
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
meso-Tetraphenylporphyrin with a pi-system extended by fusion with anthraquinone†
Mikhail A.
Filatov
*ab,
Ernesta
Heinrich
a,
Katharina
Landfester
a and
Stanislav
Baluschev
acd
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. Fax: +496131379100
bInstitute of Polymers, Bulgarian Academy of Sciences, Acad. G. Bonchev Str., block 103-A, BG - 1113 Sofia, Bulgaria. E-mail: filatov@polymer.bas.bg
cOptics and Spectroscopy Department, Faculty of Physics, Sofia University “St. Kliment Ochridski”, 5 James Bourchier, 1164 Sofia, Bulgaria
dFreiburg Institute for Advanced Studies (FRIAS), Albert-Ludwigs-Universität Freiburg, Albertstraße 19, D-79104 Freiburg, Germany
First published on 14th May 2015
Abstract
Fusion with a 9,10-anthraquinone moiety was achieved to extend porphyrin's π-system. A bridged dihydroisoindole derivative was used to prepare the corresponding meso-tetraphenyltetraanthraquinonoporphyrin (Ph4TAQP) via a thermal retro-Diels–Alder reaction. The basic optical properties of the prepared new anthraquinonoporphyrin and its complexes with Zn and Pd were studied.
Introduction
Porphyrins with aromatic rings fused to the tetrapyrrolic core, so-called π-extended porphyrins, have attracted much attention in recent years as materials for numerous applications – from biomedical sensing and imaging to organic optoelectronics.1 Metallated π-extended porphyrins are particularly important for the process of triplet–triplet annihilation photon energy upconversion (TTA-UC).2 A variety of π-extended porphyrins have been synthesized by fusing benzene,3 naphthalene,4 pyrene,5 azulene,6 anthracene,7 corannulene,8 and other aromatic moieties to the meso- and β-positions of the macrocycle. Fusion of aromatic rings to all four pyrrole residues results in particularly strong effects on the π-system, leading to enhanced light absorption and efficient emission in the near-infrared (IR-A) region of the spectrum.9First reported by Krautler and co-workers, a conjugation of naphthoquinone to a porphyrin has a remarkable effect on its properties. Particularly, resulting materials exhibit optical properties which resemble those of nanoscopic carbon materials with extended π-systems, such as graphene, graphite, and nanotubes.10 Theoretical studies of tetranaphthoquinonoporphyrin (TNQP) revealed that introduction of the carbonyl groups into the π-system results in strong alternations of bonds and a transformation of the conjugation from “benzene-type” to “butadiene-type”. Unidirectional photon-induced current associated with p–π conjugation enables light-harvesting efficiency of this kind of molecular skeleton to reach 90% in the range of 300–800 nm.11 This makes TNQPs attractive materials for panchromatic dye-sensitized solar cells. Moreover, porphyrins fused with quinone moieties are expected to exhibit interesting electrochemical properties, since they are able to accept a load of at least 8 electrons per molecule. Such materials clearly promise to expand the range of multi-electron transfer (MET) catalysts – compounds having the ability to accommodate and transfer multiple electrons to reaction substrates at one time.12
Despite promising properties, tetraquinonoporphyrins (TQP) are almost unknown because the available synthetic methods in the field of π-extended porphyrins chemistry have been very limited until recently. To the best of our knowledge, the only representative of a porphyrin directly fused with four quinone fragments was obtained by Krautler and co-workers, using the [4 + 2] cycloaddition reaction between β,β′-tetrasulfolenoporphyrin13 and an excess of benzoquinone.10
Herein we report a synthetic approach to meso-tetraphenyltetraanthraquinonoporphyrin (Ph4TAQP) based on a bridged dihydroisoindole precursor. In addition we describe the basic optical properties of the newly synthesized Ph4TAQP free-base and its metal complexes.
Results and discussion
