 7.31–7.18 (m, 13H, ArH), 5.52 (d, J = 5.2 Hz, 1H, H1), 4.85 (d, J = 10.5 Hz, 1H, CH2Ar), 4.77 (d, J = 10.5 Hz, 1H, CH2Ar), 4.54 (d, J = 11.9 Hz, 1H, CH2Ar), 4.37 (d, J = 11.9 Hz, 1H, CH2Ar), 4.36–4.34 (m, 1H, H5), 4.24 (d, J = 15.8 Hz, 1H, CH2C(O)OEt), 4.08–4.03 (m, 3H, CH2C(O)OEt, H6B, H4), 3.85–3.74 (m, 3H, OCH2CH3, H2), 3.61 (dd, J = 11.0, 1.9 Hz, 1H, H6A), 3.55 (dd, J = 9.9, 8.7 Hz, 1H, H3), 1.17 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C NMR (100 MHz; CDCl3): δ 169.6, 137.9, 137.6, 133.4, 132.2, 129.1, 128.5, 128.4, 128.2, 128.0, 127.8, 127.7, 87.2, 81.7, 79.1, 75.7, 73.4, 71.4, 70.3, 68.3, 64.1, 61.0, 14.3; MS ES [M + Na]+m/z 586.0; HRMS (TOF+) m/z calcd for C30H33N3O6SNa [M + Na]+ 586.1988, found 586.1995.
Phenyl 2-azido-3,6-di-O-benzyl-4-O-acetoxy-2-deoxy-1-thio-α-D-glucopyranoside (3)
:1). The reaction mixture was cooled back to 0 °C, acidified to pH 4 using hydrochloric acid (1 M), poured onto brine (25 mL) and extracted with ethyl acetate (3 × 40 mL). The organic layers were combined, washed with brine (25 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude product was purified by silica gel flash chromatography eluting with hexane/ethyl acetate, 4:1 to give acid 3 (199 mg, 0.37 mmol, 81%) as a colourless oil. Rf = 0.21 (EtOAc/Hex, 1:2.5); [α]D = +79.2 (c = 1.5, CHCl3); 1H NMR (400 MHz; CDCl3) δ 7.40–7.39 (m, 2H, ArH), 7.30–7.16 (m, 13H, ArH), 5.52 (d, J = 5.4 Hz, 1H, H1), 4.88 (d, J = 10.7 Hz, 1H, CH2Ar), 4.72 (d, J = 10.7 Hz, 1H, CH2Ar), 4.54 (d, J = 11.9 Hz, 1H, CH2Ar), 4.36 (d, J = 11.9 Hz, 1H, CH2Ar), 4.31–4.23 (m, 2H, H5, CH2CO2H), 4.08 (d, J = 16.7 Hz, 1H, CH2CO2H), 3.84 (dd, J = 10.2, 5.4 Hz, 1H, H2), 3.75–3.70 (m, 2H, H4, H6B), 3.57–3.51 (m, 2H, H3, H6A); 13C NMR (100 MHz, CDCl3) δ 137.2, 136.8, 132.9, 131.9, 129.0, 128.9, 128.5, 128.4, 128.2, 128.1, 127.7, 127.6, 127.5, 86.9, 80.7, 78.8, 73.3, 70.9, 69.9, 68.1, 63.9; MS NSI [M − H]−m/z 534.2; HRMS (FTMS NSI−) m/z calcd for C28H28N3O6S1 [M − H]− 534.1704, found 534.1710.
:1) then indicated the reaction was incomplete and further NaH (70 mg, 2.92 mmol) and iodoacetic acid (0.2 mL, 0.48 mmol) were added and stirring continued for 6 h at 40 °C. Upon completion (as adjudged by TLC) the reaction solution was cooled to ambient temperature, diluted with distilled H2O (15 mL) and extracted with EtOAc (4 × 10 mL). The combined organics were then washed with H2O (5 × 10 mL), dried (MgSO4) and solvent removed in vacuo. The crude acid 3 was purified by silica gel flash chromatography eluting with hexane:EtOAc, 3:1, 1:1 to afford 3 (800 mg, 1.50 mmol, 71%) as a pale orange oil. Analytical data matched those above.
:2). The reaction was diluted with diethyl ether and washed with water three times. The organic phase was dried (MgSO4) and concentrated in vacuo. The crude material was purified by silica gel flash chromatography eluting with hexane/ethyl acetate, 3:1 to yield 6 as a yellow oil (40 mg, 0.05 mmol, 42%) Rf = 0.71 (hexane/EtOAc 3:1); [α]D (c = 1.5, DCM) +24.9; 1H NMR (400 MHz; CDCl3) δ 8.31 (d, J = 8.6 Hz, 1H, NBD-ArH), 7.79–7.76 (brs, 1H, NH), 7.60–7.58 (brs, 1H, NH), 7.42–7.38 (m, 2H, ArH), 7.32–7.18 (m, 13H, ArH), 5.87 (d, J = 8.6 Hz, 1H, NBD-ArH), 5.54 (d, J = 5.5 Hz, 1H, H1), 5.01 (d, J = 10.6 Hz, 1H, CH2Ar), 4.61 (d, J = 10.6 Hz, 1H, CH2Ar), 4.48 (d, J = 11.7 Hz, 1H, CH2Ar), 4.40 (d, J = 11.7 Hz, 1H, CH2Ar), 4.23–4.19 (m, 2H, CH2C(O)NH, H6B), 4.12 (d, J = 16.3 Hz, 1H, CH2C(O)NH), 3.86 (dd, J = 10.2, 5.5 Hz, 1H, H2), 3.68–3.64 (m, 2H, H3, H5), 3.56–3.51 (m, 2H, H4, H6A), 3.10–3.07 (m, 3H, NHCH2CH2NH), 2.91–2.87 (m, 1H, HCH2CH2NH); 13C NMR (100 MHz, CDCl3) δ 172.4, 144.2, 143.9, 137.1, 136.8, 136.6, 132.9, 132.1, 129.2, 129.1, 128.9, 128.6, 128.5, 128.4, 128.3, 128.1, 128.0, 87.2, 81.1, 78.3, 76.1, 73.7, 71.4, 70.7, 68.4, 64.4, 37.5; MS ES [M + Na]+m/z 763.0; HRMS (TOF+) m/z calcd for C36H36N8O8NaS+: 763.2270, found: 763.2293; IR (neat) νmax 3280, 2922, 2104, 1663, 1579, 1294, 1260, 1103, 1047 cm−1; UV absorption λmax 489 nm, εmax 1.19 × 104 L mol−1 cm−1 (CHCl3); fluorescence emission λmax 716 nm.
