
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-specific conjugation of 8-ethynyl-BODIPY to a protein by [2 + 3] cycloaddition†
Marcel
Albrecht
a,
Andreas
Lippach
a,
Matthias P.
Exner
b,
Jihene
Jerbi
c,
Michael
Springborg
cd,
Nediljko
Budisa
b and
Gerhard
Wenz
*a
aOrganic Macromolecular Chemistry, Campus Saarbrücken C4.2, Saarland University, D-66123 Saarbrücken, Germany. E-mail: g.wenz@mx.uni-saarland.de
bDepartment of Chemistry-Biocatalysis, TU Berlin, Mueller-Breslau-Strasse 10/L1, D-10623 Berlin, Germany
cPhysical and Theoretical Chemistry, Campus Saarbrücken B2.2, Saarland University, D-66123 Saarbrücken, Germany
dSchool of Materials Science and Engineering, Tianjin University, Tianjin, 300072, People's Republic of China
First published on 7th May 2015
Abstract
We report a straightforward synthesis of 8-ethynyl-BODIPY derivatives and their potential as fluorescent labeling compounds using an alkyne–azide click chemistry approach. The ethynyl substituted BODIPY dyes at the meso-position were reacted under Cu+ catalysis and mild physiological conditions in organic and biological model systems using benzyl azide and a Barstar protein which was selectively modified by a single amino acid substituted methionine at the N-terminus (Met1) → azidohomoalanine (Aha). Conjugation with the protein and the model azide was indicated by a significant blue shift upon formation of the triazole moiety system, which allowed easy distinction between free and coupled dyes. This blue shift was rationalized by the perpendicular orientation of the triazole relative to the chromophore using time dependent density functional theory (TDDFT) calculations. A full spectroscopic and thermodynamic characterization of the protein revealed that a fluorophore was incorporated without the cross influence of protein stability and functional integrity. Furthermore, model reactions of 8-ethynyl-BODIPY derivatives with benzyl azide under copper-free conditions indicate second order kinetics with high rate constants comparable with those found for the strain-promoted azide–alkyne cycloaddition (SPAAC). In this way, we establish a unique and highly efficient method to introduce alkyne-BODIPY into a protein scaffold potentially useful for diverse applications in areas ranging from fundamental protein dynamics studies to biotechnology or cell biology.
Introduction
The visualization of enzyme and protein mediated biochemical processes is one of the most challenging tasks in the research field of life sciences. The introduction of fluorophores into proteins is the method of choice as it allows both in vitro and in vivo real time imaging by commonly used spectroscopic methods with relatively low costs and high selectivities. Fundamental strategies for the combination of fluorescent dyes with proteins include self-labeling tags in which a specific peptide sequence of the protein of interest (POI) is directly modified by the labeling compound, self-labeling protein (SLP) in which the fluorophore contains both the label and the functional moiety that are able to react with the SLP, and enzyme-mediated labeling of tags.1A breakthrough in the detection of fluorophores in living cells was achieved through the discovery of fluorescent proteins (FPs) as highly selective and stable protein tags. In this context, the green fluorescent protein (GFP) was the first example which was introduced into the POI by using efficient, genetic approaches.2 During the last few decades, based on their easy accessibility various fluorescent proteins with tunable photochemical properties have been employed in cell imaging techniques.3 However, both in in vivo and in vitro experiments the FP approach often suffers from dramatic drawbacks caused by the relatively large size of the used FPs resulting in a limitation on potential applications. To overcome size-induced side effects, various synthetic methods for the selective binding of chromophores to POIs such as modifications of cysteine containing peptides,4 metathesis reactions,5 palladium catalyzed cross-coupling reactions6 or azide/acetylene click-chemistry functionalizations7 have been established.
Based on these general considerations the site-directed and residue-specific co-translational decoration of proteins with small, easily accessible fluorophores have recently become widely used conjugation approaches in molecular and cellular biology.8 The main motivation for these experiments is that such modifications caused by addition of only a few atoms to the amino acid side chain lower the risk of compromising protein structural and functional integrity.9 Furthermore, many organic dyes have better photophysical properties and are usually more than 20 times smaller than the widely used fusion-tagged auto-fluorescent proteins.10 For biological applications, such fluorescent dyes, however, need to be fully biocompatible and meet some specific requirements such as the ability to pass through the plasma membrane and to avoid nonspecific adsorption and cross-reactivity with cellular components and structures. Furthermore, protein conjugation via genetic code engineering is in general a two-step process whereby a non-canonical amino acid (ncAA) with a chemically distinct (in an ideal case bio-orthogonal) side chain should be efficiently inserted into a target protein followed by specific coupling of the fluorescent dye.11 In addition to genetic encoding, the reactive side chain functionalities of the dye and ncAA need to be orthogonal to each other (i.e., azides only reacting with alkynes and vice versa). In particular, the copper catalyzed [2 + 3] cycloaddition of azides and alkynes provides fundamental advantages compared to other synthetic techniques as it allows a regioselective and economic combination of two reaction partners with high conversion rates under mild reaction conditions.12
In particular, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPYs) are suitable fluorescent dyes for the incorporation into proteins as they are chemically stable compounds with remarkable photophysical properties, such as high quantum yields, sharp absorption and fluorescence peaks and relatively large Stokes shifts.13 Based on these results a wide variety of synthetic approaches of protein labelling with BODIPY derivatives have been developed.14 Furthermore, several azide and ethynyl substituted BODIPY derivatives have been applied in copper catalyzed [2 + 3]-cycloadditions (the so-called click-reaction) for the synthesis and modification of cell imaging probes,15 fluorescent surfaces,16 sensor systems,17 or nanoparticles.18
In the following we describe a new class of functional BODIPYs, 8-ethynyl-BODIPYs, which show high reactivity and a significant blue shift with regard to the click-reaction. The general proof of principle of the site-specific conjugation of the ethynyl substituted BODIPYs with biologically relevant systems was illustrated using a pseudo-wild type barstar, a small ribonuclease inhibitor of 90 amino acids that is widely used for folding studies, as a model protein (Scheme 1).
