
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis of activated 2-arylazetidines via imino-aldol reaction†‡
Manas K.
Ghorai
*,
Subhomoy
Das
,
Kalpataru
Das
and
Amit
Kumar
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India. E-mail: mkghorai@iitk.ac.in
First published on 20th July 2015
Abstract
A simple and efficient synthetic route to substituted N-sulfinyl and N-sulfonyl azetidines is described involving imino-aldol reaction of ester enolates with racemic and non-racemic aldimines for obtaining β-amino esters as a key step. These β-amino esters on subsequent reduction followed by TsCl/KOH mediated cyclization produced the corresponding racemic and non-racemic azetidines with high yield and stereoselectivity.
Introduction
Azetidines are four membered nitrogen containing strained heterocycles of immense biological and pharmacological importance.1 Several racemic as well as chiral azetidines exhibit a diverse range of biological activities e.g. compounds 1–2 show inhibition properties against thrombin,1a,b3 possesses potent protein kinase C inhibitory activity,1c4–5 exert inhibitory effects against the angiotensin-converting enzyme,1d6 is potentially capable of introducing some functional groups at the 3′-nitrogen atom, such as a fluorescent or a chemiluminescent probe, an intercalator and DNA scissors (Fig. 1).1e Due to the high ring strain, the synthesis of substituted azetidines and further structural modifications still remain a challenge. However, many useful strategies have been developed for the synthesis of substituted azetidines.2 Imino-aldol reaction (addition of ester enolate to imine) is one of the prominent routes for the synthesis of β-amino esters,3 non-proteinogenic β-amino acids4a,b and β-lactam antibiotics4cvia the formation of carbon–carbon bonds.4d Lewis acid promoted reaction between a silyl enol ether and an imine is another useful method for the synthesis of β-amino esters.5 In most of the cases effective Lewis acid-activation of the aldimines is required because of poor electrophilicity of the stable N-substituted imines.6 Some other methods are also known to generate β-amino esters.7 Recently, the synthesis of 2,4-disubstituted azetidines via Lewis acid catalyzed imino-aldol reaction was reported,8 however, the substrate scope of the strategy is narrow. We have developed an efficient route to various racemic and nonracemic azetidines via imino-aldol reaction of ester enolates with N-sulfonyl or N-sulfinyl aldimines as the key step and describe our results in detail in this article.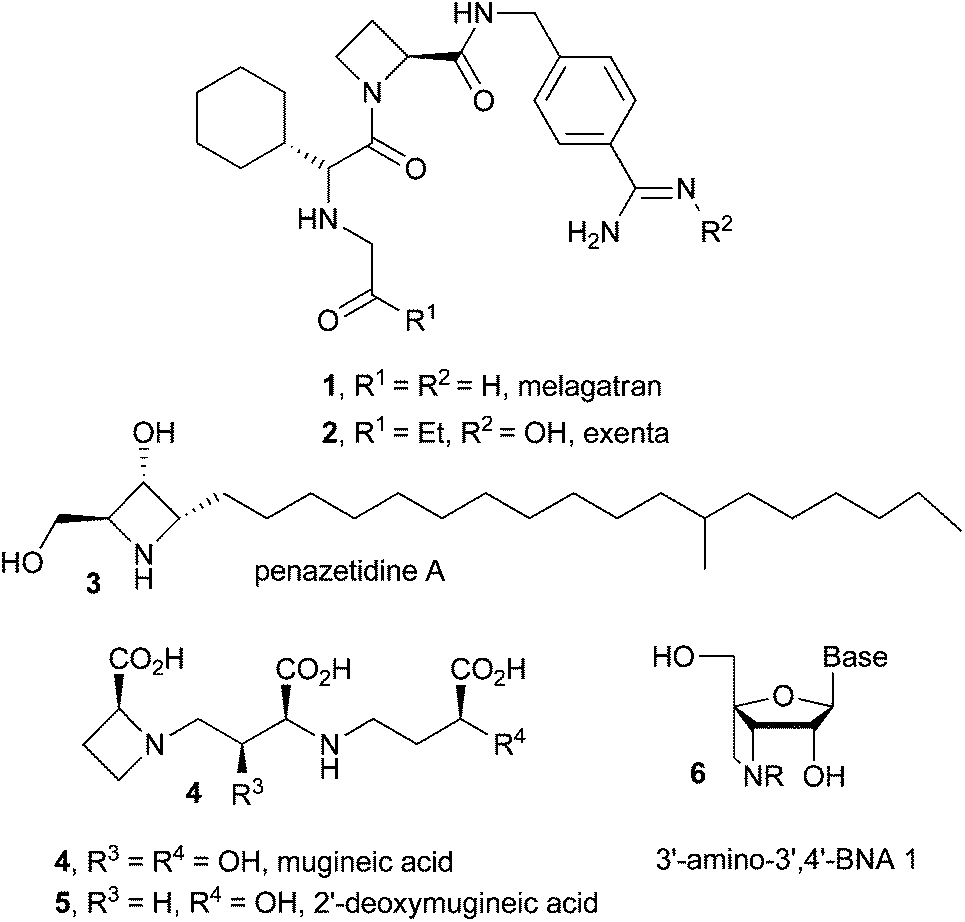 | ||
| Fig. 1 Some biologically active compounds containing azetidine. | ||
Results and discussion
We envisioned that N-sulfonyl azetidine 5 could be synthesized easily from the precursor β-amino ester derivative 3 which would be obtained through imino-aldol reaction of 1 and 2 (Scheme 1).9a We further anticipated that various chiral azetidines could be synthesized from chiral β-amino esters 10 which could easily be obtained from the imino-aldol reaction utilizing chiral sulfinimines 9 as the source of chirality.9b (Table 2) Enantiopure sulfinimines10 have been extensively used by the Davis group for the stereoselective synthesis of α-amino acids,11N-sulfinyl-cis-aziridine-2-carboxylic acids,12 taxol C-13 side chain13 and β-amino acids14 employing N-sulfinyl auxiliary as a C–N bond activating as well as the stereodirecting group.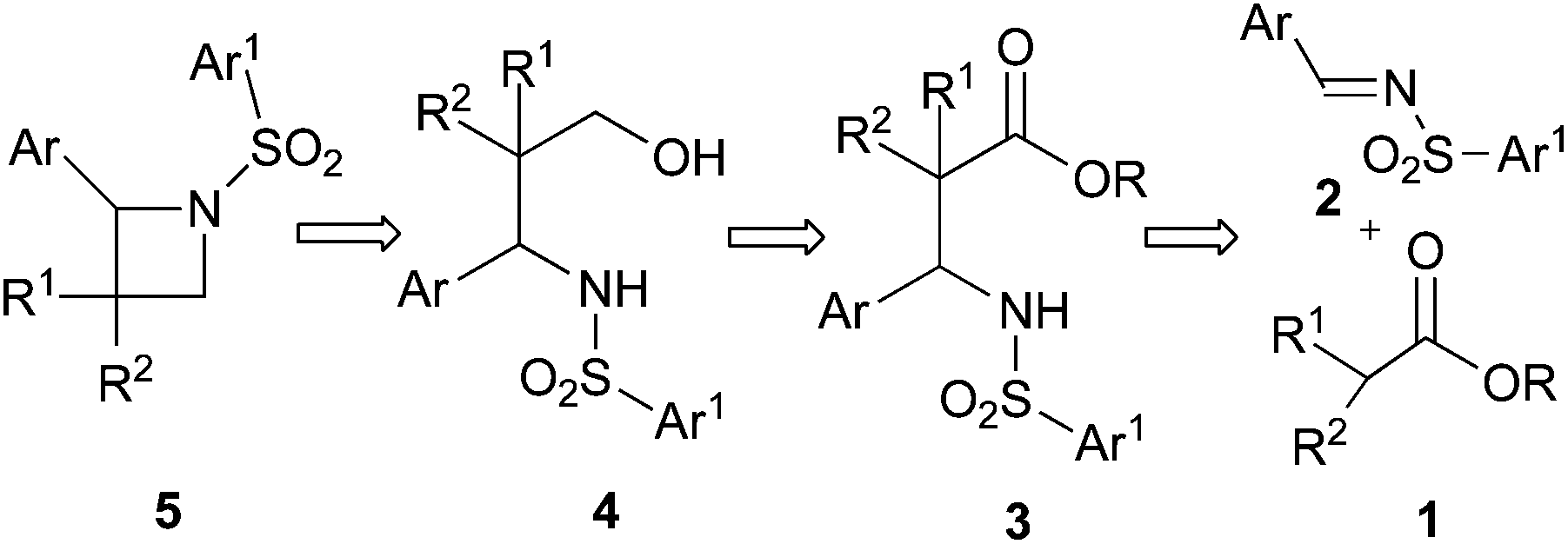 | ||
| Scheme 1 Retrosynthetic analysis for the preparation of N-sulfonyl azetidines. | ||
Initially, for the synthesis of β-amino esters a number of N-sulfonyl aldimines 2a–g (Scheme 2) were prepared in excellent yields by refluxing N-sulfonyl amine 6 and the corresponding aromatic aldehydes 7 in dry benzene in the presence of a catalytic amount of BF3·OEt2.
