
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical isolation of polygodial from Tasmannia lanceolata: a viable scaffold for synthesis†
Jeremy
Just
a,
Timothy B.
Jordan
ab,
Brett
Paull
ac,
Alex C.
Bissember
*a and
Jason A.
Smith
*a
aSchool of Physical Sciences – Chemistry, University of Tasmania, Hobart, Tasmania 7001, Australia. E-mail: Jason.Smith@utas.edu.au; Alex.Bissember@utas.edu.au; Fax: (+61) 3-6226-2858; Tel: (+61) 3-6226-2182
bAnalytical Sciences Tasmania, New Town, Tasmania, 7008, Australia
cAustralian Centre for Research on Separation Science (ACROSS), University of Tasmania, Hobart, Tasmania, 7001, Australia
First published on 10th September 2015
Abstract
Polygodial, a valuable sesquiterpene dialdehyde featuring an epimerizable stereocenter was efficiently extracted and isolated in gram-scale quantities (3.3% w/w) from Tasmannia lanceolata (Tasmanian native pepper) via a recently developed rapid pressurised hot water extraction (PHWE) technique that utilises an unmodified household espresso machine. This method was compared to the maceration of T. lanceolata under a range of conditions. Polygodial was used to achieve semi-syntheses of closely related sesquiterpene natural products drimendiol, (−)-drimenol, (+)-euryfuran, and some non-natural derivatives.
Introduction
The extraction of natural products of industrial, medicinal and biological significance from plant materials remains a subject of fundamental importance and enduring interest.1 These methods often feature specialised, custom-built apparatus and can be both time- and labour-intensive processes. Consequently, we have instituted a program of research in our laboratories that seeks to identify and utilise simple, inexpensive and readily available commercial equipment to facilitate efficient and practical extraction processes. To this end, we recently reported the development of a new efficient and straightforward method for the rapid, pressurised hot water extraction (PHWE) of shikimic acid (1) from star anise employing an unmodified household espresso machine (Scheme 1).2 This approach greatly simplified the isolation of shikimic acid and we demonstrated that purification by flash or ion-exchange chromatography was not necessary. Because of the novelty and simplicity of this new strategy, we were interested in investigating its scope and potential applications to other classes of natural products. Specifically, we wanted to determine whether this new PHWE technique was limited to the extraction of highly polar and particularly water-soluble compounds such as shikimic acid. For these reasons we investigated the utility of our new method for the extraction and isolation of the sesquiterpene dialdehyde polygodial (2) (Fig. 1) from Tasmannia lanceolata (Poir.) A.C.Sm. (Winteraceae, Tasmanian native pepper).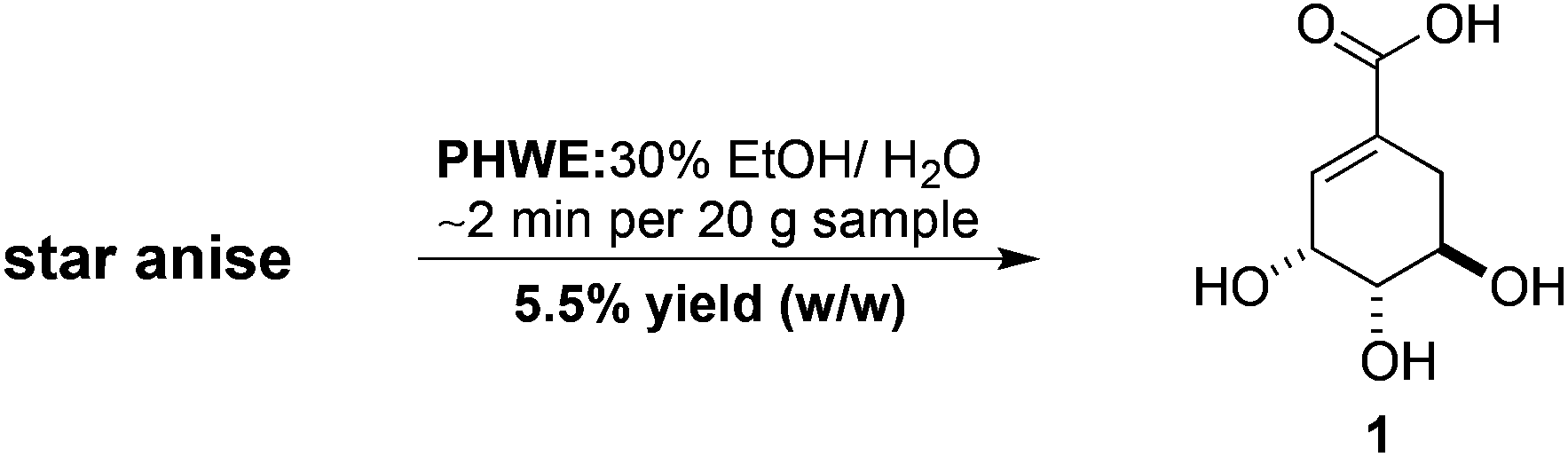 | ||
| Scheme 1 Previous work: isolation of shikimic acid (1) from star anise. | ||
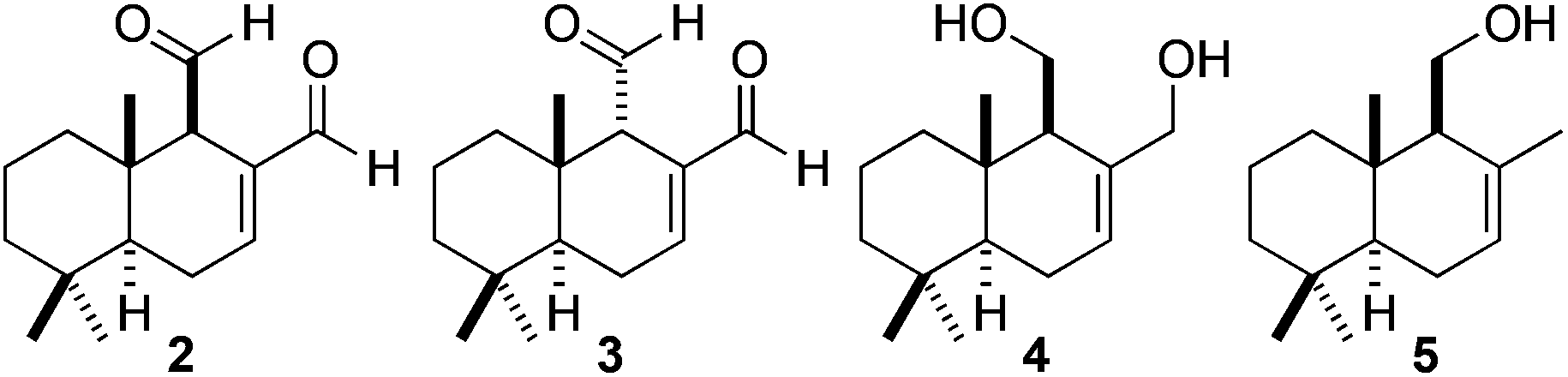 | ||
| Fig. 1 Polygodial (2), epi-polygodial (3), drimendiol (4), and (−)-drimenol (5). | ||
T. lanceolata is a shrub endemic to the woodlands and cool temperate rainforests of Tasmania and the south-eastern region of the Australian mainland.3 While polygodial (2) is found in plants in the Winteraceae,4 Polygonaceae5 and Cannellaceae6 families, it has also been found in marine sponges and molluscs.7
Interestingly, polygodial and its derivatives are known to exhibit a range of biological activity,8 including: activity as an antifouling biocide,9 a deterrent7a and anti-feedant for various insect species,7a,10 in addition to anti-leishmanial, anti-trypanosomal,11 anti-fungal,6,12 and anti-bacterial activity.13 Polygodial is much more hydrophobic than shikimic acid and contains relatively sensitive functionality.14 Indeed, sesquiterpene 2 features three contiguous stereocenters, an α,β-unsaturated aldehyde and an aliphatic aldehyde located at a potentially epimerizable stereocenter. For these reasons, we believed that this molecule represented a suitable natural product to evaluate the scope and utility of our new rapid PHWE method.
