
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic studies toward the brasilinolides: controlled assembly of a protected C1–C38 polyol based on fragment union by complex aldol reactions†
Ian
Paterson
*a,
Michael P.
Housden
a,
Christopher J.
Cordier
a,
Paul M.
Burton
a,
Friedrich A.
Mühlthau
a and
Olivier
Loiseleur
b
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: ip100@cam.ac.uk
bSyngenta Crop Protection AG, Schaffhauserstrasse, 4332 Stein, Switzerland
First published on 14th April 2015
Abstract
The brasilinolides are an architecturally complex family of 32-membered macrolides, characterised by potent immunosuppressant and antifungal properties, which represent challenging synthetic targets. By adopting a highly convergent strategy, a range of asymmetric aldol/reduction sequences and catalytic protocols were employed to assemble a series of increasingly elaborate fragments. The controlled preparation of suitable C1–C19 and C20–C38 acyclic fragments 5 and 6, containing seven and 12 stereocentres respectively, was first achieved. An adventurous C19–C20 fragment union was then explored to construct the entire carbon chain of the brasilinolides. This pivotal coupling step could be performed in a complex boron-mediated aldol reaction to install the required C19 hydroxyl stereocentre when alternative Mukaiyama-type aldol protocols proved unrewarding. A protected C1–C38 polyol 93 was subsequently prepared, setting the stage for future late-stage diversification toward the various brasilinolide congeners. Throughout this work, asymmetric boron-mediated aldol reactions of chiral ketones with aldehydes proved effective both for controlled fragment assembly and coupling with predictable stereoinduction from the enolate component.
Introduction
The polyketides represent a diverse array of structurally elaborate natural products having a wide range of biological activity, with many members having important therapeutic utility in human and veterinary medicine.1 The brasilinolides (Fig. 1), isolated by Mikami and Kobayashi and co-workers in 1996 from the pathogenic actinomycetes Nocardia brasiliensis, constitute a unique family of immunosuppressive and antifungal polyketides based on a highly hydroxylated macrolide scaffold, which presumably arises in the biosynthesis from a PKS-mediated cyclisation of a linear 38-carbon chain.2 As X-ray quality crystals of the brasilinolides proved elusive, and analysis of the 1H and 13C NMR data did not allow a full structural assignment, detailed stereochemical information was gleaned by controlled chemical degradation and spectroscopic studies of the resulting fragments. This work enabled the elucidation of the relative and absolute configuration of brasilinolide A (1), along with the congeneric brasilinolides B (2) and C (3).3 Notably, brasilinolide A (1) was reported to exhibit pronounced immunosuppressive activity in vivo (IC50 value of 0.625 μg mL−1, mouse mixed lymphocyte assay) with no significant toxicity. In addition, brasilinolide A showed antifungal activity and brasilinolide B was found to be active against a range of fungi and bacteria. Altogether, their notable biological activities, along with the low toxicity profile and favourable physicochemical properties, make further evaluation of the brasilinolides as promising lead structures of interest, with potential therapeutic applications to autoimmune diseases and fungal infections, and in the prophylaxis of organ transplant rejection.4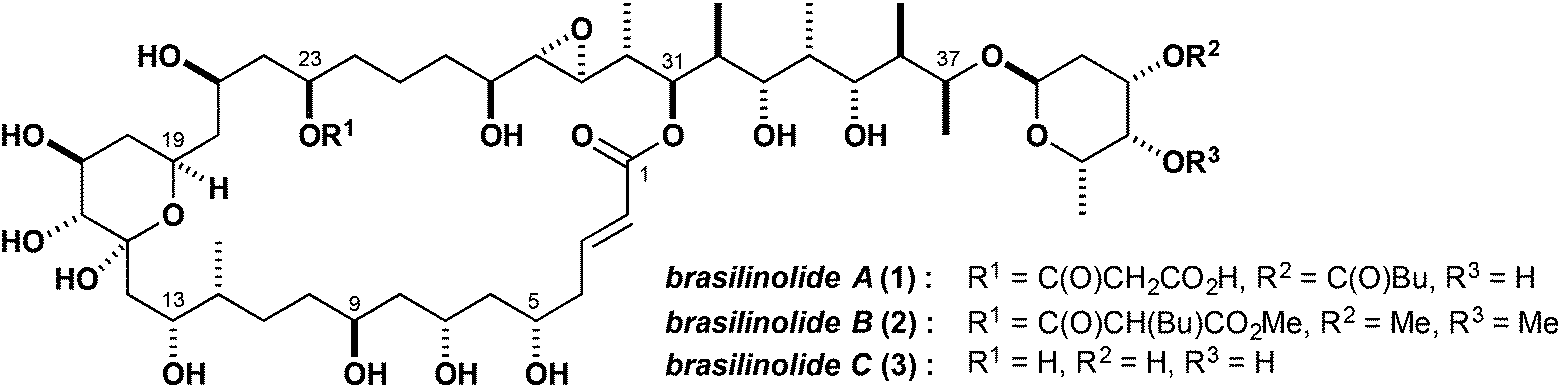 | ||
| Fig. 1 Structures of brasilinolides A–C. | ||
Each member of the brasilinolide family contains several distinctive structural features, including some 26 stereocentres, of which 16 are embedded in the macrocyclic lactone, and an elaborate propionate-derived side chain with a glycosyl appendage at C37. The signature 32-membered macrolide core itself contains a six-membered hemiacetal ring, an epoxide and a characteristic 1,3-polyol array, with an acidic 23-O-malonyl residue present in brasilinolide A (1). Herein, we report in full on progress to date toward the total synthesis of the brasilinolides, documenting the multiple selectivity and logistical issues addressed in this eventful journey.
Results and discussion
Overview of synthesis plan
Our initially devised synthesis plan for brasilinolide A (1) is outlined in Scheme 1. Such rich stereochemical features, particularly with the preponderance of secondary hydroxyl groups within the aglycon, inspired the pursuit of a strategy using asymmetric aldol chemistry5 for realising the critical fragment assembly steps to configure many of these stereocentres. Additionally, a carefully planned protecting group regime was devised for the purposes of achieving predictable stereoinduction, along with enabling the late-stage introduction of the defining brasilinolide substituents, and to ensure the necessary control in progressing the synthetic route. Building on our earlier preliminary studies,6 the highly convergent assembly of a suitably protected C1–C38 linear precursor 4 was initially targeted. Further simplification was proposed by the late-stage attachment of the polar C23-O-malonyl substituent and C37-O-glycosylation, following macrolactonisation of the C1–C38 intermediate 4 after liberation of the C31 hydroxyl group. The pivotal linear precursor 4 was therefore designed to allow for precise site-controlled functionalisation. Furthermore, it was envisaged that a high level of stereoinduction might be leveraged in its controlled aldol-based assembly from suitable ketone building blocks.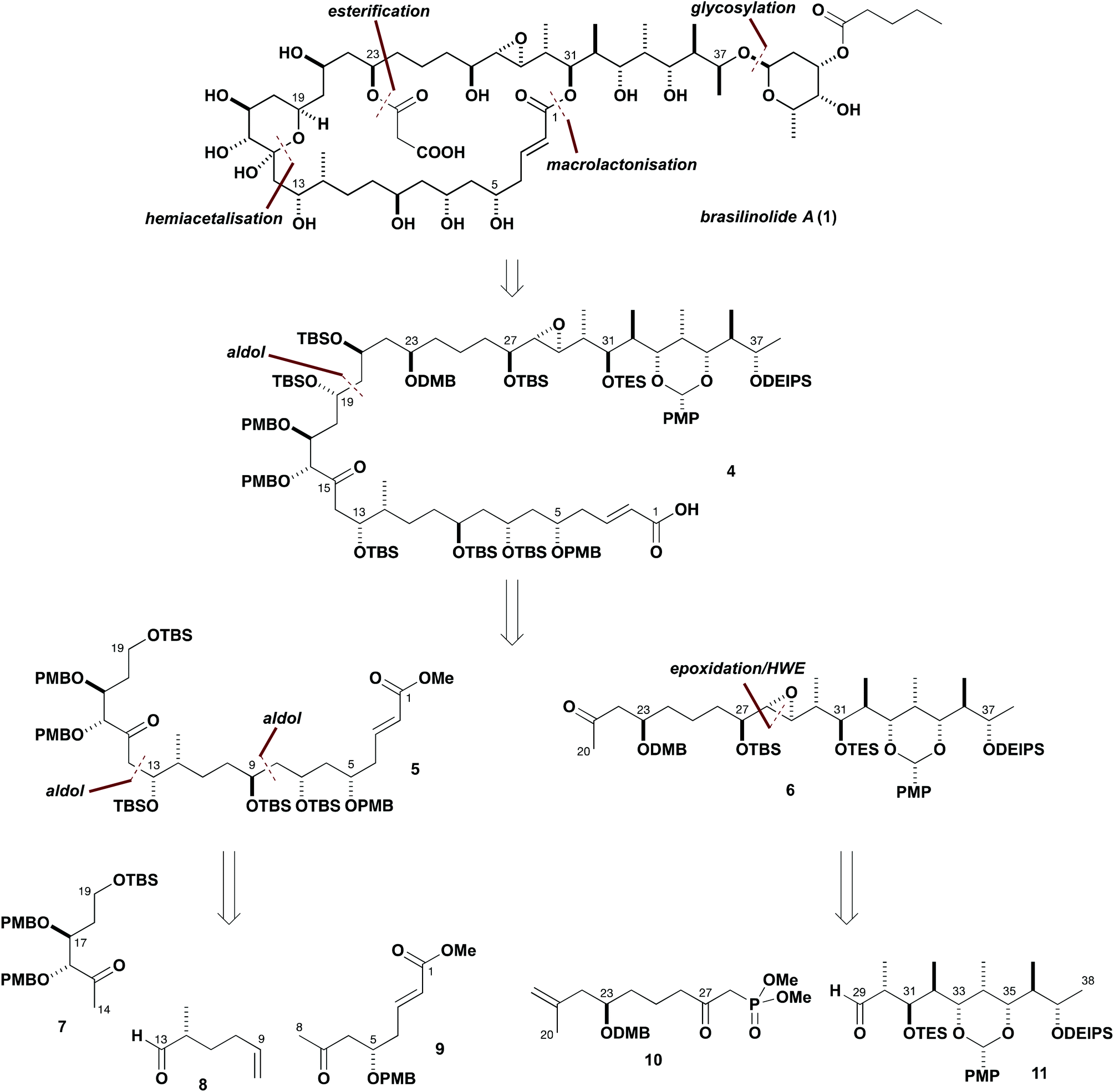 | ||
| Scheme 1 Overview of synthesis plan for brasilinolide A (1) leading back to the five chiral building blocks 7–11. | ||
Following this overall plan, 4 was disconnected at the C19–C20 bond to afford the C1–C19 fragment 5, as the corresponding southern hemisphere region, and the methyl ketone 6, corresponding to the brasilinolide northern hemisphere. It was envisaged that these two major fragments would be connected by a strategic aldol coupling between 6 and a suitable C19 aldehyde derived from 5. Further analysis of 5 suggested that favourable carbon–carbon bond scissions might be made at C8–C9 and C13–C14 to reveal three chiral building blocks 7–9 of roughly equal complexity. Examination of the C20–C38 fragment 6 suggested that a Horner–Wadsworth–Emmons (HWE) disconnection would serve to isolate the C23 stereocentre, leading back to the β-ketophosphonate 10 and the aldehyde 11. Following this synthesis plan, 4 is conveniently broken down to give five building blocks: with 7–10 having only one or two stereocentres along with the more complex polypropionate array embedded in 11.
With regard to the critical issue of controlling stereochemistry, it was envisaged that asymmetric aldol methodology developed in our group could be exploited in several situations to construct 5 and 11. As discussed in more detail later, a suitably flexible strategy to assemble and couple the five building blocks shown in Scheme 1 was devised to allow modification based on experimental findings. In two preliminary communications,6 we had previously reported synthetic routes to two major brasilinolide fragments, which now needed to be refined and optimised. This entailed some modification to the protecting groups, and scale up to enable access to larger quantities of advanced intermediates.
The C1–C19 southern fragment 5
Our initial focus was on efficiently assembling the southern hemisphere fragment 5 in Scheme 1. This contains seven oxygen-bearing stereocentres, a ketone at C15, and terminates in an enoate and TBS ether as a latent form of C19 aldehyde. Configurational analysis of 5 revealed a pivotal 1,5-anti relationship between the protected hydroxyl groups at C5 and C9, and also between C13 and C17, that would represent interesting cases for studying long-range asymmetric induction using aldol reactions of methyl ketones.7 Disconnection across the C8–C9 and C13–C14 bonds to afford β-alkoxy methyl ketones 7 and 9 was then anticipated to facilitate a useful degree of remote asymmetric induction during fragment assembly. In order to advance to exploring the projected aldol-based construction of the C1–C19 fragment 5, an efficient gram-scale preparation of the three chiral building blocks 7, 8 and 9 in high enantiomeric purity was first deemed necessary. The preparation of these building blocks is now considered in turn.As shown in Scheme 2, our initial approach towards accessing the C1–C8 methyl ketone 9 started out with the commercially available (S)-(+)-epichlorohydrin (12) as a convenient means of installing the C5 stereocentre in high configurational purity. In following this route A, epoxide opening of 12 with vinylmagnesium bromide proceeded in the presence of CuBr (10 mol%) in Et2O at −40 °C to afford an intermediate chlorohydrin; where KOH-mediated cyclisation then gave the known epoxide 13 (55%, two steps).8 Reopening of this epoxide with lithium acetylide ethylene diamine complex proceeded in DMSO to give the corresponding allylic alcohol, which was converted into its PMB ether 14 (NaH, PMBBr) in 69% overall yield. Next, the two unsaturated moieties attached to the C5 stereocentre were sequentially elaborated. Firstly, controlled oxymercuration (Hg(OAc)2 (30 mol%), PPTS) of the alkyne in wet acetone afforded the methyl ketone 15, which was followed by cross-metathesis of the olefin with methyl acrylate using Grubbs II catalyst (1 mol%) to generate enoate 9 (75%, two steps).9 This sequence afforded the enantiopure C1–C8 ketone 9 in 29% yield over six steps from (S)-(+)-epichlorohydrin (12).
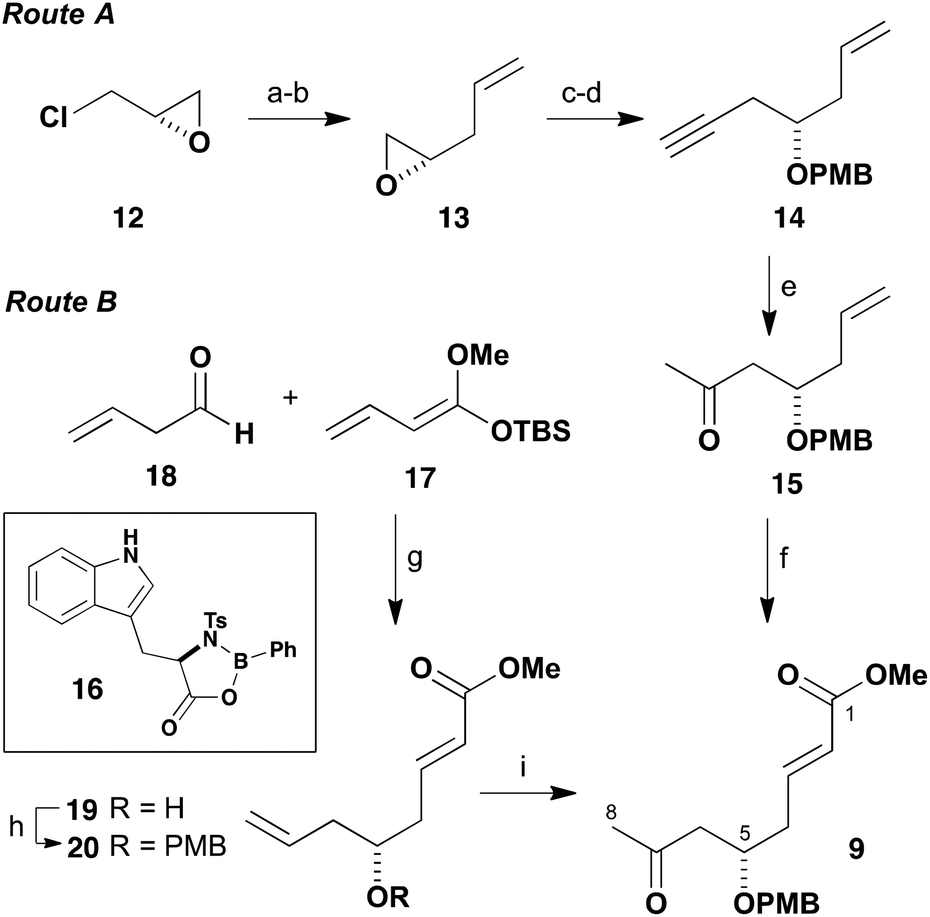 | ||
Scheme 2 Synthesis of methyl ketone 9 using either a chiral starting material or by an asymmetric vinylogous Mukaiyama aldol reaction. Reagents and conditions: (a) H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMgBr, CuBr (10 mol%) Et2O, −40 °C, 73%; (b) KOH, 75%; (c) HCCLi·(CH2NH2)2, DMSO; (d) NaH, PMBBr, THF, 69% (two steps); (e) Hg(OAc)2 (30 mol%), PPTS, H2O, acetone, 82%; (f) methyl acrylate, Grubbs II catalyst (1 mol%), CH2Cl2, 92%; (g) 16, i-PrOH, i-PrCN, 57%, >95% ee; (h) PMB-2,2,2-trichloroacetimidate, Ph3CBF4, THF, 64%; (i) PdCl2 (10 mol%), CuCl2, O2, DMF–H2O (7 CHMgBr, CuBr (10 mol%) Et2O, −40 °C, 73%; (b) KOH, 75%; (c) HCCLi·(CH2NH2)2, DMSO; (d) NaH, PMBBr, THF, 69% (two steps); (e) Hg(OAc)2 (30 mol%), PPTS, H2O, acetone, 82%; (f) methyl acrylate, Grubbs II catalyst (1 mol%), CH2Cl2, 92%; (g) 16, i-PrOH, i-PrCN, 57%, >95% ee; (h) PMB-2,2,2-trichloroacetimidate, Ph3CBF4, THF, 64%; (i) PdCl2 (10 mol%), CuCl2, O2, DMF–H2O (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 63%. :1), 63%. | ||
Although methyl ketone 9 was available in high enantiomeric purity via the foregoing route A, preliminary attempts to perform this sequence on a suitably large scale encountered a number of issues. The first three steps generate volatile compounds and the associated handling difficulties depleted yields on a larger scale. Disconnection of the δ-alkoxyenoate functionality of 9 based on a vinylogous Mukaiyama aldol reaction10 leads to an opportunity for exploring an alternative construction of this fragment using asymmetric catalysis. Following the work of Kalesse and co-workers, a complementary route B to prepare 9 was therefore developed using the D-tryptophan-derived oxazaborolidinone 16.11 In practice, a Mukaiyama aldol addition of silyl dienolate 17 to but-3-enal12 (18) in the presence of 16 afforded the required γ-adduct 19 (57%) in >95% ee. Following PMB ether formation to give 20 (PMBTCA, Ph3CBF4, 64%),13 a Wacker oxidation14 of the terminal olefin (PdCl2 (10 mol%), CuCl2, O2) then provided the methyl ketone 9 (63%). Both the above routes to 9 were then used to generate useful stocks of the planned C1–C19 southern fragment 5.
