
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A versatile approach towards multivalent saccharide displays on magnetic nanoparticles and phospholipid vesicles†
Thomas P.
Coxon
ab,
Thomas W.
Fallows
ab,
Julie E.
Gough
c and
Simon J.
Webb
*ab
aManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: S.Webb@manchester.ac.uk
bSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cSchool of Materials, University of Manchester, MSS Tower, M13 9PL, Manchester, UK. E-mail: J.Gough@manchester.ac.uk
First published on 1st September 2015
Abstract
A simple synthetic route has been devised for the production of coating agents that can give multivalent displays of saccharides on the surface of magnetite nanoparticles and phospholipid vesicles. A versatile and potentially high-throughput condensation reaction allowed the rapid synthesis of a variety of glycosylhydrazide conjugates with lipid, resorcinol or catechol termini, each in good yield and high anomeric purity. The hydrolytic stability of these adducts was assessed in D2O at different pD values using 1H-NMR spectroscopy, whilst quartz crystal microbalance with dissipation monitoring (QCM-D) confirmed that the saccharide functionality on bilayers and on nanoparticles was still available to lectins. These multivalent saccharide displays promoted nanoparticle interactions with cells, for example N-acetylglucosamine-coated nanoparticles interacted much more effectively with 3T3 fibroblasts than uncoated nanoparticles with these cells. Despite potential sensitivity to oxidation, catechol coatings on magnetite nanoparticles were found to be more stable and generate better nanoparticle interactions with fibroblasts than resorcinol coatings.
Introduction
Bionanotechnology is a rapidly developing field with exciting applications in the medical sciences.1 For example, the use of nanoscale components can enhance cancer therapies purely due to the enhanced permeability and retention (EPR) effect, which results in the accumulation of nanostructures at tumour sites where the vasculature is poorly formed.2 However, re-engineering the surface chemistry of nanoscale structures has the potential to give even more effective and specific targeting, which can be achieved by functionalisation with different classes of targeting moieties, such as peptides and proteins.3 One under-utilised class is saccharide-based, which can give coatings that selectively target sugar-binding proteins (e.g. lectins) on cell surfaces.4 This means that saccharides can be used to target cell-surface proteins, such as CD44 and the asialoglycoprotein receptor, which can be over-expressed by cancerous cells.5 Coating nanostructures with (oligo)saccharides also takes advantage of multivalency (the cluster glycoside effect6) at the surface,7 which mimics the way that cells recognise each other and their surroundings in vivo, where relatively weak individual interactions sum together to give tight binding only to the targeted cell type. Indeed multivalent displays of saccharides on the surface of nanoparticles8 and phospholipid vesicles9 have been shown to provide these nanoscale objects with the ability to target particular cell types.10 However, many reported saccharide coating procedures involve reactions on the surface of the nanostructures11 that lead to difficulty in coating characterisation, or multi-step syntheses12 that may give poor overall yields.Nanoscale magnetite is a fascinating material with exciting biomedical applications, particularly as magnetite is relatively biocompatible and most cells are unaffected by either oscillating or permanent magnetic fields. The unique properties of nanosized Fe3O4 provide several potential applications both in vitro and in vivo13 including the magnetic separation of cells or proteins that are linked to the particles, remote heating of targeted cells by alternating magnetic fields (AMFs) and use as negative contrast agents for magnetic resonance imaging (MRI).14 By functionalising magnetic nanoparticles so they target particular cell types, they could act as control elements over selected cell types, permitting targeted magnetophoresis and magnetic heating of cells and tissues. Magnetically triggered delivery of drugs from nanomaterials is another exciting prospect both for cell culture and drug delivery. Hybrid nanostructures combining magnetic nanoparticles with vesicles can release bioactive molecules and enzymes upon exposure to AMFs,15 and labelling with oligosaccharides would combine cell targeting with non-invasive triggered release of drugs.
An important barrier to progress in this area is that to obtain well characterised saccharide coatings on nanoscale objects, long and demanding syntheses are needed to obtain pure oligosaccharides with functional groups suitable for high-efficiency tagging onto the nanostructure surface. Furthermore, these coating agents need to be stable to cell culture conditions or biological fluids in vivo for periods of several weeks to allow the maximum biological effect to be exerted.
To address this problem, insight has been obtained from progress in bioorthogonal chemoligation techniques used in synthetic biology.16 Herein we report a simple synthetic route that exploits the selective reaction between hydrazides and reducing sugars (Fig. 1). The versatility of this route means that a variety of reducing sugars can be ligated to adhesive groups or lipids without the need for protecting group chemistry, greatly increasing the rate at which new coating moieties can be generated. This method can be applied to the preparation of coating molecules that can be completely characterised prior to their use for surface functionalisation.
 | ||
| Fig. 1 (a) Generalised scheme showing the condensation of hydrazides with reducing sugars to give conjugates 1a–3d. (b) Fluorescein–resorcinol conjugate 4 and fluorescein–catechol conjugate 5. | ||
Results and discussion
Synthesis of conjugates
The condensation of aldehydes and hydrazides is acid catalysed17 but the discovery that this reaction is also catalysed by aniline, which forms a Schiff base intermediate, has allowed access to dynamic combinatorial libraries under conditions compatible with many biomolecules.16,18 The aniline-catalysed formation of semicarbazones was studied by Jencks19 in the 1960s, with aniline-catalyzed hydrazone formation later studied by Dawson and co-workers in the 2000s, who showed the use of aniline gave a ∼70-fold increase in the second-order rate constant for the formation of hydrazones.18b,20Acylhydrazine adducts of simple reducing sugars were reported in the 1950s,21 with later studies of the structure and stability of the adducts revealing a preference for the ring-closed form in many cases.22 These studies suggested that a range of hydrazides should be condensable with reducing sugars under relatively mild conditions to give isolatable and characterisable saccharide conjugates.
To develop a cheap and simple method of anchoring these conjugates to iron oxide surfaces, commercially available 3,4- and 3,5-dihydroxybenzhydrazide were employed. The catechol group of the former is well known to bind well to the surface of metal oxide nanoparticles23 whilst the latter contains a resorcinol group that has been reported to bind to magnetite nanoparticles, albeit with much lower affinity than catechols.24 This resorcinol motif was investigated as an alternative to catechol as these isomeric compounds are not prone to oxidation, whereas catechol motifs have been suggested to produce some cell toxicity25 and can degrade the nanoparticle surfaces.26 To coat the surface of phospholipid vesicles with multivalent displays of saccharide, a lipid hydrazide was used that could insert into a phospholipid bilayer and project the saccharide group from the surface of the vesicle.
The reaction between hydrazides and reducing sugars was facile, requiring only a small amount of aniline16 (1 mM, equivalent to 2 mol %) as a catalyst, and these conditions were found to give crude yields generally above 70% from an 18 h reaction.27 It was found that the resorcinol–glucose adduct could be purified effectively from unreacted starting materials and byproducts by washing with hot ethanol, giving 1a as the β-anomer (>99%) in 90% yield. However, high performance liquid chromatography (HPLC) was found to be a more widely applicable purification method, although it was not possible to separate α and β anomers. Following this methodology, adducts with glucose, N-acetylglucosamine, lactose and 3′-sialyllactose (Fig. 1a) were obtained with good yield, with the equilibrated mixture containing a high percentage of the β-pyranose anomer (>85%, Table 1).28
| Adduct | Yield (%) | Anomeric purity after HPLC purification of adduct (% β) |
|---|---|---|
| 1a | 90 | >99 |
| 1b | 81 | 87 |
| 1c | 87 | 89 |
| 1d | 71 | 79 |
| 2a | 72 | 87 |
| 2b | 70 | 87 |
| 2c | 74 | 85 |
| 3a | 75 | 91 |
| 3b | 70 | 91 |
| 3c | 74 | 90 |
| 3d | 63 | 90 |
Conjugate stability
These hydrazone adducts were expected to show pH-dependent hydrolysis.29 To assess the stability of these adducts, 1H-NMR spectroscopy was used to monitor the degradation of non-oxidisable 1a in buffers at different pHs. A series of deuterated buffers were made up between pD 1 and pD 10, using sodium phosphate and deuterated trifluoroacetic acid in D2O. 1a (10 mg, 10 mM) was dissolved in each of these and 1H-NMR spectra were taken at intervals. Hydrolysis of the hydrazone bond was easily monitored at all pD values by the increase in peak integration for the anomeric peaks of free α-glucose and β-glucose, accompanied by a simultaneous reduction in the adduct β-anomer peak (Fig. 2a).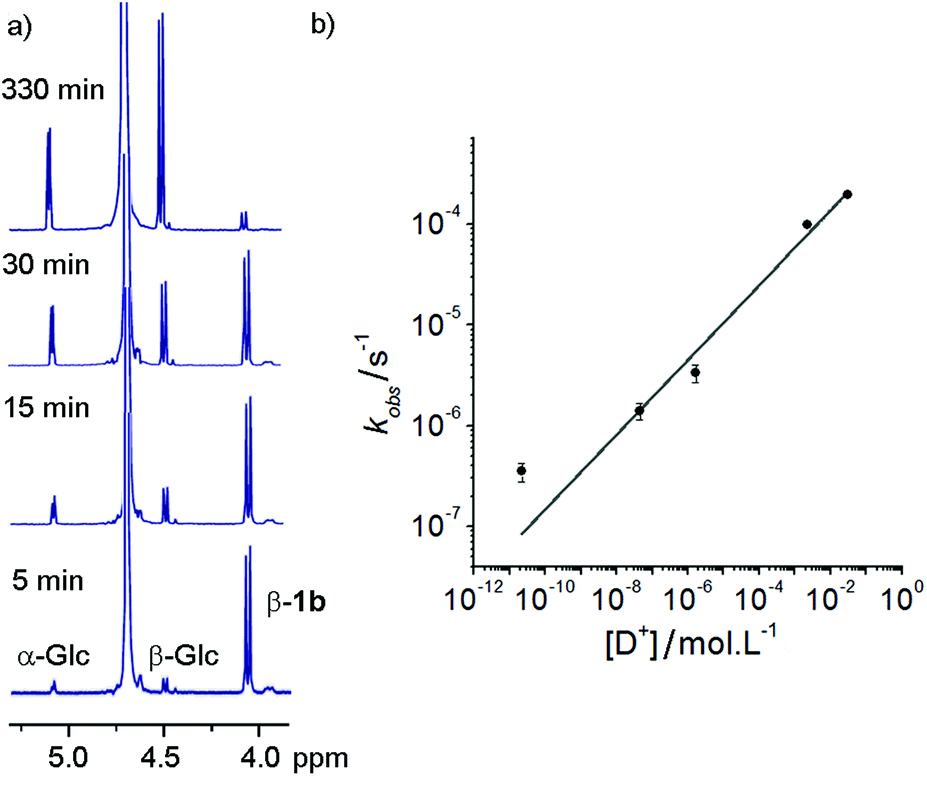 | ||
| Fig. 2 (a) Overlaid 1H-NMR spectra taken at intervals during the incubation of 1b in deuterated water at pD 1.53 at 25 °C. Indicated are the anomeric peaks for α-glucose, β-glucose and the β-adduct 1a. (b) Plot of observed rate constant values against deuteron concentration. The curve fit is to kobs = a[D+]b, where a (5 × 10−4 s−1) and b (0.37) are constants. | ||
Rapid hydrolysis was observed at low pD (pD = 1.53), with almost complete degradation of the conjugate over the course of 6 h, whereas at basic pD very little degradation was observed. Most pleasingly, at neutral pD hydrolysis was slow with more than 75% of the adduct remaining after 24 days. Furthermore no exchange of the GlcNAc from 1b with free glucose (or 1a with free GlcNAc) was observed at pD 7.6 over a period of 7 days. These properties were considered satisfactory for use in vivo or in cell culture applications.