The leaves of wild samples of T. lanceolata are known to contain polygodial in varying quantities (0.1–3% w/w).15 However, the methods used to isolate polygodial from a variety of sources by the maceration of plant material with a range of solvents,11,16 the sonication of plant material in petroleum ether,17 and percolation down columns,14,18 are generally inefficient and time-consuming processes. Moreover, in many cases more than one chromatographic purification step is required.11b,16b,c,18,19
Results and discussion
After identifying optimised conditions, we were able to perform the PHWE extraction and isolation of polygodial (2) from T. lanceolata.20 In this approach, the ground leaf material (15 g) was packed into the portafilter (sample compartment) of an espresso machine and then extracted using a 35% ethanol/water solution (200 mL).21 This procedure was repeated with further T. lanceolata samples (11 × 15 g; 180 g total). The combined extract was concentrated (to ∼1.2 L in order to remove ethanol) prior to extraction with dichloromethane. Evaporation of the solvent then provided the crude extract (6.5 g) and after flash chromatography polygodial was isolated (1.24 g, 0.7% yield w/w) (Scheme 2a).22 This entire process could be completed in ∼6 h from start to finish.23 We believe that the straightforward purification of polygodial by this approach derives from short residence times associated with this rapid PHWE method that serves to reduce the extraction of intractable green pigments which have complicated the isolation of this sesquiterpene in the past.11b,16b,c,18 | ||
| Scheme 2 Isolation of polygodial (2) from (a) T. lanceolata and (b) T. lanceolata containing high polygodial content. | ||
As part of this study, we investigated the relationship between the ethanol content of the extraction solvent and the mass of the crude T. lanceolata extract.24 In this way, we observed that the mass (and the green colour) of the PHWE extract obtained increased as the ethanol content of the extraction solvent increased (Table 1, entries 1–4).25 A comparison of the extraction efficiency of our PHWE method with maceration was then undertaken. Notably, when the latter technique was performed, either at reflux or at ambient temperature, significantly lower masses of the respective extracts were obtained (entries 5 and 6). Qualitative analysis of the 1H NMR spectra of both of these extracts suggested that these samples contained a more complex mixture of compounds, particularly in the case of the maceration performed at reflux. While performing the maceration of T. lanceolata in dichloromethane increased the mass of crude extract, 1H NMR indicated a particularly complex mixture of compounds present in this viscous dark green oil (entry 7). Therefore, even though this crude extract was twice the mass of the PHWE extract (entry 4), the amount of polygodial was similar, indicating a more selective extraction for polygodial using PHWE.
|
|
||
|---|---|---|
| Entry | EtOH/H2O (% v/v) | Mass of extracta |
| a Dried plant material (15 g) was extracted using 200 mL of solvent. b Average of two extractions. c Maceration (reflux, 1 h). d Maceration (r.t., 24 h). e Maceration using CH2Cl2 (200 mL). Value in parentheses refers to the yield (% w/w) of polygodial obtained after flash chromatography (*isolated sample is dark green in colour and 1H NMR analysis suggests the presence of significant impurities). | ||
| 1 | 0 | 87 mgb |
| 2 | 15 | 218 mgb |
| 3 | 25 | 341 mgb |
| 4 | 35 | 586 mgb (0.8) |
| 5 | 35c | 180 mg |
| 6 | 35d | 210 mg |
| 7 | —d,e | 1100 mg (1.1*) |
In addition, green pigments were unable to be separated from the sample of polygodial obtained via flash chromatography.18 Moreover, all of these maceration procedures were complicated by problems associated with the removal of plant material from the filtrate. This contrasted with our PHWE strategy, as the portafilter also served to exclude solid plant materials from the extract. In all of these experiments, the 1H NMR spectra of the respective extracts featured signals consistent with the presence of trace amounts epi-polygodial (3). Indeed, even the 1H NMR spectrum of the extract obtained from maceration at room temperature suggests the presence of small quantities of epimer 3 in the sample.26
Next, we used a Dionex ASE200 accelerated solvent extractor in order to further investigate the relationship between the ethanol content of the extraction solvent and the yield of polygodial and to also study the effect of temperature on epimerization of polygodial in more detail.24,27 Specifically, we performed experiments at temperatures between 70–130 °C and using extraction solvent mixtures between 15–35% ethanol/water (Fig. 2).28 These results indicated that the highest yield of polygodial corresponded to a temperature of 90 °C and a solvent mixture of 35% ethanol/water, which is similar to our PHWE conditions.21
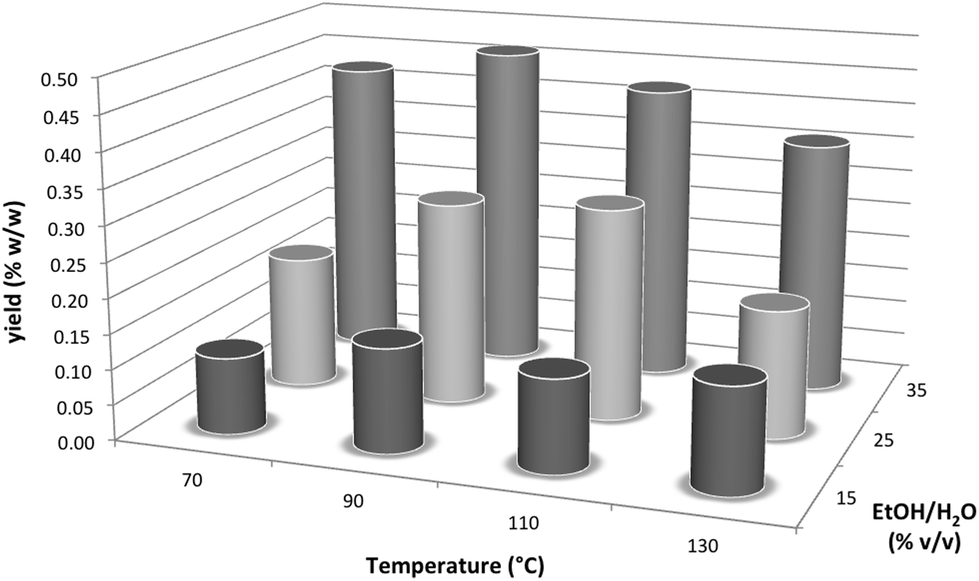 | ||
| Fig. 2 Effect of temperature and solvent composition on the yield of polygodial. Experiments were performed using a Dionex ASE200 at 500 psi (∼35 bar). Yields were determined via1H NMR spectroscopy with the aid of an internal standard. | ||
Analysis of the 1H NMR spectra of the respective extracts suggests that decomposition of polygodial also takes place at higher temperatures (≥90 °C), forming more impurities and leading to a lower combined yield of polygodial and epi-polygodial (3). Epimerization is also observed using the Dionex ASE200 at 70, 90 and 110 °C (up to a 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio of polygodial:epi-polygodial), while at 130 °C significant epimerization occurs (78:22 ratio of polygodial:epi-polygodial).29
:9 ratio of polygodial:epi-polygodial), while at 130 °C significant epimerization occurs (78:22 ratio of polygodial:epi-polygodial).29
Due to its aforementioned structural features polygodial represents a useful and versatile chiral scaffold that would be ideal for synthetic endeavors. However, the cost of polygodial ranges from ∼US$11–32 per mg from a variety of vendors and, obviously, this very high cost precludes its use as a viable starting material for complex molecule synthesis.30 With these issues in mind, we obtained samples of T. lanceolata that were specifically cultivated for their high polygodial content.31 When this plant material was extracted as before, we isolated a much higher yield of natural product 2 (1.0 g from 30 g of T. lanceolata, 3.3% w/w) (Scheme 2b). This entire process from dry leaf material to 1.0 g of polygodial could be completed within 3 h.
