
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePYRROC: the first functionalized cycloalkyne that facilitates isomer-free generation of organic molecules by SPAAC†
Corinna
Gröst
and
Thorsten
Berg
*
Institute or Organic Chemistry, University of Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: tberg@uni-leipzig.de; Fax: (+49) 341 9736599
First published on 25th February 2015
Abstract
We present the concept, synthesis, and kinetic characterization of PYRROC as the first functionalized cycloalkyne which cannot form isomers in the reaction with azides. In aqueous buffer, PYRROC displays unprecedented rate accelerations in SPAAC of three to four orders of magnitude, leading to rate constants exceeding 400 M−1 s−1.
The Huisgen 1,3-dipolar cycloaddition between azides and alkynes1 has gained tremendous attention in recent years. Catalysis by Cu(I) not only vastly accelerates the reaction, but also introduces a very high degree of regioselectivity for 1,4-triazoles over 1,5-triazoles.2,3 These favorable properties have made copper-catalyzed azide–alkyne cycloadditions (CuAAC) a centrally important reaction for the generation of bioactive molecules.4 However, copper ions are toxic to living cells, limiting the use of CuAAC in a cellular context. This limitation was overcome by strain-promoted azide–alkyne cycloaddition reactions (SPAAC),5,6 which generate triazoles in the absence of copper, allowing widespread applications related to the labeling of biomolecules. The reactivity of the first functionalized cyclooctyne OCT7 can be increased by placing fluorine in the propargylic position, or by introducing additional ring strain via sp2-hybridized centers or a fused cyclopropyl ring, as illustrated by difluorocyclooctyne (DIFO),8 dibenzocyclooctyne (DIBO),9 and bicyclononyne (BCN),10 respectively (Fig. 1a).
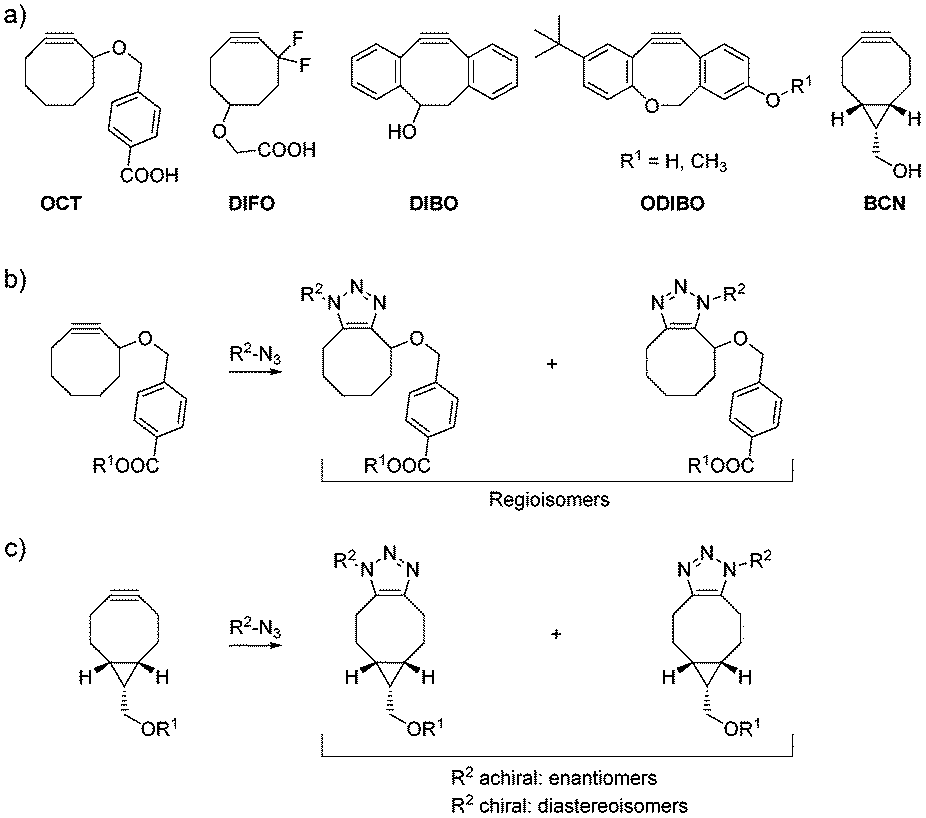 | ||
| Fig. 1 SPAAC with known functionalized cycloalkynes usually leads to the formation of isomeric products. (a) Structures of some functionalized cycloalkynes. (b) SPAAC with OCT derivatized via its carboxylic acid group leads to the formation of regioisomers. (c) SPAAC with BCN derivatized via its hydroxy group leads to the formation of enantiomers or diastereomers, depending on the absence or presence of chiral centers in the azide. | ||
