
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new multicomponent reaction: unexpected formation of derivatizable cyclic α-alkoxy isothioureas†
Fabian
Brockmeyer
,
Valentin
Morosow
and
Jürgen
Martens
*
Institut für Chemie, Carl von Ossietzky Universität Oldenburg, P. O. Box 2503, Carl-von-Ossietzky-Str. 9-11, 26111 Oldenburg, Germany. E-mail: juergen.martens@uni-oldenburg.de; Web: http://www.martens.chemie.uni-oldenburg.de
First published on 16th January 2015
Abstract
An unexpected formation of cyclic α-alkoxy isothioureas has been achieved. As is known, the heterocyclic imines 2,5-dihydro-1,3-thiazoles are convertible to bisamides with the aid of a carboxylic acid and an isocyanide (Ugi reaction). Herein, it is shown that 2,5-dihydro-1,3-thiazole S-monoxides—the respective α-sulfinyl imines—are characterized by an altered reaction behavior. In a hitherto unknown multicomponent reaction the α-sulfinyl imines react with an isocyanide under acidic conditions in an alcoholic solution to the respective α-alkoxy isothioureas in good yields. In addition to the investigations on this unexpected synthesis the regioselectivity of the acylation of the synthesized compounds is described. A rearrangement, which is accelerated by EDC and HOBt, between both possible regioisomers was found.
Introduction
For more than 60 years the 2,5-dihydro-1,3-thiazole (3-thiazoline) scaffold has been an important industrial key element in the course of the preparation of pharmaceutically active molecules.1 Furthermore, other remarkable investigations on this family of heterocycles characterize fields of research in general.2 The synthesis of 3-thiazolines was first reported by F. Asinger in 1956.3 Following a modified protocol of the Asinger reaction the cyclic imines are preparable by a four-component reaction (A 4-CR) where an α-chloro aldehyde is reacted with a carbonyl compound, ammonia, and sodium hydrosulfide.43-Thiazolines could be converted into a large number of different products by functionalizing their reactive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond.5
N double bond.5
One of the most important reactions involving imines is the Ugi three-component reaction (U 3-CR). In the U 3-CR an imine is treated with an isocyanide and a carboxylic acid to form the desired bisamide.6 As our earlier research had shown, 3-thiazolines are unproblematically employable as the imine component in the U 3-CR.7
In one of our recent studies, we had additionally investigated the chemoselective oxidation of 3-thiazolines.8 An oxidation of the sulfur atom leading to 3-thiazoline S-monoxides represents one possible and realizable reaction pathway of the oxidation of these substrates.
In order to examine the reaction behaviour of these new 3-thiazoline S-monoxides, we then tried to convert these compounds to the respective bisamides with the aid of the U 3-CR (Fig. 1). However, as presented in this paper, we observed that under the reaction conditions mentioned above, the 3-thiazoline S-monoxides react contrary to our expectations to cyclic isothioureas. The products are characterized by an alkoxy moiety next to the endocyclic nitrogen atom of the isothiourea (Fig. 2). Despite the existence of many syntheses of cyclic isothioureas,9 the class of cyclic α-alkoxy isothioureas has not yet been reported before.
 | ||
| Fig. 1 Retrosynthetic consideration of the designed synthesis route. | ||
 | ||
| Fig. 2 Unexpected three-component synthesis of cyclic isothioureas. | ||
Results and discussion
Synthesis of the precursors
First, we synthesized four known 3-thiazolines via the A 4-CR. Treatment of the respective α-chloro aldehyde with sodium hydrosulfide, an aqueous ammonia solution and acetone or cyclohexanone led to the cyclic imines 2,2,5,5-tetramethyl-2,5-dihydro-1,3-thiazole (1a),5c 5,5-diethyl-2,2-dimethyl-2,5-dihydro-1,3-thiazole (1b),7b 2,2-dimethyl-1-thia-3-azaspiro[4.5]dec-3-ene (1c),10 and 2,2-dimethyl-1-thia-4-azaspiro[4.5]dec-3-ene (1d).5cThe oxidation of 3-thiazolines 1 to sulfoxides 2 was described by us recently.8 In line with this, all four 3-thiazolines 1a–d were converted to the respective sulfoxides 2a–d with the aid of mCPBA (Fig. 3).