As shown in Scheme 3, control over the C12 methyl-bearing stereocentre in aldehyde 8 was conveniently achieved through the use of the chiral auxiliary methodology developed by Myers.15 Alkylation of the lithium enolate derived from propionamide 21 with 4-iodobut-1-ene gave the desired amide 22, which was isolated in high yield (94%) and >95:5 dr on multi-gram scale. The auxiliary was then reductively cleaved with LDA/NH3·BH3 (88%) and Swern oxidation16 of the resulting alcohol 23 gave the desired C9–C13 aldehyde 8 (97%). This scalable synthesis of 8 bearing a masked aldehyde at C9 was thus completed in three steps from 21 in 87% yield and in high enantiomeric purity (97% ee).
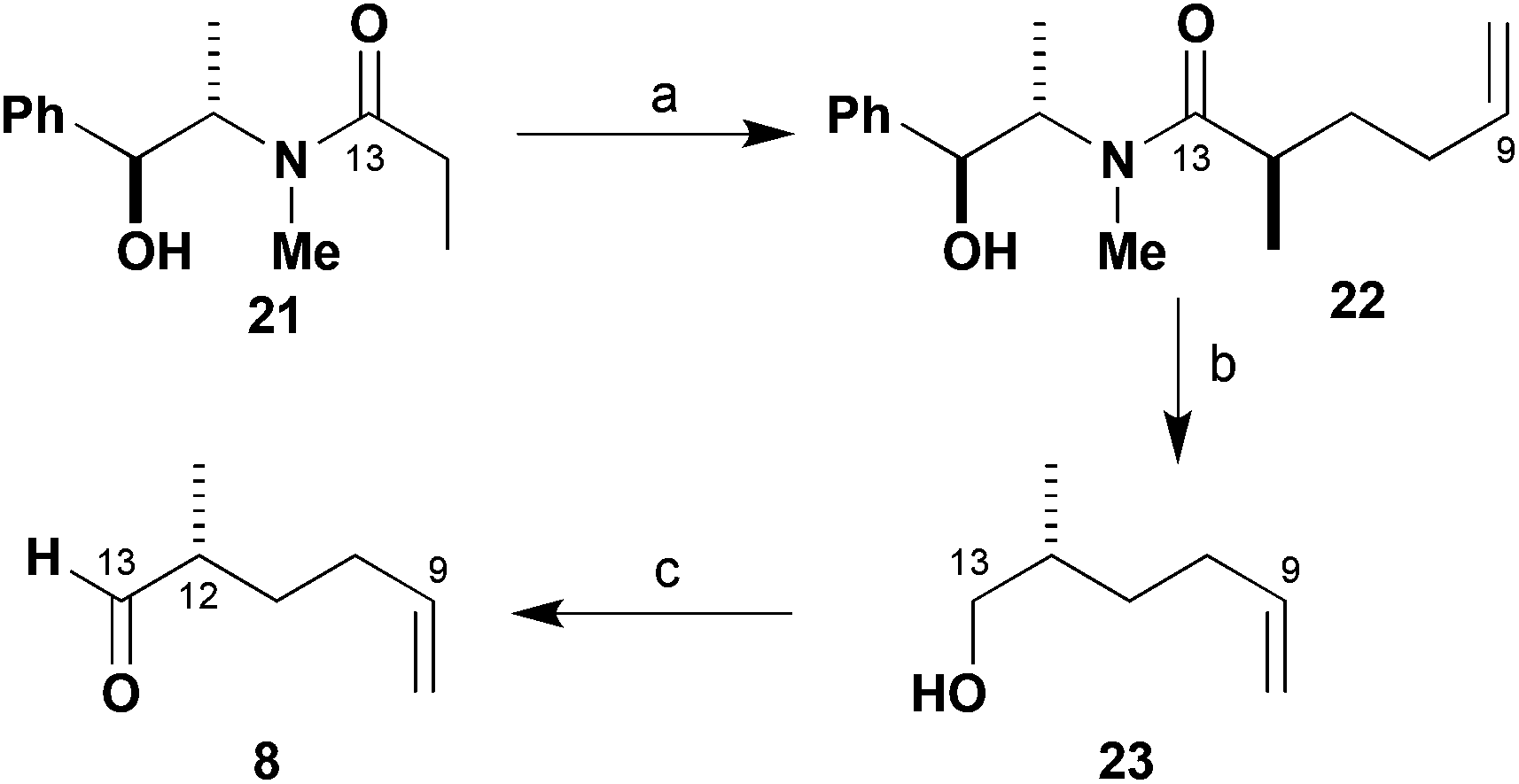 | ||
| Scheme 3 Synthesis of aldehyde 8. Reagents and conditions: (a) LDA, LiCl, THF, −78 °C; 4-iodobut-1-ene, 94%, >95:5 dr; (b) LDA, NH3·BH3, THF, 0 °C, 88%; (c) (COCl)2, DMSO, CH2Cl2; Et3N, −78 to 0 °C, 97%. | ||
As shown in Scheme 4, the synthesis of the remaining chiral building block 7 required for constructing the brasilinolide southern fragment 5 relied on asymmetric catalysis. It commenced with the TBS ether 24 prepared from but-3-en-1-ol. Ozonolysis and in situ Wittig reaction of the resulting aldehyde with Ph3PCHCOMe gave the corresponding enone 25 (69%), solely as the E-isomer.17 This was then subjected to a Sharpless asymmetric dihydroxylation,18 where a ligand screen indicated that (DHQ)2AQN (1 mol%) reliably gave chiral diol 26 with excellent enantioselectivity (91%, 97% ee). By exposure to PMBTCA in the presence of catalytic Ph3CBF4 (0.5 mol%),13 this diol was then converted into its bis-PMB ether 7 (77%). This scalable route gave methyl ketone 7 in three steps from 24 in high yield (48%) and enantiomeric purity (97% ee).
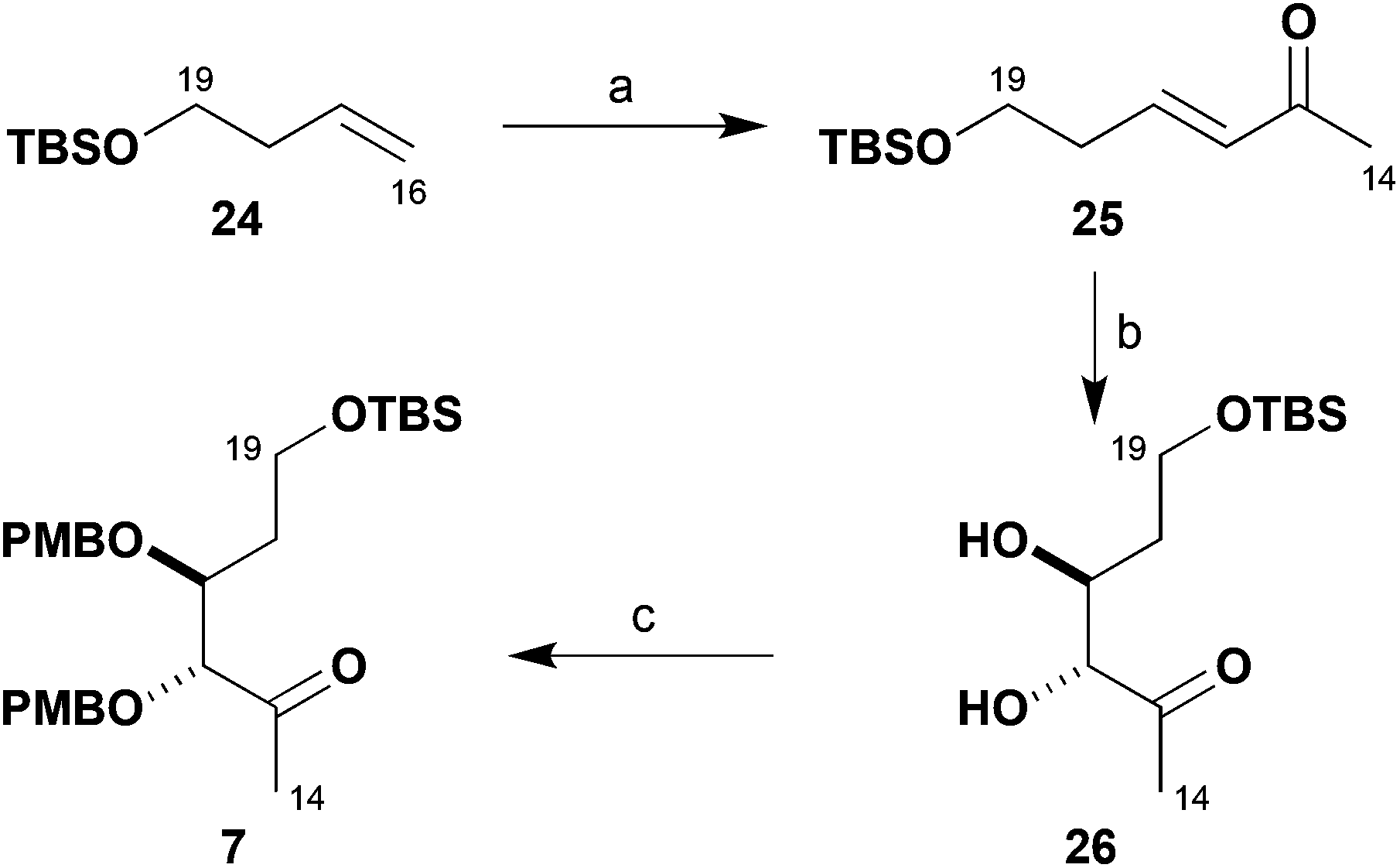 | ||
Scheme 4 Synthesis of methyl ketone 7. Reagents and conditions: (a) O3, CH2Cl2, −78 °C; Ph3PCHCOMe, 0 °C, 69%, >95:5 E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :Z; (b) K2OsO2(OH)4 (1 mol%), (DHQ)2AQN (1 mol%), K3[Fe(CN)6], K2CO3, NaHCO3, MeSO2NH2, t-BuOH–H2O (1:1), 91%, 97% ee; (c) PMB-2,2,2-trichloroacetimidate, Ph3CBF4 (0.5 mol%), THF, 77%. :Z; (b) K2OsO2(OH)4 (1 mol%), (DHQ)2AQN (1 mol%), K3[Fe(CN)6], K2CO3, NaHCO3, MeSO2NH2, t-BuOH–H2O (1:1), 91%, 97% ee; (c) PMB-2,2,2-trichloroacetimidate, Ph3CBF4 (0.5 mol%), THF, 77%. | ||
In assembling the C1–C19 southern fragment 5, we planned to use two strategic boron-mediated aldol reactions to couple together the three fragments 7, 8 and 9. With boron enolates derived from β-alkoxy methyl ketones such as 9, remote 1,5-anti stereoinduction is known to be enhanced in addition to aldehydes by employing a PMB ether.7 This facilitates aldol transition state organisation by formyl hydrogen bonding involving this ether oxygen atom. We first needed to explore the scope of this reaction to include β-alkoxy methyl ketones such as 7 that have an additional α-alkoxy stereocentre. In support of this approach, DFT calculations predicted that these aldol reactions are also likely to proceed preferentially through a bicyclic boat TS-1, where π-facial discrimination is controlled by a stabilising formyl hydrogen bond involving only the PMB ether oxygen at the β-centre and minimisation of steric interactions.19
After some optimisation, it was found that methyl ketone 7 was best treated with Bu2BOTf/i-Pr2NEt in Et2O and the resulting enolate 27 reacted with aldehyde 8 at −78 °C, leading on work-up to the isolation of adduct 28 with excellent diastereoselectivity and in high yield (>95:5 dr, 98%, Scheme 5). The newly formed stereocentre in 28 was installed with the required (13R)-configuration20 in the anticipated 1,5-anti sense relative to C17.21 As predicted in TS-1, this indicated that the β-alkoxy stereocentre was presumably responsible for controlling the π-facial selectivity of the boron enolate in this complex aldol fragment coupling. A similar aldol reaction of 7 with iso-butyraldehyde also gave the corresponding 1,5-anti adduct 29 in high diastereoselectivity, suggesting that Felkin–Anh-type 1,2-stereoinduction from the α-chiral aldehyde component 8 was not significantly contributing to the outcome.22
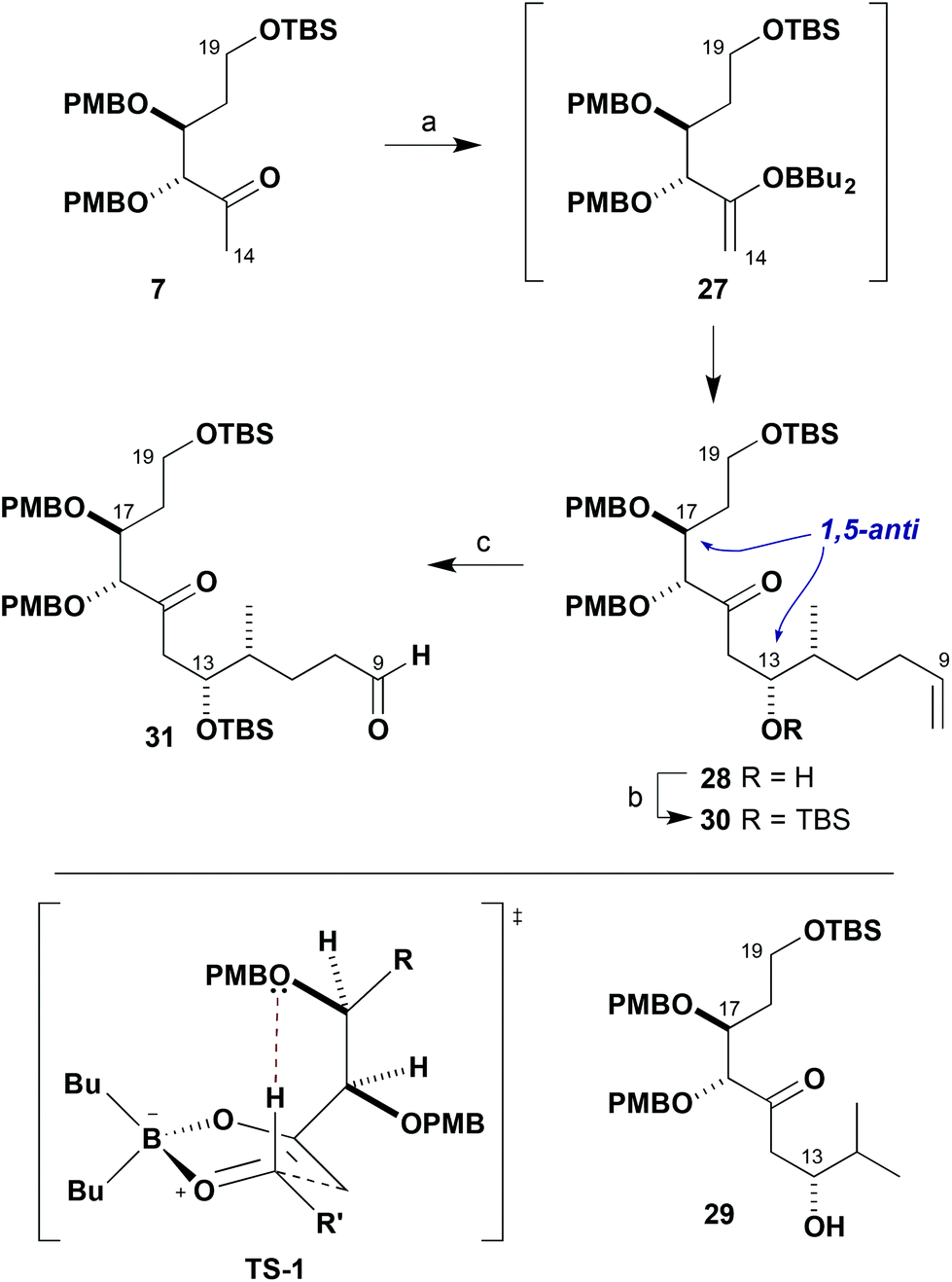 | ||
| Scheme 5 Synthesis of aldehyde 31. Reagents and conditions: (a) Ketone 7, Bu2BOTf, i-Pr2NEt, Et2O, −78 °C; aldehyde 8, Et2O, −78 °C, 98%, >95:5 dr; (b) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 98%; (c) O3, NaHCO3, Sudan Red 7B, CH2Cl2–MeOH (1:1), −78 °C, 95%. | ||
Moving forward, the resulting aldol adduct 28 was next transformed into the TBS ether 30 (TBSOTf, 2,6-lutidine) and the olefin oxidatively cleaved via ozonolysis to afford 31 (97%, two steps). This served to generate the aldehyde functionality at C9 in readiness for performing the next aldol fragment coupling with methyl ketone 9 (Scheme 6). In this case, 9 was treated with c-Hex2BCl/Et3N in Et2O and the resulting boron enolate 32 added smoothly to aldehyde 31 at −78 °C to give the β-hydroxyketone 33 (>95:5 dr, 95%). This complex aldol coupling reaction delivers equally high 1,5-anti diastereoselectivity and is considered to preferentially proceed through the related boat TS-2, again invoking a formyl hydrogen bond from a β-positioned PMB ether oxygen in the boron enolate. An Evans–Saksena23 1,3-anti reduction of 33 with Me4NBH(OAc)3 at −30 °C then served to efficiently configure the remaining stereocentre at C7, again proceeding with high diastereoselectivity (>95:5 dr, 95%). While the resulting diol 34 was protected as its cyclic di-tert-butylsilylene (Si(t-Bu)2) derivative in our previous studies,6a to facilitate progression of material on a larger scale it was decided instead to now derivatise the C7 and C9 hydroxyl groups as the bis-TBS ether 5 (TBSOTf, 2,6-lutidine, 97%). This revised approach was preferred both in terms of a substantially improved yield and to ensure that the carbon skeleton was not unduly conformationally restricted for the later macrocyclisation step. In earlier work,6a we had also explored the effect of reversing the order of the fragment couplings whereby the C8–C9 bond was forged ahead of the C13–C14 bond – however, this longer route gave a lower overall yield so was not pursued further.
