
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConsecutive three-component synthesis of (hetero)arylated propargyl amides by chemoenzymatic aminolysis–Sonogashira coupling sequence†
Sidra
Hassan
,
Anja
Ullrich
and
Thomas J. J.
Müller
*
Lehrstuhl für Organische Chemie, Institut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225, Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@uni-duesseldorf.de; Fax: +49 (0)211 8114324; Tel: +49 (0)211 8112298
First published on 27th November 2014
Abstract
A novel chemoenzymatic three-component synthesis of (hetero)arylated propargyl amides in good yields based upon Novozyme® 435 (Candida antarctica lipase B (CAL-B)) catalyzed aminolysis of methyl carboxylates followed by Sonogashira coupling with (hetero)aryliodides in a consecutive one-pot fashion has been presented. This efficient methodology can be readily concatenated with a CuAAC (Cu catalyzed alkyne azide cycloaddition) as a third consecutive step to furnish 1,4-disubstituted 1,2,3-triazole ligated arylated propargyl amides. This one-pot process can be regarded as a transition metal catalyzed sequence that takes advantage of the copper source still present from the cross-coupling step.
Introduction
Propargyl amides are particularly interesting as biologically active functionalities and as synthetic building blocks. Protein derived propargyl amides have been studied to irreversibly inhibit cysteine proteases1 while systems containing rigid propargylated aryl cores have been demonstrated to be active as structurally modified homodimeric gonadotropin releasing hormone receptor (GnRHR) antagonist dimers with rigid functionalities such as bistriazole with a hydrophilic polyethylene glycol (PEG) spacer2 or a propargylated aryl system as a spacer.3 Synthetically these compounds were prepared by coupling propargylated amino acid derivatives with the aryliodides.3 Likewise, deoxynucleoside derivatives of N-trifluoroacetyl propargyl amides were synthesized.4As synthetic building blocks, propargyl amides have been employed as monomers for the synthesis of chromophore labeled poly(N-propargyl amides) as stimulus responsive conjugated polymers.5 Optically active N-propargyl amides bearing hydroxyl groups have been synthesized and polymerized for studying their secondary structure and chiral-recognition.6 Furthermore, propargyl amides are excellent substrates in coupling–cycloisomerization sequences7 that can be expanded to three-component syntheses of blue-luminescent 5-(3-indolyl)oxazoles8 and PtCl2 induced intramolecular cyclizations of N-propargyl indole-2-carboxamides to give azepino[3,4-b]indol-1-ones.9
The combination of chemical and enzymatic transformations offers numerous opportunities for designing new syntheses. Therefore, these chemoenzymatic transformations have recently received considerable attention and many applications have been found on chiral resolution of racemic or meso substrates to furnish enantiomerically enriched chiral building blocks for organic syntheses in a catalytic fashion.10 In contrast to chemoenzymatic continuous flow processes,11 the concatenation of enzymatic and chemical catalyzed steps in a one-pot fashion thus furnishing novel types of chemoenzymatic sequences remains a major challenge. While the one-pot combination of enzymes and transition metal catalysis is dominated by dynamic kinetic resolution as an important tool in asymmetric synthesis,12 chemoenzymatic one-pot sequences involving Pd-catalyzed coupling13 or Cu-catalyzed alkyne–azide cycloaddition (CuAAC)14,15 are still in their infancy. Recently, we have disclosed a consecutive three-component sequence consisting of CAL-B (Candida antarctica lipase B) catalyzed aminolysis of methyl esters with propargyl amine furnishing propargyl amides and CuAAC to give amide ligated 1,4-disubstituted 1,2,3-triazoles in good to excellent yields. Here we communicate the first consecutive three-component syntheses of (hetero)arylated propargyl amides by chemoenzymatic aminolysis–Sonogashira coupling sequence.
Results and discussion
We have previously reported on the efficient CAL-B catalyzed aminolysis for the synthesis of propargyl amides as a milder alternative to the base catalyzed process that occurs with a remarkable chemoselectivity with α-heteroatom substituted methyl carboxylates.15 Encouraged by the excellent compatibility of CAL-B catalyzed aminolysis with copper catalysis in a single reaction vessel we set out to concatenate this aminolysis step with Sonogashira coupling to access (hetero)arylated propargyl amides in a diversity-oriented one-pot sequence.First the Sonogashira coupling of propargyl amide 3a, formed by CAL-B aminolysis of methyl ester 1a with propargylamine (2),15 and iodo benzene (4a) to furnish 3-phenylpropargyl amide 5a was optimized as a model reaction with respect to a suitable catalyst system, base, solvent and temperature (Scheme 1, Table 1).