 | ||
| Fig. 3 Synthesis of sulfoxides 2a–d. | ||
Unexpected preparation of the cyclic isothioureas
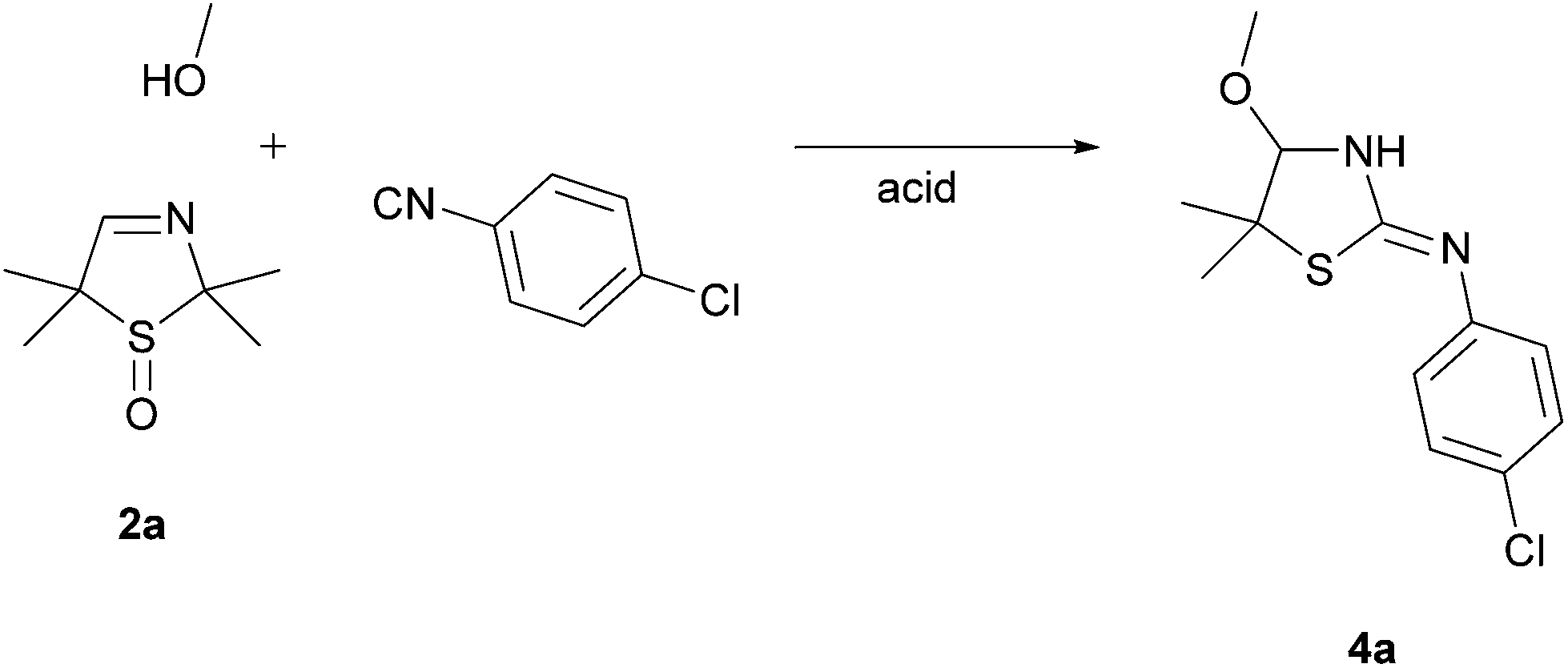
On the basis of our earlier work,7 we wanted to examine the influence of the sulfoxide moiety on the reactivity of the cyclic imine by converting the 3-thiazoline S-monoxides 2 to bisamides. To validate the applicability of unoxidized 3-thiazolines 1 in the U 3-CR, we reacted the 3-thiazoline 1a with 1-chloro-4-isocyanobenzene and benzoic acid in MeOH. The bisamide 3 was formed according to our expectations. Afterwards, the oxidized analog of 1a (3-thiazoline S-monoxide 2a) was converted analogously. It was found that after 72 h the substrate 2a was fully reacted. However, it came as a surprise that no bisamide was formed and the corresponding cyclic α-methoxy isothiourea 4a was isolated as the sole product (Scheme 1). This unexpected result implies that the reaction of the sulfoxide 2a is accompanied by a formal elimination of acetone and a recyclization realized by an addition of the isocyanide. Moreover, an addition of MeOH, which was initially applied as a solvent, to the imine bond takes place. Apart from using NMR, IR, and mass analysis, the structure of the product 4a was also verified by X-ray crystal structure analysis.11 The crystal structure reveals that the double bond is exocyclic and (Z)-configured. Contrary to our expectations, even at −40 °C in CDCl3 only the tautomer (and only one diastereomer) bearing an exocyclic double bond was observable, so that, consequently, a tautomeric equilibrium could not been proven.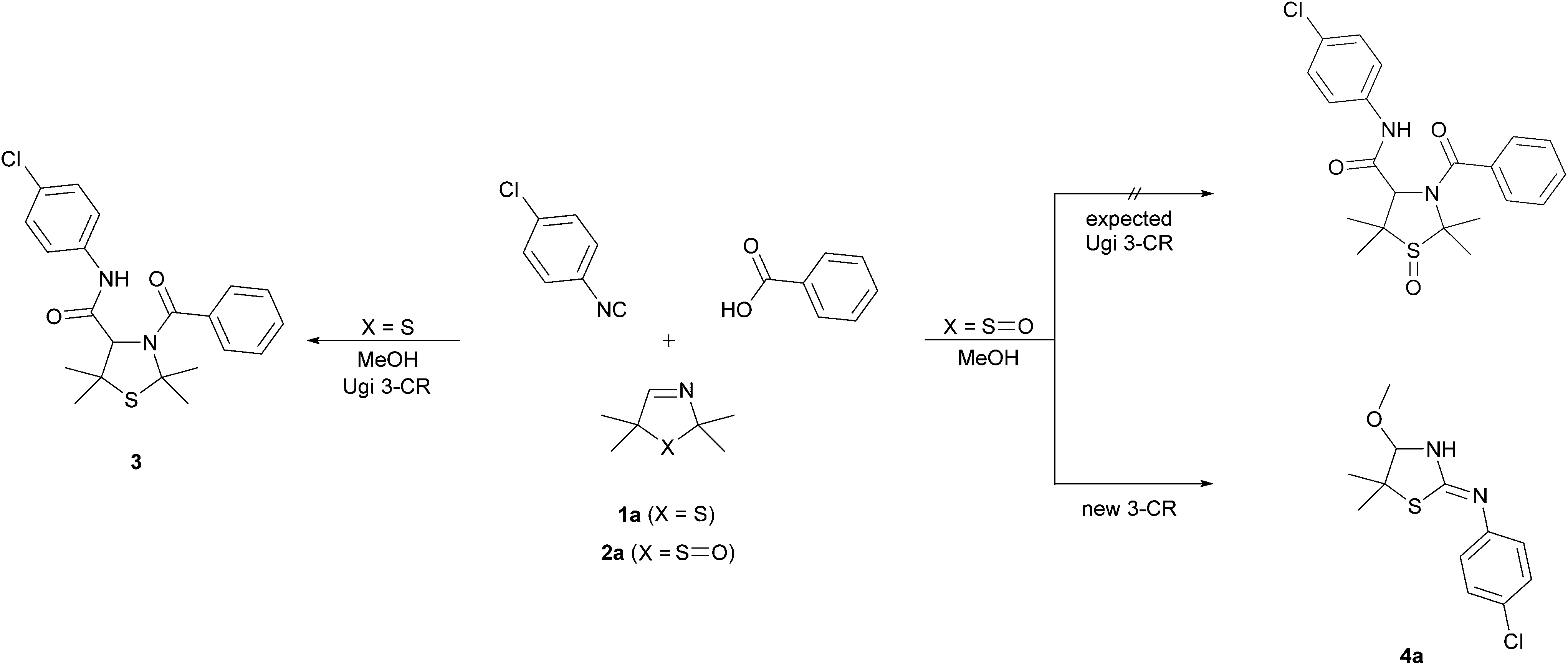 | ||
| Scheme 1 Conversion of 1a and 2a under the conditions of the Ugi reaction. | ||
