
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a flow method for the hydroboration/oxidation of olefins†
José A.
Souto
*ab,
Robert A.
Stockman
c and
Steven V.
Ley
a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: svl1000@cam.ac.uk
bDepartamento de Química Orgánica, Universidade de Vigo, Vigo, 36310, Spain. E-mail: souto@uvigo.es
cSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: robert.stockman@nottingham.ac.uk
First published on 17th February 2015
Abstract
A method for the continuous preparation of alcohols by hydroboration/oxidation of olefins using flow techniques is described. The process allows the isolation of up to 120 mmol h−1 of the desired alcohol in a very rapid manner with good functional group tolerance. The flow setup can be modified to perform a continuous extraction of the desired alcohol from the biphasic mixture produced by the reaction.
The preparation of molecular entities containing the hydroxyl functionality is a very important area of research due to the abundance of this functional group in natural products and biologically active molecules. The toolbox of chemical reactions contain many examples of procedures that accomplish this transformation, such as direct oxidation of allylic C–H bonds by selenium,1 C–H bond2 oxidation by peracids, singlet oxygen oxidation,3 treatment of diazo compounds with mineral acids,4 silane oxidation by peracids,5 ozonolysis of alkenes followed by reductive workup6 or hydroboration–oxidation tandem reaction of alkenes.7 Among them, the hydroboration–oxidation reaction has frequently been used for the insertion of hydroxyl groups either in total synthesis of natural products8 or drug discovery.9 This is mainly due to the broad availability of alkenes as starting materials and the orthogonal reactivity of this functionality to those of the polar functional groups found in many natural products.10 The reaction is highly influenced by the steric and electronic character of the alkene, and occurs with a preference towards the anti-Markownikov borane intermediate that can then be transformed into the terminal alcohol.11 Another attractive feature of the hydroboration reaction is its cis addition across the double bond, which results in a preference for the thermodynamic product when alkenes presenting two sterically differentiated faces are used.12
Despite these advantages, there are some drawbacks of this method, especially when diborane, a simple readily available reagent, is used.13 Known limitations for broader applications of the reaction, especially at a larger scale, are the high exothermicity of both steps, and lack of complete regioselectivity for initial hydroboration of the double bond or incompatibility of functional groups under either reducing or oxidative conditions. Due to the high surface area to volume ratio, flow chemistry technologies provide a practical solution to overcome some of these limitations by better mixing, more efficient heat transfer in highly exothermic reactions and improved ability to control the rate of transformation,14 compared to the batch mode. Furthermore, the possibility for continuous production of the desired product with in-line purification techniques, or for the development of a telescoped protocol offers an efficient generation of the alcohol functional group.
Our group has pioneered many advances in the use of flow protocols for organic synthesis.15 This work has led to reduced manpower requirements and accelerated reaction optimisation programmes. In an ongoing effort towards the development of improved flow processes for the synthesis of compounds of potential interest for materials, fine chemicals and pharmaceutical and agrochemical industries, we began an investigation into the development of hydroboration–oxidation flow protocol for the introduction of the hydroxyl group into olefinic skeletons.16 For this purpose a flow setup was devised and a schematic representation is depicted in Fig. 1.
 | ||
| Fig. 1 Schematic representation of our hydroboration/oxidation continuous flow setup. | ||
Using the reactor configuration shown, we began by investigating the influence of different parameters (i.e. borane source, residence time, solvent mixture) using commercially available β-pinene as a model substrate (Table 1). An initial screening of borane sources gave opposing results. We observed excellent conversion (100%) of the starting material when either a THF or DMS complex of BH3 was used. However, using the tertbutylamine complex of diborane resulted in incomplete conversion of starting materials (entries 1–3). Unfortunately, neither of these experiments could be progressed to a clean oxidation step (<10% conversion).
| Boranea | Flow A (mL min−1) | Flow B (mL min−1) | Solvent (Oxid.) | Flow C (mL min−1) | V 2 (mL) | T 2 (°C) | Conv (%) | NMR yield (%) | |
|---|---|---|---|---|---|---|---|---|---|
a 1 equiv. of borane (2.5 mmol scale).
