
Organocatalytic diastereoselective synthesis of chiral decalines through the domino Claisen–Schmidt/Henry reaction†
Adluri B.
Shashank
and
Dhevalapally B.
Ramachary
*
Catalysis Laboratory, School of Chemistry, University of Hyderabad, Hyderabad-500 046, India. E-mail: ramsc@uohyd.ernet.in
First published on 1st April 2015
Abstract
General and operative domino Claisen–Schmidt/Henry (CS/H) reaction has been revealed to obtain highly substituted chiral decalines in good yields with excellent ees and des by using push–pull enamine catalysis.
The asymmetric synthesis of functionalized bicyclic carbon frameworks is always a challenging task in synthetic chemistry. Bicyclic carbon frameworks (decalines) are found in a wide variety of polyterpenoid and steroid natural products with interesting biological activity.1 Recently, organocatalytic domino processes involving iminium and enamine activation have become useful for the synthesis of bicyclic, tricyclic and spirocyclic molecules with high ee/de.2 The domino reactions of Serebryakov 1-amino-1,3-butadiene,3 Barbas 2-amino-1,3-butadiene,4 Jørgensen/Chen trienamine5 and Ramachary/Gouverneur aminoenyne6 were well explored for the asymmetric synthesis of functionalized cyclohexanes. Although organocatalytic domino reactions were reported recently, the development of more efficient approaches in multi C–C bond formation with multiple stereogenic centers in a cascade manner is of significant interest.7
The Claisen–Schmidt reaction is one of the important C–C bond formation processes in organic chemistry which is able to provide α, β-unsaturated carbonyl compounds, which are important intermediates for the natural product synthesis.8 The Henry (nitroaldol) reaction is another powerful C–C bond forming tool for the preparation of valuable synthetic intermediates such as nitro alcohols, which can be further transformed into a number of important nitrogen and oxygen-containing compounds.9 Recently, Michael–Henry processes have been successfully demonstrated in the synthesis of substituted cyclic frameworks with multiple stereogenic centers.10 However, to the best of our knowledge, there is no report involving a domino Claisen–Schmidt/Henry reaction strategy for the selective synthesis of bicyclic decalines with three contiguous stereocenters (Scheme 1).