Due to the instability of isoindole and its π-expanded analogues,14 the formation of a fully conjugated π-system has to be performed after the formation of the porphyrin macrocycle. So far, two general synthetic methods have been employed to construct the extended porphyrin architecture: oxidative aromatization15 and thermal retro-Diels–Alder reaction.16As is shown in Scheme 1, the use of the oxidative aromatization approach for the synthesis of tetraanthraquinonoporphyrin requires the corresponding dihydroisoindole derivative (Scheme 1, route A). According to the thermal retro-Diels–Alder approach, the target molecule can be prepared from bicyclo[2.2.2]octadiene-annelated porphyrin which can undergo thermal extrusion of ethylene (route B).
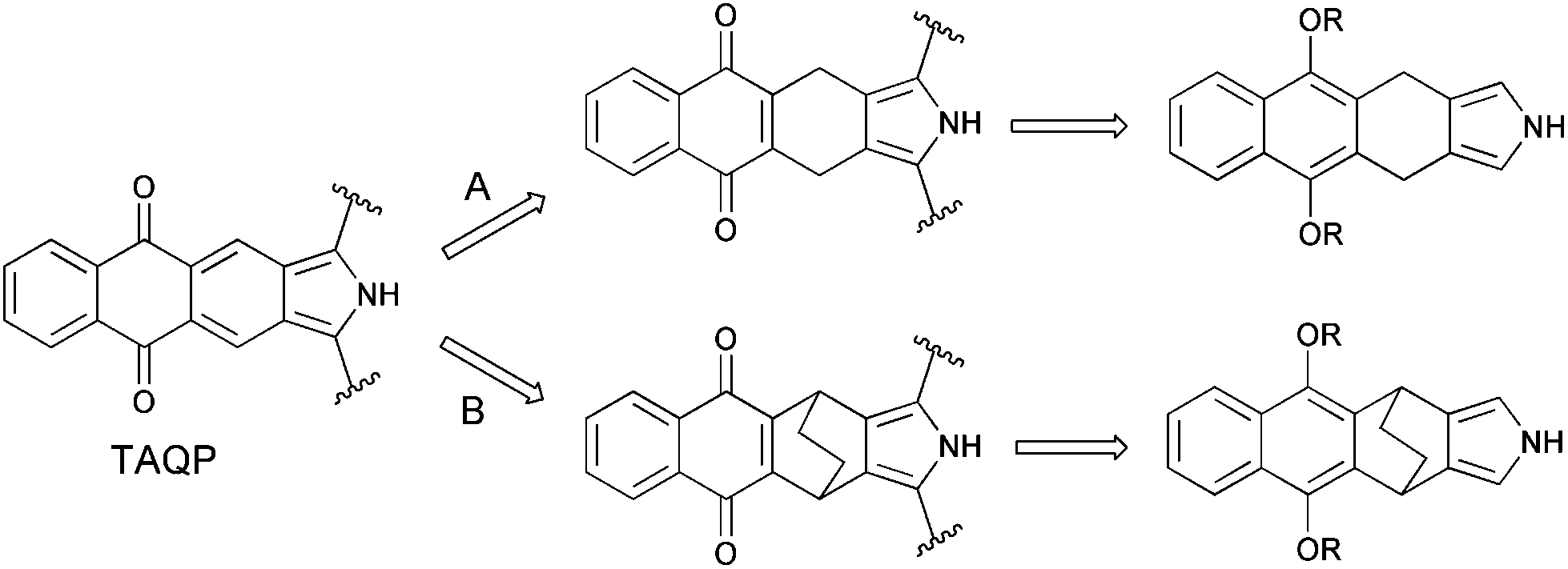 | ||
| Scheme 1 Retrosynthetical analysis of a TAQP system. | ||
A pyrrole derivative containing a naphthoquinone moiety represents a direct precursor for the synthesis of TAQP through route A. We first examined the possibility to apply directly 1,4,4a,9a-tetrahydro-anthraquinone 1 (Scheme 2) for the synthesis of the corresponding pyrrole from vinyl or allyl sulfones via a Barton–Zard reaction.17 Treatment of 1 with PhSCl, followed by oxidation with Oxone led to the chlorosulfone derivative 2. Further reaction with DBU yielded 2-phenylsulfonylanthraquinone 3, rather than the expected vinyl sulfone. An attempt to introduce 3 into Barton–Zard synthesis was unsuccessful and delivered mixture of products arising from the reduction of the quinone moiety. Thus, a protection of the reactive quinonic moiety was necessary to avoid side reactions during the pyrrole synthesis. Conversion of the quinone into corresponding hydroquinone diacetates was preferable over reductive methylation since it requires mild conditions for further deprotection.18
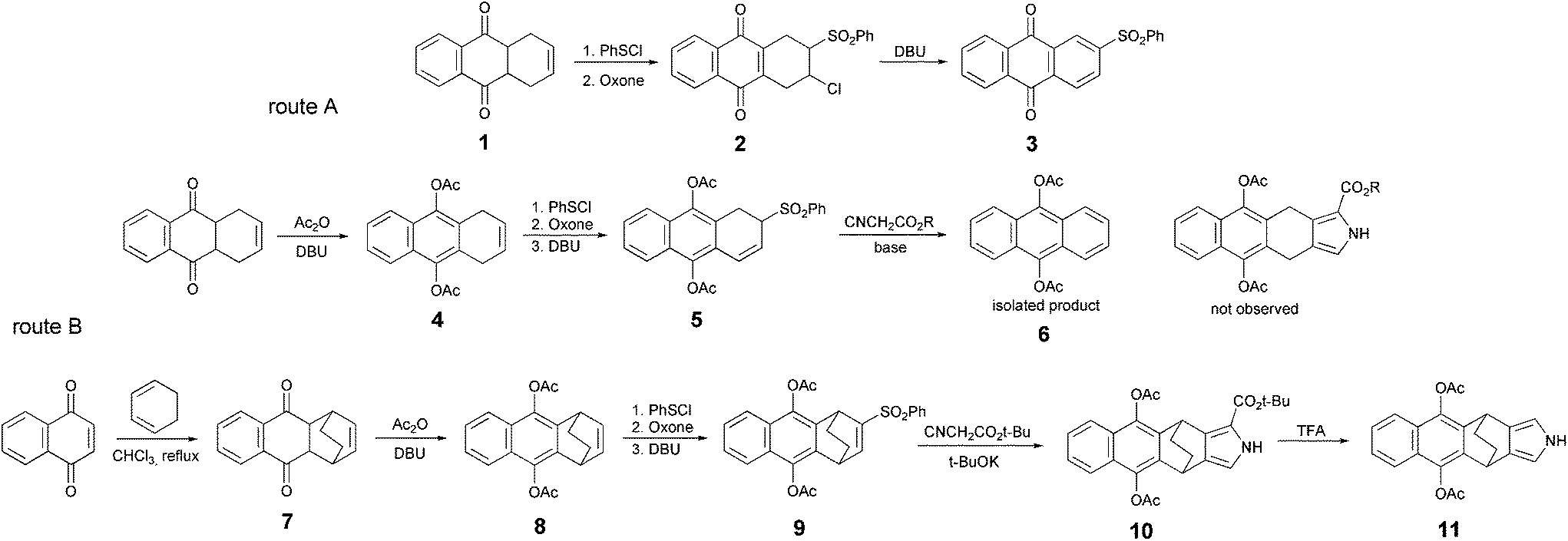 | ||
| Scheme 2 Synthesis of a TAQP pyrrole precursor. | ||
Dione 1 is known to form a deprotonated dihydronaphthoquinone irreversibly upon treatment with bases.7 Treatment of 1 with DBU and acetic anhydride provided diacetate 4. It should be noted that this procedure was found to give higher yields than previously reported aromatization of the dione ring by boiling with acetic anhydride and acetic acid in the presence of p-toluenesulfonic acid as a catalyst.19
Diacetate was then used for the preparation of allylsulfone 5, employing a previously established procedure. As expected, compound 5 was formed in good yield. However, under the conditions of Barton–Zard reaction (t-BuOK, THF, isocyanoacetate),20 no formation of the corresponding pyrrole compound was observed. Diacetoxyanthracene 6 was the only isolated product. Attempts to optimize the reaction conditions: changing the base (DBU, potassium and sodium tert-butoxides, HMDS), solvents and temperature regimes failed to deliver the target product. It is known that aromatization of cyclohexadienes can be incurred by strong bases.21 However, taking into account that a similar sulfone derivative containing butoxy-groups instead or acetoxy-groups was previously successfully used in the pyrrole synthesis,7 it is interesting that sulfone 6 behaves so differently under basic conditions, when elimination is the predominant pathway.
Thus we focused further efforts on the thermal retro-Diels–Alder approach. 1,4-Naphthoquinone was reacted with 1,3-cyclohexadiene to obtain dione precursor 7. Its acetylation gave 8, which was used for the preparation of the corresponding sulfone 9. As expected, the Barton–Zard reaction with isocyanoacetate synthesis delivered pyrrole 10.
In this case tert-butyl isocyanoacetate24 was used, since for pyrrole tert-butyl esters a decarboxylation reaction can be performed via solvolysis in neat trifluoroacetic acid. These conditions were expected to secure the hydroquinone moiety from deprotection. Indeed, treatment with TFA for 30 min delivered pyrrole 11 in good yield (68%).