A major shortcoming of SPAAC is that the degree of regioselectivity in the transition state of the reaction is usually low, resulting in the formation of isomeric products. For most functionalized cyclooctynes attached to a chemical entity R1via their respective functional groups, this yields regioisomers in similar or equal ratios (Fig. 1b). Sterically hindered DIBO derivatives offer the opportunity to tailor regioselectivity by adding sterical hindrance on one of the phenyl rings.11 For ODIBO (Fig. 1a), the formation of only a single isomer was reported from the reaction with azides.12 However, given the potential for regioisomer formation by DIBO derivatives, high regioselectivity cannot generally be guaranteed for functionalized DIBO derivatives in the reaction with any given azide. The bicyclononyne BCN10 does not form regioisomers, but either enantiomers or diastereoisomers, depending on the presence of chirality in the azide (Fig. 1c).
Whilst the formation of isomers is typically not regarded as a drawback of the cycloalkynes currently used for labeling of biomolecules by SPAAC,13 it is a prohibitive feature for a new potential application of SPAAC: isomer-free, bioorthogonal synthesis of organic molecules in the cellular environment. An isomer-free approach would facilitate the intracellular generation of large bioactive molecules with poor cell-permeability, from two smaller, more cell-permeable building blocks. Such large bioactive molecules could potentially serve as protein dimerizers,14 bivalent ligands addressing two domains within the same protein, or inhibitors of large protein–protein interfaces.15
To circumvent isomer formation in SPAAC, we propose the concept of symmetrically substituted, functionalized cycloalkynes (Fig. 2). Functionalized cycloalkynes that harbor a C2-symmetry axis, either in a preferred conformation or in a predicted conformational transition state, form uniform reaction products with azides R3-N3 regardless of the relative orientation between cycloalkyne and azide during the reaction. Key to preventing the possibility of generating isomers in SPAAC is the attachment of the substituent R1 at a nitrogen atom, which is symmetrically placed in an odd-membered ring fused at the 5,6-position of the cyclooctyne. Any substituents must be placed consistent with the outlined concept of C2-symmetry. In this communication, we report on the synthesis and kinetic characterization of the first functionalized cyclooctyne that meets these criteria. We demonstrate that the new cycloalkyne, a pyrrolocyclooctyne, is well suited for the isomer-free generation of large organic molecules in the aqueous environment, providing good yields within a short time frame.