To explore the influence of the carboxylic acid, which is not a part of the product, on the formation of the cyclic isothioureas 4, we examined the transformation of 3-thiazoline 2a with 1-chloro-4-isocyanobenzene to isothiourea 4a under different conditions by varying the type and the equivalent of the acid (Table 1). The entries 1–4 show that an acidic medium is significant for the course of the reaction and that, therefore, the amount of acid has an enormous effect on the obtained yield. The best result was received by using 1.0 equivalent of the acid (Table 1, entry 1). The acidity of benzoic acid in comparison with acetic acid, trifluoro acetic acid, and sulfuric acid seems to be optimal. Thus, the use of benzoic acid led to the highest yield. A further increase of the amount of acid did not lead to an enhanced formation of the product 4a (Table 1, entry 4).
|
|
||
|---|---|---|
| Entry | Acid | Yieldb [%] |
| a Reaction conditions: 2a (0.8 mmol), 1-chloro-4-isocyanobenzene (0.8 mmol), acid, in MeOH (5 mL), 72 h at r.t. b Isolated yield after chromatography on silica gel. | ||
| 1 | BzOH (1.0 equiv.) | 74 |
| 2 | BzOH (0.5 equiv.) | 36 |
| 3 | — | 19 |
| 4 | BzOH (1.2 equiv.) | 73 |
| 5 | AcOH (1.0 equiv.) | 27 |
| 6 | TFA (1.0 equiv.) | — |
| 7 | H2SO4 (1.0 equiv.) | — |
The observable formation of 4a in the absence of a carboxylic acid or an inorganic acid could probably be explained by the acidity of methanol. Furthermore, benzoic acid was found to be the most effective acid to convert the 3-thiazoline 2a in the presence of an isocyanide and MeOH (Table 1, entries 1, 2, and 4) in comparison with acetic acid (Table 1, entry 5), trifluoroacetic acid (Table 1, entry 6), and sulfuric acid (Table 1, entry 7).