b 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80.
c 0.5 equiv. of borane.
d 20:38:42.
e 20:60:20. :80.
c 0.5 equiv. of borane.
d 20:38:42.
e 20:60:20.
|
|||||||||
| 1 | BH3·SMe2 | 0.1 | 0.1 | H2O2–H2Ob | 0.2 | 0.02 | 25 | 100 | <10 |
| 2 | BH3·THF | 0.1 | 0.1 | H2O2–H2Ob | 0.2 | 0.02 | 25 | 100 | <10 |
| 3 | BH3·H2NR | 0.1 | 0.1 | H2O2–H2Ob | 0.2 | 0.02 | 25 | 0 | 0 |
| 4 | BH3·THF | 0.1 | 0.1 | H2O2–H2Ob | 0.2 | 0.3 | 25 | 100 | <10 |
| 5 | BH3·THF | 0.1 | 0.1 | H2O2–H2Ob | 0.6 | 0.3 | 25 | 100 | <10 |
| 6 | BH3·THF | 0.3 | 0.3 | H2O2–H2Ob | 0.6 | 2 | 25 | 100 | <10 |
| 7 | BH3·THFc | 0.3 | 0.3 | H2O2–H2Ob | 0.6 | 2 | 25 | 50 | <10 |
| 8 | BH3·THF | 0.3 | 0.3 | H2O2–H2Ob | 0.6 | 2 | 60 | 100 | <10 |
| 9 | BH3·THF | 0.3 | 0.3 | H2O2–EtOH–H2Od | 0.6 | 2 | 25 | 100 | 25 |
| 10 | BH3·THF | 0.3 | 0.3 | H2O2–EtOH–H2Od | 0.6 | 4.2 | 25 | 100 | 31 |
| 11 | BH3·THF | 0.3 | 0.3 | H2O2–EtOH–H2Od | 1.2 | 4.2 | 25 | 100 | 88 |
| 12 | BH3·THF | 0.3 | 0.3 | H2O2–EtOH–H2Od | 2 | 4.2 | 25 | 100 | 88 |
| 13 | BH3·THF | 0.3 | 0.3 | H2O2–EtOH–H2Oe | 1.2 | 4.2 | 25 | 100 | 52 |
| 14 | BH3·THF | 0.5 | 0.5 | H2O2–EtOH–H2Od | 2 | 4.2 | 25 | 100 | 89 |
| 15 | BH3·THF | 1 | 1 | H2O2–EtOH–H2Od | 4 | 4.2 | 25 | 100 | 99 |
| 16 | BH3·THF | 2 | 2 | H2O2–EtOH–H2Od | 8 | 4.2 | 25 | >99 | 99 |
Due to the less odorous nature of the THF complex, this borane source was selected for further investigations. Any initial attempt to increase the yield of the reaction either by modifying the residence time, equivalents of borane/oxidant counterpart or the reaction temperature did not lead to any improvement in the yield (entries 4–8). Pleasingly, changing the nature of the solvent mixture, by the addition of ethanol improved the outcome of the reaction allowing the isolation of the product in 25% yield (entry 9), although some blockage of the second reactor by the formation of insoluble inorganic salts was an issue. Replacement of the previous narrower reactor coil (1 mm i.d.) by a wider bore (2.4 mm i.d.) resulted in a more reliable performance and moderate 31% yield (entry 10). Raising the equivalents of the basic oxidative mixture gave the desired product in 88% yield (entry 11). Attempts to improve this result by further increasing the equivalents of the oxidising agent or the amount of ethanol in the mixture did not afford any improvement of the yield (entries 12 and 13). Surprisingly, the reaction yield was increased when the transformation was performed at high flow rates (entries 14–16) allowing the production of up to 120 mmol h−1 within 70 s of the residence time inside the reactor.17 The pumping was maintained (entry 16) for 50 min and gave 14.1 g (92% yield) of the desired material after isolation.