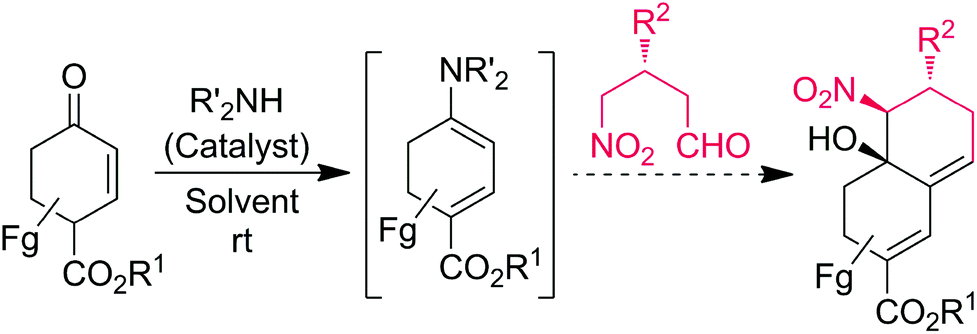 | ||
| Scheme 1 Reaction design for the diastereoselective synthesis of chiral decalines through push–pull enamine catalysis. | ||
In continuation to our interest in the development of organocatalytic domino asymmetric reactions through push–pull enamine- or dienamine-catalysis,11 herein we have designed a diastereoselective approach to the substituted chiral decalines from commercially available enones and chiral γ-nitroaldehydes through the domino Claisen–Schmidt/Henry (CS/H) reaction based on the push–pull enamine catalysis (Scheme 1). Optically pure γ-nitroaldehydes (93–95% ee), which are used as starting materials in this study were obtained from the recent discovery of List or Hayashi protocol of (S)-α,α-diphenylprolinol trimethylsilyl ether [(S)-DPPOTMS]-mediated conjugate addition of acetaldehyde to β-nitrostyrenes or nitroalkanes to α,β-unsaturated aldehydes, respectively.12,13
For the reaction optimization, we screened a few simple organocatalysts for the reaction of enone 1a with 0.5 to 1.5 equiv. of γ-nitroaldehyde (S)-2a (Table 1). Reaction of 1a with 0.5 equiv. of (S)-2a in DMSO catalyzed by 20 mol% of L-proline 3a or L-prolinol 3b did not furnish the expected product (Table 1, entries 1 and 2). Surprisingly, the same reaction in the presence of pyrrolidine 3c-catalysis furnished the product (−)-4aa in only 15% yield, but the same reaction under piperidine 3d-catalysis gave (−)-4aa in 28% yield with 92% ee and >99% de (Table 1, entries 3 and 4). To increase the CS/H reaction rate and yield, we tested the cascade reaction of 1a with (S)-2a in the presence of more basic chiral diamine catalysts (S)-3e, (R)-3e, and (S)-3f in DMSO at 25–70 °C (Table 1, entries 5–14). Interestingly, the domino reaction with 20 mol% of (S)-3e as the catalyst furnished the single isomer of (−)-4aa in 64% yield with 94% ee at 25 °C for 5 h; but the same reaction catalyzed by (S)-3f furnished (−)-4aa in reduced yield (Table 1, entries 5 and 6). There was no further improvement by changing the solvent or by