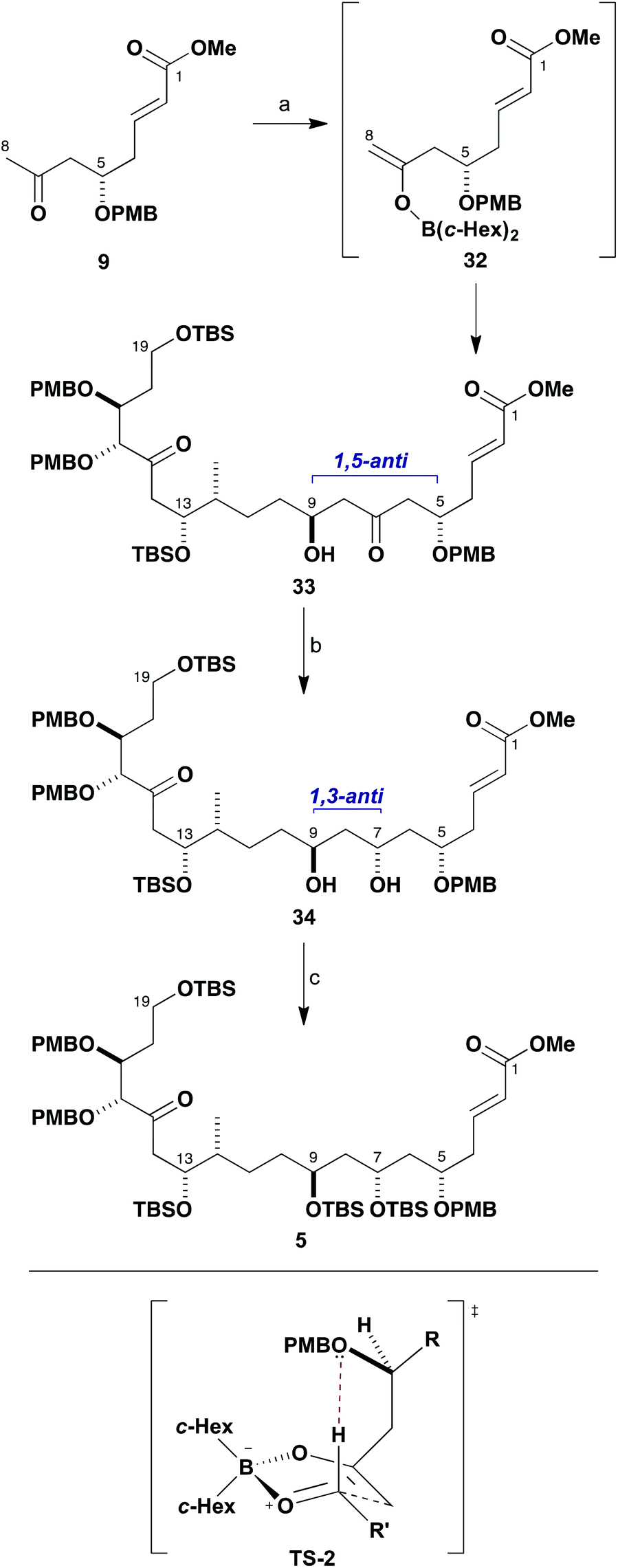 | ||
| Scheme 6 Assembly of the southern hemisphere fragment 5. Reagents and conditions: (a) Ketone 9, c-Hex2BCl, Et3N, Et2O, 0 °C; aldehyde 31, −78 to −23 °C, 95%, >95:5 dr; (b) Me4NBH(OAc)3, MeCN, AcOH (4:1), −30 °C, 95%, >95:5 dr; (c) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 97%. | ||
The fully protected C1–C19 southern fragment 5 was obtained by this improved route in a satisfying 43% overall yield in 10 steps over the longest linear sequence (16 total steps) from the alcohol precursor to 24. Implementation of this reaction sequence on multi-gram scale served to generate useful quantities of the pivotal major fragment 5 with all of the seven stereocentres installed in high configurational purity, as required for advancing synthetic efforts toward the brasilinolides.
The C20–C38 northern fragment 6
With an efficient synthesis of the C1–C19 fragment 5 in place, the construction of the C20–C38 fragment 6 was addressed next, as shown in Scheme 7. The methyl ketone 6, corresponding to the northern hemisphere of the brasilinolides, is characterised by having 11 contiguous stereocentres together with a remote oxygenated stereocentre at C23. Suitable differentiation of six hydroxyl groups is required, as well as the controlled installation of a potentially sensitive trans-epoxide moiety.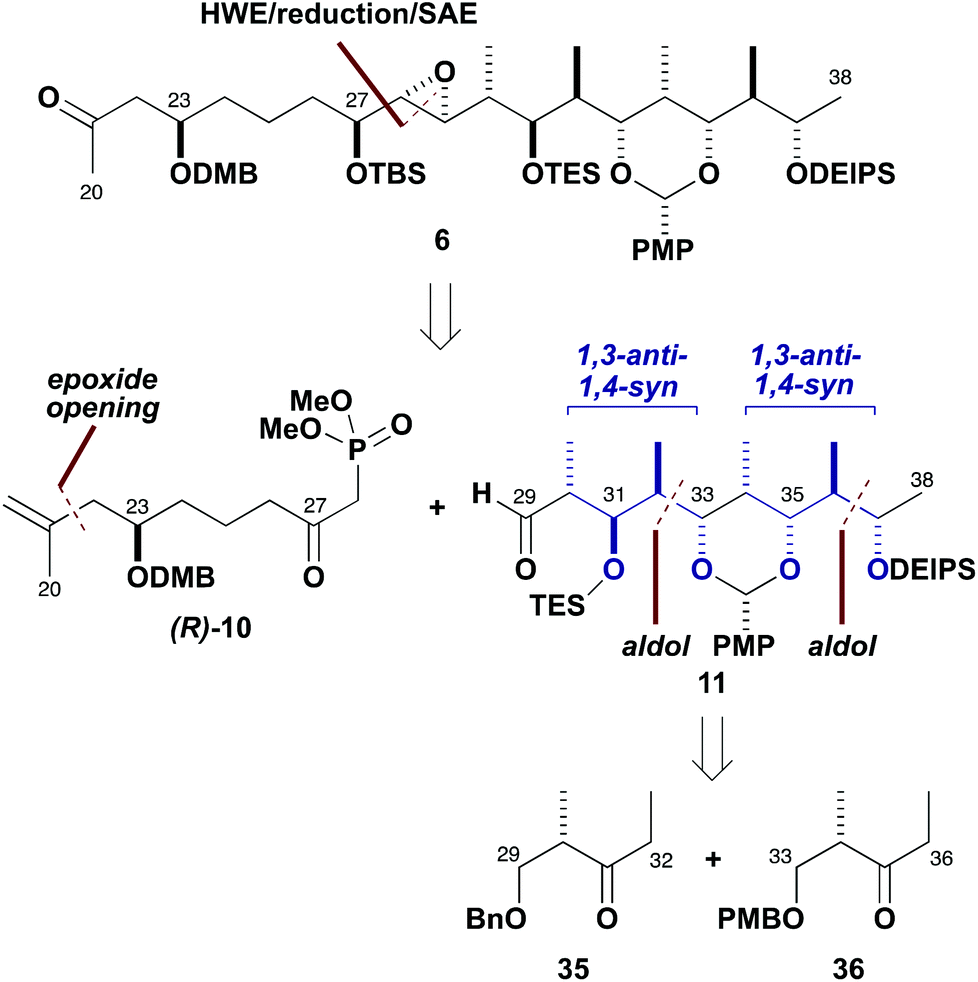 | ||
| Scheme 7 Retrosynthetic analysis of northern fragment 6. | ||
Strategically, an HWE-type disconnection across C28–C29 was proposed, leading back to the β-ketophosphonate (R)-10 (having a C21 methylene as a latent ketone) and the aldehyde 11, which contains the extended polypropionate region. Detailed configurational analysis of the aldehyde 11 reveals two similar stereotetrads with an epimeric relationship between C31 and C35. Two iterative aldol chain extensions of acetaldehyde employing the (S)-Roche ester-derived ethyl ketones 35 and 36 were chosen to install the required stereocentres associated with the C32–C33 and C36–C37 bonds via substrate-controlled induction based on aldol methodology developed in our group. The appropriate oxygenated stereocentres at C31 and C35 in 11 would then be set by controlled reductions of the corresponding ketones.
Following this plan (Scheme 8), the enantioselective preparation of the β-ketophosphonate (R)-10 was first targeted. This route relied on asymmetric catalysis through use of Jacobsen's hydrolytic kinetic resolution of terminal epoxides to configure the C23 stereocentre.24 In the event, 5-hexen-1-ol (37) was first derivatised as the TBS ether and converted into the racemic epoxide with m-CPBA. The hydrolytic resolution of this epoxide then proceeded with operational ease with (R,R)-Co(salen) pre-catalyst (0.5 mol%) to give multi-gram quantities of (R)-38 (41%) in >98% ee. Opening the epoxide (R)-38 with iso-propenylmagnesium bromide in the presence of catalytic CuI (15 mol%) then gave alcohol (R)-39 (99%). The C23 hydroxyl group was subsequently protected (NaH, DMBCl) as its DMB ether (R)-40 (82%). TBS ether cleavage of (R)-40 (PPTS, MeOH) afforded alcohol (R)-41, which was then oxidised to the aldehyde (R)-42 with Dess–Martin periodinane (84%, two steps).25 Reaction of (R)-42 with lithiated methyl dimethylphosphonate, followed by a further Dess–Martin oxidation then provided the required β-ketophosphonate (R)-10 (73%, two steps) in high enantiomeric purity.
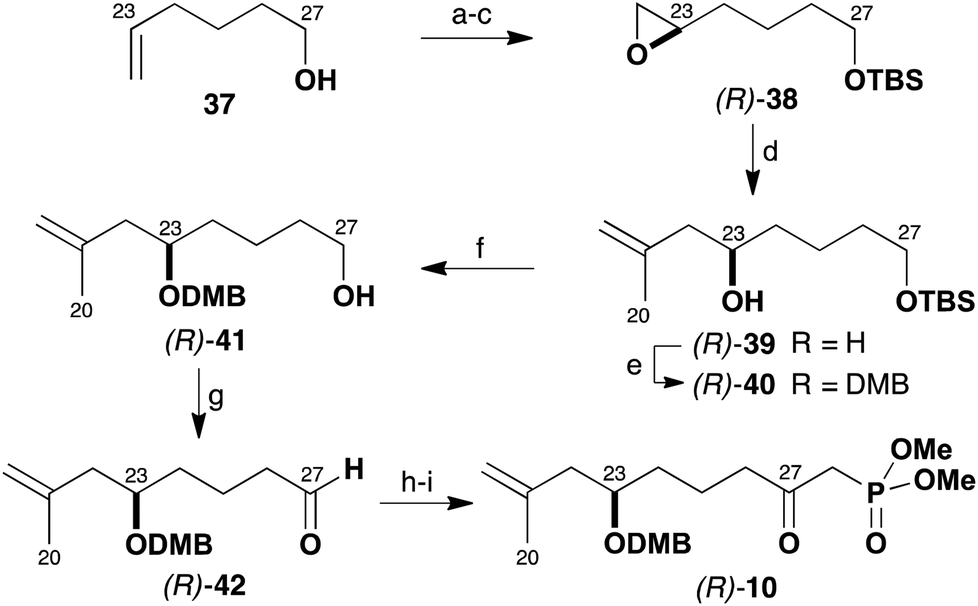 | ||
| Scheme 8 Synthesis of β-ketophosphonate (R)-10. Reagents and conditions: (a) TBSCl, imidazole, CH2Cl2, 96%; (b) m-CPBA, CH2Cl2, 96%; (c) (R,R)-Co(salen) (0.5 mol%), AcOH (1 mol%), H2O (55 mol%), THF, 41%, >98% ee; (d) iso-propenylmagnesium bromide, CuI (15 mol%), THF, −78 to −50 °C, 99%; (e) NaH, DMBCl, DMF, 82%; (f) PPTS, MeOH, 99%; (g) DMP, CH2Cl2, 85%; (h) MeP(O)(OMe)2, BuLi, THF, −78 °C; (i) DMP, CH2Cl2, 73% (two steps). | ||
With the β-ketophosphonate (R)-10 in hand, attention turned to the preparation of the C29–C38 aldehyde 11 for the planned assembly of the northern fragment 6. This started out with the known ethyl ketone 36 (Scheme 9), readily derived from (S)-Roche ester.26 We have extensively used such chiral ketones for the aldol-based construction of elaborate polypropionate sequences in a variety of complex polyketides,27 where inherently high levels of control can be attained over both their enolate geometry and π-facial selectivity in addition to the aldehyde component. Following our standard conditions, 36 was treated with c-Hex2BCl/Et3N at 0 °C in Et2O to selectively form the E-dicyclohexylboron enolate 43 and addition to acetaldehyde at −78 °C gave the expected 1,4-syn aldol adduct with high diastereoselectivity. This reaction is considered to proceed through the preferred formyl hydrogen bonded TS-3, where allylic strain is minimised. Interception of the cyclic boron intermediate 44 by preferred axial attack of an external hydride viaTS-4 also allowed the C35 stereocentre in 45 to be configured in situ. Thus, a Narasaka-type reduction of intermediate 44 with LiBH4 at −78 °C efficiently set the 1,3-syn relationship between C35 and C37 in diol 45 (88%, >95:5 dr).28 This convenient one-pot procedure efficiently installed three new stereogenic centres in excellent diastereoselectivity in 45 and proved easily scalable.
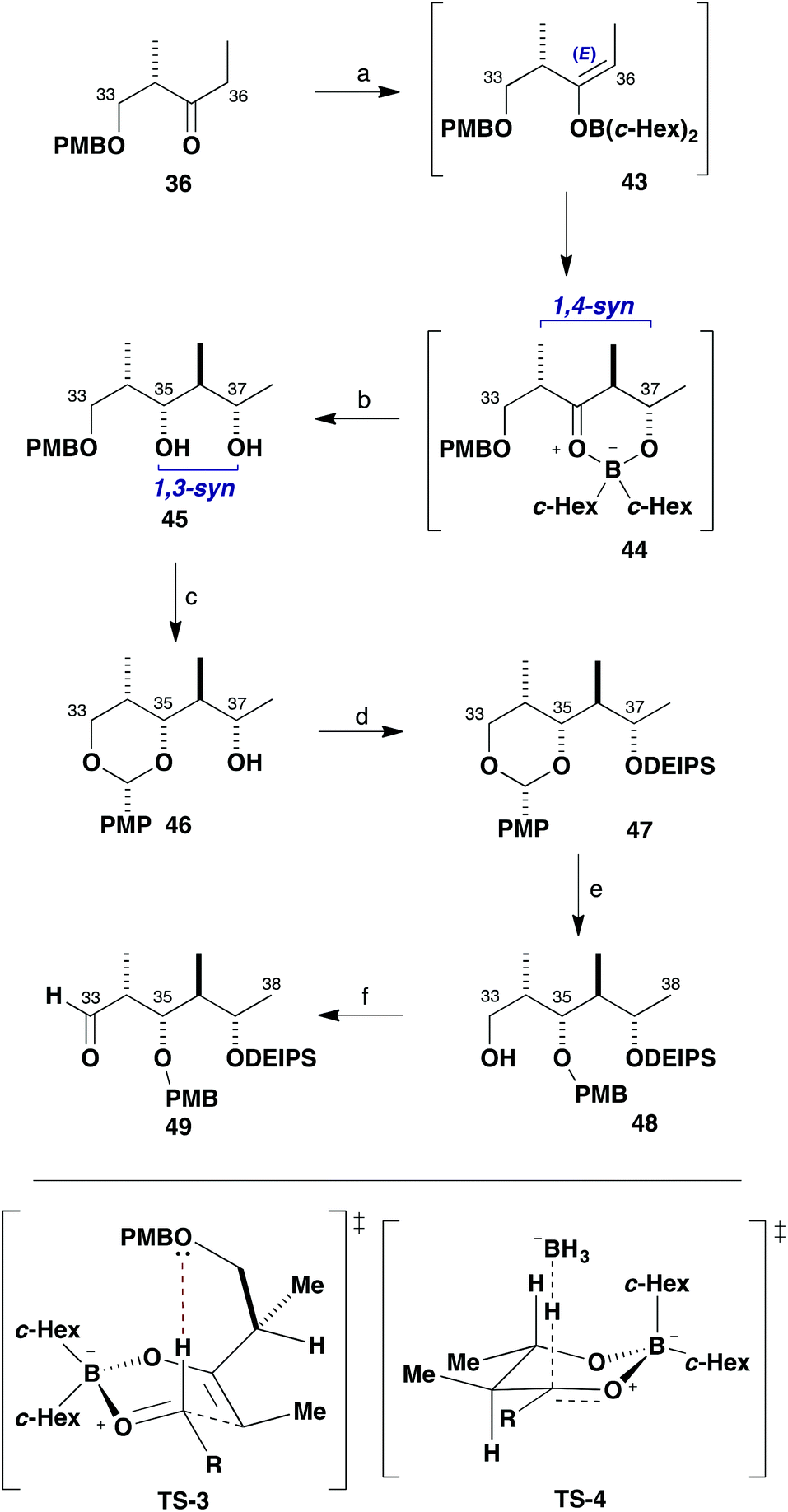 | ||
| Scheme 9 Synthesis of aldehyde 49. Reagents and conditions: (a) c-Hex2BCl, Et3N, Et2O, 0 °C; MeCHO, −78 to −20 °C; (b) LiBH4, THF, −78 °C; NaOH, MeOH, H2O2, 88%, >95:5 dr; (c) DDQ, 4 Å mol. sieves, CH2Cl2, −10 °C, 92%; (d) DEIPSCl, imidazole, CH2Cl2, 97%; (e) DIBAL, CH2Cl2, −78 °C, 93%; (f) DMP, NaHCO3, CH2Cl2, 93%. | ||
A key requirement for constructing the northern fragment 6 was that the hydroxyl stereocentres are appropriately differentiated to facilitate macrocyclisation and the sequential attachment of appendages. With this in mind, oxidation of the PMB ether 45 with DDQ under anhydrous conditions induced cyclisation to give the corresponding PMP acetal 46 (92%).29 The remaining free hydroxyl group at C37 (the future site for glycosylation) was efficiently converted into the DEIPS (Et2i-PrSi) ether 47 under standard silylation conditions (DEIPSCl, imidazole, 97%). Reductive opening of the PMP acetal, mediated by DIBAL at −78 °C, then regioselectively generated the secondary PMB ether 48 and the remaining primary alcohol was subsequently oxidised with Dess–Martin periodinane under buffered conditions to give aldehyde 49 (86%, two steps).