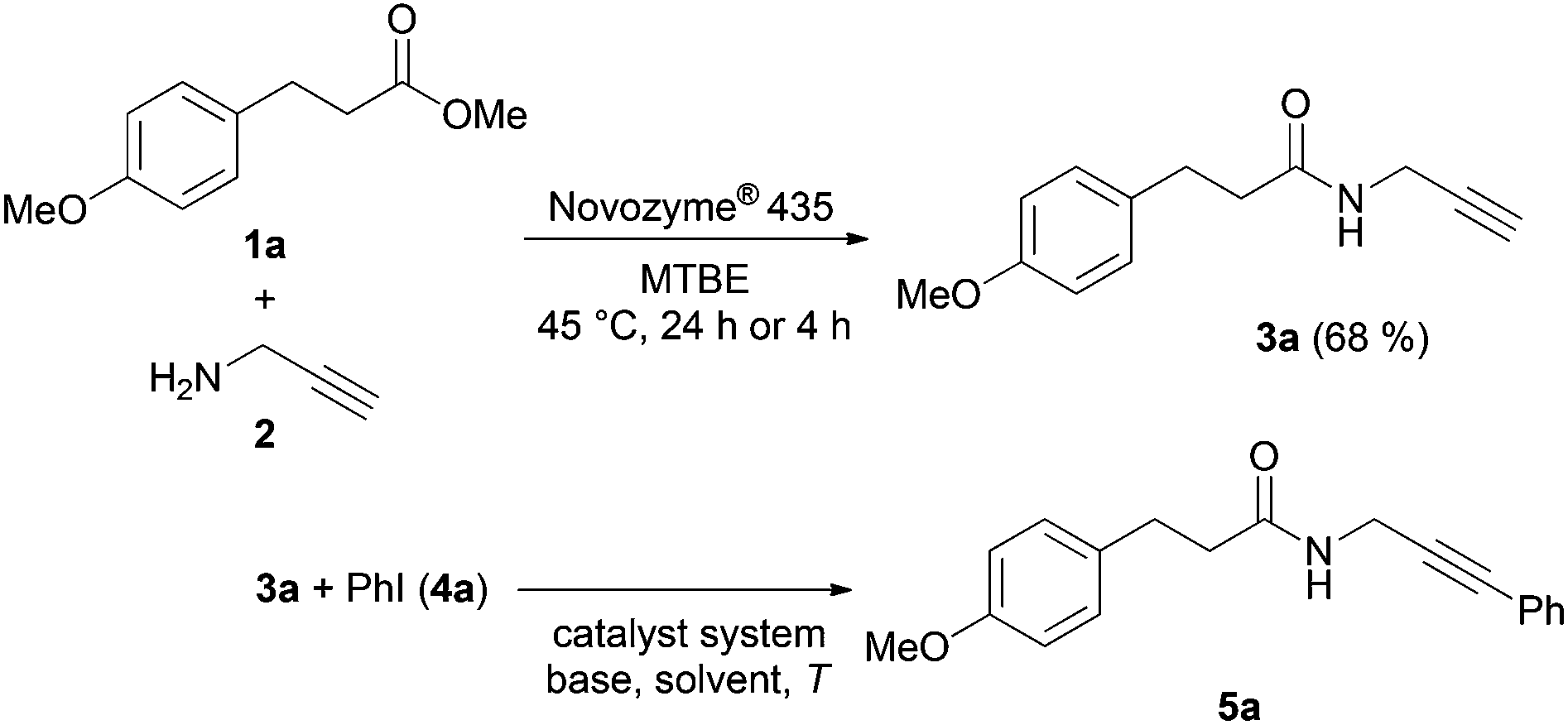 | ||
| Scheme 1 Formation of propargyl amide 3a by CAL-B catalyzed aminolysis of methyl ester 1a and the optimization of the Sonogashira coupling of propargyl amide 3a and iodo benzene (4a). | ||
| Entry | Catalyst system | Solvent | Base/additive | T, t | Yield of 5aa (%) |
|---|---|---|---|---|---|
| a Isolated yield after chromatography on silica gel. b No product formation. c IPr·HCl: 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride. d DIPEA: diisopropylethylamine. e DABCO: 1,4-diazabicyclo[2.2.2]octane. f DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. g TMG: 1,1,3,3-tetramethyl guanidine. | |||||
| 1 | 2 mol% PdCl2(PPh3)2, 4 mol% CuI | THF | NEt3 (1.0 equiv.) | rt to 50 °C, 24 h | —b |
| 2 | 2 mol% PdCl2(PPh3)2, 4 mol% CuI | 1,4-Dioxane | NEt3 (1.0 equiv.) | rt to 70 °C, 24 h | 13 |
| 3 | 2 mol% PdCl2(PPh3)2, 4 mol% CuI | 1,4-Dioxane | Pyrrolidine (1.0 equiv.) | rt to 70 °C, 24 h | 25 |
| 4 | 2 mol% PdCl2(PPh3)2, 4 mol% CuI | 1,4-Dioxane | Pyrrolidine (1.5 equiv.) | rt to 70 °C, 24 h | 33 |
| 5 | 5 mol% Pd(PPh3)4, 1 mol% CuI | DMF | Pyrrolidine/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) :4 v/v) |
45 °C, 6 h | 75 |
| 6 | 2.5 mol% Pd2(dba)3·CHCl3, 10 mol% P(2-furyl)3 | MeCN | NaOt-Bu (2.0 equiv.) | 45 °C, 24 h | —b |
| 7 | 2.5 mol% Pd2(dba)3·CHCl3, 10 mol% IPr·HClc | MeCN | NaOt-Bu (2.0 equiv.) | 45 °C, 24 h | —b |
| 8 | 5 mol% Pd(OAc)2, 10 mol% IPr·HClc | MeCN | K2CO3 (2.0 equiv.), TBAC (2.0 equiv.) | 45 °C, 24 h | —b |
| 9 | 2 mol% Pd(PPh3)4, CuI | DMF | NEt3 (1.0 equiv.) | rt, 24 h | 53 |
| 10 | 2 mol% Pd(PPh3)4, 4 mol% CuI | DMF | DIPEAd (1.5 equiv.) | 45 °C, 6 h | 67 |
| 11 | 2 mol% Pd(PPh3)4, 4 mol% CuI | DMF | Pyrrolidine (1.0 equiv.) | 45 °C, 8 h | 56 |
| 12 | 2 mol% Pd(PPh3)4, 4 mol% CuI | DMF | Pyrrolidine (1.5 equiv.) | 45 °C, 4 h | 85 |
| 13 | 2 mol% Pd(PPh3)4, 4 mol% CuI | DMF | DABCOe (1.0 equiv.) | 45 °C, 8 h | 57 |
| 14 | 2 mol% Pd(PPh3)4, 4 mol% CuI | DMF | DBUf (1.0 equiv.) | 45 °C, 8 h | 69 |
| 15 | 2 mol% Pd(PPh3)4, 4 mol% CuI | DMF | TMG (1.0 equiv.) | 45 °C, 1 h | 83 |
In comparison with standard Sonogashira conditions, propargyl amides are apparently peculiar since all Pd(II) catalyst precursors have failed to give reasonable yields (Table 1, entries 1–4). Even novel carbene ligands that have been efficiently established for Sonogashira coupling16,17 were not successful in the model reaction (Table 1, entries 7 and 8). However, Pd(PPh3)4 as the Pd(0) catalyst precursor was proven to be superior (Table 1, entries 5, 9–15).