adding a co-catalyst, but yield and ee of the reaction were increased to 70% and >99%, respectively, by taking 1.5 equiv. of (S)-2a instead of 0.5 equiv. under the (S)-3e-catalysis in DMSO at rt (entries 7–10). Reaction of the crude chiral aldehyde (S)-2a (which is obtained from the quick work up of the Hayashi method) with 1a gave the product (−)-4aa with reduced yield and ee (Table 1, entry 11). To further understand the reaction kinetics, we carried out the reaction of 1a and (S)-2a with (R)-3e as the catalyst, which gave the same enantiomer of (−)-4aa in 75% yield with 94% ee (Table 1, entry 12). In another experiment, we performed the reaction with the opposite enantiomer of γ-nitroaldehyde (R)-2a with 1a under (S)-3e-catalysis to furnish the opposite enantiomer of decalin (+)-4aa in 72% yield with 93% ee (Table 1, entry 13). Surprisingly, there was no further improvement in the yield by changing the temperature from 25 °C to 70 °C (Table 1, entry 14); and there is no reaction observed under the tert-amine, DABCO-catalysis (result not shown in Table 1). After the preliminary studies, we considered the optimization conditions to be DMSO at 25 °C using 1.5 equiv. of (S)-2a and commercially available (S)-3e as the catalyst (Table 1, entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst 3 (20 mol%) | Time (h) | Yield (%) of 4aa | ee (%) of 4aa |
| a Reactions were carried out in solvent (0.5 M) with 2.0 equiv. of 1a relative to the (S)-2a in the presence of 20 mol % of catalyst 3. b Yield refers to the column-purified product. c ee was determined by CSP-HPLC analysis. d DMF was used as the solvent. e HClO4 (20 mol%) was taken as the co-catalyst. f 1.0 equiv. of 1a was used. g 1.5 equiv. of (S)-2a was used. h Crude compound (S)-2a was used as such. i (R)-2a was used. j Reaction was performed at 70 °C. | ||||
| 1 | (S)-3a | 24 h | — | — |
| 2 | (S)-3b | 24 h | — | — |
| 3 | 3c | 24 h | 15 | n.d. |
| 4 | 3d | 24 h | 28 | 92 |
| 5 | (S)-3e | 5 h | 64 | 94 |
| 6 | (S)-3f | 5 h | 50 | 95 |
| 7d | (S)-3e | 5 h | 53 | 96 |
| 8e | (S)-3e | 24 h | — | — |
| 9f | (S)-3e | 3 h | 53 | 96 |
| 10g | (S)-3e | 6 h | 70 | >99 |
| 11g,h | (S)-3e | 5 h | 50 | 96 |