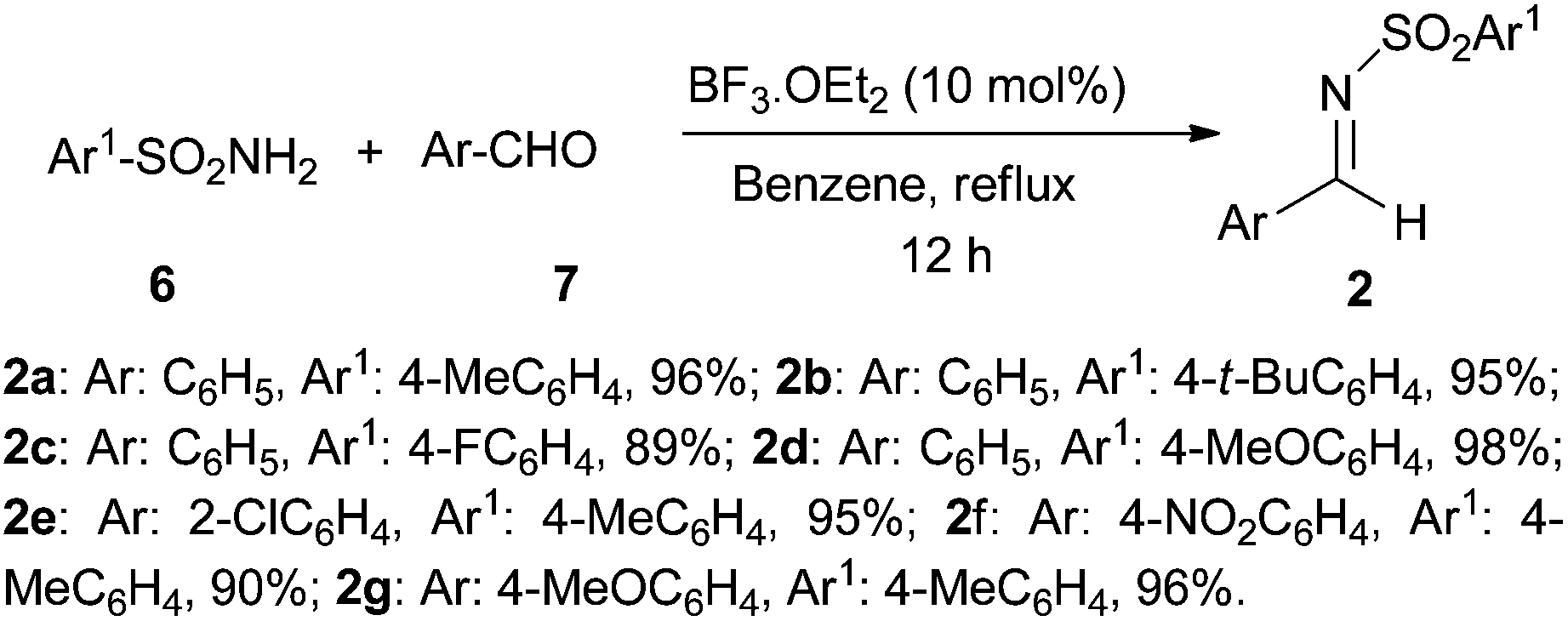 | ||
| Scheme 2 Synthesis of N-sulfonyl aldimines. | ||
Next, the ester enolate from t-butyl acetate was generated by treatment with LDA and reacted with N-tosyl phenyl aldimine 2a to afford the corresponding addition product N-tosyl-β-amino ester 3a in quantitative yield. 3a was reduced to the corresponding γ-amino alcohol 4a by treatment with LiAlH4.15 After the usual work up, the crude γ-amino alcohol 4a was treated with TsCl in the presence of excess KOH in THF under reflux conditions to produce 2-phenyl-N-tosyl azetidine 5a in excellent yield in a short period of time (Scheme 3).16 The Mitsunobu protocol16 was also found to be equally effective in the cyclization step for the synthesis of 5a from 4a, but the byproduct Ph3PO generated in this reaction made the purification process difficult.
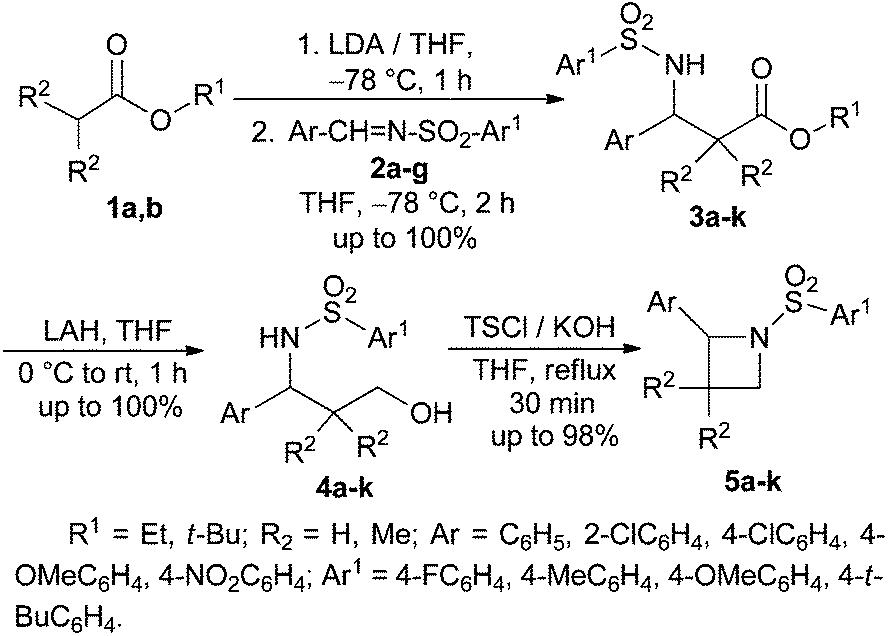 | ||
| Scheme 3 Synthesis of 2-aryl-N-sulfonylazetidines. | ||
To generalize this strategy a number of N-sulfonyl aldimines 2b–g were reacted with different ester enolates leading to the formation of N-sulfonyl-β-amino esters 3b–k in excellent yields. Reduction of 3b–k with LAH followed by cyclization of the γ-amino alcohols 4b–k using TsCl and KOH in refluxing THF afforded the corresponding azetidines 5b–k in almost quantitative yields (Table 1). All the compounds 5a–k were characterized by spectroscopic data.
| Entry | Ar | Ar1 | R1 | R2 | 3 , % yield | 4 , % yield | 5 , % yield |
|---|---|---|---|---|---|---|---|
| a All reactions were carried out with 2a–g (1.0 mmol), 1a,b (1.1 mmol), and LDA (1.1 equiv.) in dry THF under argon for 2 h at −78 °C. b Yields of isolated products (%) after column chromatographic separation. c All reactions were carried out with 3a–k (1.0 mmol), LAH (2.0 equiv.) in dry THF under argon for 1–1.5 h at 0 °C to rt. d Yields of isolated products (%) of 4a–k after column chromatographic separation. e All reactions were carried out with 4a–k (1.0 mmol), TsCl (1.1 mmol) and KOH (3.0 equiv.) in dry THF under argon for 1 h under reflux conditions. f Yield of isolated products (%) of 5a–k after column chromatographic separation. | |||||||
| 1 | C6H5 | 4-MeC6H4 | t-Bu | H | 3a | 4a | 5a |
| 100 | 100 | 98 | |||||
| 2 | 2-ClC6H4 | 4-MeC6H4 | t-Bu | H | 3b | 4b | 5b |
| 100 | 100 | 95 | |||||
| 3 | 4-NO2C6H4 | 4-MeC6H4 | t-Bu | H | 3c | 4c | 5c |
| 90 | 90 | 85 | |||||
| 4 | 4-OMeC6H4 | 4-MeC6H4 | t-Bu | H | 3d | 4d | 5d |
| 100 | 100 | 92 | |||||
| 5 | 4-ClC6H4 | 4-MeC6H4 | t-Bu | H | 3e | 4e | 5e |
| 100 | 100 | 97 | |||||
| 6 | C6H5 | 4-FC6H4 | t-Bu | H | 3f | 4f | 5f |
| 98 | 99 | 93 | |||||
| 7 | C6H5 | 4-OMeC6H4 | t-Bu | H | 3g | 4g | 5g |
| 99 | 97 | 91 | |||||
| 8 | C6H5 | 4-t-BuC6H4 | t-Bu | H | 3h | 4h | 5h |
| 97 | 98 | 90 | |||||
| 9 | C6H5 | 4-MeC6H4 | Et | Me | 3i | 4i | 5i |
| 98 | 100 | 95 | |||||
| 10 | 2-ClC6H4 | 4-MeC6H4 | Et | Me | 3j | 4j | 5j |
| 96 | 95 | 95 | |||||
| 11 | 4-OMeC6H4 | 4-MeC6H4 | Et | Me | 3k | 4k | 5k |
| 97 | 95 | 94 | |||||
The present protocol was extended to provide azetidines with substitution both at 2- and 4-positions. For this purpose, compound 3a was converted to N-tosyl-β-amino aldehyde 7 by treatment with DIBAL-H. Next, compound 7 was reacted with ethylmagnesium bromide to afford the γ-amino alcohols 8a and 8b (dr 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70) in 85% combined yield as a mixture of diastereomers. The pure diastereomers 8a and 8b were separated via flash column chromatography and they were individually reacted with tosyl chloride and KOH in THF to furnish diastereopure 2,4-disubstituted-N-tosyl azetidines 5l and 5m, respectively, in high yields (Scheme 4).