At each pD, degradation appeared to follow pseudo first order kinetics, with the data fitting an exponential decay curve (see ESI Fig. S3.2†). From these curves, observed rate constants could be obtained for each pD (Fig. 2b). A plot of log[D+] vs. log(kobs) suggests that the hydrolytic rate has an order of ∼0.37 with respect to proton concentration, within the range of values observed for the hydrolysis of hydrazones to hydrazides presented by Kalia and Raines.29
Coating stability
Although catechols are widely used for magnetite functionalisation and are believed to have some beneficial effects on the magnetic properties of the particles, they are prone to oxidation and longer term stability of such coatings under biologically relevant conditions has not been extensively studied.23,26,30 The relative stability of catechol nanoparticle coatings compared to resorcinol nanoparticle coatings was also of interest.To measure these relative stabilities, fluorescence quenching by the iron oxide surface was exploited.31 3,4- and 3,5-Dihydroxybenzhydrazide were reacted with fluorescein isothiocyanate to give fluorescein conjugates, 4 and 5 (Fig. 1b). Coating of the magnetite nanoparticles with 4 or 5 was performed by sonicating commercially available uncoated MNPs with a 10 mM solution of the desired coating molecule (4 or 5) followed by washing to give either 4-MNP or 5-MNP respectively. These coated magnetite nanoparticles (2 mg) were then incubated in a variety of buffers (20 mL) at 38 °C. At given time points, the nanoparticles were sedimented using a strong permanent magnet (0.51 T), then aliquots (2 mL) were taken from the supernatant. Increases in the supernatant fluorescence (ex. 490 nm, em. 520 nm) resulted from the release of surface-bound quenched fluorescein into solution, which could occur by the loss of Fe–OAr links (Fig. 3a).
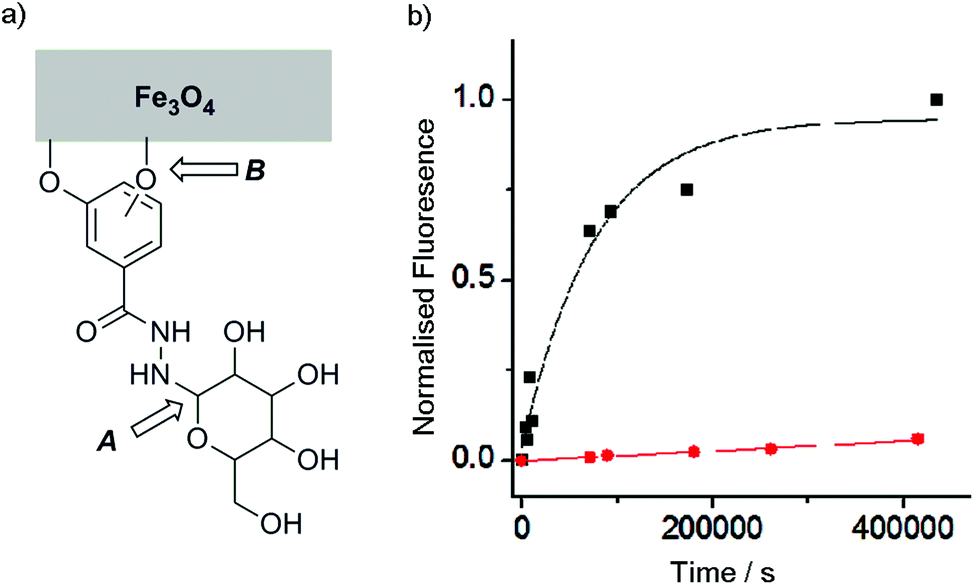 | ||
Fig. 3 (a) Schematic representation showing the two labile links in the saccharide coatings on the surface of magnetite nanoparticles (b) Plot of supernatant fluorescence against time for samples of MNPs coated with fluorescent conjugates 4 (resorcinol, ■) and 5 (catechol,  ) incubated in PBS at 38 °C. ) incubated in PBS at 38 °C. | ||
With this method, the stability of magnetite coatings were assessed in a number of different buffers at pH 7.4 (see ESI†), including cell culture media (Dulbecco's Modified Eagle Medium, DMEM), with added antibiotics and fetal bovine serum (FBS). Though it has previously been reported that FBS has a dramatic effect on magnetite coatings,32 in our studies little difference was observed between the stability in media compared to phosphate buffered saline (PBS).
Catechol coatings were found to be much more stable in PBS than the equivalent resorcinol coatings, with <10% of the loss of the 5 coating from 5-MNP after 100 h at physiological temperatures compared to >90% of the 4 coating from 4-MNP (Fig. 3). Fitting these data to first order kinetics suggests >100-fold difference in coating stability (k = 1.3 × 10−5 s−1 and <2 × 10−7 s−1 respectively), which is likely to be because the catechol hydroxyl groups can chelate to individual iron centres in magnetite26 whereas resorcinol hydroxyl groups cannot chelate the iron centres and may not be able to efficiently bridge between iron centres. Nonetheless, altering the pH to 6.5 resulted in a significant increase in the stability of the 4-MNP coating in phosphate buffer (see ESI Fig. S4.1†). These data show that for the resorcinol derivatives at neutral pH, loss of the coating molecules from the surface (B, Fig. 3a, t1/2 ∼1 day) is faster than hydrolysis of the hydrazone (A, Fig. 3a) whereas for the catechol compounds the reverse is true (t1/2 for dissociation from the surface >8 weeks).
Lectin binding
With the stability of these magnetite nanoparticle coatings confirmed, it was necessary to test that the sugar units were still available for lectin binding when bound to the magnetite surface or linked to the surface of phospholipid bilayers. To assess these interactions, Quartz Crystal Microgravimetry with Dissipation monitoring (QCM-D) was used.Having been developed for use in liquid environments, Quartz Crystal Microgravimetry (QCM) has become a very useful tool for understanding surface interactions in biology. The technique involves the use of a quartz crystal sensor with a specific resonant frequency. When a material is deposited onto the sensor surface, the increase in the effective mass of the sensor causes a reduction in its specific resonant frequency that can be related to the amount of material deposited.33 More recently, systems have been developed to include monitoring of energy dissipation (QCM-D),34 which can give information on the nature of the deposited material; a small dissipation value suggests a rigid film while larger dissipation values suggest softer, more viscoelastic films.