In our hands, a comparable batch-mode reaction (20 g scale) proceeded with similar yields and selectivity over a 4 hour reaction time period (Scheme 1). However, there are some notable advantages of the flow process. Firstly, under continuous operation, the system needs less than two hours to process the same amount of material. Secondly, in flow the malodorous BH3·DMS, which is necessary in the batch,18 can be substituted for BH3·THF; and importantly the flow reaction can be performed at room temperature. No additional cooling or heating is required,19 due to the excellent heat transfer properties of the flow equipment.
 | ||
| Scheme 1 Batch reaction for purposes of comparison. | ||
Having identified optimised reaction conditions we subjected a number of substituted alkenes to the reactor set up in order to test the scope of the new continuous flow hydroboration–oxidation protocol (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Alkene | Alcohol | Conv. (%) | NMR Yielda [%] |
|
a 2.5 mmol scale. Isolated yield between brackets.
b 98:2 regioisomeric ratio.
c 140 s residence time.
|
||||
| 1 | 1a |

|
100 | 99(92) |
| 2 | 1b |

|
100 | 97(88) |
| 3 | 1c |
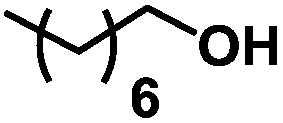
|
100 | 99(94) |
| 4 | 1d |

|
100 | 99(99) |
| 5 | 1e |
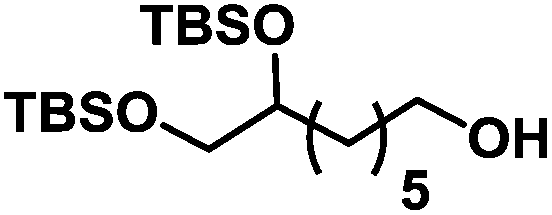
|
100 | 99(99) |
| 6 | 1f |

|
100 | 74(70) |
| 7 | 1g |

|
100 | 50 |
| 8 | 1h |

|
100 | 43(36) |
| 9 | 1i |

|
60 | 30(28) |
| 10 | 1j |

|
84 | 80(72)b |
| 11 | 1k |

|
85 | 74(65) |
| 12 | 1l |

|
39 | 34(28) |
| 13 | 75 | 70(68)c | ||

The reaction performed well for simple terminal alkenes such as 1-hexene 1b and 1-octene 1c with complete selectivity towards the functionalisation at the terminal position20 (entries 2 and 3). Tolerance towards acetal 1d and silylether 1e was noticed (entries 4 and 5).
The reaction also proceeded in good yield for more sterically hindered α-pinene 1f (entry 6). Moderate yield was observed when cycloalkene 1g was used as the substrate (entry 7). Notably, moderate yield was achieved for alcohol 2n, a challenging substrate due to the presence of a nitrile functional group, which is also susceptible to reduction. S-(−)-Limonene 1i was processed to the dioxygenated compound in modest yield of 30%21 (entry 8). Phenyl alkenes (entries 10–12) proved to have generally lower reactivity under the flow hydroboration conditions than aliphatic alkenes. Compound 1l required extended residence times in order to achieve good yields (entry 13). However, the reaction of styrene and its derivatives in flow showed higher preference towards the primary alcohol, compared to the batch procedure, with a selectivity of 92:8 for the styrene and complete anti-Markovnikoff selectivity for the other examples.22
An attractive feature of the flow procedure is the potential to perform additional steps in a telescoped fashion. In this case, we could expand on the process by incorporating an inline solvent extractor. An organic solvent phase was delivered by a fourth pump, and following appropriate mixing the extraction could be performed by a membrane separator device23 (Fig. 2).
 | ||
| Fig. 2 Schematic representation of our continuous reaction–extraction flow setup. | ||
Due to the high water miscibility of the organic solvent used in the oxidation step (THF and EtOH), the choice of the organic solvent to perform continuous extraction of this mixture was crucial. Initial attempts using diethyl ether (Et2O) as the extracting solvent were unsuccessful as a large amount of water and organic solvent passed through the membrane. When dichloromethane was used instead of Et2O, better phase separation was achieved and any residual water in the organic phase was removed easily using magnesium sulfate. At this stage, the processing scale was limited by the size of the commercial membrane separator (70 mL), thus each run was limited to 10 min.