| 12g | (R)-3e | 5 h | 75 | 94 |
| 13g,i | (S)-3e | 4 h | 72 | −93 |
| 14g,j | (S)-3e | 2 h | 57 | 98 |
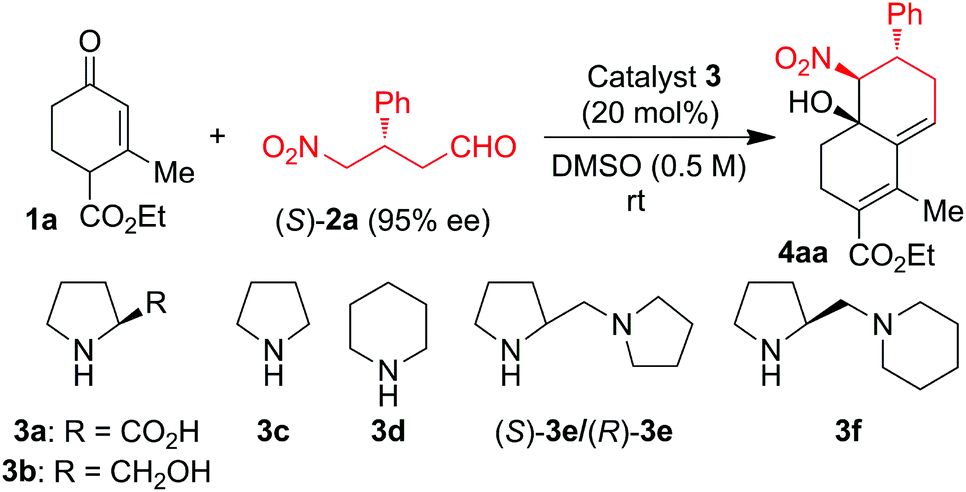
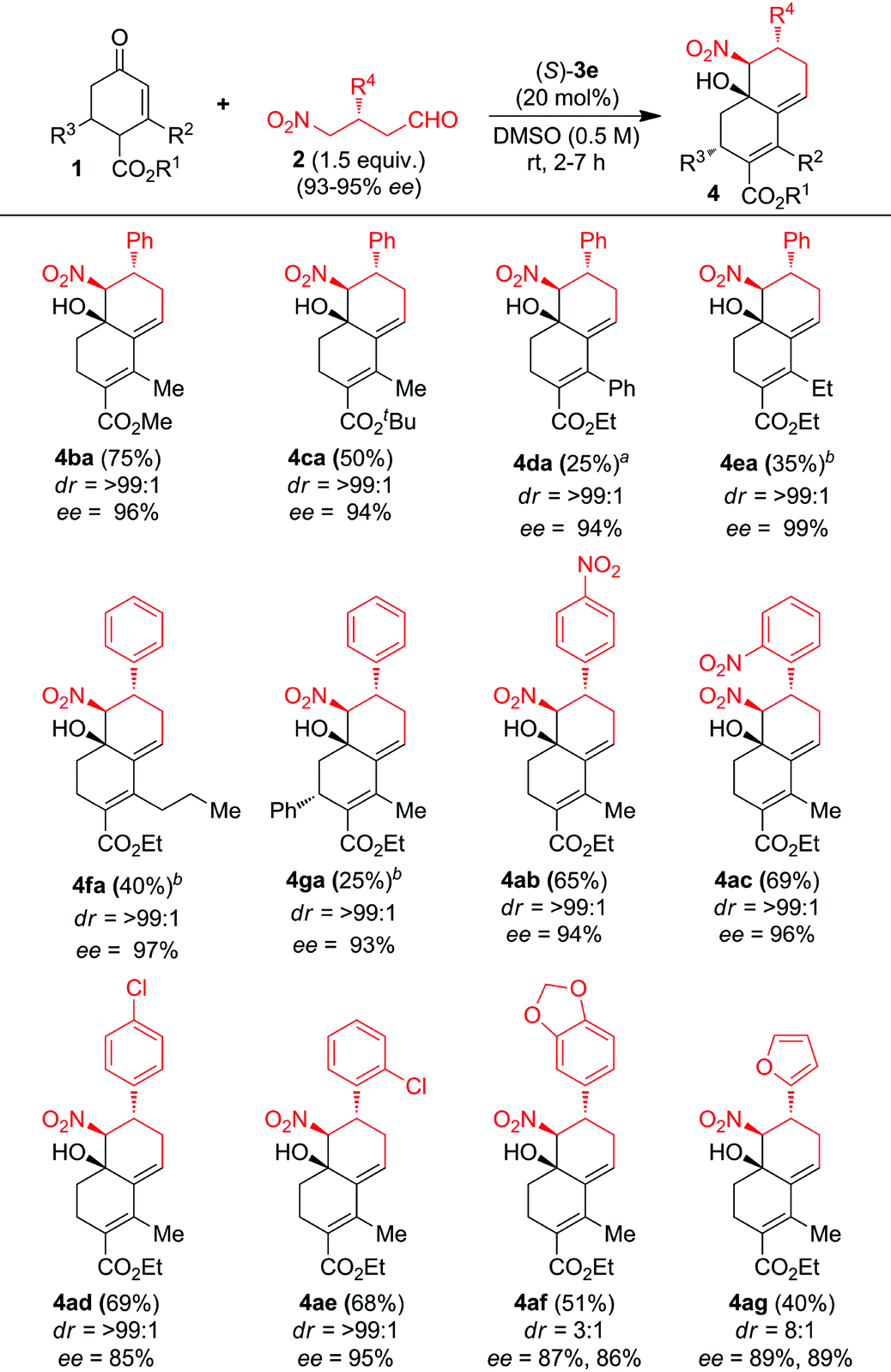
With the optimized conditions in hand, the scope and limitations of the domino CS/H reaction were investigated by using functionalized enones 1a–g and chiral γ-nitroaldehydes 2a–g (Table 2). A variety of functionalized cyclic enones 1b–g with freshly prepared chiral γ-nitroaldehyde 2a delivered the chiral decalines 4ba–ga in good to moderate yields with high ees and des (Table 2, entries 1–6). Decaline yields were reduced with the increase in the bulkiness of C-2 substitution of enones without affecting the ee and de values, for example C-2 substituted enones 1d–f furnished the expected decalines 4da–4fa in 25%, 35% and 40% yields with 94%, 99% and 97% ees and >99% de respectively (Table 2, entries 3–5). Surprisingly, the C2/C6-disubstituted enone 1g gave only one isomer of domino CS/H product 4ga in 25% yield with 93% ee and >99% de (Table 2, entry 6). The domino CS/H reaction of enone 1a with γ-nitroaldehydes bearing electron-deficient aryl substituents such as 4-nitrophenyl 2b, 2-nitrophenyl 2c, 4-chlorophenyl 2d and 2-chlorophenyl 2e gave the expected decalines 4ab–4ae in good yields with high ees and des (Table 2, entries 7–10), whereas an electron-rich aryl substituent such as 3,4-methylenedioxyphenyl 2f and a heteroaryl substituent such as furyl 2g reduced the diastereoselectivity of decalines 4af–4ag without affecting ees (Table 2, entries 11 and 12). The structure and absolute stereochemistry of the chiral CS/H products were confirmed by NMR analysis and also finally confirmed by X-ray structure analysis on (−)-4aa as shown in Fig. S1 (see the ESI†).14
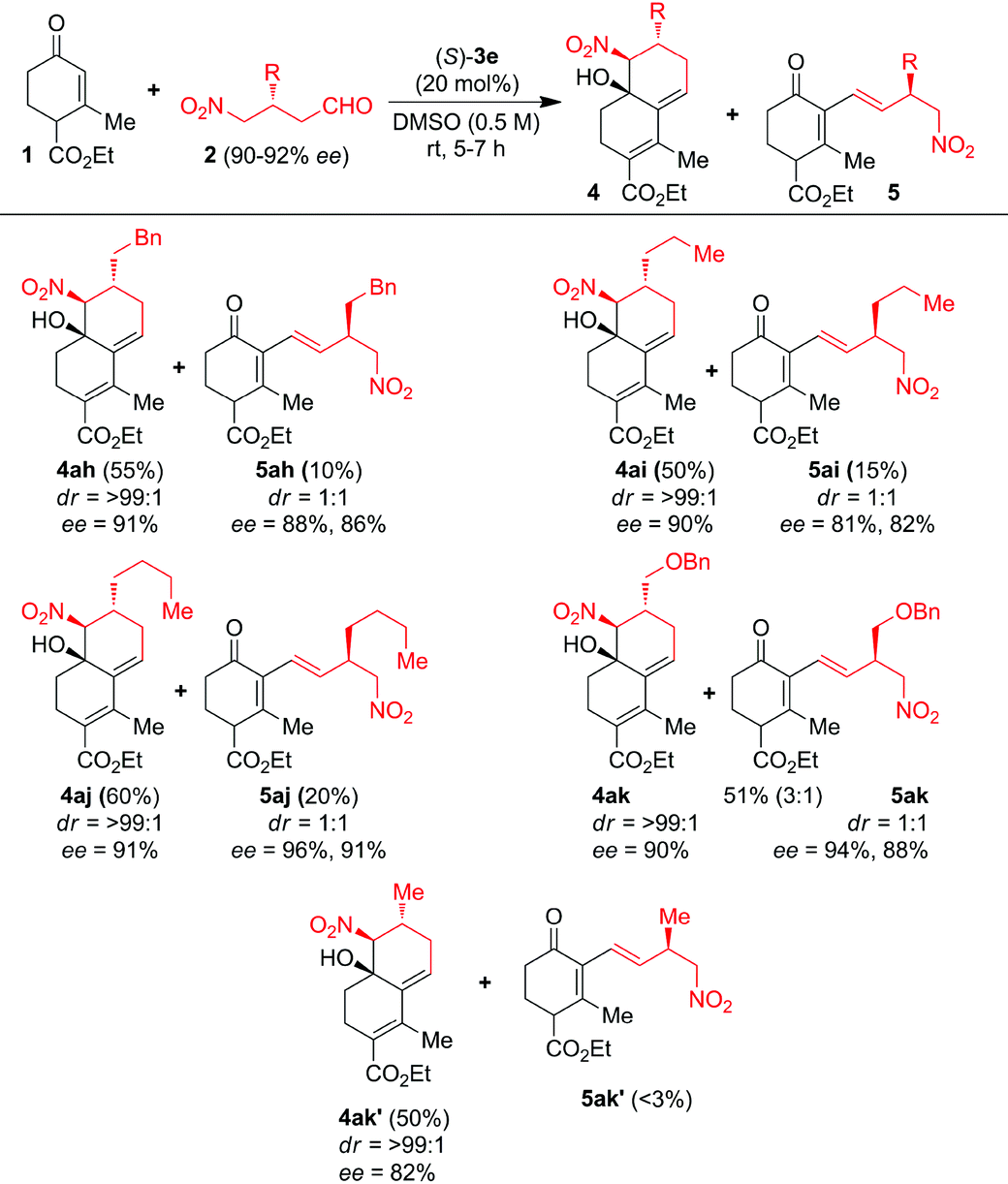
We further showed interest to screen β-alkyl-γ-nitroaldehydes 2h–k′ as substrates with 1a to investigate the electronic factors on product formation (Table 3). A series of β-alkyl-γ-nitroaldehydes 2h–k′ were reacted with 1a catalyzed by 20 mol% of (S)-3e at 25 °C for 5–7 h in DMSO. Surprisingly, in all these reactions along with CS/H products 4ah–ak′, monocyclic chiral (E)-1,3-diene products 