In order to show that simple manipulations can be directly performed on the PHWE polygodial extract before isolation, a THF solution of the crude extract was treated with LiAlH4 to provide the natural product drimendiol (4) (129 mg from 15 g of T. lanceolata, 0.8% w/w) after a simple chromatographic purification (Fig. 1).20,32
We were able to show that allylic alcohol present in drimendiol (4) can be selectively removed via hydrogenolysis to provide the natural product (−)-drimenol (5) (Fig. 1).33 Furthermore, we found that a regioselective Wittig reaction furnished diene 6 from polygodial (2). Substrate 6 was then subjected to a Diels–Alder cycloaddition affording heterocycle 7 as a single diastereoisomer (Scheme 3, top). In addition, novel annulated pyridazine 8 was efficiently prepared by the reaction of polygodial with hydrazine (Scheme 3, middle). The semi-synthesis of marine natural product (+)-euryfuran (10), found in sponges and nudibranchs,34 was also effected in 2-steps from polygodial via a reduction and acid-mediated elimination/aromatization sequence (Scheme 3, bottom).
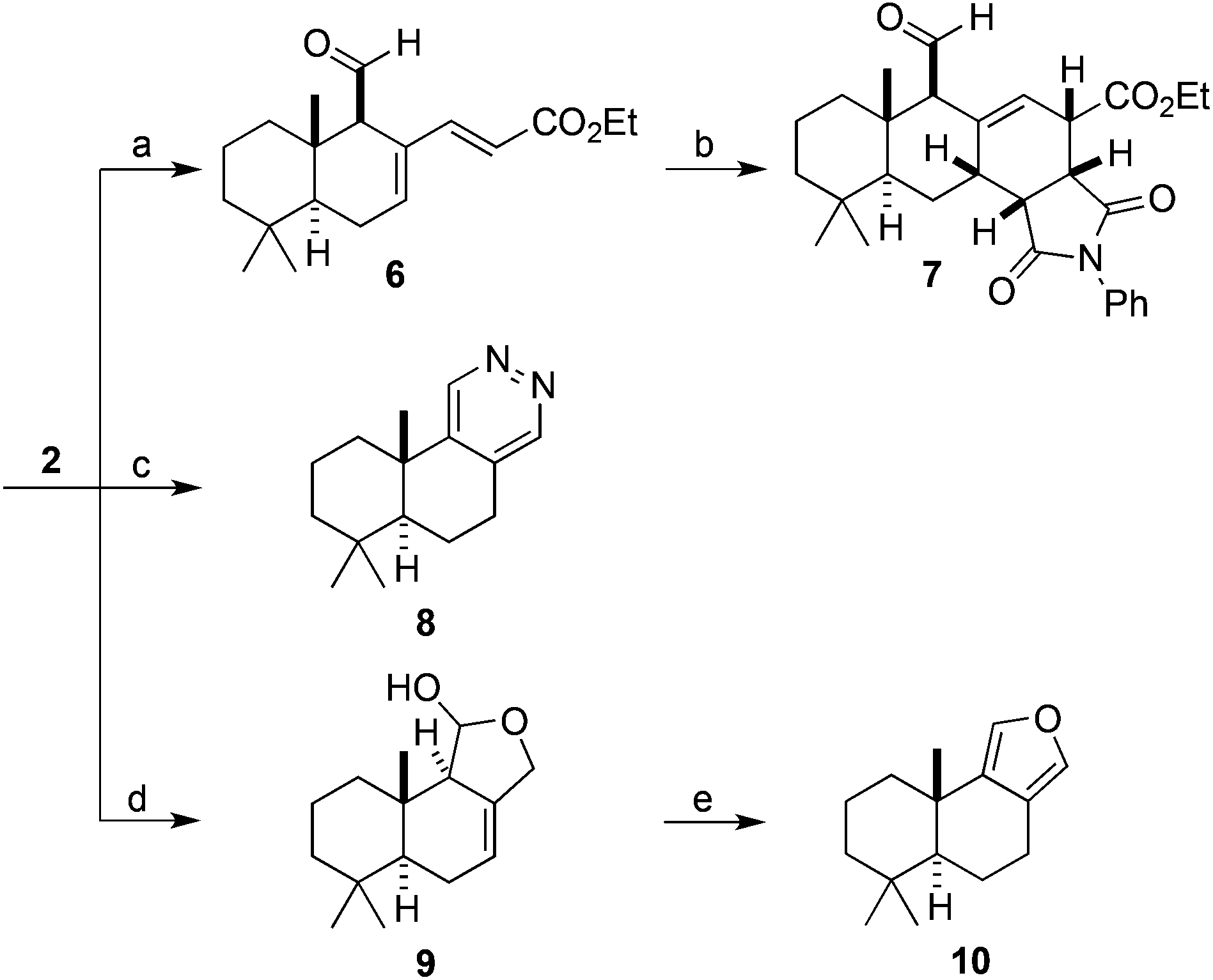 | ||
| Scheme 3 Synthesis of novel heterocycles 7 and 8 and (+)-euryfuran (10) from polygodial (2). Reaction: (a) ethyl (triphenylphosphoranylidene)acetate, 40 °C, 79% yield; (b) N-phenylmaleimide, 110 °C, 55% yield; (c) NH2NH2, 80 °C, >99% yield; (d) NaBH4, 0 °C; (e) HCl, 13% yield over two steps. | ||
Indeed, polygodial is readily accessible in our laboratories via our new PHWE technique where it can be isolated in gram-scale quantities in a short period of time. We now envision using this chiral building block to prepare more complex terpenoid-derived natural products in much the same way as the feedstock chemical (+)-sclareolide has been utilised in the past.35 Accordingly, we intend to employ polygodial to facilitate the semisynthesis of a range of targets and some possible examples are shown in Fig. 3.