Encouraged by the good performance of the system we wanted to develop a method for the continuous production of alcohols. We designed and incorporated a custom built glass extractor device (see Fig. 3) that allowed continuous extraction and collection of the organic phase for more than 30 min without issue. An analysis of the organic mixture after drying and removal of volatiles proved the isolation of the desired product in 80% yield.24
 | ||
| Fig. 3 Side and top view of an in-house gravity separating funnel and a Biotage® phase membrane separator for the continuous hydroboration/oxidation of alkenes. | ||
Conclusions
We have developed a continuous method for the hydroboration–oxidation reaction of olefins. To the best of our knowledge, this is the first report of such a continuous system. Under flow conditions, the reaction can be carried out under milder conditions and with higher selectivity than the batch reaction. Our protocol allows a continuous production of up to 120 mmol h−1 and continuous processing of the biphasic mixture produced by the reaction with a new liquid/liquid phase separation device. Investigations towards a fully telescoped functionalisation of the obtained alcohol or further derivatisation of the alkyl boranes are ongoing in our laboratory.Experimental section
1H-NMR spectra were recorded on a Bruker Avance DPX-400, with the residual solvent peak as the internal reference (CDCl3 = 7.26 ppm). 1H resonances are reported to the nearest 0.01 ppm. 13C-NMR spectra were recorded on the same spectrometer with proton decoupling, with the solvent peak as the internal reference (CDCl3 = 77.00 ppm). All 13C resonances are reported to the nearest 0.01 ppm. DEPT 135, COSY, HMQC, and HMBC experiments were used to aid structural determination and spectral assignment. The multiplicity of 1H signals are indicated as: s = singlet, d = doublet, dd = doublet of doublets, ddd = doublet of doublets of doublets, t = triplet, q = quadruplet, sext = sextet, m = multiplet, br = broad, or combinations thereof. Coupling constant (J) is quoted in Hz and reported to the nearest 0.1 Hz. Where appropriate, measures of the same coupling constant are averaged. Unless stated otherwise, reagents were obtained from commercial sources and used without purification. The removal of solvent under reduced pressure was carried out on a standard rotary evaporator. High resolution mass spectrometry (HRMS) was performed using a Waters Micromass LCT Premier™ spectrometer, using time of flight with positive ESI, or a Bruker BioApex 47e FTICR spectrometer using (positive or negative) ESI or EI at 70 eV to within a tolerance of 5 ppm of the theoretically calculated value. The flow apparatus is the Vapourtec R2 + R4 module commercialized by Vapourtec.General protocol for the hydroboration–oxidation of olefins in flow followed by regular workup (Protocol A)
A solution of alkene (2.5 mL, 1 M in THF) and a solution of BH3·THF (2.5 mL, 1 M in THF) were combined at a T-piece (each stream runs at 2.0 mL min−1) and reacted at rt in a 2 mL PFA reactor coil. The combined stream was then combined at a T-piece with an aqueous solution of NaOH (0.42 M in H2O2 (20% v/v)–H2O–EtOH 20:42:38) and reacted at rt in a 4.2 mL PFA reactor coil. To the resulting mixture, an aqueous saturated solution of NH4Cl was added, phases were separated and the organic layer extracted with Et2O (3×). Combined organic layers were washed with H2O (3×) and brine (1×), dried and evaporated. Unless otherwise stated a pure compound was isolated without further purification.
