
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation of asphaltene model compounds using a porphyrin tethered to a carboxylic acid†
Matthias
Schulze
a,
Marc P.
Lechner
a,
Jeffrey M.
Stryker
b and
Rik R.
Tykwinski
*a
aDepartment of Chemistry and Pharmacy & Interdisciplinary Center of Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Henkestraße 42, 91054 Erlangen, Germany. E-mail: rik.tykwinski@fau.de; Fax: +49-9131-85-26865; Tel: +49-9131-85-22540
bDepartment of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada
First published on 19th May 2015
Abstract
A Ni(II) porphyrin functionalized with an alkyl carboxylic acid (3) has been synthesized to model the chemical behavior of the heaviest portion of petroleum, the asphaltenes. Specifically, porphyrin 3 is used in spectroscopic studies to probe aggregation with a second asphaltene model compound containing basic nitrogen (4), designed to mimic asphaltene behavior. NMR spectroscopy documents self-association of the porphyrin and aggregation with the second model compound in solution, and a Job's plot suggests a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry for compounds 3 and 4.
:2 stoichiometry for compounds 3 and 4.
Introduction
The foreseeable exhaustion of conventional fossil fuels has resulted in an increasing focus on efficient use of alternative petroleum sources, such as heavy crude oil.1 In order to convert heavy oil to valuable fuels, however, significant upgrading is required,2 and these processes are hampered by the heaviest and most problematic component of heavy oil, the asphaltenes.3 Asphaltenes are commonly known to cause problems for a variety of practical processes, such as transportation, storage, and refining.2–4 To solve these issues, significant efforts have been expended to understand the bulk behavior of asphaltenes,2,4,5 and these efforts have even suggested that the carbon-rich and heteroatom doped composition of asphaltenes might yield materials for electronic devices.6,7 In spite of these studies, however, the general molecular structure of the asphaltenes remains to be resolved,8 and a major barrier to this goal is the formation of very stable aggregates between individual molecules that complicates many analysis techniques.9For almost a century, it has been suggested and, for the most part, accepted that asphaltenes are composed of large aromatic cores that also contain heteroaromatics and metalloporphyrins, with a molecular weight (MW) of ca. 750 g mol−1 (Fig. 1a). These large aromatic islands are then surrounded by functional groups, including alkyl groups and alkyl carboxylic acids.10–12 This paradigm gives rise to the “continental” or “island” model (more specifically the Yen–Mullins model), and it is believed that aggregation in these types of molecules is driven primarily by π–π stacking interactions.13
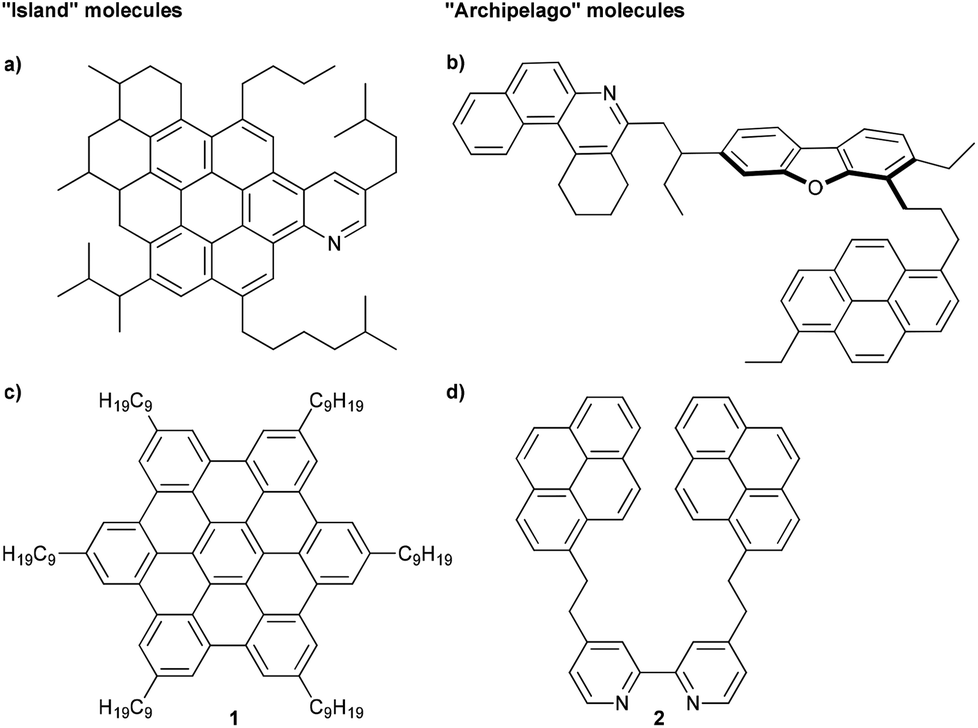 | ||
| Fig. 1 Schematic example of an (a) island asphaltene compound (MW = 756 g mol−1) from ref. 13 and (b) archipelago asphaltene compound (MW = 754 g mol−1) from ref. 8. Examples of model compounds used to study aggregation of asphaltenes, representing (c) island (MW = 1280 g mol−1)14 and (d) archipelago (MW = 613 g mol−1)15 structures. | ||
Alternatively, an archipelago model for the asphaltenes has been introduced, in which smaller aromatic islands are tethered together by alkyl chains (Fig. 1b).8 In the case of archipelago asphaltenes, it has been hypothesized that aggregation results from the cumulative effect of numerous intermolecular stabilizing forces, which might include, among others, π–π-stacking, axial coordination of metalloporphyrins, hydrogen bonding, hydrophobic interactions, and acid–base interactions.8
