
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of preorganization on the affinity of synthetic DNA binding motifs for nucleotide ligands†
S.
Vollmer
and
C.
Richert
*
Institute for Organic Chemistry, University of Stuttgart, 70569 Stuttgart, Germany. E-mail: lehrstuhl-2@oc.uni-stuttgart.de; Fax: +49 (0) 711 608 64321; Tel: +49 (0) 711 608 64311
First published on 22nd April 2015
Abstract
Triplexes with a gap in the purine strand have been shown to bind adenosine or guanosine derivatives through a combination of Watson–Crick and Hoogsteen base pairing. Rigidifying the binding site should be advantageous for affinity. Here we report that clamps delimiting the binding site have a modest effect on affinity, while bridging the gap of the purine strand can strongly increase affinity for ATP, cAMP, and FAD. The lowest dissociation constants were measured for two-strand triple helical motifs with a propylene bridge or an abasic nucleoside analog, with Kd values as low as 30 nM for cAMP in the latter case. Taken together, our data suggest that improving preorganization through covalent bridges increases the affinity for nucleotide ligands. But, a bulky bridge may also block one of two alternative binding modes for the adenine base. The results may help to design new receptors, switches, or storage motifs for purine-containing ligands.
Introduction
Nucleoside phosphates are key building blocks of the cell. Triphosphates are the monomers for both DNA and RNA synthesis.1 Adenosine monophosphate is also part of the structure of several cofactors, including acetyl-CoA, nicotinamide adenine dinucleotide (NADH/NAD+), flavin adenine dinucleotide (FADH2/FAD), and S-adenosylmethionine (SAM). Further, cyclic anhydrides of purine nucleotides are second messengers, including 3′,5′-cycloadenosine monophosphate (cAMP) and 3′,5′-cycloguanosine monophosphate (cGMP), with important functions in intracellular signal transmission and amplification.2 Binding nucleoside phosphates with high affinity and selectivity is therefore critical, and nucleotide binders are a focus of studies in analytical chemistry,3,4 medicine,5 and chemical biology.6Synthetic nucleotide binders have been constructed from a number of different compound classes. Among them are molecular tweezers,7 scorpiand receptors,8 uranyl-salophen receptors,9 pyridinium tripods,10 copper complexes,11 and phenylenediamine receptors.12 Most of these purely synthetic binders give dissociation constants in the millimolar to micromolar range. Another class of binders are nucleic acid-based binding motifs. Best known are aptamers, i.e. RNA sequences identified from sequence libraries by systematic evolution by exponential enrichment (SELEX).13 Aptamers for a number of nucleoside phosphates or cofactors, including ATP,14,15 cAMP,16 SAM,17 FAD,18,19 and GTP20 are known, and so are ribonucleopeptides21,22 that bind ATP or GTP.
Recently, designed nucleic acid-based binders for purines were reported that feature a gap in the oligopurine strand of a triplex as binding site.23–25 The designed binding motifs show low micromolar affinity for biologically important nucleoside phosphates and cofactors and bind their targets through a combination of Watson–Crick and Hoogsteen base pairing (Fig. 1). They can be immobilized on porous supports, leading to macroscopic storage devices that can reversibly bind cofactors, so that cofactor-dependent biochemical transformations can be switched on or off through modest temperature changes.24 The selectivity for different cofactors can be tuned through small changes in the sequence, and affinity for non-nucleosidic cofactors, such as folic acid can be induced.24 An RNA version of a triplex-based motif has been expressed in live cells, where it acted as a sink for cGMP, a secondary messenger.25 Triplex-based motifs have also been used for the construction of fluorescence sensors for several purines using fluorophores covalently linked to nucleobases of one of the triplex strands.26,27
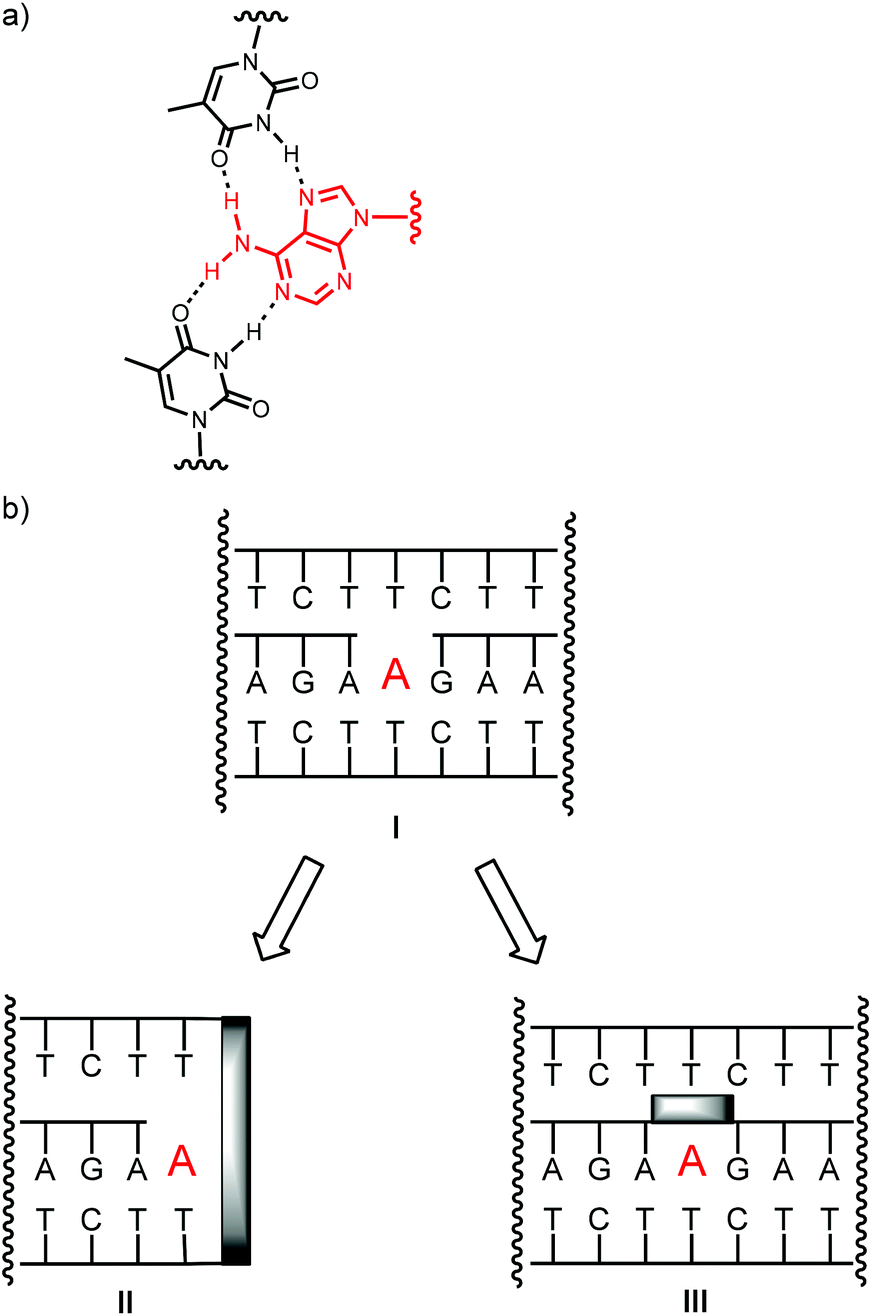 | ||
| Fig. 1 Binding adenosine through a combination of Watson–Crick and Hoogsteen base pairing. (a) Base pairing interactions, (b) strands arrangements in triple helices without (I), with a clamp (II), or with a bridge (III). | ||
Pairing between oligonucleotides is known to be accompanied by strong enthalpy-entropy compensation.28 The large entropic penalty for duplex formation can be reduced by rigidifying the strands involved. The most common way of achieving this is by clamping down on conformational flexibility through an additional covalent linkage. Locked nucleic acids (LNAs) are one well-known example for this, relying on bridges between the 2′- and 4′-positions of the riboses.29,30 Alternatively, two nucleotides may be bridged with a synthetic lock. For example, intrastrand locks were recently shown to increase the affinity and base pairing selectivity of DNA probes binding RNA target strands.31 Interstrand crosslinking with disulfide bridges,32 metal-salen complexes,33 or triazolides34 as bridges can strongly stabilize duplexes. When the synthetic lock is at the termini of two complementary strands, hairpins are formed. Here, the lock may be constructed from a stilbene diether35,36 or perylene diimide.37 Clamped constructs are also known for parallel DNA triplexes.38 Typically, the two homo-pyrimidine strands of a triplex are linked by a naphthalene diimide or perylene diimide clamp, leading to increases in free energy of formation of up to −12.3 kcal mol−1.39,40
It was therefore interesting to ask whether covalent locks in the structure of triplex-based binders for nucleotides improve pairing properties and thus target affinity. Since known triplex-based DNA motifs are at least 50 nucleotides in size, it was also desirable to prune them. The shortening of the strands was expected to labilize triplexes, further motivating our search for ways to rigidify the gap-containing, nucleotide-binding triplex structures. Here we report the synthesis of oligonucleotides that form triplex-based binding motifs with a clamp flanking the binding site (II) or a bridge connecting the termini of the oligopurine strands (III, Fig. 1b).