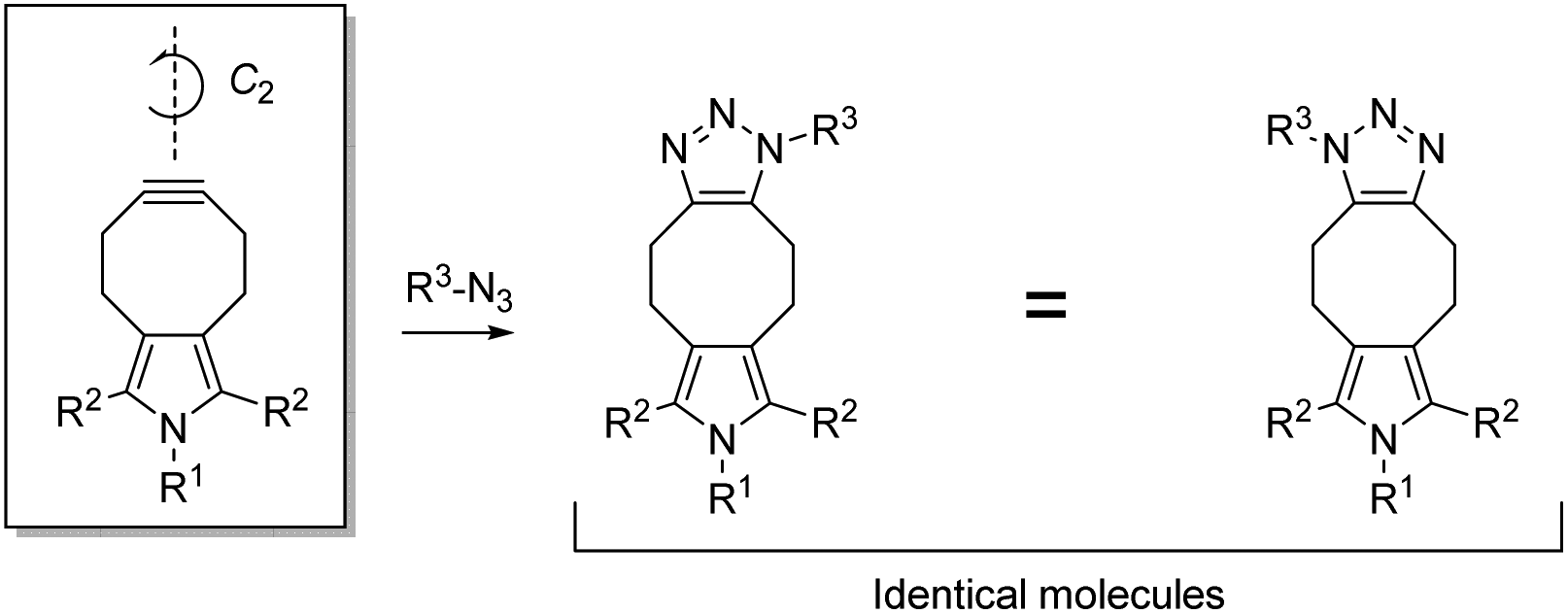 | ||
| Fig. 2 Cycloalkynes (here: pyrrolocyclooctynes) which contain a C2-symmetry axis, either in a preferred conformation or in a conformational transition state, generate chemically uniform SPAAC products. | ||
The synthesis of the pyrrolocyclooctyne started with the nitration of 1,4-cyclooctadiene with silver nitrite and TEMPO to obtain compound 1, which is subjected to the Barton–Zard reaction, leading to the formation of pyrrolocyclooctene 2 (Scheme 1).16,17 Ester hydrolysis, decarboxylation, and N-Boc protection afforded 3 in very good yield. To reduce the reactivity of 3, which is prone to decomposition, it was converted into the diester 4. In line with the literature,18 it was essential to add the solution of the 2,5-dilithiated pyrrole based on 3 to a solution of electrophile to obtain the 2,5-disubstituted pyrrole 4.19 Attempts to convert 2 into 4 directly were not successful. Bromination of the alkene moiety of 4 and removal of the protecting group led to dibromide 5, which was reacted with tert-butyl(3-iodopropoxy)dimethylsilane 6 to yield TBDMS-protected dibromide 7. The change of the N-protecting group was necessary because N-Boc protected pyrroles can undergo decomposition in the presence of strong bases used in the subsequent, two-step elimination procedure.20 Elimination of the first equivalent of hydrogen bromide from 7 was achieved by reacting 7 with 10 equivalents of KOtBu, added as a 1 M solution in THF at −10 °C. This procedure not only led to the elimination of hydrogen bromide, but also to transesterification, providing the di-tert-butylester 8 in 66% yield. A large excess of KOtBu was used because test reactions with a protecting group of similar size, 2-(trimethylsilyl)ethoxymethyl (SEM), as an N-protecting group had shown that lesser amounts of KOtBu (1 or 5 equivalents) also did not allow the isolation of the dimethylester, but instead afforded a mixture of the methyl tert-butyl ester and monocarboxylic acids. Elimination of the second equivalent of hydrogen bromide from 8 was achieved by using KOtBu in the presence of catalytic amounts of 18-crown-6, providing cycloalkyne 9 in 71% yield.21 Attempts to obtain the dimethylester derivative of 9 by using other bases such as DBU, LDA or LiOMe, or by transesterification of 9 with LiOMe, were not successful. Removal of the silyl protecting group with TBAF yielded pyrrolocyclooctyne 10 dubbed PYRROC.
