
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Putting corannulene in its place. Reactivity studies comparing corannulene with other aromatic hydrocarbons†
Stephen R. D.
George
a,
Thomas D. H.
Frith
a,
Donald S.
Thomas
*b and
Jason B.
Harper
*a
aSchool of Chemistry, University of New South Wales, UNSW Sydney, NSW 2052, Australia. E-mail: j.harper@unsw.edu.au; Fax: +61 2 9385 6141; Tel: +61 2 9385 4692
bNuclear Magnetic Resonance Facility, Mark Wainwright Analytical Centre, University of New South Wales, UNSW Sydney, NSW 2052, Australia. E-mail: donald.thomas@unsw.edu.au
First published on 21st July 2015
Abstract
A series of aromatic hydrocarbons were investigated so as to compare the reactivity of corannulene with planar aromatic hydrocarbons. Corannulene was found to be more reactive than benzene, naphthalene and triphenylene to Friedel–Crafts acylation whilst electrophilic aromatic bromination was also used to confirm that triphenylene was less reactive than corannulene and that pyrene, perylene and acenaphthene were more so. The stabilisation of a neighbouring carbocation by the various aromatic systems was investigated through consideration of the rates of methanolysis of a series of benzylic alcohols. The reactivity series was found to parallel that observed for the electrophilic aromatic substitutions and both series are supported by computational studies. As such, a reactivity scale was devised that showed that corannulene was less reactive than would be expected for an aromatic planar species of similar pi electron count.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a group of cyclic compounds consisting of fused aromatic rings that do not contain any heteroatoms; the carbon atoms are predominantly sp2 hybridised and the pi electrons of the system delocalised.1 First discovered in crude oils in the late 1800s,2 PAHs can either be a simple one ring extension of the monocyclic benzene 1 to give naphthalene 2 or form more complex polycycles such as phenanthrene 3, the tethered napthyl system acenaphthene 4, fluoranthene 5, pyrene 6 and triphenylene 7. Recently there has been significant interest3 in non-planar PAHs such as corannulene 8 and the chrysene 9,4 as potential starting points for the rational synthesis of carbon nanotubes.Reactions of these species, particularly towards electrophiles, are well studied; these reactions include the Friedel–Crafts acylation of species 1,52,53,65,778 and 8,9 and the bromination of acenaphthene 410 and pyrene 6.11 In terms of the relative reactivity of some of these species, competitive sulfonation12 showed that reactivity increased in the order benzene 1, phenanthrene 3, naphthalene 2 and, finally, pyrene 6 with the order of 1, 3 and 2 also confirmed by Gore et al.13 and Pryor et al.14 Whilst no competitive bromination studies have been undertaken, a comparison of reaction conditions allows a reactivity order to be developed. Species 1 and 2 do not react with N-bromosuccinimide at room temperature in DMF.15 The larger species 5 undergoes reaction at elevated temperature16 whilst species 4 and 6 react to completion in 215 and 2417 hours, respectively, at room temperature.
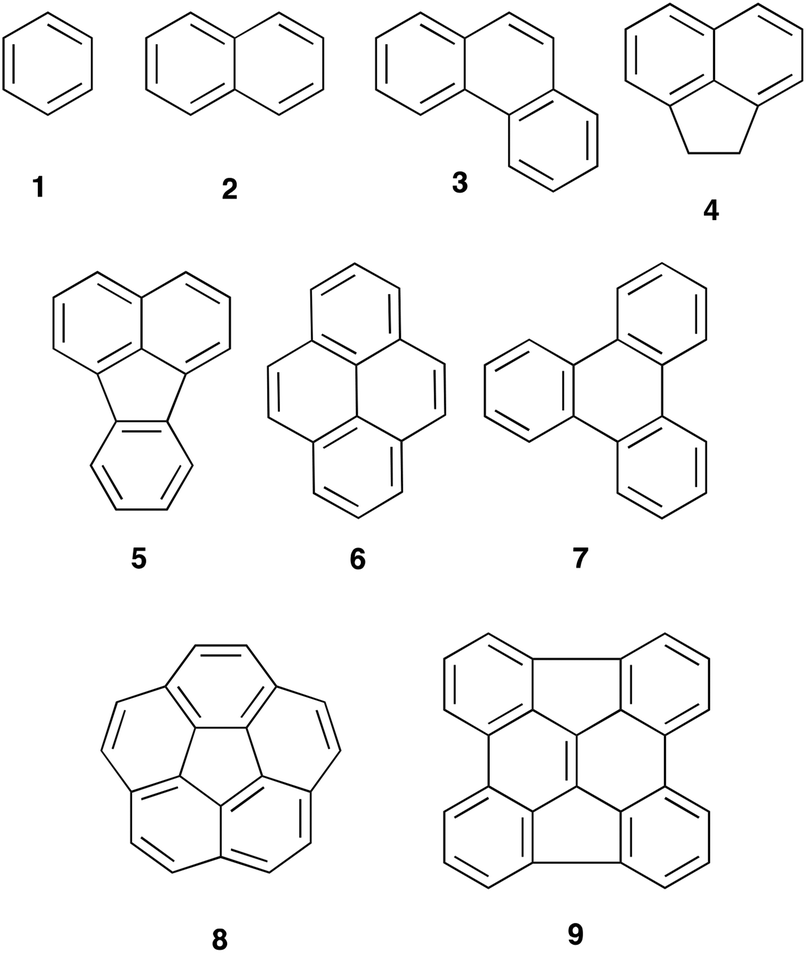
The reactivity of non-planar aromatic hydrocarbons has been considered, but typically this centres on the reactivity of a double bond with distorted geometry; this is exemplified by the work of Scott et al.,19 who showed that the reactivity of the internal double bond of the chrysene derivative 9 with nucleophiles and to cycloadditions was enhanced relative to equivalent planar systems. Reactivity at the periphery has not been considered to the same extent, with simply reports of successful electrophilic aromatic substitutions to systems such as corannulene 8 (including acylation20 and nitration21).