Much of the support for the island model comes from a “top-down” approach, i.e., the study of natural asphaltene samples by various physical and analytical methods.13 Evidence for the archipelago model using the “top-down” approach has also been reported, including ruthenium-ion-catalyzed oxidation (RICO) experiments,16–18 small-angle neutron scattering,19,20 calculations,21,22 and thermal cracking experiments.23,24 We hypothesize that a “bottom-up” approach, is paramount to understanding asphaltene aggregation on a molecular level. While the “top-down” approach looks at properties of the bulk material, a “bottom-up” study targets specific molecules with functionalities found in asphaltenes (Fig. 1c and d).25 With the exact structure of the molecules known, physical properties can be investigated at a molecular level. This tactic is quite common in supramolecular materials chemistry,26–28 for example, and should be useful toward deciphering specific intermolecular interactions related to the asphaltene aggregation, regardless of whether the continental or the archipelago motif is being considered. There are, however, surprisingly only a few efforts reported in the literature that describe the rational synthesis of compounds specifically to mimic the behavior of asphaltenes.15,29–31 Rather, existing molecules are often employed, presumably out of convenience, to emulate asphaltene molecules. For example, hexabenzocoronenes (HBCs) substituted with alkyl chains (1) have been used in studies by Gray and Müllen to represent molecules of the continental model, and such HBCs show dimer formation in organic solvents (Fig. 1c),8,14,32 while Anisimov examined the aggregation behavior of a tert-butyl substituted HBC.33 HBCs, however, have an aromatic core built from 13 fused rings, and the majority of asphaltene molecules are expected to have only 4 to 10 fused rings.34,35 Akbarzadeh et al. have used smaller ring systems (alkyl bridged pyrene model compounds) either with or without heteroatoms,29 and only structures with heteroatoms form dimers in organic solvents. In line with these results, Tan et al. have described that the 2,2′-bipyridine derivative 2 forms dimers both in solution and in the solid state (Fig. 1d),15 and it is suggested that water enhances dimer formation, presumably by the formation of hydrogen bonding, bridging the heteroatoms between two individual molecules.30 Experimental results were also supported by density functional theory and 3D-RISM-KH calculations.36,37
Studies to date with model asphaltene compounds, as briefly summarized in the preceding paragraphs, are not fully consistent with the view that asphaltene aggregation is dominated by π-stacking. Given the acknowledged structural diversity of the asphaltenes, it seems likely that intermolecular associations might be better described by multiple cooperative interactions facilitated by heteroatoms and functional groups.8 An obvious target under this premise is the interaction of an acid and a base, as expected in natural asphaltenes, for example, between pyridyl and carboxylic acid groups.12,38 To explore this concept, in the current study two model compounds have been chosen, an acid and a base (Fig. 2). A porphyrin group has been chosen as a platform for the acid moiety, namely compound 3. Porphyrins are known to exist in petroleum since the pioneering work by Treibs in 1934,39–42 and these so-called petroporphyrins are mainly observed as V(IV)O and Ni(II) species.12,43 To the best of our knowledge, however, only simple, unfunctionalized, and commercially available porphyrins have been employed to model asphaltene aggregation.44 Archipelago-type porphyrins have been used for the investigation of thermal cracking behavior,45 but there are no studies that report a synthetic strategy specifically designed for porphyrins that enable the study of asphaltene aggregation.26 It is worth noting the extensive synthetic porphyrins independently reported by Clezy and Lash to serve as model compounds for petroporphyrins, albeit without a focus on aggregation.46–48
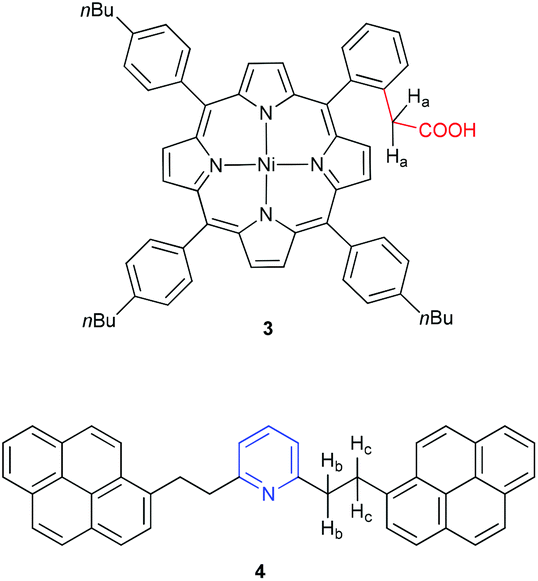 | ||
| Fig. 2 Model compounds under study to probe aggregation based on the interactions of an acid (3) and base (4).49 | ||
To explore the aggregation behavior of porphyrin 3, it is combined with a compound that features a basic nitrogen group. Namely, we have chosen archipelago model compound 4 (Fig. 2), in which the pyridyl-ring is combined with large aromatic groups appended via flexible alkyl tethers. Binding studies by NMR spectroscopy show self-association of porphyrin 3 and the formation of an aggregate between 3 and 4 in solution, demonstrating the potential of this aggregation motif for native asphaltenes.
Results and discussion
Synthesis