QCM-D can be used to study strong specific binding interactions, like biotin/streptavidin, or chemical reactivity at surfaces. In an example that combines both, Richter and co-workers ligated glycosaminoglycans with biotinylated oximes and hydrazones to give conjugates that bound to streptavidin-coated QCM-D chips. This methodology allowed them to show that glycosaminoglycan-oxime linkages were more stable towards hydrolysis.35 QCM-D has also been used to probe the weaker interactions between saccharide groups and lectins. Mouline et al.36 deposited concanavalin A onto a gold-coated quartz crystal sensor and then monitored the binding of vesicles decorated with mannosyl lipids to this surface. After performing a concentration assay, the data could be used to calculate an association constant for the interaction. A similar method was also used to generate layered aggregates of concanavalin A and mannosylated gold nanoparticles.37 Although Mouline et al. showed the adhesion of intact phospholipid vesicles to the sensor, it is also possible to deposit a simple phospholipid bilayer with desired functionality. Vesicles are deposited onto a silica-coated QCM sensor surface until they reach a critical concentration, after which they rupture to give a solid-supported bilayer.38
Two methods were employed to furnish the quartz crystal with lectins, to which saccharide-coated MNPs could bind. The first involved further functionalising gold-coated quartz sensors with 3-mercaptopropanoic acid (3-MPA). The thiol group of this molecule binds strongly to the gold surface, with the carboxylate giving it a negative charge at neutral pH that has been found to aid the formation of protein monolayers.39 The second used a clean silica sensor onto which first glycosylated phospholipid vesicles were deposited, which then burst38b to form a glycosylated bilayer on the surface. This latter method allows both the binding of lectins to lipid-saccharide conjugates in bilayers and the binding of saccharides on the surface of MNPs to be measured.
The former methodology was employed with the lectins concanavalin A (Con A) and Maackia amurensis leukoagglutinin (MAL I). The respective lectin solutions (each 0.1 mg mL−1) were flowed over 3-MPA functionalised gold surfaces. Deposition of the lectin on the surface resulted in a decrease in frequency and an increase in dissipation (Fig. 4a and b, point ii). When nanoparticles with a mismatched or no sugar coating (0.1 mg mL−1, 36 nm by DLS when bare, mismatches: 1c-coated for ConA, 1a-coated for MAL I) were flowed over the lectin functionalised chip, only small changes in frequency were observed (Fig. 4a and b, point iii). However suspensions of the matching saccharide-coated magnetite nanoparticles (glucose-coated nanoparticles 1a-MNP for ConA and sialyllactose-coated nanoparticles 1d-MNP for MAL I) gave further frequency decreases (Fig. 4a and b, point iv) when flowed over the respective lectin layers, indicating nanoparticle binding. An analogous response40 was observed for glucose-coated nanoparticles 3a-MNP for ConA, although in this case there was loss of the lectin from the surface during the flow of buffer over the chip between mis-matched (3c-MNP, Fig. 4c, point iii) and matched (3a-MNP, Fig. 4c, point iv) nanoparticles.
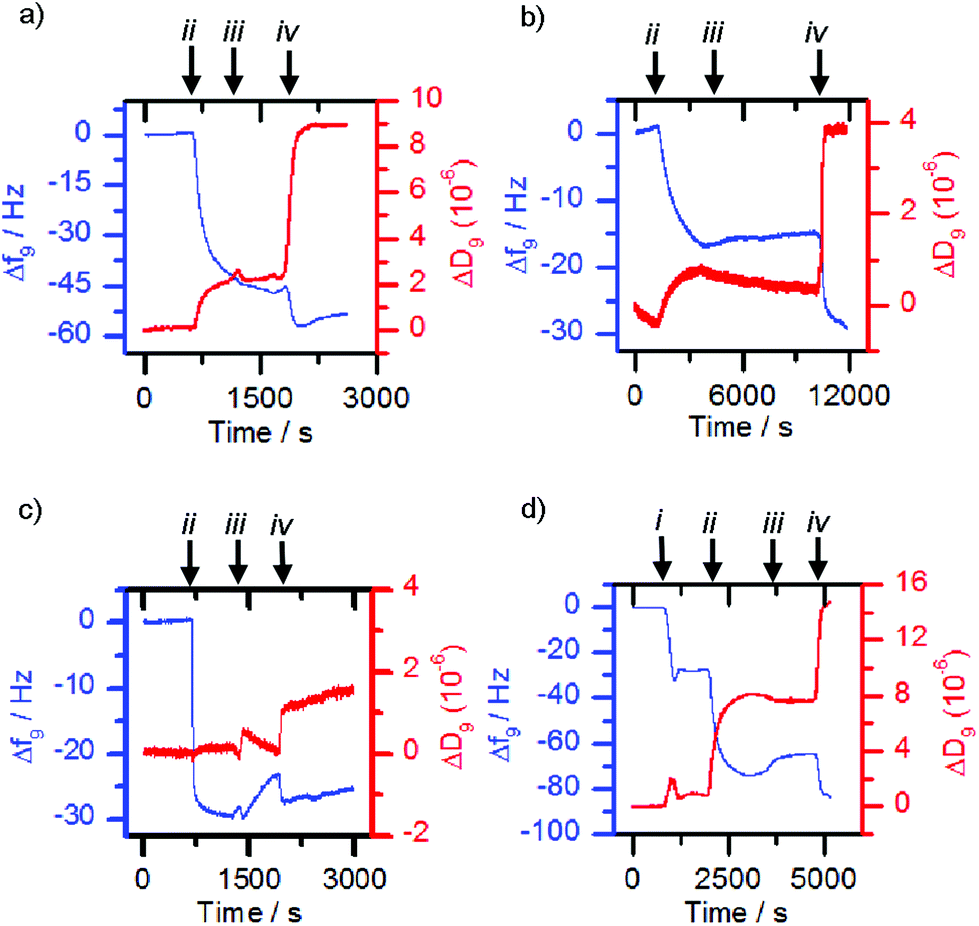 | ||
| Fig. 4 Quartz Crystal Microbalance with Dissipation monitoring (QCM-D) traces. Frequency changes over time are shown in blue while dissipation changes are shown in red. The binding of (a) 1a-MNP to concanavalin A, (b) 1d-MNP to Maackia amurensis hemagglutinin and (c) 3a-MNP to concanavalin A, was proven by first depositing the lectin onto a gold sensor functionalised with 3-mercaptopropanoic acid. The binding of (d) 1b-MNP to Wheat Germ Agglutinin required the prior deposition of a phospholipid bilayer functionalised with glycolipid 2b. Arrows (i), (ii), (iii), and (iv) on the traces indicate functionalised vesicles, lectins, mismatched nanoparticles or matched nanoparticles interacting with the chip. | ||
Although successful for Con A and MAL I, this adsorption methodology only gave poor responses for wheat germ agglutinin (WGA) and Erythrina cristagalli lectin (ECL), so the deposition of the matching bilayer was used for these lectins. The deposition of these glycosylated phospholipid bilayers was achieved by flowing sonicated DMPC vesicles (ca. 30 nm diameter) functionalised with 5% mol/mol glycolipid adduct across the sensor chip. Vesicle rupture was further encouraged by the addition of calcium and sodium ions to the 2-(4-(2-hydroxyethyl)piperazin-1-yl)ethanesulfonic acid (HEPES) buffer. For example, vesicles functionalised with 2b deposited onto the sensor surface and ruptured to form a bilayer once the critical concentration was reached, giving a characteristic QCM-D trace (Fig. 4d, point i), in which the measured frequency decreases sharply due to the deposition of intact vesicles, before increasing back to a steady value after vesicle lysis and supported bilayer formation. When the matched lectin, WGA, (0.1 mg mL−1) was then flowed across the deposited glycosylated bilayer, binding to the glycosylated bilayer was observed, indicated by a decrease in frequency (Fig. 4d, point ii). When non-functionalised or mismatched bilayers or nanoparticles were then flowed across this surface, a small frequency increase occurred (e.g.Fig. 4d, point iii addition of 1c-MNP). However when nanoparticles coated with the same sugar were then used, further deposition occurred (Fig. 4d, point iv). Analogous observations were made with vesicles functionalised with 2c, ECL and nanoparticles coated with 1c (see the ESI, Fig. S5.1†). This confirmed that any observed frequency changes were a result of specific sugar-lectin binding, rather than non-specific binding. Control experiments flowing nanoparticles over a bare sensor surface confirmed frequency changes were not simply the nanoparticles depositing directly onto the sensor.
To try and quantify the interaction between MNPs with multivalent saccharide displays and matching lectins, a UV assay was devised. Absorbance calibration curves at 280 nm were created for each lectin by dilution of stock solutions (0.1 mg mL−1, 1 mL). Resorcinol- or catechol-coated MNPs were then added in aliquots to each stock 0.1 mg mL−1 lectin solution. Each solution was stirred for 10 min before applying a permanent magnet (0.51 T, counter sunk ring N42 magnet, 50 mm diameter, 30 mm thick, 6 mm centre hole) to sediment the magnetic nanoparticles, along with any bound lectin. After each nanoparticle addition, the lectin absorbance at 280 nm was re-measured; the decrease of this absorbance allowed the amount of lectin bound per weight of MNP to be calculated. This assay was performed on Con A/1a-MNP and WGA/1b-MNP, as well as Con A/3a-MNP and WGA/3b-MNP, with each giving similar values of around 10 μmol lectin per gram of nanoparticles (see ESI S.6†). In the case of both lectins, using nanoparticles with no saccharide functionality resulted only in very slight fluctuations in the absorbance at 280 nm.