:1). The reaction solution was diluted with diethyl ether (20 mL) and washed with water (7 × 20 mL). The diethyl ether was removed in vacuo giving the crude product as a pale yellow oil. This was purified by silica gel flash chromatography, eluting with EtOAc/hexane, 4:1 to yield 7 (45 mg, 0.05 mmol, 83%) as a colourless oil. Rf = 0.5 (EtOAc/Hex, 4:1); [α]D +48.7 (c = 1.5, DCM); 1H NMR (400 MHz; CDCl3) δ 8.21 (d, J = 9.3 Hz, 1H, ArH), 8.08 (d, J = 7.1 Hz, 2H, ArH), 8.01 (d, J = 8.2 Hz, 2H, ArH), 7.94 (s, 2H, ArH), 7.90 (t, J = 7.6 Hz, 1H, ArH), 7.77 (d, J = 7.8 Hz, 1H, ArH), 7.41–7.40 (m, 3H, ArH), 7.27–7.18 (m, 12H, ArH), 6.73 (t, J = 5.8 Hz, 1H, NH), 5.51 (d, J = 5.4 Hz, 1H, H1), 5.42–5.40 (brs, 1H, NH), 4.84 (d, J = 10.5 Hz, 1H, CH2Ar), 4.61 (d, J = 10.5 Hz, 1H, CH2Ar), 4.46 (d, J = 11.9 Hz, 1H, CH2Ar), 4.36 (d, J = 11.9 Hz, 1H, CH2Ar), 4.22–4.19 (m, 1H, H5), 4.14 (d, J = 15.3 Hz, 1H, CH2C(O)NH), 3.95 (d, J = 15.3 Hz, 1H, CH2C(O)NH), 3.83 (dd, J = 10.2, 5.4 Hz, 1H, CH2Pyr), 3.72–3.48 (m, 6H, H2, H3, H4, H6ab, CH2Pyr × 1), 3.30 (t, J = 7.2 Hz, 2H, CH2C(O)), 3.13–3.02 (m, 3H, CH2NH), 2.88–2.79 (m, 1H, CH2NH), 2.18–2.12 (m, 2H, CH2 alkyl), 1.31–1.10 (m, 8H, CH2 alkyl);13C NMR (100 MHz, CDCl3) δ 172.7, 169.1, 137.3, 136.9, 135.9, 133.1, 132.1, 132.0, 131.4, 130.9, 129.9, 129.2, 128.7, 128.6, 128.4, 128.3, 128.0, 127.9, 127.8, 127.5, 127.4, 126.8, 125.9, 125.0, 124.9, 124.8, 123.4, 87.2, 81.0, 78.3, 75.9, 73.6, 71.5, 68.3, 64.2, 39.2, 38.4, 36.1, 32.8, 29.5, 27.5, 26.1; MS ES [M + Na]+m/z 926.0; HRMS (TOF+) m/z calcd for C54H58N5O6S: 904.4130, Found: 904.4081; IR (neat) νmax 3313, 3033, 2927, 2858, 2105, 1649, 1535, 1262, 1101, 1072, 1045, 1026 cm−1; UV absorption λmax1 279 nm, λmax2 329 nm, εmax1 3.42 × 104 L mol−1 cm−1, εmax2 2.69 × 104 L mol−1 cm−1 (CHCl3); fluorescence emission λmax1 327 nm, λmax2 207 nm.
Phenyl-2-azido-2-deoxy-3,6-di-O-benzyl-4-O-[(S)-2,3-bis(benzyloxy)propoxy]-1-thio-β-D-glucopyranoside (17)
To 1 (2.67 g, 5.60 mmol) was added dry DMF (30 mL) under N2 and the solution cooled to 0 °C. NaH (60% in mineral oil) (248.0 mg, 6.2 mmol) was added in two portions over 30 min while being kept under N2. 1423 (2.49 g, 6.2 mmol) in dry DMF (20 mL) was then added dropwise and the suspension allowed to warm to RT and stirred for a further 3 h. TLC analysis (3/1, hexane/EtOAc) showed the reaction to be complete and quenching was effected with aqueous NaHCO3 (1 mL). The solution was partitioned between EtOAc and H2O. The layers were separated and the organic phase washed with 1 M HCl, H2O, brine, dried (MgSO4), filtered and evaporated. The crude product was purified by flash column chromatography (EtOAc/hexane gradient 1:9) yielding 17 (3.70 g, 5.1 mmol, 90%) as a clear oil. Rf 0.71 (EtOAc/hexane 1:3); [α]D −19.1 (c = 3.0, DCM); 1H NMR (400 MHz; CDCl3) δ 7.53–7.50 (m, 2H, ArH), 7.27–7.15 (m, 23H, ArH), 4.76 (d, J = 10.5 Hz, 1H, CH2Ar), 4.73 (d, J = 10.6 Hz, 1H, CH2Ar), 4.55 (d, J = 12.0 Hz, 1H, CH2Ar), 4.51 (d, J = 12.0 Hz, 1H, CH2Ar), 4.45–4.38 (m, 4H, 2 × CH2Ar), 4.32 (d, J = 10.1 Hz, 1H, H1), 3.92–3.88 (m, 1H, H6A or H6B), 3.72–3.57 (m, 4H, H5, H6A or H6B, CH2CH[OBn]CH2OBn), 3.45–3.44 (m, 2H, CH2CH[OBn]CH2OBn), 3.38–3.32 (m, 3H, H3, H4, CH2CH[OBn]CH2OBn), 3.22 (dd, J = 10.0, 9.0 Hz, 1H, H2); 13C NMR (100 MHz; CDCl3) δ 138.5, 138.4, 138.2, 137.7, 133.7, 131.2, 129.1, 128.5, 128.4, 128.2, 128.0, 127.8, 127.7, 127.6, 127.5, 85.9, 84.9, 79.4, 78.2, 77.3, 75.8, 73.5, 73.4, 73.2, 72.4, 69.8, 68.8, 64.9; MS ES [M + NH4]+m/z 750.0; HRMS (ES-TOF+) m/z calcd for C43H49N4O6S [M + NH4]+ 749.3368, found 749.3365.