Either type of construct was found to form stable triplexes, as determined by UV-melting analysis and to bind cofactors, such as SAM, FAD, ATP, or cAMP, as determined by equilibrium filtration. Depending on the design of the oligonucleotides, micro- to nanomolar dissociation constants were found, with the tightest binding in the case of bridged triplexes and cAMP as ligand, and the weakest binding in the case of ATP.
Results
Scheme 1 shows the oligonucleotides synthesized to form triple helices of general structure II. The group of strands included all-pyrimidine strands 1–5 that can form an intermolecular triplex, and intramolecular folding motif 6 that features both the two oligopyrimidines and the oligopurine segment of a parallel triplex. The strands were prepared by automated DNA synthesis, starting from controlled pore glass (cpg) loaded with the 3′-terminal nucleoside, using a combination of conventional phosphoramidites and building blocks synthesized according to slight modifications of literature protocols. Compound 1, which features a T5 clamp, lacks modified residues and was purchased from a commercial source. Oligonucleotide 2 contains a hexaethylene glycol (HEG) linker as clamp and was prepared with phosphoramidite 8 as unnatural building block. The loop serving as clamp in 3 is a combination of a T3 segment and the pyrenyl C-nucleoside (Py), incorporated by employing phosphoramidite 9, prepared as previously reported.41,42 The pyrenyl residue can be expected to stack on the termini.41,43,44 Finally, the largest and presumably most strongly preorganizing clamp was incorporated in 5 and 6 by using the perylene diimide building block 10.39,40 Of the two strands, all-pyrimidine sequence 5 has a larger number of deoxycytidine residues, as compared to 1–4. The triple helical binding motif 6 was expected to form the desired triplex helical structure through intramolecular folding, with the PDI residue again designed to act as clamp sealing the binding site for adenosine-containing ligands. An entirely intramolecular triple helical motif is better suited for immobilization and thermal capture/release cycles without loss of strands than a bimolecular system formed by hybridization of unlinked strands.24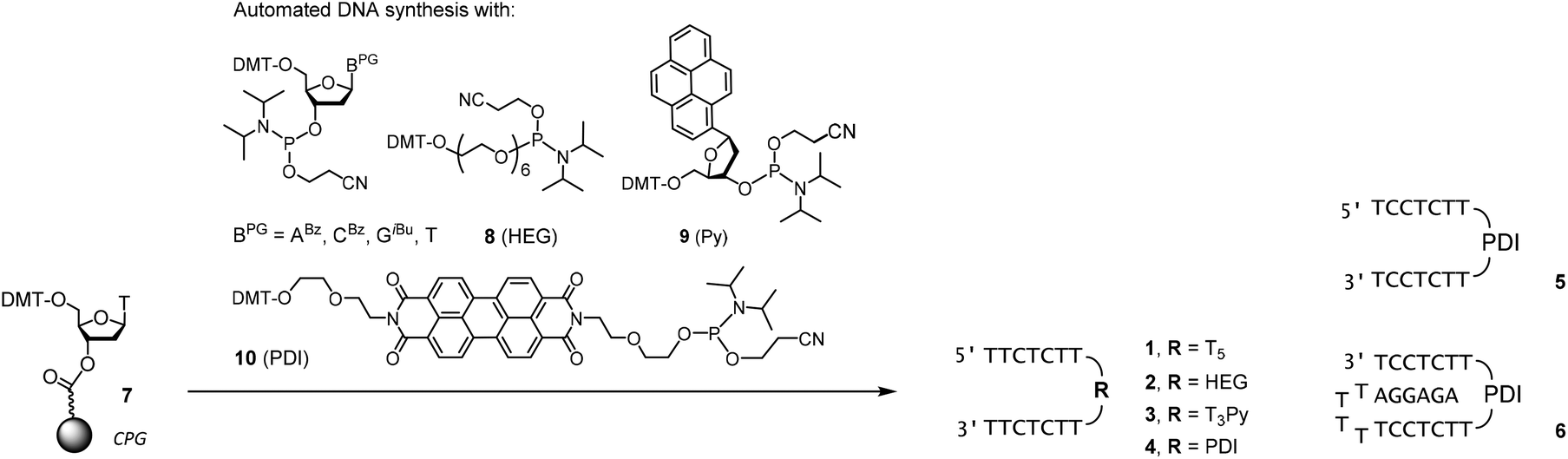 | ||
| Scheme 1 Synthesis of oligodeoxynucleotides 1–6. | ||
All six oligonucleotides (1–6) were HPLC purified and characterized by MALDI-TOF mass spectrometry. Overall yields of the oligonucleotide syntheses ranged from 3–23%, a range typical for custom-synthesized, modified DNA strands. Fig. 2 shows a representative spectrum. Other spectra can be found in the ESI (Fig. S8–S15†).