:70) in 85% combined yield as a mixture of diastereomers. The pure diastereomers 8a and 8b were separated via flash column chromatography and they were individually reacted with tosyl chloride and KOH in THF to furnish diastereopure 2,4-disubstituted-N-tosyl azetidines 5l and 5m, respectively, in high yields (Scheme 4).
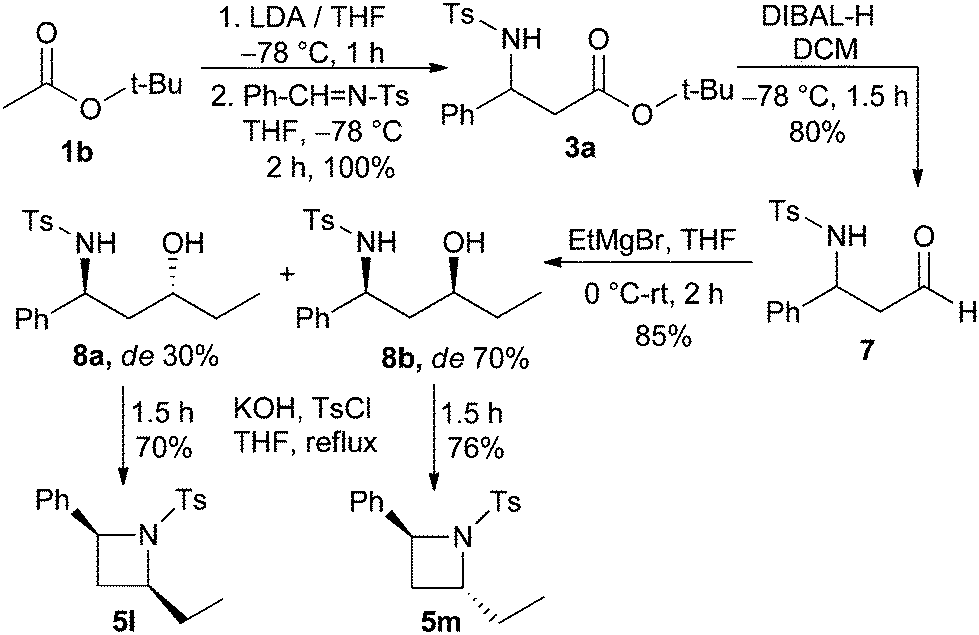 | ||
| Scheme 4 Synthesis of 2,4-disubstituted 2-phenyl-N-tosylazetidine. | ||
After the successful demonstration of our strategy for the synthesis of a variety of racemic azetidines 5a–m, we extended our protocol for the synthesis of chiral azetidines employing enantiomerically pure N-sulfinyl imines as the source of chirality. To optimize the reaction conditions for the imino aldol reaction, different bases (e.g. LDA, NaHMDS, and KHMDS) and solvents were explored to generate the enolate from the methyl acetate ester. Among the bases studied, KHMDS was found to be the best. The enolate generated from methyl acetate upon treatment with KHMDS in THF at −78 °C was reacted with (S)-(+)-N-sulfinimine109a (Ar = Ph) to produce the N-sulfinyl-β-amino ester 10a (de 84%) in 68% yield. The protocol was generalized with a number of (S)-(+)-N-sulfinimines 9b–e to produce the corresponding imino-aldol products 10b–e in 65–93% yield and dr up to >99:1 (Table 2).
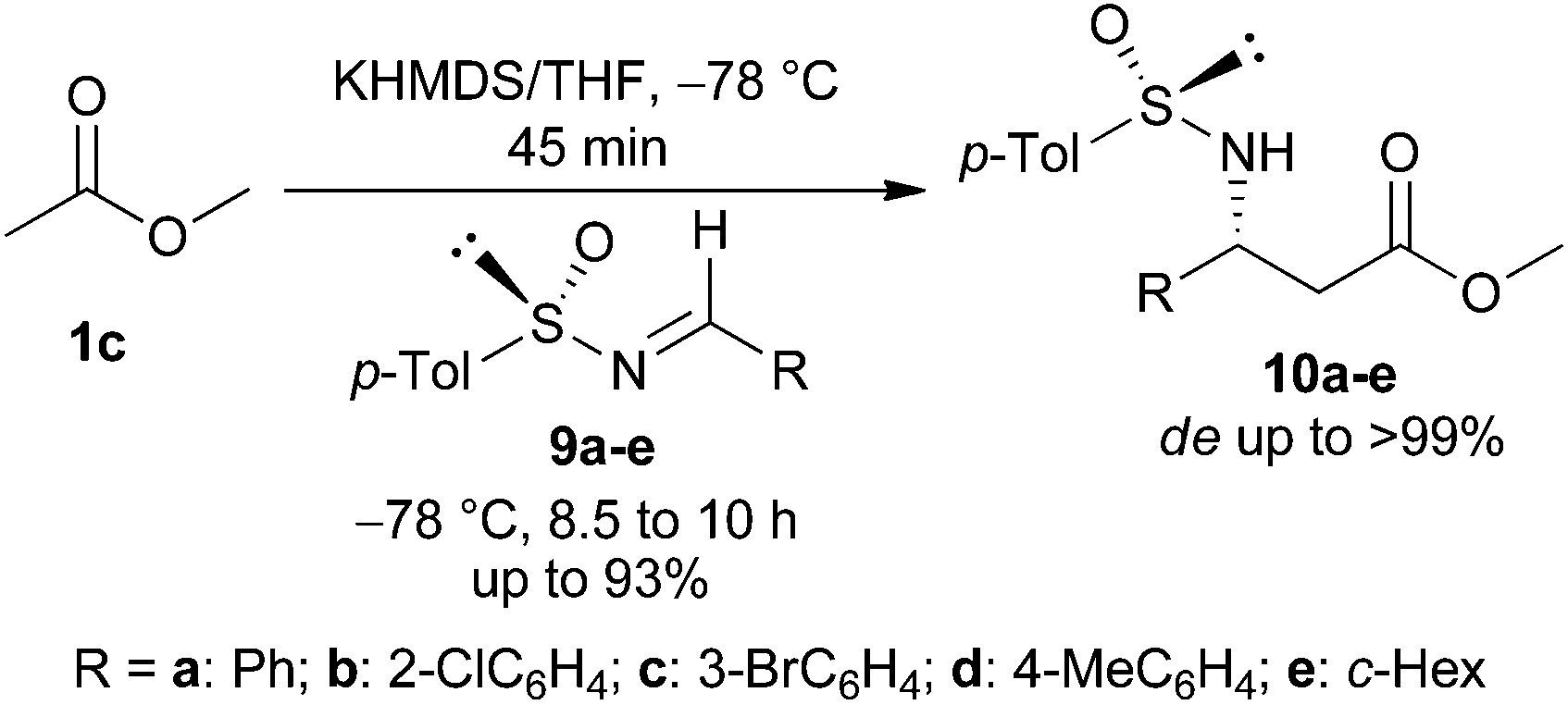
The addition product 10a was then converted into N-sulfinyl-γ-amino alcohol 11a by treatment with LAH. When 11a was reacted with tosyl chloride and KOH in THF under our optimized reaction conditions, (Ss,2R)-2-phenyl-N-sulfinyl azetidine 12a was obtained in 70% yield. Synthesis of four to six membered N-sulfinyl heterocycles via intramolecular alkylation of sulfinyl amides with alkyl halides has been reported in the literature.17 A number of enantiopure N-sulfinyl azetidines 12b–e were obtained in good yields and excellent diastereoselectivity from 11b–e following the same procedure (Table 3).