SN1 reactions adjacent to a non-planar aromatic, at the ‘benzylic’ position, have also not been considered. The corresponding data22 for planar aromatic compounds based on acetolysis of arylmethyl tosylates,‡ suggests a reactivity order similar to that described for electrophilic aromatic substitution but also highlights the importance of the site of substitution.§ Whilst the bent systems would be expected to have reduced pi orbital overlap compared to a planar counterpart and hence reduced aromatic stabilisation, which is supported by computational investigations,23 how this affects the reactivity of corannulene and its derivatives is unknown.
With the above in mind, the aim of the work described herein is to rank corannulene 8 amongst the other polycyclic aromatic hydrocarbons in terms of reactivity. In order to do this, two electrophilic aromatic substitution processes, Friedel–Crafts acylation and bromination (Scheme 1), were used to determine the reactivity of corannulene 8 in comparison to a range of commercially available planar aromatic species 1–7. Particularly, competition reactions between these species allowed an order of reactivity to be determined. Further, the ability of these species to stabilise an incipient carbocation on a carbon adjacent to the aryl system was investigated using an SN1 methanolysis pathway (Scheme 2). Each of these contrasting reactions (an electrophilic aromatic substitution and a reaction proceeding through a ‘benzylic’ carbocation) are of interest in determining the reactivity of non-planar polycyclic aromatic species relative to the related planar cases.
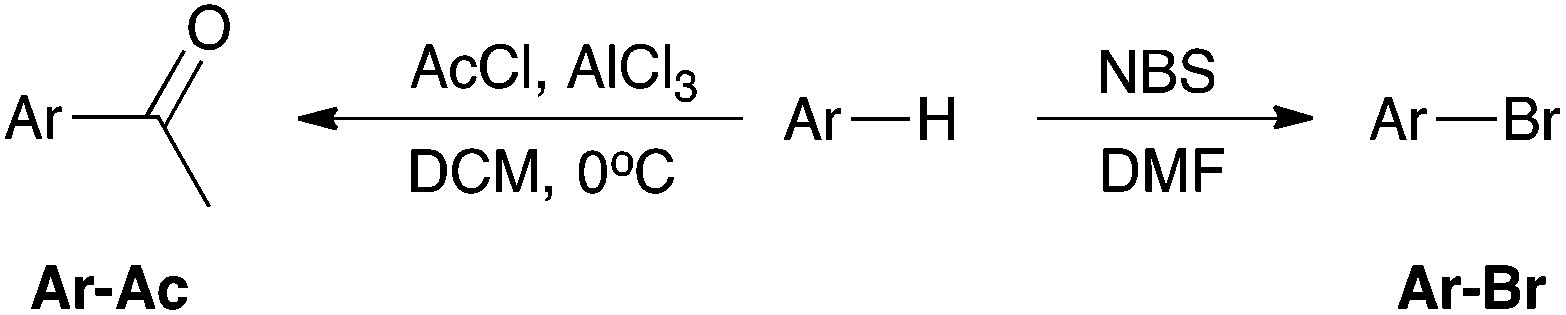 | ||
| Scheme 1 The Friedel–Crafts acylation (left) and the electrophilic bromination (right) of the desired PAHs. Ar = phenyl (1), naphthyl (2), phenanthrenyl (3), acenaphthenyl (4), fluoranthenyl (5), pyrenyl (6), triphenylenyl (7) and corannulenyl (8). | ||
 | ||
| Scheme 2 The methanolysis of benzylic alcohols 10-OH to 14-OH in acidic methanol. | ||
Experimental
All chemicals used were purchased from either Sigma-Aldrich, Alfa Aesar, Strem or Precious Metals Online and were used without further purification. Corannulene 8 was the generous gift of Prof. Jay Siegel (Tianjin University). Organic solvents used in synthesis were either used as received from Ajax Finechem or collected from a Pure Solv MD Solvent Purification System.NMR spectroscopy was performed using either a Bruker Avance 300 (300.13 MHz, 1H; 75.5 MHz, 13C), an Avance III 400 (400.13 MHz, 1H; 100.6 MHz, 13C) with a Prodigy cryoprobe cppbbo, an Avance III 500 (500.13 MHz, 1H; 125.7 MHz, 13C) with a tbi probe or an Avance III 600 (600.13 MHz, 1H; 150.9 MHz 13C). NMR spectra were processed using the Bruker TOPSPIN 3.0 software.