The choice of the base was equally important. It turned out that 1,1,3,3-tetramethyl guanidine (TMG), successfully employed in Pd-catalyzed coupling–cyclization syntheses of indoles from ortho-iodo anilines18,19 and in domino sequences involving N-propargyl sulfoximines,20 was the most favorable amidine base that not only gave good yields of propargyl amide 5a but also drastically reduced the reaction times at 45 °C (Table 1, entry 15). Moreover, it is sufficient to employ an equimolar amount of TMG to achieve full conversion. Therefore, with these conditions for the coupling step in hand, the stage was set to combine the CAL-B catalyzed aminolysis with the Sonogashira coupling in a consecutive one-pot fashion.
Upon the Novozyme® 435 catalyzed aminolysis of methyl carboxylates 1 with propargyl amine (2) in MTBE (methyl tert-butylether) at 45 °C, the formed propargyl amide 3 was subsequently reacted with (hetero)aryl iodides 4 in the presence of DMF as a cosolvent, TMG as a base and catalytic amounts of Pd(PPh3)4 and CuI at 45 °C to give 3-(hetero)arylpropargyl amides 5 in a three-component one-pot fashion in moderate to excellent yields (Scheme 2, Table 2).
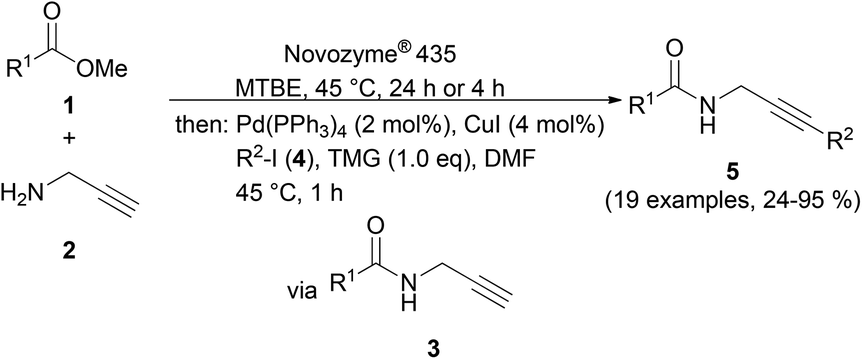 | ||