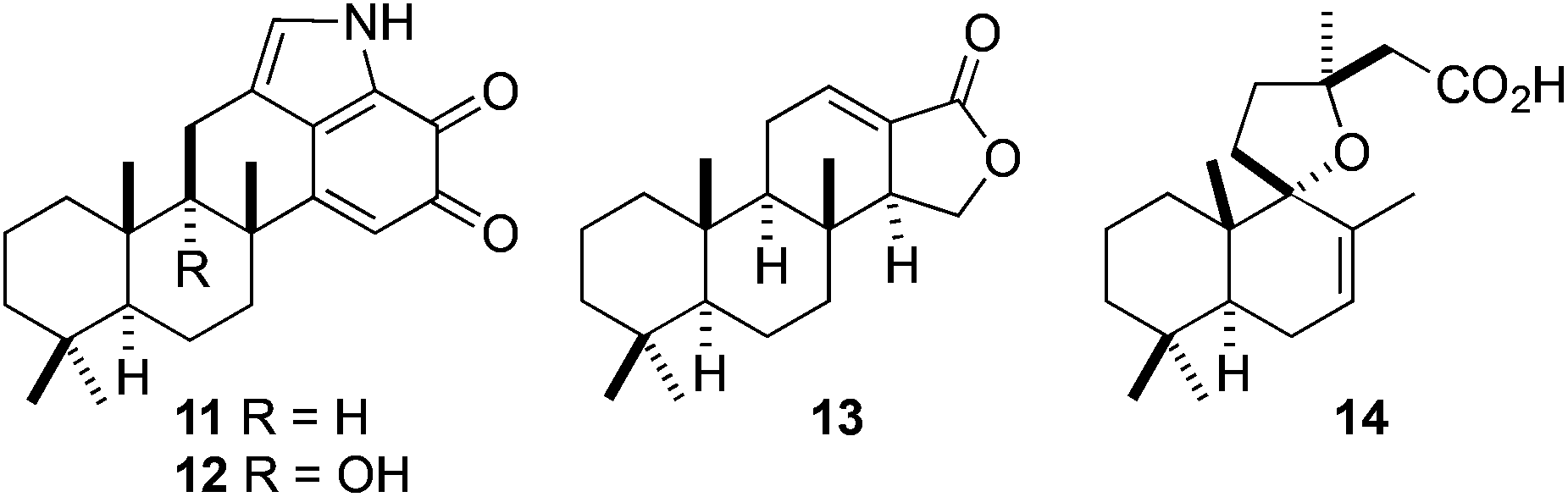 | ||
| Fig. 3 Mycoleptodiscin A (11), mycoleptodiscin B (12), isoagatholactone (13), and grindelic acid (14). | ||
Conclusions
In summary, we have extended the scope of our new method for the rapid PHWE of natural products, demonstrating that, polygodial, a relatively non-polar and hydrophobic, potentially epimerizable compound can be efficiently extracted and isolated by this technique. Our study demonstrates that this new procedure compares favourably to the maceration of T. lanceolata. Significantly, our results indicate that, for the first time, polygodial can be quickly and efficiently obtained in gram-scale quantities. Moreover, we have used polygodial to achieve semi-syntheses of closely related sesquiterpene natural products drimendiol (4) (−)-drimenol (5), and (+)-euryfuran (10). Consequently, we believe that this sesquiterpene represents a readily accessible and valuable chiral building block that can be used in target-oriented synthesis in our laboratories. In the future, we plan to employ polygodial as part of research focused on the preparation of a range of diverse natural and non-natural products.Experimental section
General experimental procedures
Plant material was ground using a Sunbeam spice/coffee bean grinder. Pressurised hot water extraction was undertaken employing a Breville Espresso Machine Model 800ES and using a Dionex ASE200 Accelerated Solvent Extractor. Unless otherwise specified, reactions were conducted with magnetic stirring in oven-dried glassware under nitrogen. Automated flash chromatography was carried out using a Reveleris® X2 Flash Chromatography System using silica gel cartridges. All NMR spectra were recorded on a Bruker NMR spectrometer operating at 400 MHz (1H) and 100 MHz (13C).Plant material
Tasmannia lanceolata leaf material was provided as a finely ground and dried sample by Diemen Pepper (http://www.diemenpepper.com). T. lanceolata containing higher polygodial content was provided as fresh leaf material from Essential Oils of Tasmania (http://www.eotasmania.com.au) and dried at 35 °C for 18 h prior to use.Polygodial (2) – extraction of T. lanceolata leaf material from Diemen Pepper (15 g)
Dried, ground T. lanceolata leaves (15 g) were mixed with sand (2 g), placed into the portafilter (sample compartment) of an espresso machine and extracted with 35% EtOH/H2O (200 mL of a hot solution). The extract was concentrated to a volume of ∼100 mL on a rotary evaporator in order to remove EtOH (50 °C water bath temperature). The ensuing mixture was extracted with CH2Cl2 (3 × 50 mL). The organic fractions were then combined, dried (MgSO4), filtered, and the solvent removed on a rotary evaporator to provide a viscous, green oil (602 mg) (see ESI, Fig. S1 and Appendix III,† for the 1H NMR spectrum of the PHWE extract). Automated gradient flash chromatography (silica; 0 → 25% EtOAc/hexane over 10 min) provided the title compound as an off-white solid (130 mg, 0.8% yield w/w).361H NMR (400 MHz, CDCl3): δ 9.53 (d, J = 4.4 Hz, 1H), 9.46 (s, 1H), 7.12 (m, 1H), 2.83 (m, 1H), 2.51 (m, 1H), 2.31 (m, 1H), 1.84 (m, 1H), 1.59–1.45 (complex m, 3H), 1.38 (td, J = 12.9, 4.5 Hz, 1H), 1.30–1.19 (complex m, 2H), 0.96 (s, 3H), 0.94 (s, 3H), 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 220.1, 193.4, 154.4, 138.5, 60.5, 49.2, 41.9, 39.8, 37.1, 33.31, 33.30, 25.4, 22.1, 18.2, 15.5.Polygodial (2) – extraction of T. lanceolata leaf material from Diemen Pepper (180 g)
Dried, ground T. lanceolata leaves (15 g) were mixed with sand (2 g), placed into the portafilter of an espresso machine and extracted with 35% EtOH/H2O (200 mL of a hot solution). This was repeated with a further 11 samples of T. lanceolata (15 g). The extracts were combined and concentrated to a volume of ∼1.2 L on a rotary evaporator in order to remove EtOH (50 °C water bath temperature). The ensuing mixture was then divided into portions of equal volume (2 × ∼600 mL) and each portion was then extracted with CH2Cl2 (4 × 100 mL). The organic fractions were then combined, dried (MgSO4), filtered, and the solvent removed on a rotary evaporator to provide a viscous, green oil (6.50 g). Automated gradient flash chromatography (silica; 0 → 25% EtOAc/hexane over 10 min) provided the title compound as an off-white solid (1.24 g, 0.7% yield w/w) (see ESI, Appendix I,† for the 1H and 13C NMR spectra of the isolated product).Polygodial (2) – extraction of T. lanceolata leaf material from essential oils of Tasmania (30 g)