The proposed mechanism for the formation of the cyclic isothioureas 4 is shown in Scheme 2. The sulfoxide 2 is protonated by the carboxylic acid. A ring-opening which is induced by a nucleophilic attack of the alcohol leads to the sulfenic acid A. The iminium ion B is formed by a substitution of the hydroxyl group by the isocyanide and a subsequent ring-closure. This ion reacts with two molecules of the alcohol under elimination of a proton to the product 4. The acetal, formed as a co-product, is hydrolyzed to the alcohol and acetone.
 | ||
| Scheme 2 Postulated mechanism. | ||
Reaction scope
Next, we investigated the generality of this unexpected synthesis of cyclic α-alkoxy isothioureas 4 by varying all three substrates (Table 2). First, the applicability of different isocyanides was studied. It was found that aryl, benzyl and alkyl isocyanides could be used for the conversion of 3-thiazolines 2. Substituents including aryl chlorides (Table 2, entries 1 and 10) and esters (Table 2, entries 5, 6, 9, and 13–15) were tolerated. Furthermore, it was determined that different alcohols, upon the condition that they can be used as a solvent, are employable reactants (Table 2, entries 7 and 8).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | R3 | R4 | Yieldb [%] |
| a Reaction conditions: 2 (1.0 equiv.), carboxylic acid (1.0 equiv.), BzOH (1.0 equiv.), in R4OH (6 mL per mmol 2), 72 h at r.t. b Isolated yield after chromatography on silica gel. | ||||||
| 1 | 2a | Me | Me | 4-Cl-Ph | Me | 4a: 74 |
| 2 | 2a | Me | Me | Ph | Me | 4b: 65 |
| 3 | 2a | Me | Me | Bn | Me | 4c: 69 |
| 4 | 2a | Me | Me | Allyl | Me | 4d: 55 |
| 5 | 2a | Me | Me | CH2COOMe | Me | 4e: 73 |
| 6 | 2a | Me | Me | (CH2)3COOEt | Me | 4f: 55 |
| 7 | 2a | Me | Me | 4-Cl-Ph | Et | 4g: 61 |
| 8 | 2a | Me | Me | Allyl | Allyl | 4h: 77 |
| 9 | 2b | Et | Et | CH2COOMe | Me | 4i: 63 |
| 10 | 2c | –(CH2)5– | 4-Cl-Ph | Me | 4j: 81 | |
| 11 | 2c | –(CH2)5– | Allyl | Me | 4k: 73 | |
| 12 | 2c | –(CH2)5– | n-Bu | Me | 4l: 71 | |