| Scheme 2 Consecutive three-component synthesis of 3-(hetero)arylpropargyl amides 5 by CAL-B catalyzed aminolysis–Sonogashira coupling sequence. | ||
| Entry | Methyl ester 1 | (Hetero)aryl iodide 4 | 3-(Hetero)aryl propargyl amide 5 |
|---|---|---|---|
| a 24 h time for CAL-B catalyzed aminolysis. b 4 h time for CAL-B catalyzed aminolysis. | |||
| 1a | R1 = p-MeOC6H4CH2CH2 (1a) | R2 = Ph (4a) |
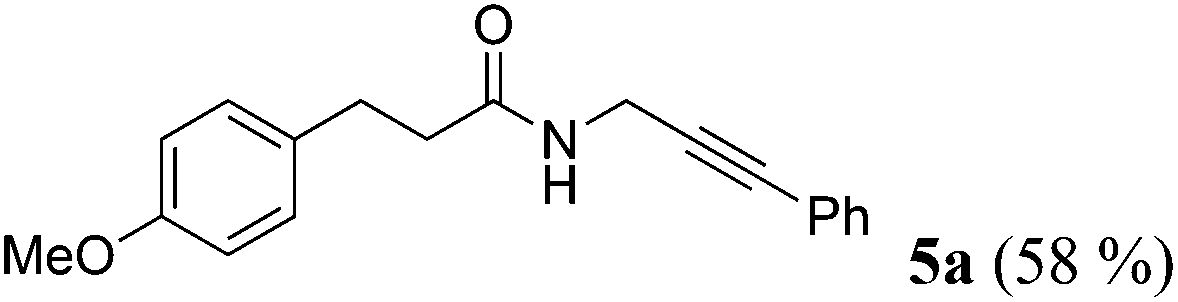
|
| 2a | R1 = C6H5CH2 (1b) | 4a |

|
| 3a | 1b | R2 = p-MeO2CC6H4 (4b) |
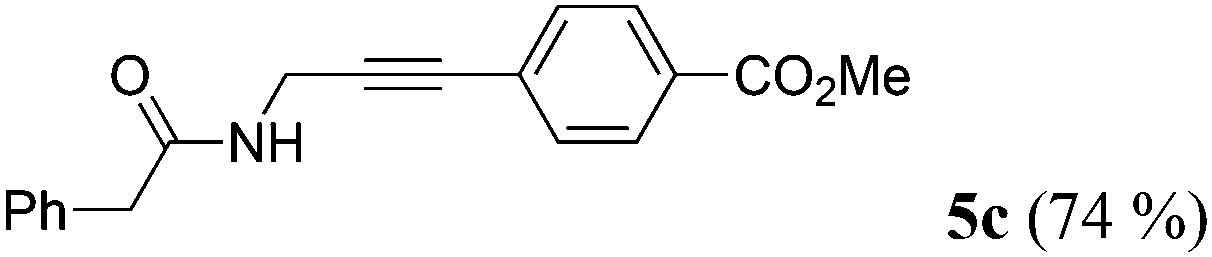
|
| 4a | R1 = C6H5CH2CH2 (1c) | 4a |

|
| 5a | R1 = E-C6H5CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH (1d) CH (1d) |
R2 = p-MeOC6H4 (4c) |
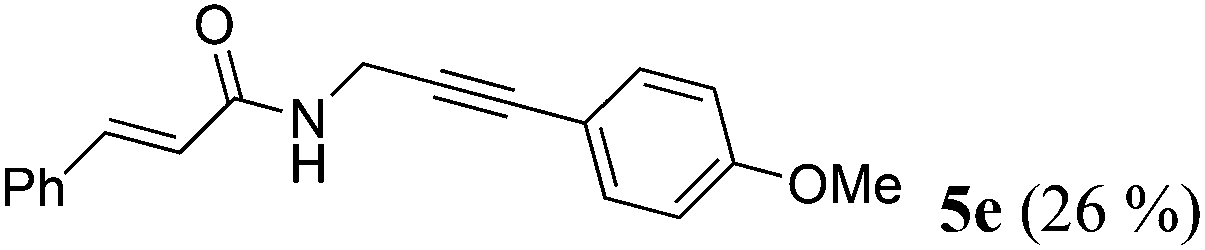
|
| 6a | R1 = C6H5C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (1e) C (1e) |
4a |
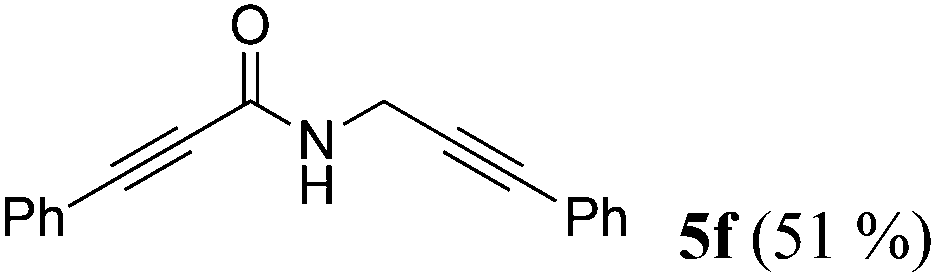
|
| 7b | R1 = C6H5OCH2 (1f) | 4a |
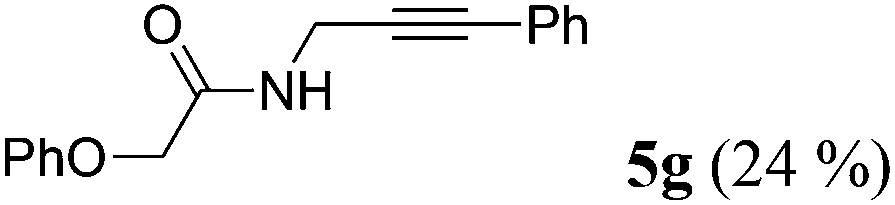
|
| 8b | R1 = C6H5NHCH2 (1g) | 4a |
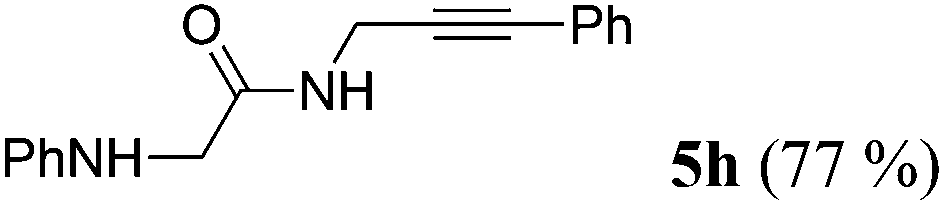
|
| 9b | R1 = C6H5SCH2 (1h) | 4a |
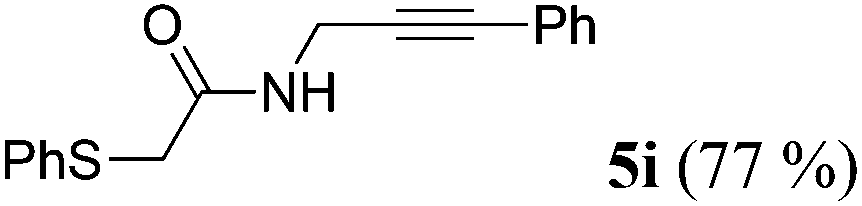
|
| 10a | R1 = p-HOC6H4CH2CH2 (1i) | 4b |
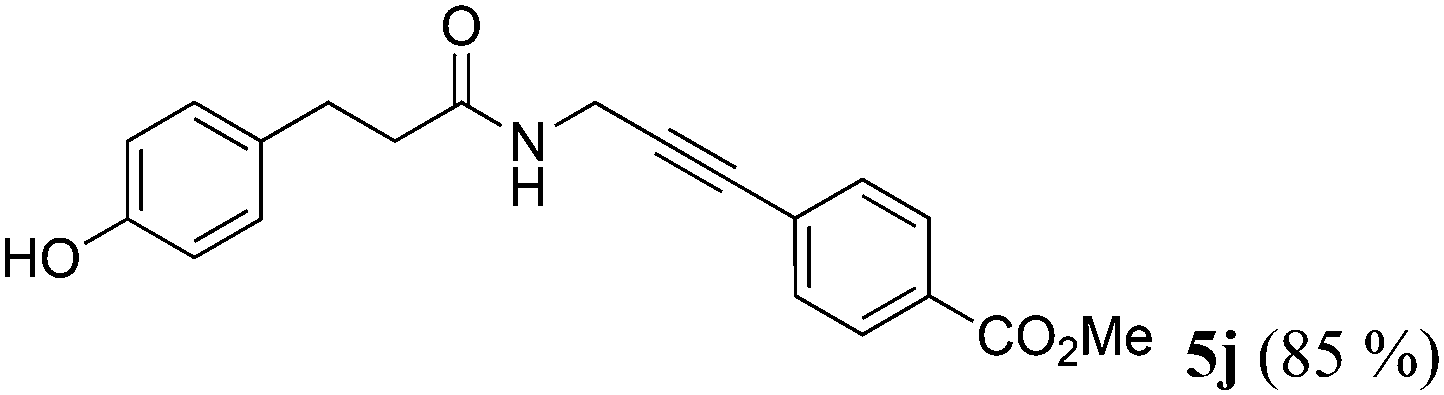
|
| 11a | 1h | 4c |

|
| 12b | R1 = (CH2)5NCH2 (1j) | R2 = p-H3CCOC6H4 (4d) |
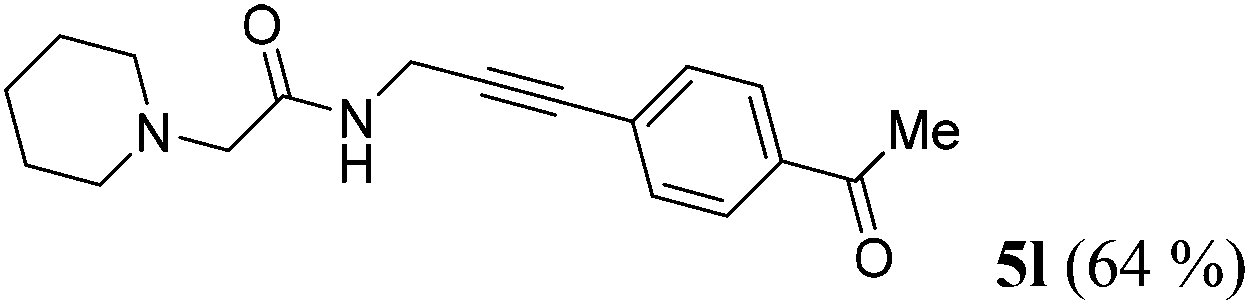
|
| 13a | R1 = Me(CH2)5CH2 (1k) | 4c |