Phenyl-2-azido-2-deoxy-3-O-benzyl-4-O-[(S)-2,3-bis(benzyloxy)propoxy]-6-O-benzoyl-1-thio-β-D-glucopyranoside (18)
To 1612 (1.83 g, 3.72 mmol) was added dry THF (20 mL) under N2 and the solution cooled to 0 °C. NaH (60% in mineral oil) (164.0 mg, 4.10 mmol) was added in two portions over 30 min. while being kept under N2. 1423 (1.81 g, 4.46 mmol) in dry THF (10 mL) was then added dropwise and the suspension allowed to warm to RT and stirred for a further 2 h. TLC analysis (3/1, hexane/EtOAc) showed the reaction to be virtually complete (Note: allowing the reaction continue for longer resulted in no further consumption of starting material and the formation of another product; suggested to be transesterification) and quenching was effected with glacial AcOH (4.1 mmol) and solvents removed in vacuo. The solution was partitioned between EtOAc and H2O. The layers were separated and the organic phase washed with 1 M HCl, H2O, brine, dried (MgSO4), filtered and evaporated. The crude product was purified by flash column chromatography (EtOAc/hexane gradient 1:9, 1:7) yielding 18 (1.26 g, 1.60 mmol, 69%) as a clear oil along with recovered starting material 18 (979 mg). Rf 0.59 (EtOAc/hexane 1:3); [α]D −19.7 (c = 3.8, DCM); 1H NMR (400 MHz; CDCl3) δ 7.97–7.94 (m, 2H, ArH), 7.56–7.52 (m, 1H, ArH), 7.45–7.38 (m, 4H, ArH), 7.30–7.11 (m, 16H, ArH), 7.02–6.98 (m, 2H, ArH), 4.76 (d, J = 10.4 Hz, 1H, CH2Ar), 4.71 (d, J = 10.4 Hz, 1H, CH2Ar), 4.71 (dd, J = 12.0, 2.0 Hz, 1H, H6A or H6B), 4.56 (d, J = 12.0 Hz, 1H, CH2Ar), 4.52 (d, J = 12.0 Hz, 1H, CH2Ar), 4.42 (d, J = 12.1 Hz, 1H, CH2Ar), 4.39 (d, J = 12.0 Hz, 1H, CH2Ar), 4.35–4.31 (m, 2H, H1, H6A or H6B), 3.99–3.98 (m, 1H, CH2CH[OBn]CH2OBn), 3.64–3.58 (m, 2H, CH2CH[OBn]CH2OBn, CH2CH[OBn]CH2OBn), 3.54–3.50 (m, 1H, H5), 3.49–3.47 (m, 2H, CH2CH[OBn]CH2OBn), 3.40 (t, J = 9.6 Hz, 1H, H3), 3.34 (t, J = 9.6 Hz, 1H, H4), 3.20 (dd, J = 10.1, 9.6 Hz, 1H, H2); 13C NMR (100 MHz; CDCl3) δ 166.0, 138.4, 138.1, 137.5, 134.2, 133.4, 130.5, 130.0, 129.8, 129.0, 128.7, 128.6, 128.5, 128.4, 128.2, 127.8, 127.7, 85.6, 84.7, 78.4, 77.4, 77.2, 76.0, 73.5, 72.5, 69.5, 64.8, 63.1; MS ES [M + Na]+m/z 768.0; HRMS (TOF+) m/z calcd for C43H49N5O6S [M + NH4]+ 763.3399, found 763.3398.
Methyl (phenyl 4-O-(2-azido-3,6-di-O-benzyl-2-deoxy-4-O-[(S)-2,3-bis(benzyloxy)propoxy]-α-D-glucopyranosyl)-2-O-benzoyl-3-O-benzyl-1-thio-α-L-idopyranoside)-uronate (20)
To 17 (500 mg, 0.68 mmol) was added acetone (5.0 mL) and the mixture cooled to 0 °C in an icebath. N-Bromosuccinimide (242 mg, 1.36 mmol) was then added. After 3 h the reaction was quenched by addition of aqueous NaHCO3 (2.0 mL) and the solution stripped. The crude mixture was purified by flash column chromatography using EtOAc/hexane (1:4, 1:3) as the eluent to yield a mixture of hemi-acetals (333 mg, 0.52 mmol, 76%) as a white foam. The mixture of anomers was used immediately. Rf 0.14 (EtOAc/hexane 1:3); MS ES: [M + Na]+m/z 663; HRMS (TOF-ES+): m/z calcd for C37H41N3O7Na [M + Na]+: 662.2837, found 662.2836. See ESI† for copies of 1H and COSY NMR spectra. To the mixture of hemi-acetals (1.8 g, 2.8 mmol) was added dry DCM (40 mL), CCl3CN (1.40 mL, 14.0 mmol) and sodium hydride 60% (11.0 mg, 0.3 mmol). The solution was stirred for 1 h. The solvent was evaporated and the crude residue purified by flash column chromatography using EtOAc/hexane (1:4 + 1% NEt3) as eluent. This yielded the desired imidate (1.70 g, 2.1 mmol, 95%) as a white foam and as a 5/1 (α/β) mixture, as determined by 1H NMR. This compound was used immediately. Rf 0.50 (EtOAc/hexane 1:3); HRMS (FTMS-NSI+): m/z calcd for C39H45N5O7 [M + NH4]+ 800.2379, found 800.2377. See ESI† for copy of 1H NMR