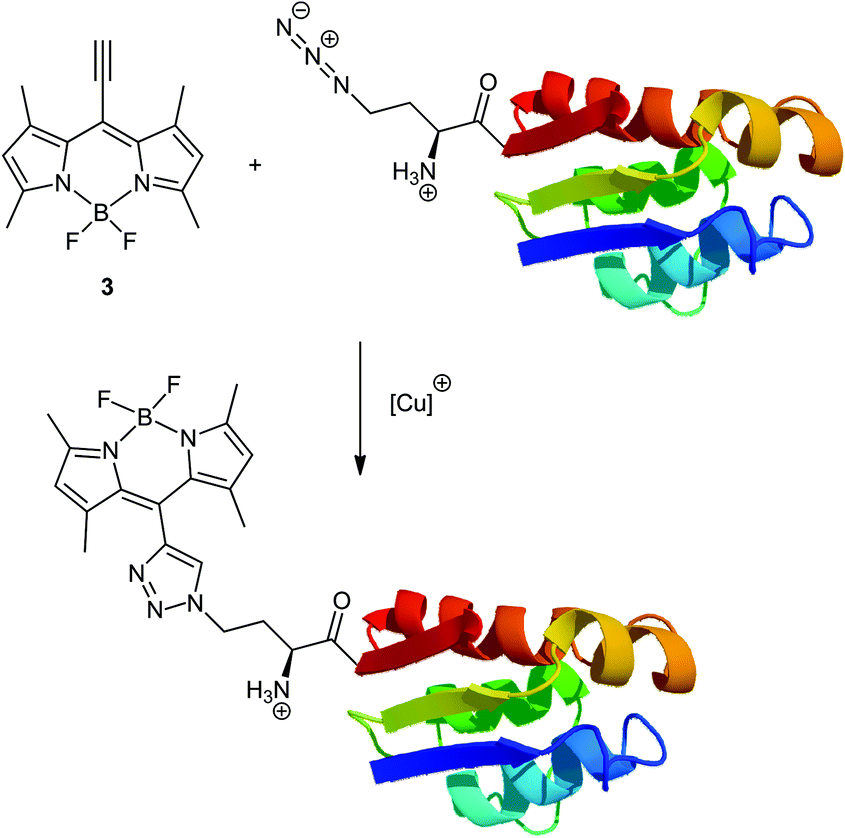 | ||
| Scheme 1 Bioorthogonal conjugation of 8-ethynyl-BODIPY 3 to pseudo-wild type barstar with azidohomoalanine at position 1. | ||
Specifically, an engineered cysteine-free ‘pseudo-wild type’ barstar (ψ-b*), Pro28Ala/Cys41Ala/ Cys83Ala with only one Met residue at the N-terminus (Met1) is used, making the incorporation of methionine analogs and subsequent coupling reaction site-specific. Its N-terminal modification can generally be expected to retain a functional protein structure while introducing novel functions.19
Results and discussion
Synthesis of 8-ethynyl BODIPY derivatives
TMS protected 8-ethynyl-BODIPYs 3/4 were synthesized starting from 3-trimethylsilylpropynal, 2,4-dimethylpyrrole and 3-ethyl-2,4-dimethylpyrrole by the condensation reaction using trifluoroacetic acid, subsequent oxidation with p-chloranil and complexation with boron trifluoride diethyl etherate (Scheme 2) as dark red solids in 17 (1) and 4% (2) yield, respectively. Due to their instability under basic conditions, standard deprotection techniques using tetrabutylammonium fluoride completely failed because complete decomposition occurred. In contrast, the mild deprotection agent AgF furnished the 8-ethynyl-BODIPYs 3/4 in yields of up to 56%. As an alternative to the literature known synthesis of 3 through Stille coupling of the corresponding 8-chloro BODIPY,20 we describe a more straightforward synthetic strategy for 8-ethynyl BODIPYs in only two steps from commercially available starting materials. | ||
| Scheme 2 Syntheses of 8-ethynyl-BODIPYs 3/4 and their conjugation to benzyl azide. | ||
Copper catalyzed model reactions
To get some primary information about their reactivities in azide–alkyne cycloadditions, model reactions of BODIPYs 3/4 with benzyl azide were performed under copper catalyzed reaction conditions. The progress of each reaction was analyzed through both 1H NMR measurements of the crude reaction mixture and through isolation of the reaction products 5/6. As expected for copper catalyzed click-reactions only the formation of the 1,4-triazole regioisomer indicated by one singlet of the triazole moiety at 7.47 (5) and 7.45 ppm (6) was observed. In general, due to the high affinity of BODIPY derivatives towards silica gel during the purification process significantly lower yields of the isolated products were obtained as determined by NMR. For example, the conjugate 5 was isolated after a reaction time of 3 d at 50 °C in 38% yield while the yield based on the NMR measurements was 52%.Photophysical properties of BODIPY derivatives
The progress of the click-reaction of the BODIPY derivatives was also observed through the changes within the absorption and fluorescence spectra since the reactive ethynyl group is conjugated with the BODIPY chromophore (Table 1 and Fig. 1). In comparison with the 8-ethynyl-BODIPY 3, the 8-triazolyl-BODIPY 5 showed hypsochromic shifts both in their absorption and emission spectra which can be explained by prevention of the conjugation between the triazole and the BODIPY moiety in 5 due to steric hindrance of coplanar alignment of triazole and the BODIPY core. This is in contrast to the literature known substituted BODIPYs in which an introduction of a triazole moiety at the pyrrole positions induces a red-shift in absorption and emission.13c Surprisingly, a similar but weaker shift was also found for triazole derivative 6 bearing no bulky methyl groups at the 1- and 7-positions of the BODIPY moiety. The quantum yields ϕ of compounds 3–6 are generally high which is typical for BODIPY derivatives.13 Interestingly, quantum yield of the methyl substituted BODIPY 5 is significantly lower than that of 6. This fact is in contrast to the literature known observation in which an increased restriction at the meso-position is connected with higher quantum yields.21 We consider that twisted internal charge transfer (TICT)22 between the BODIPY core and the triazole ring might be responsible for this abnormal spectral characteristic. Depending on the conformation of both parts of the molecule the process might be affected by the presence of the methyl groups at the BODIPY moiety at 1- and 7-positions. | ||
Fig. 1 Absorption (a) and emission (b) spectra of  8-ethynyl-BODIPY 3 and the corresponding 8-ethynyl-BODIPY 3 and the corresponding  triazole substituted BODIPY 5 in ethanol solution, excitations at 500 nm (3) and 481 nm (5), respectively. triazole substituted BODIPY 5 in ethanol solution, excitations at 500 nm (3) and 481 nm (5), respectively. | ||
| BODIPY derivatives | λ abs [nm] | λ em [nm] | Stokes shift [nm] | ϕ |
|---|---|---|---|---|
| 3 | 540 | 550 | 10 | 0.61 |
| 4 | 540 | 551 | 11 | 0.64 |
| 5 | 506 | 522 | 16 | 0.18 |
| 6 | 520 | 540 | 20 | 0.75 |
Computational analysis
