
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic effects on the substitution reactions of benzhydrols and fluorenyl alcohols. Determination of mechanism and effects of antiaromaticity†
Stephen R. D.
George
,
Timothy E.
Elton
and
Jason B.
Harper
*
School of Chemistry, University of New South Wales, UNSW Sydney, NSW 2052, Australia. E-mail: j.harper@unsw.edu.au; Fax: +61 2 9385 6141; Tel: +61 2 9385 4692
First published on 28th August 2015
Abstract
A range of substituted benzhydrols and fluorenols were prepared and subjected to acid catalysed methanolysis. Analysis of the rates of each of these processes showed correlation with Hammett σ+ parameters as is consistent with the significant build-up of positive charge adjacent to the ring. In combination with the similarity of the electronic susceptibility of the processes, these data suggest that both reactions proceed through a unimolecular rate-determining step. This shows that the effect of fusion of the phenyl systems (and hence potentially introducing an antiaromatic carbocation intermediate) is only to slow the rate of reaction rather than change the mechanism of the process.
Introduction
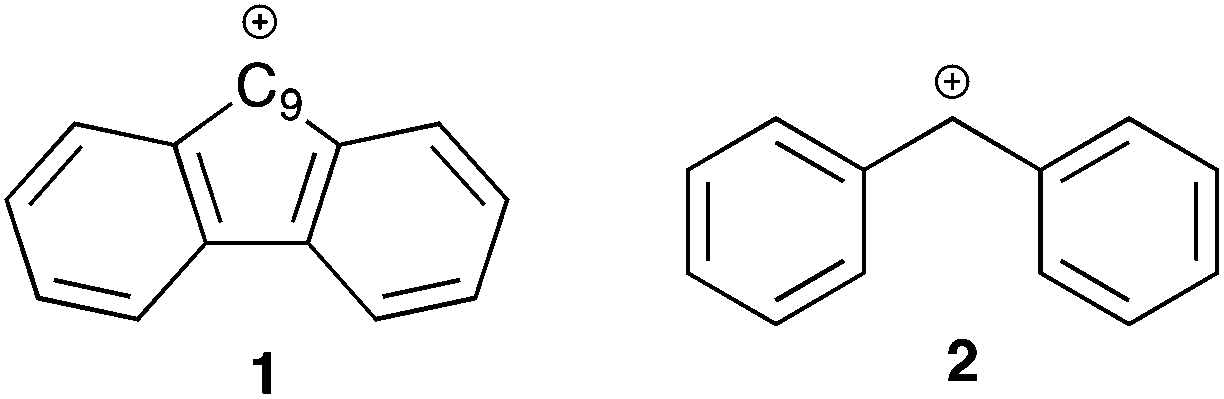
Aromaticity is a well-known (and well exploited) concept in organic chemistry that was first named in 1855 by August Wilheim Hofmann.1 In particular, aromatic systems have been noted to stabilise neighbouring carbocations through conjugation. Whilst such interactions of aromatic species are well-studied,2–5 comparatively less attention has been paid to systems with antiaromatic character and their effects on reaction outcome.6,7 A well-known example of an antiaromatic system is the fluorenyl carbocation 1.8–10 Whilst there is obviously a requirement to define an appropriate reference system, the cation 1 has been shown to