
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and antimalarial evaluation of amide and urea derivatives based on the thiaplakortone A natural product scaffold†
Brett D.
Schwartz
a,
Tina S.
Skinner-Adams
a,
Katherine T.
Andrews
a,
Mark J.
Coster
a,
Michael D.
Edstein
b,
Donna
MacKenzie
b,
Susan A.
Charman
c,
Maria
Koltun
c,
Scott
Blundell
c,
Anna
Campbell
c,
Rebecca H.
Pouwer
a,
Ronald J.
Quinn
a,
Karren D.
Beattie
a,
Peter C.
Healy
a and
Rohan A.
Davis
*a
aEskitis Institute for Drug Discovery, Griffith University, Brisbane, QLD 4111, Australia. E-mail: r.davis@griffith.edu.au; Tel: +61-7-3735-6043
bAustralian Army Malaria Institute, Brisbane, QLD 4051, Australia
cCentre for Drug Candidate Optimisation, Monash University, 381 Royal Parade, Parkville, VIC 3052, Australia
First published on 26th November 2014
Abstract
A series of amide (8–32, 40–45) and urea (33, 34, 36–39) analogues based on the thiaplakortone A natural product scaffold were synthesised and screened for in vitro antimalarial activity against chloroquine-sensitive (3D7) and chloroquine- and mefloquine-resistant (Dd2) Plasmodium falciparum parasite lines. Several analogues displayed potent inhibition of P. falciparum growth (IC50 <500 nM) and good selectivity for P. falciparum versus human neonatal foreskin fibroblast cells (selectivity index >100). Two of these compounds, 8 and 33, exhibited good aqueous solubility and metabolic stability, and when administered subcutaneously to mice (32 mg kg−1), plasma concentrations remained above 0.2 μM for at least 8 h. Both 8 and 33 were well tolerated in mice after subcutaneous administration of 32 mg kg−1 twice daily for 4 days. Using this regimen blood stage P. berghei was suppressed by 52% for 8 and 26% for 33, relative to the vehicle control.
Introduction
Malaria is an infectious disease caused by parasites belonging to the genus Plasmodium and is a major health and economic problem globally. An estimated 3.2 billion people living in tropical and subtropical regions of the world, such as Sub-Saharan Africa, Central and South America, the Middle East, India and South East Asia are at risk from this disease.1 The World Health Organisation estimated that in 2010 there were 207 million clinical cases and 627![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths due to malaria, with more than 90% of malaria cases and the majority of malaria deaths occurring in Sub-Saharan Africa.2 Despite recent data on a partially effective pre-erythrocytic stage vaccine (RTS,S),3,4 malaria prevention and treatment currently relies on small molecule drugs and vector control. It is predicted that even if the RTS,S vaccine is approved for clinical use, it will be implemented as part of a multi-pronged approach that will also include antimalarial drugs. Whilst a number of drugs are used therapeutically to treat malaria, all have now succumbed to parasite drug resistance or display reduced efficacy.5–8 Consequently, new antimalarial drugs with novel targets are urgently needed in order to combat the global problem of parasite drug resistance.
000 deaths due to malaria, with more than 90% of malaria cases and the majority of malaria deaths occurring in Sub-Saharan Africa.2 Despite recent data on a partially effective pre-erythrocytic stage vaccine (RTS,S),3,4 malaria prevention and treatment currently relies on small molecule drugs and vector control. It is predicted that even if the RTS,S vaccine is approved for clinical use, it will be implemented as part of a multi-pronged approach that will also include antimalarial drugs. Whilst a number of drugs are used therapeutically to treat malaria, all have now succumbed to parasite drug resistance or display reduced efficacy.5–8 Consequently, new antimalarial drugs with novel targets are urgently needed in order to combat the global problem of parasite drug resistance.