spectrum. To iduronic ester acceptor 1912 (833 mg, 1.78 mmol) and the above imidate (1.70 g, 2.14 mmol) was twice added dry toluene and the solvent evaporated. The residue was dried under high vacuum for 2 h and, while kept under nitrogen, dry DCM (25 mL) was added. The solution was cooled to −30 °C using a 65:35 mixture of iPrOH/H2O and a dry ice bath and TMSOTf (16 μL, 0.09 mmol) was then added. After 2 h the reaction was quenched with two drops of NEt3, solvents were removed in vacuo and flash column chromatography (EtOAc/hexane, 1:5, 1:2) as the eluent yielded 20 (1.10 g, 0.99 mmol, 55%) as a white foam, along with recovered acceptor 19 (300 mg); Rf 0.31 (EtOAc/hexane 1:3); [α]D (c = 1.0, DCM) +327.1; 1H NMR (400 MHz; CDCl3) δ 8.07–8.04 (m, 2H, ArH), 7.49–7.44 (m, 2H, ArH), 7.43–7.38 (m, 2H, ArH), 7.35–7.29 (m, 4H, ArH), 7.27–7.09 (m, 23H, ArH), 7.04–7.00 (m, 2H, ArH), 5.72 (s, 1H, H5), 5.35 (s, 1H, H4), 5.29 (d, J = 2.0 Hz, 1H, H1), 4.91 (d, J = 11.7 Hz, 1H, CH2Ph), 4.69 (d, J = 11.7 Hz, 1H, CH2Ph), 4.61 (d, J = 3.6 Hz, 1H, H1′), 4.55–4.48 (m, 2H, CH2Ph), 4.43 (s, 2H, CH2Ph), 4.38–4.31 (m, 2H, CH2Ph), 4.18 (d, J = 10.6 Hz, 1H, CH2Ph), 4.12 (t, J = 2.8 Hz, 1H, H3), 3.97 (s, 1H, H2), 3.81 (dd, J = 9.7, 3.3 Hz, 1H, H6A′), 3.71–3.63 (m, 3H, CH2Ph, CH2CH[OBn]CH2OBn), 3.61 (s, 3H, OCH3), 3.59–3.37 (m, 7H, H5′, H6B′, H4′, H3′, CH2CH[OBn]CH2OBn, CH2CH[OBn]CH2OBn,), 3.14 (dd, J = 10.0, 3.7 Hz, 1H, H2′); 13C NMR (100 MHz; CDCl3) δ 169.3, 165.6, 138.6, 138.2, 137.9, 137.8, 137.1, 135.4, 133.4, 131.5, 130.0, 129.5, 129.1, 128.8, 128.5, 128.4, 128.3, 128.2, 128.1, 127.9, 127.8, 127.6, 127.6, 127.5, 100.4, 87.0, 80.0, 78.0, 77.6, 77.4, 74.6, 73.4, 73.3, 73.1, 72.8, 72.4, 72.2, 71.6, 70.1, 69.2, 68.4, 67.6, 63.6, 52.3; MS ES [M + NH4]+m/z 1133.5; HRMS (FTMS-NSI+) m/z calcd for C64H69N4O13S [M + NH4]+ 1133.4576, found 1133.4578.
Methyl (phenyl 4-O-(2-azido-3-O-benzyl-6-O-benzoyl-2-deoxy-4-O-[(S)-2,3-bis(benzyloxy)propoxy]-α-D-glucopyranosyl)-2-O-benzoyl-3-O-benzyl-1-thio-α-L-idopyranoside)-uronate (21)
To 18 (1.26 g, 1.60 mmol) was added acetone (15.0 mL) and the mixture cooled to 0 °C in an icebath. N-Bromosuccinimide (569 mg, 3.20 mmol) was then added. After 2 h the reaction was quenched by addition of aqueous NaHCO3 (2.0 mL) and the solution stripped. The crude mixture was purified by flash column chromatography using EtOAc/hexane (1:4) as the eluent to yield the mixture of hemi-acetals (963 mg, 1.40 mmol, 87%) as a pale yellow oil. This mixture of anomers was used immediately. Rf 0.24 (EtOAc/hexane 1:3); MS ES: [M + Na]+m/z 676; HRMS (TOF-ES+): m/z calcd for C37H39N3O8Na [M + Na]+: 676.2630, found 676.2634. See ESI† for copies of 1H and COSY NMR spectra. To this mixture of hemi-acetals (963 mg, 1.40 mmol) was added dry DCM (10 mL), CCl3CN (421 μL, 4.20 mmol) and sodium hydride 60% (6.0 mg, 0.14 mmol). The solution was stirred for 2 h. The solvent was evaporated and the crude residue purified by flash column chromatography using EtOAc/hexane (1:5 + 1% NEt3) as eluent. This yielded the desired imidate (918 mg, 1.09 mmol, 77%) as a pale yellow foam and a 95/5 α/β mixture, as determined by 1H NMR. This compound was used immediately. Rf 0.57 (EtOAc/hexane 1:3). See ESI† for copy of 1H NMR spectrum. To iduronic ester acceptor 1912 (441 mg, 0.89 mmol) and the above imidate (900 mg, 1.07 mmol) was twice added dry toluene and the solvent evaporated. The residue was dried under high vacuum for 2 h and, while kept under nitrogen, dry DCM (15 mL) was added. The solution was cooled to −30 °C using a 65:35 mixture of iPrOH/H2O and a dry ice bath and TMSOTf (8 μL, 0.04 mmol) was then added. After 0.5 h the reaction was quenched with two drops