| 13 | 2c | –(CH2)5– | CH2COOMe | Me | 4m: 87 | |
| 14 | 2c | –(CH2)5– | (CH2)2COOMe | Me | 4n: 64 | |
| 15 | 2c | –(CH2)5– | (CH2)3COOMe | Me | 4o: 68 | |
| 16 | 2d | Me | Me | 4-Cl-Ph | Et | 4g: 34 |
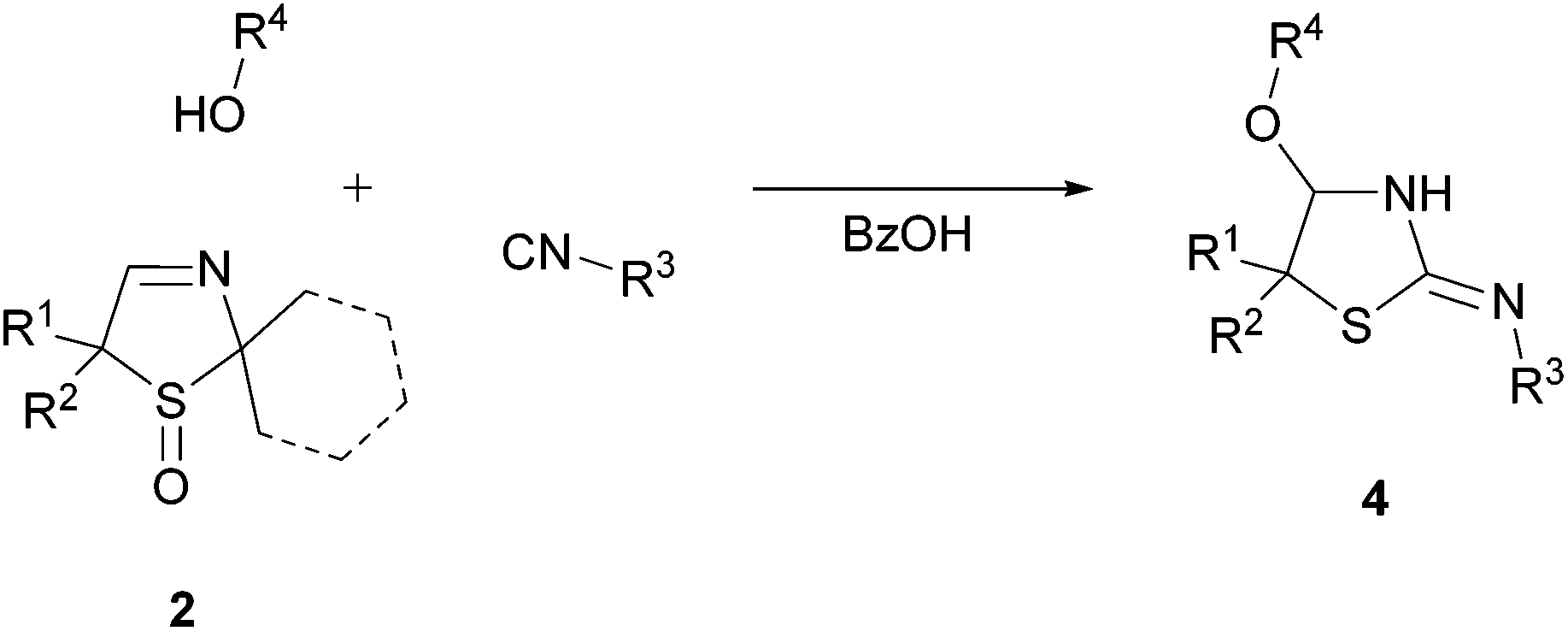
Besides model substrate 2a, three further 3-thiazoline S-monoxides 2 were examined (Table 2, entries 9–16). It turned out that compound 2c bearing a spiro-coupled cyclohexane ring at the carbon C5 gave the highest yield (up to 87%; Table 2, entry 13). Entry 16 proves that not only 3-thiazoline S-monoxides 2a–c characterized by two methyl groups located at the acetal carbon, but also 3-thiazoline S-monoxide 2d exhibiting a spiro-coupled cyclohexane ring at that carbon atom is an appropriate substrate. When the substrate 2d is used, cyclohexanone instead of acetone is the co-product.
It would therefore appear that the unexpectedly observed formation of the cyclic isothiourea 4a can be transferred to a broad range of differently substituted substrates. Additionally, the obtained products are suitable for further derivatizations.
Acylation of the synthesized isothioureas
They can be converted to N-acyl isothioureas by treating them with the respective acid and EDC12 [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide] as an activating agent in CH2Cl2. Following this route, both possible regioisomers—products 5 acylated at the exocyclic nitrogen atom and products 6 acylated at the endocyclic nitrogen atom—can be observed. Conversion of isothiourea 4j bearing an aromatic substituent at the exocyclic nitrogen atom with phenylacetic acid resulted in an excess of product 5a, which is derivatized at the exocyclic nitrogen atom (Table 3, entry 1), (5a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6a 75:25) in the crude product. Changing the solvent from CH2Cl2 to DMF or the reaction temperature from room temperature to −20 °C did not have any influence on the ratio between both regioisomers in the crude product.