Interaction of saccharide coated MNPs with cells
Many cells express saccharide binding proteins, or lectins, on their surface and their recognition properties allow cell targeting by multivalent nanostructures. The majority of animal lectins are involved in some way with immune response, although others participate in intracellular trafficking.41 Galactose-terminated saccharides have been widely used to target the asialoglycoprotein receptor on liver cells.42 Similarly, silica nanoparticles tagged with mannose have shown impressive targeting of breast cancer cell lines.43 Other monosaccharide binding proteins whose expression is altered in cancerous cells include the N-acetylglucosamine-specific receptor of the thyroid,44 and glucose transporters GLUT-1 and GLUT-3.45 GlcNAc has also previously been used to target cells that express vimentin,46 an intermediate filament protein that is expressed in 3T3 fibroblasts;47 this is a convenient cell line for initial studies.In these initial studies, Glc-coated magnetic nanoparticles were incubated with 3T3 fibroblasts to assess cellular interactions. After seeding 3T3 fibroblasts onto a 24-well plate (1 mL per well) at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and incubating for 24 h, either uncoated or 3a-coated nanoparticles were added at a concentration of 0.1 mg mL−1. Optical microscopy of the 3a-MNP with cell mixtures showed that, unlike the uncoated MNPs, almost all observable 3a-MNPs appeared to be associated with cells in some way (Fig. 5a and b), although the objects observed are likely to be conglomerates as individual nanoparticles are not visible by optical microscopy. GlcNAc-coated magnetic nanoparticles, 1b-MNPs, showed similar results with 3T3 fibroblasts (see ESI Fig. S7.1†). The absence of nanoparticles over the nuclear region suggests that they may have been taken up rather than bound to the cell surface. However, to get a more detailed assessment of cell–nanoparticle interactions, confocal fluorescence microscopy was used.
000 cells per well and incubating for 24 h, either uncoated or 3a-coated nanoparticles were added at a concentration of 0.1 mg mL−1. Optical microscopy of the 3a-MNP with cell mixtures showed that, unlike the uncoated MNPs, almost all observable 3a-MNPs appeared to be associated with cells in some way (Fig. 5a and b), although the objects observed are likely to be conglomerates as individual nanoparticles are not visible by optical microscopy. GlcNAc-coated magnetic nanoparticles, 1b-MNPs, showed similar results with 3T3 fibroblasts (see ESI Fig. S7.1†). The absence of nanoparticles over the nuclear region suggests that they may have been taken up rather than bound to the cell surface. However, to get a more detailed assessment of cell–nanoparticle interactions, confocal fluorescence microscopy was used.
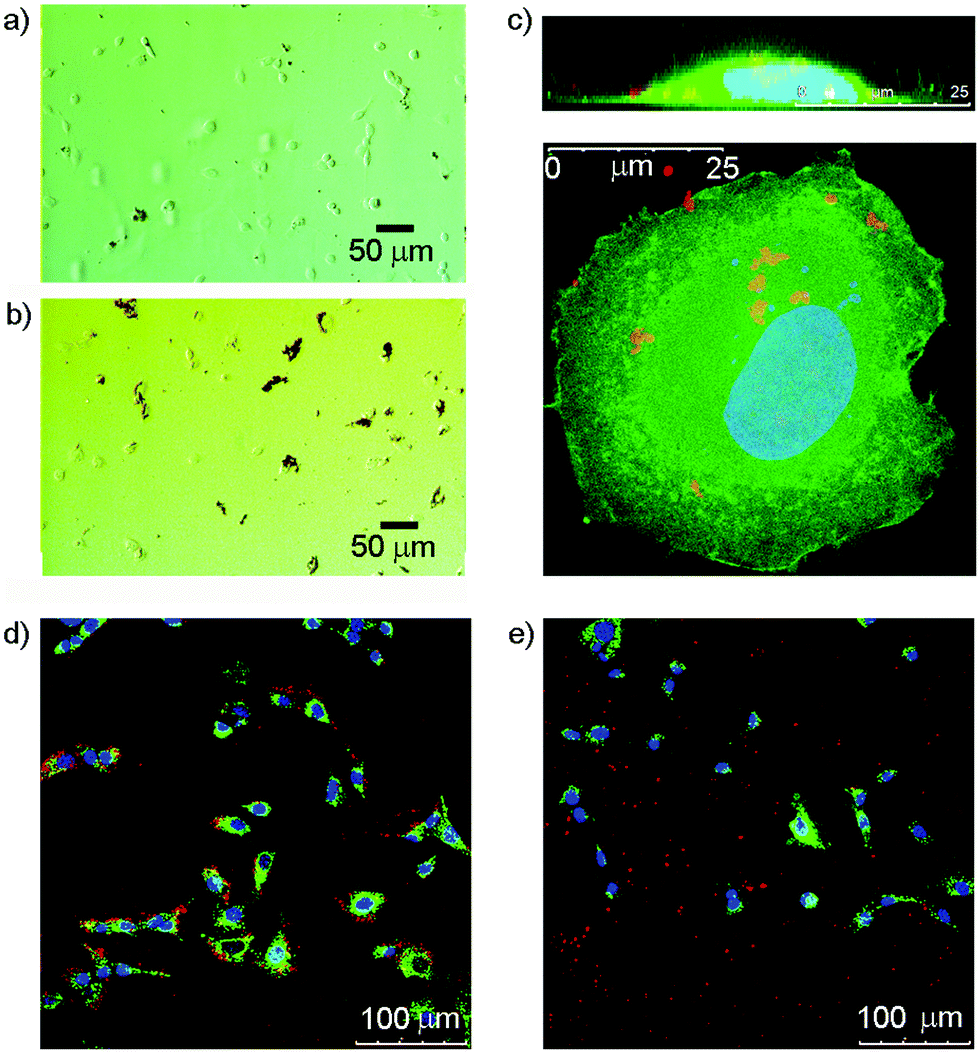 | ||
| Fig. 5 (a, b) Optical microscopy images of 3T3 fibroblasts interacting with (a) uncoated magnetite nanoparticles (b) magnetite nanoparticles coated with 3a (Glc/catechol). (c) Confocal microscopy images showing a cross-sectional (z-stack, top) view and plan view of a 3T3 fibroblast (membrane in green, nucleus in blue) interacting with 1b-MNP (red). (d, e) Flattened confocal fluorescence microscopy images showing 3T3 fibroblasts (membranes stained green, nuclei stained blue) after 3 h incubation with 0.1 mg mL−1 magnetite nanoparticles (red) coated with either (d) GlcNac/catechol 3b and dopamine–porphyrin conjugate 6 or (e) 6 alone. | ||
Both 1b-MNP and 3b-MNP were assessed for their interactions with 3T3 fibroblasts, along with MNPs with no saccharide functionality. To visualise cell–nanoparticle interactions by fluorescence microscopy, the nanoparticles were coated with both a saccharide adduct and a previously reported48 dopamine–porphyrin conjugate (N-(3,4-dihydroxyphenethyl)-4-(10,15,20-tri-p-tolylporphyrin-5-yl)benzamide 6) in a 9:1 ratio. This conjugate allowed the coated MNPs to be visualised using confocal fluorescence microscopy (ex. 420 nm, red emission at 650 nm). After seeding 3T3 fibroblasts (40000 cells per well, 1 mL per well, membranes pre-stained green with PKH67) and incubating for 24 h, nanoparticles were added at a concentration of 0.1 mg mL−1. The samples were incubated for a further 3 h before fixing with paraformaldehyde. After this, a glycine NaCl solution at pH 3 (3 × 1 mL per well) was used to remove any surface bound nanoparticles.49 Finally, the samples were mounted onto microscope slides using a mounting agent that included DAPI nuclear stain, then allowed to cure over 18 h before imaging (Fig. 5c–e).
Imaging of these mixtures of fibroblasts and nanoparticles clearly showed interactions between the cells and GlcNAc-coated nanoparticles, and suggested that MNPs lacking the saccharide coating did not interact to the same extent (Fig. 5d and e). Higher magnification imaging of single cells from this mixture, both in plan view and cross-section (z-stack), suggested some uptake of the MNPs by the cells (Fig. 5c). The red-fluorescent structures observed are too large to be individual nanoparticles and may be MNPs linked to membrane proteins located in lipid rafts or caveolae on the cell surface, proteins that include vimentin itself.50 It is also possible that those nanoparticles that have been internalised by the cell are clustered together within intracellular compartments such as endosomes or lysosomes. This would fit with literature reports that show co-localisation of internalised nanoparticles with lysosome specific dyes.51 The benefit of using multivalent displays of saccharides on MNPs became clear after the addition of 3b-MNPs (0.01 mg mL−1, estimated [3b] of 0.5 μM) to 3T3 fibroblasts that had been pre-incubated with GlcNAc (0.1 mg mL−1, 0.45 mM). Fluorescence and optical imaging showed extensive MNP–cell interactions were still present despite the presence of a 1000-fold excess of the competing ligand, free GlcNAc (see ESI†).