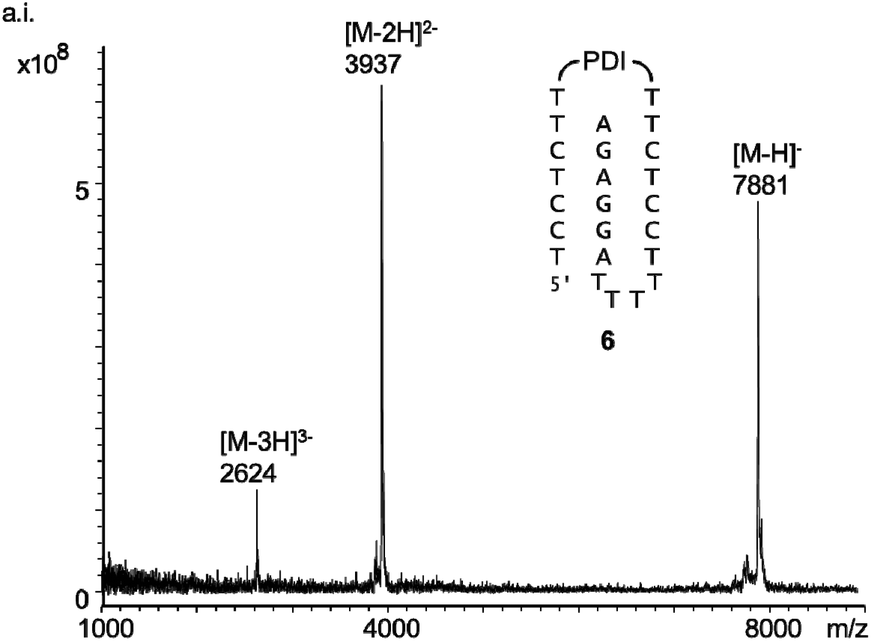 | ||
| Fig. 2 MALDI-TOF spectrum of 6 (linear negative mode). | ||
Fig. 3 shows triple helical structures expected to form upon hybridizing 1–5 with target strands 11 or 12. Either of the purine-rich strands contained a hairpin sequence to minimize frame-shifting that leads to alternative structures, with the ligand binding site blocked. Further, the increase in size caused by the hairpin facilitated separation of ligand and oligonucleotide motif in filtration assays (vide infra). The sequence of the hairpin segment was chosen to be GC-rich, in order to prevent cross hybridization with the TA-rich triple helical segment. Finally, triple helices 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 and 6 present a slightly different electrostatic profile, compared to 1–4:11, due to the additional CGC+ base triplet.
:12 and 6 present a slightly different electrostatic profile, compared to 1–4:11, due to the additional CGC+ base triplet.
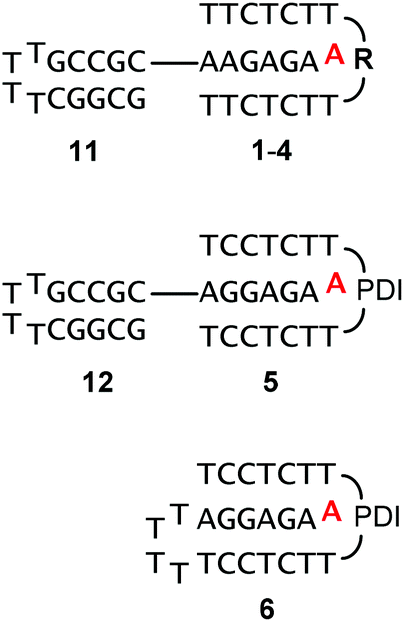 | ||
| Fig. 3 Triple-helices formed between 1–5 and oligopurine targets 11 or 12, or by intramolecular folding (6). Ligands are shown in red for clarity. See Scheme 1 for the structure of clamp residues. | ||
For all six designs, the formation of triplexes was induced by thermal hybridization, and UV-melting curves showed melting points in the range of 14–42 °C, that is above the temperature at which binding of ligands was measured (4 °C). The ability of the triple helices to bind cAMP, SAM or FAD was tested by membrane filtration, following a protocol previously described.23,24 Binding constants were determined by calculation of the fraction of bound ligands from the concentration found in the filtrate, as compared to that of a control filtration without oligonucleotide motifs. A representative set of UV/VIS-spectra for FAD is shown in Fig. 4. The dissociation constants calculated from the fractions of bound and unbound ligand are given in Table 1.
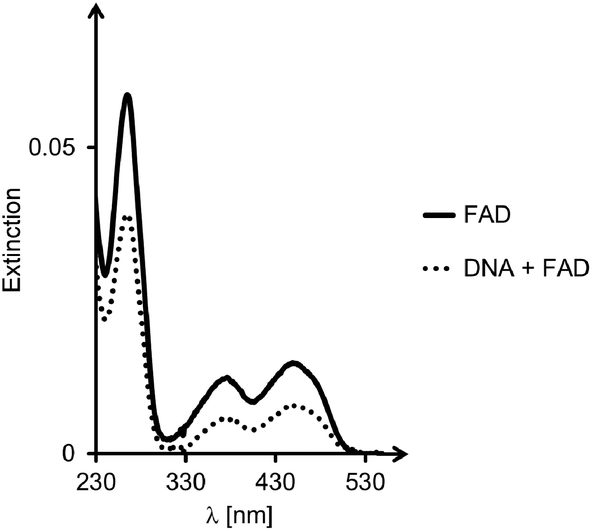 | ||
| Fig. 4 Representative UV/VIS-spectra from binding assay. Spectra of FAD solutions after exposure to 6 and filtration. The bold line is the spectrum of the reference filtration, in the absence of oligonucleotides, whereas the dotted line is from the sample containing both DNA triplex and ligand. Conditions: 10 μM DNA, 10 μM FAD, 10 mM phosphate buffer (pH = 6), 1 M NaCl; centrifugation at 4 °C, using filters with a 3 kDa size cut-off. | ||
| Sequences | Clamp | K d (cAMP) | K d (SAM) | K d (FAD) |
|---|---|---|---|---|
| a As determined by equilibrium filtration; conditions: 10 μM DNA strands, 10 μM ligand, 10 mM phosphate buffer (pH = 6), 1 M NaCl, 4 °C. | ||||
|
1:11 |
T5 | >150 | >150 | >150 |
|
2:11 |
HEG | 90 | >150 | >150 |
|
3:11 |
Py | 60 | 80 | >150 |
|
4:11 |
PDI | 40 | 50 | 9 |
|
5:12 |
PDI | >150 | 98 | 8 |
| 6 | PDI | 80 | 34 | 8 |
Triple helical system 1:11 showed no detectable binding of either of the three ligands in our assay, with dissociation constants >150 μM (Table 1). Possibly, the flexible T5-clamp does not offer a sufficient level of preorganization at the binding site, and insufficient stacking interactions for incoming adenosine phosphates. Bimolecular complex 2:11, with its shorter linker, known from similar DNA helices,45 but limited stacking capabilities, gave detectable binding for cAMP, but no measurable affinity for SAM or FAD. Pyrene-containing 3 formed a triplex with 11 that gave detectable affinity for cAMP and SAM, suggesting that its aromatic stacking moieties did attract adenosine phosphates more readily than the triplexes formed by either 1 or 2. Only when the large aromatic ring system of the perylene diimide was offered as clamp structure (4:11), did significant binding occur for any of the three ligands. Here, for FAD, with its large flavin residue, a low micromolar Kd was measured.
Perylenes have previously been reported to be excellent stacking partners for base triplets.39 Triplex 5:12, with its less stable triplex gave lower affinity for both cAMP and SAM, suggesting that preorganization is indeed important for binding nucleotide ligands tightly. Finally, 6 which forms an intramolecular triplex, showed strong affinity for all three cofactors tested, with Kd values in the range of 8–80 μM. Neither of the ligands induced a shift in the melting point, though, when added to the solution of the triplexes of this group.