Aldehyde 5 (Scheme 1) was synthesized from 1-bromo-2-(methoxymethyl)benzene as described by Ullenius and coworkers.50 Subsequently, aldehyde 5, commercial 4-n-butylbenzaldehyde (6), and pyrrole were used in a mixed porphyrin condensation with BF3·OEt2 as a Lewis-acid and EtOH as a co-catalyst, followed by oxidation with DDQ, according to procedures reported by Lindsey51 and Jux.52 From this reaction, the unsymmetrical methoxymethyl substituted porphyrin 7a and the symmetrical derivative 853 were isolated in significant yields. The desired target, porphyrin 7a, was then reacted with HBr in AcOH, according to a procedure by Jux,52 to give bromide 7b, which was used without further purification. The route of Jasinski et al. for cyanation of bromomethyl TPPs was modified slightly,54 and treatment of the crude bromide 7b with KCN in PEG 400 gave the cyanomethyl substituted porphyrin 9 in excellent yield. Hydrolysis of the nitrile 9 under acidic conditions yielded the carboxylic acid, and the free base porphyrin 10 was isolated in good yield via column chromatography. Conversion of 10 to the Ni(II) porphyrin 3 was first attempted out using Ni(acac)2 in toluene. Although quantitative metallation was achieved, purification was difficult due to presence of Ni(acac)2, which led to an isolated yield for 3 of only 31%. Changing the metallation conditions from Ni(acac)2 in toluene to Ni(OAc)2·4H2O in MeOH/CHCl3, however, afforded the desired porphyrin 3 in 95% yield. Archipelago model compound 4 was synthesized via Sonogashira reaction of 1-ethynylpyrene with 2,6-dibromopyridine and subsequent hydrogenation, as reported previously.55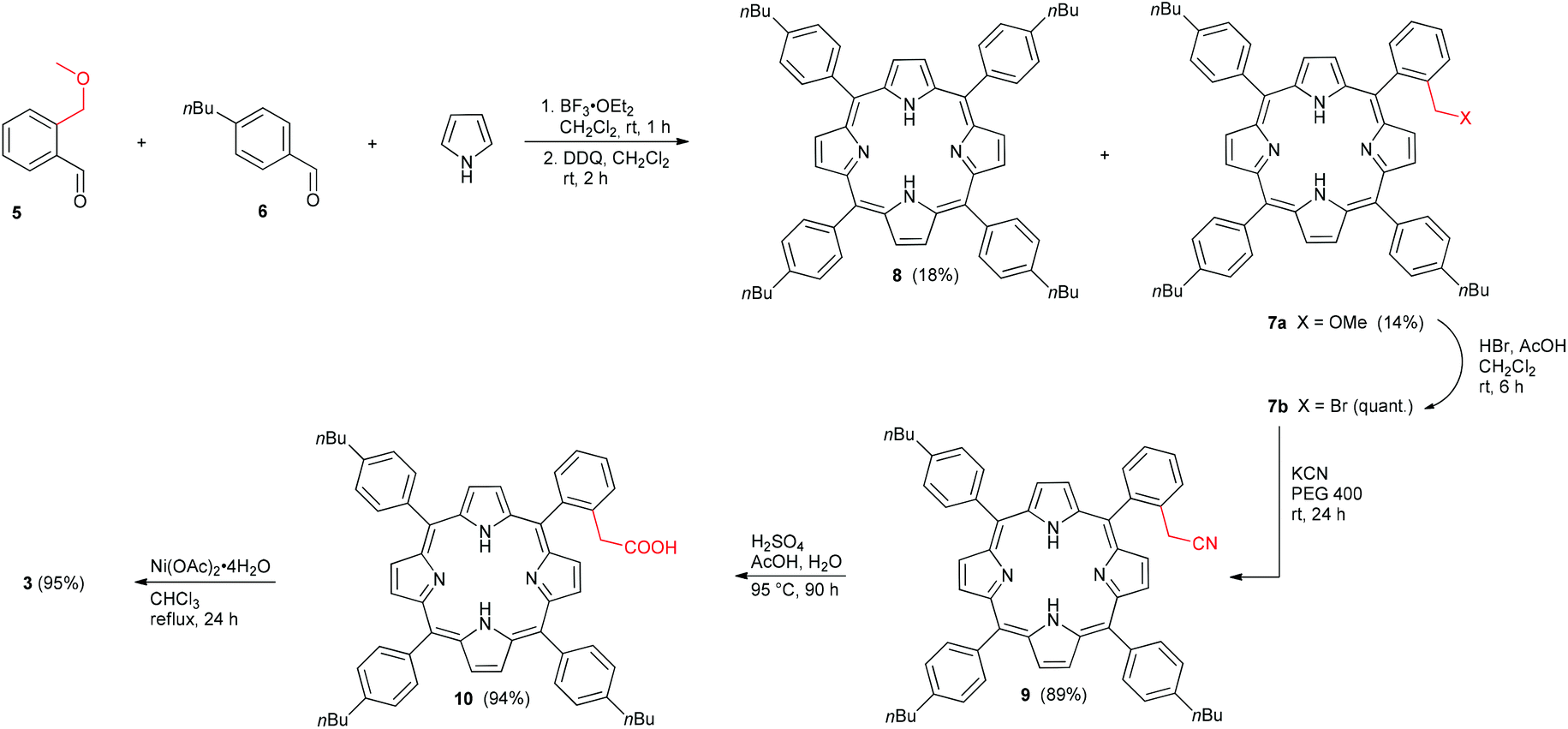 | ||
| Scheme 1 Synthesis of the Ni(II) porphyrin 3 featuring an alkyl carboxylic acid group. | ||
Aggregation in solution
Before exploring aggregation of porphyrin 3 with archipelago model compound 4, a series of control experiments were done. First, phenylacetic acid was used as a simple carboxylic acid in a NMR titration experiment with pyridine in benzene-d6 at 24 ± 2 °C (while toluene is more commonly used for aggregation studies, the symmetrical structure of benzene allows more facile analysis of 1H NMR spectra due to the reduced number of signals from the solvent). Specifically, addition of pyridine (1 to 8 equivalents) to a 2.5 mM solution of phenylacetic acid gave changes in the 1H NMR chemical shift for the signal of the methylene protons of phenylacetic acid (see ESI†). Nonlinear curve fitting for the chemical shift data with a 1:1 binding model gave an association constant Kassoc = 123 ± 12 M−1.
In a second experiment, phenylacetic acid was titrated with model compound 4,56 rather than pyridine. At first, a 2.5 mM solution of phenylacetic acid was used for the titration with 4. Limited by the solubility of 4, only up to 4 equivalents could be added. As only weak binding was observed, a second titration with a 1.25 mM solution of phenylacetic acid was performed allowing addition up to 8 equivalents of 4. Both titrations gave concentration dependent changes for the chemical shift for the signal of the methylene protons of phenylacetic acid, but the binding was quite lower compared to pyridine, with an averaged Kassoc = 24 ± 8 M−1 (see ESI†), obtained from the two titrations detailed above. In general, it is know that substitution in the 2,6-positions of the pyridyl-ring has an impact on basicity, e.g., for pyridine, 2,6-lutidine, and 2,6-diisopropylpyridine, pKa = 4.38, 5.77, and 5.34, respectively.57 Likewise, substitution also causes steric repulsion inhibiting hydrogen bonding.57 Thus, the observed decrease in Kassoc going from pyridine to model compound 4 is likely explained by a combination of steric and electronic effects, as described in detail by Kitao and Jarboe for simple pyridine derivatives.57
Turning attention to the porphyrin carboxylic acid 3, dilution titrations were first investigated by 1H NMR spectroscopy to determine self-association. It was clear from these titrations that porphyrin 3 forms a dimeric species in benzene-d6, and chemical shift analysis and fitting from multiple signals gave a dimerization constant of Kdim = 390 ± 27 M−1 (see ESI†).
Finally, a 1H NMR titration experiment was done with porphyrin 3 by addition of pyridine (1 to 8 equivalents). Fitting of the chemical shifts from multiple signals and considering Kdim = 390 ± 27 M−1 for 3, Kassoc = 178 ± 18 M−1 was determined (see ESI†), which was comparable to the binding experiment between phenylacetic acid and pyridine (vide supra).