Dried T. lanceolata leaves (15 g) were finely ground using a coffee bean grinder. This material was mixed with sand (2 g), placed into the portafilter of an espresso machine and extracted with 35% EtOH/H2O (250 mL of a hot solution). This was repeated with another sample of T. lanceolata (15 g). The extracts were combined and concentrated to a volume of ∼250 mL on a rotary evaporator in order to remove EtOH (50 °C water bath temperature). The ensuing mixture was extracted with CH2Cl2 (5 × 50 mL). The organic fractions were then combined, dried (MgSO4), filtered, and the solvent removed on a rotary evaporator to provide a viscous green oil (1.84 g) (see ESI, Appendix III,† for the 1H NMR spectrum of the PHWE extract). Automated gradient flash chromatography (silica; 0 → 25% EtOAc/hexane over 10 min) provided the title compound as an off-white solid (1.00 g, 3.3% yield w/w).Drimendiol (4)
Extraction of T. lanceolata leaf material from Diemen Pepper (15 g) was performed as described above to yield 661 mg of the crude extract containing polygodial. A mixture of this residue in THF (10 mL) was slowly added to a mixture of LiAlH4 (200 mg) in THF (10 mL) maintained at 0 °C. The reaction mixture was then warmed to r.t. and, after 1 h, THF and EtOH were added (5 mL of a 1:1 solution). The insoluble salts were dissolved by the addition of KHSO4 (80 mL of a 2 M aqueous solution). The ensuing mixture was then extracted with CH2Cl2 (3 × 50 mL), dried (MgSO4), filtered, and evaporated to dryness on a rotary evaporator to provide a dark red oil (632 mg). Flash chromatography (silica; 40 → 50% EtOAc/hexane) provided the title compound as a light-yellow oil (129 mg, 0.8% yield w/w).361H NMR (400 MHz, CDCl3): δ 5.75 (m, 1H), 4.30 (d, J = 12.3 Hz, 1H), 3.92 (d, J = 12.3 Hz, 1H), 3.85 (dd, J = 10.9, 1.8 Hz, 1H), 3.61 (dd, J = 10.7, 8.31 Hz, 1H), 2.14–1.79 (complex m, 5H), 1.57–1.35 (complex m, 4H), 1.27–1.06 (complex m, 4H), 0.85 (s, 3H), 0.84 (s, 3H), 0.72 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 137.0, 127.1, 67.3, 61.2, 54.5, 49.5, 42.1, 39.4, 35.6, 33.3, 33.0, 23.6, 22.0, 18.9, 14.6 (see ESI, Appendix II† for the 1H and 13C NMR spectra of the isolated product).
(−)-Drimenol (5)
5% Pd/C (15 mg) was added to a solution of drimendiol (4) (180 mg, 0.76 mmol) in EtOH (25 mL). The resulting suspension was maintained under H2 (1 atm) for 1.25 h then filtered through Celite™ and the solvent was removed on a rotary evaporator. The ensuing residue was purified by flash chromatography (silica; 0 → 25% EtOAc/hexane) to provide the title compound as a colourless solid (107 mg, 64% yield).371H NMR (400 MHz, CDCl3): δ 5.54 (m, 1H), 3.85 (dd, J = 11.3, 3.4 Hz, 1H), 3.73 (dd, J = 11.3, 4.9 Hz, 1H), 2.01–1.75 (complex m, 8H), 1.64–1.38 (complex m, 4H), 1.29 (br s, 1H), 1.22–1.12 (complex m, 1H), 1.06 (td, J = 13.2, 3.8 Hz, 1H), 0.89 (s, 3H), 0.86 (s, 3H), 0.85 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 133.1, 124.1, 61.0, 57.3, 42.2, 39.9, 36.1, 33.4, 33.0, 23.7, 22.1, 22.0, 18.9, 15.0; [α]22D = −12.5 (c 1.0, CHCl3).37(E)-Ethyl 3-((1R,4aS,8aS)-1-formyl-5,5,8a-trimethyl-1,4,4a,5,6,7,8,8a-octahydronaphthalen-2-yl)acrylate (6)
Ethyl (triphenylphosphoranylidene)acetate (208 mg, 0.60 mmol) was added to a solution of polygodial (2) (101 mg, 0.43 mmol) in EtOH (15 mL). The reaction was heated to 40 °C and after 2.5 h, the mixture was cooled and the solvent was removed on a rotary evaporator. The ensuing residue was purified by flash chromatography (silica; 0 → 15% EtOAc/hexane) to provide the title compound as a colourless oil (102 mg, 79% yield). IR (NaCl): ν 2928, 1713, 1627, 1368, 1311, 1712, 1040 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.46 (d, J = 4.8 Hz, 1H), 7.30 (d, J = 16.3 Hz, 1H), 6.49 (m, 1H), 5.48 (d, J = 16.3 Hz, 1H), 4.15 (q, 7.2 Hz, 2H), 2.81 (s, 1H), 2.31 (m, 1H), 2.20 (m, 1H), 1.83 (m, 1H), 1.56–1.42 (complex m, 3 H), 1.38–1.13 (complex m, 6H), 0.99 (s, 3H), 0.93 (s, 3H), 0.89 (2, 3H); 13C NMR (100 MHz, CDCl3): δ 205.4, 167.1, 146.4, 141.5, 130.5, 116.7, 62.9, 60.5, 48.8, 41.8, 40.3, 37.5, 33.3, 33.2; MS (EI): 304 (M+), 275, 259, 231, 229, 215, 103, 180, 166, 145, 123, 109, 95, 91, 81, 69, 55; HRMS (EI): calcd for C19H28O3 304.2038, found 304.2030; [α]22D = −7.6 (c 1.1, CHCl3).(3aR,4S,6R,6aS,10aS,11aS,11bR)-Ethyl 6-formyl-6a,10,10-trimethyl-1,3-dioxo-2-phenyl-2,3,3a,4,6,6a,7,8,9,10,10a,11,11a,11b-tetradecahydro-1H-naphtho[2,3-e]isoindole-4-carboxylate (7)
N-Phenylmaleimide (13.7 mg, 0.079 mmol) was added to a solution of diene 6 (22 mg, 0.072 mmol) in toluene (10 mL) and the resulting solution was then heated at reflux. After 18 h, the reaction mixture was cooled and the solvent was removed on a rotary evaporator. The ensuing residue was then purified by flash chromatography (silica; 30% EtOAc/hexane) to provide the title compound as a light-yellow oil (19 mg, 55% yield). IR (NaCl): ν 2928, 1737, 1712, 1500, 1386, 1207, 1195, 754 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.58 (d, 4.7 Hz, 1H), 7.43 (m, 2H), 7.36 (m, 1H), 7.10 (m, 2H), 5.98 (m, 1H), 4.29 (m, 2H), 3.84 (dd, J = 8.8, 6.0 Hz, 1H), 3.34 (dd, J = 8.8, 6.0 Hz, 1H), 3.27 (m, 1H), 2.74 (m, 1H), 2.52 (ddd, J = 14.4, 4.8, 1.3 Hz, 1H), 2.47 (m, 1H), 1.99 (m, 1H), 1.57–1.39 (complex m, 4H), 1.31 (t, J = 7.1 Hz, 3H), 1.30–1.19 (m, 3H), 1.10 (s, 3H), 0.98 (s, 3H), 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 202.5, 176.6, 175.7, 170.2, 137.1, 131.6, 129.4, 129.0, 126.7, 123.5, 67.3, 61.6, 48.5, 45.7, 43.6, 42.1, 41.8, 40.1, 36.8, 33.7, 33.6, 32.7, 22.4, 21.9, 18.4, 17.3, 14.3; MS (EI): 477 (M+), 462, 459, 432, 403, 402, 374, 373, 304, 174, 137, 123, 109, 91, 81, 79, 69, 55; HRMS (EI): calcd for C29H35NO5 477.25152, found 477.25083; [α]22D = +72.9 (c 0.9, CHCl3).(6aS,10aS)-7,7,10a-Trimethyl-5,6,6a,7,8,9,10,10a-octahydrobenzo[f]phthalazine (8)