At this point, another boron aldol reaction was employed for chain extension to complete the full C29–C38 backbone of fragment 11 (Scheme 10). Selective E-enolisation (c-Hex2BCl, Et3N) of ethyl ketone 3530 to give enolate 50 and subsequent reaction with aldehyde 49 proceeded smoothly to provide adduct 51 (>95:5 dr, 96%). Pleasingly, this reaction was found to be higher yielding when performed on multi-gram scale than previously observed during route investigation.6b This highly selective 1,4-syn aldol reaction is considered to proceed viaTS-5, with a stabilising formyl hydrogen bond and avoidance of allylic strain in the enolate matched with Felkin–Anh induction from the aldehyde component. It results in the controlled installation of the stereocentres at C32 and C33 with the requisite 1,2-anti configuration and C30 and C33 in a 1,4-syn relationship.
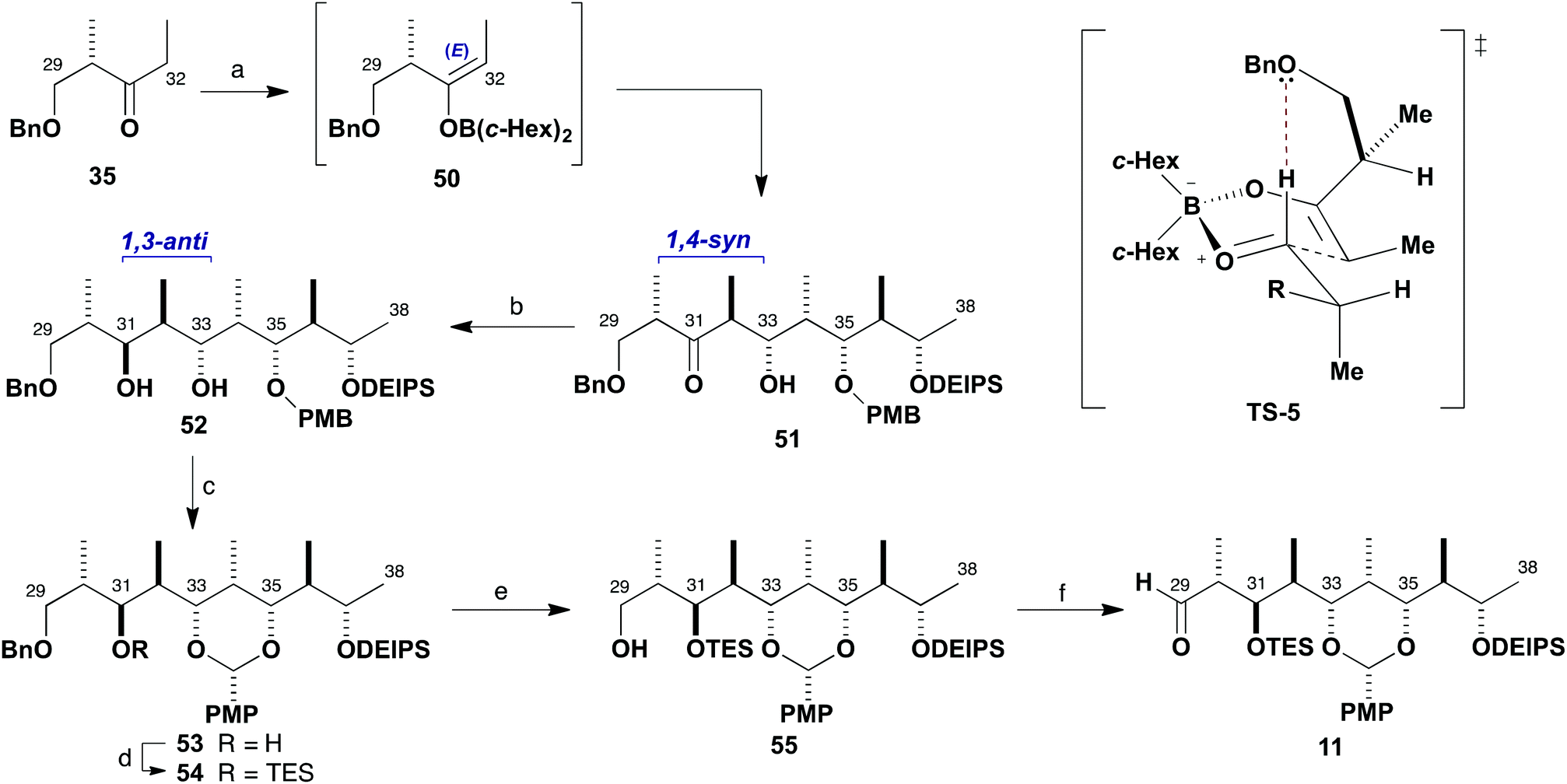 | ||
| Scheme 10 Synthesis of aldehyde 11. Reagents and conditions: (a) Ketone 35, c-Hex2BCl, Et3N, Et2O, 0 °C; aldehyde 49, Et2O, −78 to −20 °C, 96%, >95:5 dr; (b) Me4NBH(OAc)3, MeCN–AcOH (4:1), −30 °C, 91%, >95:5 dr; (c) DDQ, 4 Å mol. sieves, CH2Cl2, −10 °C, 85%; (d) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C, 99%; (e) H2, RANEY-Nickel®, EtOH, 92%; (f) DMP, NaHCO3, CH2Cl2, 92%. | ||
Directed reduction of the resulting β-hydroxyketone 51 under Evans–Saksena conditions23 using Me4NBH(OAc)3 then provided the desired 1,3-anti diol 52 with high diastereoselectivity (>95:5 dr, 91%). This reduction set the final stereogenic centre of the characteristic stereooctet spanning the C24–C38 region of the brasilinolides. Further protecting group manipulation was required at this stage to differentiate the newly formed C31 hydroxyl group, which corresponds to the site for eventual macrolactonisation. Oxidative cyclisation of the PMB ether 52 with DDQ in CH2Cl2 under anhydrous conditions proceeded cleanly and afforded the PMP acetal 53 (85%). Conversion into the TES ether 54 (TESOTf, 2,6-lutidine) was followed by selective hydrogenolysis of the benzyl ether in the presence of the PMP acetal using RANEY®-Nickel in EtOH to reveal the primary alcohol 55,31 which was oxidised using buffered Dess–Martin periodinane to give aldehyde 11 (85%, two steps).
With the β-ketophosphonate (R)-10 and the aldehyde 11 in hand, fragment union via HWE olefination was examined (Scheme 11). Following our standard conditions,32 this transformation was mediated by Ba(OH)2 in wet THF to efficiently afford enone 56 (96%) in excellent selectivity (>95:5 E:Z). Reagent-controlled CBS reduction33 of 56 to generate the required allylic alcohol 57 was found to be highly diastereoselective (88%, >95:5 dr) with the use of (R)-Me-CBS catalyst and BH3·SMe2 at −50 °C. Having installed the requisite (27S)-hydroxyl-bearing centre, the Sharpless asymmetric epoxidation of this elaborate allylic alcohol was investigated.34 In this case, the desired epoxide configuration indicated the deployment of the Ti(Oi-Pr)4–(+)-DIPT reagent system. On small scale, the Sharpless epoxidation of 57 was found to proceed to afford 58 in 59% yield, attaining an acceptable 7:1 dr, but this proved to be capricious and difficult to reproduce on a larger scale. Nevertheless, with some of the desired epoxide in hand, the synthesis of the planned northern fragment 6 (86%, two steps) was completed by TBS ether formation (TBSOTf, 2,6-lutidine) followed by a dihydroxylation–oxidative diol cleavage step (OsO4; NaIO4–SiO2) to reveal the methyl ketone functionality.35 Following this route, the preparation of the C20–C38 northern fragment 6 was achieved in 13% overall yield over 16 linear steps from ethyl ketone 36.
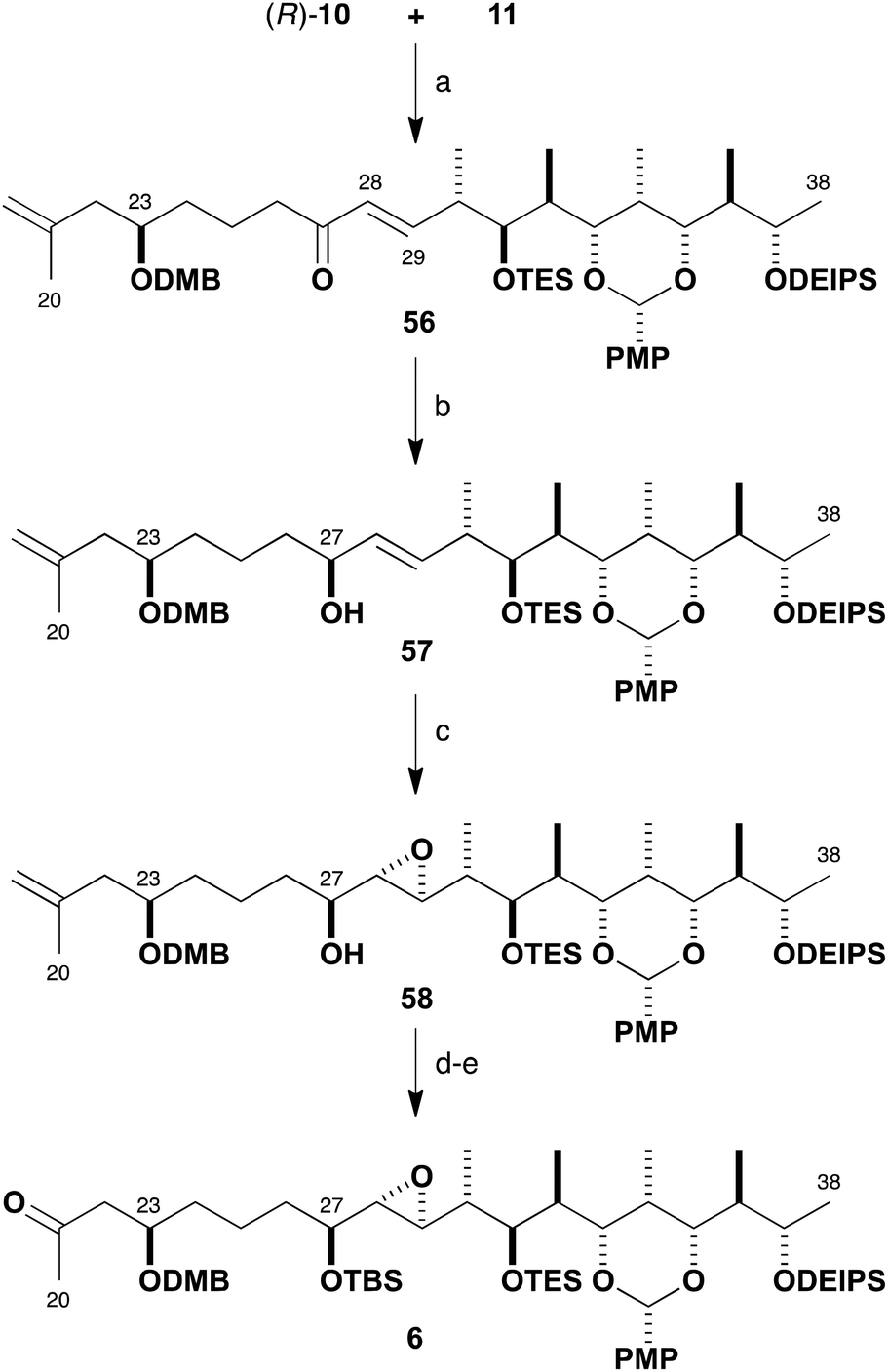 | ||
| Scheme 11 Synthesis of methyl ketone 6. Reagents and conditions: (a) Ba(OH)2, THF–H2O (40:1), 96%, >95:5 E:Z; (b) (R)-Me-CBS catalyst, BH3·SMe2, THF, −50 °C, 88%, >95:5 dr; (c) Ti(Oi-Pr)4, (+)-DIPT, t-BuOOH, 4 Å mol. sieves, CH2Cl2, −20 °C, 59%, 7:1 dr; (d) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C; (e) OsO4, NMO, THF–H2O; NaIO4–SiO2, CH2Cl2, 86% (two steps). | ||
Preliminary studies on the aldol coupling of the northern and southern fragments
With routes to the major fragments 5 and 6 developed, attention now turned to exploring the pivotal C19–C20 aldol bond construction to enable completion of the full carbon skeleton of the brasilinolides. As outlined in Scheme 12, it was initially planned that the C19 configuration could be controlled in a 1,3-anti sense relative to C17 via a Mukaiyama-type aldol bond construction.5a,36 This envisaged addition of the silyl enol ether 59 (derived from 6) to a suitable aldehyde such as 60 to be prepared from the available southern fragment 5. The anticipated β-hydroxyketone 61 resulting from this coupling would then be expected to undergo in situ hemiacetalisation at C15 to give 62. The advanced nature of both components dictated that this complex system should first be modelled by investigating the aldol coupling reactions of suitable truncated fragments, which could be readily prepared by interception of intermediates already in hand.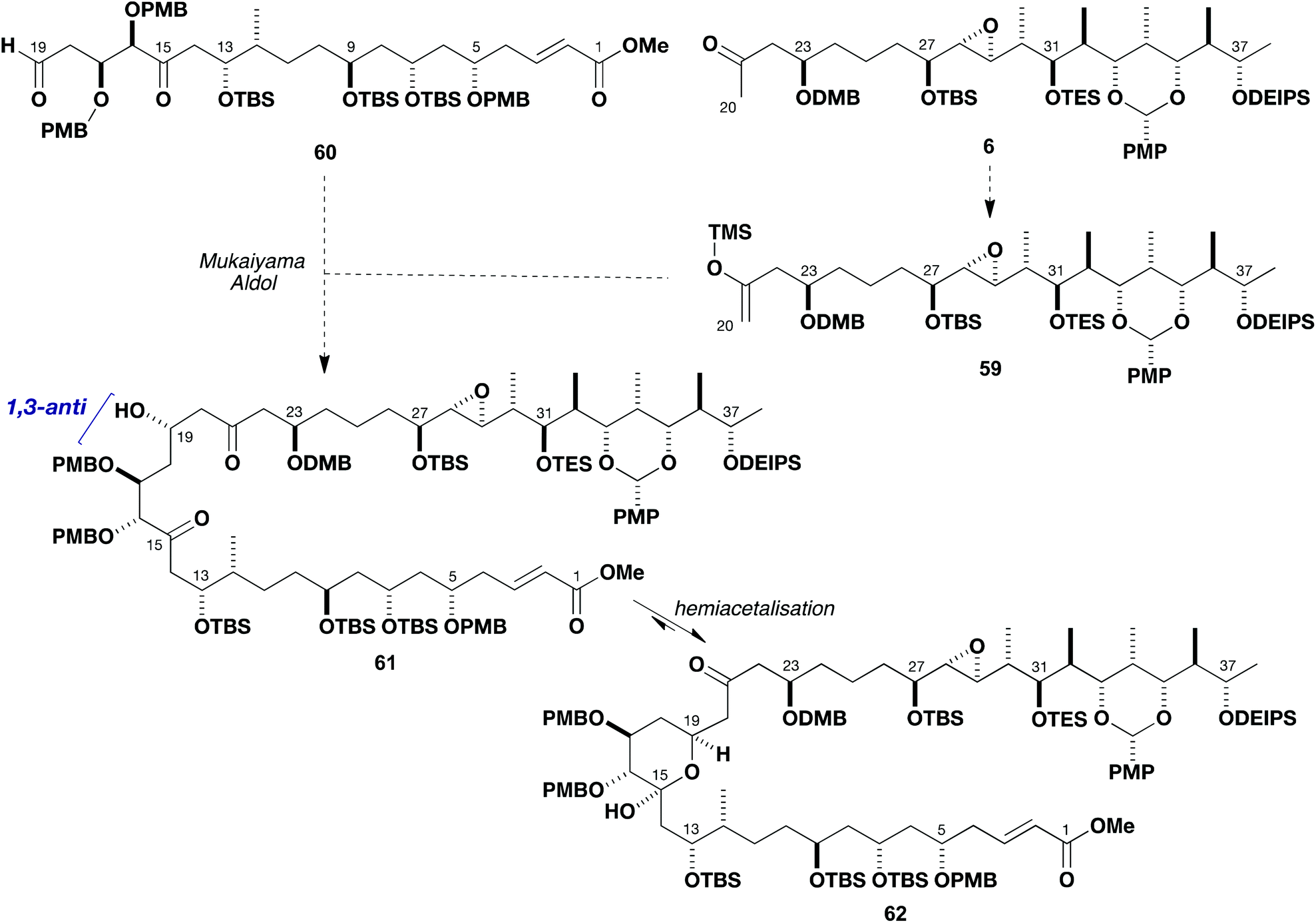 | ||
| Scheme 12 Proposed Mukaiyama-type aldol coupling between 59 and 60 to assemble the advanced C1–C38 intermediate 62. | ||