General protocol for the hydroboration–oxidation of β-pinene in batch
To a cold solution (0 °C) of β-pinene (24 mL, 148 mmol) in THF (56 mL) BH3·SMe2 (74 mL, 148 mmol, 2 M in THF) was added dropwise and the mixture was stirred at 0 °C for 1 hour. Then, maintaining the solution at 0 °C EtOH (72 mL), NaOH (80 mL, 1 M in H2O) and H2O2 (36 mL, 30% v/v in H2O) were added subsequently in a dropwise manner. The resulting mixture was stirred for 1 hour, warmed to room temperature and kept for an additional two hours at 80 °C. The reaction was then allowed to cool before quenching by the addition of a saturated aqueous solution of NH4Cl. An aqueous layer was extracted with Et2O (3×). Combined organic layers were washed with brine, dried and the solvent was evaporated. The NMR yield was determined to be 99%.General protocol for the continuous hydroboration–oxidation followed by in-line workup using the Biotage phase membrane separator (Protocol B)
A solution of alkene (1 M in THF) and a solution of BH3·THF (1 M in THF) were combined at a T-piece (each stream runs at 1.0 mL min−1) and reacted at rt in a 2 mL PFA reactor coil. The combined stream was then combined at a T-piece with an aqueous solution of NaOH (0.42 M in H2O2 (20% v/v)–H2O–EtOH 20:42:38) (4 mL min−1) and reacted at rt in a 4.2 mL PFA reactor coil. The resulting mixture was mixed at a T-piece with DCM (8 mL min−1) and the out stream dropped in a Biotage phase membrane separator collecting the organic layer from the output at the bottom.
General protocol for the continuous hydroboration–oxidation followed by in-line workup using in-house gravity separating funnel (Protocol C)
A solution of alkene (1 M in THF) and a solution of BH3·THF (1 M in THF) were combined at a T-piece (each stream runs at 1.0 mL min−1) and reacted at rt in a 2 mL PFA reactor coil. The combined stream was then combined at a T-piece with an aqueous solution of NaOH (0.42 M in H2O2 (20% v/v)–H2O–EtOH 20:42:38) (4 mL min−1) and reacted at rt in a 4.2 mL PFA reactor coil. The resulting mixture was mixed at a T-piece with DCM (8 mL min−1) and the out stream dropped in our custom built glass separator where continuous collection of the organic phase at the device output is possible by gravity separation of the aqueous-organic phase.
Acknowledgements
We are grateful to the Xunta de Galicia Government (JAS) and the EPSRC (SVL, grant no. EP/K0099494/1) for financial support.Notes and references
- (a) K. B. Sharpless and R. F. Lauer, J. Am. Chem. Soc., 1972, 94, 7154 CrossRef CAS; (b) H. P. Jensen and K. B. Sharpless, J. Org. Chem., 1975, 40, 264 CrossRef CAS.
- (a) W. Müller and H. J. Schneider, Angew. Chem., Int. Ed. Engl., 1979, 18, 407 CrossRef; (b) P. S. Traylor, D. Dolphin and T. G. Traylor, J. Chem. Soc., Chem. Commun., 1984, 279 RSC.
- (a) R. E. Ireland, S. W. Baldwin, D. J. Dawson, M. I. Dawson, J. E. Dolfini, J. Newbould, W. S. Johnson, M. Brown, R. J. Crawford, P. F. Hudrlik, G. H. Ramussen and K. K. Schmiegel, J. Am. Chem. Soc., 1970, 92, 5743 CrossRef CAS; (b) R. E. Ireland, S. W. Baldwin and S. C. Welch, J. Am. Chem. Soc., 1972, 94, 2056 CrossRef CAS; (c) E. L. Clennan and A. Pace, Tetrahedron, 2005, 61, 6665 CrossRef CAS.
- M. C. Pirrung, E. G. Rowley and C. P. Holmes, J. Org. Chem., 1993, 58, 5683 CrossRef CAS.
- (a) J. D. Sunderhaus, H. Lam and G. B. Dudley, Org. Lett., 2003, 8, 4571 CrossRef PubMed; (b) J. F. Jensen, B. Y. Svendsen, T. V. la Cour, H. L. Pedersen and M. Johannsen, J. Am. Chem. Soc., 2002, 124, 4558 CrossRef CAS PubMed; (c) T. Shimada, K. Mukaide, A. Shinohara, J. W. Han and T. Hayashi, J. Am. Chem. Soc., 2002, 124, 1584 CrossRef CAS PubMed.
- (a) H. Magari, H. Hirota and T. Takahashi, J. Chem. Soc., Chem. Commun., 1987, 1196 RSC; (b) H. Dyke, R. Sauter, P. Steel and E. J. Thomas, J. Chem. Soc., Chem. Commun., 1987, 1447 Search PubMed; (c) D. L. Boger and R. S. Coleman, J. Am. Chem. Soc., 1988, 110, 2501 CrossRef.