With pyrrole 11 in hand, we succeeded to prepare intermediate porphyrin 12 according to the conventional Lindsey condensation.22 As shown in Scheme 3, pyrrole 11 reacted with benzaldehyde in CH2Cl2 in the presence of BF3·OEt2, followed by oxidation with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) at room temperature for additional 3 hours to afford porphyrin 12 in 18% yield after purification. After further treatment of the obtained porphyrin 12 with KOH and oxidation by DDQ the resulting crude intermediate was heated at 200 °C in vacuum for 4 h. Target tetraanthraquinonoporphyrin was isolated in 65% yield after chromatographic purification and recrystallization. To our surprise, instead of the expected problems with poor solubility due to π-stacking, we observed a rather good solubility (as compared to tetranaphtho- or tetraanthraporphyrins) of the obtained product in common organic solvents (chlorohydrocarbons, aromatics, THF).
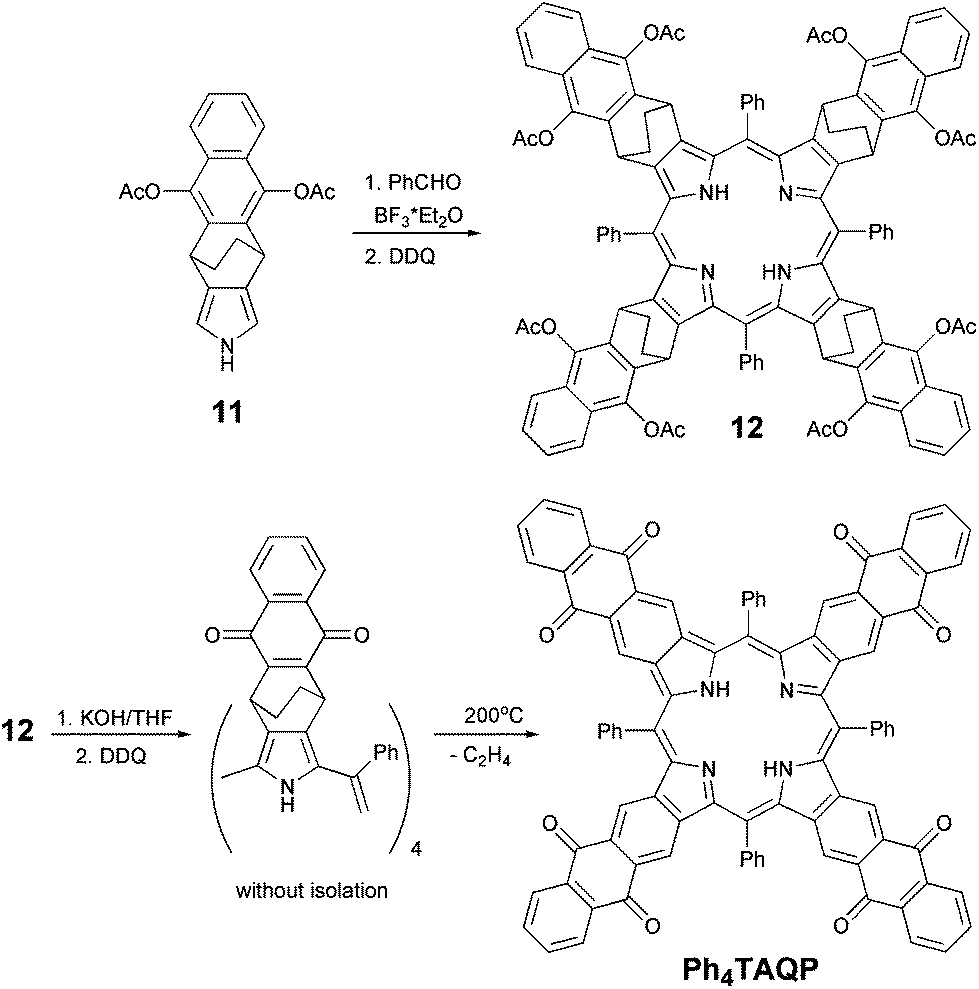 | ||
| Scheme 3 Synthesis of Ph4TAQP. | ||
The aromatization was clearly observed by the disappearance of methylene groups and the appearance of a new singlet peak in the aromatic region corresponding to eight protons on the anthraquinone rings in the 1H NMR spectrum. It is noteworthy that well-resolved 1H and 13C NMR spectra were obtained after addition of trifluoroacetic acid (TFA) which converted the porphyrin into a dication form. MALDI-TOF mass spectra gave the additional evidence for the formation of Ph4TAQP (ESI†).
The absorption and emission spectra of porphyrins 12, Ph4TAQP and its metal complexes are compared in Fig. 1. Electronic absorption spectra of 12 are similar to other tetratetraphenyl-β-octaalkylporphyrins, such as the derivatives of octaethylporphyrin (OEP) showing a Soret band at 434 nm and Q-bands at 523, 607, 675 nm in CH2Cl2 (for comparison, tetraphenyltetracyclohexenoporphyrin free base: Soret band 439 nm, Q-bands 537, 580, 606, 674 nm).20 The fluorescence spectrum of 12 is also consistent with this type of porphyrin skeleton, showing a maximum at 718 nm and a low quantum yield of emission (φfl < 0.01 in toluene, λexc = 638 nm).