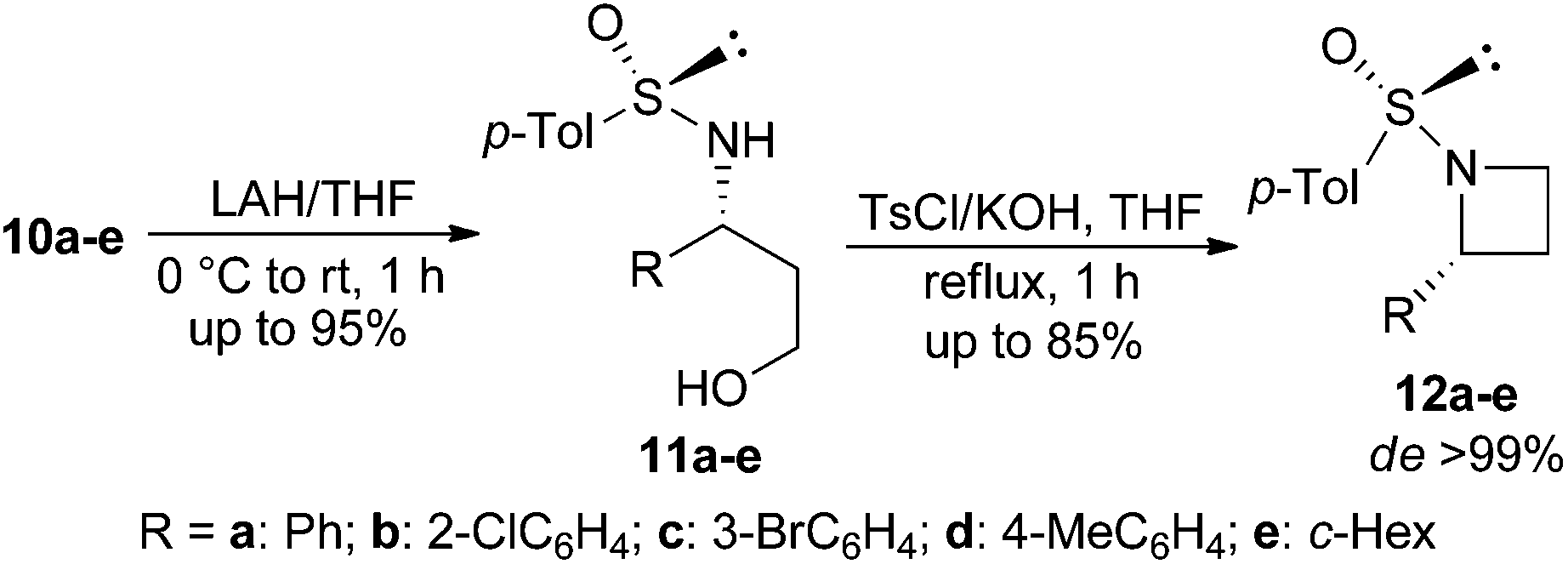
In order to ascertain the stereoselectivity (de/ee) of the products in the described imino-aldol reaction, as a representative example, β-amino ester 10a (de 84%) was converted to 2-phenyl-N-tosylazetidine (R)-5a as shown in Scheme 5. The N-sulfinyl auxiliary of 10a was removed by treatment with TFA in MeOH following a reported procedure18 (Scheme 5). The crude concentrate of the reaction mixture was treated with tosyl chloride and triethylamine in dichloromethane to produce N-tosyl-β-amino ester 13 which was reduced to N-tosyl-γ-amino alcohol (R)-4a by treatment with LAH. The usual intramolecular cyclization of (R)-4a using TsCl and excess KOH in THF under reflux conditions afforded the chiral 2-phenyl-N-tosylazetidine (R)-5a in 75% yield with 84% ee.19
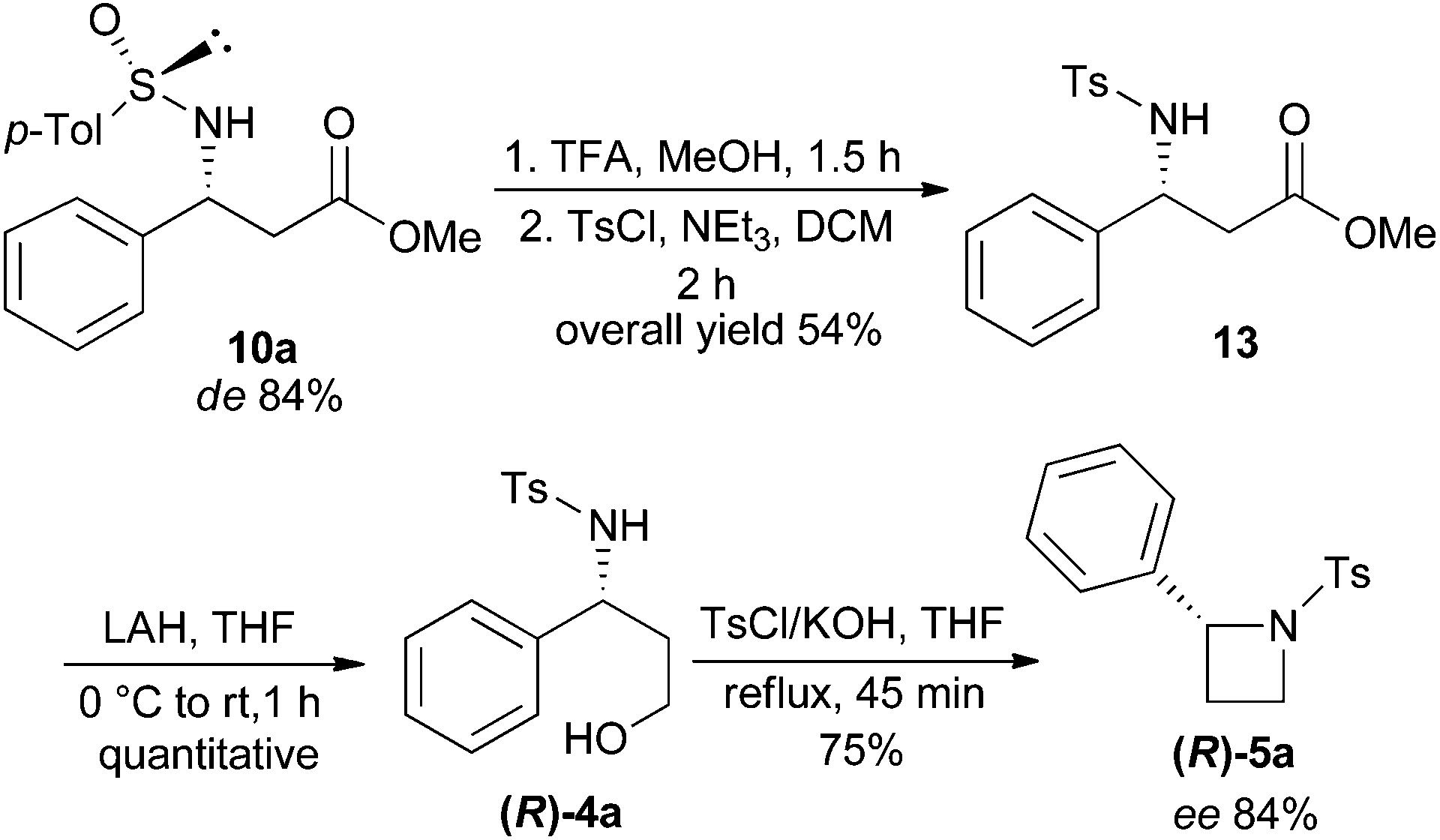 | ||
| Scheme 5 Synthesis of chiral 2-aryl-N-tosylazetidine from chiral β-amino ester. | ||
Conclusions
In summary, we have developed a simple and efficient synthetic route to a variety of 2-aryl-N-sulfonylazetidines and 2-alkyl/aryl-N-sulfinylazetidines utilizing imino-aldol reaction of ester enolates with N-sulfonyl/chiral N-sulfinyl aldimines as the key step.Experimental
General remarks
Analytical thin layer chromatography (TLC) was carried out using silica gel 60 F254 pre-coated plates. Visualization was accomplished with a UV lamp or I2 stain. Silica gel with a 230–400 mesh size was used for flash column chromatography using the combination of ethyl acetate and petroleum ether as the eluent. Unless noted, all reactions were carried out in oven-dried glassware under an atmosphere of nitrogen/argon using anhydrous solvents. Where appropriate, all reagents were purified prior to use following the guidelines of Perrin and Armerego.20 All commercial reagents were used as received. Proton nuclear magnetic resonance (1H NMR) spectra were recorded at 400 MHz/500 MHz. Chemical shifts were recorded in parts per million (ppm, δ) relative to tetramethyl silane (δ 0.00). 1H NMR splitting patterns are designated as singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), multiplet (m). Carbon nuclear magnetic resonance (13C NMR) spectra were recorded at 100 MHz/125 MHz. Mass spectra (MS) were obtained using an ESI mass spectrometer (TOF). IR spectra were recorded in KBr for solids. Melting points were determined using a hot stage apparatus and are uncorrected. Optical rotations were measured using a 2.0 mL cell with a 1.0 dm path length and are reported as [α]25D (c in g per 100 mL solvent) at 25 °C. Enantiomeric excess was determined by HPLC using a Chiralpak Cellulose 1 analytical column (detection at 254 nm).2-Phenyl-1-tosylazetidine (5a). The general method C described above was followed when 4a (100 mg, 0.32 mmol) was reacted with KOH (54 mg, 0.96 mmol) and TsCl (67 mg, 0.35 mmol) under reflux in THF for 30 min to afford 92 mg of 5a as a white solid in 98% yield; mp 104–108 °C, Rf 0.52 (40% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3062, 2982, 2936, 1595, 1466, 1394, 1354, 1340, 1302, 1280, 1246, 1217, 1174, 1112, 1088, 1067, 1018, 974, 930, 842; 1H NMR (400 MHz, CDCl3) δ 2.08–2.18 (m, 1H), 2.20–2.28 (m, 1H), 2.36 (s, 3H); 3.64–3.75 (m, 2H), 4.80 (t, J = 8.3 Hz, 1H), 7.19–7.35 (m, 7H), 7.61 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.6, 25.8, 47.2, 65.6, 126.3, 127.9, 128.4, 128.5, 129.6, 132.1, 140.5, 143.9; HRMS (ESI-TOF) calcd for C16H18NO2S (M+ + H): 288.1058, found: 288.1056.