The results obtained for 1–6 indicated that the affinity for ligands can be tuned by the choice of the clamp. But, it was also evident that for the clamp-containing triple helices, the affinity for cAMP, FAD and SAM was lower than for triplexes with a binding site in the interior of a triple helix,23–25 rather than at its terminus. This made us turn to binding motifs of general structure III (Fig. 1) and designs that feature a bridge between the oligopurine strands of the central strand. The bimolecular triple-helical structures shown in Fig. 5 were therefore pursued next. This included 13 and 14, which feature hexa- and tetranucleotide loops similar to those described in earlier work on folding motifs than can be expressed in cells.25 Shorter loops had proven poor bridges in our earlier studies,25 possibly because of conformations that interfere with binding. Also included in the group of bridged oligopurines were 15 and 16 that feature acyclic chains with a long (HEG)45 or a short linear chain (C3).26,27 The latter has the same number of carbon atoms between two phosphates as a natural ribose (shortest path along the backbone). Presumably the most rigid bridge is that of 17, with an abasic site analog (AS) replacing a natural deoxynucleoside residue. Abasic sites have been tested before in duplexes,46–48 but, to the best of our knowledge, not in triplex binding motifs. In 17, the AS-bridge leaves space for the nucleobase only. It occupies the space usually taken up by the ribose phosphate of the ligand, at least, if the ligand was to bind in the same orientation as the deoxyadenosine of an uninterrupted oligopurine chain. Finally, oligopurine 18, which features an uninterrupted strand was included as control compound, forming triplexes that do not contain a binding site.
 | ||
| Fig. 5 Triple-helices between bridged oligopurines (13–18) and oligopyrimidine 19. | ||
For all but one of the six different triplex structures, the triplex-to-duplex and duplex-to-single strands transitions coincided, so that a single sigmoidal curve was observed, even in the absence of ligands (see the left-most curve in Fig. 6a). Only for the triple helix 14:19 was the UV-melting point for the dissociation of the triplex-forming segment low enough to give a discernible second transition (see Fig. S19, ESI†). Three different adenosine phosphates were tested as ligands, namely cAMP, FAD, and ATP. The triphosphate was included because it should be a weak ligand, due to the electrostatic repulsion between the DNA backbones and the oligophosphate moiety. In the case of cAMP and 17:19, the ligand induced a significant shift in the UV-melting point (Fig. 6a and Table 2), suggesting that the ligand was bound so tightly that it remained in the binding pocket up to the temperature where the triplex-forming segment dissociated from the remaining duplex. This phenomenon has been observed previously for very stable complexes with triple helical binding motifs.23,24 Triplex 16:19 with the propylene bridge showed a similar effect, with a ΔTm of 4.4 °C in the triplex-to-duplex transition. The addition of Mg2+ (10 mM) resulted in a slight increase in the UV-melting point (+1.3 °C). When the pH was raised to a value of 7.0, melting occurred 6.0–7.5 °C lower than at pH 6.0, where triplexes containing cytosines are more stable (Table S1, ESI†). Interestingly, the drop was slightly less pronounced in the presence of cAMP as ligand.
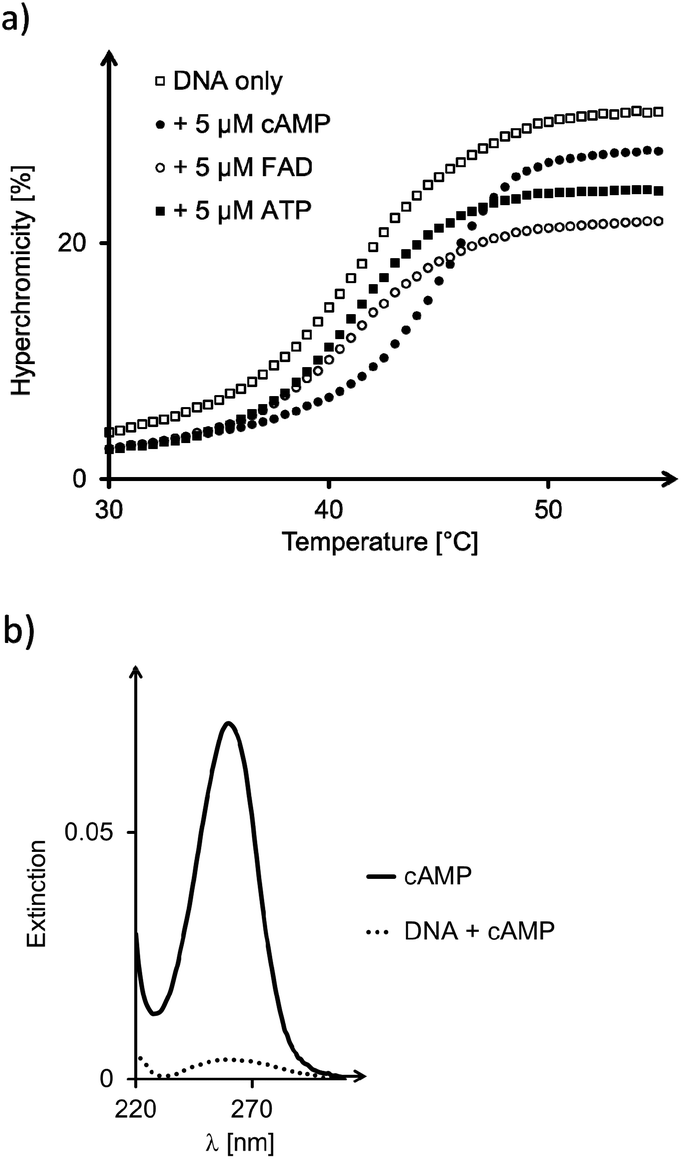 | ||
| Fig. 6 (a) UV-melting curves of triplex 17:19 alone and in the presence of cAMP, FAD, or ATP. Conditions: 1 μM DNA, 5 μM ligand, 10 mM phosphate buffer (pH = 6), 1 M NaCl, heating rate 1 °C min−1. (b) UV/VIS-spectra of filtrate of triplex 17:19 with cAMP. Conditions: 10 μM DNA, 10 μM cAMP, 10 mM phosphate buffer (pH = 6), 1 M NaCl; centrifugation at 4 °C, using filters with a cut-off of 3 kDa. | ||
| Sequences | Bridge | DNA only | ATP | cAMP | FAD |
|---|---|---|---|---|---|
| a Conditions: 1 μM DNA, 5 μM ligand, 10 mM phosphate buffer (pH = 6), 1 M NaCl, heating rate 1 °C min−1. b A single UV-transition, encompassing the triplex-to-duplex and duplex-to-single-strand transitions were observed. c Separate triplex-to-duplex and duplex-to-single-strand transitions observed, both melting points given. | |||||
|
13:19 |
T6 | 37.3 | 37.1 | 36.9 | 37.2 |
|
14:19 |
T4 | 34.0/44.0c | 34.1/44.1c | 34.6/44.4c | 35.2/44.2c |
|
15:19 |
HEG | 38.4 | 38.9 | 39.7 | 38.5 |
|
16:19 |
C3 | 40.0 | 40.6 | 44.4 | 41.1 |
|
17:19 |
AS | 40.7 | 41.9 | 45.4 | 41.9 |
|
18:19 |
A | 60.6 | 60.3 | 60.3 | 60.9 |
High affinity for adenosine phosphate ligands was also found in binding assays based on equilibrium filtration (Table 3). The group of nucleotides assayed now also included cGMP as control compound that offers a stacking surface similar to that of cAMP, but a different hydrogen bonding pattern, so as to test for the specificity of base pairing. A pair of representative absorption spectra from the binding assay involving 17:19 and cAMP are shown in Fig. 6b. It can be discerned that little unbound ligand remained in solution. Hexanucleotide-bridged 13 formed a binding motif that bound cAMP and FAD with approx. 20 μM Kd, but both ATP and cGMP too poorly to give a reliable dissociation constant (Table 3). The shorter tetranucleotide loop of 14 led to increased affinity for all complementary ligands, including ATP, but no detectable binding of cGMP. A hexaethylene glycol bridge (15) improved binding of ATP, but had a modest effect on the stability of complexes with cAMP and FAD. As with all other triplexes, no binding of cGMP was detected. Nor did the control triplex 18:19 show affinity of any of the nucleotides tested.