of NEt3, solvents were removed in vacuo and flash column chromatography (EtOAc/hexane, 1:7, 1:6, 1:5, 1:3) as the eluent yielded 21 (472 mg, 0.42 mmol, 47%) as a white foam, along with recovered acceptor 19 (123 mg). Rf 0.44 (EtOAc/hexane 1:3); [α]D (c = 0.75, DCM) +287.1; 1H NMR (400 MHz; CDCl3) δ 8.18 (dd, J = 8.4, 1.3 Hz, 2H, ArH), 8.02 (dd, J = 8.4, 1.3 Hz, 2H, ArH), 7.60–7.58 (m, 3H, ArH), 7.52–7.50 (m, 2H, ArH), 7.46–7.40 (m, 6H, ArH), 7.37–7.24 (m, 16H, ArH), 7.22–7.20 (m, 2H, ArH), 7.15 (td, J = 3.7, 1.7 Hz, 2H, ArH), 5.84 (brs, 1H, H1), 5.46 (brs, 1H, H2), 5.43 (d, J = 2.0 Hz, 1H, H5), 5.02 (d, J = 11.7 Hz, 1H, CH2Ph), 4.81–4.77 (m, 2H, CH2Ph, H6A), 4.72 (d, J = 3.6 Hz, 1H, H1′), 4.66–4.59 (m, 2H, CH2Ph), 4.57–4.49 (m, 2H, CH2Ph), 4.43 (dd, J = 12.4, 2.7 Hz, 1H, H6B), 4.25–4.22 (m, 2H, H3, CH2Ph), 4.11 (brs, 1H, H4), 4.06–4.00 (m, 2H, H5′, CH2CH[OBn]CH2OBn), 3.84–3.81 (m, 4H, OCH3, CH2Ph), 3.73–3.67 (m, 2H, CH2CH[OBn]CH2OBn), 3.62–3.60 (m, 2H, CH2CH[OBn]CH2OBn), 3.55–3.44 (m, 2H, H3′, H4′), 3.23 (dd, J = 9.9, 3.6 Hz, 1H, H2′); 13C NMR (100 MHz; CDCl3) δ 167.0, 163.7, 163.3, 136.2, 135.9, 135.4, 134.9, 133.2, 131.2, 130.9, 129.2, 127.8, 127.7, 127.4, 126.9, 126.8, 126.6, 126.3, 126.2, 126.1, 126.0, 125.9, 125.7, 125.5, 125.4, 123.1, 97.9, 84.8, 77.7, 76.5, 72.5, 71.2, 71.1, 70.5, 70.2, 69.8, 68.1, 67.5, 67.1, 66.1, 61.6, 50.3; MS ES [M + Na]+m/z 1153; HRMS (FTMS-NSI+) m/z calcd for C64H67N4O14S1 [M + NH4]+ 1147.4369, found 1147.4357.
:1); [α]D (c = 1.1, DCM) +42.9; 1H NMR (400 MHz; CDCl3) δ 8.13–8.10 (m, 3H, ArH), 8.01–7.92 (m, 11H, ArH), 7.54–7.07 (m, 116H, ArH), 5.57–5.56 (m, 5H, H1IdoA), 5.21–5.18 (m, 5H, H2IdoA), 5.10 (s, 1H, H1IdoA), 5.06 (s, 1H, H2IdoA), 4.97–4.90 (m, 6H, H1GlcN), 4.82–4.38 (m, 43H), 4.24–3.25 (m, 78H, incl. C(O)OCH3 × 6, OCH3); 13C NMR (100 MHz; CDCl3) δ 169.6, 169.4, 169.3, 167.7, 165.6, 165.2, 165.1, 138.6, 138.1, 138.0, 137.9, 137.8, 137.7, 137.5, 137.4, 133.6, 133.5, 132.3, 131.0, 130.0, 129.9, 129.8, 129.6, 129.4, 129.3, 129.2, 129.1, 129.0, 128.9, 128.8, 128.7, 128.6, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.3, 125.4, 100.3, 99.5, 99.3, 99.2, 98.2, 98.1, 98.0, 79.6, 78.3, 78.2, 78.1, 77.5, 77.4, 77.2, 76.9, 76.0, 75.6, 75.5, 75.4, 74.9, 74.4, 74.2, 74.0, 73.7, 73.6, 73.5, 73.4, 73.2, 72.5, 72.4, 71.6, 71.4, 70.7, 70.1, 68.0, 67.7, 67.4, 67.3, 67.2, 63.4, 63.3, 63.2, 63.1, 63.0, 61.7, 56.3, 52.1, 51.9, 51.8, 51.7, 51.6; (MS MALDI) m/z calcd for C264H268N18O69Na [M + Na]+ 4819.8, found 4819.8.
:95); [α]D (c = 0.5, DCM) +63.4; 1H NMR (400 MHz; CDCl3) δ 8.01–7.95 (m, 24H, ArH), 7.17–7.11 (m, 86H, ArH), 5.50–5.48 (m, 4H, H1IdoA), 5.13–5.08 (m, 4H, H2IdoA), 4.99 (brs, 1H, H1IdoA), 4.97 (brs, 1H, H2IdoA), 4.83–4.16 (m, 43H, CH2Ar × 23, H5IdoA, H1GlcN, H6ABGlcN), 4.06–4.70 (m, 21H, CH2Ar × 4, H3IdoA, H4IdoA, H5GlcN, H4GlcN × 4, CH2CH[OBn]CH2OBn), 3.65–3.56 (m, 2H, CH2CH[OBn]CH2OBn), 3.53 (s, 3H, OCH3), 3.47–3.42 (m, 14H, H3GlcN, H4GlcN × 1, CH2CH[OBn]CH2OBn C(O)OCH3 × 2), 3.25–3.04 (m, 14H, H2GlcN, C(O)OCH3 × 3); 13C NMR (100 MHz; CDCl3) δ 169.6, 169.3, 169.2, 166.0, 165.9, 165.6, 165.2, 165.1, 138.3, 138.0, 137.7, 137.5, 137.4, 137.3, 137.2, 133.7, 133.5, 133.1, 133.0, 132.5, 129.9, 129.6, 128.8, 128.7, 128.5, 128.4, 128.3, 128.2128.1, 128.0, 127.8, 127.6, 100.3, 99.1, 98.7, 98.4, 98.3, 98.3, 98.2, 98.1, 98.1, 98.0 anomeric carbons], 79.7, 78.6, 78.4, 78.3, 78.2, 77.8, 77.7, 77.6, 77.5, 77.3, 77.2, 77.0, 76.9, 76.7, 76.3, 75.9, 75.8, 75.7, 75.6, 75.4, 75.1, 75.0, 74.6, 74.5, 74.4, 74.3, 74.2, 73.9, 73.5, 72.4, 72.3, 71.7, 71.2, 70.2, 70.0, 69.9, 69.8, 69.6, 68.0, 67.1, 63.4 63.3, 63.2, 63.0, 62.3, 61.9, 61.8, 56.2, 52.3, 51.9, 51.7, 51.6, 51.6; (FTMS NSI+) m/z calcd for C223H225N17O63 [M+2NH4]2+ 2074.2457, found 2074.2422;