Authentic samples of the acylated derivatives of species 1, 2, 5, 6, 7 and 8 were prepared prior to the competition reactions described below, using equivalent methodology. The corannulene derivative 8-Ac had not been previously prepared and hence was fully characterised, whilst physical and spectral data for the remainder was consistent with that reported in literature (see ESI† for full details). The (aryl)phenylmethanols 10-OH to 14-OH were synthesised either through reduction of the commercially available parent ketone (for alcohol 10-OH), through reaction of the parent aldehyde with phenyl lithium (for alcohol 11-OH), utilising lithiation of the available aryl bromide and quenching with benzaldehyde (for alcohol 12-OH) or via a Friedel–Crafts addition of benzoyl chloride to the unsubstituted aromatic core followed by the aforementioned reduction (for alcohols 13-OH and 14-OH). The alcohols 12-OH, 13-OH and 14-OH had not been previously prepared so were fully characterised, whilst the remainder had physical and spectral data matching that reported in literature (see ESI† for full details). The corresponding methyl ethers (10-OMe to 14-OMe) were isolated after being treated with acidic methanol at reflux for 3 hours (full details can be found in the ESI†).
Acylation of polycyclic aromatic compounds – competition reactions
To a stirred suspension of aluminium(III) trichloride (51.2 mg, 384 μmol) and the two aromatic species being studied (ca. 60 μmol each) in dichloromethane (5 mL) at 0 °C under dynamic nitrogen was added acetyl chloride (8.0 μL, 112 μmol) as a solution in anhydrous dichloromethane (0.1 mL). The extents of conversion for each compound were determined through comparison of the integrations of the signals due to the starting material and the product in the 1H NMR spectrum (see ESI†). This data allowed determination of the relative rates of reaction of the two aromatic species. Each competition reaction was carried out in triplicate.Kinetic analysis of the methanolysis of alcohols 10-OH to 14-OH
A solution of sulfuric acid (98% w/w, 35 mg, 360 μmol) in deuterated methanol (0.6 mL) was generated. Of this, a portion (0.5 mL) was added to an NMR tube that contained the alcohol (28 μmol) being investigated. The reaction mixture was held at 23.8 °C in an NMR spectrometer and 1H NMR spectra taken at appropriate intervals until at least three half lives had been measured. The extent of reaction at a given time was determined through comparing the integration due to a signal corresponding to the starting material relative to the integration of a signal due to the product ether (see ESI†). This allowed calculation of observed first order rate constants for the methanolysis of the alcohols 10-OH to 14-OH under these conditions. The kinetic analysis for each alcohol was carried out in triplicate.Computational parameters
Compounds 1, 2 and 6–8 along with the corresponding non-isolable intermediates in the reaction outlined in Scheme 1 (left) were modelled using Gaussian 0324 at an RHF/6-311++G level of theory. Geometry optimized structures were obtained and energies determined. Fine grain visualization of the frontier molecular orbitals was performed using GaussView 3.0. As models for the reaction outlined in Scheme 2, the methylated derivatives S1–S5 and the corresponding benzylic cations S1+–S5+ were also treated in the same fashion.Results and discussion
Initially, Friedel–Crafts acetylation of the aromatic species 1–8 was considered. Before competition reactions could be considered, a methodology was required in which each of the species 1–8 reacted to give products readily identified using NMR spectroscopy. Using conditions similar to those described for the trimethylacetylation of corannulene 820 (addition of aromatic species to 2.5 equivalents of the preformed acetyl cation at −78 °C) allowed isolation of previously unreported monoacetylated corannulene 8-Ac. This species was readily identified using 1H NMR spectroscopy with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 noted for the relative integrations of the acetyl singlet at δ 2.82 and the signals due to the aromatic protons in the range δ 7.57–8.57. Under the same conditions, acetylation of species 1, 2, 6 and 7 also proceeded smoothly to give products readily identifiable using 1H NMR spectroscopy; acetophenone251-Ac, 1-acetylnaphthalene262-Ac-a, 2-acetylnaphthalene272-Ac-b, 1-acetylpyrene286-Ac and 2-acetyltriphenylene297-Ac) indicating that, in all cases, mono-acylation had occurred, with only reaction of naphthalene 2 giving a mixture of regioisomers. Unfortunately, under these conditions, fluoranthene 5 underwent diacetylation and reaction of phenanthrene 3 and acenaphthene 4 gave mixtures in which no product could be readily identified using 1H NMR spectroscopy. Altering either the concentration of the aromatic starting material (from 0.2 mM to 0.05 mM) or the order of addition of reagents had no effect upon this result. It is worth noting that phenanthrene 3 has been reported previously to undergo reversible Friedel–Crafts acylations to form a complex mixture of isomers,30 which may account for the complexity of the product mixture in that case.
:3 noted for the relative integrations of the acetyl singlet at δ 2.82 and the signals due to the aromatic protons in the range δ 7.57–8.57. Under the same conditions, acetylation of species 1, 2, 6 and 7 also proceeded smoothly to give products readily identifiable using 1H NMR spectroscopy; acetophenone251-Ac, 1-acetylnaphthalene262-Ac-a, 2-acetylnaphthalene272-Ac-b, 1-acetylpyrene286-Ac and 2-acetyltriphenylene297-Ac) indicating that, in all cases, mono-acylation had occurred, with only reaction of naphthalene 2 giving a mixture of regioisomers. Unfortunately, under these conditions, fluoranthene 5 underwent diacetylation and reaction of phenanthrene 3 and acenaphthene 4 gave mixtures in which no product could be readily identified using 1H NMR spectroscopy. Altering either the concentration of the aromatic starting material (from 0.2 mM to 0.05 mM) or the order of addition of reagents had no effect upon this result. It is worth noting that phenanthrene 3 has been reported previously to undergo reversible Friedel–Crafts acylations to form a complex mixture of isomers,30 which may account for the complexity of the product mixture in that case.