2-(2-Chlorophenyl)-1-tosylazetidine (5b). The general method C described above was followed when 4b (100 mg, 0.29 mmol) was reacted with KOH (48 mg, 0.87 mmol) and TsCl (61 mg, 0.32 mmol) under reflux in THF for 30 min to afford 90 mg of 5b as a white solid in 95% yield; mp 160–165 °C; Rf 0.35 (40% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3064, 3030, 2985, 2928, 2876, 1596, 1574, 1490, 1460, 1408, 1386, 1342, 1298, 1265, 1234, 1180, 1129, 1088, 1065, 1042, 1014, 976, 912, 868, 835; 1H NMR (400 MHz, CDCl3) δ 1.95–1.99 (m, 1H), 2.37–2.42 (m, 1H), 2.42 (s, 3H), 3.63–3.70 (m, 1H), 3.74–3.79 (m, 1H), 5.14 (t, J = 8.3 Hz, 1H), 7.15–7.34 (m, 5H), 7.69 (d, J = 8.3 Hz, 2H), 7.85 (dd, J = 7.8, 1.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.6, 24.9, 47.5, 62.9, 127.1, 127.9, 128.6, 128.7, 128.9, 129.8, 131.0, 131.2, 138.3, 144.2; HRMS (ESI-TOF) calcd for C16H17ClNO2S (M+ + H): 322.0669, found: 322.0666.
2-(4-Nitrophenyl)-1-tosylazetidine (5c). The general method C described above was followed when 4c (100 mg, 0.28 mmol) was reacted with KOH (47 mg, 0.84 mmol) and TsCl (59 mg, 0.31 mmol) under reflux in THF for 30 min to afford 81 mg of 5c as a white solid in 85% yield; mp 175–178 °C; Rf 0.32 (40% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3068, 3027, 2995, 2923, 2852, 1598, 1515, 1492, 1439, 1347, 1317, 1302, 1291, 1238, 1211, 1182, 1156, 1108, 1089, 1070, 1026, 1014, 957, 932, 904, 857, 820, 800; 1H NMR (400 MHz, CDCl3) δ 2.10–2.19 (m, 1H), 2.36–2.47 (m, 4H), 3.75–3.83 (m, 2H), 4.96 (t, J = 8.3 Hz, 1H), 7.34 (d, J = 7.8 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 8.17 (d, J = 5.1 Hz, 2H); 13C NMR (100 MHz CDCl3) δ 21.5, 25.5, 47.4, 64.3, 123.8, 127.0, 128.4, 129.8, 131.9, 144.5, 147.6, 147.8; HRMS (ESI-TOF) calcd for C16H17N2O4S (M+ + H): 333.0909, found: 333.0910.
2-(4-Methoxyphenyl)-1-tosylazetidine (5d). The general method C described above was followed when 4d (100 mg, 0.29 mmol) was reacted with KOH (49 mg, 0.87 mmol) and TsCl (61 mg, 0.32 mmol) under reflux in THF for 30 min to afford 87 mg of 5d as a white solid in 92% yield; mp 68–72 °C; Rf 0.40 (40% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3068, 2980, 2924, 2855, 1594, 1512, 1466, 1390, 1361, 1322, 1282, 1251, 1176, 1148, 1110, 1092, 1063, 1024, 981, 932, 895, 865, 832; 1H NMR (400 MHz, CDCl3) δ 2.08–2.21 (m, 2H), 2.35 (s, 3H), 3.59–3.69 (m, 2H), 3.70 (s, 3H), 4.70 (t, J = 8.3 Hz, 1H), 6.75–6.79 (m, 2H), 7.22–7.26 (m, 4H), 7.58 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.5, 25.9, 47.0, 55.2, 65.5, 113.8, 127.7, 128.3, 129.5, 132.2, 132.6, 143.8, 159.4; HRMS (ESI-TOF) calcd for C17H20NO3S (M+ + H): 318.1164, found: 318.1162.
2-(4-Chlorophenyl)-1-tosylazetidine (5e). The general method C described above was followed when 4e (100 mg, 0.29 mmol) was reacted with KOH (49 mg, 0.87 mmol) and TsCl (61 mg, 0.32 mmol) under reflux in THF for 30 min to afford 92 mg of 5e as a white solid in 97% yield; mp 165–170 °C; Rf 0.36 (40% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3068, 3030, 2986, 2938, 2862, 1596, 1566, 1490, 1459, 1409, 1384, 1340, 1298, 1265, 1235, 1180, 1133, 1085, 1065, 1041, 1014, 982, 911, 863, 835, 799; 1H NMR (400 MHz, CDCl3) δ 2.04–2.13 (m, 1H), 2.20–2.28 (m, 1H), 2.38 (s, 3H), 3.63–3.73 (m, 2H), 4.77 (t, J = 8.3 Hz, 1H), 7.19–7.29 (m, 6H), 7.60 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.5, 25.7, 47.2, 64.8, 127.7, 128.4, 128.6, 129.7, 132.0, 133.7, 139.1, 144.1; HRMS (ESI-TOF) calcd for C16H17ClNO2S (M+ + H): 322.0669, found: 322.0664.
1-(4-Fluorophenylsulfonyl)-2-phenylazetidine (5f). The general method C described above was followed when 4f (100 mg, 0.32 mmol) was reacted with KOH (54 mg, 0.96 mmol) and TsCl (67 mg, 0.35 mmol) under reflux in THF for 30 min to afford 88 mg of 5f as a white solid in 93% yield; mp 126–128 °C; Rf 0.32 (20% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3068, 2957, 2925, 2883, 1589, 1491, 1454, 1405, 1343, 1291, 1229, 1157, 1091, 1018, 956, 926, 841, 817; 1H NMR (400 MHz, CDCl3) δ 2.10–2.22 (m, 1H), 2.26–2.35 (m, 1H), 3.70–3.79 (m, 2H), 4.87 (t, J = 8.3 Hz, 1H), 7.08–7.34 (m, 7H), 7.70–7.73 (m, 2H); 13C NMR (100 MHz, CDCl3)δ 25.7, 47.1, 65.8, 116.1, 126.2, 128.5, 130.9, 132.1, 140.1, 164.1, 166.6; HRMS (ESI-TOF) calcd for C15H15FNO2S (M+ + H): 290.0651, found: 290.0650.
1-(4-Methoxyphenylsulfonyl)-2-phenylazetidine (5g). The general method C described above was followed when 4g (100 mg, 0.31 mmol) was reacted with KOH (52 mg, 0.93 mmol) and TsCl (65 mg, 0.34 mmol) under reflux in THF for 30 min to afford 86 mg of 5g as a white solid in 91% yield; mp 116–118 °C; Rf 0.32 (35% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3071, 3032, 3004, 2980, 2956, 2885, 2840, 1596, 1496, 1455, 1439, 1413, 1364, 1338, 1310, 1300, 1258, 1234, 1188, 1154, 1095, 1061, 1023, 956, 927, 832, 803; 1H NMR (400 MHz, CDCl3) δ 2.09–2.18 (m, 1H), 2.21–2.29 (m, 1H), 3.64–3.83 (m, 5H), 4.80 (t, J = 8.3 Hz, 1H), 6.90 (d, J = 8.0 Hz, 2H), 7.18–7.34 (m, 5H), 7.66 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 25.8, 47.0, 55.6, 65.5, 114.1, 126.3, 127.1, 127.9, 128.4, 130.5, 140.6, 163.3; HRMS (ESI-TOF) calcd for C16H18NO3S (M+ + H): 304.1007, found: 304.1007.
1-(4-tert-Butylphenylsulfonyl)-2-phenylazetidine (5h). The general method C described above was followed when 4h (100 mg, 0.28 mmol) was reacted with KOH (47 mg, 0.84 mmol) and TsCl (59 mg, 0.31 mmol) under reflux in THF for 30 min to afford 85 mg of 5h as a white solid in 90% yield; mp 122–124 °C; Rf 0.38 (20% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3063, 3032, 2951, 1596, 1476, 1401, 1361, 1340, 1311, 1293, 1269, 1230, 1202, 1160, 1114, 1090, 1066, 1024, 958, 926, 834, 810; 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 9H), 2.10–2.27 (m, 2H), 3.70–3.74 (m, 2H), 4.84 (t, J = 8.3 Hz, 1H), 7.18–7.34 (m, 5H), 7.43 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 25.8, 31.1, 35.2, 47.0, 65.6, 125.9, 126.3, 127.9, 128.3, 128.4, 140.5, 156.8; HRMS (ESI) calcd for C19H24NO2S (M+ + H): 330.1528, found: 330.1526.
3,3-Dimethyl-2-phenyl-1-tosylazetidine (5i). The general method C described above was followed when 4i (100 mg, 0.30 mmol) was reacted with KOH (50 mg, 0.90 mmol) and TsCl (63 mg, 0.33 mmol) under reflux in THF for 30 min to afford 90 mg of 5i as a white solid in 95% yield; mp 132–134 °C; Rf 0.38 (20% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3087, 3062, 3033, 2966, 2924, 2876, 1596, 1492, 1458, 1391, 1376, 1342, 1298, 1275, 1232, 1180, 1157, 1088, 1052, 1029, 1017, 975, 909, 869, 816, 802, 763; 1H NMR (400 MHz, CDCl3) δ 0.69 (s, 3H), 0.90 (s, 3H), 2.39 (s, 3H), 3.35–3.41 (m, 2H), 4.46 (s, 1H), 7.18–7.29 (m, 7H), 7.62 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.6, 22.9, 27.2, 36.0, 60.6, 74.5, 126.3, 127.6, 128.0, 128.5, 129.5, 131.9, 137.0, 143.9; HRMS (ESI-TOF) calcd for C18H22NO2S (M+ + H): 316.1371, found: 316.1370.