| Sequences | Bridge | ATP | cAMP | FAD | cGMP |
|---|---|---|---|---|---|
| a As determined by equilibrium filtration; conditions: 10 μM DNA strands, 10 μM ligand, 10 mM phosphate buffer (pH = 6), 1 M NaCl, 4 °C. | |||||
|
13:19 |
T6 | >150 | 20 | 16 | >150 |
|
14:19 |
T4 | 129 | 8 | 10 | >150 |
|
15:19 |
HEG | 29 | 2.8 | 15 | >150 |
|
16:19 |
C3 | 2.8 | 0.04 | 1.4 | >150 |
|
17:19 |
AS | 5 | 0.03 | 1.8 | >150 |
|
18:19 |
A | >150 | >150 | >150 | >150 |
Very tight binding of cAMP was found for both 16:19 and 17:19. Here, dissociation constants below 50 nM were determined, which is close to the detection limit of our absorption-based assay. The strongest complex was found for cAMP and 17:19, with a Kd of 30 nM. Tight binding was confirmed independently by analyzing the complex retained by the filter via HPLC (Fig. S25, ESI†). For both ATP and FAD, the triplex with the propylene chain strand gave the lowest Kd value, though, suggesting that it is the better binding motif for these larger ligands.
Discussion
Several aspects of the current results are interesting. The fact that neither of the triplexes bound guanine-containing cGMP and that control triplex 18:19 lacking a binding site did not show affinity for any of the ligands tested suggests that binding does indeed occur by a combination of Watson–Crick and Hoogsteen binding. Further, preorganization appears to be advantageous for tight binding, both on the side of the triplex motif and on the side of the ligand, as suggested by the low dissociation constant measured for the complex of 17:19 and cAMP. In this triple helical motif, the entire structure but the nucleobase itself was retained from a continuous triple helix. The cyclic AMP is also significantly more rigid than adenosine monophosphate, as the annelation freezes out the conformational flexibility of the ring.
Earlier studies of DNA and RNA triplexes binding cGMP had led to a hexanucleotide loop as preferred linker.25 Our structure search now led to much shorter linkers, with a backbone close to that of a single nucleotide residue but lacking the adenine base (16 and 17) as the preferred binders, with the highest affinity for the ligands tested. The HEG bridge of 15 has a length between that of the hexa- and tetranucleotide loops of 13 and 14, and the short linkers that were modeled after a single nucleotide.
Naïvely, the observation that bridges with the backbone length of a natural nucleotide (C3, AS) should allow the binding of an adenine phosphate ligands at all, is surprising, as the backbone of the triplex and the ribose phosphate should get into a steric conflict (Fig. 7, binding mode A). However, a more careful inspection of possible binding modes reveals that there is a second, alternative binding orientation, with the nucleobase flipped by 180° (binding mode B in Fig. 7).49 The abasic site-bridged triplex has a more rigid, cyclic linker structure, and can be expected to be slightly better preorganized towards ligand binding than the C3-containing analogue with its acyclic backbone. However the abasic site-containing triplex will almost certainly suffer the steric clash that prevents binding mode A, resulting in an entropically less favorable situation. This may explain why 16 produces the better binders for two of the three adenosyl ligands tested, as compared to 17. It is unclear whether the propylene chain of the C3-bridge fully suppresses binding mode A, or allows it, with slightly distorted backbone geometry in the linker between the oligopurine segments. We are currently initiating a project aimed at elucidating the full three-dimensional structure of C3-bridged triplex with nucleotide ligands bound in order to shed light on this.
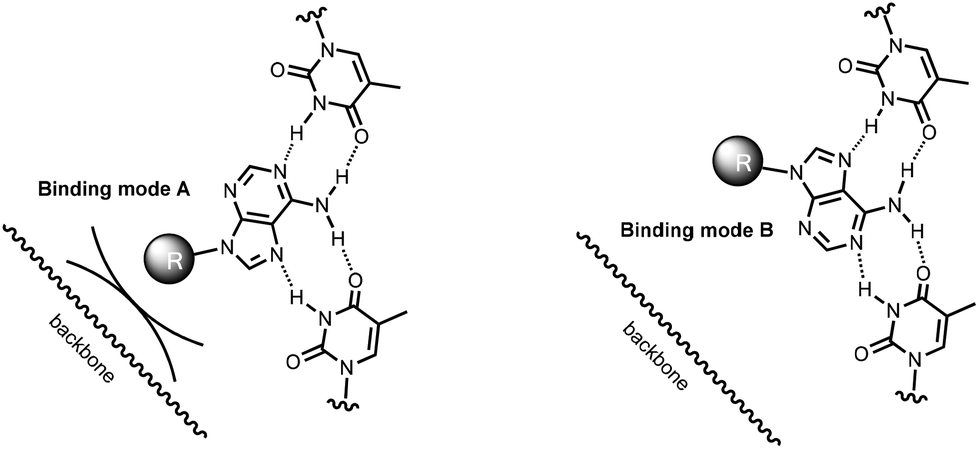 | ||
| Fig. 7 Two orientations allow an adenosyl ligand to bind via a combination of Watson–Crick and Hoogsteen base pairing. Binding mode A differs from binding mode B by a 180° flip of the ligand and avoids a steric clash of the ribose phosphate (abbreviated as a ball) and the linker backbone of the oligopurine strand (shown as a wobbly line). | ||
Further, one may ask whether anything can be learned from the stability of the triplex motifs prior to ligand binding (and thus the level of preorganization) and the stability of the desired complexes with our ligands. The UV-melting points listed in Table 2 allow a measure of how well the triplex-forming strand or segment is anchored on the underlying duplex regions. “Carving out” the binding site destabilizes the triplex thermally, with the single melting transitions shifted to lower temperatures by at least 20 °C for all binding motifs, compared to control triplex 18:19 (third column of Table 2). Starting with the constructs featuring oligonucleotide loops (13 and 14), melting points then gradually increase, with HEG-linked triplex 15:19 melting slightly higher and those featuring the C3 or abasic site moiety giving melting points that are higher still (16:19 and 17:19). A plot of the triplex melting points against the dissociation constants of complexes with the three ligands tested (Fig. 8) shows a rough correlation. A close numerical correlation would have been unreasonable to expect, as this level of analysis ignores alternative binding modes (Fig. 7) and thus possible conformations of the triplex that may be stable but are unproductive.