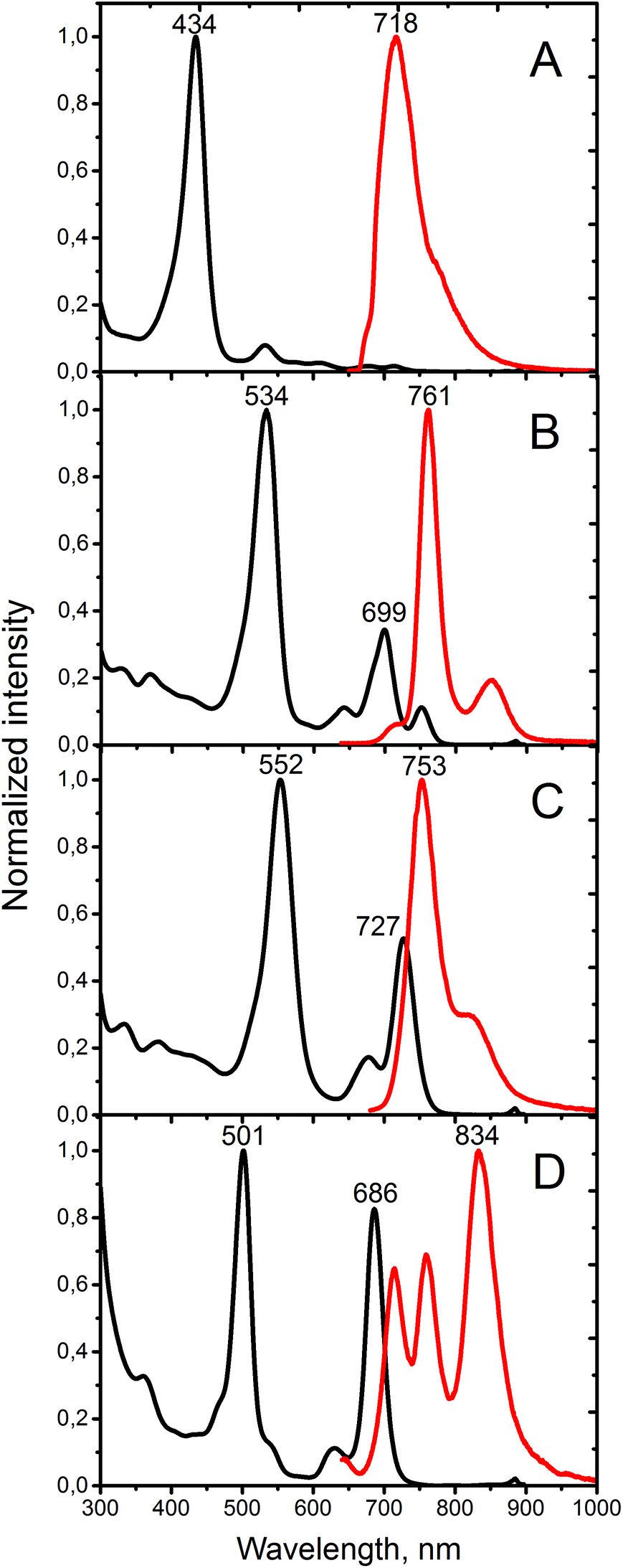 | ||
| Fig. 1 Absorption (black) and emission (red lines) of (A) porphyrin 12, (B) Ph4TAQP free base, (C) Ph4TAQP-Zn, and (D) Ph4TAQP-Pd. Solvent: toluene. | ||
Ph4TAQP exhibits strongly red-shifted Soret and Q-bands (Fig. 1B). The vibronic structure in the Q-band region is well-resolved. The lowest energy Q-band (752 nm) is red-shifted by 77 nm relative to the corresponding transition of the porphyrin 12 due to the effect of extended π-conjugation. At the same time, intensification of Q-bands is taking place – the maximum absorption ratio of the Q-band to the Soret band is enhanced from 0.09 (in 12) to 0.35. The free-base shows much stronger emission (φfl = 0.08) than the parent compound 12, with a small Stokes shift (9 nm). Metal insertion has a profound effect on optical properties. The absorption spectra of Zn and Pd-complexes are shown in Fig. 1C and D. Very strong blue-shift by 66 nm upon palladium insertion and 25 nm upon zinc insertion are observed for the lowest energy Q-band. Both complexes show relatively strong emission (φem = 0.11 and 0.06 for Zn and Pd-complexes respectively). The emission of Ph4TAQP shows multiple maxima that may be associated either with excimer formation or formation of charge-transfer excited states. Solutions of Ph4TAQP and its metal complexes do not decompose noticeably when exposed to daylight for several days, indicating good photostability compared to other π-extended porphyrins.23
Comparison of the absorption spectra of Ph4TAQPPd with those of palladium(II) tetraphenyltetrabenzo- and tetraphenyltetranaphthoporphyrins (Ph4TBPPd and Ph4TNPPd respectively, Fig. 2) demonstrates the effect of anthraquinone fusion on the porphyrin core with respect to annelation of extra benzo-rings. The strong effect on the energies of S1 and S2 states of the molecule is manifested by the pronounced red shift of the Soret and Q-bands. While in the case of Ph4TBPPd and Ph4TNPPd the Soret band is shifted only by 20–30 nm with respect to parent palladium(II) tetraphenylporphyrin, fusion of anthracenes causes 100 nm red shift. Nevertheless, a “spectral window” between the Soret and Q-bands allows for the application of Ph4TAQPPd as a sensitizer for the TTA-UC process that will be reported in a separate study.
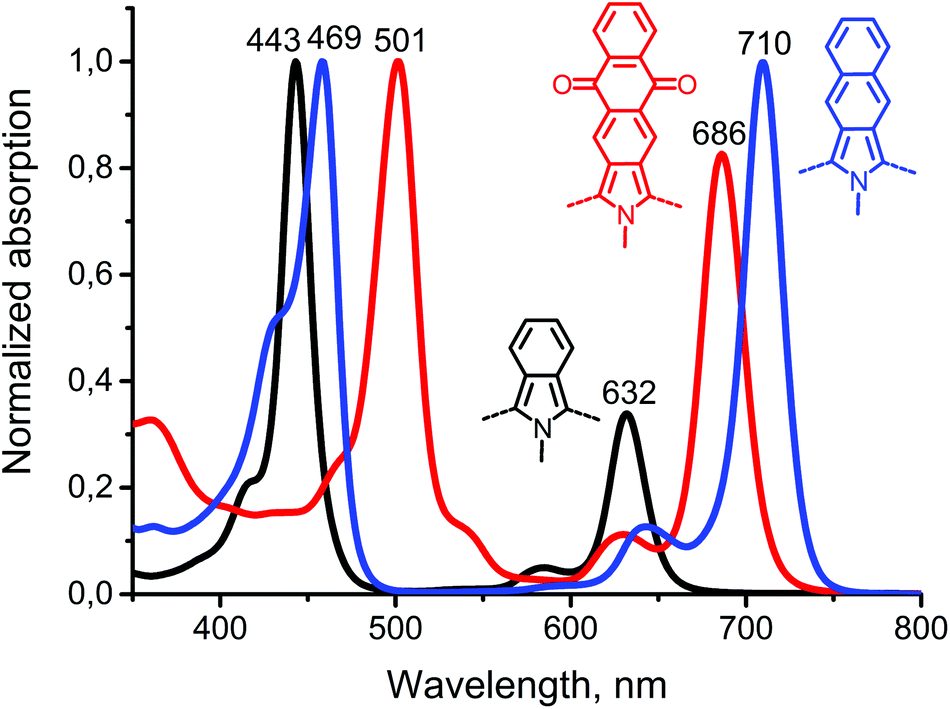 | ||
| Fig. 2 UV-Vis absorption spectra of Ph4TBPPd (black), Ph4TAQPPd (red) and Ph4TNPPd (blue). | ||
Conclusions
Two approaches towards the synthesis of TAQP were explored: the one based on the hydroisoindole precursor and bridged dihydroisoindole. The latter was found to be suitable for the synthesis of a target compound using the Barton–Zard reaction. The strategy based on oxidative aromatization of the dihydroisoindole precursor failed to deliver the target compound due to side reactions in the course of pyrrole synthesis. The optical properties of Ph4TAQP indicate electronic features that call for theoretical studies, as well as for better characterization using photophysical and electrochemical experiments. Indeed, new quinonoporphyrins are expected to exhibit interesting electrochemical properties as a result of the directly conjugated porphyrin and quinone moieties. Such materials appear to be of interest in photon energy conversion systems and in other applications. We relay a detailed discussion of the photophysical properties of variously substituted TAQP for a separate study.Experimental
1,4,4a,9a-Tetrahydroanthraquinone7 and tert-butyl isocyanoacetate24 were prepared according to published synthetic protocols. DBU, thiophenol, bis(benzonitrile)palladium(II) chloride, DDQ, N-chlorosuccinimide, Oxone, 1,4-naphthoquinone, trifluoroacetic acid, benzaldehyde, boron trifluoride etherate and extra dry THF were purchased from Sigma-Aldrich. The handling of all air/water sensitive materials was carried out using standard high vacuum techniques. All solvents and reagents were obtained from commercial sources and used as received. Where mixtures of solvents were used, ratios are reported by volume. Column chromatography was carried out on silica gel 60 at normal pressure. NMR spectra were recorded on Bruker DPX 250, Bruker AC300 NMR and Bruker Avance 500 spectrometers, with the solvent proton or carbon signal as an internal standard. Elemental analysis was carried out using a Foss Heraeus Vario EL. Electronic absorption spectra were recorded on a Perkin Elmer Lambda 25 instrument. MALDI-TOF spectra were recorded on a Bruker Reflex spectrometer III instrument using dithranol as a matrix. Melting points were determined on a Büchi hot stage apparatus and are uncorrected. Emission spectra were recorded using a Fluoromax-2 instrument. Emission quantum yields of the compounds were measured relative to the fluorescence of free-base tetraphenylporphyrin (φfl = 0.11)25 in deoxygenated toluene.2-Benzenesulfonyl-3-chloro-1,2,3,4-tetrahydro-anthra-quinone 2