In recent efforts to discover new antimalarial leads from nature, a high-throughput antimalarial screening campaign using a pre-fractionated natural product library9,10 identified four novel antimalarial thiazine-derived alkaloids, thiaplakortones A–D (1–4; Fig. 1), from the Australian marine sponge Plakortis lita.11 Thiaplakortone A (1) was the most active natural product with IC50 values of 51 and 6.6 nM against chloroquine-sensitive (3D7) and multi-drug resistant (Dd2) P. falciparum malaria parasite lines, respectively.11
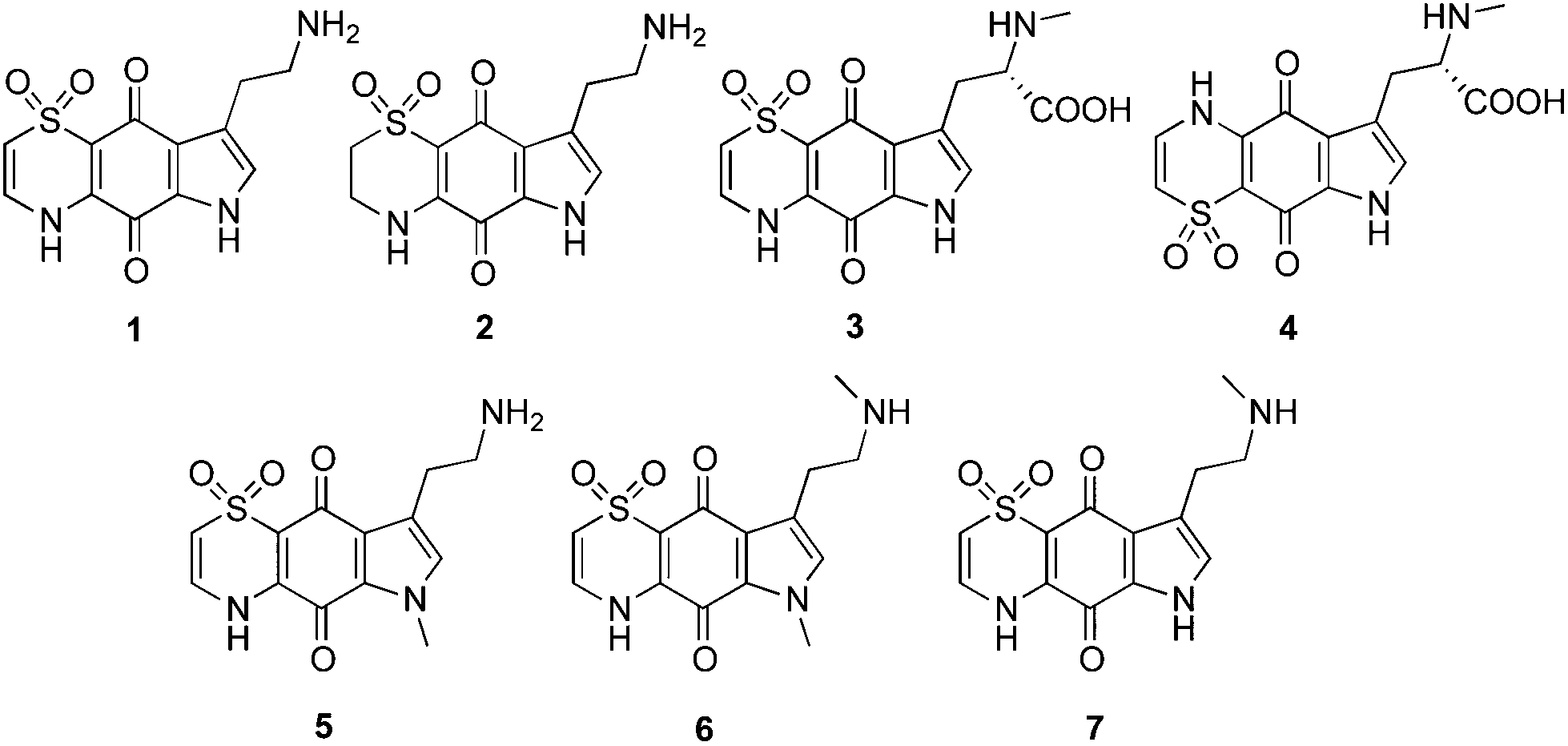 | ||
| Fig. 1 Chemical structures of the natural products thiaplakortones A–D (1–4) and previously synthesised methyl analogues (5–7). | ||
The first total synthesis of 1, along with a series of mono- and di-methyl analogues (5–7), has been reported and some preliminary structure–activity relationships ascertained.12 While in vivo toxicity effects for several of the synthetic compounds indicated potential liabilities associated with this structure class, the limited number of analogues investigated made it difficult to assess their true potential as antimalarial leads. To more thoroughly explore this compound class we generated a series of amide and urea analogues based on the thiaplakortone A scaffold. Herein we report the synthesis of these compounds (8–46) along with their in vitro antimalarial and cytotoxicity activities, in vivo tolerability, ADME profiles and antimalarial efficacy in a murine malaria model.