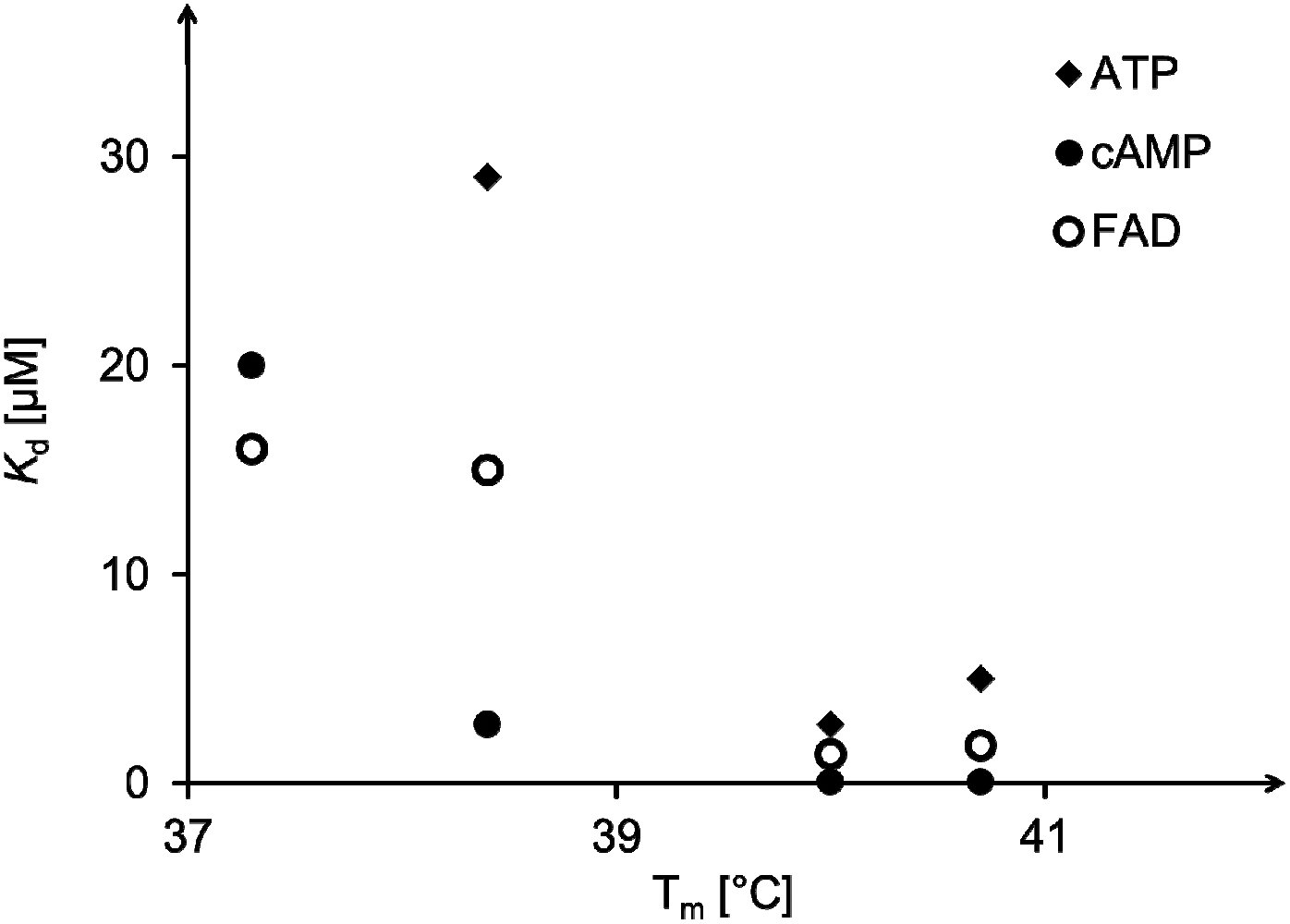 | ||
| Fig. 8 Correlation between triplex melting temperature in the absence of a ligand and dissociation constants of complexes formed with ATP, cAMP or FAD as ligands. See Tables 2 and 3 for details. | ||
Some unproductive conformations that may exist are shown in cartoon format in Fig. 9. In either design (terminal binding site or interior binding site), a blocked state can readily be imagined. For clamp-containing structures, the stacking moiety may stack directly on the terminal base triplet (state V), thus preventing the ligand from binding. In bridged systems, a well-stacking or well preorganizing linker can be expected to favor the productive, open state (VII), making the blocked state, with the complementary thymidines bulged out (VIII), less likely, and thus preorganizing the binding motif toward a complex with the ligands. Unless in doing so the linker blocks one binding mode of the adenine base (Fig. 7), it will most probably favor strong binding, both on enthalpic and entropic grounds. This may, at least in part, explain why abasic site-bridged triplex 17:19 forms such a stable complex with cAMP.
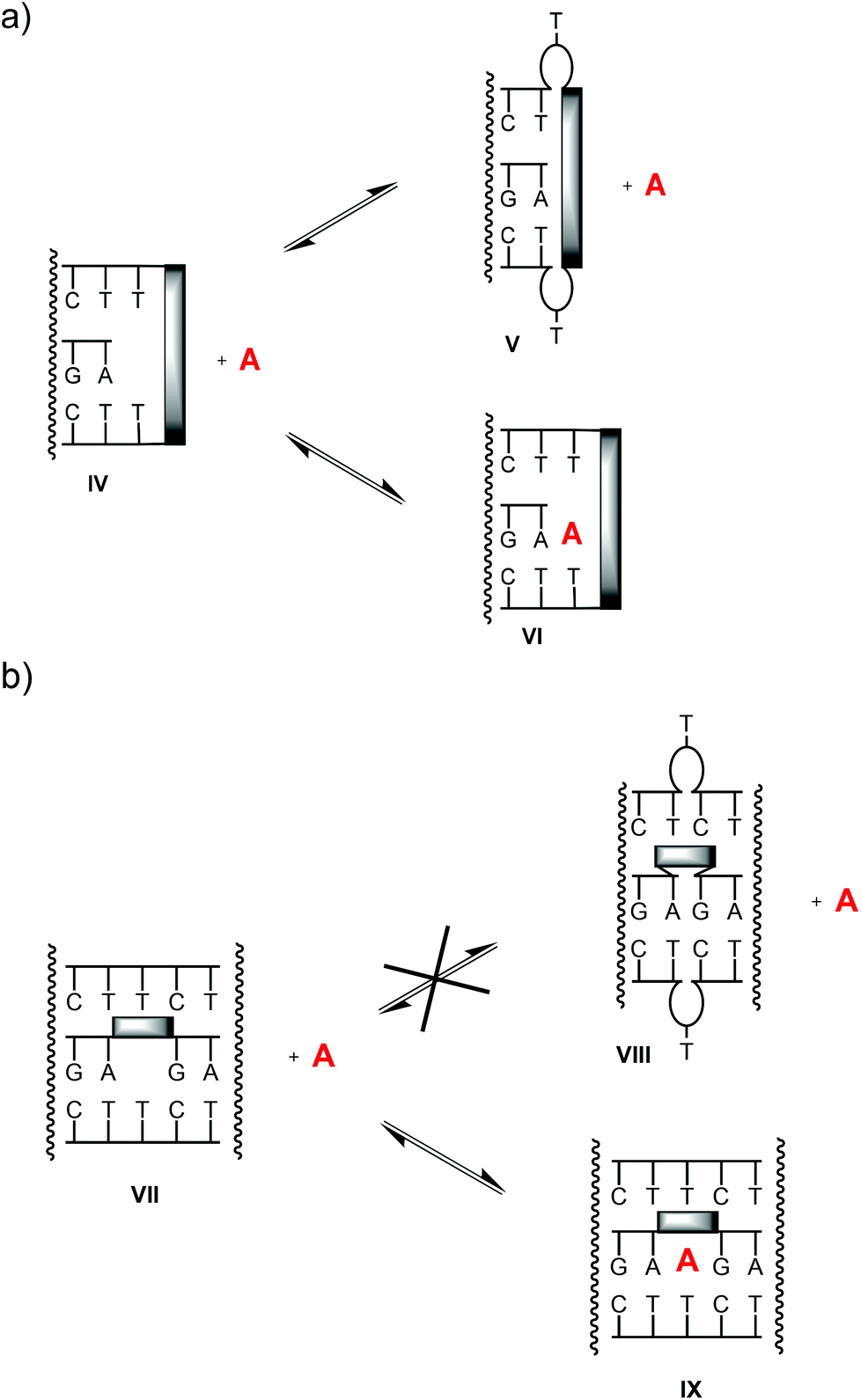 | ||
| Fig. 9 Possible equilibria between triplex motifs with productive, accessible binding sites (states IV, VII), blocked binding sites (V, VIII) and complexes with ligand A (VI, IX). | ||
Conclusion
Among the two different designs of triplex-based DNA binding motifs tested, the one with a lock bridging the oligopurine strands of two triplex segments was found to bind adenosine-containing ligands more tightly than the one with a binding site at the terminus of a single triple helix. Among the ligands tested, cAMP gave the lowest dissociation constants, lower than those of the larger ligands with much more total stacking surface (FAD) or electrostatically more favorable ligands (SAM). Further, the tightest complex was found for a triplex with a short bridge with a sterically demanding and conformationally restricted abasic nucleotide analog (17:19:cAMP, Kd = 30 nM). This level of affinity rivals that of riboswitches binding nucleobases by engulfing them in the interior of their folded structure.50 In our case, thermal stability of ligand-free triplexes correlates roughly with binding affinity towards ATP, FAD and cAMP. These results point to preorganization as an important factor in the binding of adenosine phosphates by triplex-based DNA binding motifs that can engage in Watson–Crick and Hoogsteen base pairing.