be substantially destabilised by antiaromaticity through both experimental11 and computational means.12 Whilst not exceptionally unstable, it is destabilised relative to the corresponding non-fused carbocation 2 by ca. 6 kcal mol−1.13 This leads to the central questions of this study, namely what influence does the antiaromatic character of the fluorenyl carbocation have on the reactivity (and possible available mechanistic pathways) at the 9-position of the parent fluorene? Furthermore, what effects does the antiaromatic nature of the system have on the transmission of electronic effects throughout the molecule?
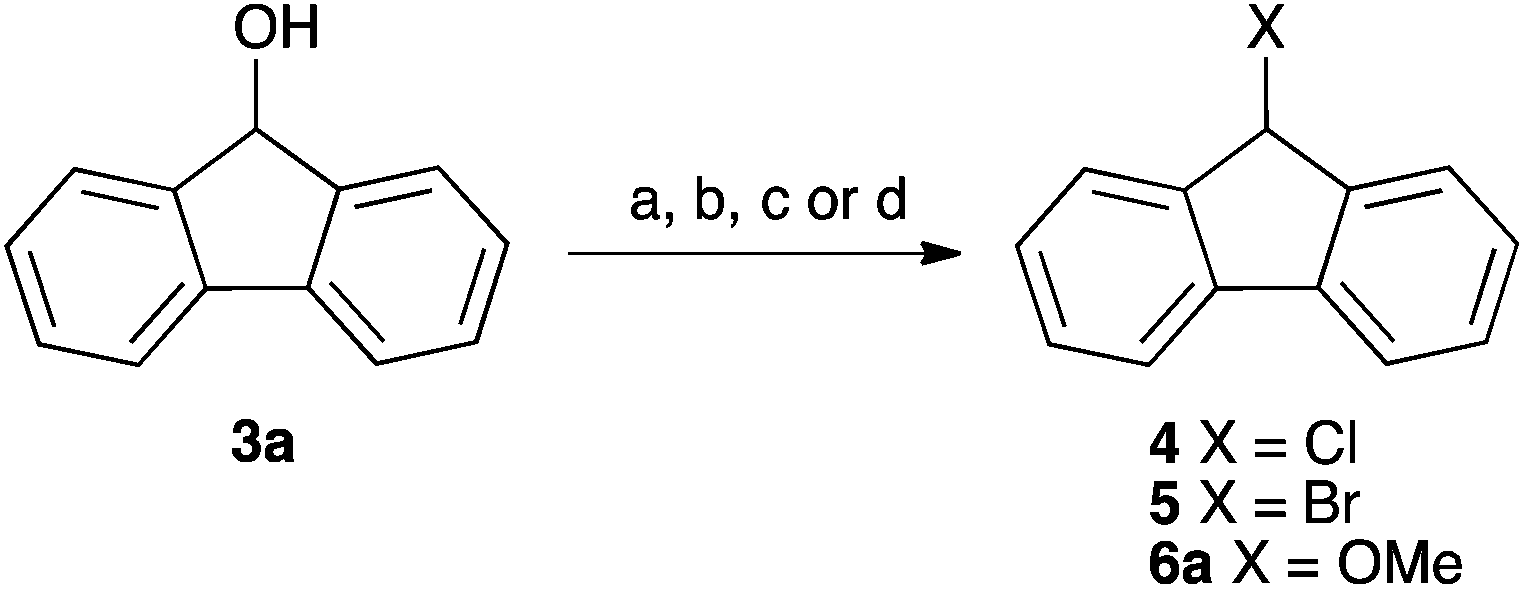 | ||
| Scheme 1 Some example substitution reactions of fluorenol 3a. (a) PBr3;14 (b) POCl3;15 (c) HCl, Δ;16 (d) MeOH, H+, Δ.17,18 | ||
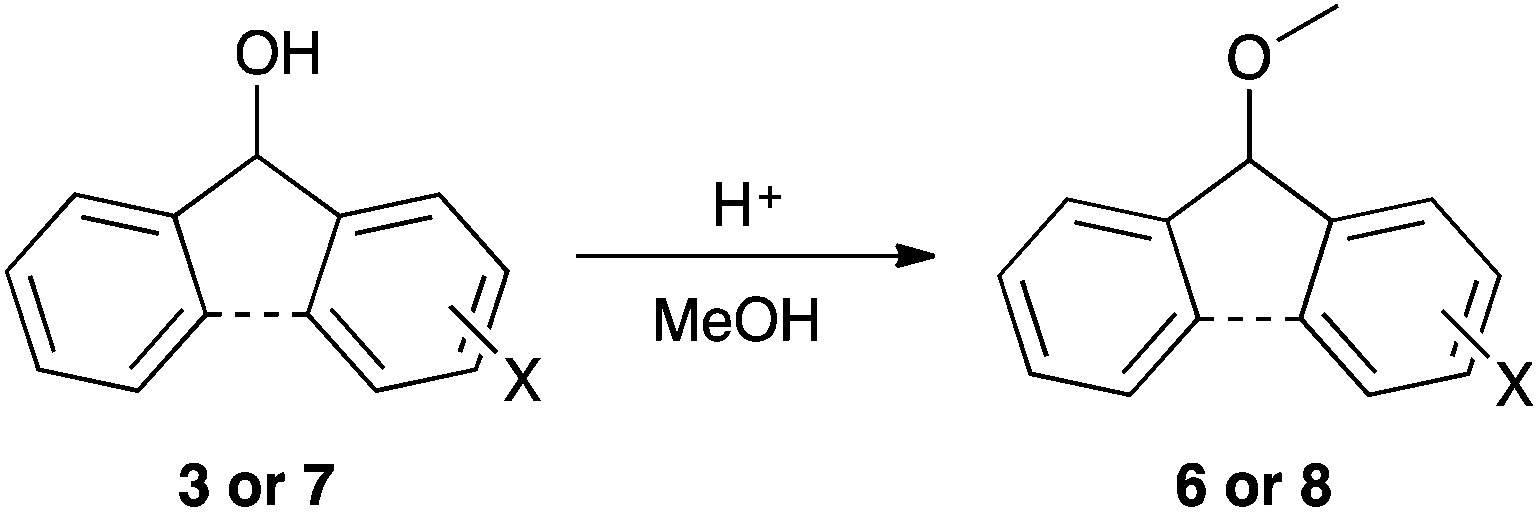 | ||
| Scheme 2 The methanolysis of the fluorenols 3 and benzhydrols 7 to the corresponding methyl ethers 6 and 8, respectively. | ||
Results and discussion
Before the kinetic studies could be performed on the desired substituted benzhydrols 7 and fluorenols 3, they needed to be synthesised. The substituted benzhydrols 7 were obtained through either (a) reduction of the appropriate benzophenones 9a and 9b, which were commercially available, with lithium aluminium hydride (to give the unsubstituted 7a and the p-methylated 7b) or (b) the lithiation of an appropriate aryl bromide 10c–e with nbutyl lithium at −78 °C in tetrahydrofuran followed by nucleophilic addition to benzaldehyde (Scheme 3, Table 1). Product identification was achieved through 1H NMR spectroscopy (taking particular note of the methine signals at δ ca. 6) and comparison of physical and spectral data with that reported.19–25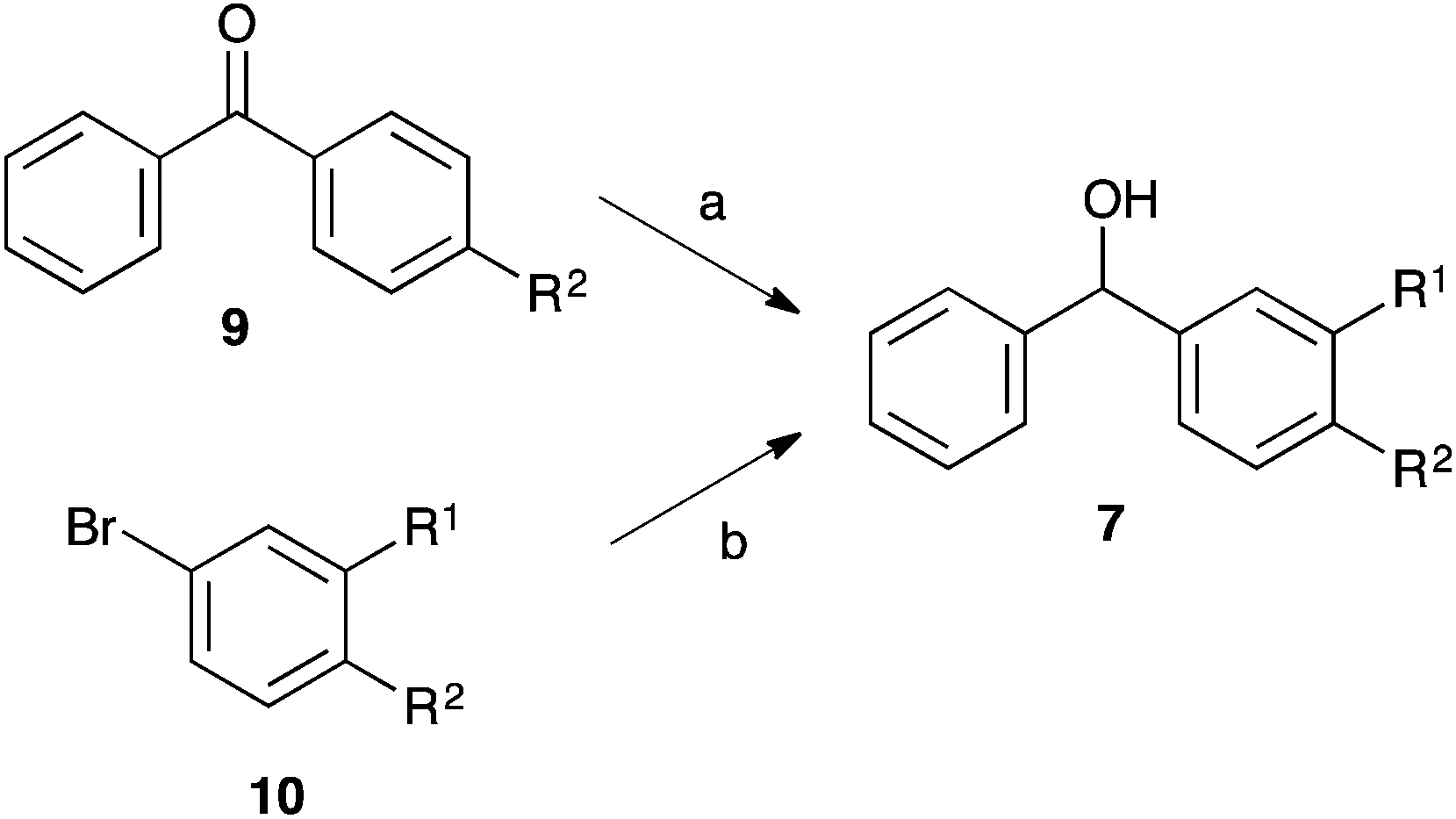 | ||