Results and discussion
Chemistry
With the synthetic route for the lead molecule thiaplakortone A previously optimised,12 suitable quantities of the natural product were available to commence more extensive structure–activity investigations. To address the possible toxicity associated with the primary amino group of thiaplakortone A, initial analogues focused on replacement of this motif with a series of amides and ureas.The amide series (8–32) was accessed through established procedures, by either exposure of thiaplakortone A to acid anhydrides, acyl chlorides or amide formation under standard HBTU protocols (Scheme 1).13 On scale-up, methods using HBTU were avoided, since yields were poor and purification was not trivial. These shortcomings were addressed by reaction of 1 with carbonyl imidazoles,14 that allowed for enhanced yields and undemanding purifications. During the course of these studies, single crystal X-ray structures were obtained on the hydrochloride salt of the natural product 1 and its propyl amide analogue 9. These data represent the first X-ray structures of this class and confirmed the previously reported NMR-based assignment11 of the thiazine ring regiochemistry (Fig. 2).
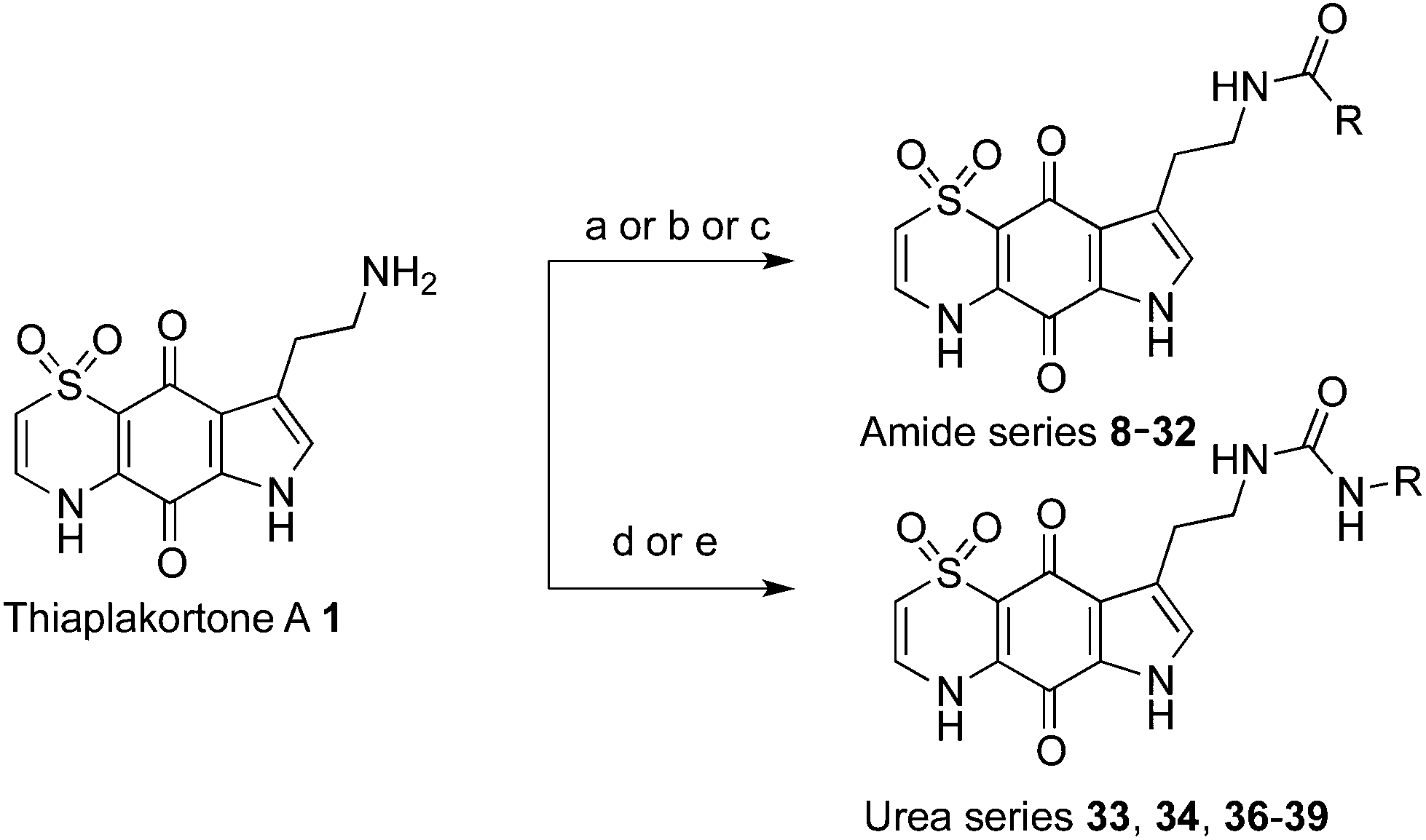 | ||
| Scheme 1 Synthesis of thiaplakortone A amide and urea analogues. Reagents and conditions: (a) acid anhydride, pyridine, dioxane, rt; (b) HBTU, DIPEA, carboxylic acid, DMF; (c) (i) carbonyl diimidazole, carboxylic acid, THF, reflux; (ii) DIPEA, DMF, rt; (d) DIPEA, isocyanate, DMF, 0 °C; (e) carbamoyl imidazole, DIPEA, DMF, rt. | ||
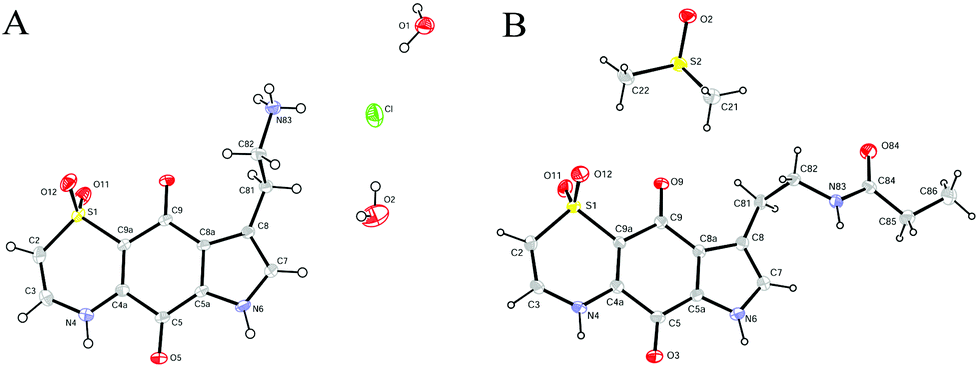 | ||
| Fig. 2 ORTEP diagrams for (A) the hydrochloride salt of thiaplakortone A (1) and (B) the propylamide analogue (9). | ||
Synthesis of analogues (33–34, 36–39) in the urea series was accomplished by treatment of thiaplakortone A with commercially available isocyanates. In the case of compounds 37 and 39 these molecules were synthesised using their respective carbamoyl imidazoles (Scheme 1).15,16
The indole-N-alkylated acetamides 40–45 were accessed from either indole-N-alkylation of Boc-protected thiaplakortone A (35)12 followed by subsequent deprotection and acetamide formation with N-acetyl imidazole or from direct alkylation of the acetamide 8 (Scheme 2). Compound 40 was accessed from acetylation of the previously reported N-methyl analogue 512 with N-acetylimidazole.