2-(2-Chlorophenyl)-3,3-dimethyl-1-tosylazetidine (5j). The general method C described above was followed when 4j (100 mg, 0.27 mmol) was reacted with KOH (46 mg, 0.81 mmol) and TsCl (57 mg, 0.30 mmol) under reflux in THF for 30 min to afford 90 mg of 5j as a white solid in 95% yield; mp 137–139 °C; Rf 0.47 (25% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3065, 3026, 2987, 2961, 2927, 2881, 1595, 1571, 1493, 1461, 1438, 1371, 1343, 1304, 1290, 1270, 1235, 1203, 1168, 1138, 1087, 1064, 1047, 1016, 975, 905, 864, 849, 819, 763; 1H NMR (400 MHz, CDCl3) δ 0.71 (s, 3H), 0.96 (s, 3H), 2.40 (s, 3H), 3.34 (d, J = 7.6 Hz, 1H), 3.45 (d, J = 7.6 Hz, 1H), 4.89 (s, 1H), 7.13–7.27 (m, 3H), 7.32 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 7.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.6, 22.9, 27.6, 36.3, 60.7, 71.6, 126.7, 128.5, 128.6, 128.9, 129.5, 129.7, 131.9, 135.4, 144.1; HRMS (ESI-TOF) calcd for C18H21ClNO2S (M+ + H): 350.0982, found: 350.0982.
2-(4-Methoxyphenyl)-3,3-dimethyl-1-tosylazetidine (5k). The general method C described above was followed when 4k (100 mg, 0.27 mmol) was reacted with KOH (46 mg, 0.82 mmol) and TsCl (57 mg, 0.30 mmol) under reflux in THF for 30 min to afford 90 mg of 5k as a white solid in 94% yield; mp 141–143 °C; Rf 0.32 (25% ethyl acetate/petroleum ether); IR νmax (KBr, cm−1) 3069, 2960, 2927, 2862, 2836, 1594, 1515, 1462, 1392, 1372, 1335, 1303, 1253, 1177, 1157, 1109, 1089, 1065, 1037, 982, 933, 898, 873, 840, 812, 765; 1H NMR (400 MHz, CDCl3) δ 0.78 (s, 3H), 0.94 (s, 3H), 2.47 (s, 3H), 3.39 (d, J = 7.3 Hz, 1H), 3.46 (d, J = 7.3 Hz, 1H), 3.79 (s, 3H), 4.45 (s, 1H), 6.86–6.89 (m, 2H), 7.24–7.27 (m, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.5, 22.8, 27.0, 36.2, 55.2, 60.5, 74.4, 113.6, 127.1, 127.6, 128.5, 129.2, 129.5, 143.8, 159.2; HRMS (ESI-TOF) calcd for C19H24NO3S (M+ + H): 346.1477, found: 346.1474.
(2S,4S)-2-Ethyl-4-phenyl-1-tosylazetidine (5l). The general method C described above was followed when 7a (100 mg, 0.30 mmol) was reacted with KOH (50 mg, 0.90 mmol) and TsCl (63 mg, 0.33 mmol) under reflux in THF for 1.5 h to afford 66 mg of 5l as a white solid in 70% yield; mp 130–132 °C; Rf 0.36 (25% ethyl acetate/petroleum ether); IR νmax (cm−1, KBr) 3032, 2972, 2929, 2876, 1599, 1493, 1453, 1388, 1337, 1305, 1250, 1218, 1186, 1157, 1085, 1042, 1011, 978, 952, 819, 755; 1H NMR (400 MHz, CDCl3) δ 0.84 (t, J = 7.3 Hz, 3H), 1.67–1.77 (m, 2H), 1.83–1.88 (m, 1H), 2.34–2.39 (m, 4H), 3.77–3.80 (m, 1H), 4.61 (t, J = 8.3 Hz, 1H), 7.19–7.33 (m, 7H), 7.59 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 8.4, 21.5, 28.7, 31.6, 61.2, 62.1, 126.3, 127.8, 128.3, 128.4, 129.5, 132.7, 140.8, HRMS (ESI-TOF) calcd for C18H22NO2S (M+ + H): 316.1371, found: 316.1373.
(2R,4S)-2-Ethyl-4-phenyl-1-tosylazetidine (5m). The general method C described above was followed when 7b (100 mg, 0.30 mmol) was reacted with KOH (50 mg, 0.90 mmol) and TsCl (63 mg, 0.33 mmol) under reflux in THF for 1.5 h to afford 72 mg of 5m as a white solid in 76% yield; Low melting solid; Rf 0.34 (25% ethyl acetate/petroleum ether); IR νmax (cm−1, KBr) 3032, 2972, 2929, 2876, 1599, 1493, 1453, 1388, 1337, 1305, 1250, 1218, 1186, 1157, 1085, 1042, 1011, 978, 952, 819, 755; 1H NMR (400 MHz, CDCl3) δ 0.84 (t, J = 7.5 Hz, 3H), 1.35 (m, 1H), 1.69–1.77 (m, 1H), 2.14–2.24 (m, 2H), 2.27 (s, 3H), 4.25–4.31 (m, 1H), 5.11 (t, J = 7.8 Hz, 1H), 6.98 (d, J = 8.3 Hz, 2H), 7.09–7.23 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 8.5, 21.4, 26.7, 30.9, 64.7, 127.1, 127.3, 127.9, 128.2, 129.0, 137.0, 138.9, 142.5; HRMS (ESI-TOF) calcd for C18H22NO2S (M+ + H): 316.1371, found: 316.1373.
(Ss,2R)-(+)-2-Phenyl-1-(p-tolylsulfinyl)azetidine (12a). The general method C described above was followed when 11a (100 mg, 0.35 mmol) was reacted with KOH (50 mg, 1.05 mmol) and TsCl (73 mg, 0.39 mmol) under reflux in THF for 1 h to afford 66 mg of 12a as a white solid in 70% yield; mp 56–58 °C; Rf 0.45 (30% ethyl acetate in petroleum ether); [α]25D = +257.1 (c 0.375 in CH2Cl2); IR νmax (KBr, cm−1) 2956, 2878, 1597, 1492, 1452, 1091, 1066, 812; 1H NMR (500 MHz, CDCl3) δ 2.22–2.28 (m, 1H), 2.40 (s, 3H), 2.43–2.49 (m, 1H), 2.73–2.75 (m, 1H), 3.96–4.02 (m, 1H), 5.29–5.33 (m, 1H), 7.21–7.59 (m, 9H); 13C NMR (125 MHz, CDCl3) δ 21.5, 26.6, 37.5, 61.7, 125.7, 126.7, 128.0, 128.7, 129.6, 139.7, 141.2, 141.6; HRMS (ESI-TOF) calcd for C16H18NOS (M+ + H) 272.1109, found 272.1109.
(Ss,2R)-(+)-2-(2-Chlorophenyl)-1-(p-tolylsulfinyl)azetidine (12b). The general method C described above was followed when 11b (100 mg, 0.31 mmol) was reacted with KOH (52 mg, 0.93 mmol) and TsCl (65 mg, 0.34 mmol) under reflux in THF for 1 h to afford 77 mg of 12b as a white solid in 82% yield; mp 70 °C; Rf 0.23 (30% ethyl acetate in petroleum ether); [α]25D = +87.7 (c 0.65 in CH2Cl2); IR νmax (KBr, cm−1) 2968, 2873, 1592, 1469, 1089, 1065, 1036, 824, 754; 1H NMR (400 MHz, CDCl3): δ 2.04–2.13 (m, 1H), 2.43 (s, 3H), 2.63–2.71 (m, 1H), 2.74–2.79 (m, 1H), 4.04–4.10 (m, 1H), 5.64 (t, J = 8.6 Hz, 1H), 7.24– 7.38 (m, 4H), 7.65 (d, J = 8.0 Hz, 2H), 7.83 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 21.4, 25.6, 37.6, 58.8, 125.5, 126.9, 127.1, 128.6, 129.5, 129.6, 131.9, 139.3, 139.7, 141.3; HRMS (ESI-TOF) calcd for C16H17ClNOS (M+ + H) 306.0719, found 306.0719.