| Scheme 3 The synthesis of the substituted benzhydrols 7. (a) LiAlH4, THF, 0 °C; (b) (i) nBuLi, (ii) benzaldehyde, THF, −78 °C to r.t. | ||
| Compound | R1 | R2 | Yield (%) |
|---|---|---|---|
| From substituted benzophenones 9 | |||
| 7a | H | H | 95 |
| 7b | H | CH3 | 79 |
| From substituted bromobenzenes 10 | |||
| 7c | CH3 | H | 49 |
| 7d | H | CF3 | 87 |
| 7e | OCH3 | H | 53 |
The fluorenols 3 were prepared through the corresponding fluorenones, themselves generated from the appropriate biphenyl carboxylic acids. A range of commercially available starting materials 11–14 were used to access the required substituted biphenyl carboxylic acids 15 (Scheme 4, Table 2) with all pathways utilising Suzuki coupling chemistry.26 Again, 1H NMR spectroscopy and melting point comparisons were used to confirm the identity of the products,27–30 with only the dimethylated species 15g requiring full characterisation due to its novelty.
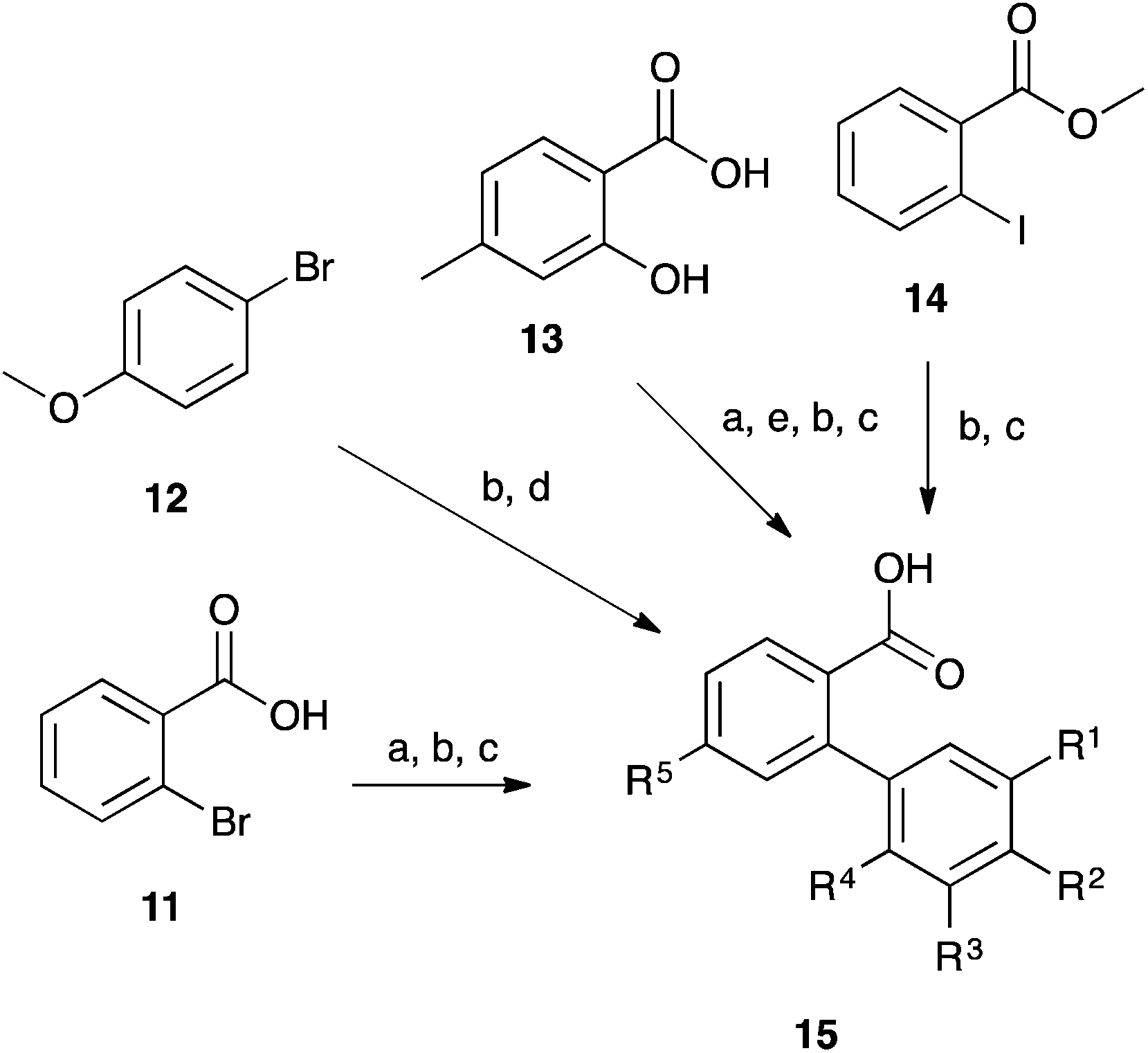 | ||
| Scheme 4 The formation of the substituted biphenyl carboxylic acids 15. (a) cat. H2SO4, MeOH, Δ; (b) ArB(OH)2, SPhos, Pd(OAc)2, Na2CO3, CH3CN/H2O, Δ; (c) NaOH, EtOH, Δ, then acid work-up; (d) KMnO4, Py/H2O, Δ; (e) Tf2O, NEt3, DCM, 0 °C. | ||
| No. | R1 | R2 | R3 | R4 | R5 | Yield (%) |
|---|---|---|---|---|---|---|
| From 2-bromobenzoic acid 11 | ||||||
| 15c | H | CH3 | H | H | H | 23 |
| 15d | H | CF3 | H | H | H | 70 |
| From 4-bromoanisole 12 | ||||||
| 15e | H | OCH3 | H | H | H | 92 |
| From 4-methylsalicylic acid 13 | ||||||
| 15f | H | H | H | H | CH3 | 72 |
| From methyl 2-iodobenzoate 14 | ||||||
| 15g | CH3 | H | H | CH3 | H | 78 |
| 15h | H | H | H | CH3 | H | 50 |