Whilst the above results gave a clear indication of which systems could be directly monitored using 1H NMR spectroscopy, and which conditions could be readily used, they also highlighted a problem with this reaction in terms of determining absolute rate constants; the reactions went to completion in under an hour, which, when considered alongside the heterogeneous nature of the aluminium catalyst, made monitoring the individual reaction rates very difficult.
As an alternative, competition reactions were used in which two aromatic species were placed in the same reaction vessel with ca. 0.5 equivalents (relative to the total amount of competing substrates) of acetyl chloride and ca. 3 equivalents of aluminium trichloride. The extents of conversion of the two systems were monitored using 1H NMR spectroscopy and this data converted into a ratio of the rate constants of the two species present (Scheme 3, Chart 1).31 As described previously, this is a particularly robust method in which the data is independent of the ratio of the starting materials or the extent of reaction.
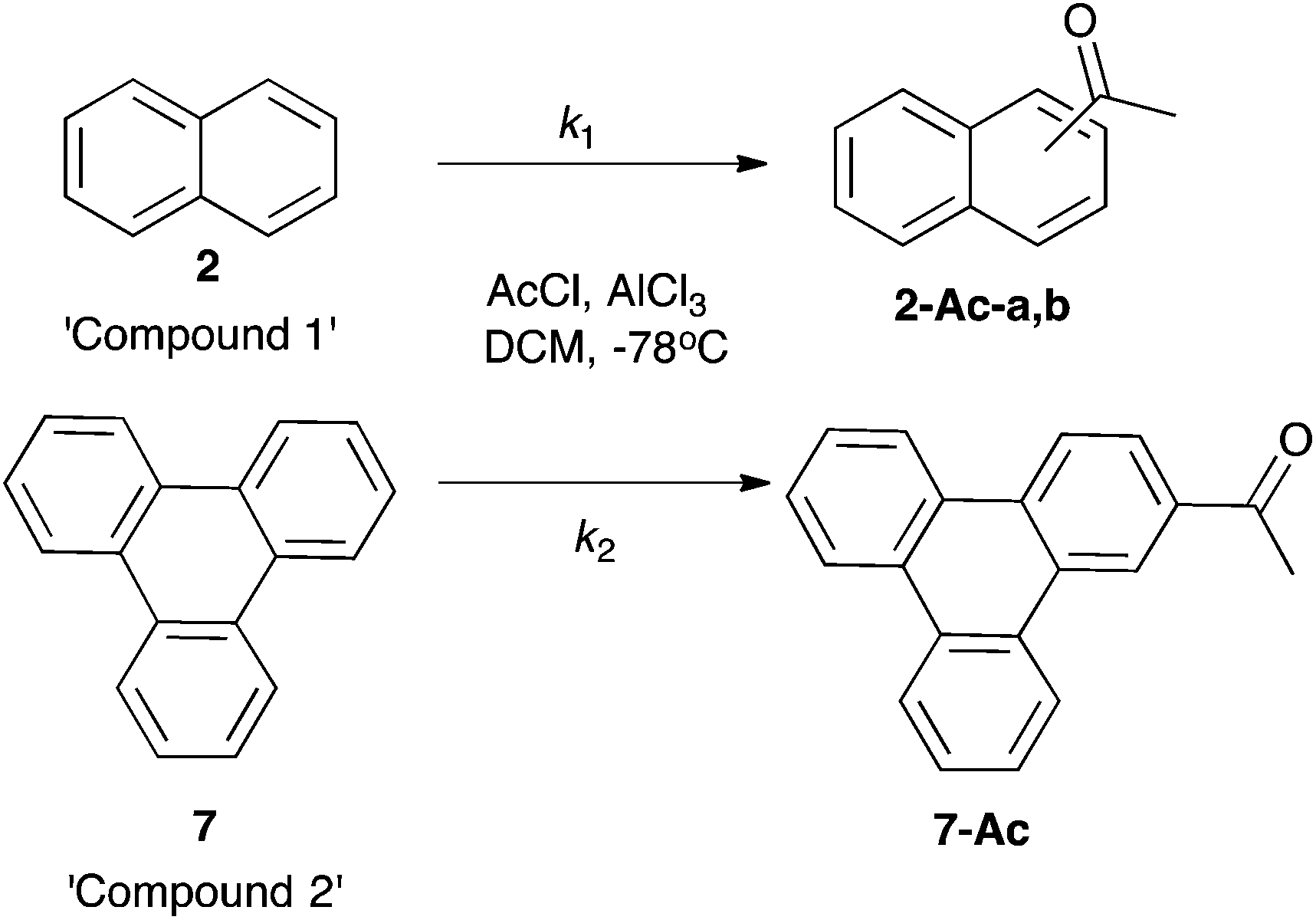 | ||
| Scheme 3 An example competition reaction between naphthalene 2 and triphenylene 7. | ||
 | ||
| Chart 1 A measure of the ratio of the respective reaction rates as calculated from their extents of conversion as per Yau et al.31 | ||
Using this technique, the five systems (1, 2 and 6–8) that mono-acetylate to give products readily identified using NMR spectroscopy were investigated using competition experiments. The resulting 1H NMR spectra were compared to authentic samples of the starting materials and products and the resulting ratio of rate constants tabulated below (Table 1).