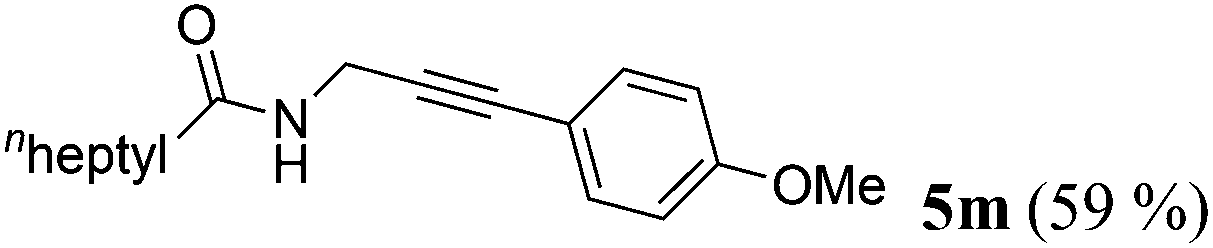
|
| 14a | R1 = F3CCONHCH2 (1l) | 4a |

|
| 15b | R1 = ClCH2 (1m) | 4b |

|
| 16a | R1 = 2-furyl (1n) | R2 = 2-thienyl (4e) |

|
| 17a | R1 = 2-thienyl (1o) | 4e |
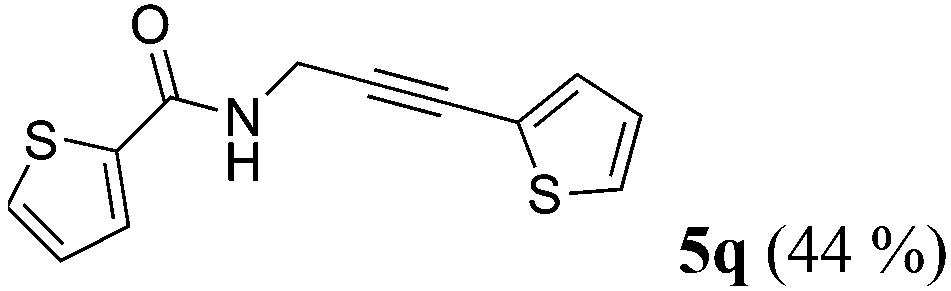
|
| 18a | 1n | R2 = 5-OHC-2-furyl (4f) |
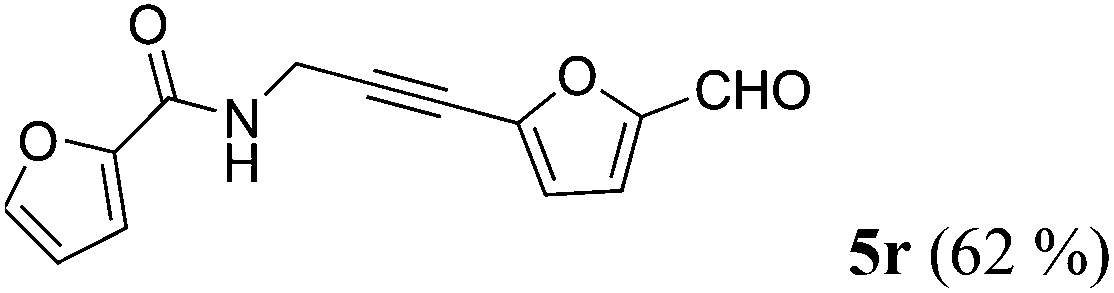
|
| 19b | R1 = p-(MeO2CCH2CH2)C6H4OCH2 (1p) | 4f |
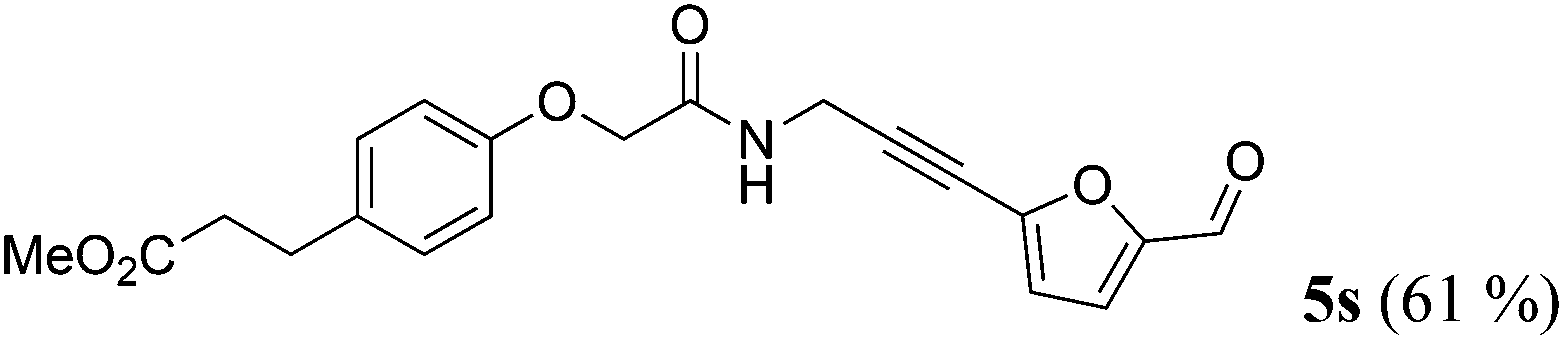
|
Taking into account the already established substrate scope of the Novozyme® 435 catalyzed aminolysis15 the general substitution pattern is equally well accepted in this chemoenzymatic sequence. Moreover, also a free phenol moiety in substrate 1i is well tolerated furnishing the propargyl amide 5j in remarkably high yield (Table 2, entry 10). The same holds true for methyl chloroacetate (1m), which is transformed to give the corresponding propargyl amide 5o in excellent yield (Table 2, entry 15). Most importantly, the chemoselectivity of the aminolysis of the more electrophilic methyl carboxylate in substrate 1p underlines the superiority of applying CAL-B as a catalyst in the sequence. However, it should be noted that 2-iodopyridine and 1,3-diiodobenzene failed to undergo the Sonogashira step under the optimized conditions.