Attempts were made to quantify these cell–nanoparticle interactions from the fluorescence images by counting the number of red pixels from nanoparticles in each image. Though at best semi-quantitative, pixel counting is an established technique in fluorescence microscopy that has been used previously to evaluate cell–nanoparticle interactions.52 In each case, only those red pixels that appeared to be within the boundaries of a cell, and therefore associated in some manner, were counted. Table 2 also gives values for the percentage of red pixels that were determined not to have interacted with cells in each case. The green fluorescent membrane stain, PKH67, was used to help determine cell boundaries.
| Sample | Average red pixels/cell | % Unassociated |
|---|---|---|
| 1b-MNP | 13 | 36 |
| 3b-MNP | 61 | 10 |
| Control | 4 | 80 |
This experiment gave an indication that nanoparticles coated with the catechol-GlcNAc coating, 3b, are much more effective than both resorcinol-GlcNAc (1b) coated and uncoated MNPs. Not only were fewer MNPs not associated with cells in this sample, a measure that could be affected by the efficiency of washing or the hydrophobicity of different nanoparticle coatings, but there were more associated nanoparticles within the boundaries of each cell. These data show a three-fold increase in cell association for nanoparticles that have resorcinol-based coating molecule 1b compared to control nanoparticles with no saccharide functionality. However those nanoparticles with catechol-GlcNAc coating 3b showed even better cell association, with a fifteen-fold increase compared to controls, confirming that catechol-based coatings are more effective for cellular targeting than the resorcinol equivalents.
Conclusions
A simple, versatile synthetic route has been developed that allows several saccharide coatings for magnetite nanoparticles to be produced rapidly and with good anomeric purity. These coatings have proven to be sufficiently stable for use in cell culture and have shown specific binding to lectins. Therefore, they have potential for use in cell sorting or targeted hyperthermia applications. Initial studies with cells suggest improved interactions when magnetite nanoparticles have saccharide functionalisation, which may indicate the specific binding of these saccharides to cell-surface lectins. These studies also suggest that despite sensitivity to oxidation, catechol-based coatings are considerably more effective than their resorcinol-based equivalents.These targeted magnetite nanoparticles could have applications in drug delivery or hyperthermia therapies, both of which can benefit from active targeting. The inclusion of a fluorescent tag also allows the possibility of multi-modal imaging for diagnostics, using both fluorescence and magnetic resonance imaging, and theranostic applications. Ongoing studies seek to expand the repertoire of saccharides appended to MNPs and to investigate chemoenzymatic methods of increasing the complexity of the oligosaccharide coatings.
Experimental procedures
General materials
Reagents were purchased from Sigma-Aldrich Co. Ltd, Dorset, UK with the exception of 3,4-dihydroxybenzhydrazide which was purchased from Alfa Aesar, Lancashire, UK and Fluorochem, Derbyshire, UK. All lectins were purchased from Vector Labs with the exception of Wheat Germ Agglutinin which was purchased from Sigma-Aldrich Co. Ltd., Dorset, UK. Magnetite nanoparticles (nanopowder, <50 nm (TEM), ≥98% trace metals basis) were purchased from Sigma-Aldrich Co. Ltd., Dorset, UK. N-(3,4-Dihydroxyphenethyl)-4-(10,15,20-tri-p-tolylporphyrin-5-yl)benzamide 6 was synthesised as per literature procedures.48Reversed phase HPLC purification and analysis was performed using an Agilent 110 series system with an Agilent Eclipse XDB-C18 (9.4 × 250 mm) column. Nanoparticle sonication was performed at 20 kHz using a Sonics VCX130PB Ultrasonic processor with a stepped micro tip (3 mm diameter, 136 mm length). Electrospray mass spectrometry was carried out using a Micromass LCT instrument using a Waters 2790 separations module with electrospray ionization and TOF fragment detection. Fluorescence spectra were taken on a Perkin-Elmer LS55 Fluorimeter. UV spectra were taken using a Jasco V-660 spectrometer. QCM-D was performed using a Q-Sense E4 with an Ismatec IPC-N 4 peristaltic pump or a Q-Sense E1 system with accompanying Q-Soft data acquisition software. NMR spectra were taken in deuterated solvents using either a Brüker 400 Avance spectrometer with broadband probe or a Brüker 500 MHz Avance III spectrometer with QCP-F cryoprobe equipped with z-gradients. Chemical shift values are reported in ppm referenced to residual non-deuterated solvent and relative to tetramethylsilane. Coupling constants are reported in Hertz (Hz). Multiplicities are reported using the following notations: singlet (s), doublet (d), doublet of doublets (dd), triplet (t) and multiplet (m). Where necessary, COSY, DEPT135 and HMQC experiments were used to aid peak assignment.
Fluorescence images were taken using a Zeiss Axio Imager A1 fluorescence microscope with a Canon Powershot G6 digital camera attached. Confocal microscopy was performed using a Leika TCS SP5. Where necessary, pixel and cell counting analysis was carried out using ImageJ.
General procedure for the synthesis of conjugates
Saccharide (0.3 mmol) and hydrazide (0.3 mmol) were dissolved in methanol with aniline (6 mL of 1 mM stock solution). The reaction was allowed to reflux overnight under a N2 atmosphere. After this time, the reaction was allowed to cool before removal of the solvent under reduced pressure. Purification was achieved by HPLC, using a gradient method shifting linearly over 1 h (from 5% to 50% THF in water for compounds 1a–1d and 3a–3d, and from 50% to 95% THF in water for compounds 2a–2c). The eluent was monitored by UV-visible spectroscopy and the product was collected and freeze-dried to give a white powder.:87 mixture of α/β-anomers in methanol-d4). 1H NMR (400 MHz, CD3OD): δ 6.73 (d, J = 2.2 Hz, 2H, ArH-2), 6.43 (t, J = 2.2 Hz, 1H, ArH-4), 4.66 (d, J = 4.8 Hz, 0.13H, CH-1α′), 4.08 (d, J = 9.5 Hz, 0.87H, CH-1β′), 3.88 (dd, J1 = 12.0 Hz, J2 = 2.2 Hz, 1H, CH-6a′), 3.80 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H, CH-3′), 3.63 (dd, J1 = 12.0 Hz, J2 = 6.4 Hz, 1H, CH-6b′), 3.50 (dd, J1 = 10.0 Hz, J2 = 8.3 Hz, 1H, CH-2′), 3.22–3.31 (m, 2H, CH-4′, CH-5′), 2.09 (s, 3H, COCH3). 13C NMR (101 MHz, CD3OD): δ 175.3, 167.0, 160.0 (2C), 135.4, 107.1 (2C), 106.8, 91.2, 79.2, 76.4, 72.3, 63.0, 54.7, 23.3. HRMS (ES+): m/z calcd for [C15H22N3O8]+ 372.1407, found 372.1406. Anal. calcd for C15H21N3O8 (+2H2O): C, 44.23; H, 6.19; N, 10.32; found C, 44.02; H, 6.22, N, 10.08.
:89 mixture of α/β-anomers in methanol-d4). 1H NMR (400 MHz, CD3OD): δ 6.71 (d, J = 2.2 Hz, 2H, ArH-2), 6.44 (t, J = 2.2 Hz, 1H, ArH-4), 4.63 (d, J = 5.0 Hz, 0.11H, CH-1α′), 4.34 (d, J = 7.2 Hz, 1H, CH-1′′), 4.06 (d, J = 8.8 Hz, 0.89H, CH-1β′), 3.97 (dd, J1 = 11.6 Hz, J2 = 2.2 Hz, 1H, CH-6a′), 3.76–3.87 (m, 3H, CH-4′′, CH-6′′), 3.70 (dd, J1 = 11.5 Hz, J2 = 4.7 Hz, 1H, CH-6b′), 3.44–3.65 (m, 6H, CH-3′-4′-5′, CH-2′′-3′′-5′′), 3.28–3.35 (MeOH peak obscuring CH-2′). 13C NMR (101 MHz, CD3OD): δ 170.8, 160.0 (2C), 136.1, 106.9 (2C), 106.6, 105.1, 92.1, 80.4, 77.7, 77.1, 76.6, 74.8, 72.5, 72.2, 70.3, 62.5, 62.1. HRMS (ES+): m/z calcd for [C19H29N2O13]+ 493.1670, found 493.1671. Anal. calcd for C19H28N2O13 (+ 3.5H2O): C, 41.08; H, 6.35; N, 5.04; found C, 40.94; H, 6.54, N, 4.93.
:79 mixture of α/β-anomers in methanol-d4). 1H NMR (400 MHz, CD3OD): δ 6.59 (d, J = 2.2 Hz, 2H, ArH-2), 6.32 (t, J = 2.2 Hz, 1H, ArH-4), 4.51 (d, J = 5.3 Hz, 0.21H, CH-1α′), 4.29 (d, J = 7.8, 1H, CH-1′′), 3.95 (m, 1.79H, CH-1β′, 16-CH), 3.88 (dd, J1 = 11.8 Hz, J2 = 2.0 Hz, 1H, CH-6a′), 3.33–3.83 (m, 17H, CH-2′-3′-4′-5′, CH-6b′, CH-2′′-3′′-4′′-5′′-6′′, 14-CH, 15-CH, 17–19-CH), 2.76 (ddd, J1 = 12.0 Hz, J2 = 2.0 Hz, J3 = 2.0 Hz, 1H, 13-CHa), 1.91 (s, 3H, 20-CH), 2.76 (ddd, J1 = 11.0 Hz, J2 = 9.4 Hz, J3 = 3.6 Hz, 1H, 13-CHb). 13C NMR (101 MHz, CD3OD): δ 175.5, 174.9, 169.2, 160.0 (2C), 136.1, 107.0 (3C), 105.1, 101.1, 91.8, 80.7, 77.7, 77.1, 76.6, 76.0, 75.0, 73.0, 72.1, 70.8, 70.1, 69.4, 69.0, 64.6, 62.8, 62.0, 54.0, 42.2, 22.6. HRMS (ES+): m/z calcd for [C30H45N3O21Na]+ 806.2438, found 806.2421. Anal. calcd for C30H44N3O21Na (+ 2.5H2O): C, 41.48; H, 5.92; N, 4.84; found C, 41.49; H, 5.76, N, 4.54.