| Compound 1 | Compound 2 | Ratio of rates (k1/k2) |
|---|---|---|
| a This assumes a detection limit for the extent of conversion of either compound of 5%. | ||
| Benzene 1 | Naphthalene 2 | <0.02a |
| Naphthalene 2 | Pyrene 6 | <0.02a |
| Naphthalene 2 | Corannulene 8 | <0.02a |
| Pyrene 6 | Triphenylene 7 | >50a |
| Pyrene 6 | Corannulene 8 | >50a |
| Triphenylene 7 | Corannulene 8 | 0.29(2) |
The first result to note is the complete selectivity under the reaction conditions with naphthalene 2 reacting whilst benzene 1 did not. This result was not unexpected as the enhanced reactivity of naphthalene 2 compared to benzene 1 towards electrophilic aromatic substitution, due to the lower decrease in the aromatic stabilisation energy on reaction, is well noted in the literature.12,32
With one ‘end’ of the reactivity scale identified (in this case the lower), it became of interest to find the most reactive of the aromatic species being considered. Pyrene 6 showed complete selectivity regardless of the competition partner chosen, was the most reactive of the systems chosen to the Friedel–Crafts acylation, likely due to the significant amount of delocalisation of positive charge possible in the acylated intermediate which is a phenalenyl cation.
To find the order of reactivity of naphthalene 2, triphenylene 7 and corannulene 8, the two former species were tested against corannulene 8. The resulting 1H NMR spectra indicated that corannulene 8 was the most reactive of the three species (as its extent of conversion was higher than either of the other two species) and, through the use of Chart 1, had a rate constant that was 3.50(20) times greater than triphenylene 7 and at least 50 times greater than naphthalene 2.
The acylation results clearly showed that (with respect to reaction with the acetyl cation) corannulene 8 reacts at a faster rate than triphenylene 7, naphthalene 2 and benzene 1 and at a slower rate than pyrene 6. The order of pyrene 6, naphthalene 2 and benzene 1 in this reactivity series is in good agreement with the sulfonation12 results previously mentioned and is not unreasonable given the increasing number of pi electrons which may be involved in delocalisation of the positive charge in the intermediates. The three larger systems 6–8 have comparable numbers of pi electrons, so comparison of their reactivity demonstrates the importance of the structure of the systems. The significant reactivity of triphenylene 7 and corannulene 8 is perhaps not overly surprising considering the larger potential pi orbital conjugation though there are fewer resonance forms in which Kekule sextets are disrupted in the corannulene 8 case suggesting curvature may negate (to some extent) the energetic cost of such disruption. The significantly greater reactivity of the pyrene in these cases can be correlated to the increased extent of delocalisation in the phenalenyl type intermediate.
In order to further understand this order of reactivity, computational studies were carried out. The aromatic species 1, 2 and 6–8 along with the corresponding acylated cationic intermediates were modelled at RHF/6-311++G level of theory and their energies determined in the gas phase (see Table S3† for full details). As the relative energies of the intermediates can be correlated to the relative energies of the transition state leading to them, these data allowed the activation barriers for the acylation of aromatic species 1, 2 and 6–8 to be ranked (see Fig. S1†). These were found to parallel the order of reactivity outlined in Table 1, though the subtle difference between triphenylene 7 and corannulene 8 was not reproduced.
Investigation of the frontier molecular orbitals of the intermediates in all of these cases shows significant delocalisation across the whole of the molecule in each case; this is particularly notable in the pyrene 6 (see Table S4†), once again consistent with the phenalenyl cation. The one exception is the LUMO in the triphenylene case, which is localised on the ring that is being substituted. Examination of the Mulliken charges (Table S5†) further supports the delocalisation of charge in the intermediates and further highlights the localisation of charge on one ring in the triphenylene case.
Confirmation of the reactivity order observed for acylation was highly desirable and, as such, electrophilic bromination using N-bromosuccinimide was examined; this reagent has been used extensively for bromination of a range of PAHs.16,33
The reaction of corannulene 8 with N-bromosuccinimide over 24 hours resulted in a complex mixture of species as noted using 1H NMR spectroscopy (see ESI† for conditions and analysis). Importantly a large amount of unreacted corannulene 8 was present at the end of the reaction (ca. 50% as determined using 1H NMR analyses).¶ The small extent of conversion demonstrates that corannulene 8 is far less reactive than pyrene 6 which, under the same conditions, resulted in a greater than 80% isolated yield of the mono-brominated product 8-Br after 24 hours, as is consistent with literature reports.15 Triphenylene 7, benzene 1 and naphthalene 2 did not react with N-bromosuccinimide under the same conditions (with the latter two cases being consistent with literature reports15) whilst acenaphthene 4 gave the monobrominated product 5-bromoacenaphthene 4-Br in 3 hours in an 84% yield. The extent of conversion of phenanthrene 3 was ca. 38% after 24 h to a complex mixture of brominated isomers, indicating that it has comparable reactivity to corannulene 8 under these conditions.