 | ||
| Scheme 1 Synthesis of PYRROC (10). | ||
In order to investigate the reaction kinetic of PYRROC (10), the second order rate constant of its reaction with benzyl azide was determined using kinetic NMR experiments. The 1H-NMR of the reaction between PYRROC (10) and benzyl azide shows only one peak for the benzylic protons of the resulting triazole, confirming that PYRROC indeed forms only a single compound in SPAAC with benzyl azide (ESI Fig. S1a and b†). PYRROC displayed a second-order rate constant k of 0.060 ± 0.004 M−1 s−1 in CD3CN (ESI Fig. S1c and d†). This value is between the rate constant of DIBO9 (k = 0.057 M−1 s−1 in CD3OD), and DIFO8 (k = 0.076 M−1 s−1 in CD3CN) (Fig. 1a), both of which have proven their utility in a large number of biological studies. Thus, PYRROC reacts approximately six times faster than the 3,4-benzannulated cyclooctyne MOBO (k = 0.0095 M−1 s−1 in CD3CN),22 but four times slower than the 5,6-benzannulated cyclooctyne COMBO (k = 0.24 M−1 s−1 in CD3CN).20 The lower reactivity of PYRROC compared to COMBO confirms theoretical predictions that fusion of a 5-membered ring to the 5,6-position of cyclooctyne is less beneficial for reactivity than fusion of a 6-membered ring.23
We envision the application of symmetrically functionalized cycloalkynes such as PYRROC in the bioorthogonal, isomer-free generation of organic molecules in the cellular milieu. To test the suitability of PYRROC for this type of application in a manner as general as possible, we coupled 10 to the fluorophore BODIPY-FL24,25 by activating the primary hydroxyl group as a p-nitrophenylcarbonate, and reacting 11 with the primary amine 12 (Scheme 2). In the course of this study, fluorophore-labeled PYRROC 13 was subjected to SPAAC with three fluorophore azides with substantially different chemical structures: 5-TAMRA azide (14), BODIPY-TMR azide (15), and 3-azido-7-hydroxycoumarin (16) (Scheme 