Finally, we have combined the chemoenzymatic aminolysis–Sonogashira coupling with a terminal CuAAC step furnishing propargyl amide functionalized 1-aryl 4-benzyl 1,2,3-triazoles 7 in good yields (Scheme 3). Commencing with the CAL-B catalyzed aminolysis, the propargyl amides 3 are reacted with p-iodo[(trimethylsilyl)ethynyl]benzene (4i) to give the TMS-protected 3(p-ethynyl)phenyl propargyl amides 8, which are rapidly desilylated by potassium fluoride to furnish the ethynyl derivatives 9. The presence of the Sonogashira cocatalyst CuI and benzyl azide (6) terminates the sequence by a CuAAC giving rise to the formation of functionalized 1,2,3-triazoles 7 in the sense of a transition metal catalyzed one-pot sequence.21
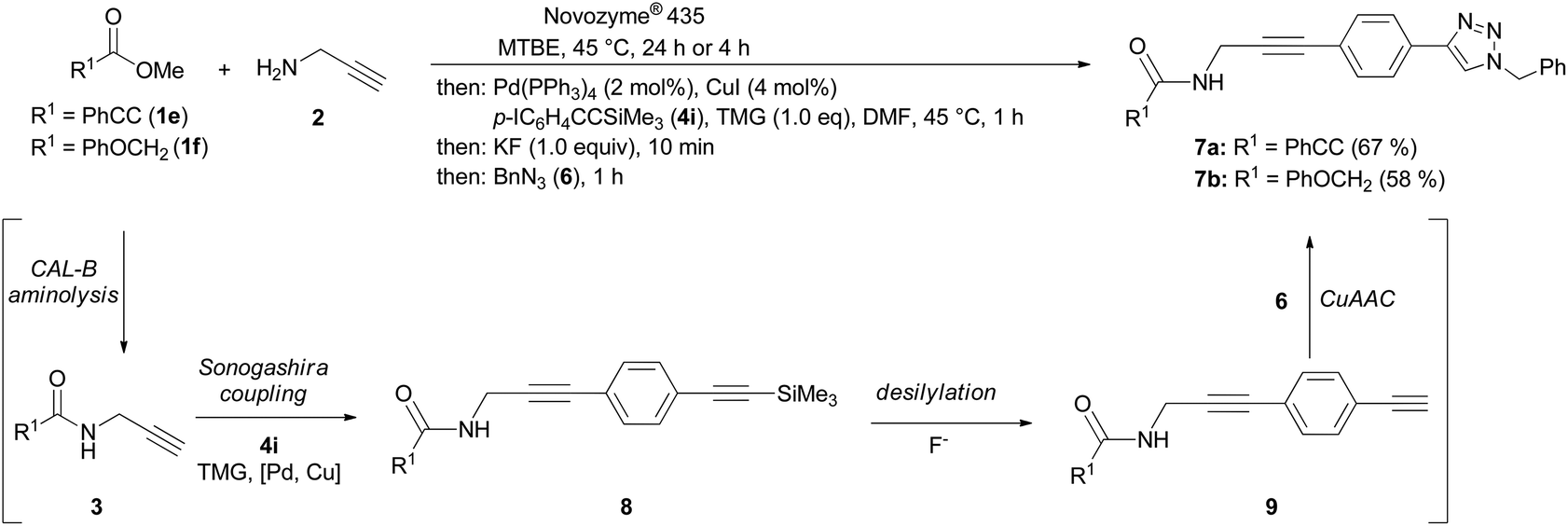 | ||
| Scheme 3 Consecutive four-component synthesis of 3-(4-1,2,3-triazolyl)phenyl propargyl amides 7 by CAL-B catalyzed aminolysis–Sonogashira coupling-CuAAC sequence. | ||
Conclusions
In conclusion, we have disclosed a novel chemoenzymatic one-pot synthesis of 3-(hetero)arylpropargyl amides 5 by CAL-B catalyzed aminolysis–Sonogashira coupling sequence. This combination of enzyme-metal catalyzed methodology is well suited for application to more sophisticated peptides and aryl halides as a bioorganic tool for the efficient generation of peptidomimetics in a one-pot fashion. Furthermore, the potential to concatenate CuAAC as a third step opens up new avenues for the rapid alignment of various functionalities in the sense of diversity-oriented synthesis of complex chromophores.22 Studies directed to further expand and develop this chemoenzymatic sequence are currently underway.Experimental
Chemoenzymatic three-component synthesis of methyl 4-(3-(3-(4-hydroxyphenyl)propanamido)prop-1-yn-1-yl) benzoate (5j) (typical procedure)
To a solution of propargylamine (2) (55 mg, 1.00 mmol) in dry MTBE (2.0 mL) in a screw-cap Schlenk vessel, methyl p-hydroxy dihydrocinnamate (1i) (216 mg, 1.20 mmol) and Novozyme® 435 (108 mg, 50% w/w of substrate 1i) were successively added and the reaction was allowed to shake in an incubating shaker at 45 °C for 24 h. After the complete conversion (monitored by TLC), DMF (2.0 mL) was added to the reaction mixture which was then flushed with argon for 15 min. Then methyl p-iodobenzoate (4b) (262 mg, 1.00 mmol), TMG (115 mg, 1.00 mmol), Pd(PPh3)4 (23 mg, 0.02 mmol), and CuI (8 mg, 0.04 mmol) were successively added to the reaction mixture under argon and the reaction was allowed to shake at 45 °C for 1 h. The reaction mixture was filtered to remove the enzyme beads. Then, brine (5.0 mL) was added to the filtrate, followed by extraction with ethyl acetate (3 × 10.0 mL). The combined organic layers were dried with anhydrous Na2SO4 and after column chromatography on silica gel (n-hexane/ethyl acetate), 288 mg (85%) of analytically pure compound 5j was obtained as a colorless solid, Mp 151 °C.