5ah–ak′ were isolated in minor quantities through the Claisen–Schmidt/isomerization (CS/I) reaction (Table 3). The CS/H products 4ah–ak′ were furnished in good to excellent ees and des whereas the CS/I products 5ah–ak′ were furnished in good to excellent ees with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Interestingly, formation of monocyclic chiral (E)-1,3-diene 5ak′ is very poor from the domino reaction of 1a with simple (R)-3-methyl-4-nitrobutanal 2k′ under the (S)-3e-catalysis. These results suggest that the sterically crowded β-alkyl group in 2h–k′ is responsible for the formation of major/minor isomers of CS intermediates, from which the minor trans-isomer is converted into the CS/I product.
:1 dr. Interestingly, formation of monocyclic chiral (E)-1,3-diene 5ak′ is very poor from the domino reaction of 1a with simple (R)-3-methyl-4-nitrobutanal 2k′ under the (S)-3e-catalysis. These results suggest that the sterically crowded β-alkyl group in 2h–k′ is responsible for the formation of major/minor isomers of CS intermediates, from which the minor trans-isomer is converted into the CS/I product.
With its applications in mind, we explored the utilization of CS/H products 4 in the synthesis of chiral terpenoid type compounds 6–11via reduction, oxidation and organocatalytic reductive coupling (OrgRC) reactions (Scheme 2). Interestingly, CS/H reaction of 1a–b with functionally rich chiral γ-nitroaldehyde (−)-2l in DMSO at 25 °C under (S)-3e-catalysis for 48 h furnished the terpenoid-type tricyclic products 6 and 7 in good yields with 1:1.5 to 1:2 dr. In both the cases cis-isomer 7 was formed in major compared to trans-isomer 6. The OrgRC15 reaction of the dienal 8al (which is obtained after high-yielding ester reduction followed by oxidation of (−)-7al) with Meldrum's acid and Hantzsch ester in CH3CN (0.5 M) at 25 °C for 24 h furnished the chiral terpenoid-type product (−)-9al in 60% yield. High-yielding reduction of chiral decaline (−)-4aa with 2.5 equiv. of DIBAL-H in dry DCM at 0–25 °C for 1.0 h furnished the allylic alcohol, which on oxidation with mCPBA furnished regioselectively a single isomer of epoxide (−)-11aa in 64% overall yield (Scheme 