The absorption and emission energies of the ethynyl- and triazole-modified BODIPY derivatives in the ground state in the ethanol environment were calculated using time dependent DFT (TDDFT). The calculations were performed with the Gaussian09 suite of programs.23 The molecular orbitals were visualized using GaussView05. All geometries were optimized by the B3LYP24,25 method with the standard 6-311G* basis set. We optimized the structures of the energetically lowest singlet and triplet states using the latter as an approximation to that of the second-lowest singlet state.While the ethynyl substituted BODIPYs 3 and 4 revealed free rotation of the acetylene moiety, the triazole ring of BODIPY 5 shows restricted rotation due to the interaction of the heterocyclic ring with the methyl groups attached to the BODIPY moiety at 1- and 7-positions. In the most stable conformation of 5, the triazole ring is oriented perpendicular to the BODIPY unit, as shown in Fig. 2. As expected, in BODIPY derivative 6, with lacking methyl groups at positions 1 and 7, the triazole ring and the BODIPY unit adopt an energetically favored conformation in which both rings are located in a more planar structure with a relatively small torsion angle to each other.
 | ||
| Fig. 2 Calculated HOMOs (top) and LUMOs (bottom) of BODIPY derivatives 5 (left) and 6 (right). | ||
Subsequently, using TDDFT calculations we determine the excitation spectra for BODIPYs 3/4 and 5/6 starting from the electronic ground state. Thereby the structure of the energetically lowest triplet state is used as an approximation to that of the first excited singlet state. Relevant data are specified in Table 2.
| Compound | Calc. λabs [nm] | Exp. λabs [nm] |
|---|---|---|
| 3 | 482 | 540 |
| 4 | 476 | 540 |
| 5 | 458 | 506 |
| 6 | 455 | 520 |
In general, the wavelengths of the calculated absorption maxima were lower than the experimental data, but the calculated hypsochromic shifts from 482 to 458 nm (Δλ = 24 nm) and 476 to 455 nm (Δλ = 21 nm) for the transformations of BODIPYs 3 to 5 and 4 to 6, respectively, are in good agreement with the experimental hypsochromic shifts of 34 nm (3 → 5) and 20 nm (4 → 6). The high shift for the sterically hindered methyl derivatives 3/5 can be explained through the disruption between the frontier orbitals of the triazole and BODIPY moieties in compound 5. Due to the reduced rotation barrier between the triazole and BODIPY in the non-methylated compound 6 the interconnection of the frontier orbitals is stronger than that in 5 as shown in Fig. 2.
Copper-free cycloadditions
Surprisingly, the regioselective triazole formation was also observed under copper free reaction conditions. As for the copper catalyzed click-reaction of BODIPY 3 the sole formation of the 1,4-regioisomer was evidenced by a single signal for the triazole ring at 7.47 ppm. The reaction kinetics could be followed through UV/vis measurements. A mixture of BODIPY 3 (25 μM), and benzyl azide (15 mM) in ethanol/water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was incubated in a 10 mm quartz cuvette at 25 °C. The reaction progress was visualized by a characteristic decrease in the absorption maximum of the starting material 3 at 540 nm and a simultaneous increase in the absorption band of product 5 at 506 nm (Fig. 3). Furthermore, the appearance of an isosbestic point at 521 nm indicates the formation of one defined species with high conversion rates without the formation of any side products. Similar results were obtained for the copper-free click reaction of BODIPY 4 to the corresponding triazole substituted BODIPY 6.
:1 v/v) was incubated in a 10 mm quartz cuvette at 25 °C. The reaction progress was visualized by a characteristic decrease in the absorption maximum of the starting material 3 at 540 nm and a simultaneous increase in the absorption band of product 5 at 506 nm (Fig. 3). Furthermore, the appearance of an isosbestic point at 521 nm indicates the formation of one defined species with high conversion rates without the formation of any side products. Similar results were obtained for the copper-free click reaction of BODIPY 4 to the corresponding triazole substituted BODIPY 6.
 | ||
| Fig. 3 UV/vis spectra during the reaction of BODIPY 3 (25 μM) and benzyl azide (15 mM) with BODIPY 5 under copper free reaction conditions in water/ethanol (1:2 v/v). | ||
The apparent pseudo first order rate constants were determined from the decay of the absorption at 540 nm of a 250 μM solution of 3 with various concentrations of excess benzyl azide. Clear second order kinetics was found after plotting the apparent rate constants as a function of the benzyl azide concentration. The obtained second order rate constant k2 3.56 × 10−3 M−1 s−1 was even in the same order of magnitude like those found for the strain-promoted azide–alkyne cycloaddition (SPAAC), which usually exhibit values between 3.0 × 10−3 and 4 M−1 s−1.26 The observed extraordinary reactivity of alkyne 3 may be rationalized by its conjugation with the electron deficient BODIPY core. In addition compared to the conversion of 3 to triazole 5 a ten times smaller rate constant of 3.0 × 10−4 M−1 s−1 for the reaction of ethynyl BODIPY 4 with benzyl azide was observed under copper-free coupling conditions. The presence of an intensive peak at 509 nm in the normalized absorption spectrum of 4 indicates an increased aggregate formation of BODIPY 4 in aqueous ethanol solution which inhibits the triazole formation (ESI Fig. S13†).