General method for saponification of oligosaccharides 24 and 25
Decasaccharide 25 (71.4 mg, 17.4 μmol) was dissolved in THF (1 mL) and MeOH (0.5 mL). The solution was cooled to 0 °C and LiOH (22.0 mg, 522.5 μmol) in H2O (0.5 mL) added dropwise, whereupon the solution turned a faint yellow colour. Stirring was continued for 5 h at this temperature, at room temperature overnight and quenched with 1 M HCl (520 μL). Solvents were removed in vacuo and the crude white solid purified by silica gel flash chromatography, eluting with DCM/MeOH, 95/5, 90/10 to give the decasaccharide acid (31.0 mg, 10.0 μmol, 60%) as a white solid. Rf 0.41 (MeOH/DCM 1:9); 1H NMR (400 MHz; Acetic acid, d4) δ 7.36–6.97 (m, 60H, ArH), 5.22–5.15 (m, 4H, H1IdoA), 5.03 (s, 3H, H1GlcN), 4.99–4.98 (m, 2H, H1GlcN), 4.87 (s, 1H, H1IdoA), 4.81–4.80 (m, 4H), 4.73 (s, 1H), 4.69–4.60 (m, 8H), 4.56–4.51 (m, 8H), 4.47–4.39 (m, 4H), 4.36 (d, J = 2.9 Hz, 2H), 4.33–4.25 (m, 4H), 4.13 (s, 1H), 4.04–4.01 (m, 5H), 3.96–3.77 (m, 21H), 3.69–3.40 (m, 30H), 3.33 (brs, 4H, includes OCH3); HRMS (FTMS NSI−) m/z calcd for C148H165N15O53 [M − 2H]2− 1500.0344, found 1500.0334.
Reaction of compound 24 (350 mg, 73.0 μmol) following the above procedure yielded the product dodecasaccharide acid (235 mg, 57.0 μmol, 79%). Rf 0.49 (MeOH/DCM 1:9); 1H NMR (400 MHz; MeOD) δ 7.56–7.16 (m, 100H, ArH), 5.32–5.30 (brs, 6H, H1IdoA), 5.14–5.12 (brs, 6H, H1GlcN), 4.78–3.44 (m, 60H, H2GlcN, H3GlcN, H4GlcN, H5GlcN, H6abGlcN, H2IdoA, H3IdoA, H4IdoA, H5IdoA); HRMS (FTMS NSI−) m/z calcd for C216H229N18O63 [M − 3H]3− 1360.8428, found 1360.8405.
General method for O-sulfation and hydrogenation of oligosaccharides
The above decasaccharide acid (31.0 mg, 10.0 μmol) was dissolved in dry pyridine (1 mL) under N2 at RT. SO3·Py complex (66 mg, 414 μmol) was added and the suspension stirred 50 °C for 5 h then at RT overnight. A white precipitate formed from the pale yellow solution after 2 h. TLC analysis (EtOAc/Pyr/H2O/AcOH, 6/5/3/1) showed no starting material remained and one product spot had formed at lower Rf. The reaction was quenched by addition of NaHCO3 (102 mg) in H2O (0.5 mL) and the solvents removed in vacuo (co-evaporationg with toluene). The crude white paste was then purified by silica gel flash chromatography, eluting with DCM/MeOH, 9/1, 4/1. The residue obtained was dissolved in MeOH/DCM (2 mL, v/v), stirred with Amberlite IR120+-Na+ resin for 12 h, filtered and solvent removed in vacuo to give target O-sulfated species (24.0 mg, 5.8 μmol, 56%) as a white solid which was used immediately in the next step. Rf 0.37 (MeOH/DCM/NH4OH 6:14:2). This material (11.1 mg, 2.7 μmol) was dissolved in MeOH (0.5 mL) and THF (0.5 mL) at RT. NaHCO3 (5 mg, 54 μmol) in H2O (0.5 mL) was added along with Pd(OH)2/C (10 mg) and the system purged with H2. Stirring was continued at 50 °C for 4 d. The suspension was then filtered through Celite® and solvents removed in vacuo. The residue was dissolved in water and desalted by passage through a Sephadex G-20 resin using water as eluent to reveal the free sugar (4.6 mg, 1.6 μmol, 59%) as a clear glass. 1H NMR (400 MHz; D2O) δ 5.07.5.05 (m, 3H, H1IdoA), 5.98–4.92 (m, 4H, H1GlcN), 4.88 (s, 1H, H1IdoA), 4.81–4.78 (m, 4H, H5IdoA), 4.36 (s, 1H, H5IdoA), 4.22–4.05 (m, 20H, H2IdoA, H3IdoA, H6GlcN), 3.99–3.70 (m, 13H, H4IdoA, H5GlcN, 3H from NRE tether), 3.65–3.42 (m, 11H, H3GlcN, H4GlcN, 2H from NRE tether), 3.28 (s, 3H, OCH3), 3.27–3.19 (m, H4GlcN), 2.69–2.59 (m, 5H, H2GlcN); HRMS (FTMS NSI−) m/z calcd for C64H99N5O83S10 [M − 6H]6− 430.8487, found 430.8491, calcd for C64H100N5O83S10 [M − 5H]5− 517.2198, found 517.2206.