- (a) J. V. B. Kanth and H. C. Brown, J. Org. Chem., 2001, 66, 5359 CrossRef CAS PubMed; (b) J. M. Clay and E. Vedejs, J. Am. Chem. Soc., 2005, 127, 5766 CrossRef CAS PubMed; (c) P. K. Patra, K. Nishide, K. Fuji and M. Node, Synthesis, 2004, 1003 CAS.
- (a) H. Takanaka, K. Kawai, K. Fujiwara and A. Murai, Tetrahedron, 2002, 58, 10017 CrossRef; (b) N. Buschmann, A. Rueckert and S. Blechert, J. Org. Chem., 2002, 67, 4325 CrossRef CAS PubMed; (c) S. Liras, M. P. Allen and J. Blake, Org. Lett., 2001, 3, 3483 CrossRef CAS PubMed; (d) M. J. Bassindal, P. Hamley and J. P. A. Harrity, Tetrahedron Lett., 2001, 42, 9055 CrossRef.
- (a) R. E. Dolle, S. J. Schmidt, K. F. Erhard and L. I. Kruse, J. Am. Chem. Soc., 1989, 111, 278 CrossRef CAS; (b) D. H. B. Ripin, W. Cai and S. J. Brenek, Tetrahedron Lett., 2000, 41, 5817 CrossRef CAS; (c) D. H. B. Ripin, S. Abele, W. Cai, T. Blumenkopf, J. M. Casavant, J. L. Doty, M. Flanagan, C. Koecher, K. W. Laue, K. McCarthy, C. Meltz, M. Munchhoff, K. Pouwer, B. Shah, J. Sun, J. Teixeira, T. Vries, D. A. Whipple and G. Wilcox, Org. Process Res. Dev., 2003, 7, 115 CrossRef CAS.
- (a) H. C. Brown and B. C. Subba Rao, J. Am. Chem. Soc., 1956, 78, 5694 CrossRef CAS; (b) H. C. Brown and B. C. Subba Rao, J. Org. Chem., 1957, 22, 1136 CrossRef CAS.
- H. C. Brown and G. Zweifer, J. Am. Chem. Soc., 1960, 4708 CrossRef CAS.
- (a) H. C. Brown, Organic Syntheses via Boranes, Wiley-Interscience, New York, 1975, vol. 1. Reprinted by Aldrich Chemical Co Search PubMed; (b) J. L'Homme and G. Ourisson, Tetrahedron, 1968, 24, 3201 CrossRef.
- Diborane itself reacts slowly with alkenes and alkynes but its reactivity is critically enhanced by complexation with Lewis bases (a) D. T. Hurd, J. Am. Chem. Soc., 1948, 70, 2053 CrossRef CAS; (b) H. C. Brown and B. C. Subba Rao, J. Am. Chem. Soc., 1956, 78, 2582 CrossRef CAS; (c) H. C. Brown and B. C. Subba Rao, J. Am. Chem. Soc., 1959, 81, 6423 CrossRef CAS.
- (a) F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478 CrossRef CAS PubMed; (b) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583 RSC; (c) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849 RSC; (d) V. Hessel, I. V. Gursel, Q. Wang, T. Noel and J. Lang, Chem. Eng. Technol., 2012, 35, 1184 CrossRef CAS.