Conjugation with the protein
An azide-labeled protein Aha-ψ-b* was expressed in yields of approximately 50% (5 mg L−1) compared to ψ-b* (10 mg L−1).27 This is significantly better than yields of proteins containing unnatural amino acids incorporated via suppression-based methods, which typically do not exceed 20% of the wild-type protein.28 Aha-ψ-b* was purified to >80% with single ion exchange chromatography, as determined by SDS-PAGE, which was sufficient for chemical modification. Conjugation of alkyne-BODIPY 3 and Aha-ψ-b* occurred with 50% total yield after desalting, during which the purity increased to >90% (see Fig. 4A and B). Electrospray mass-spectrometric analysis (ESI-MS) of Aha-ψ-b* clearly revealed a quantitative or near-quantitative level of replacement without detectable amounts of the parent protein as the contaminant (see Fig. 4C). While the yield is sufficient for most experimental setups, it should be noted that such conjugations usually exhibit efficiencies of approx. 80% (see Li et al., 201029 for an example). However, as there is no pronounced mass peak corresponding to the starting material Aha-ψ-b*, it can be concluded that the low yield is caused by denaturation of proteins and losses during desalting, and can potentially be increased by optimization of reaction conditions.
 | ||
| Fig. 4 SDS-PAGE gels of expression of Aha-ψ-b* (A), dito of ψ-b*, Aha-ψ-b* and BODIPY-Aha-ψ-b* with and without Coomassie staining (B), ESI-MS spectra (C) and thermal stability profiles (D) of Aha-ψ-b* and BODIPY-Aha-ψ-b*, respectively. | ||
It can be seen in Fig. 4D that the conjugate exhibits a slightly decreased thermal stability (the melting point is lowered by 7 °C). However, this effect may be reduced by optimization of expression and reaction conditions.
The expression of the corresponding protein samples was induced with 0.5 mM of Aha and 1 mM of IPTG at OD600 = 0.7. After 4 h at 30 °C, cells were harvested. A sample of uninduced and induced culture (corresponding to OD600 = 1 in 1 mL) was applied to a 17% SDS-PAGE gel to determine ψ-b* expression. The induced culture shows a distinct band at the expected migration length (M(Aha-ψ-b*) = 10247.9 Da, indicated by the arrow). Protein conjugation was analyzed through standard expression techniques. The samples were run on a 17% tris-glycine SDS-gel. Bands were visualized at 365 nm (Fig. 4B, right) with Coomassie staining (Fig. 4B, left). Only the band corresponding to BODIPY-Aha-ψ-b* exhibits fluorescence without staining (Fig. 4B, right). Furthermore, success in protein conjugation was controlled using ESI-MS measurements of Aha-ψ-b* and BODIPY-Aha-ψ-b*. The mass spectrum of Aha-ψ-b* shows a single peak at 10246.8 Da (theoretical mass 10247.9 Da), the spectrum of BODIPY-Aha-ψ-b* exhibits a pronounced peak at 10518 Da (theoretical mass 10520 Da). To obtain further the same information about their thermal stability samples of modified and unmodified proteins were thermally denatured from 25 °C (Aha-ψ-b*) or 40 °C (BODIPY-Aha-ψ-b*) to 95 °C. Circular dichroism was monitored at 222 nm (characteristic minimum for α-helices). The melting point only slightly dropped from 62.2 °C for Aha-ψ-b* to 55.2 °C for BODIPY-Aha-ψ-b*.
From the UV/vis and fluorescence spectroscopy data shown in Fig. 5 it can be concluded that conjugation with alkyne-BODIPY 3 endows Aha-ψ-b* with a distinct fluorescence that is significantly blue-shifted compared to the unconjugated BODIPY. These results are comparable with those found for the model reactions (see Fig. 1).
 | ||
| Fig. 5 Absorption (A) and emission (B) spectra of coupling partners and conjugates. | ||
As fluorescence tagging of proteins is very desirable, several attempts have been made to modify amino acids that have been installed via suppression techniques,30,31 or to directly incorporate fluorescent amino acids.32 However, a full spectroscopic characterization of proteins with installed fluorophores is rarely pursued, probably due to the low amounts of substances that can be typically obtained by stop-codon suppression.
In the absorption spectra both BODIPY-Aha-ψ-b* and Aha-ψ-b* show a peak at 280 nm, as expected for proteins (see Fig. 5A). BODIPY-Aha-ψ-b*, however, shows an additional peak at 508 nm, which is in correspondence with conjugated BODIPY and clearly differs from the spectrum of unconjugated BODIPY 3, which shows a broad maximum between 540 and 570 nm. Similar results were observed in the corresponding emission spectra of all coupling components. The fluorescence was determined with an excitation wavelength of 500 nm. A background spectrum of Tris buffer at pH 8 is also shown. Aha-ψ-b* shows a minimal emission signal due to light scattering. BODIPY-Aha-ψ-b* exhibits a maximum at the expected wavelength (522 nm) that is blue-shifted from the emission of unconjugated BODIPY 3 at 550 nm.
Self-aggregation of BODIPY 3 was demonstrated by the appearance of a UV/vis absorption band at 509 nm. With an increasing content of an organic solvent this band decreased and an increasing absorption of the free dye at 540 nm appeared (ESI Fig. S12†). Significant amounts of the free dye are necessary for the copper-free coupling reaction. The model reactions already revealed that a content of 66 vol% of ethanol in water is required for a sufficient coupling rate. On the other hand this solvent is highly non-polar for the Barstar protein leading to its denaturation. The copper-catalyzed click-reaction was successful, because it is much faster and requires much lower concentrations of the free dye than the copper-free coupling. Currently we are looking for ethynyl-BODIPY derivatives with improved solubility in water which would allow conjugation of proteins under copper-free physiological conditions.