:6a 75:25) in the crude product. Changing the solvent from CH2Cl2 to DMF or the reaction temperature from room temperature to −20 °C did not have any influence on the ratio between both regioisomers in the crude product.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Methoda | R1 | R2 | R3 | R4 | R5 | Ratiob5:6 |
Yieldc [%] |
| a Reaction conditions: (A) 4 (1.0 equiv.), carboxylic acid (1.2 equiv.), EDC (1.3 equiv.), in CH2Cl2 (8 mL per mmol 4), 10 h at r.t.; (B) carboxylic acid (1.0 equiv.), carboxylic acid (1.2 equiv.), EDC (1.3 equiv.), HOBt (1.3 equiv.), in CH2Cl2 (8 mL per mmol 4), 2 h at r.t.; all reaction times were optimized with a view to the highest overall yield. b Ratio was determined by 1H NMR spectroscopy of the crude product. c Isolated yield after chromatography on silica gel. | |||||||||
| 1 | 4j | A | –(CH2)5– | 4-Cl-Ph | Me | Bn | 75:25 |
5a: 20; 6a: 55 | |
| 2 | 4j | B | –(CH2)5– | 4-Cl-Ph | Me | Bn | 0:100 |
5a: —; 6a: 83 | |
| 3 | 4a | B | Me | Me | 4-Cl-Ph | Me | Bn | 0:100 |
5b: —; 6b: 88 |
| 4 | 4a | B | Me | Me | 4-Cl-Ph | Me | Et | 0:100 |
5c: —; 6c: 78 |
| 5 | 4l | A | –(CH2)5– | n-Bu | Me | Bn | 40:60 |
5d: 22; 6d: 41 | |
| 6 | 4l | B | –(CH2)5– | n-Bu | Me | Bn | 40:60 |
5d: 28; 6d: 56 | |
| 7 | 4n | A | –(CH2)5– | (CH2)2COOMe | Me | CH2N(Ph)Fmoc | 41:59 |
5e: 18; 6e: 21 | |
| 8 | 4n | B | –(CH2)5– | (CH2)2COOMe | Me | CH2N(Ph)Fmoc | 43:57 |
5e: 23; 6e: 28 | |

After isolation by column chromatography, the ratio was shifted to 5a:6a 27:73. To explain this shift of the ratio between 5a and 6a during the isolation, we postulated that a rearrangement of product 5, once formed by an exocyclic acylation, to 6 is possible. The existence of this rearrangement was proven by a control experiment. Isolated 5a was kept in CDCl3 solution at room temperature without convection. As determined by 1H NMR analysis, the rearrangement to 6a was fully completed after 24 d (Fig. S1, ESI†). In a repetition of this experiment using additional EDC the rearrangement was already completed after 9 d (Fig. S2, ESI†). Thus, the rearrangement between both regioisomers is accelerated by EDC.
By using hydroxybenzotriazole (HOBt), which is mainly used as the additional activating agent to suppress the racemization of chiral substrates during peptide syntheses,13 in addition to EDC, we improved the outcome of the acylation reaction. Moreover, the use of HOBt in the conversion of 4j with phenylacetic acid led to the formation of only one regioisomer (6a) after 2 h (Table 3, entry 2). In line with this, the rearrangement of isolated 5a in CDCl3 under the addition of EDC and HOBt took less than 20 min (Fig. S3, ESI†). This clarifies that the rearrangement between both regioisomers is accelerated more strongly by a combination of EDC and HOBt than by the mere use of EDC. Entries 3 and 4 of Table 3 verify the regiospecific acylation of cyclic isothioureas 4 bearing an aromatic substrate at the exocyclic nitrogen atom within the reaction time, when HOBt is added to the reaction mixture.
A conversion of the isothiourea 4l characterized by an aliphatic substituent at the exocyclic nitrogen atom led mainly to product 6d (5d:6d 40:60 in crude product; Table 3, entry 5; coupling agent: exclusive EDC).
The rearrangement of 5d is very slow. Monitoring the rearrangement of isolated 5d in CDCl3 at room temperature without the influence of EDC and HOBt we ascertained that only half the amount of substrate 5d was rearranged to 6d after 115 d (Fig. S4, ESI†). In the conversion of 4l, the addition of HOBt only had an influence on the overall yield (raised from 63% to 85%; Table 3, entry 6) and not on the product ratio. These observations were confirmed by the conversion of substrate 4n, which is also characterized by an aliphatic substituent at the exocyclic nitrogen atom, with Fmoc-N-phenylglycine (Table 3, entries 7 and 8).
When a substrate is converted which bears an aromatic substituent at the exocyclic nitrogen atom the acylation at this exocyclic nitrogen atom is kinetically more preferred than the acylation at the endocyclic nitrogen atom. This is probably due to the mesomeric stabilization of the cationic intermediate stage. On the other hand, the thermodynamic stability of the product which is characterized by an exocyclic double bond and an acyl moiety at the endocyclic nitrogen atom in comparison to the regioisomer is enhanced by the aromatic substituent, because an exocyclic double bond results in a conjugated π-system. This results in the observed selectivities and speeds of rearrangements.
Conclusions
In summary, 3-thiazoline S-monoxides, which are easily preparable by an Asinger 4-CR and a subsequent oxidation, were converted to cyclic α-alkoxy isothioureas. The investigated conversion of the substrates with isocyanides and alcohols under acidic conditions opens unexpected access to the novel class of cyclic α-alkoxy isothioureas in good yields. This procedure may be useful for a general conversion of S,N-acetals to the corresponding isothioureas.The derivatization of the synthesized cyclic isothioureas by an acylation leads to two different regioisomeric products. The kinetic product, which is acylated at the exocyclic nitrogen atom, rearranges to the thermodynamic more stable second regioisomer, which is acylated at the endocyclic nitrogen atom. The selectivity and the speed of the rearrangement depend tremendously on the type of the substituent. EDC and HOBt were found to accelerate the rearrangement of N-acyl isothioureas.