As shown in Scheme 13, the truncated methyl ketone (R)-63 was easily accessed from the previously described intermediate (R)-40. Thus dihydroxylation of olefin (R)-40 (OsO4, NMO) and oxidative glycol cleavage with NaIO4–SiO2 provided (R)-63 (86%). The model aldehyde 64 was prepared from the β-hydroxyketone 29 obtained in exploratory studies (Scheme 5) for the southern fragment synthesis. In this case, the secondary alcohol was first protected as the TBS ether (TBSOTf, 2,6-lutidine) and the ketone reduced (NaBH4). Then, the primary TBS ether was cleaved (PPTS, MeOH) and both hydroxyl groups oxidised under Swern conditions to give the δ-ketoaldehyde 64 (74%, four steps). This sequence served to circumvent deleterious cyclisation–elimination pathways, which can occur in the oxidative manipulation of 1,5-hydroxyketones.37
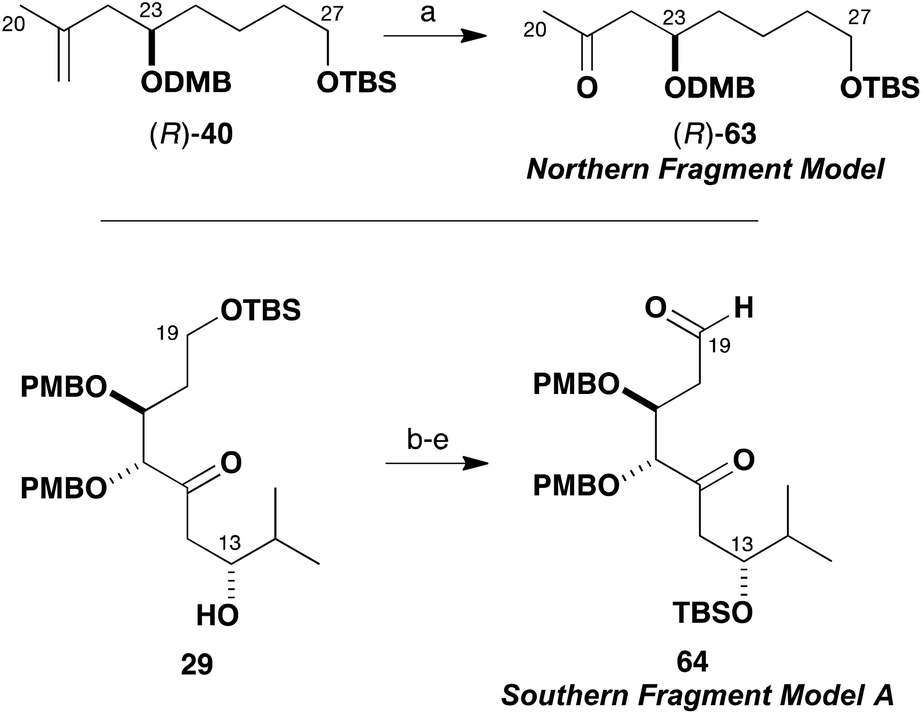 | ||
| Scheme 13 Preparation of model fragments (R)-63 and 64. Reagents and conditions: (a) OsO4, N-morpholine-N-oxide, THF–H2O; NaIO4–SiO2, 86%; (b) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 95%; (c) NaBH4, MeOH, 99%; (d) PPTS, MeOH, 85%; (e) (COCl)2, DMSO, CH2Cl2, −78 °C; Et3N, 93%. | ||
Given the β-oxygenation at C17 in the aldehyde component 64, it was anticipated that addition of the silyl enol ether (R)-65 derived from (R)-63 would install the C19 hydroxyl stereocentre in a 1,3-anti sense (Scheme 14). The predicted 1,3-stereoinduction in such Mukaiyama aldol reactions of β-alkoxyaldehydes to 66 invokes an acyclic transition state based on the Evans polar model.38 In support of this plan, a similar Mukaiyama aldol coupling (Scheme 15) of silyl enol ether 67 with the δ-ketoaldehyde 68 mediated by BF3·OEt2 at −98 °C gave the hemiacetal adduct 70 (cyclised version of 69) as a single diastereomer, which served as a key step in our total synthesis of auriside A (71).39
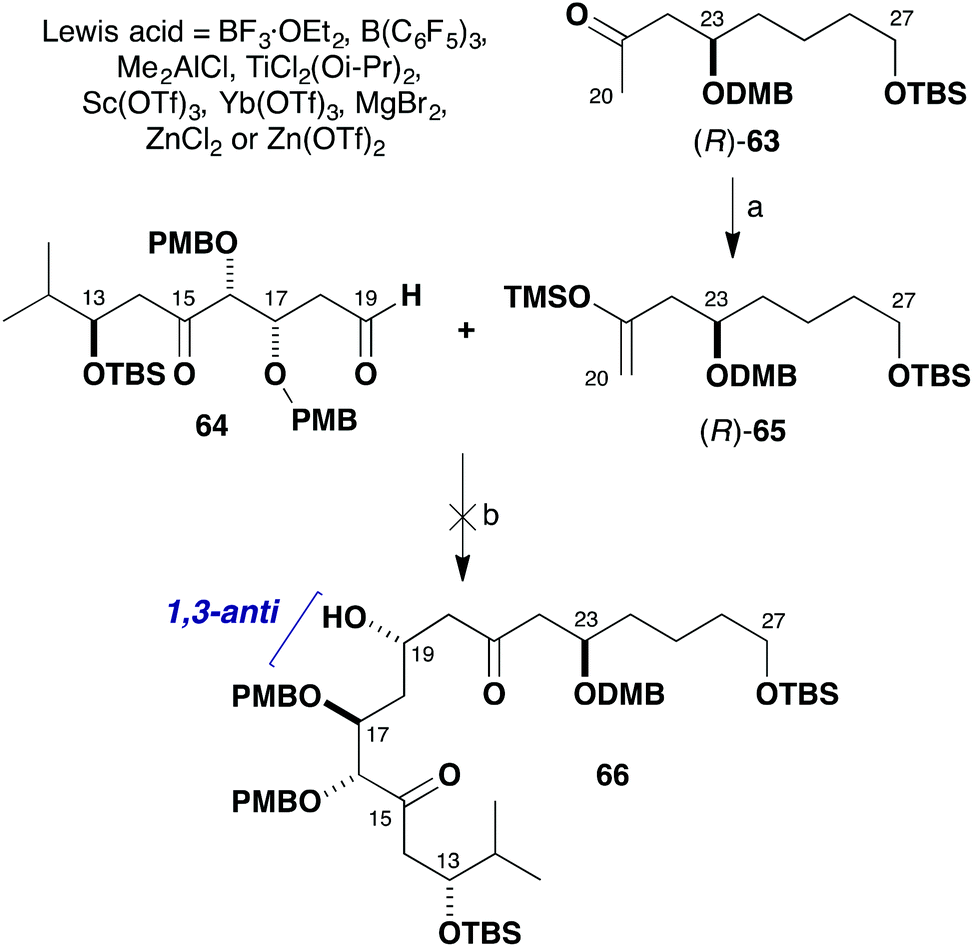 | ||
| Scheme 14 Attempted Mukaiyama aldol addition between (R)-65 and 64. Reagents and conditions: (a) LiHMDS, THF, −78 °C; TMSCl·Et3N, 90%; (b) Lewis acid, CH2Cl2, −98 to −78 °C. | ||
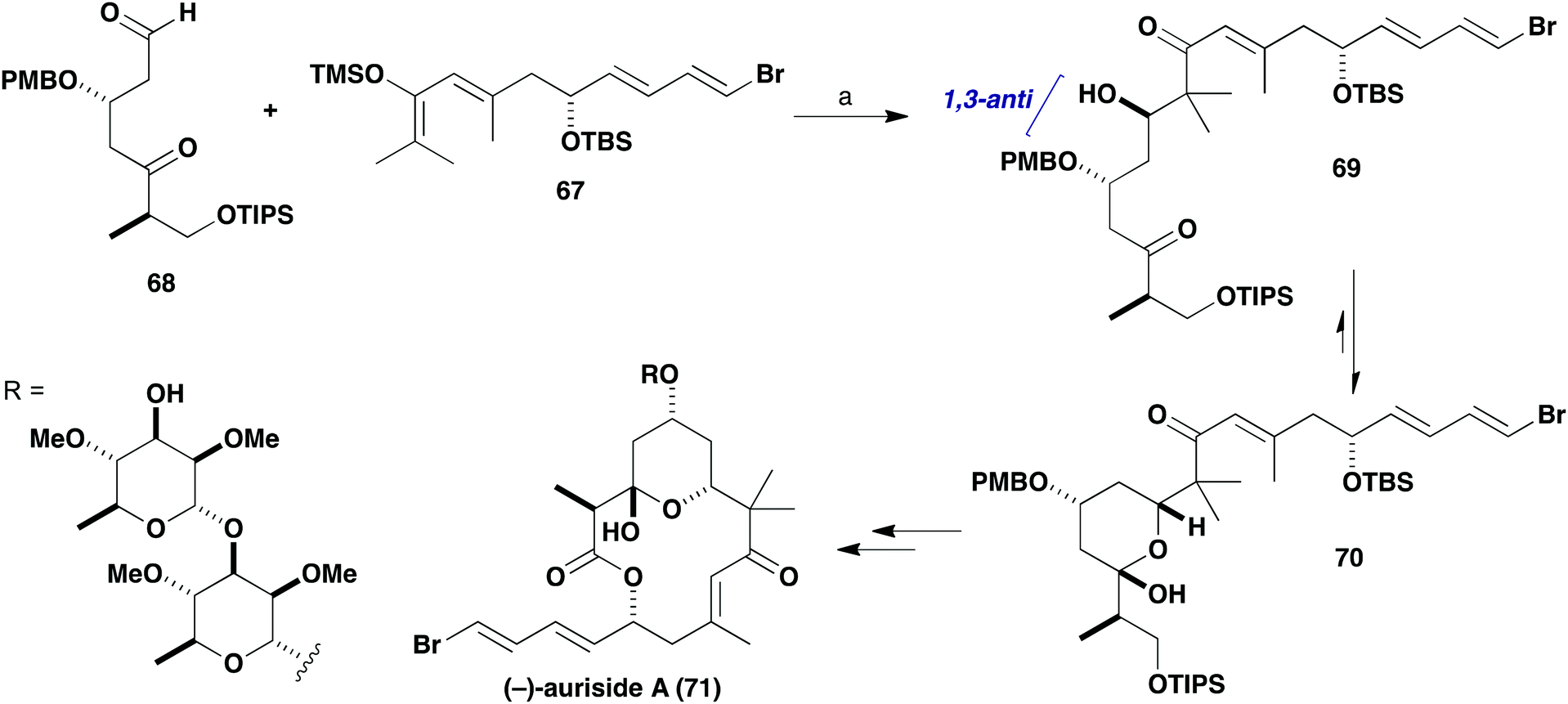 | ||
| Scheme 15 Fragment coupling using a Mukaiyama aldol reaction in the synthesis of (−)-auriside A (71). Reagents and conditions: (a) BF3·OEt2, CaH2, CH2Cl2, −98 °C, 10 min, 66%, single diastereomer. | ||
However, although the aldol reaction of silyl enol ether (R)-65 using BF3·OEt2 at −78 °C was found to proceed smoothly with simple aldehydes, many attempts to form the requisite C19–C20 bond in the more elaborate aldehyde 64 failed. As indicated in Scheme 14, the use of BF3·OEt2, B(C6F5)3, Me2AlCl, TiCl2(Oi-Pr)2, Sc(OTf)3 or Yb(OTf)3 all led to decomposition of the δ-ketoaldehyde component 64. Disappointingly, screening further Lewis acids such as MgBr2, ZnCl2 or Zn(OTf)2 also did not affect the desired aldol addition but served to only cleave the silyl enol ether to afford the corresponding methyl ketone (R)-63. In all of these cases, no product-derived material was observed in the crude 1H NMR spectra.
As the Mukaiyama aldol approach failed in this situation due to the unanticipated sensitivity of 64 to Lewis acids, it was proposed that the desired construction of the C19–C20 bond might be realised through the much milder conditions associated with a boron-mediated aldol reaction (Scheme 16). By comparison with the known reactivity of such β-alkoxy methyl ketones, the newly formed C19 hydroxyl centre likely would preferentially form with the undesired configuration due to the inherent 1,5-anti induction from the (23R)-stereocentre.
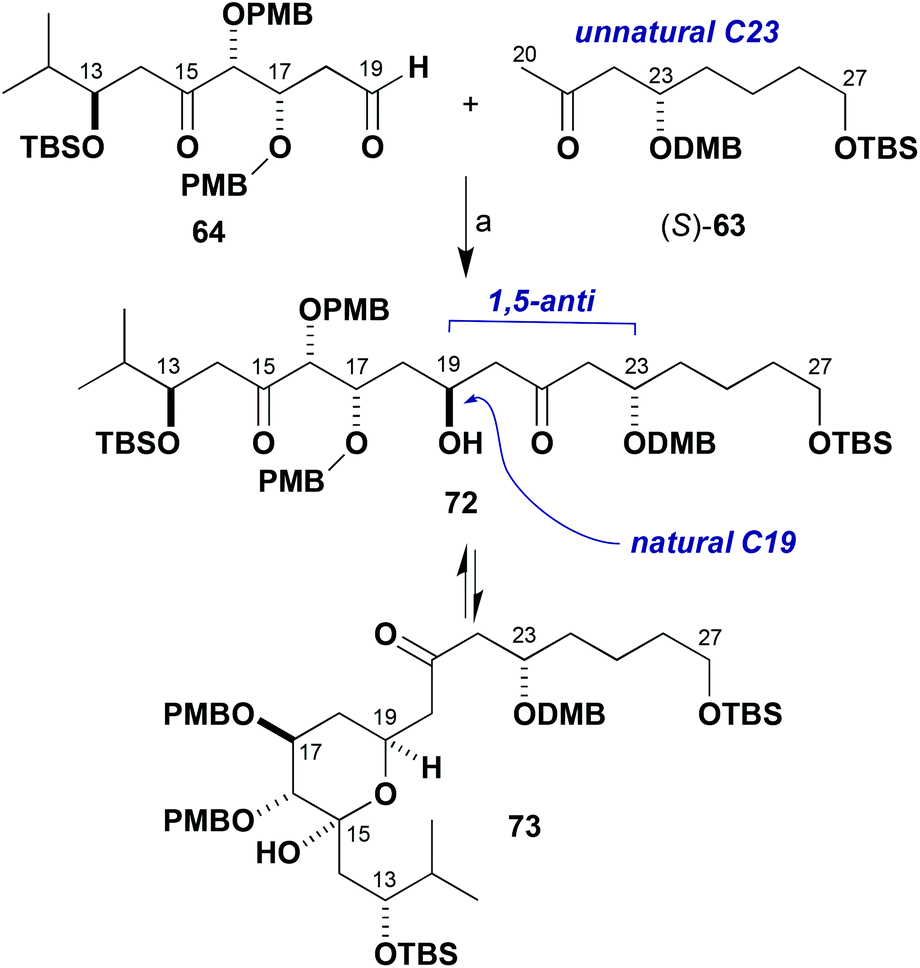 | ||
| Scheme 16 Model boron-mediated aldol reaction between (S)-63 and 64. Reagents and conditions: (a) Ketone (S)-63, c-Hex2BCl, Et3N, Et2O, 0 °C, 1 h; aldehyde 64, −78 to −20 °C. | ||
As the stereocentre at C23 was anticipated to control the installation of the C19 hydroxyl group in a 1,5-anti sense,7,21 it was decided to consider inverting the configuration of the enolate component to achieve the requisite diastereoselectivity in the coupling step and this might help simplify the NMR analysis. This revised strategy was adopted having noted that a late-stage installation of an ester appendage at C23 might be performed with inversion of configuration under Mitsunobu conditions instead of the previously anticipated direct esterification suggested earlier in Scheme 1. Therefore, model studies using the enantiomeric methyl ketone (S)-63 were pursued next, where this was easily prepared by changing the enantiomer of the Jacobsen catalyst used in the established route (Scheme 8). In the event, enolisation of methyl ketone (S)-63 under standard conditions (c-Hex2BCl, Et3N) and addition to 64 resulted in, for the first time, evidence of adduct formation in the crude 1H NMR spectrum. The NMR analysis, however, was complicated due to the presumed equilibrium formed between the β-hydroxyketone 72 and the cyclic hemiacetal form 73 and diastereomers thereof. These observations were taken as a promising indication that further investigations of these types of model systems would yield the desired outcome to be deployed in the real fragment coupling.
It was also apparent from several attempted manipulations of the mixture of 72 and 73 that in the full system (cf.Scheme 12) clean formation of the C15 hemiacetal product would be difficult to realise. Faced with these problems, it was proposed that masking the C15 ketone as an alkene might help attain more control in this critical fragment coupling step. Previously prepared ketone 30 allowed for such exploratory investigations to be performed (Scheme 17). It was found to be possible to methylenate ketone 30 under both Takai–Lombardo40 and Nysted reagent41 protocols to give 74 (94% and 74% yields, respectively). Selective cleavage of the primary TBS ether (HF·pyr) then gave alcohol 75 (96%), which was oxidised to give the revised model aldehyde 76 (95%) with Dess–Martin periodinane in readiness for further fragment coupling studies (Tables 1 and 2).