1H NMR δH (300 MHz, CD2Cl2) 8.06 (2H, m), 7.97 (2H, m), 7.79–7.59 (5H, m), 5.03 (1H, q, J = 3.3 Hz), 4.05 (1H, m), 3.42–2.92 (4H, m). 13C NMR δC (75 MHz, CD2Cl2) 184.04, 140.57, 140.45, 139.49, 135.24, 134.59, 134.56, 133.21, 130.52, 129.97, 127.01, 126.94, 62.80, 51.53, 30.72, 20.30. Anal. calcd for C20H17ClO4S: C, 61.77; H, 4.41; found: C, 61.23; H, 4.65.2-Benzenesulfonyl-anthraquinone 3
1H NMR δH (300 MHz, CD2Cl2) 8.76 (1H, t, J = 1.2 Hz), 8.4 (2H, t, J = 1.1 Hz), 8.28 (2H, m), 8.09–8.03 (2H, m), 7.90–7.82 (2H, m), 7.64–7.53 (2H, m). 13C NMR δC (75 MHz, CD2Cl2) 182.06, 148.44, 142.38, 137.36, 135.57, 135.41, 135.37, 134.94, 134.73, 134.61, 134.57, 133.18, 130.62, 129.26, 129.16, 128.02, 127.05. Anal. calcd for C20H12O4S: C, 68.95; H, 3.47; found: C, 68.32; H, 3.72.9,10-Diacetoxy-1,4-dihydro-anthracene 4
The title compound was prepared following a modified literature procedure.26 1,8-Diazabicycloundec-7-ene (10.5 mL, 70 mmol) was added to a stirred solution of 1,4,4a,9a-tetrahydroanthraquinone (6.36 g, 30 mmol) and THF (100 mL) at room temperature. The mixture was cooled in an ice bath and acetic anhydride (8.5 mL, 90 mmol) was added dropwise over a period of 10 min and the resulting solution was stirred for 2 hours. Then diethyl ether (100 mL) was added to precipitate the product. The solid so formed was filtered and washed with ether (50 mL) to give 8.44 g (95%) of the product as a white powder (m.p. 255–257 °C, lit. 256–258 °C).261H NMR δH (300 MHz, CD2Cl2) 7.75 (2H, m), 7.51 (2H, m), 5.95 (2H, m), 3.37 (4H, br. s), 2.49 (6H, s). 13C NMR δC (75 MHz, CD2Cl2) 169.57, 142.2, 126.97, 126.52, 125.59, 123.54, 121.62, 25.0, 20.96.9,10-Diacetoxy-2-benzenesulfonyl-1,2-dihydro-anthracene 5
The title compound was prepared following a modified literature procedure.27 Thiophenol (2 mL, 2.2 g, 20 mmol) was added dropwise to a suspension of N-chlorosuccinimide (2.67 g, 20 mmol) in CH2Cl2 (20 mL) under cooling in an ice bath. The mixture was stirred for 1 h at r.t. and the resulting orange solution was added dropwise to a stirred solution of 9,10-diacetoxy-1,4-dihydro-anthracene (5.92 g, 20 mmol) in CH2Cl2 (150 mL) at 0 °C. The mixture was stirred at room temperature for 2 h and then evaporated in a vacuum. The residue was dissolved in methanol (60 mL) and a suspension of Oxone (12.3 g, 20 mmol) in water (30 mL) was added under vigorous stirring. The mixture was stirred at room temperature for 2 days, diluted with water (100 mL) and extracted with CH2Cl2. The combined organic layers were dried with Na2SO4 and evaporated to dryness. The resulting solid was dissolved in CH2Cl2 (50 mL), and DBU (3 mL, 20 mmol) was added dropwise over a period of 10 min at 0 °C. The mixture was stirred for 1 h at room temperature, washed with water, dried with Na2SO4 and evaporated in a vacuum. The solid residue was recrystallized from MeOH to give 6.1 g (70%) of the title compound as a white powder (m.p. 155–157 °C). 1H NMR δH (300 MHz, CD2Cl2) 7.69 (4H, m), 7.51 (2H, m), 7.3 (3H, d, J = 6.9 Hz), 6.84 (1H, dd, J = 9.9 Hz), 6.18 (1H, dd, J = 9.9 Hz), 4.09 (1H, m), 3.48 (1H, m), 3.11 (1H, m), 2.49 (3H, s), 2.46 (3H, s). 13C NMR δC (75 MHz, CD2Cl2) 169.17, 136.76, 134.33, 129.88, 129.11, 128.06, 128.0, 127.54, 127.43, 127.06, 122.41, 122.09, 121.85, 121.72, 121.51, 23.1, 20.93. Anal. calcd for C24H20O6S: C, 66.04; H, 4.62; found: C, 66.32; H, 4.85.1,2,3,4,4a,9a-Hexahydro-1,4-etheno-anthraquinone 7