- For selected publications: (a) A. Polyzos, M. O'Brien, T. Pugaard-Petersen, I. R. Baxendale and S. V. Ley, Angew. Chem., Int. Ed., 2011, 50, 1190 CrossRef CAS PubMed; (b) C. F. Carter, M. D. Hopkin, A. Burke, J. G. Goode, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 765 RSC; (c) D. L. Browne, B. J. Deadman, R. Ashe, I. R. Baxendale and S. V. Ley, Org. Process Res. Dev., 2011, 15, 693 CrossRef CAS; (d) T. Brodmann, P. Koos, A. Metzger, P. Knochel and S. V. Ley, Org. Process Res. Dev., 2012, 16, 1102 CrossRef CAS; (e) J. C. Pastre, D. L. Browne, M. O'Brien and S. V. Ley, Org. Process Res. Dev., 2013, 17, 1183 CrossRef CAS; (f) P. R. D. Murray, D. L. Browne, J. C. Pastre, C. Butters, D. Guthrie and S. V. Ley, Org. Process Res. Dev., 2013, 17, 1192 CrossRef CAS; (g) J. A. Newby, D. W. Blaylock, P. M. Witt, J. C. Pastre, M. K. Zacarova, S. V. Ley and D. L. Browne, Org. Process Res. Dev., 2014, 18, 1211 CrossRef CAS; (h) T. Ouchi, C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Process Res. Dev., 2014, 18, 1560 CrossRef CAS; (i) S. Newton, C. F. Carter, C. M. Pearson, L. C. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915 CrossRef CAS PubMed; (j) R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 144 CrossRef CAS PubMed; (k) D. N. Tran, C. Battilocchio, S.-B. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120 RSC.
- Despite the broad applicability of hydroboration/oxidation reaction in batch chemistry we are not aware of any reported procedures overcoming this classical transformation in flow.
- The residence time, under optimised conditions, for the hydroboration step is 50 seconds and 20 seconds for the oxidation reaction.
- For its THF complex the borane intermediate of β-pinene which shows significant dissociation into triisopinocanphenyldiborane and α-pinene has been reported. H. C. Brown and A. W. Moerikofer, J. Am. Chem. Soc., 1962, 84, 1478 CrossRef CAS.
- P. K. Patra, K. Nishide, K. Fuji and M. Node, Synthesis, 2004, 1003 CAS.
- A detailed study in the batch hydroboration/oxidation of terminal olefins has shown a 94:6 preference for the terminal alcohol. See ref. 11 for details.
- Initial terpene and mixed monooxygenated compounds were also observed in the crude mixture.
- Batch hydroboration/oxidation of styrenes generally affords the primary alcohol in 80:20 regioisomeric ratio. See ref. 11 for details.
- We used the Biotage® phase separator to perform the continuous separation of the organic and aqueous layers obtained after hydroboration–oxidation reaction. http://www.biotage.com/product-page/phase-separators.
- A regular workup of the aqueous solution left in the phase separator, following the general protocol described above allows the recovery of the remaining 20% of the product.
- H. C. Brown and A. W. Moerikofer, J. Am. Chem. Soc., 1962, 84, 1478 CrossRef CAS.
- M. Azri, A. Rani, N. Borduas, V. Colquhoun, R. Hanley, H. Johnson, S. Larger, P. D. Lickiss, V. Llopis-Mestre, S. Luu, M. Mogstad, P. Oczipka, J. R. Sherwood, T. Welton and J.-Y. Xing, Green Chem., 2014, 16, 1282 RSC.
- J. Julis and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8615 CrossRef CAS PubMed.
- L. G. Meimetis, D. E. Williams, N. R. Mawji, C. A. Banuelos, A. A. Lal, J. J. Park, A. H. Tien, J. Garcia-Fernandez, N. J. de Voogd, M. D. Sadar and R. J. Andersen, J. Med. Chem., 2012, 55, 503 CrossRef CAS PubMed.
- L. Pehlivan, E. Métay, O. Boyron, P. Demonchaux, G. Mignani and M. Lemaire, Eur. J. Org. Chem., 2011, 4687 CrossRef CAS.
- L. R. Reddy, B. Hu, M. Prashad and K. Prasad, Org. Lett., 2008, 10, 133 CrossRef PubMed.
- D. C. Harrowven, D. D. Pascoe, D. Demurtas and H. O. Bourne, Angew. Chem., Int. Ed., 2005, 44, 1221 CrossRef CAS PubMed.
- B. S. Bodnar and P. F. Vogt, J. Org. Chem., 2009, 74, 2598 CrossRef CAS PubMed.
- L. L. W. Cheung, G. Vasapollo and H. Alper, Adv. Synth. Catal., 2012, 354, 2019 CrossRef CAS.
- I. N. Redwan and M. Grøtli, J. Org. Chem., 2012, 77, 7071 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob00170f |
| This journal is © The Royal Society of Chemistry 2015 |