Experimental
General
Ligands cAMP, SAM, and ATP as well as the reagents were from Sigma-Aldrich/Fluka (Deisenhofen, Germany), Acros (Geel, Belgium), or Fluorochem (Karlsruhe, Germany). Unmodified oligonucleotides and strand 15–17 were used obtained from Biomers (Ulm, Germany) in HPLC-purified form and were without further purification. Flavin adenine dinucleotide was obtained from Alfa Aesar (Karlsruhe, Germany), cGMP was from Carbosynth (Compton, Berkshire, UK). The reagents 2-cyanoethyl N,N-diisopropylchlorophosphoramidite and 17-O-(4,4′-dimethoxytrityl)-hexaethyleneglycol-1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphor-amidite were from ChemGenes (Wilmington, USA). All chemicals used for automated solid-phase DNA synthesis were from Proligo (Hamburg, Deutschland), including the unmodified phosphoramidites of deoxynucleosides, DCI activator (0.25 M 4,5-dicyanoimidazol in acetonitrile), the cpg loaded with the first deoxynucleoside (pore size 1000 Å, 27–32 μmol g−1 loading), amidite diluent (abs. acetonitrile, ≤90 ppm water), tetrazole solution (0.45 M in abs. acetonitrile), cap B (THF/pyridine/1-methylimidazole 80:10:10 v/v/v), cap A (THF/tert-butylphenoxyacetic anhydride 100:5 v/v), oxidizer solution (0.1 M iodine in THF/pyridine/water 77:21:2 v/v/v), and deblock solution (trichloroacetic acid/dichloromethane 3:97 v/v).
:1:1, v/v/v) to yield 0.41 g (4.7 mmol, 25%) of compound 20 as a red solid. TLC, dichloromethane/methanol/trimethylamine, 98:1:1, v/v/v: Rf = 0.28. The spectroscopic data were in agreement with the data reported in literature.39
Synthesis of modified DNA strands 2–6
Modified oligonucleotides were synthesized on a 8909 Expedite synthesizer (Perseptive Biosystems Foster City, CA) with Expedite Workstation as software (version 2.5) according to the 1 μmol standard protocol. For modified phosphoramidites (8–10) 0.1 M solutions in acetonitrile were used, and the coupling times were extended to 440 s. All strands were deprotected and cleaved from solid support by treatment with aqueous ammonia at 55 °C for 16 h. Crudes were purified by reversed-phase HPLC on a Dynamax System (Rainin) with an EC 250/4.6 Nucleosil 120-5 C4 (Macherey-Nagel) column and a gradient of acetonitrile in 0.1 M triethylammonium acetate buffer, pH 7.0. Yields were determined by UV-absorption at 260 nm.Binding constants via equilibrium filtration
Dissociation constants were determined using an established equilibrium filtration assay.23,24 For this, solutions of the oligonucleotides and ligands (10 μM each) in 10 mM phosphate buffer, 1 M NaCl, pH = 6.0 were prepared, together with a reference sample containing ligand only in buffer. For annealing, solutions (510 μL) were heated to 55 °C and allowed to cool to r.t. in 3 h. After 30 min at r.t., samples were cooled to 4 °C and kept at this temperature overnight. The solutions were then loaded on Amicon ultracentrifugal filter units from Millipore with a molecular weight cutoff of 3 kDa and centrifuged for 1 h at 14000g and 4 °C. The filtrates (450 μL) were diluted with water (150 μL), and the absorption was measured in the range of 600–220 nm. The concentration of ligand in the filtrate (unbound ligand) was calculated according to Lambert-Beer's law with the following extinction coefficients: ε260(cAMP) = 12300 M−1 cm−1, ε260(ATP) = 15400 M−1 cm−1, ε260(SAM) = 15400 M−1 cm−1, ε450(FAD) = 11300 M−1 cm−1, ε254(cGMP) = 13600 M−1 cm−1.
UV melting curves
The UV melting curves were measured on a Lamda 25 spectrophotometer (Perkin Elmer) with peltier-based temperature control. Sample were 1 μM oligonucleotides in 10 mM phosphate buffer, 1 M NaCl, pH = 6.0 with or without 5 μM ligand. Absorption was measured at 260 nm in the temperature range between 5 °C and 85 °C with a heating and cooling rate of 1 °C min−1. For each sample, two heating and cooling curves were recorded. The melting points were calculated as the extrema of the first derivation of the heating curves using UV-WinLab (Perkin Elmer).MALDI-TOF mass spectrometry
The MALDI-TOF mass spectra were measured on a Bruker Reflex IV or Microflex spectrometer in linear negative mode with an external calibration. For sample preparation 0.5 μL of the oligonucleotide solution (10−6–10−8 M in water) were put on an MTP AnchorChip™ target (Bruker) and evaporated to dryness at 5 mbar. Then, 0.5 μL of a matrix/comatrix solution (0.3 M 2,4,6-trihydroxyacetophenone monohydrate in ethanol/0.1 M diammonium citrate in water; 2:1 v/v) were pipetted onto the spot and allowed to dry at ambient pressure. The target was then introduced into the mass spectrometer.
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG, grant no. RI 1063/13-1 to C.R.). The authors thank A. Göckel for discussions, and H. Griesser, Dr E. Kervio and M. Kramer for a review of the manuscript.Notes and references
- A. Kornberg and T. A. Baker, DNAReplication, University Science Books, Sausalito, California, 2nd edn, 2005 Search PubMed.
- S. Fukumoto, H. Koyama, M. Hosoi, K. Yamakawa, S. Tanaka, H. Morii and Y. Nishizawa, Circ. Res., 1999, 85, 985–991 CrossRef CAS.
- Y. Xiao, L. Guo, X. Jiang and Y. Wang, Anal. Chem., 2013, 85, 3198–3206 CrossRef CAS PubMed.
- J. Adachi, M. Kishida, S. Watanabe, Y. Hashimoto, K. Fukamizu and T. Tomonaga, J. Proteome Res., 2014, 13, 5461–5470 CrossRef CAS PubMed.
- G. Haskó, J. Linden, B. Cronstein and P. Pacher, Nat. Rev. Drug Discovery, 2008, 7, 759–770 CrossRef PubMed.
- D. Kalia, G. Merey, S. Nakayama, Y. Zheng, J. Zhou, Y. Luo, M. Guo, B. T. Roembke and H. O. Sintim, Chem. Soc. Rev., 2013, 42, 305–341 RSC.