The interactions between model compounds 3 and 4 were then explored, using an analogous set of conditions for both the titration and 1H NMR chemical shift analysis. Namely, the concentration of porphyrin 3 was kept constant at 2.5 mM, and the concentration of model compound 4 was varied from a minimum of 1.25 mM to a maximum of 10 mM (this was the solubility limit of 4). The signals of the two methylene groups of 4 (Hb and Hc, see Fig. 2) shifted upfield from 3.86/3.34 (Hc/Hb) to 3.78/3.29 (Hc/Hb) ppm upon addition of 0.5 equivalents 4 to the porphyrin solution (Fig. 3), whereas further addition of 4 resulted in less significant downfield shifts. The signal of the methylene group protons of 3 (Ha, see Fig. 2) shifted downfield upon addition of 4, from 3.20 ppm to a maximum of 3.43 ppm, after addition of four equivalents of 4.
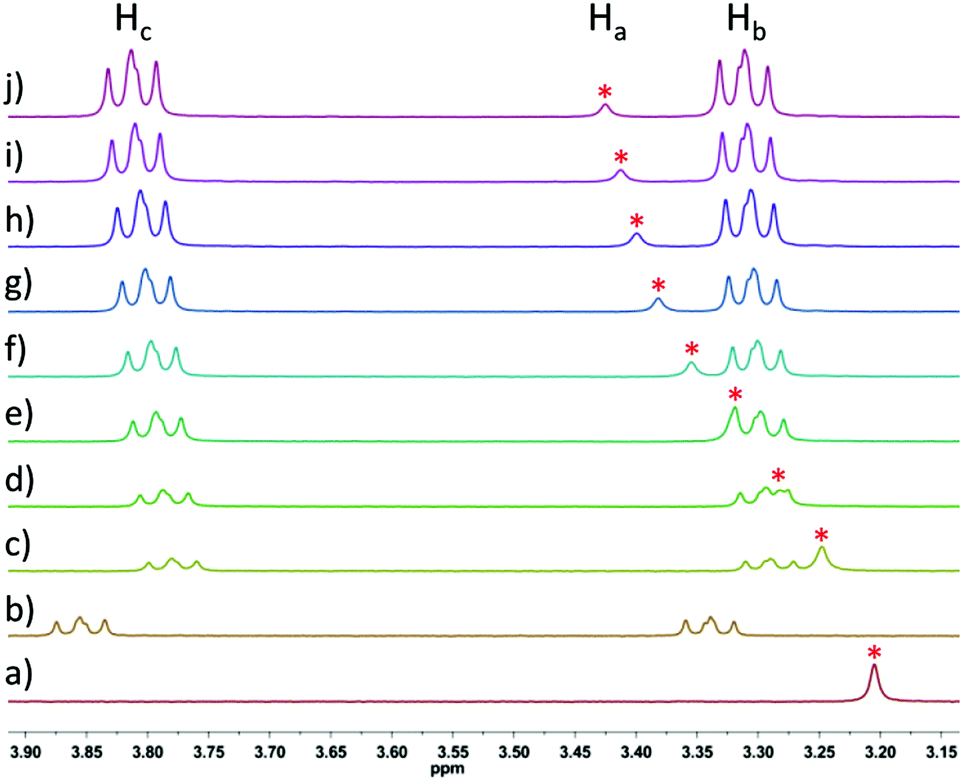 | ||
| Fig. 3
1H NMR titration of 3 (2.5 mM constant) with increasing amounts of 4 in benzene-d6; (a) 3 (2.5 mM); (b) 4 (1.25 mM); (c) 3:4 = 2:1; (d) 3:4 = 1:1; (e) 3:4 = 1:1.5; (f) 3:4 = 1:2; (g) 3:4 = 1:2.5; (h) 3:4 = 1:3; (i) 3:4 = 1:3.5; (j) 3:4 = 1:4; * = signal of the methylene group protons Ha of 3; see Fig. 2 for proton assignment. | ||
The observed relationship between chemical shift and concentration is consistent with expected acid–base interactions between the carboxylic acid functionality of 3 and the basic nitrogen of 4. The chemical shifts for multiple signals upon titration of 3 with 4 were fit to a 1:1 binding model, suggesting a Kassoc = 316 ± 32 M−1 (Fig. 4). The 1:1 binding model is also supported by the presence of a weak signal for a 1:1 complex between 3 and 4 in the high resolution APPI mass spectrum (see ESI†).
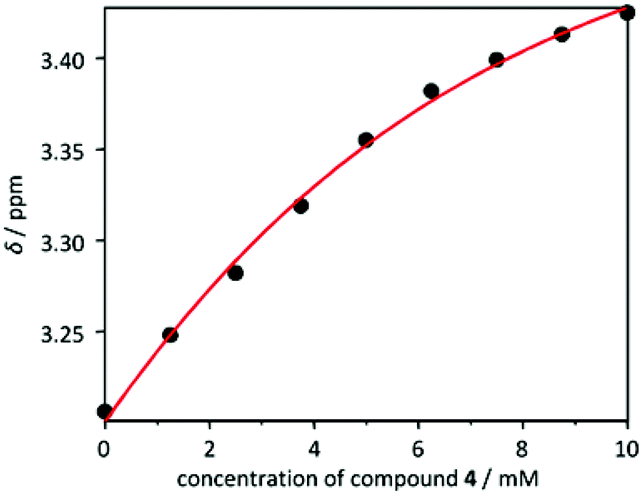 | ||
| Fig. 4 Concentration dependent chemical shifts for the signal of the methylene group protons of porphyrin 3 upon titration with 4 and fitting to a 1:1 binding model. | ||
To further investigate the nature of the complex formed between 3 and 4 in solution, Job's method of continuous variation was used.58,59 Interestingly, the maximum is observed in the Job's plot at a mole fraction of 0.3 for 3, suggesting a 1:2 complex between the porphyrin acid 3 and model compound 4, respectively. Nonlinear curve fitting, considering both binding events, namely self-association of 3 (Kdim = 390 ± 27 M−1) and a 1:2 binding model, results in an overall association constant of 1.23 ± 0.1 × 106 M−2.60
The formation of a 1:2 complex between model compounds 3 and 4 would be in line with the results of Tan et al., who report formation of a dimer for the ethano-linked pyrene substituted 2,2′-bipyridine derivative 2 (in CDCl3),15 which shares a structure similar to that of 4. It has also been shown that dimerization of 2 is enhanced by the addition of water, which had the effect of forming an intermolecular bridge between bipyridyl groups.30 It is, thus, reasonable that the carboxylic acid functionality of porphyrin 3 might mediate similar interactions between 4, by bridging between two pyridyl-units, as in the role of water for the dimerization of 2. The suggested stoichiometry from the Job's plot must be interpreted with caution, however, due to the self-association of porphyrin 3. Thus, it is not possible at this point to unambiguously declare the nature of the interaction between 3 and 4.