(Ss,2R)-(+)-2-(3-Bromophenyl)-1-(p-tolylsulfinyl)azetidine (12c). The general method C described above was followed when 11c (100 mg, 0.27 mmol) was reacted with KOH (46 mg, 0.81 mmol) and TsCl (57 mg, 0.30 mmol) under reflux in THF for 1 h to afford 81 mg of 12c as a white solid in 85% yield; mp 89–92 °C; Rf 0.35 (30% ethyl acetate in petroleum ether); [α]25D = +135.5 (c 0.62 in CH2Cl2); IR νmax (KBr, cm−1) 2921, 2852, 1593, 1426, 1359, 1092, 1030, 810; 1H NMR (400 MHz, CDCl3, ppm): δ 2.13–2.19 (m, 1H), 2.36 (s, 3H), 2.65–2.68 (m, 1H), 3.89–3.96 (m, 1H), 5.19 (t, J = 8.0 Hz, 1H), 7.24–7.46 (m, 5H); 7.58 (d, J = 8.3 Hz, 2H), 7.67 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 21.4, 26.4, 37.5, 60.8, 122.8, 125.3, 125.6, 128.6, 129.6, 130.2, 131.0, 139.3, 141.3, 143.9; HRMS (ESI-TOF) calcd for C16H17BrNOS (M+ + H) 350.0214, found 350.0214.
(Ss,2R)-(+)-2-p-Tolyl-1-(p-tolylsulfinyl)azetidine (12d). The general method C described above was followed when 11d (100 mg, 0.33 mmol) was reacted with KOH (56 mg, 0.99 mmol) and TsCl (69 mg, 0.36 mmol) under reflux in THF for 1 h to afford 71 mg of 12d as a colorless liquid in 75% yield; Rf 0.37 (30% ethyl acetate in petroleum ether); [α]25D = +208.4 (c 0.475 in CH2Cl2); IR νmax (neat, cm−1) 2924, 2855, 1598, 1459, 1362, 1176, 1099, 809, 719; 1H NMR (400 MHz, CDCl3): δ 2.16–2.28 (m, 2H), 2.33 (s, 3H), 2.44 (s, 3H), 3.67–3.72 (m, 1H), 3.75–3.79 (m, 1H), 4.80 (t, J = 8.3 Hz, 1H), 7.14 (d, J = 8.0 Hz, 2H), 7.29–7.33 (m, 4H), 7.68 (d, J = 8.3 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 21.3, 21.7, 26.0, 47.3, 65.8, 126.4, 128.6, 129.3, 129.7, 132.0, 137.7, 137.9, 144.0; HRMS (ESI-TOF) calcd for C17H20NOS (M + H)+ 286.1266, found 286.1266.
(Ss,2R)-(+)-2-Cyclohexyl-1-(p-tolylsulfinyl)azetidines (12e). The general method C described above was followed when 11e (100 mg, 0.34 mmol) was reacted with KOH (57 mg, 1.02 mmol) and TsCl (71 mg, 0.37 mmol) under reflux in THF for 1 h to afford 64 mg of 12e as a colorless liquid in 81% yield; Rf 0.42 (30% ethyl acetate in petroleum ether); [α]25D = +65.0 (c 0.2 in CH2Cl2); IR νmax (neat, cm−1) 3484, 2925, 2852, 1597, 1492, 1448, 1397, 1378, 1350, 1302, 1263, 1236, 1177, 1163, 1092, 1068, 1047, 1017, 994, 974, 940, 922, 889, 811; 1H NMR (400 MHz, CDCl3): δ 0.88–1.04 (m, 2H), 1.18–1.28 (m, 3H), 1.57–1.79 (m, 5H), 1.89–1.99 (m, 2H), 2.05–2.09 (m, 1H), 2.37 (s, 3H), 2.60 (td, J = 8.9 Hz, 3.9 Hz, 1H), 3.86 (dd, J = 16.9 Hz, 9.2 Hz, 1H), 4.01 (dd, J = 16.3 Hz, 8.2 Hz, 1H), 7.25 (d, J = 7.8 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 21.1, 21.4, 25.8, 26.5, 27.8, 29.0, 37.2, 44.3, 64.7, 125.6, 129.5, 140.2, 141.0; HRMS (ESI-TOF) calcd for C16H24NOS (M + H)+ 278.1579, found 278.1572.
(R)-2-Phenyl-1-tosylazetidine ((R)-5a). The general method C described above was followed when (R)-4a (100 mg, 0.32 mmol) was reacted with KOH (54 mg, 0.96 mmol) and TsCl (67 mg, 0.35 mmol) under reflux in THF for 45 min to afford 70 mg of (R)-5a as a white solid in 75% yield; mp 104–108 °C, Rf 0.52 (40% ethyl acetate/petroleum ether); [α]25D = +254.8 (c 0.25 in CH2Cl2); IR νmax (KBr, cm−1) 3062, 2982, 2936, 1595, 1466, 1394, 1354, 1340, 1302, 1280, 1246, 1217, 1174, 1112, 1088, 1067, 1018, 974, 930, 842, 822, 766; 1H NMR (400 MHz, CDCl3) δ 2.08–2.18 (m, 1H), 2.20–2.28 (m, 1H), 2.36 (s, 3H); 3.64–3.75 (m, 2H), 4.80 (t, J = 8.3 Hz, 1H), 7.19–7.35 (m, 7H), 7.61 (d, J = 8.0 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 21.6, 25.8, 47.2, 65.6, 126.3, 127.9, 128.4, 128.5, 129.6, 132.1, 140.5, 143.9; HRMS (ESI-TOF) calcd for C16H18NO2S (M+ + H): 288.1058, found: 288.1056; ee 84%. The enantiomeric excess was determined by chiral HPLC analysis (Cellulose 1), n-hexane/i-propanol = 90
:10, flow rate = 1.0 mL min−1, tR (2) = 11.35 min (major).
Acknowledgements
M.K.G. is grateful to DST and IIT Kanpur, India, for financial support. S.D. thanks U.G.C., India for a research fellowship.Notes and references
- (a) B. I. Ericksson, S. Carlsson, M. Halvarsson, B. Risberg and C. Mattsson, Thromb. Haemostosis, 1997, 78, 1404 Search PubMed; (b) I. Kirk, (Astrazeneca AB), PCT Int. Appl, WO 2000041716, 2000 Search PubMed; Chem. Abstr. 2000 123 99559 Search PubMed; (c) D.-G. Liu and G.-Q. Lin, Tetrahedron Lett., 1999, 40, 337 CrossRef CAS; (d) T. Shioiri, Y. Hamada and F. Matsuura, Tetrahedron, 1995, 51, 3939 CrossRef CAS; (e) S. Obika, J.-i. Andoh, M. Onoda, O. Nakagawa, A. Hiroto, T. Sugimoto and T. Imanishi, Tetrahedron Lett., 2003, 44, 5267 CrossRef CAS; (f) T. Takemoto, K. Nomoto, S. Fushiya, R. Ouchi, G. Kusano, H. Hikino, S.-i. Takagi, Y. Matsuura and M. Kakudo, Proc. Jpn. Acad. Ser. B, 1978, 54, 469 CrossRef CAS; (g) S. Singh, G. Crossaley, S. Ghosal, Y. Lefievre and M. W. Pennington, Tetrahedron Lett., 2005, 46, 1419 CrossRef CAS PubMed; (h) K. Isono, K. Asahi and S. Suzuki, J. Am. Chem. Soc., 1969, 91, 7490 CrossRef CAS; (i) T. Akihisa, S. Mafune, M. Ukiya, Y. Kimura, K. Yasukawa, T. Suzuki, H. Tokuda, N. Tanabe and T. Fukuoka, J. Nat. Prod., 2004, 67, 479 CrossRef CAS PubMed; (j) D. Jin, S. Takai, M. Yamada, M. Sakaguchi and M. Miyazaki, Life Sci., 2002, 71, 437 CrossRef CAS; (k) E. Ziemińska, A. Stafiej and J. W. Łazarewicz, Neurochem. Int., 2003, 43, 481 CrossRef; (l) Y. Ohfune, M. Tomita and K. Nomoto, J. Am. Chem. Soc., 1981, 103, 2409 CrossRef CAS; (m) Q. Cheng, H. Kiyota, M. Yamaguchi, T. Horiguchi and T. Oritani, Bioorg. Med. Chem. Lett., 2003, 13, 1075 CrossRef CAS; (n) A. W. Bannon, M. W. Decker, M. W. Holladay, P. Curzon, D. Donnelly-Roberts, P. S. Puttfarcken, R. S. Bitner, A. Diaz, A. H. Dickenson, R. D. Porsolt, M. Williams and S. P. Arneric, Science, 1998, 279, 77 CrossRef CAS; (o) K. J. af Forselles, J. Root, T. Clarke, D. Davey, K. Aughton, K. Dack and N. Pullen, Br J. Pharmacol., 2011, 164, 1847 CrossRef CAS PubMed; (p) K. Lu, Y. Jiang, B. Chen, E. M. Eldemenky, G. Ma, M. Packiarajan, G. Chandrasena, A. D. White, K. A. Jones, B. Li and S.-P. Hong, Bioorg. Med. Chem. Lett., 2011, 11, 5310 CrossRef PubMed.