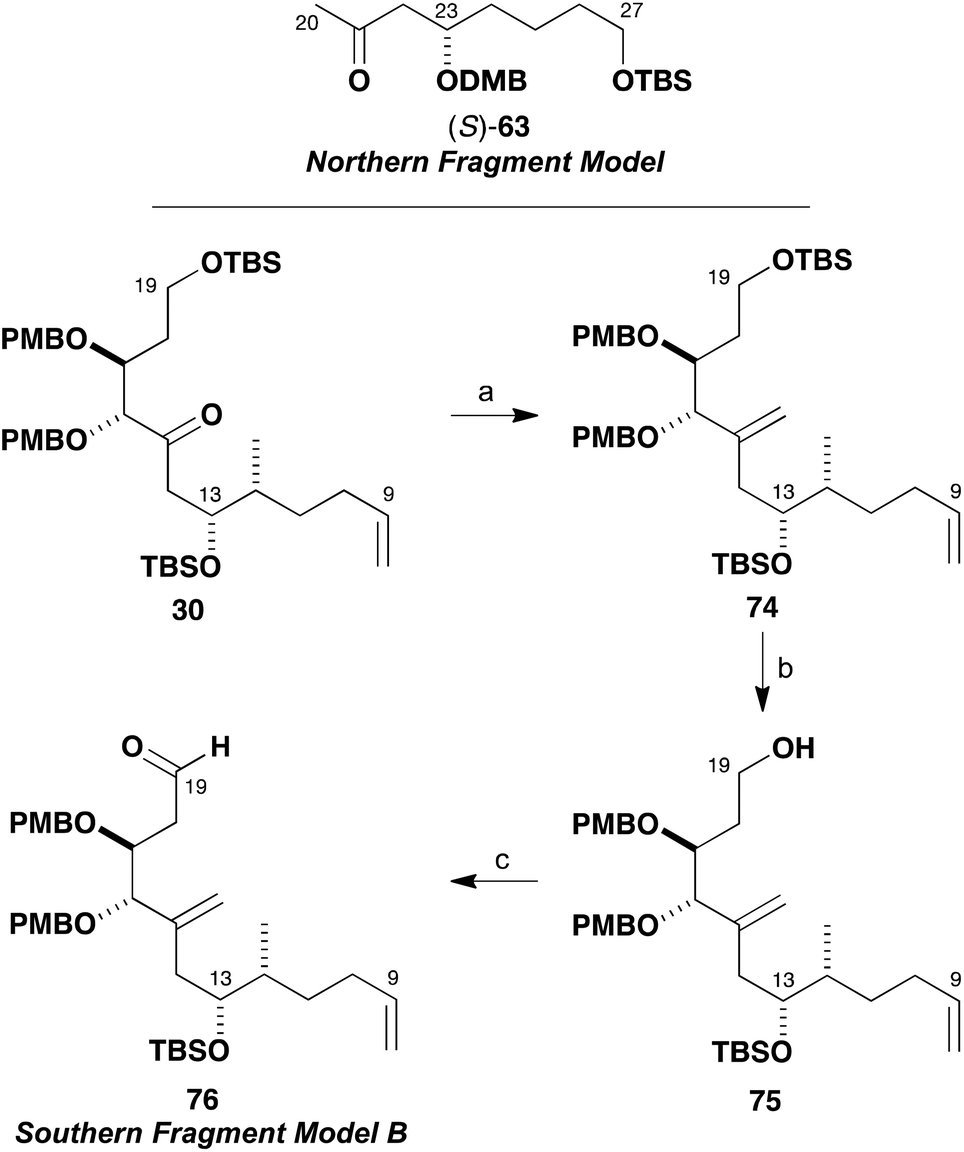 | ||
| Scheme 17 Preparation of aldehyde 76. Reagents and conditions: (a) Nysted reagent (cyclo-dibromodi-μ-methylene[μ-(tetrahydrofuran)]trizinc), TiCl4, THF, 94% or PbI2, CH2I2, Zn, THF, 74%; (b) HF·pyr, pyr, THF, 96%; (c) DMP, NaHCO3, CH2Cl2, 95%. | ||
| Entry | Lewis acid | Base | Enolisation temp. (Time) | Reaction temp. (Time) | Solvent | Crude dr 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :78 :78 |
|---|---|---|---|---|---|---|
| 1 | c-Hex2BCl | Et3N | −78 °C (1 h) | −78 °C (3 h) → −23 °C (16 h) | Et2O | 3:1 |
| 2 | Bu2BOTf | i-Pr2NEt | −78 °C (1 h) | −78 °C (3 h) | Et2O | 3.5:1 |
| 3 | 9-BBNOTf | i-Pr2NEt | −78 °C (1 h) | −78 °C (1 h) | Et2O | 4:1 |
| 4 | 9-BBNCl | i-Pr2NEt | −78 °C (1 h) | −78 °C (3 h) | Et2O | 4:1 |
| 5 | 9-BBNCl | i-Pr2NEt | −78 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Et2O | 4.8:1 |
| 6 | (−)-Ipc2BCl | Et3N | 0 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Et2O | 2.5:1 |
| 7 | (−)-Ipc2BCl | Et3N | 0 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Pentane | 2:1 |
| 8 | (−)-Ipc2BCl | Et3N | 0 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | CH2Cl2 | 1:1 |
| 9 | (+)-Ipc2BCl | Et3N | 0 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Et2O | 1:2 |
| Entry | Lewis acid | Base | Enolisation temp. (Time) | Reaction temp. (Time) | Solvent | Crude dr 79:80 |
|---|---|---|---|---|---|---|
| 1 | c-Hex2BCl | Et3N | −78 °C (1 h) | −78 °C (3 h) → −23 °C (16 h) | Et2O | 5:1 |
| 2 | (+)-Ipc2BCl | Et3N | −78 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Et2O | 17:1 |
| 3 | (−)-Ipc2BCl | Et3N | −78 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Et2O | 1:1.5 |
Enolisation of methyl ketone (S)-63 (epi-C23 series) and subsequent aldol addition to aldehyde 76 was first performed in Et2O at low temperature with a variety of achiral boron reagents (Scheme 18, Table 1, entries 1–5). As anticipated, there was some selectivity for the desired 1,5-anti product 77, although not as high as previously observed in these types of reaction. There was a modest increase in diastereoselectivity to a more promising 4.8:1 dr (entry 5), when using 9-BBNCl/i-Pr2NEt. In this complex aldol coupling, it was conjectured that the aldehyde component 76 has a π-facial bias in the opposing sense to that desired and 1,3-syn to the C17 stereogenic centre. This would appear to violate the bias for 1,3-anti stereoinduction observed in simple systems according to the Evans polar model.38 There is, however, some evidence to suggest that additional stereochemical features and steric bulk in the γ-position of β-alkoxyaldehydes can disrupt this model by way of a gearing effect, such as that described by Sulikowski and co-workers.42
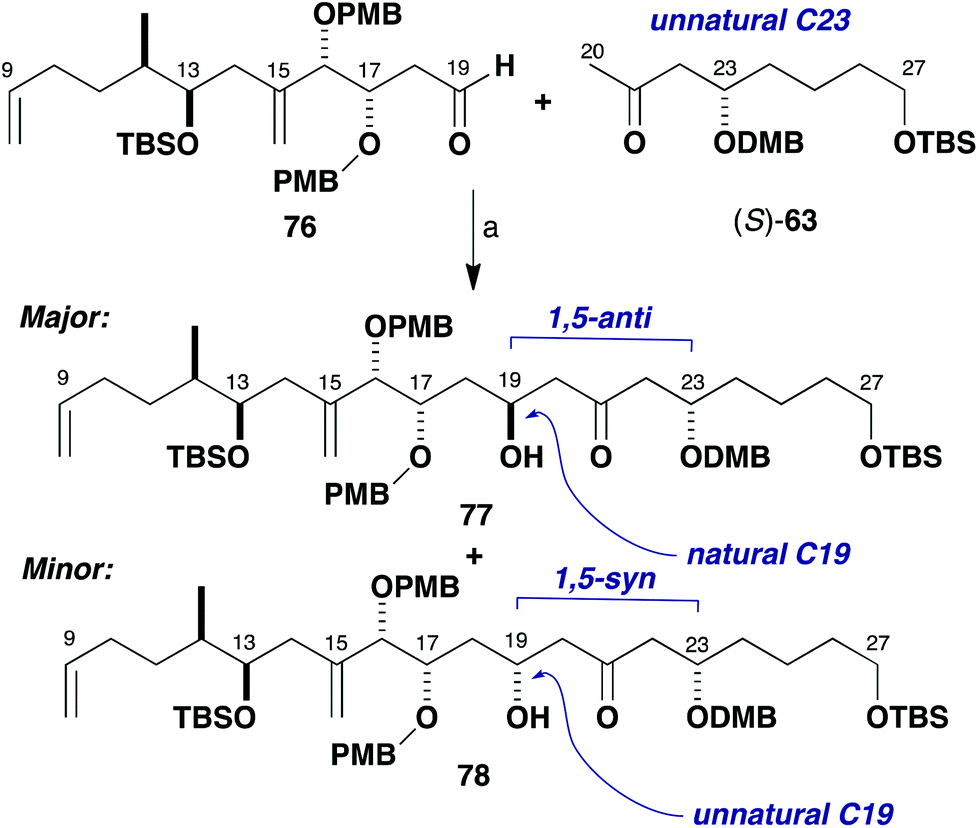 | ||
| Scheme 18 Model aldol coupling between (S)-63 and 76. Reagents and conditions: (a) Ketone (S)-63, L2BX, R3N; aldehyde 76, conditions in Table 1. | ||
Having achieved a promising level of selectivity for setting the desired C19 configuration in 77, switching to a chiral boron reagent ((−)-Ipc2BCl) was expected to help reinforce the enolate π-facial preference (entries 6–8).43 Unfortunately, the product 77 was only observed to form with moderate diastereoselectivity (2.5:1 dr, 77:78) and changing the reaction solvent to pentane or CH2Cl2 was also found to give no improvement. This reduced diastereoselectivity was further observed by reaction with the enantiomeric (+)-Ipc2BCl (entry 9), whereby the chiral reagent reversed the sense of stereoinduction to the undesired C19 configuration.
The unexpected sense of π-facial bias of aldehyde 76 towards boron-mediated aldol additions prompted further investigation of this reaction. Further studies were performed with the methyl ketone (R)-63 (natural C23 configuration as in the brasilinolides). This was anticipated to expand the data set for the effects of stereochemical bias from the enolate, aldehyde and boron reagent (Scheme 19, Table 2). Reaction of aldehyde 76 with the dicyclohexylboron enolate of (R)-63 proceeded in 5:1 dr for the 1,5-anti product 79 corresponding to the unnatural configuration at C19 (entry 1). Comparison to the corresponding result with (S)-63 (Table 1, entry 1) shows an increase in 1,5-anti selectivity, indicating a small matched effect between the enolate and aldehyde. Indeed, reinforcement of this substrate-based stereoinduction with the appropriate (+)-Ipc2BCl reagent (Table 2, entry 2) now achieved excellent levels of diastereoselectivity (17:1 dr, 79:80) in favour of the 1,5-anti product. Unsurprisingly, when the enantiomeric (−)-Ipc2BCl was employed to help favour the 1,5-syn product 80 with the desired C19 configuration (entry 3), the diastereoselectivity was poor (1:1.5 dr, 79:80, 1,5-anti:syn) as the chiral reagent is tasked with overriding the inherent substrate reactivity.
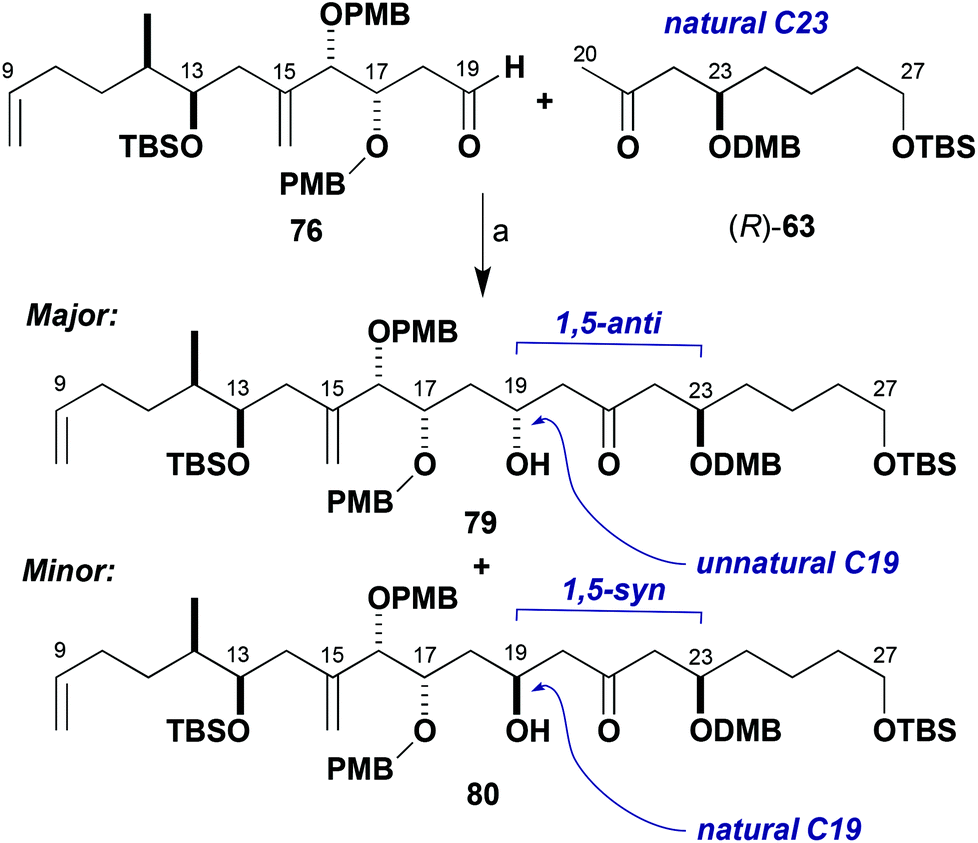 | ||
| Scheme 19 Model aldol coupling between (R)-63 and 76. Reagents and conditions: (a) Ketone (R)-63, L2BX, R3N; aldehyde 76, conditions in Table 2. | ||
This preliminary investigation of the unexpectedly complex C19–C20 aldol coupling step gave a helpful steer as to how to best evolve our strategy to advance the synthesis of the brasilinolides. It was concluded that it would be highly challenging to achieve excellent diastereoselectivity for the correct C19 configuration using boron-mediated aldol reactions. The completely matched reactivity between all components was identified as the scenario where (R)-63 (natural C23 configuration), aldehyde 76 (facial preference 1,3-syn to C17) and (+)-Ipc2BCl were used to give product 79 with the incorrect C19 configuration (Table 2, entry 2). Although as discussed previously, the C23 stereocentre could potentially be inverted at a later stage on esterification, selective inversion of C19 would be considerably more problematic. Having established a set of workable, albeit unoptimised, conditions for the formation of the C19–C20 bond with the C15-methylene model substrate 76, it was important in going forward to able to transfer this chemistry to the real system. Disappointingly, the C15-methylene functionality proved to be unreactive towards standard oxidation conditions to regenerate the required ketone presumably due to steric congestion. This shortcoming necessitated a change in strategy for masking the C15 ketone in the southern fragment as discussed below.
Elaboration of 5 to the advanced southern fragment 81
Based on the foregoing findings, a C15-acetoxy aldehyde 81 (Scheme 20) was proposed as the revised southern fragment to be used in the pivotal C19–C20 aldol coupling step. It was envisaged that the C15 ketone could subsequently be reinstalled by oxidation after selective cleavage of the acetate. Given the already high level of molecular complexity of the northern and southern fragments, it was advantageous that any reduction of the C15 ketone moiety was performed in a highly stereocontrolled fashion. In practice, reduction of 5 with Zn(BH4)2 at low temperature proceeded cleanly to afford alcohol 82 with high diastereoselectivity (99%, >95:5 dr).44 As expected from the reaction proceeding by a chelation pathway, this was assigned as the C15,C16-anti stereoisomer with the (15S)-configuration. Acetylation then gave ester 83 in 97% yield.
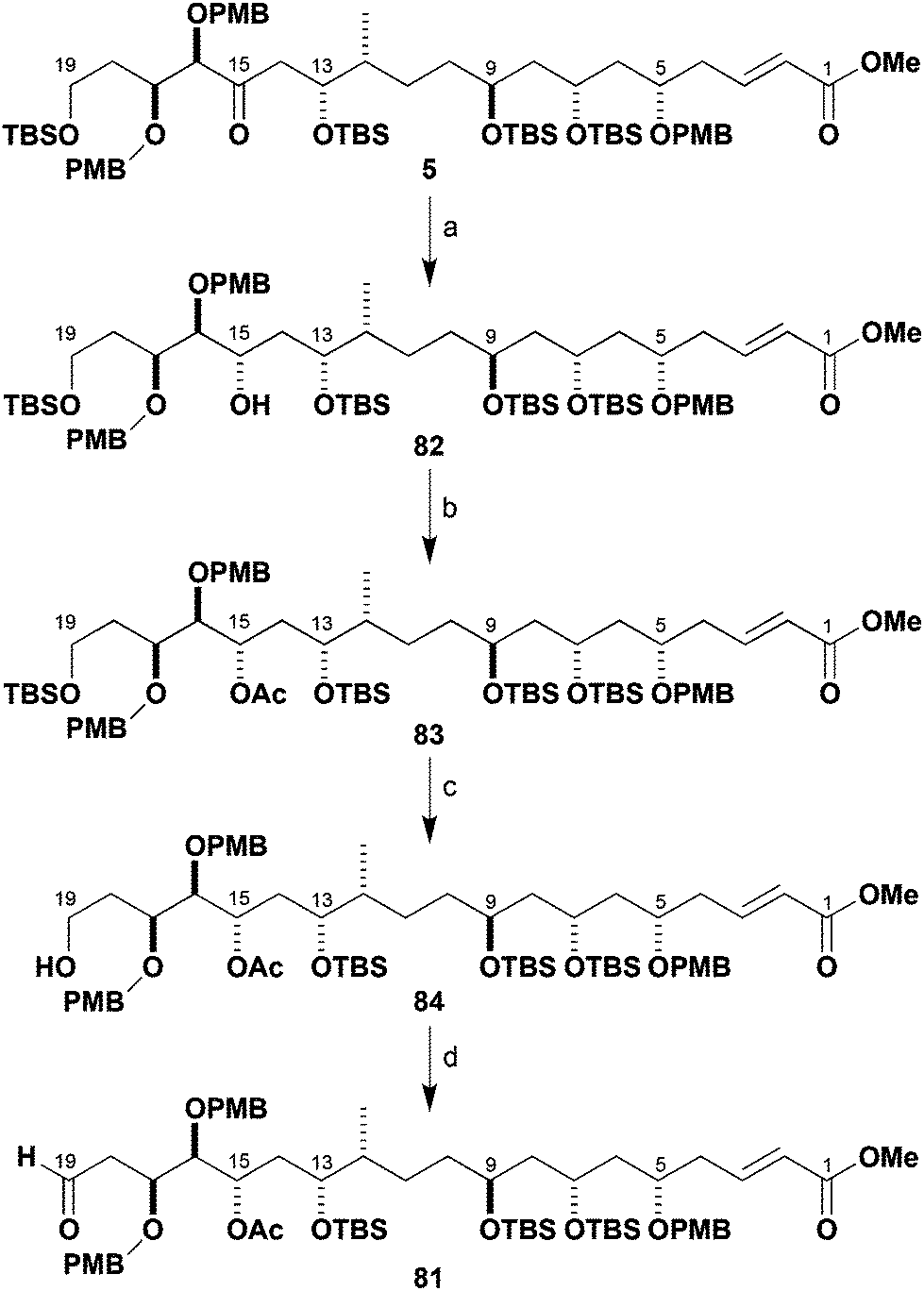 | ||
| Scheme 20 Preparation of C1–C19 aldehyde 81. Reagents and conditions: (a) Zn(BH4)2, Et2O, −78 to 0 °C, 99%, >95:5 dr; (b) Ac2O, Et3N, DMAP, CH2Cl2, 97%; (c) TBAF, AcOH, THF, 90% (brsm); (d) DMP, NaHCO3, CH2Cl2, 98%. | ||
Selective desilylation of 83 (TBAF, AcOH) at C19 to give 84 was followed by oxidation with Dess–Martin periodinane to afford aldehyde 81 (89%, two steps). This high yielding sequence gave the required stereodefined C15-acetoxy aldehyde 81 in 37% overall yield over 14 steps from 24.