2). Surprisingly, treatment of the chiral decalines (−)-4aa and (−)-4ba with 1.4 equiv. of TBHP under 2 mol% of VO(acac)2 in DCM at 25 °C for 14 h furnished regioselectively single isomers of epoxides (−)-10aa and (−)-10ba in each 30% yield with >99% ee. The structure and regiochemistry of the products 6–11 were confirmed by NMR analysis and also finally confirmed by X-ray structure analysis on 7bl as shown in Fig. S2 (see the ESI†).14
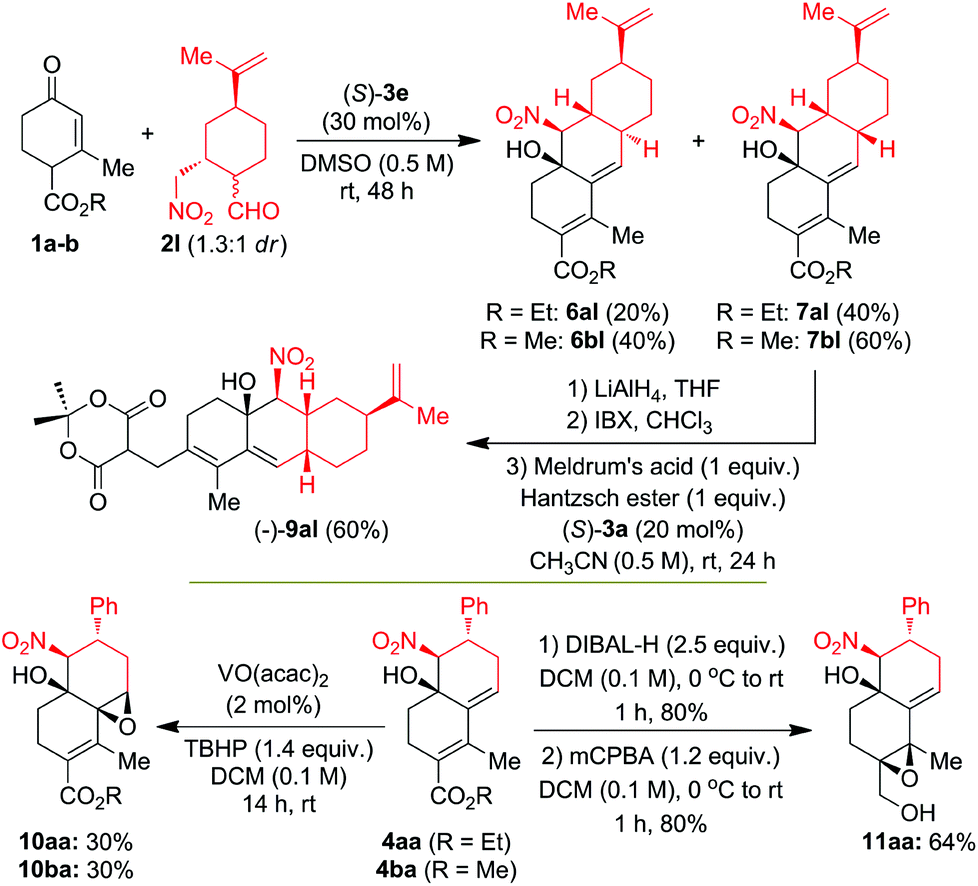 | ||
| Scheme 2 Applications of domino CS/H chiral products. | ||
Even though additional studies are needed to securely elucidate the mechanism of CS/H and CS/I reactions through (S)-3e-catalysis, the domino reaction proceeds by a stepwise manner between the in situ generated push–pull enamines 12 with γ-nitroaldehydes 2 (Scheme 3). First, reaction of catalyst (S)-3e with 1a generates a chiral push–pull enamine 12 through iminium formation. The chiral γ-nitroaldehyde 2 reacts with the in situ generated 12 to furnish the Claisen–Schmidt intermediates 13 as the major [cis-13] and minor [trans-13] isomers due to the steric hindrance between diamine and β-aryl/alkyl groups. Further in situ treatment of 13 with diamine (S)-3e, major cis-13 isomer transforms into the single isomer of decaline 4via intramolecular Henry reaction. Based on the crystal structure studies, we can rationalize that the re-face of the nucleophilic carbon attacks the si-face of the electrophilic carbon in an intramolecular manner. The formation of chiral (E)-1,3-dienes 5 from minor trans-13 isomer can be explained by using steric hindrance-induced (S)-3e-catalyzed isomerization (see full details in Scheme S1, ESI†).
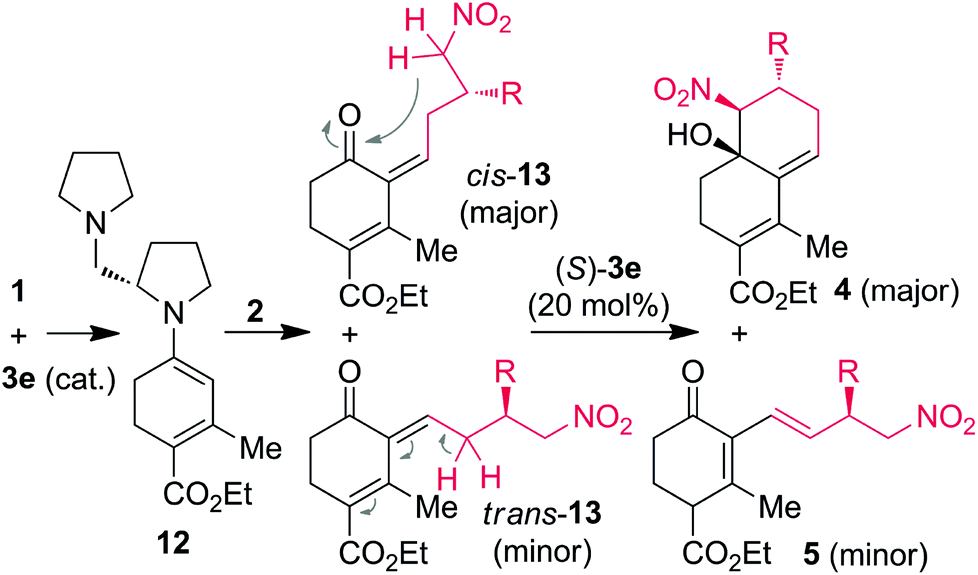 | ||
| Scheme 3 Proposed reaction mechanism. | ||
In summary, we have developed a domino Claisen–Schmidt/Henry process for the synthesis of terpenoid-type chiral decalines with three-contiguous stereocenters. This novel CS/H reaction proceeds in good yields with high enantio- and diastereoselectivity through push–pull enamine catalysis. Furthermore, we have demonstrated the application of chiral CS/H products in the synthesis of highly substituted terpenoid-type compounds.