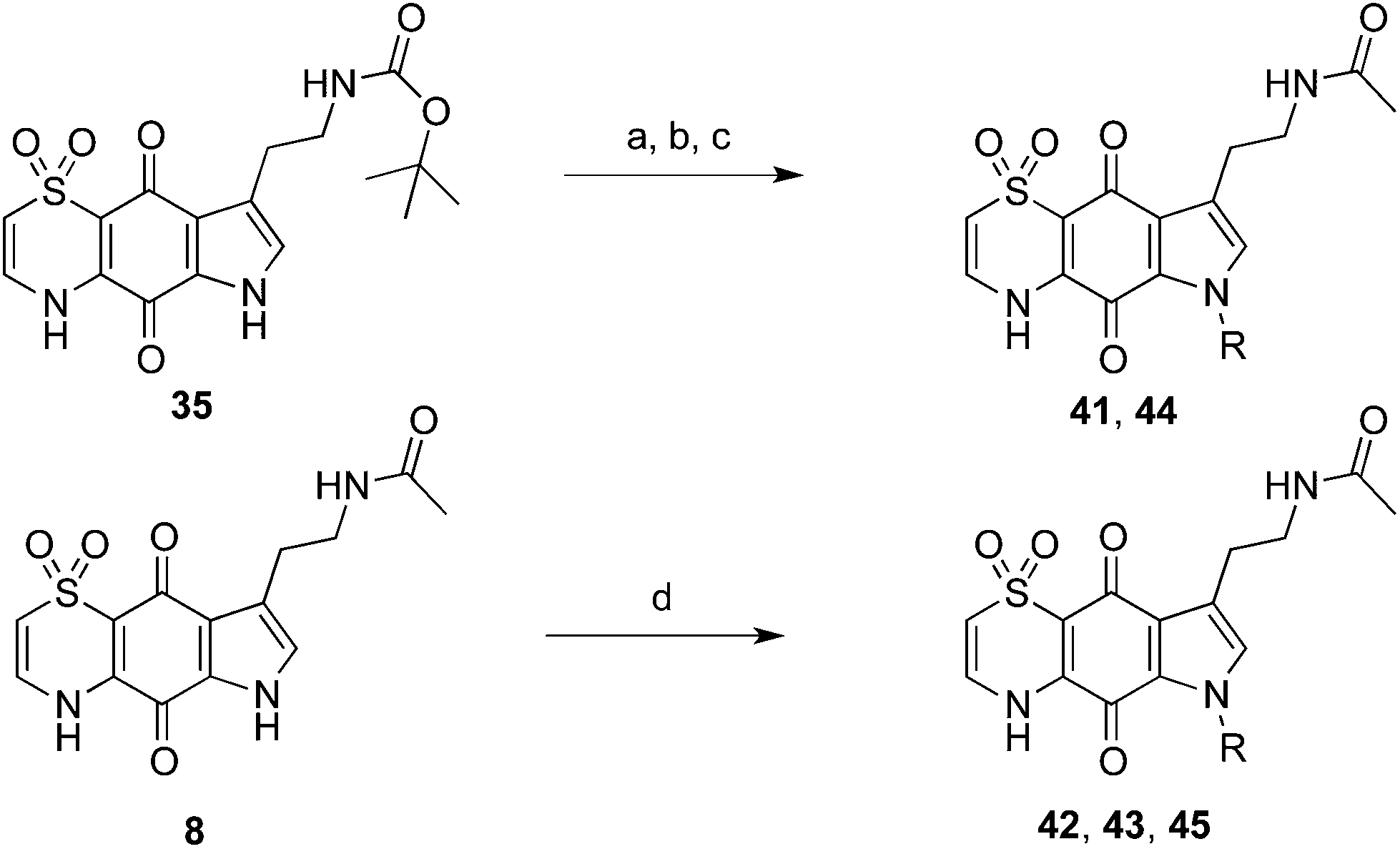 | ||
| Scheme 2 Synthesis of thiaplakortone A acetamide analogues. Reagents and conditions: (a) KOH, DMSO, alkyl bromide, rt; (b) (i) HCl/dioxane, 0 °C to 40 °C; (ii) NH3(aq.) (c) N-acetylimidazole, DMF; (d) KOH, DMSO, alkyl bromide, rt. | ||
In vitro antimalarial activity and mammalian cell cytotoxicity
Synthetic analogues 8–45 were all evaluated for in vitro antimalarial activity against the chloroquine-sensitive 3D7 and the multidrug-resistant Dd2 P. falciparum lines. To compare the selectivity of the compounds for malaria parasites versus normal mammalian cells, cytotoxicity tests were carried out using the human neonatal foreskin fibroblast (NFF) cell line. All biological data, including selectivity indices (SI) are detailed in Tables 1–3.|
|
Mean IC50 ± SD (nM) | SId | ||||
|---|---|---|---|---|---|---|
| 3D7a | Dd2b | NFFc | 3D7 | Dd2 | ||
| a 3D7 = P. falciparum chloroquine-sensitive line. b Dd2 = P. falciparum multi-drug-resistant line. c NFF = neonatal foreskin fibroblast cells. d SI = selectivity index = NFF cell-line IC50/P. falciparum IC50. e CQ = chloroquine (positive control). | ||||||