Conclusions
We have synthesized a new metalloporphyrin bearing an alkyl carboxylic acid (3), and specific aspects of asphaltene aggregation have been studied using this porphyrin. NMR spectroscopic analyses document the self-dimerization of the porphyrin carboxylic acid 3 in benzene-d6, as well as aggregation of 3 with an asphaltene model compound that bears a basic pyridine group (i.e., archipelago 4). A Job's plot analysis between 3 and 4 suggests a 1:2 complex, but the exact nature of the association cannot be categorically established, due mainly to the self-association (self-dimerization) of porphyrin acid 3.
While the stoichiometry of the 3/4 complex remains unresolved, this study does clearly show that the interactions between 3 and 4 are stronger than those of the individual model compound with either a simple base or acid, respectively. That is to say that the Kassoc determined for 3 with pyridine (178 ± 18 M−1) and 4 with phenylacetic acid (24 ± 8 M−1) are weaker than for 3 and 4 together, regardless of whether a 1:1 or 1:2 complex is formed. Based on our results, we hypothesize that the enhanced aggregation observed for 3 and 4 originates from the presence of additional intermolecular forces beyond the expected acid–base interactions (e.g., π-stacking, hydrogen bonding), as suggested in the archipelago model.8 Further analysis by both experiment and theoretical calculations are underway.
Experimental section
General
All chemicals and solvents were used as received from commercial suppliers. 1-Ethynylpyrene was prepared according to literature.61 For the aldehyde synthesis, dry THF was distilled from sodium/benzophenone and DMF “with molecular sieve” (water <50 ppm) from Acros Organics was used. Solvent ratios are given as volume:volume. NMR spectra were recorded on a Bruker Avance 300 operating at 300 MHz (1H NMR) and 75 MHz (13C NMR), or a Bruker Avance 400 operating at 400 MHz (1H NMR) and 100 MHz (13C NMR), or a Jeol GX 400 operating at 100 MHz (13C NMR) at rt, if not otherwise specified. Signals were referenced to residual solvent peaks (δ in parts per million (ppm) 1H: CDCl3, 7.26 ppm; C6D6, 7.16 ppm; 13C: CDCl3, 77.0 ppm; C6D6, 128.0 ppm). Coupling constants were assigned as observed. Mass spectra were recorded from Bruker micro TOF II (ESI) and Bruker maxis 4G (APPI) instruments. IR spectra were recorded on a Varian 660-IR spectrometer. UV-vis spectra were recorded on a Varian Cary 5000 UV-vis spectrophotometer (λ in nm; ε in M−1 cm−1). TLC analysis was carried out with TLC plates from Macherey-Nagel (ALUGRAM® SIL G/UV254) and visualized by UV-light of 254 nm or 366 nm. Silica Gel 60 M (0.04–0.063 mm) for column chromatography was purchased from Macherey-Nagel.
:1) afforded compound 4 as a yellow solid (0.366 g, 73%). Mp 186–187 °C. Rf = 0.18 (CHCl3/hexanes 4:1). IR (ATR) 3037 (w), 2924 (w), 1708 (w), 1579 (m), 1453 (m) cm−1; 1H NMR (300 MHz, CDCl3) δ 8.35 (d, J = 9.3 Hz, 2H), 8.14 (d, J = 7.6 Hz, 4H), 8.11–7.94 (m, 10H), 7.83 (d, J = 7.8 Hz, 2H), 7.32 (t, J = 7.7 Hz, 1H), 6.80 (d, J = 7.7 Hz, 2H), 3.88–3.83 (m, 4H), 3.48–3.43 (m, 4H); 1H NMR (400 MHz, C6D6) δ 8.38 (d, J = 9.2 Hz, 2H), 7.97–7.90 (m, 8H), 7.85–7.73 (m, 8H), 6.89 (t, J = 7.6 Hz, 1H), 6.51 (d, J = 7.6 Hz, 2H), 3.87–3.83 (m, 4H), 3.36–3.32 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 160.4, 135.7, 131.4, 130.9, 129.9, 128.7, 127.5, 127.34, 127.31, 126.6, 125.8, 125.01, 124.97, 124.9, 124.8, 124.7, 123.4, 120.8, 39.8, 33.6 (one signal coincident or not observed); 13C NMR (100 MHz, C6D6) δ 161.1, 136.6, 136.3, 132.0, 131.5, 130.5, 129.3, 128.6, 127.0, 126.0, 125.8, 125.7, 125.2, 125.1, 123.9, 120.5, 40.7, 33.9 (three signals coincident or not observed); 13C–1H HSQC (400 MHz, C6D6, selected correlations) δ 136.3 ↔ 6.89; 120.5 ↔ 6.51; 40.7 ↔ 3.36–3.32; 33.9 ↔ 3.87–3.83; 13C–1H HMBC (400 MHz, C6D6, selected correlations) δ 161.1 ↔ 6.89, 6.51, 3.87–3.83, 3.36–3.32; 136.6 ↔ 8.38, 7.97–7.90, 3.87–3.83, 3.36–3.32; 120.5 ↔ 6.51, 3.36–3.32; 40.7 ↔ 6.51, 3.87–3.83; 33.9 ↔ 3.36–3.32. APPI HRMS m/z calcd for C41H30N ([M + H]+) 536.2373, found 536.2374.
:1) and extracted with Et2O (2 × 75 mL). The combined organic layers were washed with H2O (3 × 150 mL) and dried over MgSO4. After filtration and removal of the solvent, the residue was dissolved in CH2Cl2 and filtered through a plug of silica gel. Evaporation of the solvent and drying in vacuo afforded the product 5 (0.695 g, 62%) as a yellow oil. Rf = 0.49 (hexanes/CH2Cl2 1:1). IR (ATR) 3067 (vw), 2983 (w), 2927 (w), 2874 (w), 2822 (w), 2736 (w), 1691 (s) 1599 (m), 1574 (m), 1451 (m) cm−1; 1H NMR (300 MHz, CDCl3) δ 10.12 (s, 1H), 7.78–7.76 (m, 1H), 7.56–7.48 (m, 2H), 7.39 (td, J = 7.3 Hz, 1.4 Hz, 1H), 4.78 (s, 2H), 3.39 (s, 3H); 13C (75 MHz, CDCl3) δ 192.4, 140.5, 133.5, 133.0, 132.0, 127.7, 127.4, 71.5, 58.2. ESI HRMS m/z calcd for C9H11O2 ([M + H]+) 151.0754, found 151.0753.