2). The reactions were carried out in phosphate-buffered saline (PBS) to mimic the salt concentrations in a cell. The kinetics of SPAAC were analyzed via FRET (fluorescence resonance energy transfer). To allow for quantitative analysis of the reaction kinetics, each reaction was carried out in parallel with a standard curve mimicking the composition of the reaction mixture at various degrees of conversion of the reactants to the triazole products. This required prior preparation and isolation of the triazoles 17–19 on a preparative scale to allow for characterization of their fluorescence properties.
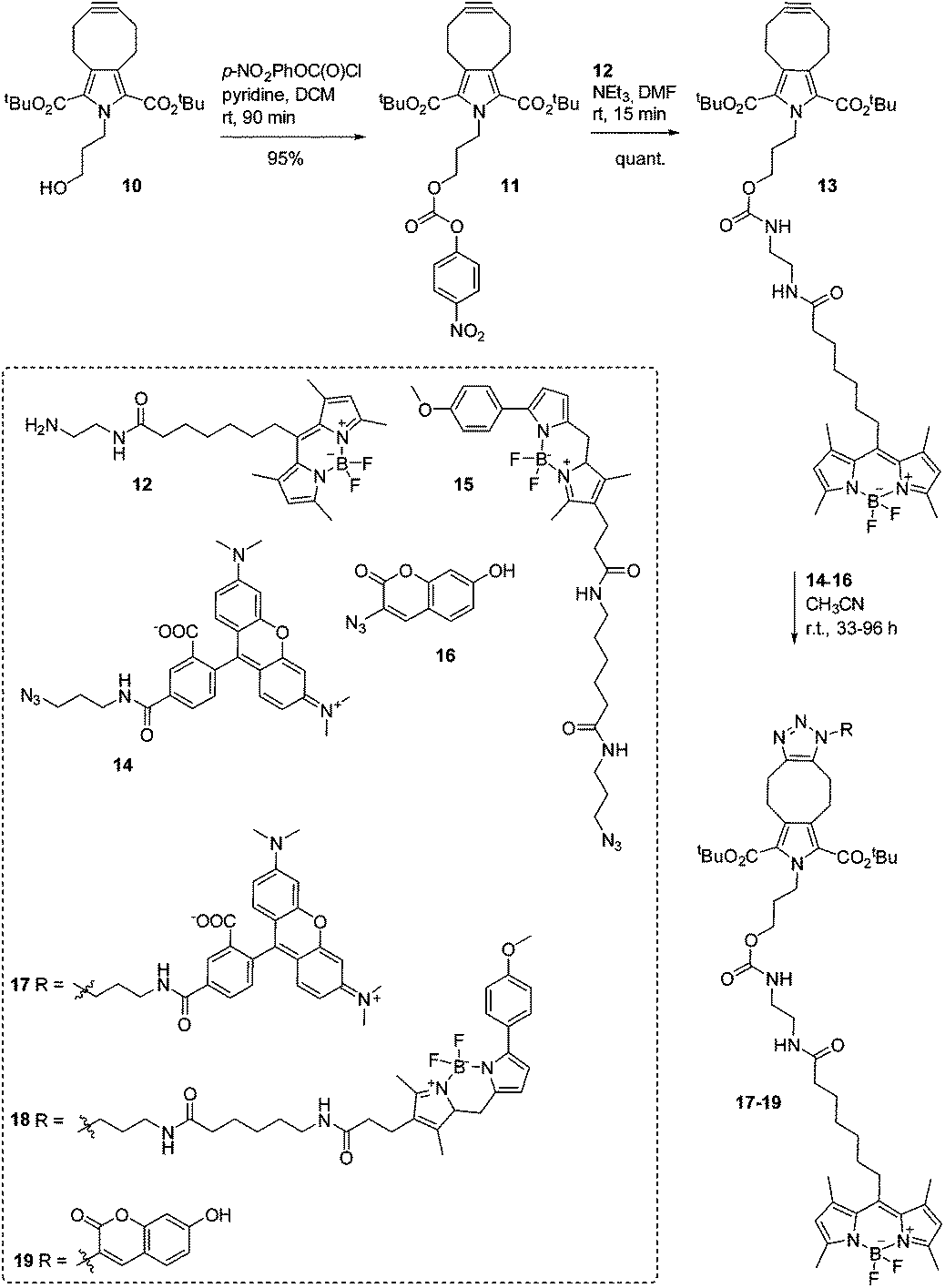 | ||
| Scheme 2 Synthesis of fluorophophore-labeled triazoles 17–19. | ||
The reaction between PYRROC-BODIPY-FL (13) and 5-TAMRA azide (14) resulting in triazole 17 was found to proceed with a second-order rate constant of 13.9 ± 0.3 M−1 s−1 (ESI Fig. S2a–d†), which is more than 200-fold faster than the rate constant for the reaction between PYRROC (10) and benzyl azide in acetonitrile. Using 10 μM of both PYRROC-BODIPY-FL (13) and TAMRA azide (14) in PBS, 74 ± 8% of 13 was converted to the triazole 17 after 6 h (ESI Fig. S2e†). At 50 μM of both reactants, 95 ± 1% conversion of 13 was achieved after only 2 h.
The reaction between BODIPY-FL-labeled PYRROC (13) and BODIPY-TMR azide (15) in PBS was monitored by the decrease in intensity of the BODIPY-FL emission and the concomitant increase of intensity of the BODIPY-TMR emission by FRET (Fig. 3a and b). Using the reactants at a concentration of only 3 μM in PBS, the reaction proceeded with a second-order rate constant of 234 ± 2 M−1 s−1 (Fig. 3c and ESI Fig. S3c†). 88% of triazole 18 was formed after 4 h, and more than 90% after 24 h (Fig. 3d). This indicates that the approach is well suited to generating organic molecules in high yields at low micromolar concentrations in the aqueous environment.