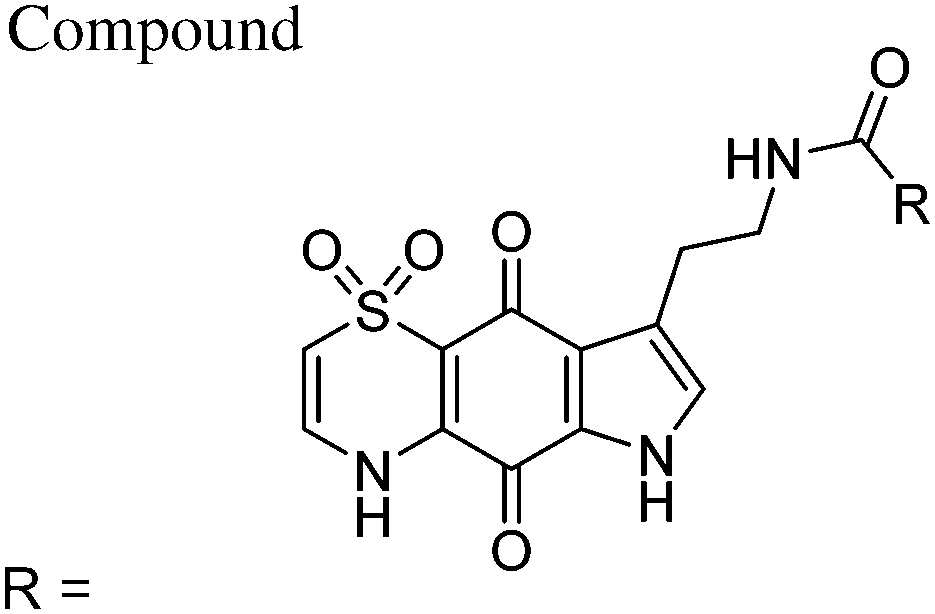
| 8 | Methyl | 472 ± 102 | 182 ± 17.8 | 46400 ± 12700 |
98 | 255 |
| 9 | Ethyl | 703 ± 177 | 420 ± 50 | 47000 ± 12000 |
67 | 112 |
| 10 | n-Propyl | 439 ± 158 | 373 ± 41 | 29000 ± 15000 |
66 | 78 |
| 11 | c-Propyl | 1139 ± 249 | 783 ± 63 | 37600 ± 2070 |
33 | 48 |
| 12 | i-Propyl | 537 ± 3 | 473 ± 129 | 76800 ± 15200 |
143 | 162 |
| 13 | n-Butyl | 644 ± 261 | 525 ± 98 | 37000 ± 16000 |
57 | 70 |
| 14 | i-Butyl | 537 ± 68 | 565 ± 64 | 43600 ± 16000 |
81 | 77 |
| 15 | Cyclopentylmethyl | 604 ± 27 | 408 ± 54 | 24500 ± 3900 |
40 | 60 |
| 16 | Phenyl | 582 ± 99 | 330 ± 71 | 40800 ± 2000 |
70 | 124 |
| 17 | 2-Fluorophenyl | 445 ± 37 | 478 ± 80 | 36100 ± 17100 |
81 | 76 |
| 18 | 3-Fluorophenyl | 1260 ± 137 | 1120 ± 329 | 40400 ± 6275 |
34 | 37 |
| 19 | 3-Bromophenyl | 1423 ± 320 | 888 ± 101 | 34900 ± 6550 |
24 | 39 |
| 20 | 4-Fluorophenyl | 1290 ± 253 | 805 ± 153 | 77300 ± 18380 |
60 | 96 |
| 21 | 2,5-Difluorophenyl | 1300 ± 144 | 986 ± 218 | 32724 ± 4633 |