Conclusion
8-Ethynyl-BODIPY 3 is a very promising new fluorescence label which allows site-specific conjugation with and without copper catalysis. The induced blue shift provides a simple test for the success of the coupling which can be monitored using optical spectroscopy techniques. As an example for its potential as a fluorescent probe the protein Aha-ψ-b* was successfully conjugated with alkyne-BODIPY 3via a co-translationally introduced Aha. While only specific in the absence of methionine codons other than the start codon, this constitutes a general protein modification technique, as internal methionines are rarely structurally or functionally relevant. Based on these facts the designed BODIPYs 3/4 are promising labeling components in various biological systems under copper free conditions with direct control over the formation of the resulting bioconjugates.Experimental
Synthesis of ethynyl-BODIPYs
A mixture of 3-trimethylsilylpropynal (1.76 mL, 12 mmol) and 2,4-dimethylpyrrole (2.47 mL, 24 mmol) dissolved in 50 mL dry dichloromethane was treated with a catalytic amount (3 drops) of trifluoroacetic acid. After stirring at RT for 18 h p-chloranil (2.95 g, 12 mmol) was added and the resulting mixture was stirred at RT for 4 h. Then triethylamine (6.1 mL, 44 mmol) was added and stirring was continued for an additional 45 min. After addition of boron trifluoride diethyl etherate (6.0 mL, 48 mmol) the mixture was stirred for 1 h. The solvent was evaporated under reduced pressure and the crude reaction product was dissolved in a mixture of water and diethyl ether. The aqueous phase was separated and extracted with diethyl ether (2×). The combined organic fractions were dried over magnesium sulfate and purified by column chromatography (SiO2, n-hexane/dichloromethane 1:1), trimethylsilyl-ethynyl-BODIPY 1: a dark red solid (702 mg, 17%), 1H NMR (CDCl3): δ = 6.07 (s, 2H, pyrrole-H), 2.53 (s, 6H, methyl-H), 2.45 (s, 6H, methyl-H), 0.30 (s, 9H, TMS-H) ppm. HRMS: calc. for C16H19N2BF2Si: 316.1379; found: 316.1385.
2 was synthesized according to the procedure for the preparation of 1 using 2-methylpyrrole as the starting material: trimethylsilyl-ethynyl-BODIPY 2: a dark red solid (151 mg, 4%), 1H NMR (CDCl3): δ = 7.13 (d, J = 4.1 Hz, 2H, pyrrole-H), 6.25 (d, J = 4.1 Hz, 2H, pyrrole-H), 2.60 (s, 6H, methyl-H), 0.31 (s, 9H, TMS-H) ppm. 13C NMR (CDCl3): δ = 157.9, 150.6, 136.5, 128.6, 119.3, 109.1, 97.8, 14.9, −0.5 ppm. HRMS: calc. for C18H23N2BF2Si: 344.1692; found: 344.1689.
3: A mixture of trimethylsilyl-ethynyl-BODIPY 1 (172 mg, 0.5 mmol) and silver(I) fluoride (96 mg, 0.75 mmol) dissolved in 5 mL dry methanol was stirred under exclusion of light at RT for 24 h. After the addition of hydrochloric acid (1 M, 2.25 mL, 2.25 mmol) and stirring for an additional 10 min the mixture was filtered. Water was added and the mixture was extracted with ethyl acetate (3×). The organic phase was washed with a saturated sodium chloride solution and dried over magnesium sulfate. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography (SiO2, n-hexane/ethyl acetate 10:1), ethynyl-BODIPY 3: a red solid (93 mg, 54%), 1H NMR (CDCl3): δ = 6.08 (s, 2H, pyrrole-H), 3.92 (s, 1H, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 2.54 (s, 6H, methyl-H), 2.46 (s, 6H, methyl-H) ppm. 13C NMR (CDCl3): δ = 155.1, 142.6, 133.3, 121.0, 110.0, 94.4, 79.2, 15.4, 14.6 ppm. IR (ATR): 3265, 2922, 2108, 1540, 1466, 1404, 1306, 1192, 1048, 976, 801, 704 cm−1. HRMS: calc. for C15H15N2BF2: 272.1296; found: 272.1286.
CH), 2.54 (s, 6H, methyl-H), 2.46 (s, 6H, methyl-H) ppm. 13C NMR (CDCl3): δ = 155.1, 142.6, 133.3, 121.0, 110.0, 94.4, 79.2, 15.4, 14.6 ppm. IR (ATR): 3265, 2922, 2108, 1540, 1466, 1404, 1306, 1192, 1048, 976, 801, 704 cm−1. HRMS: calc. for C15H15N2BF2: 272.1296; found: 272.1286.
4: A mixture of trimethylsilyl-ethynyl-BODIPY 2 (385 mg, 1.22 mmol) and silver(I) fluoride (232 mg, 1.83 mmol) dissolved in 10 mL dry methanol was stirred under exclusion of light at RT for 24 h. After the addition of hydrochloric acid (1 M, 5.50 mL, 5.50 mmol) and stirring for an additional 10 min the mixture was filtered. Water was added and the mixture was extracted with ethyl acetate (3×). The organic phase was washed with a saturated sodium chloride solution and dried over magnesium sulfate. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography (SiO2, dichloromethane), ethynyl-BODIPY 4: a red solid (167 mg, 56%), 1H NMR (CDCl3): δ = 7.15 (d, J = 4.1 Hz, 2H, pyrrole-H), 6.27 (d, J = 4.1 Hz, 2H, pyrrole-H), 3.64 (s, 1H, CCH), 2.61 (s, 6H, methyl-H) ppm. 13C NMR (CDCl3): δ = 158.5, 136.7, 128.8, 120.5, 119.7, 88.5, 77.0, 15.0 ppm. IR (ATR): 3253, 2106, 1560, 1429, 1342, 1265, 1223, 1198, 1073, 998, 934, 779, 716 cm−1. HRMS: calc. for C13H11N2BF2: 244.0983; found: 244.0982.