Reaction of the above dodecasaccharide acid (170.0 mg, 42.0 μmol) following the above O-sulfation procedure yielded the target O-sulfated species (120.0 mg, 25.0 μmol, 60%) which was used immediately in the next step. Rf 0.69 (MeOH/DCM, 1:5); FTMS (NSI−) m/z calcd for C216H226N18O81S6 [M − 6H]6− 760.8746, found 760.8747. Reaction of this compound (65.0 mg, 13.5 μmol) following the above hydrogenation procedure yielded the free sugar (30.0 mg, 13.4 μmol, quant.). 1H NMR (400 MHz; D2O) δ 5.05–5.03 (brs, 5H, H1IdoA), 4.91–4.88 (m, 7H, H1GlcN × 6, H1IdoA), 4.78 (brs, 5H, H5IdoA), 4.36 (s, 1H, H5IdoA), 4.20 (brs, 5H, H2IdoA), 4.11–4.02 (m, 7H, H3IdoA × 6, H2IdoA), 3.95–3.87 (m, 6H, H4IdoA), 3.76–3.68 (m, 19H, H5GlcN H6ABGlcN, 1H NRE tether), 3.56–3.45 (15H, H3GlcN H4GlcN × 5, 4H NRE tether), 3.29 (s, 3H, OMe), 3.23–3.17 (m, 1H, H4GlcN), 2.59–2.55 (m, 6H, H2GlcN); HRMS (FTMS-NSI−) m/z calcd for C76H118N6O81S6 [M − 6H]6− 433.7276, found 433.7262, calcd for C76H119N6O81S6 [M − 5H]5− 520.6746, found 520.6732.
General method for N-sulfation of oligosaccharides
The decasaccharide free sugar (4.0 mg, 1.4 μmol) was dissolved in H2O (0.5 mL) at RT. NaHCO3 (10.1 mg, 120 μmol) and SO3·pyr complex (9.5 mg, 60.0 μmol) were then added and the suspension stirred at room temperature. Further additions of both reagents were made at 1 h, 3 h, 8 h, 20 h and 24 h. After 28 h solvent was removed in vacuo to reveal a crude white solid which was purified by passage through a Sephadex G-20 resin using water as eluent. The target material (4.6 mg, 1.4 μmol, 90%) was isolated as a clear glass. This material was used immediately in the periodate cleavage step.Reaction of the dodecasaccharide free sugar (30.0 mg, 10.8 μmol) following the above procedure yielded the product (35.0 mg, 9.1 μmol, 92%). 1H NMR (400 MHz; D2O) δ 5.21–5.14 (m, 11H, H1IdoA, H1GlcN), 4.90 (brs, 1H, H1IdoA), 4.75–4.73 (m, 5H, H5IdoA), 4.32 (s, 1H, H5IdoA), 4.21–4.19 (m, 6H, H2IdoA), 4.11–4.09 (m, 6H, H3IdoA), 3.95–3.45 (m, 41H, H4IdoA, H5GlcN H6ABGlcN, H3GlcN H4GlcN, NRE tether, 5H), 3.29 (s, 3H, OMe), 3.13–3.09 (m, 6H, H2GlcN); HRMS (FTMS-NSI−) m/z calcd for C76H118N6O99S12 [M − 6H]6− 513.6844, found 513.6840, C76H117N6O99S12 [M − 7H]6− 440.1570, found 440.1567.
General method for periodate cleavage of oligosaccharide NRE tether 26 and 27
The N-sulfated decasaccharide (4.6 mg, 1.4 μmol) was dissolved in H2O (200 μL) at room temperature. NaIO4 (0.4 mg, 1.5 μmol) was added and the solution shaken in a 2.0 mL Eppendorf tube for 24 h. The reaction solution was diluted up to 1.0 mL with H2O and passed through a Sephadex G-20 resin using water as eluent. The target material 27 (4.3 mg, 1.3 μmol, 95%) was isolated as a white glass after freeze-drying. 1H NMR (600 MHz; D2O) δ 5.27–2.24 (m, 5H, H1GlcN), 5.08 (brs, 4H, H1IdoA), 4.88 (s, 1H, H1IdoA), 4.78 (brs, 4H, H5IdoA), 4.39 (brs, 1H, H5IdoA), 4.21–4.00 (m, 23H), 3.89–3.77 (m, 11H), 3.67–3.65 (m, 6H), 3.26 (s, 3H, OCH3), 3.24–3.21 (m, 5H, H2GlcN); HRMS (FTMS NSI−) m/z calcd for C63H95N5O82S10 [M − 6H]6− 425.5110, found 425.5115.Reaction of the N-sulfated dodecasaccharide (30.0 mg, 8.6 μmol) following the above procedure yielded the product 26 (26.3 mg, 7.6 μmol, 88%). 1H NMR (800 MHz; D2O) δ showed a complex mixture of cyclic acetal forms. See ESI† for copies of 800 MHz 1D and 2D NMR spectra; HRMS (FTMS-NSI−) m/z calcd for C75H112N6O98S12 [M − 8H]8− 381.0066, found 381.0082.
Acknowledgements
MRC (G0601746 and G902173) and CRUK (C2075/A9106). The EPSRC National Mass Spectrometry Centre, Swansea, Dr Matthew Cliff (MIB NMR Facility) and Mr. Mark Howard (early D-GlcN work) are also thanked.Notes and references
- (a) R. Sasisekharan, Z. Shriver, G. Venkataraman and U. Narayanasami, Nat. Rev. Cancer, 2002, 2, 521–528 CrossRef CAS PubMed; (b) B. Casu, A. Naggi and G. Torri, Matrix Biol., 2010, 29, 442–452 CrossRef CAS PubMed; (c) J. Bishop, M. Schuksz and J. D. Esko, Nature, 2007, 446, 1030–1037 CrossRef CAS PubMed; (d) P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed; (e) F. Baleux, L. Loureiro-Morais, Y. Hersant, P. Clayette, F. Arenzana-Seisdedos, D. Bonnaffé and H. Lortat-Jacob, Nat. Chem. Biol., 2009, 5, 743–748 CrossRef CAS PubMed.
- (a) Y. Xu, S. Masuko, M. Takieddin, H. Xu, R. Liu, J. Jing, S. A. Mousa, R. J. Linhardt and J. Liu, Science, 2011, 334, 498–501 CrossRef CAS PubMed; (b) Y. Xu, C. Cai, K. Chandarajoti, P.-H. Hsieh, L. Li, T. Q. Pham, E. M. Sparkenbaugh, J. Sheng, N. S. Key, R. Pawlinski, E. N. Harris, R. J. Linhardt and J. Liu, Nat. Chem. Biol., 2014, 10, 248–250 CrossRef CAS PubMed; (c) G. Tiruchinapally, Z. Yin, M. El-Dakdouki, Z. Wang and X. Huang, Chem. – Eur. J., 2011, 17, 10106–10112 CrossRef CAS PubMed.