Having obtained the required substituted biphenyl benzoic acids 15, Friedel–Crafts chemistry was used to cyclise the systems to the corresponding substituted fluorenones 16, which were subsequently reduced with lithium aluminium hydride to give the desired fluorenols 3 (Scheme 5, Table 3). With the unsubstituted fluorenol 3a being the only previously fully characterised species,31,32 all of the remaining substituted fluorenols 3 were fully characterised to ensure identity.
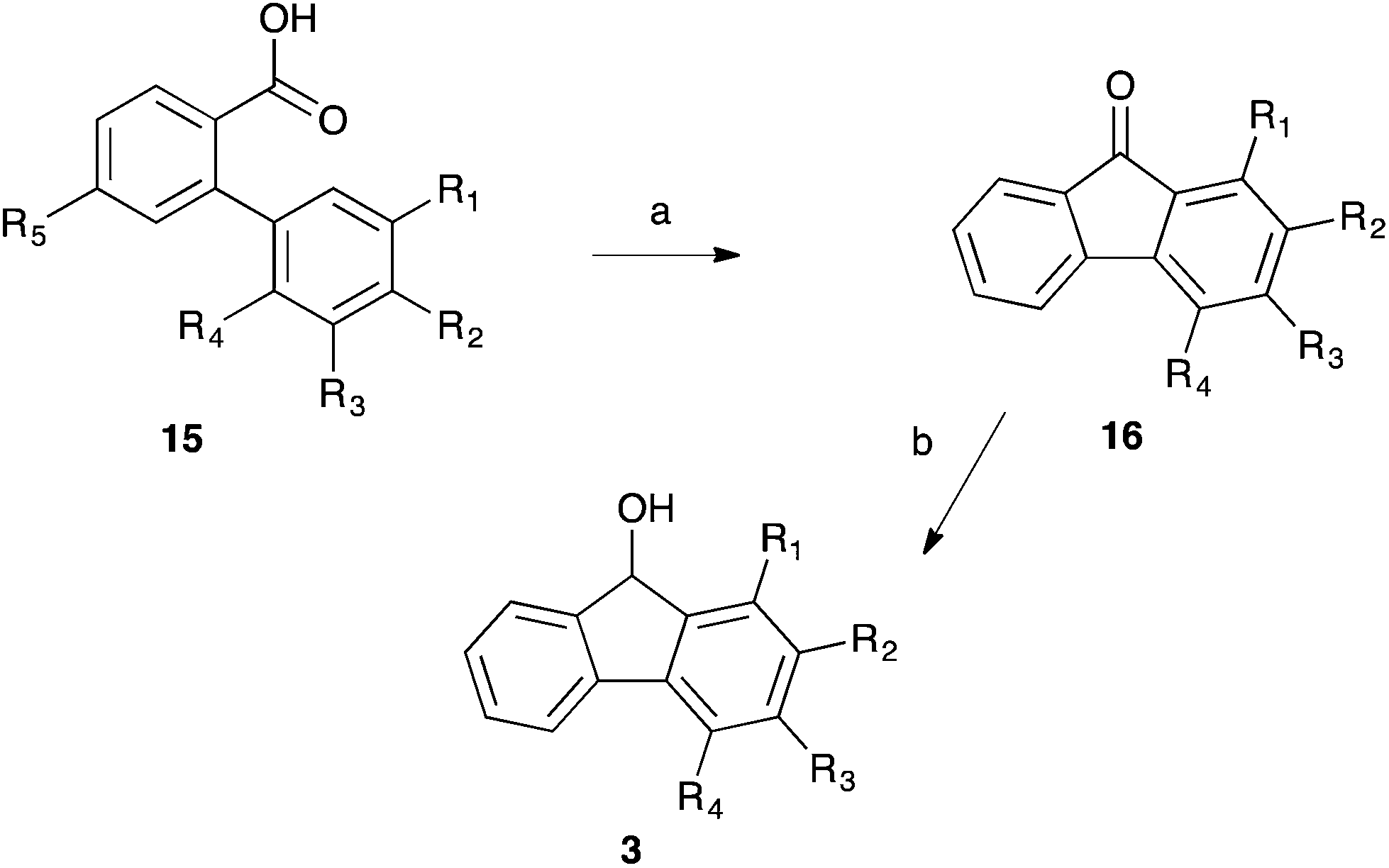 | ||
| Scheme 5 The synthesis of the required fluorenols 3 through Friedel–Crafts ring closure and subsequent lithium aluminium hydride reduction. (a) (i) SOCl2, (ii) AlCl3, DCM, 0 °C; (b) LiAlH4, THF, 0 °C. | ||
| No. | R1 | R2 | R3 | R4 | Yield (%) |
|---|---|---|---|---|---|
| a Due to the symmetry of the system (i.e. R5 is equivalent to R3 after ring closure), compound 3f is the same as compound 3b and is therefore not shown in the table. | |||||
| 3a | H | H | H | H | 99 |
| 3b | H | H | CH3 | H | 89 |
| 3c | H | CH3 | H | H | 13 |
| 3d | H | CF3 | H | H | 31 |
| 3e | H | OCH3 | H | H | 74 |
| 3g | CH3 | H | H | CH3 | 91 |
| 3h | H | H | H | CH3 | 87 |
A further fluorenol 3i was prepared through reaction of a substituent on a fluorenone. This species was prepared through oxidation of 3-methylfluorenone 16h followed immediately by esterification in acidic methanol and subsequent selective reduction using sodium borohydride (Scheme 6). Again, this species is novel and was fully characterised.
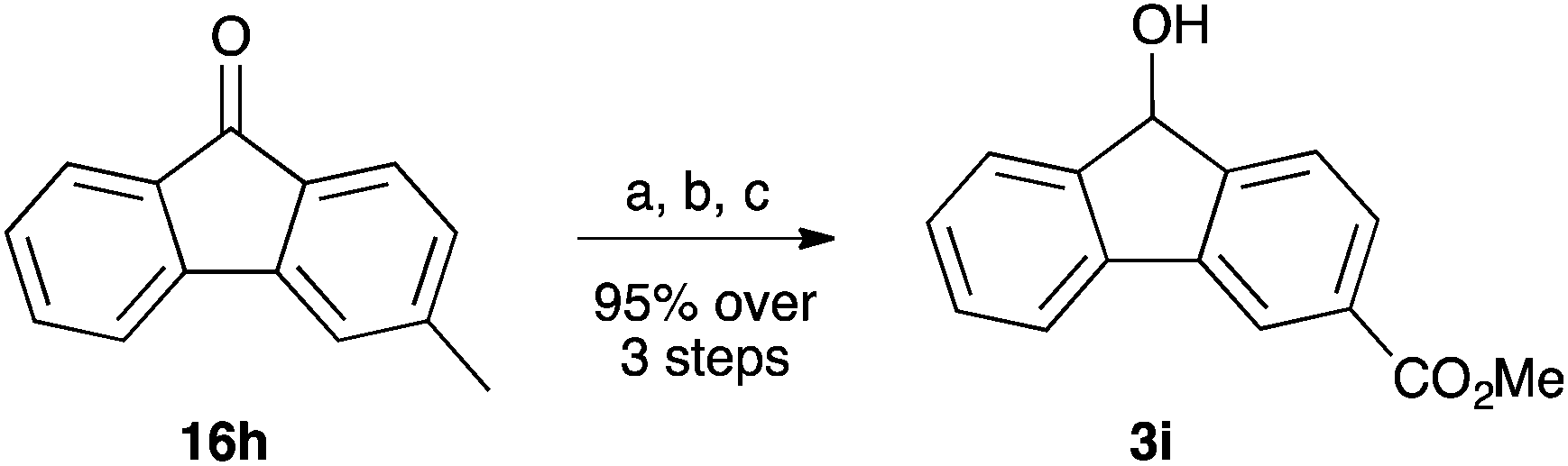 | ||
| Scheme 6 The conversion of 3-methylfluorenone 16h to the methyl ester 3i. (a) KMnO4, Py/H2O, Δ; (b) cat. H2SO4, MeOH, Δ; (c) NaBH4, MeOH, 0 °C. | ||
With the necessary species in hand, kinetic experiments were carried out to determine the observed first order rate constant (kobs) for the methanolysis of the benzhydrols 7.‡ Determined in a manner analogous to described previously using 1H NMR spectroscopy,33 the observed rate constant is a function of the extent of protonation of the alcohol and the acid concentration. As also detailed previously, variation in the former can be considered negligible and all changes in the observed rate constant attributed to changes in the substrate affecting the stabilisation of the intermediate carbocation.