Copper(I) catalyzed click-reaction
5/6: A mixture of the corresponding ethynyl-BODIPY (0.145 mmol), benzyl azide (19 mg, 0.145 mmol), copper(II) sulfate pentahydrate (4 mg, 0.015 mmol) and sodium ascorbate (9 mg, 0.045 mmol) in 10 mL dimethyl sulfoxide and 0.1 mL water was stirred at 50 °C for 3 d. Dichloromethane was added and the organic phase was successively washed with a saturated sodium chloride solution and water. The mixture was dried over magnesium sulfate and the crude reaction product was purified by column chromatography (SiO2, n-hexane/ethyl acetate 4:1 → 2:1).
Triazole substituted BODIPY 5, a dark red solid (22 mg, 38%), 1H NMR (CDCl3): δ = 7.47 (s, 1H, triazole-H), 7.38 (m, 3H, phenyl-H), 7.32 (m, 2H, phenyl-H), 5.96 (s, 2H, pyrrole-H), 5.64 (s, 2H, benzyl-H), 2.53 (s, 6H, methyl-H), 1.39 (s, 6H, methyl-H) ppm. 13C NMR (CDCl3): δ = 156.6, 142.7, 140.8, 134.5, 132.3, 129.2, 129.1, 128.0, 123.2, 121.4, 110.0, 54.6, 14.6, 14.2 ppm. IR (ATR): 2923, 1729, 1545, 1507, 1467, 1405, 1307, 1180, 1154, 1044, 971, 816, 711, 697 cm−1. HRMS: calc. for C22H22N5BF2: 405.1936; found: 405.1942.
Triazole substituted BODIPY 6, a dark red solid (18 mg, 33%), 1H NMR (CDCl3): δ = 7.75 (s, 1H, triazole-H), 7.42 (m, 3H, phenyl-H), 7.35 (m, 2H, phenyl-H), 7.10 (d, J = 4.2 Hz, 2H, pyrrole-H), 6.27 (d, J = 4.2 Hz, 2H, pyrrole-H), 5.64 (s, 2H, benzyl-H), 2.63 (s, 6H, methyl-H) ppm. 13C NMR (CDCl3): δ = 158.2, 141.8, 133.9, 133.5, 129.9, 129.4, 129.2, 128.2, 125.7, 119.6, 110.0, 54.5, 15.0 ppm. IR (ATR): 2923, 1735, 1551, 1455, 1261, 1181, 1002, 860, 763, 709 cm−1. HRMS: calc. for C20H18N5BF2: 377.1623; found: 377.1617.
Copper free click-reaction
5: A mixture of the corresponding ethynyl-BODIPY (0.145 mmol) and benzyl azide (19 mg, 0.145 mmol) in 10 mL dimethyl sulfoxide and 0.1 mL water was stirred at 50 °C for 3 d. Dichloromethane was added and the organic phase was successively washed with a saturated sodium chloride solution and water. The mixture was dried over magnesium sulfate and the crude reaction product was purified by column chromatography (SiO2, n-hexane/ethyl acetate 4:1 → 2:1).
Triazole substituted BODIPY 5, a dark red solid (14 mg, 24%). The spectroscopic data are identical to those obtained using the copper(I) catalyzed approach.
Triazole substituted BODIPY 6, a dark red solid (21 mg, 35%). The spectroscopic data are identical to those obtained using the copper(I) catalyzed approach.
Expression and purification of ψ-b* and Aha-ψ-b*
The proteins ψ-b* and Aha-ψ-b* were modified as described in the previously described protocol.33 Details are provided in the ESI†.Cycloaddition on a model protein
Copper-induced cycloaddition was carried out as follows: 0.2 μg μL−1 Aha-ψ-b* (or ψ-b* as a negative control), 0.02 μg μL−1 alkyne-BODIPY 3, 1 mM CuSO4, 2 mM TCEP and 2 mM BCA were mixed in PBS at pH 8 (137 mM NaCl, 2.68 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4) in a total volume of 30 μL. A spatula tip of powdered copper was added. The reaction mixture was incubated at 37 °C for 4 h with agitation. Several reaction supernatants were combined and purified on a Zeba Desalt Spin Column (Pierce Biotechnology, Rockford, USA) equilibrated with 50 mM Tris-HCl at pH 8.Analysis of protein–dye conjugates
MS analysis of full length proteins was performed on an LTQ-FT Ultra mass spectrometer (Thermo Scientific) coupled online to an Ultimate 3000 HPLC Instrument (Thermo Scientific). Desalting was carried out with MassPREP Online Desalting Cartridges (Waters). Briefly, proteins were loaded in 1% formic acid and eluted in a 5 min gradient from 6 to 95% acetonitrile, 1% formic acid. Spectra were acquired in full scan mode with a resolution of 200000 at m/z 400 and afterwards deconvoluted with the software Promass (Thermo Scientific) using basic deconvolution default settings.
The melting curves of Aha-ψ-b* before and after conjugation were measured at a concentration of 20 μM by monitoring the changes in residual ellipticity (i.e. unfolding) at 220 nm. The protein solutions were heated from 25 °C or 40 °C to 95 °C with a rate of 30 °C h−1 in 110-QS Hellma quartz cells with an optical path length of 0.1 cm. These experiments were performed on a Jasco J-715 dichrograph equipped with a Peltier-type FDCD and water bath attachment (model PTC-423S/15 and F250).
Fluorescence spectra of samples were recorded on a LS 55 (PerkinElmer Life Sciences, Boston, USA) with an excitation/emission slit of 5 nm. The concentrations of the samples were 0.2 μM for BODIPY-Aha-ψ-b* and Aha-ψ-b*, and 1 μM for BODIPY. The proteins were excited at 500 nm and the fluorescence was measured in the range of 510–700 nm at 20 °C. At least three spectra were accumulated for each sample.