:87 mixture of α/β-anomers in methanol-d4). 1H NMR (400 MHz, CD3OD): δ 4.52 (d, J = 5.0 Hz, 0.13H, CH-1α′), 4.04 (s, 2H, COCH2O), 3.87 (d, J = 9.0 Hz, 0.87H, CH-1β′), 3.83 (dd, J1 = 11.5 Hz, J2 = 2.3 Hz, 1H, CH-6a′), 3.50–3.71 (m, 13H, CH-6b′, OCH2CH2O), 3.42 (t, J = 6.4 Hz, 2H, OCH2R), 3.35 (dd, J1 = 8.6 Hz, J2 = 8.6 Hz, 1H, CH-3′), 3.17–3.30 (m, MeOH obscuring CH-5′), 3.13 (m, 2H, CH-2′, CH-4′), 1.53 (m, 2H, RCH2CH3), 1.24 (m, 30H, RCH2CH2R), 0.86 (t, J = 7.0 Hz, 3H, RCH2CH3). 13C NMR (101 MHz, CD3OD): δ 175.4, 92.7, 79.2, 78.4, 73.7, 72.6, 72.4, 72.1, 71.6 (3C), 71.5, 71.4, 71.2, 62.9, 33.1, 30.8 (12C), 30.5, 27.3, 23.8, 14.5. HRMS (ES+): m/z calcd for [C32H65N2O10]+ 637.4634, found 637.4644. Anal. calcd for C32H65N2O10 (+H2O): C, 58.69; H, 10.16; N, 4.28; found C, 58.65; H, 9.98, N, 4.35.
:87 mixture of α/β-anomers in methanol-d4). 1H NMR (400 MHz, CD3OD): δ 4.62 (d, J = 5.0 Hz, 0.13H, CH-1α′), 4.01 (s, 2H, COCH2O), 3.95 (d, J = 9.6 Hz, 0.87H, CH-1β′), 3.79 (dd, J1 = 12.0 Hz, J2 = 1.8 Hz, 1H, CH-6a′), 3.50–3.70 (m, 15H, CH-4′, CH-5′, CH-6b′, OCH2CH2O), 3.42 (m, 3H, CH-3′, OCH2R), 3.18–3.31 (m, MeOH obscuring CH-2′), 1.97 (s, 3H, COCH3), 1.52 (m, 2H, RCH2CH3), 1.24 (m, 30H, RCH2CH2R), 0.85 (t, J = 7.0 Hz, 3H, RCH2CH3). 13C NMR (101 MHz, CD3OD): δ 175.2, 175.0, 91.2, 79.2, 76.4, 72.6, 72.4, 72.3, 71.6 (3C), 71.5, 71.4, 71.2, 63.1, 54.7, 33.1, 30.8 (12C), 30.5, 27.2, 23.8, 23.2, 14.5. HRMS (ES+): m/z calcd for [C34H68N3O10]+ 678.4899, found 678.4889. Anal. calcd for C34H67N3O10 (+2.5H2O): C, 56.49; H, 10.04; N, 5.81; found C, 56.35; H, 10.41, N, 5.68.
:85 mixture of α/β-anomers in methanol-d4). 1H NMR (400 MHz, CD3OD): δ 4.61 (d, J = 5.1 Hz, 0.15H, CH-1α′), 4.36 (d, J = 7.6 Hz, 1H, CH-1′′), 4.10 (s, 2H, COCH2O), 3.99 (d, J = 8.9 Hz, 0.85H, CH-1β′), 3.94 (dd, J1 = 12.0 Hz, J2 = 2.3 Hz, 1H, CH-6a′), 3.76–3.87 (m, 4H, CH-6b′, CH-4′′, CH-6′′), 3.63–3.75 (m, 12H, OCH2CH2O), 3.53–3.63 (m, 5H, CH-4′, CH-5′, CH-2′′, CH-3′′, CH-5′′), 3.46–3.52 (m, 3H, CH-3′, OCH2R), 3.27 (t, J = 8.9 Hz, 1H, CH-2′), 1.59 (m, 2H, RCH2CH3), 1.31 (m, 30H, RCH2CH2R), 0.92 (t, J = 6.8 Hz, 3H, RCH2CH3). 13C NMR (101 MHz, CD3OD): δ 175.5, 105.1, 92.9, 80.4, 77.8, 77.1, 76.4, 74.8, 72.6, 72.5, 72.4, 72.2, 71.5 (3C), 71.4 (2C), 71.2, 70.3, 63.1, 62.3, 33.1, 30.8 (12C), 30.5, 27.2, 23.8, 14.5. HRMS (ES+): m/z calcd for [C38H75N2O15]+ 799.5162, found 799.5122. Anal. calcd for C38H74N2O15 (+3H2O): C, 53.50; H, 9.45; N, 3.28; found C, 53.46; H, 9.80, N, 2.91.
:91 mixture of α/β anomers in methanol-d4). 1H-NMR (400 MHz, CD3OD): δ 7.27 (d, J = 2.2 Hz, 1H, ArH-2), 7.20 (dd, J1 = 8.3 Hz, J2 = 2.2 Hz, 1H, ArH-5), 6.81 (d, J = 8.3 Hz, 1H, ArH-6), 4.58 (d, J = 4.9 Hz, 0.09H, CH-1α′), 3.97 (d, J = 8.8 Hz, 0.91H, CH-1β′), 3.91 (dd, J1 = 11.6 Hz, J2 = 2.3 Hz, 1H, CH-6a′), 3.63 (dd, J1 = 12.1 Hz, J2 = 6.6 Hz, 1H, CH-6b′), 3.42 (dd, J1 = 8.6 Hz, J2 = 8.6 Hz, 1H, CH-3′), 3.28–3.34 (m, MeOH obscuring CH-5′), 3.22 (m, 2H, CH-2′, CH-4′). 13C-NMR (101 MHz, CD3OD): δ 170.5, 150.6, 146.4, 125.3, 121.0, 115.9, 115.6, 92.4, 79.0, 78.3, 72.6, 71.8, 63.0. HRMS (ES+): m/z calcd for [C13H18N2O8Na]+ 353.0961, found 353.0975. Anal. calcd for C13H18N2O8 (+H2O): C, 44.83; H, 5.79; N, 8.04; found C, 44.71; H, 6.19; N, 7.82.
:91 mixture of α/β anomers in methanol-d4). 1H-NMR (400 MHz, CD3OD): δ 7.29 (d, J = 2.2 Hz, 1H, ArH-2), 7.22 (dd, J1 = 8.3 Hz, J2 = 2.2 Hz, 1H, ArH-6), 6.81 (d, J1 = 8.3 Hz, 1H, ArH-5), 4.54 (d, J = 4.9 Hz, 0.09H, CH-1α′), 4.05 (d, J = 9.5 Hz, 0.91H, CH-1β′), 3.85 (dd, J1 = 12.0 Hz, J2 = 2.0 Hz, 1H, CH-6a′), 3.79 (dd, J1 = 9.8 Hz, J2 = 9.8 Hz, 1H, CH-3), 3.61 (dd, J1 = 12.0 Hz, J2 = 6.4 Hz, 1H, CH-6b′), 3.20–3.33 (m, MeOH peak obscuring CH-2′, CH-4′, CH-5′), 2.07 (s, 3H, COCH3). 13C-NMR (101 MHz, CD3OD): δ 175.3, 168.8, 150.5, 146.4, 125.3, 120.7, 116.2, 115.7, 91.3, 79.2, 76.4, 72.3, 63.1, 54.7, 23.2. HRMS (ES+): m/z calcd for [C15H21N3O8Na]+ 394.1226, found 394.1221. Anal. calcd for C15H21N3O8 (+ THF + 4.5H2O): C, 43.59; H, 7.12; N, 8.03; found C, 43.99; H, 6.69; N, 7.89.