The methanolysis of benzhydrol 7a has been reported previously at 23.8 °C, with an observed rate constant of 6.16(6) × 10−5 s−1.33 The same process was undertaken with the series of substituted benzhydrols 7b–7e and the rate constants are shown in Table 4 (which is ordered by the electronic nature of the substituent). Note that for the case with the extremely electron-withdrawing substituent (the trifluoromethyl derivative 7e), as the reaction was very slow, it was carried out in a temperature controlled water bath rather than in situ in the spectrometer.
| Alcohol | k obs/10−5 s−1 |
|---|---|
| 7b | 142(16) |
| 7c | 14(3) |
| 7a | 6.16(0.6)33 |
| 7d | 4(1) |
| 7e | 0.0206(0.0012) |
As can be seen in the data presented in Table 4, there is a significant decrease in the rate constant with increasing electron-withdrawing ability. Whilst the Hammett plot of these data against σ values34 gave a good correlation (R2 = 0.974, Fig. S1†) unsurprisingly, given the anticipated significant build-up of positive charge as the reaction proceeds, the correlation improves significantly when σ+ values35 are used (R2 = 0.999, Fig. 1). This, in combination with the very large electronic susceptibility in this case (a reaction constant, ρ+, of −4.15 ± 0.07), is consistent with a significant amount of charge build-up in the transition state as would be anticipated for a process that proceeds through an SN1 mechanism.§ The good correlation across all of the substituents considered is also consistent with no change in mechanism across the range of substrates.