- R. Schwörer, O. Zubkova, J. E. Turnbull and P. C. Tyler, Chem. – Eur. J., 2013, 19, 6817–6823 CrossRef PubMed.
- G. J. Miller, S. U. Hansen, E. Avizienyte, G. Rushton, C. Cole, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2013, 4, 3218–3222 RSC.
- (a) S. U. Hansen, G. J. Miller, G. C. Jayson and J. M. Gardiner, Org. Lett., 2013, 15, 88–91 CrossRef CAS PubMed; (b) G. C. Jayson, S. U. Hansen, G. J. Miller, C. L. Cole, G. Rushton, E. Avizienyte and J. M. Gardiner, Chem. Commun., 2015, 51, 13846–13849 RSC.
- (a) S. Maza, M. Mar Kayser, G. Macchione, J. López-Prados, J. Angulo, J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2013, 11, 3510–3525 RSC; (b) J. L. de Paz, E. A. Moseman, C. Noti, L. Polito, U. H. von Andrian and P. H. Seeberger, ACS Chem. Biol., 2007, 11, 735–744 CrossRef PubMed.
- (a) S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2013, 20, 34–41 CrossRef PubMed; (b) Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. de Vries, J. Glushka, J. C. Paulson and G. J. Boons, Science, 2013, 341, 379–383 CrossRef CAS PubMed; (c) J. E. Hudak, H. H. Yu and C. R. Bertozzi, J. Am. Chem. Soc., 2011, 133, 16127–16135 CrossRef CAS PubMed.
- (a) A. Guerry, J. Bernard, E. Samain, E. Fleury, S. Cottaz and S. Halila, Bioconjugate Chem., 2013, 24, 544–549 CrossRef CAS PubMed; (b) F. Mallevre, A. Roget, T. Minon, Y. Kervella, D. Ropartz and M. C. Ralet, Bioconjugate Chem., 2013, 24, 1264–1269 CrossRef CAS PubMed.
- (a) J. L. de Paz, C. Noti and P. H. Seeberger, J. Am. Chem. Soc., 2006, 128, 2766–2767 CrossRef CAS PubMed; (b) J. Etxebarria, S. Serna, A. Beloqui, M. Martin-Lomas and N.-C. Reichardt, Chem. – Eur. J., 2013, 19, 4776–4785 CrossRef CAS PubMed; (c) R. Šardzík, G. T. Noble, M. T. Weissenborn, A. Martin, S. J. Webb and S. L. Flitsch, Beilstein J. Org. Chem., 2010, 6, 699–703 CrossRef PubMed; (d) C. Cai, K. Edgar, J. Liu and R. J. Linhardt, Carbohydr. Res., 2013, 372, 30–34 CrossRef CAS PubMed; (e) I. Sittel, A.-T. Tran, D. Benito-Alifonso and M. C. Galan, Chem. Commun., 2013, 49, 4217–4219 RSC; (f) C. Zong, A. Venot, O. Dhamele and G.-J. Boons, Org. Lett., 2013, 15, 342–345 CrossRef CAS PubMed.
- M. Shyam, S. M. Rele, S. Suri, S. S. Iyer, S. Subramanian Baskaran, L. Elliot and E. L. Chaikof, J. Org. Chem., 2004, 69, 9159–9170 CrossRef PubMed.
- S. U. Hansen, G. J. Miller, C. Cole, G. Rushton, E. Avizienyte, G. C. Jayson and J. M. Gardiner, Nat. Commun., 2013, 4, 2016 Search PubMed.
- S. U. Hansen, G. J. Miller, M. Baráth, K. R. Broberg, E. Avizienyte, M. Helliwell, J. Raftery, G. C. Jayson and J. M. Gardiner, J. Org. Chem., 2012, 77, 7823–7843 CrossRef CAS PubMed.
- K. A. Allen, R. L. Brown, G. Norris, P. C. Tyler, D. K. Watt and O. V. Zubkova, Carbohydr. Res., 2010, 345, 1831–1841 CrossRef CAS PubMed.
- S. Hagihara, A. Miyazaki, I. Matsuo, A. Tatami, T. Suzuki and Y. Ito, Glycobiology, 2007, 17, 1070–1076 CrossRef CAS PubMed.
- C. L. Johnson and Z. Guo, J. Carbohydr. Chem., 2013, 32, 301–323 CrossRef CAS.
- J. Lindberg, P. Strålfors and P. Konradsson, Tetrahedron, 2002, 58, 4245–4248 CrossRef CAS.
- S. Koshida, Y. Suda, Y. Fukui, J. Ormsby, M. Sobel and S. Kusumoto, Tetrahedron Lett., 1999, 40, 5725–5728 CrossRef CAS.
- P. J. Garegg, I. Kvarnström, A. Niklasson, G. Niklasson and S. C. T. Svensson, J. Carbohydr. Chem., 1993, 12, 933–953 CrossRef CAS.
- A. Cotte, B. Bader, J. Kuhlmann and H. Waldmann, Chem. – Eur. J., 1999, 5, 922–936 CrossRef CAS.
- K. C. Etika, F. D. Jochum, M. A. Cox, P. Schattling, P. Theato and J. C. Grunlan, Macromolecules, 2010, 43, 9447–9453 CrossRef CAS.
- Recent effective methodology for aldehyde conjugation using various labelling reagents has been described, see: S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2013, 20, 34–41 CrossRef PubMed.
- J. F. Tocanne, H. M. Verheij, J. A. F. Op Den Kamp and L. L. M. Van Deenen, Chem. Phys. Lipids, 1974, 13, 389–392 CrossRef CAS PubMed.
- A. Esswein, H. Rembold and R. R. Schmidt, Carbohydr. Res., 1990, 200, 287–305 CrossRef CAS PubMed.
- See ESI pages 4–5† for details of gram-scale synthesis.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental procedures not contained in the main text and also 1H, 13C, COSY, HMQC/HSQC NMR and MS data for compounds 2–27. CCDC 998097–998099. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01706h |
| ‡ Data not shown. |
| This journal is © The Royal Society of Chemistry 2015 |