:90 mixture of α/β anomers in methanol-d4). 1H-NMR (400 MHz, CD3OD): δ 7.27 (d, J = 2.2 Hz, 1H, ArH-2), 7.20 (dd, J1 = 8.3 Hz, J2 = 2.2 Hz, 1H, ArH-6), 6.81 (d, J1 = 8.3 Hz, 1H, ArH-5), 4.60 (d, J = 5.3 Hz, 0.10H, CH-1α′), 4.32 (d, J = 7.6 Hz, 1H, CH-1′′), 4.03 (d, J = 8.9 Hz, 0.90H, CH-1β′), 3.95 (dd, J1 = 11.7 Hz, J2 = 2.0 Hz, 1H, CH-6a′), 3.74–3.85 (m, 3H, CH-4′′, CH-6′′), 3.68 (dd, J1 = 11.5 Hz, J2 = 4.6 Hz, 1H, CH-6b′), 3.42–3.62 (m, 6H, CH-3′-4′-5′, CH-2′′, CH-3′′, CH-5′′), 3.25–3.36 (MeOH peak obscuring CH-2′). 13C-NMR (101 MHz, CD3OD): δ 171.2, 150.1, 145.9, 125.3, 120.8, 115.9, 115.5, 104.9, 91.8, 80.3, 77.8, 77.3, 76.7, 74.5, 72.9, 72.4, 70.5, 63.0, 62.0. HRMS (ES+): m/z calcd for [C19H29N2O13]+ 493.1670, found 493.1672. Anal. calcd for C19H28N2O13 (+ 3H2O): C, 41.76; H, 6.27; N, 5.13; found C, 42.15; H, 6.13; N, 5.02.
:90 mixture of α/β-anomers in methanol-d4). 1H-NMR (400 MHz, CD3OD): δ 7.29 (d, J = 2.2 Hz, 1H, ArH-2), 7.22 (dd, J1 = 8.3 Hz, J2 = 2.2 Hz, 1H, ArH-6), 6.82 (d, J1 = 8.3 Hz, 1H, ArH-5), 4.50 (d, J = 5.1 Hz, 0.10H, CH-1α′), 4.39 (d, J = 7.8 Hz, 1H, CH-1′′), 4.07 (m, 1.90H, CH-1β′, 16-CH), 3.98 (dd, J1 = 11.8 Hz, J2 = 2.0 Hz, 1H, CH-6a′), 3.40–3.94 (m, 17H, CH-2′-3′-4′-5′, CH-6b′, CH-2′′-3′′-4′′-5′′-6′′, 14–15-CH, 17–19-CH), 2.86 (ddd, J1 = 11.9 Hz, J2 = 2.2 Hz, J3 = 2.0 Hz, 1H, 13-CHa), 2.01 (s, 3H, 20-CH) 1.74 (ddd, J1 = 9.6 Hz, J2 = 9.0 Hz, J3 = 2.3 Hz, 1H, 13-CHb). 13C-NMR (101 MHz, CD3OD): δ 175.5, 175.1, 169.9, 150.0, 142.4, 129.1, 120.6, 116.0, 115.6, 105.1, 101.4, 91.8, 80.6, 77.6, 77.1, 76.6, 75.9, 75.0, 73.1, 72.0, 70.8, 70.1, 69.3, 69.0, 64.6, 62.8, 61.9, 54.0, 42.1, 22.7. HRMS (ES+): m/z calcd for [C30H45N3O21Na]+ 806.2438, found 806.2484. Anal. calcd for C30H44N3O21Na (+ 4.5H2O): C, 40.64; H, 6.02; N, 4.74; found C, 40.65; H, 5.68; N, 4.50.
General nanoparticle coating procedure
Magnetite nanoparticles (10 mg) were suspended in methanol (5 mL) by probe sonication for 5 minutes. To this suspension was added the desired coating molecule, X (X = any of 1a to 3d, 0.1 mmol). The sample was sonicated for a further 45 minutes to give X-MNP. Remaining coating material was removed by centrifugation, supernatant removal and methanol washing (3 × 10 mL). Finally, the coated nanoparticles were resuspended in milli-Q filtered water (1 mL) and used immediately. For fluorescence microscopy applications, an adaptation of this coating procedure was used, sonicating magnetite nanoparticles (10 mg) in methanol (5 mL) with the desired hydrazone coating molecule along with N-(3,4-dihydroxyphenethyl)-4-(10,15,20-tri-p-tolylporphyrin-5-yl)benzamide (total 10 mg, ratio of 9:1 hydrazone:porphyrin).
General vesicle preparation procedure
A lipid film was produced by adding DMPC (14 mg, 20 μmol) to the appropriate glycolipid 2b or 2c (5% mol/mol) in chloroform and then removing the solvent by rotary evaporation. After adding HEPES buffer, the lipid film was resuspended by vortex mixing and the resulting multilamellar vesicles were probe sonicated for 20 minutes to form small unilamellar vesicles (shown to be 28 ± 2 nm diameter by dynamic light scattering, see ESI†).38bAcknowledgements
TPC and TWF would like to thank the North-West Nanoscience Doctoral Training Centre, EPSRC grant EP/G03737X/1 and EPSRC grant EP/K039547/1 for equipment funding. Additional research data supporting this publication are available from the eScholar repository at http://dx.doi.org/10.15127/1.272343. We thank Biolin Scientific (UK) for the generous loan of a Q-Sense E4 instrument and Dr Chris Blanford for the use of a Q-Sense E1 instrument. We would also like to thank Mr Simon McAdams for TEM imaging and Dr Louise Carney for assistance with cell culture.Notes and references
- (a) R. de la Rica, D. Aili and M. M. Stevens, Adv. Drug Delivery Rev., 2012, 64, 967–978 CrossRef CAS PubMed; (b) P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed; (c) G. Y. Tonga, K. Saha and V. M. Rotello, Adv. Mater., 2014, 26, 797–802 CrossRef PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 145–160 CrossRef PubMed.
- (a) Y. Ling, K. Wei, Y. Luo, X. Gao and S. Zhong, Biomaterials, 2011, 32, 7139–7150 CrossRef CAS PubMed; (b) H. Mok, O. Veiseh, C. Fang, F. M. Kievit, F. Y. Wang, J. O. Park and M. Zhang, Mol. Pharm., 2010, 7, 1930–1939 CrossRef CAS PubMed; (c) W. Lu, G. Zhang, R. Zhang, L. G. Flores II, Q. Huang, J. G. Gelovani and C. Li, Cancer Res., 2010, 70, 3177–3188 CrossRef CAS PubMed; (d) M. Mahmoudi, S. Sant, B. Wang, S. Laurent and T. Sen, Adv. Drug Delivery Rev., 2011, 63, 24–46 CrossRef CAS PubMed.
- (a) J. Blanchfield and I. Toth, Curr. Med. Chem., 2004, 11, 2375–2382 CrossRef CAS PubMed; (b) D. Benito-Alifonso, S. Tremel, B. Hou, H. Lockyear, J. Mantell, D. J. Fermin, P. Verkade, M. Berry and M. C. Galan, Angew. Chem., Int. Ed., 2014, 53, 810–814 CrossRef CAS PubMed; (c) Y. Lee, H. Lee, Y. B. Kim, J. Kim, T. Hyeon, H. W. Park, P. B. Messersmith and T. G. Park, Adv. Mater., 2008, 20, 4154–4157 CAS; (d) A. Robinson, J. M. Fang, P. T. Chou, K. W. Liao, R. M. Chu and S. J. Lee, ChemBioChem, 2005, 6, 1899–1905 CrossRef CAS PubMed; (e) Y. Yang, M. Yu, T. T. Yan, Z. H. Zhao, Y. L. Sha and Z. J. Li, Bioorg. Med. Chem., 2010, 18, 5234–5240 CrossRef CAS PubMed.
- (a) R. J. Stockert, Physiol. Rev., 1995, 75, 591–609 CAS; (b) A. Raz, L. Meromsky and R. Lotan, Cancer Res., 1986, 46, 3667–3672 CAS; (c) V. M. Platt and F. C. Szoka Jr., Mol. Pharmaceutics, 2008, 5, 474–486 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- (a) C. Tassa, J. L. Duffner, T. A. Lewis, R. Weissleder, S. L. Schreiber, A. N. Koehler and S. Y. Shaw, Bioconjugate Chem., 2010, 21, 14–19 CrossRef CAS PubMed; (b) M. Durka, K. Buffet, J. Iehl, M. Holler, J. F. Nierengarten and S. P. Vincent, Chem. – Eur. J., 2012, 18, 641–651 CrossRef CAS PubMed; (c) M. Marradi, M. Martin-Lomas and S. Penades, Adv. Carbohydr. Chem. Biochem., 2010, 64, 211–290 CrossRef CAS PubMed; (d) S. Cecioni, V. Oerthel, J. Iehl, M. Holler, D. Goyard, J.-P. Praly, A. Imberty, J.-F. Nierengarten and S. Vidal, Chem. – Eur. J., 2011, 17, 3252–3261 CrossRef CAS PubMed; (e) X. Wang, E. Matei, L. Deng, O. Ramström, A. M. Gronenborn and M. Yan, Chem. Commun., 2011, 47, 8620–8622 RSC; (f) G. T. Noble, K. P. Liem, S. L. Flitsch and S. J. Webb, Org. Biomol. Chem., 2009, 7, 5245–5254 RSC.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- S. Kawakami and M. Hashida, J. Controlled Release, 2014, 190, 542–555 CrossRef CAS PubMed.
- H. Zhang, Y. Ma and X. L. Sun, Med. Res. Rev., 2010, 30, 270–289 CrossRef CAS PubMed.
- M. Moros, B. Hernáez, E. Garet, J. T. Dias, B. Sáez, V. Grazú, A. González-Fernández, C. Alonso and J. M. de la Fuente, ACS Nano, 2012, 6, 1565–1577 CrossRef CAS PubMed.