- H. Y. Kuchelmeister and C. Schmuck, Chem. – Eur. J., 2011, 17, 5311–5318 CrossRef CAS PubMed.
- (a) J. González-García, S. Tomić, A. Lopera, L. Guijarro, I. Piantanida and E. García-España, Org. Biomol. Chem., 2015, 13, 1732–1740 RSC; (b) M. Inclán, M. T. Albelda, E. Carbonell, S. Blasco, A. Bauzá, A. Frontera and E. García-España, Chem. – Eur. J., 2014, 20, 3730–3741 CrossRef PubMed.
- A. D. Cort, G. Forte and L. Schiaffino, J. Org. Chem., 2011, 76, 7569–7572 CrossRef PubMed.
- K. Ghosh, S. S. Ali, A. R. Sarkar, A. Samadder, A. R. Khuda-Bukhsh, I. D. Petsalakis and G. Theodorakopoulos, Org. Biomol. Chem., 2013, 11, 5666–5672 CAS.
- E. Kataev, R. Arnold, T. Rüffer and H. Lang, Inorg. Chem., 2012, 51, 7948–7950 CrossRef CAS PubMed.
- K. Ghosh and I. Saha, Org. Biomol. Chem., 2012, 10, 9383–9392 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- M. Sassanfar and J. W. Szostak, Nature, 1993, 364, 550–553 CrossRef CAS PubMed.
- M. Barbu and M. N. Stojanovic, ChemBioChem, 2012, 13, 658–660 CrossRef CAS PubMed.
- M. Koizumi and R. R. Breaker, Biochemistry, 2000, 39, 8983–8992 Search PubMed.
- D. H. Burke and L. Gold, Nucleic Acids Res., 1997, 25, 2020–2024 CrossRef CAS PubMed.
- P. Burgstaller and M. Famulok, Angew. Chem., Int. Ed. Engl., 1994, 33, 1084–1087 CrossRef PubMed.
- C. T. Lauhon and J. W. Szostak, J. Am. Chem. Soc., 1995, 117, 1246–1257 CrossRef CAS.
- Z. Huang and J. W. Szostak, RNA, 2003, 9, 1456–1463 CrossRef CAS.
- S. Nakano, T. Mashima, A. Matsugami, M. Inoue, M. Katahira and T. Morii, J. Am. Chem. Soc., 2011, 133, 4567–4579 CrossRef CAS PubMed.
- S. Nakano, M. Fukuda, T. Tamura, R. Sakaguchi, E. Nakata and T. Morii, J. Am. Chem. Soc., 2013, 135, 3465–3473 CrossRef CAS PubMed.
- C. Kröner, M. Röthlingshöfer and C. Richert, J. Org. Chem., 2011, 76, 2933–2936 CrossRef PubMed.
- C. Kröner, A. Göckel, W. Liu and C. Richert, Chem. – Eur. J., 2013, 19, 15879–15887 CrossRef PubMed.
- C. Kröner, M. Thunemann, S. Vollmer, M. Kinzer, R. Feil and C. Richert, Angew. Chem., 2014, 126, 9352–9356 ( Angew. Chem., Int. Ed. , 2014 , 53 , 9198–9202 ) CrossRef PubMed.
- M. Patel, A. Dutta and H. Huang, Anal. Bioanal. Chem., 2011, 400, 3035–3040 CrossRef CAS PubMed.
- Q. Zhang, Y. Wang, X. Meng, R. Dhar and H. Huang, Anal. Chem., 2013, 85, 201–207 CrossRef CAS PubMed.
- M. S. Searle and D. H. Williams, Nucleic Acids Res., 1993, 21, 2051–2056 CrossRef CAS PubMed.
- S. K. Singh, P. Nielsen, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455–456 RSC.
- A. A. Koshkin, P. Nielsen, M. Meldgaard, V. K. Rajwanshi, S. K. Singh and J. Wengel, J. Am. Chem. Soc., 1998, 120, 13252–13253 CrossRef CAS.
- C. Prestinari and C. Richert, Chem. Commun., 2011, 47, 10824–10826 RSC.
- N. C. Chaudhuri and E. T. Kool, J. Am. Chem. Soc., 1995, 117, 10434–10442 CrossRef CAS PubMed.
- K. V. Gothelf, A. Thomsen, M. Nielsen, E. Cló and R. S. Brown, J. Am. Chem. Soc., 2004, 126, 1044–1046 CrossRef CAS PubMed.
- A. H. El-Sagheer, R. Kumar, S. Findlow, J. M. Werner, A. N. Lane and T. Brown, ChemBioChem, 2008, 9, 50–52 CrossRef CAS PubMed.
- F. D. Lewis, X. Liu, Y. Wu, S. E. Miller, M. R. Wasielewski, R. L. Letsinger, R. Sanishivili, A. Joachimiak, V. Tereshko and M. Egli, J. Am. Chem. Soc., 1999, 121, 9905–9906 CrossRef CAS.
- F. D. Lewis, Y. Wu and X. Liu, J. Am. Chem. Soc., 2002, 124, 12165–12173 CrossRef CAS PubMed.
- M. Hariharan, Y. Zheng, H. Long, T. A. Zeidan, G. C. Schatz, J. Vura-Weis, M. R. Wasielewski, X. Zuo, D. M. Tiede and F. D. Lewis, J. Am. Chem. Soc., 2009, 131, 5920–5929 CrossRef CAS PubMed.
- N. T. Thuong and C. Hélène, Angew. Chem., Int. Ed. Engl., 1993, 32, 666–690 CrossRef PubMed.
- S. Bevers, S. Schutte and L. W. McLaughlin, J. Am. Chem. Soc., 2000, 122, 5905–5915 CrossRef CAS.
- N. Rahe, C. Rinn and T. Carell, Chem. Commun., 2003, 2120–2121 RSC.
- K. M. Guckian, B. A. Schweitzer, R. X.-F. Ren, C. J. Sheils, P. L. Paris, D. C. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 1996, 118, 8182–8183 CrossRef CAS PubMed.
- S. Hainke, S. Arndt and O. Seitz, Org. Biomol. Chem., 2005, 3, 4233–4238 CAS.
- R. X.-F. Ren, N. C. Chaudhuri, P. L. Paris, S. Rumney and E. T. Kool, J. Am. Chem. Soc., 1996, 118, 7671–7678 CrossRef CAS PubMed.
- S. Egetenmeyer and C. Richert, Chem. – Eur. J., 2011, 17, 11813–11827 CrossRef CAS PubMed.
- S. Rumney and E. T. Kool, J. Am. Chem. Soc., 1995, 117, 5635–5646 CrossRef CAS PubMed.
- H. Huang and M. M. Greenberg, J. Org. Chem., 2008, 73, 2695–2703 CrossRef CAS PubMed.
- T. Takada, Y. Otsuka, M. Nakamura and K. Yamana, Chem. – Eur. J., 2012, 18, 9300–9304 CrossRef CAS PubMed.
- T. Takada, K. Yamaguchi, S. Tsukamoto, M. Nakamura and K. Yamana, Analyst, 2014, 139, 4016–4021 RSC.
- H. Huang and P. C. Tlatelpa, Chem. Commun., 2015, 51, 5337–5339 RSC.
- M. Mandal, B. Boese, J. E. Barrick, W. C. Winkler and R. R. Breaker, Cell, 2003, 113, 577–586 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: MALDI-TOF mass spectra of modified oligonucleotides, additional UV/VIS-spectra from filtration assays, HPLC traces, UV-melting points, and additional UV-melting curves. See DOI: 10.1039/c5ob00508f |
| This journal is © The Royal Society of Chemistry 2015 |