:1) to afford the porphyrin 8 (0.430 g, 18%) as the first fraction and the porphyrin 7a (0.445 g, 14%) as the second fraction as purple solids. Mp 226–228 °C. Rf = 0.59 (hexanes/CH2Cl2 1:1). UV-vis (CH2Cl2) λmax (ε) 419 (569000), 516 (21000), 550 (9830), 590 (6480), 646 (5150) nm; IR (ATR) 3314 (w), 2951 (m), 2923 (m), 2858 (m), 1465 (m), 1346 (m) cm−1; 1H NMR (300 MHz, CDCl3) δ 8.92–8.89 (m, 6H), 8.73 (d, J = 4.8 Hz, 2H), 8.20–8.09 (m, 7H), 7.97 (d, J = 7.2 Hz, 1H), 7.85 (td, J = 7.6 Hz, J = 1.2 Hz, 1H), 7.66 (td, J = 7.5 Hz, J = 1.2 Hz, 1H), 7.60–7.57 (m, 6H), 4.15 (s, 2H), 2.99 (t, J = 7.7 Hz, 6H), 2.89 (s, 3H), 1.94 (pent, J = 7.6 Hz, 6H), 1.62 (sext, J = 7.4 Hz, 6H), 1.13 (t, J = 7.3 Hz, 9H), −2.66 (br s, 2H); 13C NMR (75 MHz, CDCl3) δ 142.3, 140.19, 140.17, 139.5, 139.3, 134.6, 134.0, 131.1 (br), 130.7 (br), 128.6, 126.73, 126.69, 126.3, 125.5, 120.6, 120.3, 116.9, 72.7, 58.1, 35.7, 33.8, 22.6, 14.2 (12 signals coincident or not observed). APPI HRMS m/z calcd for C58H59N4O ([M + H]+) 827.4683, found 827.4713.
:1). IR (ATR) 3314 (w), 3020 (vw), 2951 (w), 2923 (m), 2854 (m), 1466 (w), 1347 (w) cm−1; 1H NMR (300 MHz, CDCl3) δ 8.89 (s, 8H), 8.14 (d, J = 8.0 Hz, 8H), 7.57 (d, J = 8.0 Hz, 8H), 2.97 (t, J = 7.7 Hz, 8H), 1.98–1.88 (m, 8H), 1.61 (sext, J = 7.4 Hz, 8H), 1.12 (t, J = 7.3 Hz, 12H), −2.71 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 142.3, 139.5, 134.6, 130.9 (br), 126.7, 120.1, 35.7, 33.8, 22.6, 14.1 (1 signal coincident or not observed). ESI HRMS m/z calcd for C60H63N4 ([M + H]+) 839.5047, found 839.5053.
:1)) dried in vacuo and used without further purification. In PEG 400 (12 mL), the crude product 7b was dissolved, KCN (0.452 g, 6.94 mmol) was added, and the mixture was stirred at rt for 24 h. CH2Cl2 (100 mL) and H2O (100 mL) were added, the organic phase was separated and washed with H2O (4 × 100 mL). After drying over MgSO4, the mixture was filtered and the solvent removed. The residue was purified by column chromatography (silica gel, gradient hexanes/CH2Cl2 2:1 → 0:1) to afford the product 9 (51 mg, 89%) as a purple solid. Mp 180–182 °C. Rf = 0.41 (hexanes/CH2Cl2 1:1). UV-vis (CH2Cl2) λmax (ε) 419 (546000), 516 (21100), 552 (10000), 591 (6070), 646 (5440) nm; IR (ATR) 3314 (w), 2923 (m), 2855 (w), 1465 (m), 1346 (m) cm−1; 1H NMR (300 MHz, CDCl3) δ 8.90–8.88 (m, 6H), 8.61 (d, J = 4.8 Hz, 2H), 8.17–8.08 (m, 7H), 7.98 (d, J = 7.2 Hz, 1H), 7.87 (td, J = 7.6 Hz, J = 1.3 Hz, 1H), 7.73 (t, J = 6.9 Hz, 1H), 7.57 (d, J = 8.1 Hz, 6H), 3.39 (s, 2H), 2.97 (t, J = 7.7 Hz, 6H), 1.92 (pent, J = 7.6 Hz, 6H), 1.60 (sext, J = 7.4 Hz, 6H), 1.10 (t, J = 7.3 Hz, 9H), −2.73 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 142.4, 141.3, 139.3, 139.0, 134.6, 134.5, 134.3, 132.0, 131.2 (br), 129.3, 127.5, 126.8, 126.72, 126.65, 121.1, 120.6, 117.8, 115.0, 35.6, 33.8, 22.8, 22.6, 14.1 (12 signals coincident or not observed). ESI HRMS m/z calcd for C58H56N5 ([M + H]+) 822.4530, found 822.4513.
:1, 150 mL), and H2O (50 mL). The solution was dried over MgSO4 and filtered. The solvent was removed and the residue was purified by column chromatography (silica gel, CH2Cl2/EtOAc 9:1) to afford the product 10 (37 mg, 94%) as a purple solid. Mp 255–258 °C. Rf = 0.58 (CH2Cl2/acetone 19:1). UV-vis (THF) λmax (ε) 418 (567000), 514 (22500), 549 (10800), 592 (6220), 647 (4550) nm; IR (ATR) 3314 (w), 3021 (w), 2953 (m), 2924 (m), 2855 (m), 1707 (m), 1467 (m), 1347 (m) cm−1; 1H NMR (300 MHz, CDCl3) δ 8.86 (d, J = 4.9 Hz, 2H), 8.83 (d, J = 4.8 Hz, 2H), 8.77 (d, J = 4.8 Hz, 2H), 8.61 (d, J = 4.8 Hz, 2H), 8.14–8.00 (m, 7H), 7.76–7.68 (m, 1H), 7.58 (t, J = 8.9 Hz, 4H), 7.49 (d, J = 7.4 Hz, 2H), 7.36 (d, J = 6.8 Hz, 2H), 3.37 (s, 2H), 2.97 (t, J = 7.7 Hz, 2H), 2.85 (t, J = 7.6 Hz, 4H), 1.97–1.78 (m, 6H), 1.67–1.47 (m, 6H), 1.13–1.03 (m, 9H), −2.74 (br s, 2H); 13C NMR (100 MHz, CDCl3) δ 175.4, 142.3, 142.2, 141.9, 139.5, 139.2, 135.4, 134.6, 134.51, 134.46, 134.41, 131.1 (br), 129.3, 128.6, 126.7, 126.6, 125.5, 120.6, 120.3, 116.7, 38.9, 35.7, 35.6, 33.8, 33.7, 22.63, 22.58, 14.14, 14.10 (6 signals coincident or not observed). ESI HRMS m/z calcd for C58H57N4O2 ([M + H]+) 841.4476, found 841.4476; m/z calcd for C58H56N4NaO2 ([M + Na]+) 863.4296, found 863.4314.