We thank DST (New Delhi) for financial support. ABS thanks CSIR, New Delhi for his research fellowship.
Notes and references
- (a) V. Singh, S. R. Iyer and S. Pal, Tetrahedron, 2005, 61, 9197 CrossRef CAS PubMed; (b) T. Tokoroyama, Synthesis, 2000, 611 CrossRef CAS PubMed; (c) M. A. Varner and R. B. Grossman, Tetrahedron, 1999, 55, 13867 CrossRef CAS; (d) G. Mehta and V. Singh, Chem. Rev., 1999, 99, 881 CrossRef CAS PubMed; (e) G. Mehta and A. Srikrishna, Chem. Rev., 1997, 97, 671 CrossRef CAS PubMed; (f) A. T. Merritt and S. V. Ley, Nat. Prod. Rep., 1992, 9, 243 RSC.
- For recent reviews on organocatalytic domino reactions, see: (a) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed; (b) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237 CrossRef CAS PubMed; (c) D. B. Ramachary and S. Jain, Org. Biomol. Chem., 2011, 9, 1277 RSC; (d) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (e) X. Yu and W. Wang, Org. Biomol. Chem., 2008, 6, 2037 RSC; (f) D. Enders, C. Grondal and M. R. M. Huttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS PubMed.
- For selected examples on Serebryakov dienamine catalysis, see: (a) L. Albrecht, G. Dickmeiss, F. C. Acosta, C. Rodríguez-Escrich, R. L. Davis and K. A. Jørgensen, J. Am. Chem. Soc., 2012, 134, 2543 CrossRef CAS PubMed; (b) N. Utsumi, H. Zhang, F. Tanaka and C. F. Barbas III, Angew. Chem., Int. Ed., 2007, 46, 1878 CrossRef CAS PubMed; (c) S. Bertelsen, M. Marigo, S. Brandes, P. Dinér and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973 CrossRef CAS PubMed; (d) A. G. Nigmatov and E. P. Serebryakov, Russ. Chem. Bull., 1993, 42, 213 CrossRef.
- For selected examples on Barbas dienamine catalysis, see: (a) X. Yin, Y. Zheng, X. Feng, K. Jiang, X.-Z. Wei, N. Gao and Y.-C. Chen, Angew. Chem., Int. Ed., 2014, 53, 6245 CrossRef CAS PubMed; (b) M. P. Lalonde, M. A. McGowan, N. S. Rajapaksa and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 1891 CrossRef CAS PubMed; (c) X. Feng, Z. Zhou, R. Zhou, Q.-Q. Zhou, L. Dong and Y.-C. Chen, J. Am. Chem. Soc., 2012, 134, 19942 CrossRef CAS PubMed; (d) L.-Y. Wu, G. Bencivenni, M. Mancinelli, A. Mazzanti, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7196 CrossRef CAS PubMed; (e) D. B. Ramachary, Y. V. Reddy and B. V. Prakash, Org. Biomol. Chem., 2008, 6, 719 RSC; (f) Y. Yamamoto, N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5962 CrossRef CAS PubMed; (g) D. B. Ramachary, N. S. Chowdari and C. F. Barbas III, Angew. Chem., Int. Ed., 2003, 42, 4233 CrossRef CAS PubMed.
- (a) B. S. Donslund, K. S. Halskov, L. A. Leth, B. M. Paz and K. A. Jørgensen, Chem. Commun., 2014, 50, 13676 RSC; (b) X. Feng, Z. Zhou, C. Ma, X. Yin, R. Li, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 14173 CrossRef CAS PubMed; (c) I. Kumar, P. Ramaraju and N. A. Mir, Org. Biomol. Chem., 2013, 11, 709 RSC; (d) Z.-J. Jia, H. Jiang, J.-L. Li, B. Gschwend, Q.-Z. Li, X. Yin, J. Grouleff, Y.-C. Chen and K. A. Jørgensen, J. Am. Chem. Soc., 2011, 133, 5053 CrossRef CAS PubMed.