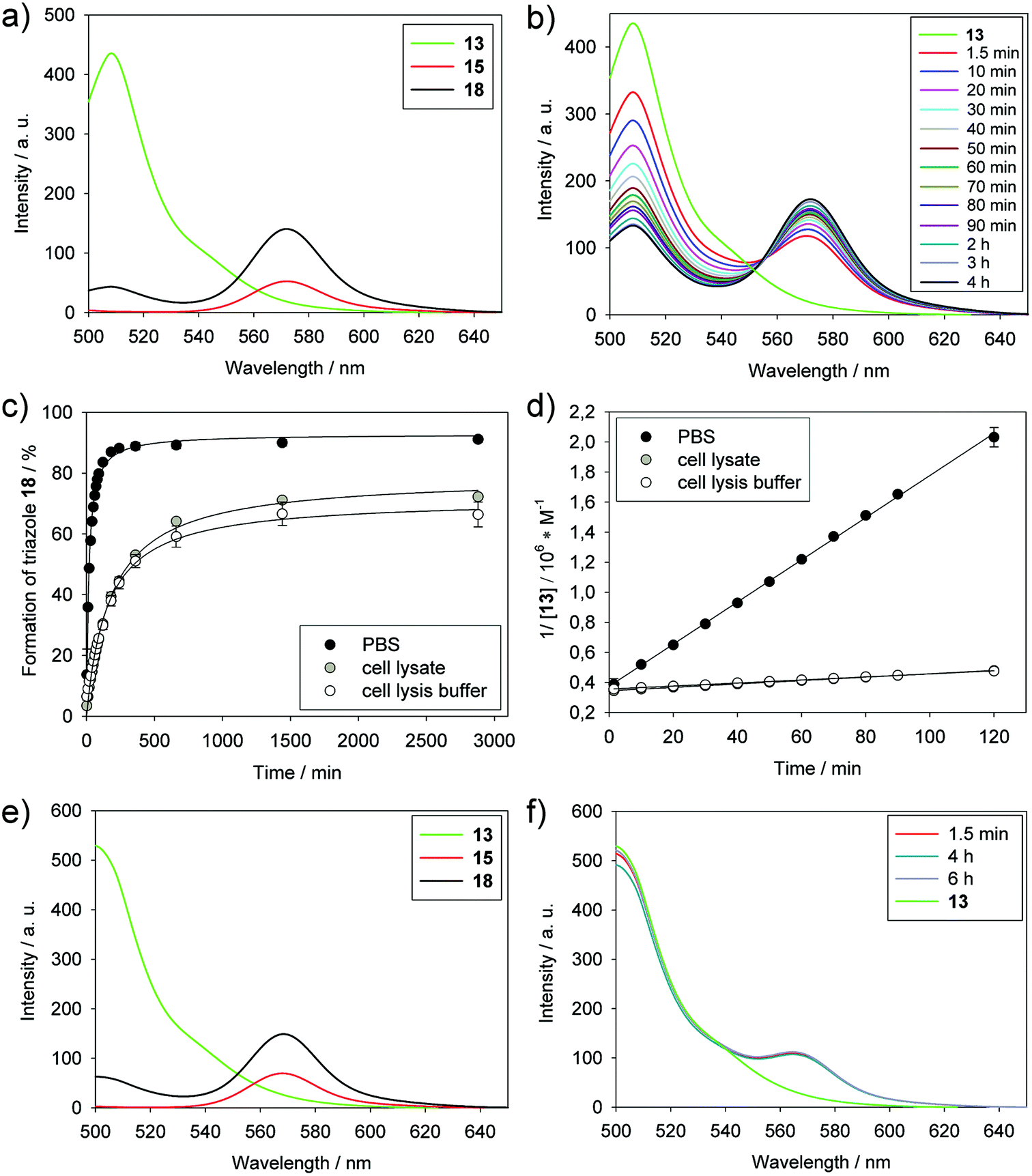 | ||
| Fig. 3 Kinetics of the reaction between BODIPY-FL-labeled PYRROC (13) and BODIPY-TMR azide (15) followed by FRET. (a) Fluorescence spectra of 13, 15, and the triazole product 18 (3 μM each) in PBS. Excitation wavelength: 485 nm. a. u.: arbitrary units. (b) Time course of the reaction after addition of 15 to 13 (initial final concentrations of 13 and 15: 3 μM each). (c) Determination of the second-order rate constant of 13 in reaction with 15 in PBS, cell lysis buffer, and cell lysates (initial concentrations of 13 and 15: 3 μM each). (d) Determination of the conversion of 13 and 15 to 18 in PBS, cell lysis buffer, and cell lysates (initial concentrations of 13 and 15: 3 μM). (e) Fluorescence spectra of 13, 15, and 18 (3 μM each) in acetonitrile. Excitation wavelength: 485 nm. (f) Time course of the reaction after addition of 15 to 13 (initial concentrations of 13 and 15: 3 μM each). | ||
In order to analyze whether PYRROC is suitable for SPAAC in the cellular milieu, we carried out the reaction in K562 lysates. At a concentration of 3 μM of BODIPY-FL-PYRROC (13) and 3 μM BODIPY-TMR azide (15), the second-order rate constant in cell lysates was determined as 17 ± 0.3 M−1 s−1, which is slightly more than one order of magnitude slower than the reaction in PBS (Fig. 3c and ESI Fig. S3†). The slower reaction in cell lysates as compared to PBS could be due to an effective reduction of the concentration of 13 by non-specific reactivity with thiols, or non-covalent interactions with cell components. A more trivial potential reason is the higher viscosity of the cell lysis buffer, which, unlike PBS, contains 10% of glycerol, and thus reduces the rate of reaction. To investigate the effect of the cell lysis buffer on the reaction rate, the reaction was carried out in cell lysis buffer without cell components. The rate constant of 16 ± 0.3 M−1 s−1 indicate that the reduced rate in cell lysates is caused by the components of the buffer, and not by unspecific reaction with thiols. Despite the reduced rates, more than 70% conversion was achieved in cell lysates after 24 h (Fig. 3d). These data place PYRROC into a promising position to mediate bioorthogonal reactions in the cellular milieu.