Revisiting the northern fragment
One of the concerns in our planned synthesis of the brasilinolides was the potential reactivity of the C28–C29 epoxide moiety in the northern hemisphere. In the context of the structure determination studies performed on brasilinolide C (3), it was demonstrated that the epoxide was prone to intramolecular nucleophilic opening by pendant hydroxyl groups.3b Fortunately, this had not been a problem to date, where presumably the adjacent protecting groups shielded the epoxide even towards exposure to Lewis acids such as dibutylboron triflate. An unexpected hurdle, however, was the reliable installation of the epoxide itself in acceptable yield, diastereoselectivity, and on a reasonable scale. At the time, Sharpless asymmetric epoxidation of the allylic alcohol 57 (Scheme 11) did not appear to satisfy all these requirements and a screen of reaction conditions and other hydroxyl-directed electrophilic epoxidation protocols was performed to no avail.With the exploration of alternative electrophilic epoxidation methods on 57 proving unrewarding, it was proposed that the enone 85 (Scheme 21) could be employed as a substrate for nucleophilic epoxidation. This enone was prepared by an analogous route to that shown in Scheme 11 for the corresponding C23-epi compound 56 but now using the (23S)-β-ketophosphonate (S)-10 for the HWE step.20 Serendipitously, epoxidation of 85 with t-BuOOLi was found to be effective at −78 °C to afford 86 in both high diastereoselectivity (30:1 dr) and yield (91%). The configuration at C28 and C29, however, was subsequently determined to be epimeric to that required for the brasilinolides.20 Given this unexpectedly high selectivity and the enhanced scalability of the transformation, it was decided at this stage to go forward to advance the chemistry required for constructing the brasilinolides, chiefly examination of the major fragment coupling and further manipulation. The greater availability of the advanced intermediate 86 with the opposite epoxide configuration was considered to be a good opportunity for embarking on this intelligence gathering exercise.
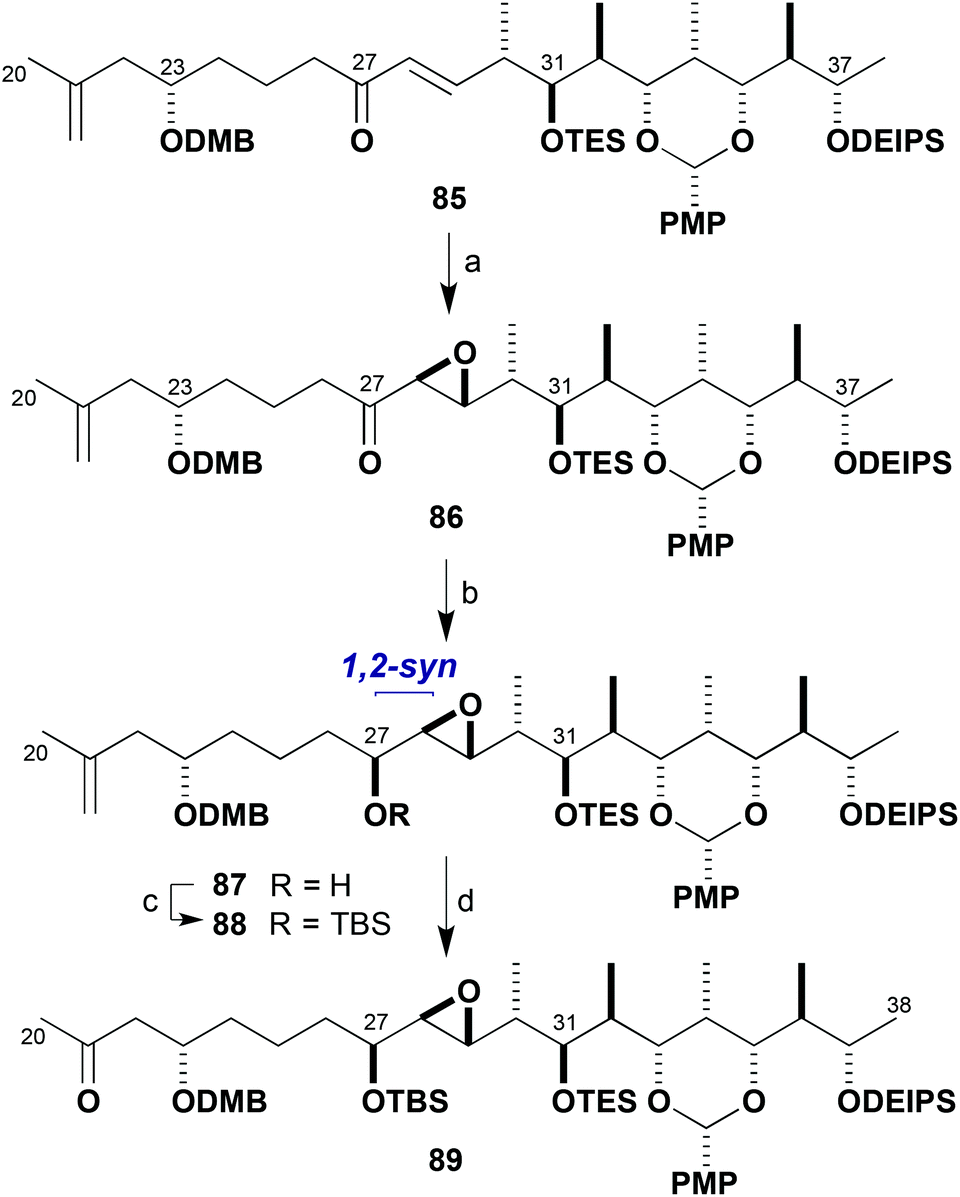 | ||
| Scheme 21 Synthesis of ketone 89. Reagents and conditions: (a) t-BuOOH, BuLi, THF, −78 to −20 °C, 91%, 30:1 dr; (b) DIBAL, t-BuOMe, −98 °C, 83%, 10:1 dr; (c) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 91%; (d) OsO4, N-methylmorpholine-N-oxide, THF–H2O; NaIO4–SiO2, CH2Cl2, 88%. | ||
As shown in Scheme 21, stereocontrolled reduction of ketone 86 was affected by treatment with DIBAL in t-BuOMe at −98 °C to give the alcohol 87 (83%) with 10:1 dr for the natural C27 configuration (Mosher ester analysis20). Alcohol 87 was protected as the TBS ether 88 (TBSOTf, 2,6-lutidine) and oxidative cleavage of the alkene (OsO4, NaIO4–SiO2) afforded the methyl ketone 89 (80%, two steps). With this modified route established, sufficient quantities of a full northern fragment mimic 89 were now generated to develop suitable conditions for achieving the complex aldol coupling with the full southern aldehyde 81.
Realising the aldol coupling of the northern and southern fragments
With useful stocks of the major fragments now in hand, their planned aldol union to assemble an advanced C1–C38 intermediate for the brasilinolides could be studied and any pitfalls identified. From experience gained earlier of boron-mediated aldol reactions using truncated systems, the desired 1,5-anti selective addition reaction between 81 and 89 was much anticipated (Scheme 22). At the outset, preliminary screenings of standard conditions using c-Hex2BCl/Et3N were found to give a modest 2:1 dr in favour of the 1,5-anti product 90 (Table 3, entry 1). Use of (−)-Ipc2BCl as the matched chiral boron reagent proceeded with an improved 3:1 dr in favour of the desired C19 diastereomer (entry 2) but in low conversion. By contrast, use of (+)-Ipc2BCl resulted in negligible diastereoselectivity for this transformation (entry 3). While all of these exploratory reactions gave a disappointingly low conversion, both starting materials were recoverable (Table 3).
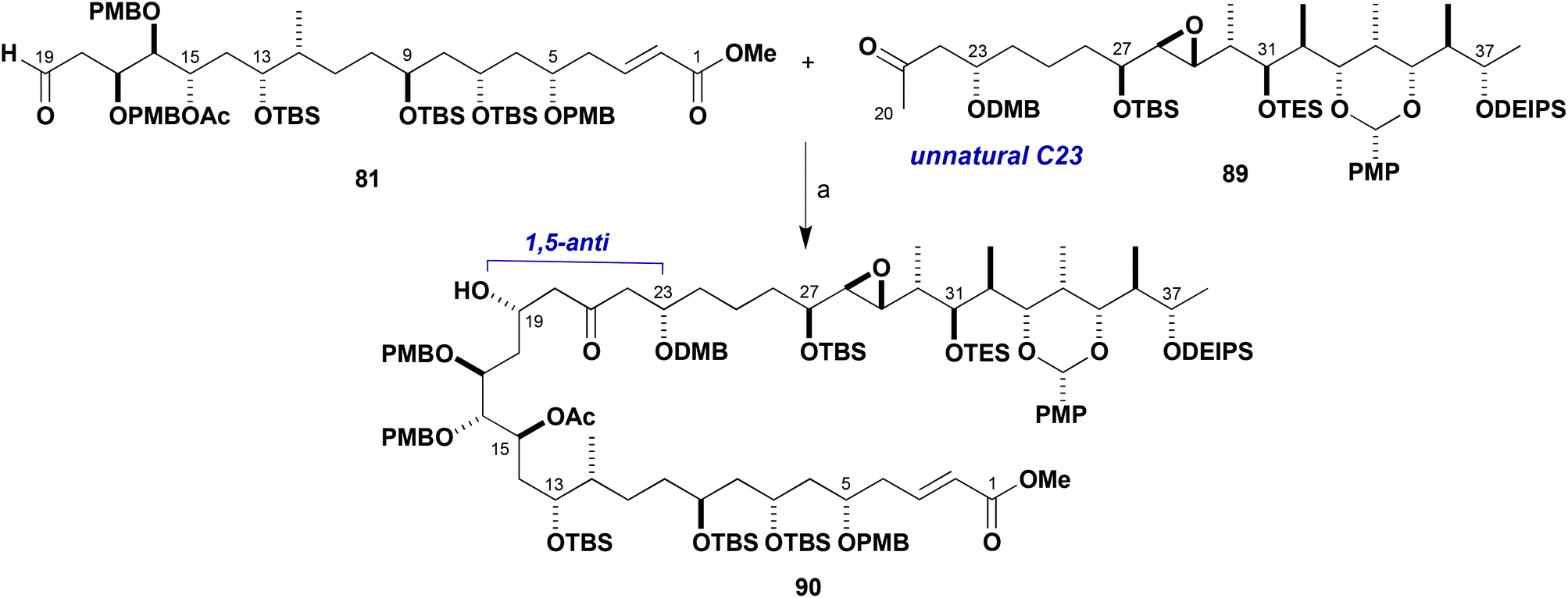 | ||
| Scheme 22 Aldol coupling of fragments 81 and 89. Reagents and conditions: (a) Ketone 89, L2BX, R3N; aldehyde 81, conditions in Table 3. | ||
| Entry | Lewis acid | Base | Enolisation temp. (Time) | Reaction temp. (Time) | Solvent | Isolated yield (%) | dr 1,5-anti:syn |
|---|---|---|---|---|---|---|---|
| 1 | c-Hex2BCl | Et3N | −78 °C (1 h) | −78 °C (3 h) → −23 °C (16 h) | Et2O | 8 | 2:1 |
| 2 | (−)-Ipc2BCl | Et3N | 0 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Et2O | 5 | 3:1 |
| 3 | (+)-Ipc2BCl | Et3N | 0 °C (1 h) | −78 °C (3 h) → −20 °C (1 h) | Et2O | 4 | 1:1.2 |
| 4 | Bu2BOTf | i-Pr2NEt | −78 °C (1 h) | −78 °C (4 h) | Et2O | 32 | 2.5:1 |
| 5 | Bu2BOTf | i-Pr2NEt | −78 °C (1 h) | −98 °C (1 h) → −78 °C (4 h) | Et2O | 48 (95% brsm) | 3.5:1 |
| 6 | Bu2BOTf | i-Pr2NEt | −78 °C (0.5 h) | −98 °C (1 h) → −78 °C (4 h) | Et2O | 44 | 3:1 |
| 7 | Bu2BOTf | i-Pr2NEt | −50 °C (0.5 h) | −98 °C (1 h) → −78 °C (4 h) | Et2O | <5 | — |
| 8 | Bu2BOTf | i-Pr2NEt | −78 °C (1 h) | −98 °C (1 h) → −78 °C (4 h) | Pentane | 23 | 2.5:1 |
With a view to enhancing the reactivity of the boron enolate, the Lewis acid was changed to Bu2BOTf (entries 4–8). Conducting the aldol reaction at −78 °C led to a 32% isolated yield of a separable 2.5:1 mixture of adducts in favour of the desired C19 configuration in 90 with good recovery of unreacted ketone 89 and aldehyde 81 (entry 4). Use of the more reactive dibutylboron enolate was compatible with performing the reaction at a lower temperature (−98 °C), resulting in both higher diastereoselectivity and isolated yield (entry 5), with both starting materials also recoverable. This provided the aldol adducts in 95% yield brsm with a workable 3.5:1 dr in favour of 90. This would prove to be the preferred set of conditions for carrying out this challenging aldol fragment union, despite varying different reaction parameters (entries 6–8).
Further elaboration to a C1–C38 advanced intermediate
With access to the configurationally pure aldol product 90 representing the full C1–C38 carbon skeleton, studies into its manipulation to afford an advanced brasilinolide intermediate were commenced (Scheme 23). Hydroxyl-directed reduction of the ketone 90 under Evans–Saksena conditions23 proceeded smoothly to give the 1,3-anti diol 91 (>95:5 dr, 85%). This transformation represented the controlled installation of the remaining C21 stereocentre. The hydroxyl groups were then both derivatised as TBS ethers (TBSOTf, 2,6-lutidine) to give the fully protected intermediate 92 (98%).
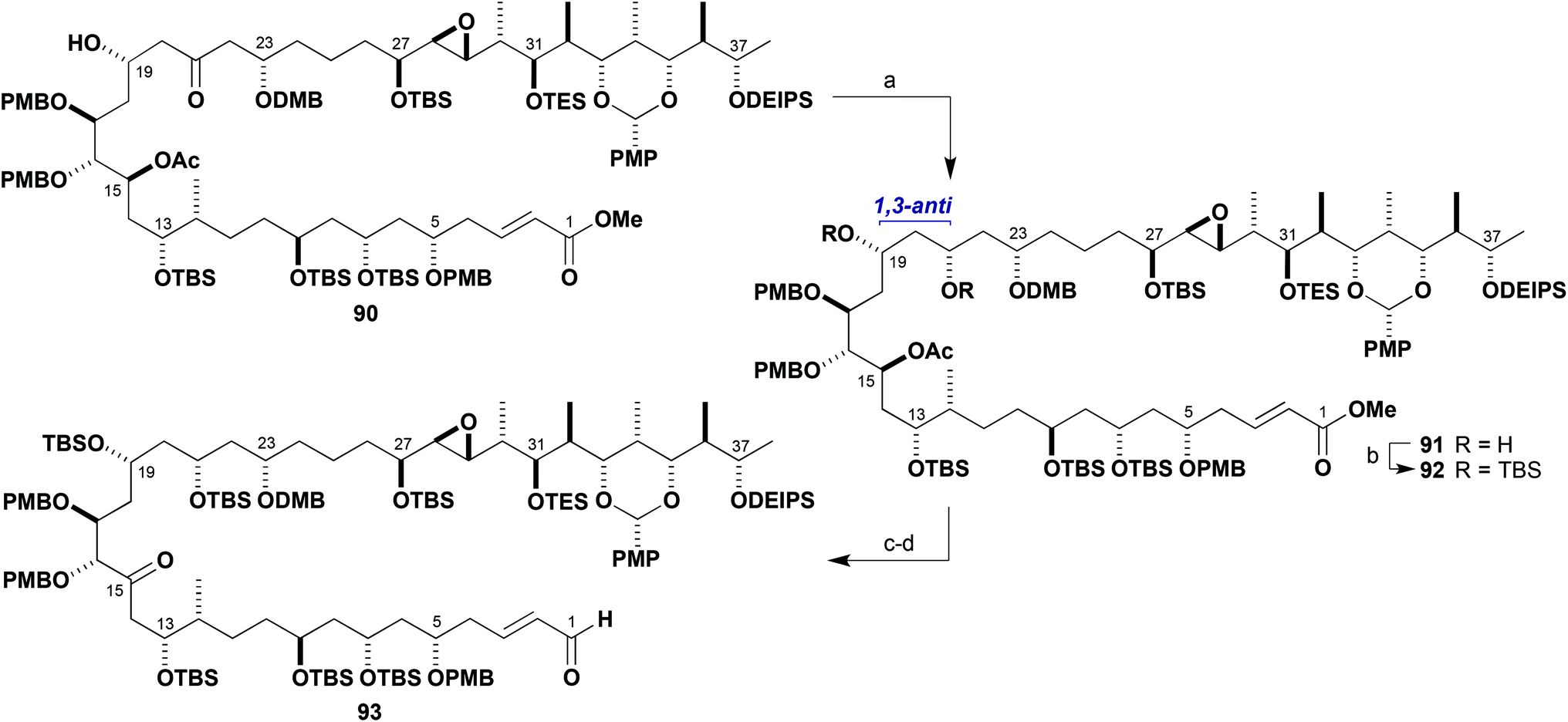 | ||
| Scheme 23 Preparation of the advanced C1–C38 coupled product 93. Reagents and conditions: (a) Me4NBH(OAc)3, MeCN–AcOH (4:1), −30 °C, 85%, >95:5 dr; (b) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 98%; (c) DIBAL, CH2Cl2, −78 °C; (d) DMP, NaHCO3, CH2Cl2, 90% (2 steps). | ||
We now required to saponify the acetate group at C15 in 92 with concomitant generation of the free carboxylic acid at C1 (Scheme 23). However, a screen of standard reagents (e.g. LiOH) led to hydrolysis of the methyl ester but without achieving the acetate cleavage. Gratifyingly, reduction of 92 with DIBAL at −78 °C successfully cleaved the acetate and also reduced the ester to an inconsequential mixture of allylic alcohol and enal. The crude mixture of these products was subjected to Dess–Martin oxidation to afford solely enal 93 (90%, two steps), also reintroducing the required ketone oxidation level at C15.45 While this has the wrong configuration for the epoxide, it is anticipated that this sequence should be directly applicable to the desired epoxide series.