- (a) L. Kiss, S. Mangelinckx, F. Fülöp and N. De Kimpe, Org. Lett., 2007, 9, 4399 CrossRef CAS PubMed; (b) F. Couty, G. Evano, M. Vargass-Sanchez and G. Bouzas, J. Org. Chem., 2005, 70, 9028 CrossRef CAS PubMed; (c) D. Enders and J. Gries, Synthesis, 2005, 3508 CrossRef CAS; (d) W. V. Branbandt, G. Verniest, D. De Smaele, G. Duvey and N. De Kimpe, J. Org. Chem., 2006, 71, 7100 CrossRef PubMed; (e) L.-G. Meng, P. Cai, Q. Guo and S. Xue, J. Org. Chem., 2008, 73, 8491 CrossRef CAS PubMed; (f) M. Sivaprakasam, F. Couty, O. David, J. Marrot, R. Sridhar, B. Srinivas and K. R. Rao, Eur. J. Org. Chem., 2007, 5734 CrossRef CAS PubMed; (g) H. Lu and C. Li, Org. Lett., 2006, 8, 5365 CrossRef CAS PubMed; (h) M. Tiecco, L. Testaferri, A. Temperini, R. Terlizzi, L. Bagnoli, F. Marini and C. Santi, Org. Biomol. Chem., 2007, 5, 3510 RSC; (i) E. Banide, V. L. de Talancé, G. Schmidt, H. Lubin, S. Comesse, L. Dechoux, L. Hamon and C. Kadouri-Puchot, Eur. J. Org. Chem., 2007, 4517 CrossRef CAS PubMed; (j) F. Couty and D. Prim, Tetrahedron: Asymmetry, 2002, 13, 2619 CrossRef CAS; (k) N. H. Cromwell and B. Phillips, Chem. Rev., 1979, 79, 331 CrossRef CAS; (l) M. K. Ghorai, A. Kumar and S. Halder, Tetrahedron, 2007, 63, 4779 CrossRef CAS PubMed; (m) B. Alcaide, P. Almendros, C. Aragoncillo and R. Rodríguez-Acebes, J. Org. Chem., 2001, 66, 5208 CrossRef CAS PubMed; (n) M. Bouazaoui, J. Martinez and F. Cavelier, Eur. J. Org. Chem., 2009, 2729 CrossRef CAS PubMed; (o) P. Pérez-Faginas, M. T. Aranda, M. T. García-López, R. Snoeck, G. Andrei, J. Balzarini and R. González-Muñiz, Bioorg. Med. Chem., 2011, 19, 1155 CrossRef PubMed; (p) J. Shimokawa, T. Harada, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2011, 133, 17634 CrossRef CAS PubMed; (q) S. Dekeukeleire, M. D'hooghe, K. W. Törnroos and N. De Kimpe, J. Org. Chem., 2010, 75, 5934 CrossRef CAS PubMed; (r) A. Rai and L. D. S. Yadav, Org. Biomol. Chem., 2011, 9, 8058 RSC; (s) A. Feula, L. Male and J. S. Fossey, Org. Lett., 2010, 12, 5044 CrossRef CAS PubMed; (t) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS PubMed; (u) L. Menguy and F. Couty, Tetrahedron: Asymmetry, 2010, 21, 2385 CrossRef CAS PubMed; (v) Y. Ye, H. Wang and R. Fan, Org. Lett., 2010, 10, 2802 CrossRef PubMed; (w) S. T. M. Orr, S. Cabral, D. P. Fernando and T. Makowski, Tetrahedron Lett., 2011, 52, 3618 CrossRef CAS PubMed; (x) V. K. Yadav, B. D. Narhe and V. Sriramurthy, Org. Biomol. Chem., 2012, 10, 4390 RSC; (y) T. Biswas, J. P. Mukherjee and S. K. Chattopadhyay, Tetrahedron: Asymmetry, 2012, 23, 1416 CrossRef CAS PubMed; (z) C.-B. Miao, C.-P. Dong, M. Zhang, W.-P. Ren, Q. Meng, X.-Q. Sun and H.-T. Yang, J. Org. Chem., 2013, 78, 4329 CrossRef CAS PubMed.
- (a) P. G. Cozzi, B. Di Simone and A. Umani-Ronchi, Tetrahedron Lett., 1996, 37, 1691 CrossRef CAS; (b) T. P. Tang and J. A. Ellman, J. Org. Chem., 2002, 67, 7819 CrossRef CAS PubMed; (c) A. R. Maolanon, J. S. Villadsen, N. J. Christensen, C. Hoeck, T. Friis, P. Harris, C. H. Gotfredsen, P. Fristrup and C. A. Olsen, J. Med. Chem., 2014, 57, 9644 CrossRef CAS PubMed.
- (a) A. V. Sivakumar, G. S. Babu and S. V. Bhat, Tetrahedron: Asymmetry, 2001, 12, 1095 CrossRef CAS; (b) E. Juaristi, D. Quintana and J. Escalante, Aldrichimica Acta, 1994, 27, 3 CAS; (c) F. H. van der Steen, H. Kleijn, G. J. P. Britovsek, J. T. B. H. Jastrzebski and G. van Koten, J. Org. Chem., 1992, 57, 3906 CrossRef CAS; (d) T. Kambara, M. A. Hussein, H. Fujieda, A. Iida and K. Tomioka, Tetrahedron Lett., 1998, 39, 9055 CrossRef CAS.
- (a) S. Kobayashi, M. Araki and M. Yasuda, Tetrahedron Lett., 1995, 36, 5773 CrossRef CAS; (b) C. Bellucci, P. G. Cozzi and A. Umani-Ronchi, Tetrahedron Lett., 1995, 36, 7289 CrossRef CAS.
- (a) S. Saito, K. Hatanaka and H. Yamamoto, Tetrahedron, 2001, 57, 875 CrossRef CAS; (b) G. A. Molander and P. J. Stengel, J. Org. Chem., 1995, 60, 6660 CrossRef CAS; (c) O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- (a) J. d'Angelo and J. Maddaluno, J. Am. Chem. Soc., 1986, 108, 8112 CrossRef; (b) D. Gray, C. Concellón and T. Gallagher, J. Org. Chem., 2004, 69, 4849 CrossRef CAS PubMed; (c) S. G. Davies and O. Ichihara, Tetrahedron Lett., 1998, 39, 6045 CrossRef CAS; (d) A. A. Kudale, P. Anaspure, F. Goswami and M. Voss, Tetrahedron Lett., 2014, 55, 7219 CrossRef CAS PubMed; (e) L. Thander, K. Sarkar and S. K. Chattopadhyay, Tetrahedron: Asymmetry, 2009, 20, 1213 CrossRef CAS PubMed.
- B. Das, P. Balasubramanyam, B. Veeranjaneyulu and G. C. Reddy, J. Org. Chem., 2009, 74, 9505 CrossRef CAS PubMed.
- (a) M. K. Ghorai, K. Das, A. Kumar and A. Das, Tetrahedron Lett., 2006, 47, 5393 CrossRef CAS PubMed; (b) The concept and initial results are described in the Ph.D. thesis of Dr. K. Das submitted to IIT Kanpur (India) in 2007.
- F. A. Davis, Y. Zhang, Y. Andemichael, T. Fang, D. L. Fanelli and H. Zhang, J. Org. Chem., 1999, 64, 1403 CrossRef CAS.
- F. A. Davis, R. E. Reddy and P. S. Portonovo, Tetrahedron Lett., 1994, 35, 9351 CrossRef CAS.
- (a) F. A. Davis, P. Zhou and G. V. Reddy, J. Org. Chem., 1994, 59, 3243 CrossRef CAS; (b) F. A. Davis and P. Zhou, Tetrahedron Lett., 1994, 35, 7525 CrossRef CAS.
- F. A. Davis, R. T. Reddy and R. E. Reddy, J. Org. Chem., 1992, 57, 6387 CrossRef CAS.
- J. Jiang, K. K. Schumacher, M. M. Joullie, F. A. Davis and R. E. Reddy, Tetrahedron Lett., 1994, 35, 2121 CrossRef CAS.
- (a) F. A. Davis, Y. Zhang and G. Anilkumar, J. Org. Chem., 2003, 68, 8061 CrossRef CAS PubMed; (b) E. J. Thomas and C. F. Vickers, Tetrahedron: Asymmetry, 2009, 20, 970 CrossRef CAS PubMed; (c) F. A. Davis and Y. Zhang, Tetrahedron Lett., 2009, 50, 5205 CrossRef CAS PubMed.
- M. K. Ghorai, K. Das and A. Kumar, Tetrahedron Lett., 2007, 48, 2471 CrossRef CAS PubMed.
- (a) Q. Chen, J. Li and C. Yuan, Synthesis, 2008, 2986 CAS; (b) J. A. Burkhard, C. Guérot, H. Knust and E. M. Carreira, Org. Lett., 2012, 14, 66 CrossRef CAS PubMed.
- F. A. Davis, R. E. Reddy and J. M. Szewczyk, J. Org. Chem., 1995, 60, 7037 CrossRef CAS.
- After purification, the 1H NMR spectrum of (R)-5a was compared with racemic 2-phenyl-N-tosylazetidine 5a and ee of (R)-5a was found to be 84% as determined by HPLC analysis using chiral column. Please see ESI† for details.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn, 1988 Search PubMed.
Footnotes |
| † Dedicated to Prof. Tarun Kumar Sarkar on his 67th birthday. |
| ‡ Electronic supplementary information (ESI) available: Copies of 1H, and 13C NMR spectra and HPLC chromatogram. See DOI: 10.1039/c5ob01140j |
| This journal is © The Royal Society of Chemistry 2015 |