The higher reactivity of corannulene 8 and pyrene 6 compared to benzene 1 and naphthalene 2 for electrophilic bromination is consistent with all previous data. The lack of reactivity of triphenylene 7 is particularly surprising indicating that the threshold reactivity required for reaction in this case lies between triphenylene 7 and corannulene 8. The much higher reactivity of pyrene 6 and acenaphthene 4 as compared to corannulene 8 is also an expected result, particularly when one considers the donating effect of the ethane bridge in acenaphthene 4. The high reactivity of phenanthrene 3 is the most contradictory result, as literature precedent indicates reactivity comparable to naphthalene 2.
Clearly, further consideration of the systems was required and, as discussed in the Introduction, an SN1 methanolysis pathway (Scheme 4) was also considered. Once again, the difference between these two reactions (an electrophilic aromatic substitution and a process which proceeds through a benzylic carbocation) should be noted but this provides further insight into the reactivity of curved polycyclic systems relative to their planar counterparts. The reactivities of the derivatives 10-OH to 14-OH allowed the effectiveness of the various PAHs at stabilising developing adjacent positive charges to be probed. The reaction was simple to perform and monitor in situ using 1H NMR spectroscopy allowing rate constants to be determined. It is worth noting that the observed first order rate constant (kobs) is a function of the first-order rate constant for the formation of the intermediate cation (k1), the concentration of acid ([H+]) and the extent of protonation of the alcohol as determined by the position of equilibrium (K) (Scheme 4, Chart 2).
 | ||
| Scheme 4 The methanolysis of an alcohol, which proceeds through a protonated alcohol and subsequent carbocation intermediate. | ||
 | ||
| Chart 2 The observed rate constant (kobs) expressed as a function of the first-order rate constant (k1), the acid concentration ([H+]) and the pre-equilibrium constant (K). | ||
Given the acid concentration ([H+]) is controlled through the methodology, it is important to consider whether the equilibrium constant (K) will vary for the different alcohols. Whilst it would be simplest to assume that the value for K does not change, any significant alteration of this value between the alcohols would invalidate any comparison of kobs between the systems. The easiest way to consider whether the position of equilibrium would vary with the structure of the alcohol might be to consider any changes in the pKa values of the corresponding protonated species. Unfortunately, these values have not been reported for the protonated forms of the alcohols of interest. Irrespective, any effects of changing the structure of the alcohol upon the pKa value of the protonated form of that alcohol (and ultimately the position of the equilibrium) would be expected to be significantly less than any effects upon the stabilisation of the intermediate carbocation (due to its increased proximity to the adjacent aromatic system) and affect the observed rate in the same way. Therefore, any changes in the observed rate constant can be attributed mainly to changes in the stabilisation of the intermediate carbocation, so comparing values for the observed rate constants is reasonable.
The observed rate constants for the methanolysis of the benzylic alcohols are presented in Table 2. In all cases the reaction proceeded irreversibly to give the corresponding methyl ester.
| Alcohol | k obs/10−5 s−1 |
|---|---|
| Benzhydrol 10-OH | 6.16(0.6) |
| (2-Naphthyl)phenylmethanol 11-OH | 21.0(1.4) |
| (9-Phenanthrenyl)phenylmethanol 12-OH | 19.3(1.9) |
| (Triphenylenyl)phenylmethanol 13-OH | 33.8(7.9) |
| (Corannulenyl)phenylmethanol 14-OH | 38.6(2.7) |
The observed rate constant for the methanolysis of 2-naphthyl-1-phenyl methanol 11-OH is ca. 3.5 times greater than that for benzhydrol 10-OH. The effect is similar to that described previously in the acetolysis of arylmethyl tosylates,22 though the magnitude of the change is less; this may either be the result of a different degree of charge build-up or steric interactions between the two aromatic systems limiting orbital overlap in this case. Irrespective, it is also consistent with the results described above for electrophilic aromatic substitution and is due to the lower decrease in aromatic stabilisation energy on delocalisation of the incipient positive charge and the greater pi system over which it may delocalised.
The rate constant for the reaction of the phenanthrene derivative 12-OH was found to be the same as for the naphthalene derivative 11-OH, indicating that the phenanthrenyl system was able to stabilise the intermediate carbocation only to a similar extent as the naphthyl system. This is somewhat surprising given the significant rate enhancement seen between 2-naphthyl and 9-phenanthrenyl systems in the acetolysis of arylmethyl tosylates, though this may be the result of the greater potential steric interactions between the aryl systems in this case. It is also consistent with a similar overall number of pi electrons in the molecule.
Methanolysis of the triphenylene derivative 13-OH gave a rate constant ca. 5 times faster than the parent benzhydrol 10-OH indicating that the triphenylene 7 core stabilised the incipient carbocation to a greater degree than either naphthalene 2 or phenanthrene 3, a result consistent with the previously studied Friedel–Crafts reactions and the increased size of the pi system.
Lastly, the rate constant for the methanolysis of (corannulenyl)phenylmethanol 14-OH was found to be the same as for the triphenylene derivative 13-OH and hence greater than the other cases considered, indicating that corannulene is the (equally) most effective cation at stabilising adjacent incipient positive charges. This result is consistent with the similar rate constants found in the Friedel–Crafts competition reactions and the similar sizes of the pi systems.