Hydrazine hydrate (150 mg, 3.00 mmol) was added to a solution of polygodial (2) (52 mg, 0.22 mmol) in EtOH (10 mL). The reaction mixture was heated at reflux for 1.5 h then cooled and the mixture was concentrated on a rotary evaporator to provide the title compound as a colourless solid (51 mg, >99% yield). IR (NaCl): ν 2999, 2959, 2947, 2926, 2866, 1559, 1458, 1422, 1383, 1175, 997, 909, 733 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.99 (s, 1H), 8.80 (s, 1H), 2.91 (dd, J = 18.5, 6.7 Hz, 1H), 2.80 (ddd, J = 18.5, 11.1, 7.5 Hz, 1H), 2.34 (d, J = 12.5 Hz, 1H), 2.00 (dd, J = 13.7, 7.5 Hz, 1H), 1.85–1.62 (complex m, 3H), 1.53 (m, 1H), 1.41 (td, J = 13.0, 4.0 Hz, 1H) 1.30–1.19 (complex m, 5H), 0.97 (s, 3H), 0.94 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 152.3, 149.1, 148.4, 135.7, 49.6, 41.4, 37.1, 36.5, 33.6, 33.2, 27.0, 24.3, 21.6, 18.8, 17.9; MS (EI): 230 (M+), 215, 161, 159, 147, 145, 133, 119, 108, 91; HRMS (EI): calcd for C15H22N2 230.17830, found 230.17887; [α]22D = +16.9 (c 0.7, CHCl3).(+)-Euryfuran (10)
NaBH4 (27 mg, 0.71 mmol) was added to a solution of polygodial (2) (83 mg, 0.35 mmol) in EtOH (20 mL) at 0 °C. After 0.33 h H2O (30 mL) was added and the resulting mixture was extracted with CH2Cl2 (3 × 30 mL) to provide a pale-yellow oil (74 mg). This residue was subjected to flash chromatography (silica; 0 → 30% EtOAc/hexane over 10 min) to afford lactol 9 (21 mg) which still contained impurities. HCl (2 M, 2 drops) was then added to a solution of lactol 9 in CH2Cl2 (5 mL). After 18 h, the solvent was removed on a rotary evaporator and the ensuing residue was purified by flash chromatography (silica; hexane) to provide the title compound as a pale-yellow oil (10 mg, 0.046 mmol, 13% yield over two steps).381H NMR (400 MHz, CDCl3): δ 7.08 (d, J = 1.5 Hz, 1H), 7.05 (q, J = 1.5 Hz, 1H), 2.77 (dd, J = 16.4, 6.4 Hz, 1H), 2.50 (dddd, J = 16.4, 12.0, 7.4, 1.8 Hz, 1H), 1.97 (m, 1H), 1.81 (dd, J = 13.5, 7.4 Hz, 1H), 1.74–1.44 (complex m, 5H), 1.30–1.24 (complex m, 2H), 1.21 (s, 3H), 0.94 (s, 3H), 0.91 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 137.6, 137.1, 120.1, 51.7, 42.2, 39.6, 34.0, 33.7, 33.3, 25.1, 21.7, 20.6, 19.24, 19.18; [α]22D = +17.0 (c 0.2, CHCl3).38Maceration of T. lanceolata leaf material from Diemen Pepper (Table 1, entry 5)
Dried, ground T. lanceolata leaves (15 g) were added to 35% EtOH/H2O (200 mL) and the ensuing mixture heated at reflux for 1 h. The mixture was then cooled to r.t. then filtered through Celite™. The resulting green mixture was then extracted with CH2Cl2 (2 × 100 mL). The organic fractions were combined, dried (MgSO4), filtered, and the solvent removed on a rotary evaporator to provide a viscous, dark green oil (180 mg) (see ESI, Appendix IV,† for the 1H NMR spectrum of the maceration extract).Maceration of T. lanceolata leaf material from Diemen Pepper (Table 1, entry 6)
Dried, finely ground T. lanceolata leaves (15 g) were stirred in 35% EtOH/H2O (200 mL) for 24 h. The mixture was then filtered through Celite™ and the ensuing green mixture was then extracted with CH2Cl2 (2 × 100 mL). The organic fractions were combined, dried (MgSO4), filtered, and the solvent removed on a rotary evaporator to provide a viscous, dark green oil (210 mg) (see ESI, Appendix IV,† for the 1H NMR spectrum of the maceration extract).Maceration of T. lanceolata leaf material from Diemen Pepper (Table 1, entry 7)
Dried, finely ground T. lanceolata leaves (15 g) were stirred in CH2Cl2 (200 mL) for 24 h. The mixture was then filtered through Celite™ and the filtrate was then concentrated on a rotary evaporator to provide a viscous, dark green residue (1.10 g) (Fig. S2, ESI;† see ESI, Appendix V,† for the 1H NMR spectrum of the maceration extract). Automated gradient flash chromatography (silica; 0 → 25% EtOAc/hexane over 10 min) provided the title compound as a dark green oil (164 mg, 1.1% yield w/w) (see ESI, Fig. S2 and Appendix V,† for the 1H NMR spectrum of the isolated product).Dionex ASE200 extraction experiments performed at 500 psi (Fig. 2, ESI: Table S1†)