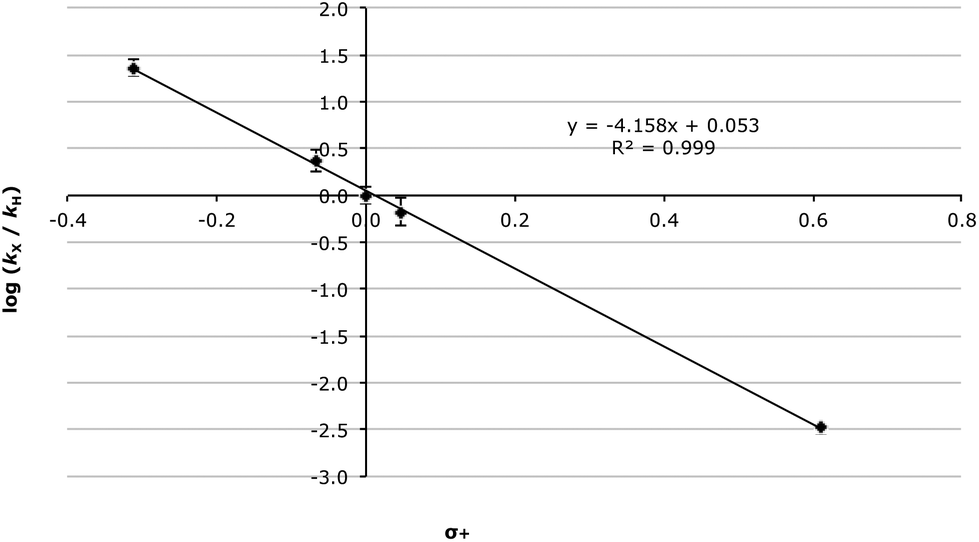 | ||
| Fig. 1 Hammett plot (using σ+ values) for the methanolysis of the benzhydrols 7 at 23.8 °C with errors calculated through standard means.36 | ||
Initial studies on the corresponding fluorenols 3 under the same conditions as were used for the benzhydrols 7 showed that the fluorenols 3 did not react to any observable extent in a reasonable time frame. As such, the reactions were repeated at 60.0 °C where the reaction rates allowed more practical assessment of reaction progress (with half lives in the order of hours) to give the rate constants shown in Table 5 (once again, ordered by electronic nature of the substituent). It is worth noting that in comparison the methanolysis of benzhydrol 7a proceeds with a rate constant of 5.44(30) × 10−3 s−1 under these conditions.
| Alcohol | k obs/10−5 s−1 |
|---|---|
| a Due to the symmetry of the system (i.e. R5 is equivalent to R3 after ring closure), compound 3f is the same as compound 3b and is therefore not shown in the table. | |
| 3g | 348(20) |
| 3b | 56(11) |
| 3h | 11.7(0.6) |
| 3c | 5.73(0.23) |
| 3a | 3.14(0.30) |
| 3e | 2.36(0.24) |
| 3i | 0.0246(0.0025) |
| 3d | 0.0448(0.0020) |
Once again, there is a clear trend in these data, with marked decreases in the rate constant on increasing the extent of electron withdrawing ability of the substituent. Quantification through a Hammett plot is more complicated in this case as several assumptions must be made. In all cases, the effect of the substituents is assumed to be unaffected by the fusion to the adjacent ring; whilst a reasonable and necessary assumption, its limitations are demonstrated by comparing the rates of species 3h and 3c. Alcohol 3g is further problematic as the effects of the two substituents is simply assumed to be additive (reasonable given they are both electron donating) whilst a substituent at the 1-position is assumed to have a sigma value the same as that at the 3-position.¶
Even taking into account these complications, the Hammett analysis is fruitful. For the data presented in Table 5, using the Hammett σ values34 gave a reasonable correlation (R2 = 0.949, Fig. S2†) though, again, this improves significantly when σ+ values35 are used (R2 = 0.980, Fig. 2).
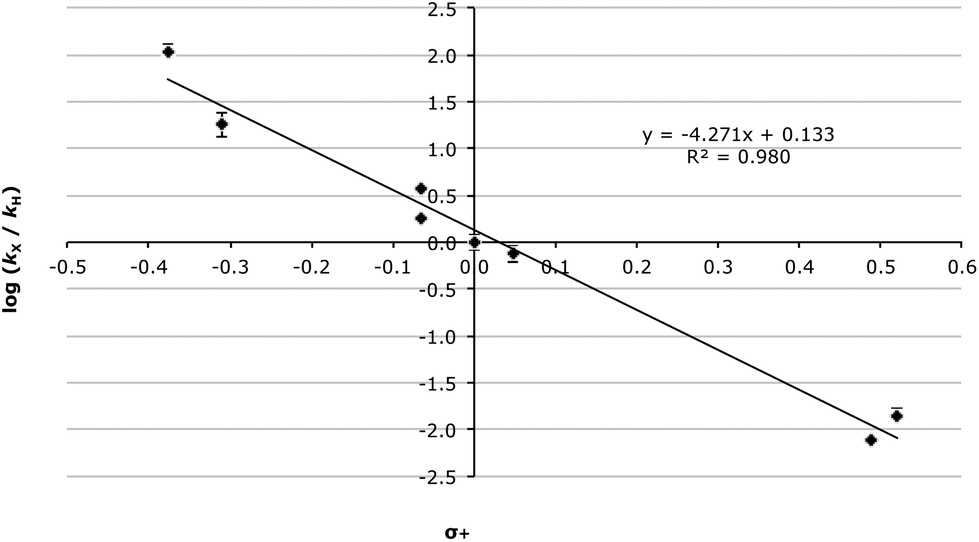 | ||
| Fig. 2 Hammett plot (using σ+ values) for the methanolysis of the fluorenols 3 at 60.0 °C with errors calculated through standard means.36 | ||
There are several key points to take away from the correlation presented in Fig. 2. Initially the fact that the correlation with σ+ values, which represent the enhanced resonance properties of the substituents, is better than with σ values is consistent with significant carbocation character developing adjacent to the ring. Once again, this is supported by the magnitude and sign of the slope (−4.27 ± 0.25). Further, the linearity of the data indicates no change in mechanism over the range of species studied.