A mixture of 1,4-naphthoquinone (10 g, 63 mmol), 1,3-cyclohexadiene (9.5 mL, 100 mmol) and 2,6-di-tert-butylphenol (0.05 g, 0.24 mmol) was dissolved in CHCl3 and refluxed for 24 h under argon. The resulting mixture was evaporated in a vacuum and the residue was recrystallized from EtOH to give 12.7 g (85%) of the title compound as a white powder (m.p. 83–85 °C). 1H NMR δH (300 MHz, CD2Cl2) 7.98 (2H, m), 7.69 (2H, m), 6.13 (2H, m), 3.3 (2H, m), 3.21 (2H, t, J = 1.3 Hz), 1.78 (2H, m), 1.38 (2H, m). 13C NMR δC (75 MHz, CD2Cl2) 198.12, 181.74, 151.07, 136.28, 134.47, 134.33, 134.24, 133.83, 133.0, 127.14, 126.59, 50.99, 36.26, 34.72, 25.44, 25.11. Anal. calcd C, 80.65; H, 5.92; O, 13.43; found: C, 80.12; H, 6.04.9,10-Acetoxy-1,2,3,4-tetrahydro-1,4-etheno-anthracene 8
The title compound was obtained according to the procedure described for 4. Yield: 90%. White powder with m.p. 232–233 °C. 1H NMR δH (300 MHz, CD2Cl2) 7.8 (2H, m), 7.5 (2H, m), 6.55 (2H, m), 4.08 (2H, m), 2.52 (2H, m), 1.58 (4H, s). 13C NMR δC (75 MHz, CD2Cl2) 170.01, 137.63, 135.28, 134.36, 126.61, 126.37, 121.80, 34.95, 24.94, 21.03. Anal. calcd for C20H18O4: C, 74.52; H, 5.63; found: C, 74.87; H, 5.85.9,10-Diacetoxy-12-benzenesulfonyl-1,2,3,4-tetrahydro-1,4-etheno-anthracene 9
The title compound was obtained according to the procedure described for 5. Yield: 65%. White powder with m.p. 213–214 °C. 1H NMR δH (300 MHz, CD2Cl2) 7.81 (3H, m), 7.69 (1H, m), 7.62 (1H, m), 7.51 (5H, m), 4.34 (2H, m), 2.52 (3H, s), 2.42 (3H, s), 1.65 (4H, m). 13C NMR δC (75 MHz, CD2Cl2) 169.80, 169.50, 147.74, 144.23, 139.86, 138.46, 138.14, 134.08, 131.21, 131.18, 129.84, 128.32, 127.23, 127.15, 126.75, 126.56, 122.06, 121.93, 36.5, 35.59, 25.60, 24.81, 21.0, 20.86. Anal. calcd for C26H22O6S: C, 67.52; H, 4.79; found: C, 67.89; H, 5.04.5,10-Diacetoxy-4,11-etheno-2H-naphtho[2,3-f]isoindole-1-carboxylic acid tert-butyl ester 10
The title compound was obtained according to a previously published general procedure.24 Yield: 78%. White powder with m.p. 186–187 °C. 1H NMR δH (300 MHz, CD2Cl2) 8.62 (1H, br. s), 7.78 (2H, m), 7.5 (2H, m), 6.7 (1H, d, J = 2.7 Hz), 4.93 (1H, m), 4.42 (1H, m), 2.54 (3H, s), 2.53 (3H, s), 1.77 (4H, m), 1.61 (9H, s). 13C NMR δC (75 MHz, CD2Cl2) 169.93, 169.87, 161.40, 138.45, 137.96, 134.75, 134.21, 132.88, 129.02, 126.83, 126.56, 126.55, 121.86, 117.12, 114.22, 81.06, 32.54, 32.29, 28.84, 28.51, 27.20, 26.59, 21.14, 21.06. Anal. calcd for C27H27NO6: C, 70.27; H, 5.90; N, 3.03; found: C, 69.89; H, 6.14; N, 2.87.5,10-Diacetoxy-4,11-etheno-2H-naphtho[2,3-f]isoindole 11
Compound 10 (1 g, 2.2 mmol) was dissolved in TFA (30 mL), and the solution was stirred for 30 min under Ar at room temperature. After the addition of CH2Cl2 (50 mL), the mixture was washed with water, then with 10% solution of Na2CO3, dried with Na2SO4 and evaporated in a vacuum. The residue was passed through a layer of silica using CH2Cl2 as the eluent. The solvent was evaporated to give 0.53 g (68%) of the title compound as a gray solid (m.p. 130–132 °C). 1H NMR δH (300 MHz, CD2Cl2) 7.76 (2H, m), 7.47 (2H, m), 6.58 (2H, d, J = 2.4 Hz), 4.41 (2H, t, J = 1.3 Hz), 2.53 (6H, s), 1.75 (4H, m). 13C NMR δC (75 MHz, CD2Cl2) 169.96, 137.94, 135.36, 126.91, 126.64, 126.53, 121.82, 109.94, 32.28, 31.05, 27.65, 21.07. Anal. calcd for C22H19NO4: C, 73.12; H, 5.30; N, 3.88; found: C, 72.65; H, 5.14; N, 3.47.Porphyrin 12
5,10-Diacetoxy-4,11-etheno-2H-naphtho[2,3-f]isoindole (0.3 g, 0.83 mmol) was dissolved in CH2Cl2 (83 mL) freshly distilled from CaH2, and benzaldehyde (0.088 g, 0.83 mmol) was added. The mixture was stirred under nitrogen for 10 min in the dark at room temperature. BF3·Et2O (10 μL, 0.083 mmol) was added in one portion, and the mixture was stirred for an additional 2 h. DDQ (0.141 g, 0.62 mmol) was added followed by additional stirring for 2 h. The resulting mixture was washed with aqueous Na2SO3, dried over Na2SO4 and concentrated in a vacuum. The residue was purified on a silica gel column (eluent CH2Cl2, then CH2Cl2–HOAc, green band collected). Additional purification by recrystallization from CH2Cl2–Et2O delivered the title product (67 mg, 18%) as a dark-green powder. 1H NMR δH (300 MHz, CD2Cl2–TFA) 8.96–6.85 (36H, m), 4.74–4.17 (8H, m), 3.22–2.74 (24H, m), 2.15–1.84 (16H, m). 13C NMR δC (75 MHz, CD2Cl2–TFA) 170.26, 137.89, 135.43, 134.96, 132.05, 127.12, 126.98, 126.66, 126.27, 121.71, 120.45, 110.42, 32.59, 31.16, 27.34, 21.45. UV/vis (CH2Cl2) λmax (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε): 434 (5.2), 523 (4.21), 607 (3.93), 675 (3.84). MALDI-TOF: m/z found 1791.61, calcd for [M+] C116H86N4O16 1791.60. Anal. calcd for C92H46N4O8: C, 77.75; H, 4.84, N, 3.13; found: C, 78.58; H, 5.36; N, 3.41.