:1, 50 mL), and H2O (50 mL). The organic layer was dried over MgSO4 and filtered. After removal of the solvent, the residue was purified by column chromatography (silica gel, gradient CH2Cl2/acetone 1:0 → 15:1) to yield the product 3 (15.0 mg, 95%) as a red-purple solid. Mp 256–259 °C. Rf = 0.81 (CH2Cl2/acetone 19:1). UV-vis (CH2Cl2) λmax (ε) 415 (312000), 528 (22100) nm; IR (ATR) 3023 (w), 2953 (s), 2926 (s), 2859 (m), 1709 (m), 1460 (m), 1352 (m) cm−1; 1H NMR (300 MHz, CDCl3) δ 8.76 (d, J = 5.0 Hz, 2H), 8.73 (d, J = 5.0 Hz, 2H), 8.68 (d, J = 5.0 Hz, 2H), 8.51 (d, J = 4.9 Hz, 2H), 7.96–7.82 (m, 7H), 7.69–7.63 (m, 1H), 7.55 (t, J = 6.9 Hz, 2H), 7.48 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.2 Hz, 4H), 3.24 (s, 2H), 2.90 (t, J = 7.7 Hz, 2H), 2.82 (t, J = 7.7 Hz, 4H), 1.91–1.74 (m, 6H), 1.62–1.44 (m, 6H), 1.09–1.01 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 175.2, 142.9, 142.8, 142.4, 142.34, 142.27, 140.6, 138.1, 138.0, 134.9, 133.9, 133.64, 133.59, 132.5, 132.21, 132.17, 131.5, 129.3, 128.6, 126.9, 126.8, 125.7, 119.3, 119.1, 115.8, 38.6, 35.6, 35.5, 33.8, 33.7, 22.6, 22.5, 14.10, 14.07 (1 signal coincident or not observed). ESI HRMS m/z calcd for C58H54N4NaNiO2 ([M + Na]+) 919.3493, found 919.3486; m/z calcd for C58H53N4Na2NiO2 ([M − H + 2Na]+) 941.3312, found 941.3303.
Acknowledgements
We are grateful for financial support from the Institute for Oil Sands Innovation at the University of Alberta, the Natural Sciences and Engineering Research Council of Canada, and the Graduate School Molecular Science (GSMS) at FAU. We thank Dr. Alexander Scherer for his assistance, as well as Prof. Jürgen Schatz and Dr. Max von Delius for helpful discussions regarding assessment of the binding constants.Notes and references
- K. Aleklett, M. Höök, K. Jakobsson, M. Lardelli, S. Snowden and B. Söderbergh, Energy Policy, 2010, 38, 1398–1414 CrossRef.
- M. R. Gray and W. C. McCaffrey, Energy Fuels, 2002, 16, 756–766 CrossRef CAS.
- I. Gawel, D. Bociarska and P. Biskupski, Appl. Catal., A, 2005, 295, 89–94 CrossRef CAS.
- N. Haji-Akbari, P. Teeraphapkul and H. S. Fogler, Energy Fuels, 2014, 28, 909–919 CrossRef CAS.
- S. Chiaberge, G. Guglielmetti, L. Montanari, M. Salvalaggio, L. Santolini, S. Spera and P. Cesti, Energy Fuels, 2009, 23, 4486–4495 CrossRef CAS.
- R. R. Chianelli, K. Castillo, V. Gupta, A. M. Qudah, B. Torres and R. E. Abujnah, Asphaltene based photovoltaic devices. US Patent2014/0234026, 2014 Search PubMed.
- S. M. Hashmi, K. X. Zhong and A. Firoozabadi, Soft Matter, 2012, 8, 8778–8785 RSC.
- M. R. Gray, R. R. Tykwinski, J. M. Stryker and X. Tan, Energy Fuels, 2011, 25, 3125–3134 CrossRef CAS.
- M. P. Hoepfner and H. S. Fogler, Langmuir, 2013, 29, 15423–15432 CrossRef CAS PubMed.
- T. E. Havre and J. Sjöblom, Colloids Surf., A, 2003, 228, 131–142 CrossRef CAS.
- S. B. Jaffe, H. Freund and W. N. Olmstead, Ind. Eng. Chem. Res., 2005, 44, 9840–9852 CrossRef CAS.
- J. G. Speight, Oil Gas Sci. Technol., 2004, 59, 467–477 CAS.
- O. C. Mullins, H. Sabbah, J. Eyssautier, A. E. Pomerantz, L. Barré, A. B. Andrews, Y. Ruiz-Morales, F. Mostowfi, R. McFarlane, L. Goual, R. Lepkowicz, T. Cooper, J. Orbulescu, R. M. Leblanc, J. Edwards and R. N. Zare, Energy Fuels, 2012, 26, 3986–4003 CrossRef CAS.
- F. Rakotondradany, H. Fenniri, P. Rahimi, K. L. Gawrys, P. K. Kilpatrick and M. R. Gray, Energy Fuels, 2006, 20, 2439–2447 CrossRef CAS.
- X. Tan, H. Fenniri and M. R. Gray, Energy Fuels, 2008, 22, 715–720 CrossRef CAS.
- Z. Wang, W. Liang, G. Que and J. Qian, Pet. Sci. Technol., 1997, 15, 559–577 CrossRef CAS.
- O. P. Strausz, P. Peng and J. Murgich, Energy Fuels, 2002, 16, 809–822 CrossRef CAS.
- O. P. Strausz, T. W. Mojelsky, E. M. Lown, I. Kowalewski and F. Behar, Energy Fuels, 1999, 13, 228–247 CrossRef CAS.
- P. M. Spiecker, K. L. Gawrys, C. B. Trail and P. K. Kilpatrick, Colloids Surf., A, 2003, 220, 9–27 CrossRef CAS.
- K. L. Gawrys, G. A. Blankenship and P. K. Kilpatrick, Langmuir, 2006, 22, 4487–4497 CrossRef CAS PubMed.