These data allow the effect of ring fusion of the benzhydrols 7 to give the fluorenols 3 on reactivity to be considered. The goodness of fit with enhanced resonance Hammett sigma parameters in the both cases, and the slopes of said correlations, is consistent with significant build-up of positive charge adjacent to the phenyl ring in the transition state for the fluorenols 3. This is consistent with both reactions proceeding through an SN1 mechanism.|| The similarities in the electronic susceptibilities of the two processes (same given measurement uncertainties) shows that whilst the fusion of the carbon backbone may significantly decrease the overall ability of the system to stabilise the intermediate carbocation (as demonstrated by the relative rates of the two processes shown in Scheme 2 and summarised in Tables 1 and 2), it is has a very minor effect upon the extent to which the electronic nature of the substituents affects reaction outcome.
Conclusions
The above results demonstrate that whilst introducing anti-aromatic character into a carbocation intermediate may dramatically affect the rate of reaction, it does not necessarily change the mechanism of reaction where a viable alternative pathway is not available. Further, it demonstrates that the transmission of electronic effects in antiaromatic systems are comparable to those in aromatic systems. This has implications for the reactivity of larger polycyclic hydrocarbons with antiaromatic character.Experimental
All chemicals used were purchased from either Sigma-Aldrich, Alfa Aesar, Strem, Boron Molecular or Precious Metals Online and were used without further purification. Organic solvents used in synthesis were either used as received from Ajax Finechem or collected from a Pure Solv MD Solvent Purification System.NMR spectroscopy was performed using either a Bruker Avance 300 (300.13 MHz, 1H; 75.5 MHz, 13C), an Avance III 400 (400.13 MHz, 1H; 100.6 MHz, 13C) with a Prodigy cryoprobe cppbbo, an Avance III 500 (500.13 MHz, 1H; 125.7 MHz, 13C) with a tbi probe or an Avance III 600 (600.13 MHz, 1H; 150.9 MHz 13C). NMR spectra were processed using the Bruker TOPSPIN 3.0 software.
Full synthetic procedures can be found in the ESI† as per the reaction schemes shown throughout the Results and Discussion. The carboxylic acid intermediate 16g as well as the alcohols 3b–3i had not been previously prepared so were fully characterised, whilst the remainder had physical and spectral data matching that reported in literature (see ESI† for full details).
The corresponding methyl ethers (6a–c and 6e–h and 8a–e) were isolated after being treated with acidic methanol at reflux for 3 hours (full details of the preparation, along with appropriate characterisation, can be found in the ESI†).
Kinetic analysis of the methanolysis of alcohols 3 and 7
A solution of sulfuric acid (98% w/w, 35 mg, 360 μmol) in deuterated methanol (0.6 mL) was generated. Of this, a portion (0.5 mL) was added to an NMR tube that contained the alcohol (28 μmol) being investigated. The reaction mixture was held at either 23.8 °C or 60 °C in an NMR spectrometer or temperature controlled water bath and 1H NMR spectra taken at appropriate intervals until at least three half lives had been measured. The extent of reaction at a given time was determined through comparing the integration due to a signal corresponding to the starting material relative to the integration of a signal due to the product ether (see ESI†). This allowed calculation of observed first order rate constants for the methanolysis of the alcohols under these conditions. The kinetic analysis for each alcohol was carried out in triplicate.Acknowledgements
SRDG acknowledges the support of the Australian government through the receipt of an Australian Postgraduate Award. JBH acknowledges financial support from the Australian Research Council Discovery Project Funding Scheme (Project DP130102331). The authors would like to acknowledge the NMR Facility within the Mark Wainwright Analytical Centre at the University of New South Wales for NMR support.Notes and references
- A. W. Hofmann, Ann. Ch. Pharm., 1855, XCVII, 197 Search PubMed.
- R. A. McClelland, in Organic Reaction Mechanisms 2009, John Wiley & Sons, Ltd, 2011, pp. 257–273 Search PubMed.
- D. H. Aue, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 487–508 CrossRef CAS PubMed.
- M. Pittelkow, J. B. Christensen and T. I. Solling, Org. Biomol. Chem., 2005, 3, 2441–2449 CAS.
- P. Buzek, P. v. R. Schleyer and S. Sieber, Chem. Unserer Zeit, 1992, 26, 116–128 CrossRef CAS PubMed.
- A. D. Allen, J. D. Colomvakos, O. S. Tee and T. T. Tidwell, J. Org. Chem., 1994, 59, 7185–7187 CrossRef CAS.
- G. W. Cowell and A. Ledwith, J. Chem. Soc. B, 1967, 695–697 RSC.
- N. S. Mills and S. P. McClintock, Chem. Commun., 2012, 48, 8099–8101 RSC.
- H. Jiao, P. v. R. Schleyer, Y. Mo, M. A. McAllister and T. T. Tidwell, J. Am. Chem. Soc., 1997, 119, 7075–7083 CrossRef CAS.
- C. Do, J. Hatfield, S. Patel, D. Vasudevan, C. Tirla and N. S. Mills, J. Org. Chem., 2011, 76, 181–187 CrossRef CAS PubMed.
- M. C. Courtney, A. C. MacCormack and R. A. M. O'Ferrall, J. Phys. Org. Chem., 2002, 15, 529–539 CrossRef CAS PubMed.