ε): 434 (5.2), 523 (4.21), 607 (3.93), 675 (3.84). MALDI-TOF: m/z found 1791.61, calcd for [M+] C116H86N4O16 1791.60. Anal. calcd for C92H46N4O8: C, 77.75; H, 4.84, N, 3.13; found: C, 78.58; H, 5.36; N, 3.41.
Ph4TAQP free base
Porphyrin 12 (50 mg) was dissolved in THF (10 mL) and a solution of KOH (0.25 g) in EtOH (5 mL) was added. The mixture was stirred at room temperature for 12 h, then concentrated HCl (1 ml) was added and the solution was evaporated in a vacuum. The residue was washed several times with CH2Cl2 to separate soluble porphyrin from the inorganic solid, the resulting solution was dried with Na2SO4 and filtered. DDQ (0.188 g, 0.83 mmol) was then added and the mixture was stirred for 6 h. The resulting mixture was washed with aqueous Na2SO3, dried over Na2SO4 and concentrated in a vacuum. The residual solid was heated in a vacuum oven at 200 °C for 4 h. Then it was dissolved in CH2Cl2 and purified on a silica gel column (eluent CH2Cl2, then CH2Cl2–THF, purple band collected). Additional purification by repetitive precipitation from CH2Cl2–Et2O delivered the title product (24 mg, 65%) as a purple powder. 1H NMR δH (500 MHz, C2D2Cl4–TFA) 8.69 (8H, m), 8.44 (8H, s), 8.38 (4H, m), 8.29–8.17 (16H, m), 7.83 (8H, m), 4.05 (4H, br. s). 13C NMR δC (125 MHz, C2D2Cl4–TFA) 181.73, 142.53, 138.04, 135.86, 135.52, 134.56, 133.53, 133.12, 133.11, 130.39, 127.33, 124.70, 117.37. UV/vis (CH2Cl2) λmax (logε): 534 (5.18), 642 (4.22), 699 (4.72), 752 (4.22). MALDI-TOF: m/z found 1355.33, calcd for [M+] C92H46N4O8 1355.33. Anal. calcd for C92H46N4O8: C, 82.75; H, 3.47, N, 4.20; found: C, 83.57; H, 3.98; N, 4.74.
Ph
4
TAQP-Pd was obtained in 75% yield after heating a mixture of the free-base porphyrin, excess PdCl2(PhCN)2 (2 eq.) and Et3N (10 eq.) in benzonitrile at 160 °C for 0.5–3 h (control by UV-Vis spectroscopy), with subsequent filtration through a layer of silica (eluent CH2Cl2) and evaporation of the filtrate. UV/vis (CH2Cl2) λmax (logε): 501 (5.05), 629 (4.11), 686 (4.97). MALDI-TOF: m/z found 1439.2361, calcd for [M+] C92H44N4O8Pd 1439.22.
Ph
4
TAQP-Zn was obtained in 90% yield after the treatment of a free-base in THF with an excess of Zn(OAc)2·2H2O, followed by subsequent precipitation with MeOH, filtration and drying in a vacuum. UV/vis (CH2Cl2) λmax (logε): 552 (5.12), 677 (4.35), 727 (4.84). MALDI-TOF: m/z found 1397.24, calcd for [M+] C92H44N4O8Zn 1397.24.
Acknowledgements
M.A.F. acknowledges POLINNOVA project (FP7-REGPOT-2012-2013-1) for the financial support. Financial support from the Bulgarian Science Fund (DFNI E 02/11 – SunStore-project) and the European Commission FCFP FRIAS COFUND Fellowship Programme (FP7-MCA-609305) for S.B. are highly appreciated.Notes and references
- (a) S. M. Borisov, G. Nuss, W. Haas, R. Saf, M. Schmuck and I. Kilmant, J. Photochem. Photobiol., A, 2009, 201, 128 CrossRef CAS; (b) C. Wohnhaas, V. Mailänder, M. Dröge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422 CrossRef CAS PubMed; (c) R. Kumar, T. Y. Ohulchanskyy, I. Roy, S. K. Gupta, C. Borek, M. E. Thompson and P. N. Prasad, ACS Appl. Mater. Interfaces, 2009, 1, 1474 CrossRef CAS PubMed; (d) M. D. Perez, C. Borek, P. I. Djurovich, E. I. Mayo, R. R. Lunt, S. R. Forrest and M. E. Thompson, Adv. Mater., 2009, 21, 1517 CrossRef CAS; (e) T. N. Singh-Rachford, A. Haefele, R. Ziessel and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164 CrossRef CAS PubMed; (f) L. H. Hutter, B. J. Müller, K. Koren, S. M. Borisov and I. Klimant, J. Mater. Chem. C, 2014, 2, 7589 RSC; (g) S. M. Borisov, C. Larndorfer and I. Klimant, Adv. Funct. Mater., 2012, 22, 4360 CrossRef CAS; (h) T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83, 8756 CrossRef CAS PubMed; (i) B. J. Muller, T. Burger, S. M. Borisov and I. Klimant, Sens. Actuators, B, 2015, 216, 527 CrossRef.
- (a) S. Baluschev, T. Miteva, V. Yakutkin, Y. S. Avlasevich, K. Müllen, G. Nelles, S. Chernov, S. Aleshchenkov, A. Cheprakov, A. Yasuda and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693 CrossRef CAS PubMed; (b) C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446 CrossRef CAS; (c) A. J. Svagan, D. Busko, Y. Avlasevich, G. Glasser, S. Baluschev and K. Landfester, ACS Nano, 2014, 8, 8198 CrossRef CAS PubMed.