1H NMR (300 MHz, DMSO-d6): δ = 2.35 (t, 3J = 7.9 Hz, 2 H), 2.72 (t, 3J = 7.9 Hz, 2 H), 3.85 (s, 3 H), 4.13 (d, 3J = 5.5 Hz, 2 H), 6.64 (d, 3J = 8.4 Hz, 2 H), 6.99 (d, 3J4,3 = 8.4 Hz, 2 H), 7.54 (d, 3J = 8.5 Hz, 2 H), 7.94 (d, 3J = 8.5 Hz, 2 H), 8.38 (t, 3J = 5.5 Hz, 1 H), 9.14 (s, 1 H). 13C NMR (75 MHz, DMSO-d6): δ = 28.5 (CH2), 30.1 (CH2), 37.2 (CH2), 52.3 (CH3), 80.7 (Cquat), 90.6 (Cquat), 115.0 (CH), 127.1 (Cquat), 129.0 (CH), 129.1 (Cquat), 129.3 (CH), 131.2 (Cquat), 131.6 (CH), 155.4 (Cquat), 165.6 (Cquat), 171.3 (Cquat). EI-MS (m/z (%)): 337 (M+, 13), 336 ([M − H]+, 22), 230 (C13H12NO3+, 100), 188 (C11H10NO2+, 19), 120 (C8H8O+, 20), 107 (C7H7O+, 52). IR (ATR) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] = 3385 (m), 3291 (m), 2966 (m), 2951 (w), 2924 (w), 2845 (w), 1705 (s), 1632 (s), 1601 (w), 1533 (m), 1516 (s), 1435 (m), 1362 (w), 1344 (m), 1288 (m), 1279 (s), 1259 (m), 1225 (s), 1198 (m), 1175 (m), 1103 (m), 1011 (w), 966 (w), 862 (m), 831 (w), 816 (m), 766 (s), 684 (m), 640 (w). Anal. calcd for C20H19NO4 (337.4): C 71.20, H 5.68, N 4.15; Found: C 70.96, H 5.38, N 4.07.
[cm−1] = 3385 (m), 3291 (m), 2966 (m), 2951 (w), 2924 (w), 2845 (w), 1705 (s), 1632 (s), 1601 (w), 1533 (m), 1516 (s), 1435 (m), 1362 (w), 1344 (m), 1288 (m), 1279 (s), 1259 (m), 1225 (s), 1198 (m), 1175 (m), 1103 (m), 1011 (w), 966 (w), 862 (m), 831 (w), 816 (m), 766 (s), 684 (m), 640 (w). Anal. calcd for C20H19NO4 (337.4): C 71.20, H 5.68, N 4.15; Found: C 70.96, H 5.38, N 4.07.
Chemoenzymatic four-component synthesis of N-(3-(4-(1-benzyl-1H-1,2,3-triazol-4-yl)phenyl)prop-2-yn-1-yl)-2-phenoxy acetamide (7b) (typical procedure)
To a solution of propargylamine (2) (55 mg, 1.00 mmol) in dry MTBE (2.0 mL) in a screw-cap Schlenk vessel, methyl 2-phenoxyacetate (1f) (199 mg, 1.20 mmol) and Novozyme® 435 (100 mg, 50% w/w of substrate 1f) were successively added and the reaction was allowed to shake in an incubating shaker at 45 °C for 4 h. After the complete conversion (monitored by TLC), DMF (2.0 mL) was added to the reaction mixture, which was then flushed with argon for 15 min. Then (4-iodophenyl)ethynyl trimethylsilane (4i) (300 mg, 1.00 mmol), TMG (115 mg, 1.00 mmol), Pd(PPh3)4 (23 mg, 0.02 mmol), and CuI (8 mg,0.04 mmol) were successively added to the reaction mixture under argon and the reaction was allowed to shake at 45 °C for 1 h. Then potassium fluoride (58 mg, 1.00 mmol) was added to the reaction mixture and after 10 min benzyl azide (6) (133 mg, 1.00 mmol) was added. After 1 h of shaking in the incubating shaker the reaction mixture was filtered to remove the enzyme beads. Then, brine (5.0 mL) was added to the filtrate followed by extraction with ethyl acetate (3 × 10.0 mL). The combined organic layers were dried with anhydrous Na2SO4 and after column chromatography on silica gel (n-hexane/ethyl acetate) 245 mg (58%) of analytically pure compound 7b was obtained as a yellow solid, Mp 147 °C.Yellow solid. Mp 147 °C. 1H NMR (300 MHz, d6-DMSO): δ = 4.21 (d, 3J = 5.6 Hz, 2 H), 4.54 (s, 2 H), 5.65 (s, 2 H), 6.97–7.00 (m, 3 H), 7.28–7.39 (br m, 7 H), 7.45 (d, 3J = 8.4 Hz, 2 H), 7.59–7.62 (m, 1 H), 7.86 (d, 3J = 8.4 Hz, 2 H), 8.69 (s, 1 H).13C NMR (75 MHz, d6-DMSO): δ = 28.5 (CH2), 53.1 (CH2), 66.8 (CH2), 81.3 (Cquat), 87.7 (Cquat), 114.7 (CH), 121.2 (CH), 121.5 (Cquat), 122.1 (CH), 125.3 (CH), 127.9 (CH), 128.2 (CH), 129.5 (CH), 130.7 (Cquat), 132.0 (CH), 135.9 (Cquat), 145.9 (Cquat), 157.6 (Cquat), 167.7 (Cquat). MALDI-MS: m/z = 423 ([M + H]+). IR (ATR) [cm−1] = 3034 (w), 2922 (w), 2910 (w), 2856 (w), 1662 (s), 1598 (w), 1587 (w), 1519 (m), 1489 (s), 1456 (w), 1435 (w), 1409 (w), 1350 (w), 1286 (w), 1242 (s), 1226 (s), 1170 (w), 1080 (w), 1060 (w), 1047 (w), 1028 (w), 1018 (w), 1001 (w), 835 (m), 798 (m), 756 (s), 721 (s), 657 (w), 603 (w). Anal. calcd for C26H22N4O2 (422.5): C 73.92, H 5.25, N 13.26; Found: C 74.06, N 5.32, H 13.47.