Absorption spectra of samples were recorded on a Lambda 35 (PerkinElmer Life Sciences, Boston, USA) UV/vis spectrophotometer in the range of 250–700 nm at 20 °C.
Analysis of the model reactions
All NMR spectra including 1H, 13C, H/H-COSY and C/H-COSY were recorded at room temperature by using a Bruker 400 MHz Ultrashield Plus BioSpin Spectrometer Magnet System (1H: 400 MHz, 13C: 100.6 MHz). The chemical shifts are given in parts per million (ppm) in relation to the corresponding solvent signal.IR spectra of solid samples were recorded on a Bruker Tensor 27 FT-IR-Spectrometer equipped with a Golden Gate Diamond ATR accessory using the software OPUS by Bruker.
UV kinetics were performed using an Evolution 220 UV/vis Spectrophotometer (Thermo Scientific), 1 mM quartz glass cuvettes (Hellma 110-QS) and ultrapure solvents.
The fluorescence measurements and the determination of the quantum yields were performed according to the literature known procedures established by Jung et al., 2014.13c
Acknowledgements
The authors wish to thank the following persons: Dr Patrick Durkin for synthesis of Aha; Nina Bach und Katja Bäuml from the group of Professor Sieber for mass spectroscopy; Anett Pfeiffer for initial protein expression and conjugation experiments. Blandine Boßmann is gratefully acknowledged for performing the fluorescence measurements. Furthermore, we would like to thank Dennis Meier and Ronny Heisel for their synthetic support for preparing various BODIPY derivatives. Many thanks also to Peter Wiater for his support concerning the DFT calculations. The calculations were carried out at C3M-Saar at Saarland University.Notes and references
- For recent reviews in the field of tag-mediated protein labeling see: (a) M. J. Hinner and K. Johnsson, Curr. Opin. Biotechnol., 2010, 21, 766–776 CrossRef CAS PubMed; (b) J. M. Chalker, G. J. L. Bernardes and B. G. Davies, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed; (c) C. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784–792 CrossRef CAS PubMed; (d) D. W. Romanini and V. W. Cornish, Nat. Chem., 2012, 4, 248–250 CrossRef CAS PubMed; (e) Y. Takaoka, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed., 2013, 52, 4088–4106 Search PubMed; (f) D. Jung, K. Min, J. Jung, W. Jang and Y. Kwon, Mol. BioSyst., 2013, 9, 862–872 RSC.
- R. Heim, D. C. Prasher and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12501–12504 CrossRef CAS.
- (a) I. V. Yampolsky, A. A. Kislukhin, T. T. Amatov, D. Shcherbo, V. K. Potapov, S. Lukyanoy and K. A. Lukyanov, Bioorg. Chem., 2008, 36, 96–104 CrossRef CAS PubMed; (b) R. M. Wachter, J. L. Watkins and H. Kim, Biochemistry, 2010, 49, 7417–7427 CrossRef CAS PubMed; (c) J. Conyard, M. Kondo, I. A. Heisler, G. Jones, A. Badrigde, L. M. Tolbert, K. M. Solntsev and S. R. Meech, J. Phys. Chem. B, 2011, 115, 1571–1577 CrossRef CAS PubMed; (d) B. L. Grigorenko, A. V. Nemukhin, I. V. Polykov and A. I. Krylov, J. Phys. Chem. Lett., 2013, 4, 1743–1747 CrossRef CAS PubMed.
- (a) H. Nonaka, S. Fujishima, S. Uchinomiya, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 9301–9309 CrossRef CAS PubMed; (b) A. Tirat, F. Freuler, T. Stettler, L. M. Mayr and L. Leder, Int. J. Biol. Macromol., 2006, 39, 66–76 CrossRef CAS PubMed.
- (a) Y. A. Lin and B. G. Davis, Beilstein J. Org. Chem., 2010, 6, 1219–1228 CrossRef CAS PubMed; (b) Y. A. Lin, J. M. Chalker and B. G. Davis, J. Am. Chem. Soc., 2010, 132, 16805–16811 CrossRef CAS PubMed.
- (a) V. Böhrsch and C. P. R. Hackenberger, ChemCatChem, 2010, 2, 243–245 CrossRef; (b) J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed; (c) N. Li, R. K. V. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed.
- (a) E. Kaya, M. Vrabel, C. Deiml, S. Prill, V. S. Fluxa and T. Carrel, Angew. Chem., Int. Ed., 2012, 51, 4466–4469 CrossRef CAS PubMed; (b) Y. Kim, S. H. Kim, D. Ferracane, J. A. Katzenellenbogen and C. M. Schroeder, Bioconjugate Chem., 2012, 23, 1891–1901 CrossRef CAS PubMed; (c) N. W. Nairn, K. D. Shanebeck, A. Wang, T. J. Graddis, M. P. VanBrunt, K. C. Thornton and K. Grabstein, Bioconjugate Chem., 2012, 23, 2087–2097 CrossRef CAS PubMed; (d) J. Szychowski, A. Mahdavi, J. J. L. Hodas, J. D. Bagert, J. T. Ngo, P. Landgraf, D. C. Dieterich, E. M. Schuman and D. A. Tirrell, J. Am. Chem. Soc., 2010, 132, 18351–18360 CrossRef CAS PubMed; (e) J. T. Ngo and D. A. Tirrell, Acc. Chem. Res., 2011, 44, 677–685 CrossRef CAS PubMed; (f) K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- J. H. Bae, M. Rubini, G. Jung, G. Wiegand, M. H. J. Seifert, M. K. Azim, J.-S. Kim, A. Zumbusch, T. A. Holak, L. Moroder, R. Huber and N. Budisa, J. Mol. Biol., 2008, 328, 1071–1081 Search PubMed.
- L. Merkel, M. Schauer, G. Antranikan and N. Budisa, ChemBioChem, 2010, 11, 1505–1507 CrossRef CAS PubMed.