- (a) O. S. Finikova, S. Y. Chernov, A. V. Cheprakov, M. A. Filatov, S. A. Vinogradov and I. P. Beletskaya, Dokl. Chem., 2003, 391, 222 CrossRef CAS; (b) M. A. Filatov, A. V. Cheprakov and I. P. Beletskaya, Eur. J. Org. Chem., 2007, 3468 CrossRef CAS; (c) M. A. Filatov, A. Y. Lebedev, S. A. Vinogradov and A. V. Cheprakov, J. Org. Chem., 2008, 73, 4175 CrossRef CAS PubMed.
- (a) O. S. Finikova, S. E. Aleshchenkov, R. P. Brinas, A. V. Cheprakov, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2005, 70, 4617 CrossRef CAS PubMed; (b) M. A. Filatov and A. V. Cheprakov, Tetrahedron, 2011, 67, 3559 CrossRef CAS; (c) E. R. Ranyuk, M. A. Filatov, A. D. Averin, A. V. Cheprakov and I. P. Beletskaya, Synthesis, 2012, 393 CAS; (d) J. M. Manley, T. J. Roper and T. D. Lash, J. Org. Chem., 2005, 70, 874 CrossRef CAS PubMed; (e) X.-Z. Jiang, C.-X. Cai, J.-T. Liu and H. Uno, Org. Biomol. Chem., 2012, 10, 3110 RSC.
- V. Gandhi, M. L. Thompson and T. D. Lash, Tetrahedron, 2010, 66, 1787 CrossRef CAS.
- K. Kurotobi, K. S. Kim, S. B. Noh, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 3944 CrossRef CAS PubMed.
- (a) M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva, K. Landfester, S. E. Aleshchenkov and A. V. Cheprakov, J. Org. Chem., 2012, 77, 11119 CrossRef CAS PubMed; (b) H. Yamada, D. Kuzuhara, T. Takahashi, Y. Shirnizu, K. Uota, T. Okujima, H. Uno and N. Ono, Org. Lett., 2008, 10, 2947 CrossRef CAS PubMed.
- H. Boedigheimer, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2010, 75, 2518 CrossRef CAS PubMed.
- (a) A. V. Cheprakov, in The Synthesis of π-Extended Porphyrins. Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2011, vol. 13 Search PubMed; (b) N. Ono, H. Yamada and T. Okujima, in Synthesis of Porphyrins Fused with Aromatic Rings. Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2011, vol. 2 Search PubMed.
- S. Banala, T. Ruhl, K. Wurst and B. Krautler, Angew. Chem., Int. Ed., 2009, 48, 599 CrossRef CAS PubMed.
- D. Qi and J. Jiang, Int. J. Quantum Chem., 2013, 113, 2605 CrossRef CAS.
- H. Tributsch, Electrochim. Acta, 2007, 2302 CrossRef CAS.
- (a) S. Banala, K. Wurst and B. Krautler, J. Porphyrins Phthalocyanines, 2014, 18, 115 CrossRef CAS; (b) S. Banala, R. G. Huber, T. Müller, M. Fechtel, K. R. Liedl and B. Kräutler, Chem. Commun., 2012, 48, 4359 RSC; (c) B. Kräutler, C. S. Sheehan and A. Rieder, Helv. Chim. Acta, 2000, 83, 583 CrossRef.
- (a) R. Bonnet and R. F. C. Brown, J. Chem. Soc., Chem. Commun., 1972, 393 RSC; (b) J. Bornstein, J. E. Shields and D. E. Remy, J. Chem. Soc., Chem. Commun., 1972, 1149 RSC.
- A. V. Cheprakov and M. A. Filatov, J. Porphyrins Phthalocyanines, 2009, 13, 291 CrossRef CAS.
- (a) S. Ito, N. Ochi, T. Murashima, N. Ono and H. Uno, Chem. Commun., 2000, 893 RSC; (b) T. Okujima, T. Kikkawa, H. Nakano, H. Kubota, N. Fukugami, N. Ono, H. Yamada and H. Uno, Chem. – Eur. J., 2012, 18, 12854 CrossRef CAS PubMed; (c) Y. Tomimori, T. Okujima, T. Yano, S. Mori, N. Ono, H. Yamada and H. Uno, Tetrahedron, 2011, 67, 3187 CrossRef CAS; (d) H. Uno, H. Uoyama, C. Chenxin, H. Tahara, Y. Shimizu, H. Hagiwara, Y. Hanasaki, H. Yamada and T. Okujima, Heterocycles, 2010, 80 Search PubMed; (e) H. Uoyama, T. Takiue, K. Tominaga, N. Ono and H. Uno, J. Porphyrins Phthalocyanines, 2009, 13, 122 CrossRef CAS.
- (a) D. P. Arnold, L. Burgess-Dean, J. Hubbard and M. Abdur Rahman, Aust. J. Chem., 1994, 969 CrossRef CAS; (b) Y. Abel, E. Haake, G. Haake, W. Schmidt, D. Struve, A. Walter and F. P. Montforts, Helv. Chim. Acta, 1998, 81, 1978 CrossRef CAS.
- B. P. Bandgar, L. S. Uppalla, A. D. Sagar and V. S. Sadavarte, Tetrahedron Lett., 2001, 42, 1163 CrossRef CAS.
- J. J. Stromich, A. K. Weber, Y. R. Mirzaei, M. D. Caldwell and D. E. Lewis, Bioorg. Med. Chem. Lett., 2010, 20, 1928 CrossRef CAS PubMed.
- O. S. Finikova, A. V. Cheprakov, I. P. Beletskaya, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2004, 69, 522 CrossRef CAS PubMed.
- H. Pines and W. M. Stalick, Base-catalyzed reactions of hydrocarbons and related compounds, Academic Press, New York, 1977, p. 483 Search PubMed.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearny and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827 CrossRef CAS.
- M. A. Filatov, E. Heinrich, D. Busko, I. Z. Ilieva, K. Landfester and S. Baluschev, Phys. Chem. Chem. Phys., 2015, 17, 6501 RSC.
- B. H. Novak and T. D. Lash, J. Org. Chem., 1998, 63, 3998 CrossRef CAS.
- P. G. Seybold and M. Gouterman, J. Mol. Spectrosc., 1969, 31, 1 CrossRef CAS.
- S. R. Angle and W. Yang, J. Am. Chem. Soc., 1990, 112, 4524 CrossRef CAS.
- P. B. Hopkins and P. L. Fuchs, J. Org. Chem., 1978, 43, 1208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and mass-spectroscopy data. See DOI: 10.1039/c5ob00884k |
| This journal is © The Royal Society of Chemistry 2015 |