Once again, computational studies were carried out to support this observed order of reactivity. In this case, rather than model the starting materials 10-OH to 14-OH (and the corresponding benzylic cations) the methylated derivatives of species 1, 2 and 6–8 (compounds S1–S5, see ESI†) along with the corresponding benzylic cations were considered; this was anticipated to not only reduce computational cost but also focus on the extent to which the (poly)cyclic aromatic substituent stabilised the developing positive charge. The relative energies of the methyl derivatives S1–S5 and their corresponding benzylic cations (Table S6, Fig. S2†) generally paralleled the order of reactivity seen for species 10-OH to 14-OH. The differences in relative energies were much smaller than observed for the acylation of species 1, 2 and 6–8, as would be expected based on the similarities in observed rates. Note that the calculated data suggests that the phenanthrene derivative 12-OH would react faster than the naphthalene derivative 11-OH (though they are observed to be the same experimentally) and that the triphenylene derivative 13-OH is as reactive under these conditions as the naphthalene derivative 12-OH (it is faster, just outside experimental error) and less reactive than the corrannulene derivative 14-OH (same within uncertainty). The latter may simply be the result of the larger uncertainty in the rate constant in that case.
The frontier molecular orbitals of the species S1–S5 and the corresponding benzylic cations were also examined (Table S7†). As for the previous case, there was significant delocalisation of these orbitals across the whole molecule in almost every example, with the LUMO in the triphenylene case being (again) a notable exception. Once again, the Mulliken charges (Table S8†) support significant delocalisation of the charge and the importance of not disrupting Kekule sextets in cases such as triphenylene.
Given both the experimental and the computational data, the larger rate constant for methanolysis for the corannulenyl case 14-OH as compared to (say) the naphthyl case 11-OH may be expected simply due to the size of the available conjugation pathway, any contribution to the larger observed rate constant due to either the curvature of the corannulenyl portion of the molecule or its rapid inversion at room temperature cannot be differentiated.
Conclusions
Utilising Friedel–Crafts reaction conditions, the order of reactivity for species 1, 2, 6, 7 and 8 to electrophilic substitution was readily determined. Electrophilic bromination reactions further confirmed this data whilst including the substituted naphthalene 4 in overall order. Importantly, corannulene 8 was found to be more reactive than benzene 1, naphthalene 2 and triphenylene 7 and less reactive than pyrene 6 and acenaphthene 4. Comparison of the rate constants for acylation suggests that corannulene 8 was most similar to triphenylene 7 in terms of stability of the intermediate. The order of reactivity was paralleled in computational studies.SN1 methanolysis of the benzylic alcohols 10-OH to 14-OH was successfully performed, and showed the greater stabilising ability (of an incipient carbocation) of corannulene 8 compared to naphthalene 2 and benzene 1, as well as confirming the similarity between triphenylene 7 and corannulene 8 noted above. Once again, computational studies were consistent with these outcomes.
The overall goal of this study was to compare corannulene 8 with other common planar PAHs in order to construct a reactivity scale. This has been achieved with the most notable outcome being the similar results for triphenylene 7 and corannulene 8.
Acknowledgements
SRDG acknowledges the support of the Australian government through the receipt of an Australian Postgraduate Award. JBH acknowledges financial support from the Australian Research Council Discovery Project Funding Scheme (Project DP130102331). The authors would like to acknowledge the NMR Facility within the Mark Wainwright Analytical Centre at the University of New South Wales for NMR support.Notes and references
- J. C. Fetzer, Polycyclic Aromat. Compd., 2007, 27, 143–162 CrossRef CAS PubMed.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1872, 5, 12 CrossRef PubMed.
- C. B. Larsen and N. T. Lucas, Chem. N.Z., 2012, 76, 49–55 CAS.
- L. T. Scott, M. M. Boorum, B. J. McMahon, S. Hagen, J. Mack, J. Blank, H. Wegner and A. d. Meijere, Science, 2002, 22, 1500–1503 CrossRef PubMed.
- H. Sharghi, M. Jokar, M. M. Doroodmand and R. Khalifeh, Adv. Synth. Catal., 2010, 352, 3031–3044 CrossRef CAS PubMed.
- E. Mosettig and J. van de Kamp, J. Am. Chem. Soc., 1930, 52, 3704–3720 CrossRef CAS.
- J.-H. Li, D. V. Ferraris, P. W. Kletzly, W. Li, E. Y. Wang, A. D. Xing, W. Xu and J. Zhang, Preparation of symmetrically disubstituted aromatic compounds and pharmaceutical compositions for the inhibition and/or modulation of human poly(ADP-ribose) glycohydrolase (PARG) activity, WO2002057211A1, Guilford Pharmaceuticals Inc., USA, 2002, 92 pp Search PubMed.
- M. J. Tanga, R. F. Davis and E. J. Reist, J. Heterocycl. Chem., 1987, 24, 39–41 CrossRef CAS PubMed.
- S. Nishida, Y. Morita, A. Ueda, T. Kobayashi, K. Fukui, K. Ogasawara, K. Sato, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2008, 130, 14954–14955 CrossRef CAS PubMed.
- G. Felix, M. Laguerre, J. Dunogues and R. Calas, J. Org. Chem., 1982, 47, 1423–1427 CrossRef CAS.
- R. S. Kathayat and N. S. Finney, J. Am. Chem. Soc., 2013, 135, 12612–12614 CrossRef CAS PubMed.
- A. R. Katritzky, M. S. Kim, D. Fedoseyenko, K. Widyan, M. Siskin and M. Francisco, Tetrahedron, 2009, 65, 1111–1114 CrossRef CAS PubMed.