Experimental
Synthesis of (RS)-(Z)-2-(4-chlorophenylimino)-4-methoxy-5,5-dimethyl-1,3-thiazolidine (4a) as a representative example for the synthesis of all cyclic isothioureas 4
The sulfoxide 2a (717 mg, 4.50 mmol) was dissolved in 9 mL methanol. 1-Chloro-4-isocyanobenzene (619 mg, 4.50 mmol), dissolved in 9 mL methanol, and benzoic acid (550 mg, 4.50 mmol), dissolved in 9 mL methanol, were added dropwise. The reaction mixture was vigorously stirred for 72 h at r.t. The solvent was removed on a rotary evaporator. Purification of the crude product by column chromatography on silica gel (EtOAc, Rf = 0.63) afforded 4a (902 mg, 74%) as a colorless solid, mp 115–117 °C (from CH2Cl2–n-hexane); IR (ATR):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 3111, 3051, 2960, 2897, 2827, 1636, 1584, 1484, 1437, 1368, 1341, 1158, 1144, 1063, 843, 824 cm−1; 1H NMR (500.1 MHz, CDCl3): δ 1.46, 1.52 [6 H, 2s, C(CH3)2], 3.29 (3 H, s, OCH3), 4.34 (1 H, s, NCH), 6.94–6.95 [2 H, m, 2 m-CHAr(Cl)], 7.22–7.24 [2 H, m, 2 o-CHAr(Cl)] ppm; 13C NMR (125.8 MHz, CDCl3): δ 22.61, 30.66 [C(CH3)2], 55.30 (OCH3), 57.04 [C(CH3)2], 95.10 (NCH), 123.48 [2 m-CHAr(Cl)], 128.88 (ClCAr), 129.12 [2 o-CHAr(Cl)], 148.41 (NCAr), 162.82 (CN) ppm; MS (CI-sector): m/z 271.2 (M + H+, 100%); HRMS (CI-sector): found 271.0673; calc. for C12H16ClN2OS [M + H]+ 271.0672.
3111, 3051, 2960, 2897, 2827, 1636, 1584, 1484, 1437, 1368, 1341, 1158, 1144, 1063, 843, 824 cm−1; 1H NMR (500.1 MHz, CDCl3): δ 1.46, 1.52 [6 H, 2s, C(CH3)2], 3.29 (3 H, s, OCH3), 4.34 (1 H, s, NCH), 6.94–6.95 [2 H, m, 2 m-CHAr(Cl)], 7.22–7.24 [2 H, m, 2 o-CHAr(Cl)] ppm; 13C NMR (125.8 MHz, CDCl3): δ 22.61, 30.66 [C(CH3)2], 55.30 (OCH3), 57.04 [C(CH3)2], 95.10 (NCH), 123.48 [2 m-CHAr(Cl)], 128.88 (ClCAr), 129.12 [2 o-CHAr(Cl)], 148.41 (NCAr), 162.82 (CN) ppm; MS (CI-sector): m/z 271.2 (M + H+, 100%); HRMS (CI-sector): found 271.0673; calc. for C12H16ClN2OS [M + H]+ 271.0672.
Single crystals of 4a were recrystallized from CH2Cl2 and n-hexane, mounted in inert oil and transferred to the cold gas stream of the diffractometer.
Crystal structure determination of 4a
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 6165 reflections measured, 6165 unique (Rint = 0.0000) which were used in all calculations. The final wR(F2) was 0.1262 (all data).
, Z = 2, 6165 reflections measured, 6165 unique (Rint = 0.0000) which were used in all calculations. The final wR(F2) was 0.1262 (all data).
Acknowledgements
The authors gratefully acknowledge Marc Schmidtmann for X-ray crystallography.Notes and references
- W. Keim and H. Offermanns, Angew. Chem., 2007, 119, 6116–6120 ( Angew. Chem. Int. Ed. , 2007 , 46 , 6010–6013 ) CrossRef.
- (a) A. Dömling and I. Ugi, Angew. Chem., 1993, 105, 634 ( Angew. Chem. Int. Ed. Engl. , 1993 , 32 , 563 ) CrossRef; (b) J. S. Elmore, D. S. Mottram, M. Enser and J. D. Wood, J. Agric. Food Chem., 1997, 45, 3603–3607 CrossRef CAS; (c) A. Dömling, I. Ugi and E. Herdtweck, Acta Chem. Scand., 1998, 52, 107 CrossRef PubMed; (d) X. Fernandez, R. Fellous, L. Lizzani-Cuvelier, M. Loiseau and E. Duñach, Tetrahedron Lett., 2001, 42, 1519–1521 CrossRef CAS.