Dried, ground T. lanceolata leaves (∼7 g; Diemen Pepper sample) and Celite™ (∼3 g) were packed into a Dionex ASE200 extraction cell (22 mL). Experiments were performed using 15, 25, and 35% EtOH/H2O extraction solutions at 70, 90, and 110 °C, respectively. The following instrument-specific parameters were kept constant across all experiments:- Pressure 500 psi
- Preheat time: 0 min
- Heat time: 5 min
- Static time: 5 min
- Flush: 60%
- Cycles: 2
- Purge: 90 s
- Total extraction time: ∼20–25 min
- Overall extraction volume: ∼35–40 mL
Dionex ASE200 extraction experiments performed at 1500 psi (ESI: Fig. S3 and Table S2†)
Dried, ground T. lanceolata leaves (∼7 g; Diemen Pepper sample) and Celite™ (∼3 g) were packed into a Dionex ASE200 extraction cell (22 mL). Experiments were performed using 15, 25, and 35% EtOH/H2O extraction solutions at 70, 90, 110, and 150 °C. The following instrument-specific parameters were kept constant across all extractions:- Pressure 1500 psi
- Preheat time: 0 min
- Heat time: 5 min (7 min for 150 °C run)
- Static time: 5 min
- Flush: 60%
- Cycles: 2
- Purge: 90 s
- Total extraction time: ∼20–25 min
- Overall extraction volume: ∼35–40 mL
Representative work-up procedure for each extract obtained from each Dionex ASE200 extraction experiment
The extract was concentrated to a volume of ∼20 mL on a rotary evaporator to in order to remove EtOH (40 °C water bath temperature). The ensuing mixture was then extracted with CH2Cl2 (3 × 20 mL) and the organic fractions were combined, dried (MgSO4) and filtered. 1,3,5-Trimethoxybenzene (internal standard; 10.0 mL of a 1.20 M solution in EtOH) was added and the solvent removed on a rotary evaporator to provide a green residue. The 1H NMR spectrum (CDCl3) of the extract thus obtained enabled yield of polygodial to be determined. The 1H NMR spectrum of the extract thus obtained enabled the ratio of polygodial to epi-polygodial to be estimated by integration of the two aldehyde signals (9.53 & 9.45 ppm, and 9.84 & 9.40 ppm) respectively relative to the singlet (6.08 ppm) corresponding to 1,3,5-trimethoxybenzene.39 For representative 1H NMR spectra see ESI (Appendix V†).Acknowledgements
The authors acknowledge the School of Physical Sciences – Chemistry for financial support and the Central Science Laboratory at University of Tasmania for access to NMR spectroscopy services. J. J. thanks the Australian Government for an Australian Postgraduate Award. The authors thank Diemen Pepper and Essential Oils of Tasmania for generously providing samples of Tasmannia lanceolata.Notes and references
- (a) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2011, 75, 311 CrossRef PubMed; (b) D. J. Newman and L.-A. Giddings, Phytochem. Rev., 2014, 13, 123 CrossRef CAS; (c) A. Bauer and M. Brönstrup, Nat. Prod. Rep., 2014, 31, 35 RSC.
- (a) J. Just, B. J. Deans, W. J. Olivier, B. Paull, A. C. Bissember and J. A. Smith, Org. Lett., 2015, 17, 2428 CrossRef CAS PubMed; (b) For a recent review on PHWE, see: C. C. Teo, S. N. Tan, J. W. H. Yong, C. S. Huw and E. S. Ong, J. Chromatogr., A, 2010, 1217, 2484 CrossRef CAS PubMed; (c) For a review on natural product isolation including PHWE see: F. Bucar, A. Wube and M. Schmid, Nat. Prod. Rep., 2013, 30, 525 RSC.
- C. Read and R. Menary, J. Essent. Oil Res., 2011, 13, 348 CrossRef.
- M. C. Mecchi and J. H. G. Lago, Nat. Prod. Res., 2013, 27, 1927 CrossRef CAS PubMed.
- Y. Asakawa, A. Ludwiczuk, L. Harinantenaina, M. Toyota, M. Nishiki, A. Bardon and K. Nii, Nat. Prod. Commun., 2012, 7, 685 CAS.
- I. Kubo and M. Taniguchi, J. Nat. Prod., 1988, 51, 22 CrossRef CAS.
- (a) N. Prota, H. J. Bouwmeester and M. A. Jongsma, Pest Manage. Sci., 2014, 70, 682 CrossRef CAS PubMed; (b) M. S. Butler and R. J. Capon, Aust. J. Chem., 1993, 46, 1255 CrossRef CAS.
- The phytochemistry and chemotherapeutic potential of T. lanceolata has been studied extensively. For a recent review see: I. E. Cock, Phcog. Commn., 2014, 3, 1 Search PubMed.
- P. L. Cahill and J. M. Kuhajek, Biofouling, 2014, 30, 1035 CrossRef CAS PubMed.
- (a) V. Caprioli, G. Cimino, R. Colle and M. Gavagnin, J. Nat. Prod., 1987, 50, 146 CrossRef CAS PubMed; (b) Y. Asakawa, G. W. Dawson, D. C. Griffiths, J. Y. Lallemand, S. V. Ley, K. Mori, A. Mudd, M. Pezechk-Leclaire, J. A. Pickett, H. Watanabe, C. M. Woodcock and Z. Zhong-Ning, J. Chem. Ecol., 1988, 14, 1845 CrossRef CAS PubMed.
- (a) V. D. Claudino, K. C. da Silva, V. Cechinel Filho, R. A. Yunes, F. Delle Monache, A. Giménez, E. Salamanca, D. Gutierrez-Yapu and A. Malheiros, Mem. Inst. Oswaldo Cruz, 2013, 108, 140 CrossRef CAS PubMed; (b) D. S. Corrêa, A. G. Tempone, J. Q. Reimão, N. N. Taniwaki, P. Romoff, O. A. Fávero, P. Sartorelli, M. C. Mecchi and J. H. G. Lago, Parasitol. Res., 2011, 109, 231 CrossRef PubMed.
- (a) I. Kubo, K. Fujita and S. H. Lee, J. Agric. Food Chem., 2001, 49, 1607 CrossRef CAS PubMed; (b) R. F. McCallion, A. L. Cole, J. R. Walker, J. W. Blunt and M. H. Munro, Planta Med., 1982, 44, 134–138 CrossRef CAS PubMed.