- W. C. Herndon and N. S. Mills, J. Org. Chem., 2005, 70, 8492–8496 CrossRef CAS PubMed.
- T. L. Amyes, J. P. Richard and M. Novak, J. Am. Chem. Soc., 1992, 114, 8032–8041 CrossRef CAS.
- J. Wang, W. Wan, H. Jiang, Y. Gao, X. Jiang, H. Lin, W. Zhao and J. Hao, Org. Lett., 2010, 12, 3874–3877 CrossRef CAS PubMed.
- K. Venkatesan, S. Dhivya, J. Rethavathi and N. Srinivasan, J. Pharm. Res. Opin., 2012, 2, 191–196 CAS.
- E. Gaillard, M. A. Fox and P. Wan, J. Am. Chem. Soc., 1989, 111, 2180–2186 CrossRef CAS.
- O. Toussaint, P. Capdevielle and M. Maumy, Tetrahedron, 1984, 40, 3229–3233 CrossRef CAS.
- P. Wan and E. Krogh, J. Chem. Soc., Chem. Commun., 1985, 1207–1208 RSC.
- C. S. Marvel and N. A. Hansen, Org. Synth., 1928, 8, 24–25 CrossRef CAS.
- A. G. Davies, J. Kenyon, B. J. Lyons and T. A. Rohan, J. Chem. Soc., 1954, 3474–3479 RSC.
- K. Suzuki, T. Arao, S. Ishii, Y. Maeda, K. Kondo and T. Aoyama, Tetrahedron Lett., 2006, 47, 5789–5792 CrossRef CAS PubMed.
- V. J. Shiner and C. J. Verbanic, J. Am. Chem. Soc., 1957, 79, 369–373 CrossRef CAS.
- X. Jia, L. Fang, A. Lin, Y. Pan and C. Zhu, Synlett, 2009, 495–499 CAS.
- C.-T. Lee and B. H. Lipshutz, Org. Lett., 2008, 10, 4187–4190 CrossRef CAS PubMed.
- M. Kuriyama, R. Shimazawa, T. Enomoto and R. Shirai, J. Org. Chem., 2008, 73, 6939–6942 CrossRef CAS PubMed.
- S. R. D. George, L. T. Scott and J. B. Harper, Polcycl. Aromat. Comp., 2015 DOI:10.1080/10406638.2015.1046609.
- D. N. Korolev and N. A. Bumagin, Tetrahedron Lett., 2006, 47, 4225–4229 CrossRef CAS PubMed.
- Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574–2577 CrossRef CAS PubMed.
- N. C. Deno, J. Am. Chem. Soc., 1950, 72, 4057–4059 CrossRef CAS.
- C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132, 14006–14008 CrossRef CAS PubMed.
- Z. Hou, K. Takamine, O. Aoki, H. Shiraishi, Y. Fujiwara and H. Taniguchi, J. Org. Chem., 1988, 53, 6077–6084 CrossRef CAS.
- K. Hattori, H. Sajiki and K. Hirota, Tetrahedron, 2001, 57, 4817–4824 CrossRef CAS.
- S. R. D. George, T. D. H. Frith, D. S. Thomas and J. B. Harper, Org. Biomol. Chem., 2015, 13, 9035–9041 CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 1958, 80, 4979–4987 CrossRef CAS.
- D. C. Baird, Experimentation: An Introduction to Measurement Theory and Experiment Design, Pearson Education, New York, 1994 Search PubMed.
- H. H. Jaffé, Chem. Rev., 1953, 53, 191–271 CrossRef.
- T. Fujita and T. Nishioka, in Progress in Physical Organic Chemistry, ed. R. W. Taft, John Wiley & Sons, New York, 1976, pp. 49–89 Search PubMed.
- C. Li, P. Ross, J. R. Szulejko and T. B. McMahon, J. Am. Chem. Soc., 1996, 118 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Hammett plots of the data presented in Tables 1 and 2 against σ values; Hammett plot of the data presented in Table 2 showing the effect of modifying the effective σ+ value for the ortho-methyl substituent; general reduction of the ketones 9a,b using lithium aluminium hydride; general reaction of benzaldehyde with in situ generated aryl lithium from the aryl bromides 10c–e; general method for the esterification of benzoic acids; general Suzuki coupling to give methyl biphenyl benzoates; general deprotection of methyl benzoates; formation of the substituted biphenyl-2-carboxylic acids 15g,h; preparation of 2-(4′-methoxy)benzoic acid 15e; general method for the ring closure of the benzoic acids 15 to the fluorenones 16; preparation of compounds 16i and 3i; general method for the formation of the fluorenols 3; general method for the formation of the methyl ethers 6 and 8; kinetic analysis, including all rate data, for the methanolysis of species 3 and 7; 1H and 13C NMR spectra for all novel compounds. See DOI: 10.1039/c5ob01637a |
| ‡ In determining kobs, no assumption is made on the order of the reaction. If the process is first order with respect to the protonated alcohol species, then kobs = k1, whilst if the process is bimolecular involving the methanol nucleophile, then kobs = k2[methanol] (the latter concentration term is constant). In either case, subsequent analysis allows determination of reaction order. |
| § Values for related process that proceed through an SN1 reaction mechanism, such as the reaction of (diphenyl)methyl chlorides with alcohols, have reaction constants in range −2 to −5; the value depends on the exact nature of the reagents, along with the temperature and the solvent. For an early review in which this is shown, see the work of Jaffé.37 |
| ¶ No attempt was made at more complicated analysis, such as that by Fujita and Nishioka,38 though it should be noted that the field effects of a methyl substituent would result in the assumptions used here underestimating the electron donating effect of an ortho-methyl substituent. Increasing the electron donating effect (corresponding to decreasing the effective substituent constant) for alcohol 3h does improve the correlation (Fig. S3†), with no significant effect on the observed reaction constant. |
| || It is also possible that one or both processes proceed through a bimolecular mechanism with an extremely ‘open’ or ‘loose’ transition state,39 though this is considered the less likely of the two possibilities. |
| This journal is © The Royal Society of Chemistry 2015 |