- F. Asinger, Angew. Chem., 1956, 68, 413 CrossRef CAS.
- J. Martens, H. Offermanns and P. Scherberich, Angew. Chem., 1981, 93, 680–683 ( Angew. Chem. Int. Ed. Engl. , 1981 , 20 , 668 ) CrossRef CAS.
- For examples, see: (a) K. Harada, in The chemistry of the carbon-nitrogen double bond, ed. S. Patai, John Wiley & Sons, London, 1992, ch. 6, pp. 255–298 Search PubMed; (b) H. Gröger, Y. Saida, H. Sasai, K. Yamaguchi, J. Martens and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 3089–3103 CrossRef; (c) R. Sail, J. Kopf and P. Köll, Tetrahedron: Asymmetry, 2000, 11, 423–433 CrossRef; (d) T. Stalling, F. Brockmeyer, D. Kröger and A. Schwäblein, Z. Naturforsch., B: Chem. Sci., 2012, 67, 1045–1055 CrossRef CAS; (e) A. Sehlinger, T. Stalling, J. Martens and M. A. R. Meier, Macromol. Chem. Phys., 2014, 215, 412–420 CrossRef CAS.
- (a) I. Ugi, R. Meyer, U. Fetzer and C. Steinbrückner, Angew. Chem., 1959, 71, 386 Search PubMed; (b) I. Ugi, Angew. Chem., 1962, 74, 9–22 ( Angew. Chem. Int. Ed. Engl. , 1962 , 1 , 8–21 ) CrossRef CAS; (c) I. Ugi and E. Wischhöfer, Chem. Ber., 1962, 95, 136–140 CrossRef CAS.
- For examples, see: (a) J. Kintscher and J. Martens, Synthesis, 1992, 837–838 CrossRef CAS; (b) M. Hatam, D. Tehranfar and J. Martens, Synth. Commun., 1995, 25, 1677–1688 CrossRef CAS; (c) F. Brockmeyer, D. Kröger, T. Stalling, P. Ullrich and J. Martens, Helv. Chim. Acta, 2012, 95, 1857–1870 CrossRef CAS.
- F. Brockmeyer and J. Martens, ChemSusChem, 2014, 7, 2441–2444 CrossRef CAS PubMed.
- (a) A. Hirashima, H. Tarui and M. Eto, Biosci., Biotechnol., Biochem., 1994, 58, 1206–1209 CrossRef CAS; (b) R. F. Ambartsumova, M. G. Levkovich, E. G. Milgrom and N. D. Abdullaev, Chem. Heterocycl. Compd., 1997, 33, 112–117 CrossRef CAS; (c) B. R. Dixon, C. M. Bagi, C. R. Brennan, D. R. Brittelli, W. H. Bullock, J. Chen, W. L. Collibee, R. Dally, J. S. Johnson, H. C. E. Kluender, W. F. Lathrop, P. Liu, C. A. Mase, A. M. Redman, W. J. Scott, K. Urbahns and D. J. Wolanin, US Pat, 6353006 B1, 2002 Search PubMed; (d) H. Kai, Y. Morioki, T. Murashi, K. Morita, S. Shinonome, H. Nakazato, K. Kawamoto, K. Hanasaki, F. Takahashi, S. i. Mihara, T. Arai, K. Abe, H. Okabe, T. Baba, T. Yoshikawa and H. Takenaka, Bioorg. Med. Chem. Lett., 2007, 17, 4030–4034 CrossRef CAS PubMed; (e) N. Lui, K. Jelich, M. Littmann, K. Lorenz and B. Lecorre, WO Pat, 2009033583 A1, 2009 Search PubMed.
- K. Drauz, H. G. Koban, J. Martens and W. Schwarze, Liebigs Ann. Chem., 1985, 448–452 CrossRef CAS.
- The X-ray crystal structure is shown in the ESI.† CCDC 1037548 contains the supplementary crystallographic data.
- EDC is a common coupling agent. For another example, see: M. J. Niphakis, B. J. Turunen and G. I. Georg, J. Org. Chem., 2010, 75, 6793–6805 CrossRef CAS PubMed.
- W. König and R. Geiger, Chem. Ber., 1997, 103, 788–798 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for all isolated new compounds. 1H and 13C NMR spectra of all isolated new compounds as well as 1H NMR spectra (stacked) of the monitored rearrangement of 5a and 5d. CCDC 1037548. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob02608j |
| This journal is © The Royal Society of Chemistry 2015 |