Reaction of BODIPY-FL-PYRROC (13) with 3-azido-7-hydroxycoumarin (16) (both at 1 μM in PBS) was characterized by a second-order rate constant of 903 ± 23 M−1 s−1 (ESI Fig. S4†). This represents the highest rate constant of a SPAAC reaction reported to date, and is approximately 20-fold higher than the previously highest rate constant of 45.1 M−1 s−1.12 To analyze the influence of the BODIPY-FL substituent of PYRROC on the rate constants, we took advantage of the fact that the quantum yield of 16 increases strongly after reacting with alkynes to form triazoles. Thus, the rate of reaction between unsubstituted PYRROC 10 and 16 can also be followed by fluorescence techniques, and can thus be compared to the rate of reaction between 13 and 16. Under otherwise identical assay conditions, the reaction of 16 with unlabeled PYRROC (10) proceeded with a less than twofold lower rate constant (k = 492 ± 43 M−1 s−1, ESI Fig. S5†). These data indicate that while BODIPY-FL does contribute to the high rate constants of 13 in PBS to some extent, the majority of the high reaction rates observed in the kinetic experiments is inherent to PYRROC.
The rate constants observed with PYRROC in SPAAC in aqueous solutions are three to four orders of magnitude higher than the rate constants measured in the standard reaction of PYRROC with benzyl azide in acetonitrile. Similar rate accelerations have been observed for Diels–Alder reactions in water compared to nonpolar solvents.26,27 They are typically rationalized by a hydrophobic effect, leading to pre-association of the organic molecules in order to minimize their exposure to water, and reduction of the hydrophobic surface area in the course of the reaction.28 The difference in rate constants in the reactions of 13 with the aliphatic azides 14 and 15 can be rationalized by the increased hydrophobicity of 15. However, the higher rate constants obtained with 16 compared to 14 and 15 cannot be explained with its hydrophobicity; instead, it might reflect a higher intrinsic reactivity of 16 caused by the conjugation of the azide moiety with the phenolic system, which is partially deprotonated at the neutral pH of PBS.
Rate enhancements in SPAAC reactions as a result of shifting to a more polar solvent or solvent mixture have also previously been reported,10,12,29 but the effect has not been so pronounced. This is presumably because the SPAAC reaction rate determinations have been carried out in partly aqueous solutions only.