- (a) D. B. Ramachary, C. Venkaiah and P. M. Krishna, Org. Lett., 2013, 15, 4714 CrossRef CAS PubMed; (b) D. B. Ramachary, C. Venkaiah and R. Madhavachary, Org. Lett., 2013, 15, 3042 CrossRef CAS PubMed; (c) D. B. Ramachary, C. Venkaiah and P. M. Krishna, Chem. Commun., 2012, 48, 2252 RSC; (d) F. Silva, M. Sawicki and V. Gouverneur, Org. Lett., 2006, 8, 5417 CrossRef CAS PubMed.
- (a) G. Talavera, E. Reyes, J. L. Vicario and L. Carrillo, Angew. Chem., Int. Ed., 2012, 51, 4104 CrossRef CAS PubMed; (b) D. Enders, M. R. M. Huttl, J. Runsink, C. Raabe and B. Wendt, Angew. Chem., Int. Ed., 2007, 46, 467 CrossRef CAS PubMed; (c) C.-L. Cao, X.-L. Sun, Y.-B. Kang and Y. Tang, Org. Lett., 2007, 9, 4151 CrossRef CAS PubMed; (d) D. Enders, M. R. M. Huttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861 CrossRef CAS PubMed; (e) D. B. Ramachary and C. F. Barbas III, Chem. – Eur. J., 2004, 10, 5323 CrossRef CAS PubMed.
- (a) A. R. Martin, K. Mohanan, L. Toupet, J. J. Vasseur and M. Smietana, Eur. J. Org. Chem., 2011, 3184 CrossRef CAS PubMed; (b) D. B. Ramachary, K. Ramakumar, A. Bharanishashank and V. V. Narayana, J. Comb. Chem., 2010, 12, 855 CrossRef CAS PubMed.
- (a) T. Mandal, S. Samanta and C.-G. Zhao, Org. Lett., 2007, 9, 943 CrossRef CAS PubMed; (b) H. Li, B. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 732 CrossRef CAS PubMed; (c) T. Marcelli, R. N. S. van der Haas, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 929 CrossRef CAS PubMed.
- (a) C. C. J. Loh, I. Atodiresei and D. Enders, Chem. – Eur. J., 2013, 19, 10822 CrossRef CAS PubMed; (b) M. Tsakos, M. R. J. Elsegood and C. G. Kokotos, Chem. Commun., 2013, 49, 2219 RSC; (c) B. Tan, Y. Lu, X. Zeng, P. J. Chua and G. Zhong, Org. Lett., 2010, 12, 2682 CrossRef CAS PubMed; (d) M. Rueping, A. Kuenkel and R. Frohlich, Chem. – Eur. J., 2010, 16, 4173 CrossRef CAS PubMed; (e) B. Tan, P. J. Chua, X. Zeng, M. Lu and G. Zhong, Org. Lett., 2008, 10, 3489 CrossRef CAS PubMed; (f) E. Reyes, H. Jiang, A. Milelli, P. Elsner, R. G. Hazell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2007, 46, 9202 CrossRef CAS PubMed.
- (a) D. B. Ramachary and K. Ramakumar, Eur. J. Org. Chem., 2011, 2599 CrossRef CAS PubMed; (b) D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865 CrossRef CAS PubMed.
- (a) P. Garcia-Garcia, A. Ladepeche, R. Halder and B. List, Angew. Chem., Int. Ed., 2008, 47, 4719 CrossRef CAS PubMed; (b) H. Gotoh, H. Ishikawa and Y. Hayashi, Org. Lett., 2007, 9, 5307 CrossRef CAS PubMed.
- (a) S. Anwar, H. J. Chang and K. Chen, Org. Lett., 2011, 13, 2200 CrossRef CAS PubMed; (b) D. Enders, A. A. Narine, T. R. Benninghaus and G. Raabe, Synlett, 2007, 1667 CrossRef CAS PubMed.
- CCDC 1038959 for (−)-4aa and CCDC 1038960 for (−)-7bl contain the supplementary crystallographic data for this paper.
- (a) R. Madhavachary and D. B. Ramachary, Eur. J. Org. Chem., 2014, 7317 CrossRef CAS PubMed; (b) D. B. Ramachary and Y. V. Reddy, J. Org. Chem., 2010, 75, 74 CrossRef CAS PubMed; (c) D. B. Ramachary and M. Kishor, J. Org. Chem., 2007, 72, 5056 CrossRef CAS PubMed; (d) D. B. Ramachary, M. Kishor and G. Babul Reddy, Org. Biomol. Chem., 2006, 4, 1641 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data (1H NMR, 13C NMR, HRMS and HPLC) for all new compounds. CCDC 1038959 and 1038960. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob00562k |
| This journal is © The Royal Society of Chemistry 2015 |