Acknowledgements
The authors cordially thank CLIB Graduate Cluster for the financial support (scholarship of S.H.) and the Fonds der Chemischen Industrie.Notes and references
- C. Arkona and J. Rademann, Angew. Chem., Int. Ed., 2013, 52, 2 CrossRef PubMed.
- K. M. Bonger, R. J. B. H. N. van der Berg, L. H. Heitman, A. P. IJzerman, J. Oosterom, C. M. Timmers, H. O. Overkleeft and G. A. van der Marel, Bioorg. Med. Chem., 2007, 15, 4841 CrossRef CAS PubMed.
- K. M. Bonger, R. J. B. H. N. van den Berg, A. D. Knijnenburg, L. H. Heitman, A. P. IJzerman, J. Oosterom, C. M. Timmers, H. S. Overkleefta and G. A. van der Marel, Bioorg. Med. Chem., 2008, 16, 3744 CrossRef CAS PubMed.
- N. K. Garg, C. C. Woodroofe, C. J. Lacenere, S. R. Quake and B. M. Stoltz, Chem. Commun., 2005, 4551 RSC.
- R. Nomura, K. Yamada and T. Masuda, Chem. Commun., 2002, 478 RSC.
- F. Sanda, T. Fujii, J. Tabei, M. Shiotsuki and T. Masuda, Macromol. Chem. Phys., 2008, 209, 112 CrossRef CAS.
- (a) E. Merkul and T. J. J. Müller, Chem. Commun., 2006, 4817 RSC; (b) E. Merkul, O. Grotkopp and T. J. J. Müller, Synthesis, 2009, 502 CAS; (c) E. Merkul, C. Boersch, W. Frank and T. J. J. Müller, Org. Lett., 2009, 11, 2269 CrossRef CAS PubMed.
- O. Grotkopp, A. Ahmad, W. Frank and T. J. J. Müller, Org. Biomol. Chem., 2011, 9, 8130 CAS.
- M. Gruit, A. P. Davtyan and M. Beller, Org. Biomol. Chem., 2011, 9, 1148–1159 CAS.
- For a recent monograph, see e.g. Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, 3rd edn, 2012 Search PubMed.
- For coupled chemo(enzymatic) reactions in continuous flow, see e.g. R. Yuryev, S. Strompen and A. Liese, Beilstein J. Org. Chem., 2011, 7, 1449 CrossRef CAS PubMed.
- For reviews, see e.g. (a) O. Pàmies and J.-E. Bäckvall, Chem. Rev., 2003, 103, 3247 CrossRef PubMed; (b) J. H. Lee, K. Han, M.-J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999 CrossRef CAS; (c) P. Hoyos, V. Pace and A. R. Alcántara, Adv. Synth. Catal., 2012, 354, 2585 CrossRef CAS.
- E. Burda, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2008, 47, 9551 CrossRef CAS PubMed.
- A. Cuetos, F. R. Bisogno, I. Lavandera and V. Gotor, Chem. Commun., 2013, 49, 2625 RSC.
- S. Hassan, R. Tschersich and T. J. J. Müller, Tetrahedron Lett., 2013, 54, 4641 CrossRef CAS PubMed.
- A. Saito, K. Iimura and Y. Hanzawa, Tetrahedron Lett., 2010, 51, 1471 CrossRef CAS PubMed.
- Y.-M. Pan, F.-J. Zheng, H.-X. Lin and Z.-P. Zhan, J. Org. Chem., 2009, 74, 3148 CrossRef CAS PubMed.
- M. C. Fagnola, I. Candiani, G. Visentin, W. Cabri, F. Zarini, N. Mongelli and A. Bedeschi, Tetrahedron Lett., 1997, 38, 2307 CrossRef CAS.
- A. L. Smith, G. I. Stevenson, C. J. Swain and J. L. Castro, Tetrahedron Lett., 1998, 39, 8317 CrossRef CAS.
- R. F. Schumacher, A. Honraedt and C. Bolm, Eur. J. Org. Chem., 2012, 3737 CrossRef CAS.
- T. J. J. Müller, Top. Organomet. Chem., 2006, 19, 149 CrossRef.
- T. J. J. Müller and D. M. D'Souza, Pure Appl. Chem., 2008, 80, 609 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, and 1H and 13C NMR spectra of compounds 5 and 7. See DOI: 10.1039/c4ob02386b |
| This journal is © The Royal Society of Chemistry 2015 |