- I. Kubo, K. Fujita, S. H. Lee and T. J. Ha, Phytother. Res., 2005, 19, 1013 CrossRef CAS PubMed.
- Polygodial reportedly decomposes slowly in air at ambient temperature, see: C. S. Barnes and J. W. Loder, Aust. J. Chem., 1962, 15, 322 CrossRef CAS.
- C. Read and R. Menary, J. Essent. Oil Res., 2011, 13, 348 CrossRef.
- See, for example: (a) V. C. Filho, V. Schlemper, A. R. S. Santos, T. R. Pinheiro, R. A. Yunes, G. L. Mendes, J. B. Calixto and F. D. Monache, J. Ethnopharmacol., 1998, 62, 223 CrossRef; (b) T. M. de A. Alves, F. L. Ribeiro, H. Kloos and C. L. Zani, Mem. Inst. Oswaldo Cruz, 2001, 96, 831 CrossRef; (c) M. C. Mecchi and J. H. G. Lago, Nat. Prod. Res., 2013, 27, 1927 CrossRef CAS PubMed; (d) O. M. Muñoz, J. D. Maya, J. Ferreira, P. Christen, J. S. Martin, R. López-Muñoz, A. Morello and U. Kemmerling, Z. Naturforsch., C: Biosci., 2013, 68, 198 CrossRef.
- V. A. Dragar, S. M. Garland and R. C. Menary, J. Agric. Food Chem., 1998, 46, 3210 CrossRef CAS.
- A. Malheiros, V. C. Filho, C. B. Schmitt, R. A. Yunes, A. Escalante, L. Svetaz, S. Zacchino and F. D. Monache, J. Pharm. Pharmaceut. Sci., 2005, 8, 335 CAS.
- We have certainly encountered these problems when performing maceration extractions of T. lanceolata in the past: R. M. Deans, M. G. Gardiner, J. Horne, A. C. Hung, C. J. T. Hyland, J. Just, J. A. Smith and J. Yin, Asian J. Org. Chem., 2014, 3, 1193 CrossRef CAS.
- Diemen Pepper (http://www.diemenpepper.com) provided samples of T. lanceolata.
- A Breville espresso machine model 800ES was used. These systems are available for ∼US$300 from retailers such as Amazon. In order to aid dispersion, sand (2 g) was added to the ground T. lanceolata leaf material in the portafilter. The temperature of water observed in espresso machines is reported to be up to 96 °C (with pressures of ∼9 bar). See: G. Caprioli, M. Cortese, G. Cristalli, F. Maggi, L. Odello, M. Ricciutelli, G. Sagratini, V. Sirocchi, G. Tomassoni and S. Vittori, Food Chem., 2012, 135, 1127 CrossRef CAS PubMed.
- 1H NMR analysis indicated that epi-polygodial (3) was not present in the isolated sample of polygodial. When we performed the extraction of only 15 g of leaf material we obtained a comparable yield (130 mg, 0.8% w/w).
- Purification of polygodial was performed using a Reveleris® X2 automated flash chromatography system. In this way, gradient elution provided the isolation of the natural product in a 10 min program.
- The same bulk sample of finely milled T. lanceolata leaf supplied by Diemen Pepper was used in this study.
- The ethanol content was not increased beyond 35% in order to minimise the flammability of the solvent mixture.
- We believe that it is, perhaps, more likely that epimerization occurs while the extract is being concentrated on a rotary evaporator (water bath temperature: 50 °C) , rather than during maceration.
- The Dionex ASE200 is an accelerated solvent extractor that enables PHWE studies to be performed. However, it should be noted that, unlike an espresso machine, it is not a flow extraction system. Consequently, a direct comparison between the two systems cannot be undertaken.
- The lowest pressure setting on a Dionex ASE200 is 500 psi (∼35 bar). Very similar results were obtained when these experiments were performed at 1500 psi (∼100 bar) using a Dionex ASE200. Samples of T. lanceolata were extracted over ∼20–25 min.
- Extractions performed with 35% ethanol/water using a Dionex ASE200 system operating at 500 psi. The epimerization of polygodial (2) to epi-polygodial (3) was determined via1H NMR spectroscopy with the aid of an internal standard. More detailed data is provided in the Experimental section.
- This is based upon a recent search using Scifinder Scholar (8th May, 2015). The shortest synthetic route to polygodial is a 7-step sequence from β-cyclocitral: D. A. Hollinshead, S. C. Howell, S. V. Ley, M. Mahon, N. M. Radcliffe and P. A. Worthington, J. Chem. Soc., Perkin Trans. 1, 1983, 1579 RSC.
- Essential Oils of Tasmania (http://www.eotasmania.com.au) provided samples of T. lanceolata containing high polygodial content.
- Drimendiol has been isolated from plants of the Drimys (Winteraceae) genus and some fungal species: (a) M. Gaviria, C. Quijano, J. Pino and S. Madriñán, Chem. Biodiversity, 2011, 8, 532 CrossRef CAS PubMed; (b) X.-Y. Yang, T. Feng, J.-H. Ding, Z.-H. Li, Y. Li, Q.-Y. Fan and J.-K. Liu, Nat. Prod. Bioprospect., 2013, 3, 154 CrossRef CAS.
- For a review of the syntheses of (−)-drimenol, its utility as a synthetic precursor see: M. Cortéz, V. Delgado, C. Saitz and V. Armstrong, Nat. Prod. Commun., 2011, 6, 477 Search PubMed.
- (a) S. H. Grode and J. H. Cardellina, J. Nat. Prod., 1984, 47, 76 CrossRef CAS; (b) J. E. Hochlowski, R. P. Walker, C. Ireland and D. J. Faulkner, J. Org. Chem., 2002, 47, 88 CrossRef.
- For a recent example of the use of (+)-sclareolide to facilitate complex molecule synthesis see: (a) D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432 CrossRef CAS PubMed. For recent reviews on the total synthesis of terpenoid-derived natural products see: (b) M. A. González, Tetrahedron, 2008, 64, 445 CrossRef; (c) E. C. Cherney and P. S. Baran, Isr. J. Chem., 2011, 51, 391 CrossRef CAS PubMed; (d) M. J. Schnermann and R. A. Shenvi, Nat. Prod. Rep., 2015, 32, 543 RSC.
- B. Rodríguez, N. Zapata, P. Medina and E. Viñuela, Magn. Reson. Chem., 2005, 43, 82 CrossRef PubMed.
- S. Urban and R. J. Capon, J. Nat. Prod., 1996, 59, 900 CrossRef CAS.
- T. Nakano, M. A. Maillo and A. Rojas, J. Chem. Soc., Perkin Trans. 1, 1987, 2137 RSC.
- 1H NMR spectra obtained using a 30 degree pulse width and a 10 s relaxation time.
Footnote |
| † Electronic supplementary information (ESI) available: Photos of extracts and pure compounds, data from ASE experiments, NMR spectra for extracts and pure compounds. See DOI: 10.1039/c5ob01573a |
| This journal is © The Royal Society of Chemistry 2015 |