- L. Lartigue, C. Innocenti, T. Kalaivani, A. Awwad, M. del Mar Sanchez Duque, Y. Guari, J. Larionova, C. Guérin, J. L. G. Montero, V. Barragan-Montero, P. Arosio, A. Lascialfari, D. Gatteschi and C. Sangregorio, J. Am. Chem. Soc., 2011, 133, 10459–10472 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS PubMed.
- (a) A. Samanta and B. J. Ravoo, Angew. Chem., Int. Ed., 2014, 53, 12946–12950 CrossRef CAS PubMed; (b) D. Li, W. Y. Teoh, J. J. Gooding, C. Selomulya and R. Amal, Adv. Funct. Mater., 2010, 20, 1767–1777 CrossRef CAS; (c) S. Cheong, P. Ferguson, K. W. Feindel, I. F. Hermans, P. T. Callaghan, C. Meyer, A. Slocombe, C.-H. Su, F.-Y. Cheng, C.-S. Yeh, B. Ingham, M. F. Toney and R. D. Tilley, Angew. Chem., Int. Ed., 2011, 50, 4206–4209 CrossRef CAS PubMed; (d) C. Blanco-Andujar, D. Ortega, P. Southern, Q. A. Pankhurst and N. T. K. Thanh, Nanoscale, 2015, 7, 1768–1775 RSC.
- (a) A. Booth, I. C. Pintre, Y. Lin, J. E. Gough and S. J. Webb, Phys. Chem. Chem. Phys., 2015, 17, 15579–15588 RSC; (b) F. de Cogan, A. Booth, J. E. Gough and S. J. Webb, Angew. Chem., Int. Ed., 2011, 50, 12290–12293 CrossRef CAS PubMed; (c) F. de Cogan, A. Booth, J. E. Gough and S. J. Webb, Soft Matter, 2013, 9, 2245–2253 RSC.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 9963–9965 CrossRef CAS PubMed.
- R. L. E. Furlan, Y.-F. Ng, S. Otto and J. K. M. Sanders, J. Am. Chem. Soc., 2001, 123, 8876–8877 CrossRef CAS PubMed.
- (a) V. T. Bhat, A. M. Caniard, T. Luksch, R. Brenk, D. J. Campopiano and M. F. Greaney, Nat. Chem., 2010, 2, 490–497 CrossRef CAS PubMed; (b) A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef CAS.
- A. Dirksen and P. E. Dawson, Bioconjugate Chem., 2008, 19, 2543–2548 CrossRef CAS PubMed.
- (a) B. Helferich and H. Schirp, Chem. Ber., 1951, 84, 469–471 CrossRef CAS; (b) B. Helferich and H. Schirp, Chem. Ber., 1953, 86, 547–556 CrossRef CAS.
- (a) A. V. Gudmundsdottir, C. E. Paul and M. Nitz, Carbohydr. Res., 2009, 344, 278–284 CrossRef CAS PubMed; (b) K. Bailey and A. G. Butterfield, Can. J. Chem., 1981, 59, 641–646 CrossRef CAS; (c) B. Bendiak, Carbohydr. Res., 1997, 304, 85–90 CrossRef CAS PubMed; (d) Y. Takeda, Carbohydr. Res., 1979, 77, 9–23 CrossRef CAS.
- X. Fan, L. Lin, J. L. Dalsin and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 15843–15847 CrossRef CAS PubMed.
- G. Absalan, M. Asadi, S. Kamran, L. Sheikhian and D. M. Goltz, J. Hazard. Mater., 2011, 192, 476–484 CrossRef CAS PubMed.
- N. Schweigert, A. J. B. Zehnder and R. I. L. Eggen, Environ. Microbiol., 2001, 3, 81–91 CrossRef CAS PubMed.
- A. K. L. Yuen, G. A. Hutton, A. F. Masters and T. Maschmeyer, Dalton Trans., 2012, 41, 2545–2559 RSC.
- In the absence of catalyst, ∼5% reaction was observed after 24 h.
- No change in the anomeric ratio of 1a was observed upon dissolution in D2O, although the ratio changed to 78% β-anomer after 460 h in d6-DMSO.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- M. D. Shultz, J. U. Reveles, S. N. Khanna and E. E. Carpenter, J. Am. Chem. Soc., 2007, 129, 2482–2487 CrossRef CAS PubMed.
- C.-J. Yu, S.-M. Wu and W.-L. Tseng, Anal. Chem., 2013, 85, 8559–8565 CrossRef CAS PubMed.
- S. L. Saville, R. C. Stone, B. Qi and O. Thompson Mefford, J. Mater. Chem., 2012, 22, 24909–24917 RSC.
- K. A. Marx, Biomacromolecules, 2003, 4, 1099–1120 CrossRef CAS PubMed.
- D. Johannsmann, Phys. Chem. Chem. Phys., 2008, 10, 4516–4534 RSC.
- D. Thakar, E. Migliorini, L. Coche-Guerente, R. Sadir, H. Lortat-Jacob, D. Boturyn, O. Renaudet, P. Labbe and R. P. Richter, Chem. Commun., 2014, 50, 15148–15151 RSC.
- Z. Mouline, E. Mahon, E. Gomez, V. Barragan-Montero, J. L. Montero and M. Barboiu, Chem. Commun., 2014, 50, 731–733 RSC.
- E. Mahon, Z. Mouline, M. Silion, A. Gilles, M. Pintaela and M. Barboiu, Chem. Commun., 2013, 49, 3004–3006 RSC.
- (a) E. Reimhult, F. Höök and B. Kasemo, Langmuir, 2003, 19, 1681–1691 CrossRef CAS; (b) B. Seantier, C. Breffa, O. Félix and G. Decher, J. Phys. Chem. B, 2005, 109, 21755–21765 CrossRef CAS PubMed.
- M. Tominaga, M. Ohtani and I. Taniguchi, Phys. Chem. Chem. Phys., 2008, 10, 6928–6934 RSC.
- The smaller response for these catechol coated nanoparticles was ascribed to the use of a Q-Sense E1 instrument for these measurements; the response for the analogous resorcinol coated nanoparticles also became smaller when using this instrument.
- D. C. Kilpatrick, Biochim. Biophys. Acta, 2002, 1572, 187–197 CrossRef CAS.
- R. Duncan, J. Kopeček, P. Rejmanová and J. B. Lloyd, Biochim. Biophys. Acta, 1983, 755, 518–521 CrossRef CAS.
- M. Gary-Bobo, Y. Mir, C. Rouxel, D. Brevet, I. Basile, M. Maynadier, O. Vaillant, O. Mongin, M. Blanchard-Desce, A. Morère, M. Garcia, J. O. Durand and L. Raehm, Angew. Chem., Int. Ed., 2011, 50, 11425–11429 CrossRef CAS PubMed.
- V. Thibault, O. Blanck, J. Courageot, C. Pachetti, C. Perrin, A. de Mascarel and R. Miquelis, Endocrinology, 1993, 132, 468–476 CrossRef CAS PubMed.
- A. Krzeslak, K. Wojcik-Krowiranda, E. Forma, P. Jozwiak, H. Romanowicz, A. Bienkiewicz and M. Brys, Pathol. Oncol. Res., 2012, 18, 721–728 CrossRef CAS PubMed.
- H. Ise, S. Kobayashi, M. Goto, T. Sato, M. Kawakubo, M. Takahashi, U. Ikeda and T. Akaike, Glycobiology, 2010, 20, 843–864 CrossRef CAS PubMed.
- N. Du, H. Cong, H. Tian, H. Zhang, W. Zhang, L. Song and P. Tien, J. Virol., 2014, 88, 5816–5833 CrossRef PubMed.
- T. Coxon, A. Almond, J. E. Gough and S. J. Webb, MRS Proc., 2014, 1688, mrss14-1688-y08-13, DOI:10.1557/opl.2014.433.
- S. Kameyama, M. Horie, T. Kikuchi, T. Omura, A. Tadokoro, T. Takeuchi, I. Nakase, Y. Sugiura and S. Futaki, Biopolymers, 2007, 88, 98–107 CrossRef CAS PubMed.
- (a) R. R. Sprenger, R. D. Fontijn, J. van Marle, H. Pannekoek and A. J. G. Horrevoets, Biochem. J., 2006, 400, 401–410 CrossRef CAS PubMed; (b) K. Uehara and A. Uehara, Anat. Rec., 2010, 293, 2034–2043 CrossRef CAS PubMed.
- A. Villanueva, M. Cañete, A. G. Roca, M. Calero, S. Veintemillas-Verdaguer, C. J. Serna, M. Del Puerto Morales and R. Miranda, Nanotechnology, 2009, 20, 115103 CrossRef PubMed.
- (a) A. Erten, W. Wrasidlo, M. Scadeng, S. Esener, R. M. Hoffman, M. Bouvet and M. Makale, Nanomedicine, 2010, 6, 797–807 CrossRef CAS PubMed; (b) C. Gottstein, G. Wu, B. J. Wong and J. A. Zasadzinski, ACS Nano, 2013, 7, 4933–4945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cell culture procedures, additional lectin binding data and copies of NMR spectra. See DOI: 10.1039/c5ob01591j |
| This journal is © The Royal Society of Chemistry 2015 |