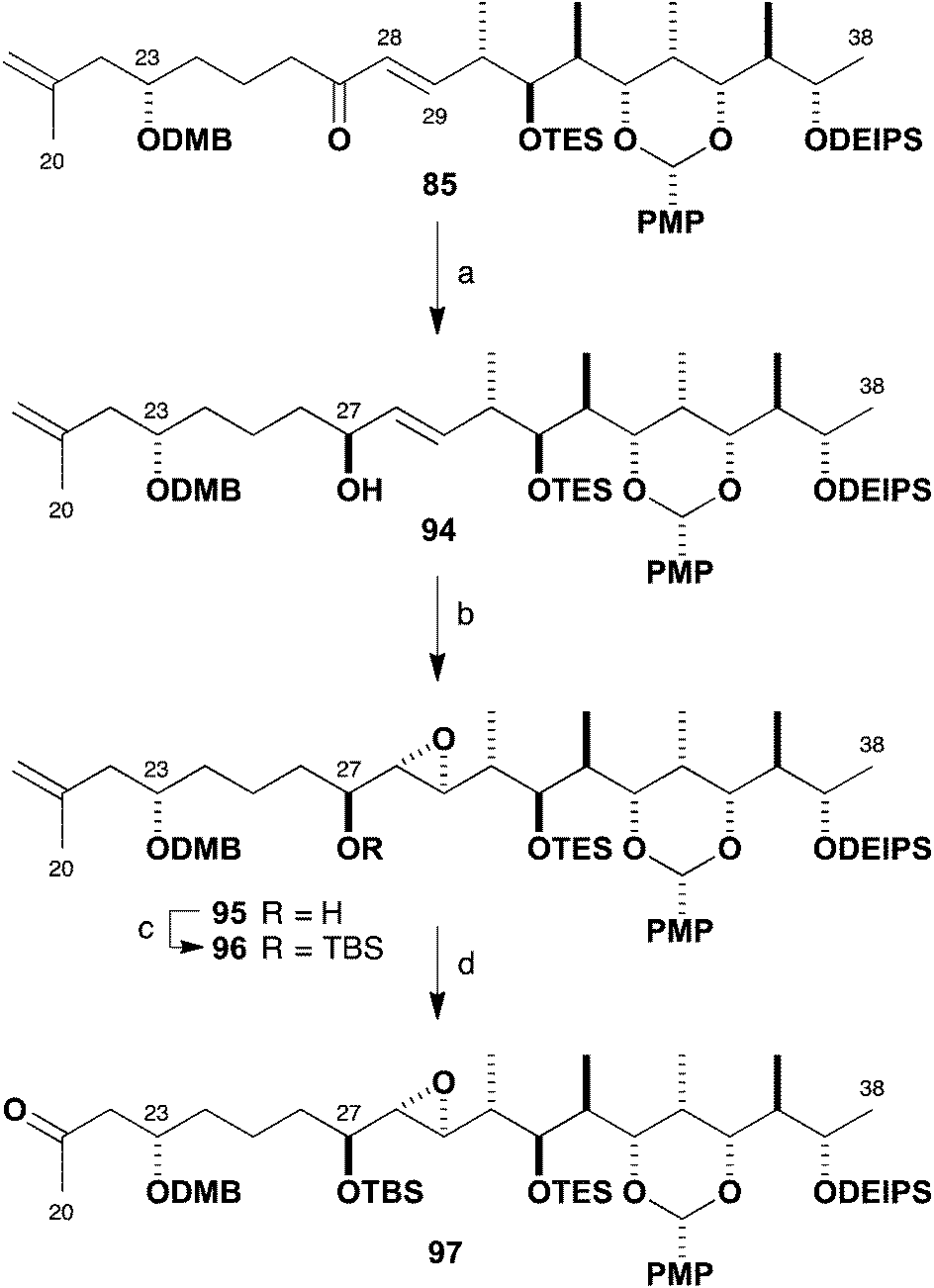
As a prelude to these future efforts, we returned to earlier intermediates to address the incorrectly configured epoxide (Scheme 24). Reagent-controlled CBS reduction of enone 85 to generate allylic alcohol 94 proceeded smoothly at −50 °C (88%, >95:5 dr) as had been observed with the epi-C23 compound 56. After some experimentation, it was found that Sharpless asymmetric epoxidation directed by the (27S)-hydroxyl-bearing stereocentre could be affected using a Ti(Oi-Pr)4–(+)-DET reagent system to give 95 in high yield (86%) but only moderate diastereoselectivity (2:1 dr 95:87). Nonetheless, this reproducible transformation now gave ready access to useful amounts of 95 possessing the requisite epoxide configuration. Thus epoxide 95 was converted into TBS ether 96 (TBSOTf, 2,6-lutidine, 86%) and finally to the revised northern fragment 97 by a dihydroxylation–oxidative diol cleavage step (OsO4; NaIO4–SiO2, 85%), in readiness for repeating the aldol coupling with aldehyde 81 (cf.Scheme 22).
 | ||
| Scheme 24 Preparation of C20–C38 methyl ketone 97. Reagents and conditions: (a) (R)-Me-CBS catalyst, BH3·SMe2, THF, −50 °C, 88%, >95:5 dr; (b) Ti(Oi-Pr)4 (10 mol%), (+)-DIPT (15 mol%), t-BuOOH, 4 Å mol. sieves, CH2Cl2, −25 °C, 86%, 2:1 dr; (c) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 86%; (d) OsO4, NMO, THF–H2O; NaIO4–SiO2, CH2Cl2, 85%. | ||
Conclusions and outlook
The brasilinolides are an architecturally complex family of polyhydroxylated 32-membered macrolides, characterised by their potent immunosuppressant and antifungal properties. In work directed toward their total synthesis, a range of asymmetric aldol/reduction sequences and catalytic protocols were employed to assemble a series of increasingly elaborate fragments. By adopting the highly convergent strategy outlined in Scheme 1, the preparation of the required C1–C19 and C20–C38 acyclic fragments 5 (10 steps, 43%) and 6 (16 steps, 13% from 36), containing seven and 12 stereocentres respectively, was first achieved. An adventurous fragment union by controlled C19–C20 bond formation was then extensively explored to construct the entire carbon chain of the brasilinolides. Notably, the use of boron-mediated aldol reactions of chiral ketones proved efficient on multiple occasions to assemble and then couple key fragments, with predictable installation of the required configuration via either 1,4- or 1,5-stereoinduction from the enolate component. In particular, this chemistry proved crucial for achieving the complex aldol coupling between methyl ketone 89 and aldehyde 81 to give adduct 90, where Mukaiyama-type aldol protocols had earlier proved unrewarding in model studies. Following the controlled manipulation of various intermediates derived from 90, the preparation of the protected C1–C38 polyol 93 (21 steps from 36 in the longest linear sequence) was then achieved, thus setting the stage for future efforts directed at late-stage diversification to access the various brasilinolide congeners.From our perspective, the controlled assembly of the highly oxygenated and stereochemically elaborate molecular architecture of the brasilinolides serves to test the limits of chemical synthesis. Moreover, this adventurous endeavour underscores the multiple logistical and selectivity issues that are regularly faced in tackling the synthesis of such promising polyketide natural products of biological and medicinal importance.1b,d,46 In future work, it is anticipated that we can build on the supportive findings reported here to further progress a unified approach to the construction of the brasilinolides.
Experimental
Full experimental and characterisation details are provided in the ESI.†Acknowledgements
We thank the EPSRC (EP/F025734/1) and Syngenta for support, the Isaac Newton–Mays Wild Research Fellowship at Downing College (M.P.H.), the Herchel Smith Postdoctoral Fellowships Fund at Cambridge (C.J.C.) and the Deutsche Akademie der Naturforser Leopoldina (F.A.M.; BMBF-LPD 9901/8-148) for additional funding, and the EPSRC National Mass Spectrometry Centre (Swansea) for mass spectra.Notes and references
- (a) C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed; (b) S. M. Dalby and I. Paterson, Curr. Opin. Drug Discovery Dev., 2010, 13, 777 CAS; (c) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS PubMed; (d) I. Paterson and K.-S. Yeung, Chem. Rev., 2005, 105, 4237 CrossRef PubMed; (e) K.-S. Yeung and I. Paterson, Angew. Chem., Int. Ed., 2002, 41, 4632 CrossRef CAS PubMed.
- (a) H. Shigamori, Y. Tanaka, K. Yazawa, Y. Mikami and J. Kobayashi, Tetrahedron, 1996, 52, 9031 CrossRef; (b) Y. Tanaka, H. Komaki, K. Yazawa, Y. Mikami, A. Nemoto, T. Tojyo, K. Kadawaki, H. Shigemori and J. Kobayashi, J. Antibiot., 1997, 50, 1036 CrossRef CAS.
- (a) Y. Mikami, H. Komaki, T. Imai and K. Yazawa, J. Antibiot., 2000, 53, 70 CrossRef CAS; (b) K. Komatsu, M. Tsuda, Y. Tanaka, Y. Mikami and J. Kobayashi, J. Org. Chem., 2004, 69, 1535 CrossRef CAS PubMed.
- For a review of the chemistry and biology of immunosuppressive natural products, see: J. Mann, Nat. Prod. Rep., 2001, 18, 417 RSC.
- For a selection of reviews on modern aldol chemistry, see: (a) S. B. J. Kan, K. K.-H. Ng and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 9097 CrossRef CAS PubMed; (b) Modern Methods in Stereoselective Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2013 Search PubMed; (c) L. M. Geary and P. G. Hultin, Tetrahedron: Asymmetry, 2009, 20, 131 CrossRef CAS PubMed; (d) Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) I. Paterson, F. A. Mühlthau, C. J. Cordier, M. P. Housden, P. M. Burton and O. Loiseleur, Org. Lett., 2009, 11, 353 CrossRef CAS PubMed; (b) I. Paterson, P. M. Burton, C. J. Cordier, M. P. Housden, F. A. Mühlthau and O. Loiseleur, Org. Lett., 2009, 11, 693 CrossRef CAS PubMed.
- (a) I. Paterson, K. R. Gibson and R. M. Oballa, Tetrahedron Lett., 1996, 37, 8585 CrossRef CAS; (b) D. A. Evans, P. J. Coleman and B. Côté, J. Org. Chem., 1997, 62, 788 CrossRef CAS; (c) I. Paterson and L. A. Collett, Tetrahedron Lett., 2001, 42, 1187 CrossRef CAS; (d) D. A. Evans, B. Côté, P. J. Coleman and B. T. Connell, J. Am. Chem. Soc., 2003, 125, 10893 CrossRef CAS PubMed; (e) L. C. Dias and A. M. Aguilar, Chem. Soc. Rev., 2008, 37, 451 RSC.
- (a) A. De Camp Schuda, P. H. Mazzocchi, G. Fritz and T. Morgan, Synthesis, 1986, 309 CrossRef PubMed; (b) S. A. Burova and F. E. McDonald, J. Am. Chem. Soc., 2004, 126, 2495 CrossRef CAS PubMed.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS PubMed.
- For a selection of reviews on the vinylogous aldol reaction, see: (a) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076 CrossRef CAS PubMed; (b) M. Kalesse, in Topics in Current Chemistry, ed. J. Mulzer, Springer, Berlin, 2005, pp. 43–76 Search PubMed; (c) S. E. Denmark, J. R. Heemstra, Jr. and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682 CrossRef CAS PubMed.
- S. Simsek, M. Horzella and M. Kalesse, Org. Lett., 2007, 9, 5637 CrossRef CAS PubMed.
- (a) M. T. Crimmins, S. J. Kirincich, A. J. Wells and A. L. Choy, Synth. Commun., 1998, 28, 3675 CrossRef CAS; (b) V. V. Samoshin, D. E. Gremyachinskiy, L. L. Smith, I. V. Bliznets and P. H. Gross, Tetrahedron Lett., 2002, 43, 6329 CrossRef CAS.
- (a) R. E. Ireland, J. Liu and T. D. Roper, Tetrahedron, 1997, 53, 13221 CrossRef CAS; (b) I. Paterson, M. J. Coster, D. Y.-K. Chen, J. L. Acena, J. Bach, L. Keown and T. Trieselmann, Org. Biomol. Chem., 2005, 3, 2420 RSC.
- G. Kresze, A. Maschke, R. Albrecht, K. Bederke, H. P. Patzschke, H. Smalla and A. Trede, Angew. Chem., Int. Ed. Engl., 1962, 1, 80 CrossRef PubMed.
- A. G. Myers, B. H. Yang, H. Chen, L. McKinstry, D. J. Kopecky and J. L. Gleason, J. Am. Chem. Soc., 1997, 119, 6496 CrossRef CAS.
- (a) K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651 CrossRef CAS; (b) A. J. Mancuso, S. L. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480 CrossRef CAS.
- Y.-S. Hon, L. Lu, R.-C. Chang, S.-W. Lin, P.-P. Sun and C.-F. Lee, Tetrahedron, 2000, 56, 9269 CrossRef CAS.
- H. C. Kolb, M. S. Van Nieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS.
- We thank Dr Robert Paton for carrying out DFT transition state calculations on closely related model systems, which indicated that TS-1 was preferred. (a) R. S. Paton and J. M. Goodman, J. Org. Chem., 2008, 73, 1253 CrossRef CAS PubMed; (b) R. S. Paton and J. M. Goodman, Org. Lett., 2006, 8, 4299 CrossRef CAS PubMed.
- See the ESI† for details.
- For representative applications of the 1,5-anti boron-mediated aldol reaction, see: (a) I. Paterson, D. J. Wallace and K. R. Gibson, Tetrahedron Lett., 1997, 38, 8911 CrossRef CAS; (b) I. Paterson and M. Tudge, Tetrahedron, 2003, 59, 6833 CrossRef CAS; (c) I. Paterson, M. E. Di Francesco and T. Kühn, Org. Lett., 2003, 5, 599 CrossRef CAS PubMed; (d) I. Paterson, M. J. Coster, D. Y.-K. Chen, K. R. Gibson and D. J. Wallace, Org. Biomol. Chem., 2005, 3, 2410 RSC.
- For a review of asymmetric aldol reactions using boron enolates, see: C. J. Cowden and I. Paterson, Org. React., 1997, 51, 1 CAS.
- D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560 CrossRef CAS.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- (a) I. Paterson, J. M. Goodman and M. Isaka, Tetrahedron Lett., 1989, 30, 7121 CrossRef CAS; (b) I. Paterson, G. J. Florence, K. Gerlach, J. P. Scott and N. Sereinig, J. Am. Chem. Soc., 2001, 123, 9535 CrossRef CAS PubMed.
- For representative applications of the 1,4-syn boron-mediated aldol reaction, see: (a) I. Paterson, M. Donghi and K. Gerlach, Angew. Chem., Int. Ed., 2000, 39, 3315 CrossRef CAS; (b) I. Paterson, R. D. M. Davies, A. C. Heimann, R. Marquez and A. Meyer, Org. Lett., 2003, 5, 4477 CrossRef CAS PubMed; (c) I. Paterson, K. Ashton, R. Britton, G. Cecere, G. Chouraqui, G. J. Florence, H. Knust and J. Stafford, Chem. – Asian J., 2008, 3, 367 CrossRef CAS PubMed; (d) I. Paterson, M. Razzak and E. A. Anderson, Org. Lett., 2008, 10, 3295 CrossRef CAS PubMed; (e) I. Paterson, A. D. Findlay and C. Noti, Chem. – Asian J., 2009, 4, 594 CrossRef CAS PubMed; (f) I. Paterson and T. Paquette, Org. Lett., 2010, 12, 2158 CrossRef CAS PubMed; (g) I. Paterson, L. J. Gibson and S. B. J. Kan, Org. Lett., 2010, 12, 5530 CrossRef CAS PubMed.
- (a) I. Paterson and M. V. Perkins, Tetrahedron, 1996, 52, 1811 CrossRef; (b) I. Paterson, V. A. Steadman, M. D. McLeod and T. Trieselmann, Tetrahedron, 2011, 67, 10119 CrossRef CAS PubMed.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 889 CrossRef CAS.
- I. Paterson, R. D. Norcross, R. A. Ward, P. Romea and M. A. Lister, J. Am. Chem. Soc., 1994, 116, 11287 CrossRef CAS.
- K. Horita, T. Yoshioka, T. Tanaka, Y. Oikawa and O. Yonemitsu, Tetrahedron, 1986, 42, 3021 CrossRef CAS.
- I. Paterson, K.-S. Yeung and J. B. Smaill, Synlett, 1993, 774 CrossRef CAS.
- (a) E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925 CrossRef CAS; (b) E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986 CrossRef CAS.
- (a) Y. Gao, R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765 CrossRef CAS; (b) R. A. Johnson and K. B. Sharpless, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 1993 Search PubMed.
- Y.-L. Zhong and T. K. M. Shing, J. Org. Chem., 1997, 62, 2622 CrossRef CAS.
- T. Mukaiyama, K. Banno and K. Narasaka, J. Am. Chem. Soc., 1974, 96, 7503 CrossRef CAS.
- I. Paterson, E. A. Anderson, S. M. Dalby, J. H. Lim, P. Maltas, O. Loiseleur, J. Genovino and C. Moessner, Org. Biomol. Chem., 2012, 10, 5861 CAS.
- D. A. Evans, M. J. Dart, J. L. Duffy and M. G. Young, J. Am. Chem. Soc., 1996, 118, 4322 CrossRef CAS.
- I. Paterson, G. J. Florence, A. C. Heimann and A. C. Mackay, Angew. Chem., Int. Ed., 2005, 44, 1130 CrossRef CAS PubMed.
- (a) L. Lombardo, Tetrahedron Lett., 1982, 23, 4293 CrossRef CAS; (b) S. Matsubara, T. Mizuno, T. Otake, M. Kobata, K. Utimoto and K. Takai, Synlett, 1998, 1369 CrossRef CAS PubMed.
- S. Matsubara, M. Sugihara and K. Utimoto, Synlett, 1998, 313 CrossRef CAS.
- V. P. Ghidu, J. Wang, B. Wu, Q. Liu, A. Jacobs, L. J. Marnett and G. A. Sulikowski, J. Org. Chem., 2008, 73, 4949 CrossRef CAS PubMed.
- (a) I. Paterson, M. A. Lister and C. K. McClure, Tetrahedron Lett., 1986, 27, 4787 CrossRef CAS; (b) I. Paterson and J. M. Goodman, Tetrahedron Lett., 1989, 30, 997 CrossRef CAS; (c) I. Paterson, J. M. Goodman, M. A. Lister, R. C. Schumann, C. K. McClure and R. D. Norcross, Tetrahedron, 1990, 46, 4663 CrossRef CAS.
- T. Oishi and T. Nakata, Acc. Chem. Res., 1984, 17, 338 CrossRef CAS.
- The acid at C1 could be introduced via Pinnick oxidation of 94 (ca. 90% yield by 1H NMR analysis). B. S. Bal, W. E. Childers, Jr. and H. W. Pinnick, Tetrahedron, 1981, 37, 2091 CrossRef CAS.
- For recent work from our group on other complex polyketides, see: (a) I. Paterson, P. Maltas and E. A. Anderson, Pure Appl. Chem., 2013, 85, 1113 CrossRef; (b) S. M. Dalby, J. Goodwin-Tindall and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 6517 CrossRef CAS PubMed; (c) I. Paterson, S. J. Fink, L. Y. W. Lee, S. J. Atkinson and S. B. Blakey, Org. Lett., 2013, 15, 3118 CrossRef CAS PubMed; (d) I. Paterson, K.-H. Ng, S. Williams, D. C. Millican and S. M. Dalby, Angew. Chem., Int. Ed., 2014, 53, 2692 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00498e. All data accompanying this publication are directly available within the publication and the ESI. |
| This journal is © The Royal Society of Chemistry 2015 |