- J. M. Sheremata, M. R. Gray, H. D. Dettman and W. C. McCaffrey, Energy Fuels, 2004, 18, 1377–1384 CrossRef CAS.
- J. Murgich, J. A. Abanero and O. P. Strausz, Energy Fuels, 1999, 13, 278–286 CrossRef CAS.
- R. I. Rueda-Velásquez, H. Freund, K. Qian, W. N. Olmstead and M. R. Gray, Energy Fuels, 2013, 27, 1817–1829 CrossRef.
- A. Karimi, K. Qian, W. N. Olmstead, H. Freund, C. Yung and M. R. Gray, Energy Fuels, 2011, 25, 3581–3589 CrossRef CAS.
- A. Scherer, F. Hampel, M. R. Gray, J. M. Stryker and R. R. Tykwinski, J. Phys. Org. Chem., 2012, 25, 597–606 CrossRef CAS.
- S. Tanaka, M. Shirakawa, K. Kaneko, M. Takeuchi and S. Shinkai, Langmuir, 2005, 21, 2163–2172 CrossRef CAS PubMed.
- F. Sguerra, V. Bulach and M. W. Hosseini, Dalton Trans., 2012, 41, 14683–14689 RSC.
- Y. K. Kryschenko, S. R. Seidel, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2003, 125, 5193–5198 CrossRef CAS PubMed.
- K. Akbarzadeh, D. C. Bressler, J. Wang, K. L. Gawrys, M. R. Gray, P. K. Kilpatrick and H. W. Yarranton, Energy Fuels, 2005, 19, 1268–1271 CrossRef CAS.
- X. Tan, H. Fenniri and M. R. Gray, Energy Fuels, 2009, 23, 3687–3693 CrossRef CAS.
- J. Wang, N. van der T. Opedal, Q. Lu, Z. Xu, H. Zeng and J. Sjöblom, Energy Fuels, 2012, 26, 2591–2599 CrossRef CAS.
- H. Sabbah, A. E. Pomerantz, M. Wagner, K. Müllen and R. N. Zare, Energy Fuels, 2012, 26, 3521–3526 CrossRef CAS.
- B. Breure, D. Subramanian, J. Leys, C. J. Peters and M. A. Anisimov, Energy Fuels, 2013, 27, 172–176 CrossRef CAS.
- Y. Ruiz-Morales and O. C. Mullins, Energy Fuels, 2007, 21, 256–265 CrossRef CAS.
- Y. Ruiz-Morales, X. Wu and O. C. Mullins, Energy Fuels, 2007, 21, 944–952 CrossRef CAS.
- L. M. da Costa, S. Hayaki, S. R. Stoyanov, S. Gusarov, X. Tan, M. R. Gray, J. M. Stryker, R. Tykwinski, J. W. de M. Carneiro, H. Sato, P. R. Seidl and A. Kovalenko, Phys. Chem. Chem. Phys., 2012, 14, 3922–3934 RSC.
- L. M. da Costa, S. R. Stoyanov, S. Gusarov, X. Tan, M. R. Gray, J. M. Stryker, R. Tykwinski, J. W. de M. Carneiro, P. R. Seidl and A. Kovalenko, Energy Fuels, 2012, 26, 2727–2735 CrossRef CAS.
- S. Mitra-Kirtley, O. C. Mullins, J. van Elp, S. J. George, J. Chen and S. P. Cramer, J. Am. Chem. Soc., 1993, 115, 252–258 CrossRef CAS.
- A. Treibs, Liebigs Ann. Chem., 1934, 509, 103–114 CrossRef CAS.
- A. Treibs, Liebigs Ann. Chem., 1934, 510, 42–62 CrossRef CAS.
- A. Treibs, Liebigs Ann. Chem., 1935, 517, 172–196 CrossRef CAS.
- A. Treibs, Angew. Chem., 1936, 49, 682–686 CrossRef CAS.
- G. P. Dechaine and M. R. Gray, Energy Fuels, 2010, 24, 2795–2808 CrossRef CAS.
- C.-X. Yin, X. Tan, K. Müllen, J. M. Stryker and M. R. Gray, Energy Fuels, 2008, 22, 2465–2469 CrossRef CAS.
- S. D. Cardozo, M. Schulze, R. R. Tykwinski and M. R. Gray, Energy Fuels, 2015, 29, 1494–1502 CrossRef CAS.
- P. S. Clezy, Aust. J. Chem., 1991, 44, 1163–1193 CrossRef CAS.
- T. D. Lash, Energy Fuels, 1993, 7, 166–171 CrossRef CAS.
- B. E. Smith and T. D. Lash, Tetrahedron, 2010, 66, 4413–4422 CrossRef CAS.
- The assignment of Hb and Hc was possible by 2D NMR spectroscopic measurements, primarily using long range correlations of Hb and Hc to the carbons of the pyridyl ring (see ESI†).
- B. Christenson, G. Hallnemo and C. Ullenius, Tetrahedron, 1991, 47, 4739–4752 CrossRef CAS.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef CAS.
- N. Jux, Org. Lett., 2000, 2, 2129–2132 CrossRef CAS PubMed.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CAS.
- S. Jasinski, E. A. Ermilov, N. Jux and B. Röder, Eur. J. Org. Chem., 2007, 1075–1084 CrossRef CAS.
- A. H. Alshareef, A. Scherer, X. Tan, K. Azyat, J. M. Stryker, R. R. Tykwinski and M. R. Gray, Energy Fuels, 2012, 26, 1828–1843 CrossRef CAS.
- No evidence for self-association was found (see ESI†).
- T. Kitao and C. H. Jarboe, J. Org. Chem., 1967, 32, 407–410 CrossRef CAS.
- K. Hirose, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193–209 CrossRef CAS.
- E. J. Olson and P. Bühlmann, J. Org. Chem., 2011, 76, 8406–8412 CrossRef CAS PubMed.
- S. Natesan, T. Wang, V. Lukacova, V. Bartus, A. Khandelwal and S. Balaz, J. Chem. Inf. Model., 2011, 51, 1132–1150 CAS.
- M. Hissler, A. Harriman, A. Khatyr and R. Ziessel, Chem. – Eur. J., 1999, 5, 3366–3381 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of compounds 3–10, 2D NMR spectra of compound 4, as well as experimental details and NMR data of the aggregation studies. See DOI: 10.1039/c5ob00836k |
| This journal is © The Royal Society of Chemistry 2015 |