- S. van de Linde, M. Heilemann and M. Sauer, Annu. Rev. Phys. Chem., 2012, 63, 519–540 CrossRef CAS PubMed.
- S. Lepthien, L. Merkel and N. Budisa, Angew. Chem., Int. Ed., 2010, 49, 5446–5450 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Medal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (c) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC; (d) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS PubMed; (e) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed; (b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed; (c) M. Wirtz, A. Grüter, P. Rebmann, T. Dier, D. A. Volmer, V. Huch and G. Jung, Chem. Commun., 2014, 50, 12694–12697 RSC.
- (a) S. L. Niu, C. Massif, G. Ulrich, R. Ziessel, P.-Y. Renard and A. Romieu, Org. Biomol. Chem., 2011, 9, 66–69 RSC; (b) T. Kobayashi, T. Komatsu, M. Kamiya, C. Campos, M. González-Gaitán, T. Terai, K. Hanaoka and T. Nagano, J. Am. Chem. Soc., 2012, 134, 11153–11160 CrossRef CAS PubMed; (c) K. E. Wilke, S. Francis and E. E. Carlson, J. Am. Chem. Soc., 2012, 134, 9150–9153 CrossRef CAS PubMed; (d) J.-J. Lee, S.-C. Lee, D. Zhai, Y.-H. Ahn, H. Y. Yeo, Y. L. Tan and Y.-T. Chang, Chem. Commun., 2011, 47, 4508–4510 RSC; (e) R. Weinstein, J. Kanter, B. Friedman, L. G. Ellies, M. E. Baker and R. Y. Tsien, Bioconjugate Chem., 2013, 24, 766–771 CrossRef PubMed.
- (a) X. Zhang, Y. Xiao, J. Qi, J. Qu, B. Kim, X. Yue and K. B. Belfield, J. Org. Chem., 2013, 78, 9153–9160 CrossRef CAS PubMed; (b) T. Uppal, N. V. S. D. K. Bhupathiraju and M. G. H. Vicente, Tetrahedron, 2013, 69, 4687–4693 CrossRef CAS; (c) C. W. Cunningham, A. Mukhopadhyay, G. H. Lushington, B. S. J. Blagg, T. E. Prisinzano and J. P. Krise, Mol. Pharm., 2010, 7, 1301–1310 CrossRef CAS PubMed.
- A. M. Hansen, A. L. Sewell, R. H. Pedersen, D.-L. Long, N. Gadegaard and R. Marquez, Tetrahedron, 2013, 69, 8527–8533 CrossRef CAS.
- S. Ast, T. Fischer, H. Müller, W. Mickler, M. Schwichtenberg, K. Rurack and H.-J. Holdt, Chem. – Eur. J., 2013, 19, 2990–3005 CrossRef CAS PubMed.
- (a) Q. M. Kainz, A. Schätz, A. Zöpfl, W. J. Stark and O. Reiser, Chem. Mater., 2011, 23, 3606–3613 CrossRef CAS; (b) S. Zhang, Z. Li, S. Samarajeewa, G. Sun, C. Yang and K. L. Wooley, J. Am. Chem. Soc., 2011, 133, 11046–11049 CrossRef CAS PubMed.
- R. Golbik, G. Fischer and A. R. Fersht, Protein Sci., 1999, 8, 1505–1514 CrossRef CAS PubMed.
- H. Wang, M. G. H. Vicente, F. R. Fronczek and K. M. Smith, Chem. – Eur. J., 2014, 20, 5064–5074 CrossRef CAS PubMed.
- S. Zhui, J. Zhang, G. Vegesna, F.-T. Luo, S. A. Green and H. Lui, Org. Lett., 2011, 13, 438–441 CrossRef PubMed.
- Z. Lou, S. Yang, P. Li, P. Zhou and K. Han, Phys. Chem. Chem. Phys., 2014, 16, 3749–3756 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc, Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- (a) J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS PubMed; (b) M. Chigrinova, C. S. McKay, L.-P. B. Beaulieu, K. A. Udachin, A. M. Beauchemin and J. P. Pezacki, Org. Biomol. Chem., 2013, 11, 3436–3441 RSC; (c) S. V. Orski, G. R. Sheppard, S. Arumugan, R. M. Arnold, V. V. Popik and J. Locklin, Langmuir, 2012, 28, 14693–14702 CrossRef CAS PubMed; (d) M. F. Debets, J. S. Prins, D. Merkx, S. S. van Berkel, F. L. van Delft, J. C. M. van Hest and F. P. J. T. Rutjes, Org. Biomol. Chem., 2014, 12, 5031–5037 RSC; (e) C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199–9208 CrossRef CAS PubMed.
- L. Merkel, H. S. G. Beckmann, V. Wittmann and N. Budisa, ChemBioChem, 2008, 9, 1220–1224 CrossRef CAS PubMed.
- J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. W. Zhang and P. G. Schultz, Science, 2003, 301, 964–967 CrossRef CAS PubMed.
- X. Li, T. Fekner and M. K. Chan, Chem. – Asian J., 2010, 5, 1765–1769 CrossRef CAS PubMed.
- Y. J. Lee, B. Wu, J. E. Raymond, Y. Zeng, X. Q. Fang, K. L. Wooley and W. R. S. Liu, ACS Chem. Biol., 2013, 8, 1664–1670 CrossRef CAS PubMed.
- Y. M. Li, M. Pan, Y. T. Li, Y. C. Huang and Q. X. Guo, Org. Biomol. Chem., 2013, 11, 2624–2629 CAS.
- M. J. Schmidt and D. Summerer, Angew. Chem., Int. Ed., 2013, 52, 4690–4693 CrossRef CAS PubMed.
- Y. Ma, H. Biava, R. Contestabile, N. Budisa and M. L. di Salvo, Molecules, 2014, 2, 1004–1022 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedure for barstar biosynthesis, 1H and 13C NMR spectra and UV-vis spectra of 3. See DOI: 10.1039/c5ob00505a |
| This journal is © The Royal Society of Chemistry 2015 |