- P. H. Gore, C. K. Thadani, S. Thorburn and M. Yusuf, J. Chem. Soc. C: Org., 1971, 2329–2330 RSC.
- W. A. Pryor, G. J. Gleicher, J. P. Cosgrove and D. F. Church, J. Org. Chem., 1984, 49, 5189–5194 CrossRef CAS.
- R. H. Mitchell, Y.-H. Lai and R. V. Williams, J. Org. Chem., 1979, 44, 4733–4735 CrossRef CAS.
- T. Morofuji, A. Shimizu and J.-i. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 7259–7262 CrossRef CAS PubMed.
- J.-Y. Hu, X. Feng, N. Seto, J.-H. Do, X. Zeng, Z. Tao and T. Yamato, J. Mol. Struct., 2013, 1035, 19–26 CrossRef CAS PubMed.
- I. B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, Academic Press, 2nd edn, 1971 Search PubMed.
- H. E. Bronstein and L. T. Scott, J. Org. Chem., 2007, 73, 88–93 CrossRef PubMed.
- N. Niamnont, N. Kimpitak, K. Wongravee, P. Rashatasakhon, K. K. Baldridge, J. S. Siegel and M. Sukwattanasinitt, Chem. Commun., 2013, 49, 780–782 RSC.
- H. Boedigheimer, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2010, 75, 2518–2527 CrossRef CAS PubMed.
- A. Streitwieser, Jr., H. A. Hammond, R. H. Jagow, R. M. Williams, R. G. Jesaitis, C. J. Chang and R. Wolf, J. Am. Chem. Soc., 1970, 92, 5141–5150 CrossRef.
- M. A. Dobrowolski, A. Ciesielski and M. K. Cyranski, Phys. Chem. Chem. Phys., 2011, 13, 20557–20563 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.05, Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- R. J. Abraham, M. Mobli and R. J. Smith, Magn. Reson. Chem., 2003, 41, 26–36 CrossRef CAS.
- J. Ruan, X. Li, O. Saidi and J. Xiao, J. Am. Chem. Soc., 2008, 130, 2424–2425 CrossRef CAS PubMed.
- J. Zhao, C. O. Hughes and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 7436–7437 CrossRef CAS PubMed.
- J. Weng, Q. Mei, Q. Ling, Q. Fan and W. Huang, Tetrahedron, 2012, 68, 3129–3134 CrossRef CAS PubMed.
- T. T. Jayanth and C.-H. Cheng, Chem. Commun., 2006, 894–896 RSC.
- F. Fernandez, G. Gomez, C. Lopez and A. Santos, J. Prakt. Chem., 1989, 331, 15–21 CrossRef CAS PubMed.
- H. M. Yau, A. K. Croft and J. B. Harper, Chem. Commun., 2012, 48, 8937–8939 RSC.
- Y. Fujiwara, R. Asano, I. Moritani and S. Teranishi, J. Org. Chem., 1976, 41, 1681–1683 CrossRef CAS.
- H. Eguchi, H. Kawaguchi, S. Yoshinaga, A. Nishida, T. Nishiguchi and S. Fujisaki, Bull. Chem. Soc. Jpn., 1994, 67, 1918–1921 CrossRef CAS.
- W. C. Herndon and N. S. Mills, J. Org. Chem., 2005, 70, 8492–8496 CrossRef CAS PubMed.
- G. P. Ford and P. S. Herman, J. Mol. Struct., 1991, 236, 269 CrossRef.
- F. T. Ogata, M. Nojima and S. Kusabayashi, J. Am. Chem. Soc., 1981, 103, 1145–1153 CrossRef CAS.
- T. J. Seiders, E. L. Elliott, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 1999, 121, 7804–7813 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of the acylated derivatives of species 1, 2 and 6–8, the alcohols 10-OH to 14-OH and the ethers 10-OCH3 to 14-OCH3; conditions used for the bromination of corannulene 8; spectra of all novel compounds; details of the competitive acylation reactions involving species 1, 2 and 6–8; kinetic analyses of the methanolysis of the alcohols 10-OH to 14-OH; energies of the species 1, 2 and 6–8 and the corresponding acylated intermediates 1-Int, 2-Inta,b and 6-Int to 8-Int; reaction coordinate showing relative energies for the process shown in Scheme 1 (left); frontier molecular orbitals for the species 1-Int, 2-Inta,b and 6-Int to 8-Int; charge densities for the species 1-Int, 2-Inta,b and 6-Int to 8-Int; energies of the methylated species S1–S5 and the corresponding acylated intermediates S1+–S5+; diagram showing relative energies of S1–S5 and S1+–S5+; frontier molecular orbitals for the species S1+–S5+; charge densities for the species S1+–S5+. See DOI: 10.1039/c5ob01215e |
| ‡ There is also some computational34,35 and experimental36 data for the relative stability of carbocations adjacent to various planar aromatic species, with larger planar systems general giving the more stable carbocation, though the importance of the site of substitution is further demonstrated. |
| § The symmetry of corannulene 8 means that issues of the site of substitution do not arise with this non-planar aromatic. |
| ¶ It is worth noting that the monobromination of corannulene has been reported using bromine/iron(III) bromide at low temperatures.37 |
| This journal is © The Royal Society of Chemistry 2015 |