In order to validate the aqueous buffer as the underlying cause of the high reaction rates, we carried out the reaction between 13 and 15 in acetonitrile and followed it by analysis of the fluorescence spectra. Using the same concentrations (3 μM) of cycloalkyne 13 and azide 15, no significant reaction was observed after 6 h (Fig. 3e and f). In contrast, the reaction in PBS had shown significant progress after just 90 seconds, and was virtually complete after only 4 h (Fig. 3b). This demonstrates that the aqueous buffer is a major contributor for the high reaction constants in SPAAC.
In conclusion, we have presented PYRROC as the first functionalized cycloalkyne that cannot form isomers in SPAAC. The design concept of PYRROC facilitates a new field of application for SPAAC: the creation of uniform organic molecules, with a molecular weight that is prohibitively high for cell permeability, in the cellular environment. PYRROC displays unprecedented reaction rates in the reaction with azides in aqueous systems, and is suitable for derivatization at the hydroxy group. The absence of charged chemical moieties in PYRROC is likely to contribute to good cell permeability of its conjugates with organic molecules. No limitations with respect to the selection of azides have been observed to this point. We believe that the concept of symmetrically substituted cycloalkynes, as exemplified by PYRROC, will strongly influence future research in the fields of bioorthogonal chemistry, chemical biology, and medicinal chemistry. Applications of PYRROC in cell-based assays will be reported in due course.
This work was supported by the Deutsche Forschungsgemeinschaft (BE4572/2-1). We extend our thanks to Lothar Hennig for support with the NMR-based kinetic analysis, Angela Berg for experimental support, and Ralf Hoffmann and Thole Züchner for providing access to their fluorescence spectrometer.
Notes and references
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- G. Wittig and A. Krebs, Chem. Ber., 1961, 94, 3260–3275 CrossRef CAS.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- N. E. Mbua, J. Guo, M. A. Wolfert, R. Steet and G. J. Boons, ChemBioChem, 2011, 12, 1912–1921 CrossRef CAS PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. Hendriks, F. P. Rutjes, J. C. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- F. Starke, M. Walther and H. J. Pietzsch, ARKIVOC, 2010, 350–359 CrossRef CAS.
- C. D. McNitt and V. V. Popik, Org. Biomol. Chem., 2012, 10, 8200–8202 CAS.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. T. Dirks, F. P. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- A. Rutkowska and C. Schultz, Angew. Chem., Int. Ed., 2012, 51, 8166–8176 CrossRef CAS PubMed.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS PubMed.
- S. Maity, S. Manna, S. Rana, T. Naveen, A. Mallick and D. Maiti, J. Am. Chem. Soc., 2013, 135, 3355–3358 CrossRef CAS PubMed.
- D. H. R. Barton and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1985, 1098–1100 RSC.
- A. Fürstner, H. Krause and O. R. Thiel, Tetrahedron, 2002, 58, 6373–6380 CrossRef.
- T. J. Donohoe, C. E. Headley, R. P. Cousins and A. Cowley, Org. Lett., 2003, 5, 999–1002 CrossRef CAS PubMed.
- B. R. Varga, M. Kallay, K. Hegyi, S. Beni and P. Kele, Chemistry, 2012, 18, 822–828 CrossRef CAS PubMed.
- C. Antony-Mayer and H. Meier, Chem. Ber., 1988, 121, 2013–2018 CrossRef CAS.
- E. M. Sletten, H. Nakamura, J. C. Jewett and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 11799–11805 CrossRef CAS PubMed.
- K. Chenoweth, D. Chenoweth and W. A. Goddard 3rd, Org. Biomol. Chem., 2009, 7, 5255–5258 CAS.
- I. A. Boldyrev and J. G. Molotkovsky, Russ. J. Bioorg. Chem., 2006, 32, 78–83 CrossRef CAS.
- N. Elumalai, A. Berg, K. Natarajan, A. Scharow and T. Berg, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201410672.
- S. Otto, W. Blokzijl and J. B. F. N. Engberts, J. Org. Chem., 1994, 59, 5372–5376 CrossRef CAS.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- S. Otto and J. B. Engberts, Org. Biomol. Chem., 2003, 1, 2809–2820 CAS.
- X. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, kinetic experiments, and methods. See DOI: 10.1039/c5ob00212e |
| This journal is © The Royal Society of Chemistry 2015 |