25 | 33 |
| 22 | 2,6-Difluorophenyl | 1090 ± 39 | 821 ± 185 | 73290 ± 4955 |
67 | 89 |
| 23 | 5-Fluoro-2-morpholinophenyl | 1210 ± 128 | 1630 ± 819 | 82500 ± 841 |
68 | 51 |
| 24 | 2-(2-Fluorophenyl)acetyl | 1300 ± 176 | 909 ± 188 | 72120 ± 8015 |
55 | 79 |
| 25 | 2-Fluoro-5-(trifluoromethyl)phenyl | 1957 ± 203 | 1490 ± 167 | 42792 ± 5587 |
22 | 29 |
| 26 | 3-Chloro-4-methoxyphenyl | 925 ± 227 | 619 ± 66 | 39600 ± 13740 |
43 | 64 |
| 27 | 2-Hydroxy-4-methoxyphenyl | 1412 ± 160 | 1089 ± 230 | 35400 ± 2500 |
25 | 32 |
| 28 | 3-(4-Methoxyphenyl)propyl | 1633 ± 275 | 848 ± 235 | 61300 ± 4300 |
38 | 72 |
| 29 | Isoquinolin-1-yl | 824 ± 136 | 728 ± 203 | >100000 |
>121 | >137 |
| 30 | Furan-3-yl | 1079 ± 618 | 509 ± 308 | 63600 ± 6000 |
59 | 125 |
| 31 | (S)-5-Oxopyrrolidin-2-yl | 1120 ± 380 | 700 ± 215 | 14000 ± 2000 |
13 | 20 |
| 32 | 1H-Indol-2-yl | 886 ± 324 | 378 ± 24 | 33900 ± 4100 |
38 | 90 |
| CQ | 12 ± 4 | 109 ± 60 | 46000 ± 14000 |
3833 | 422 | |
|
|
Mean IC50 ± SD (nM) | SId | ||||
|---|---|---|---|---|---|---|
| 3D7a | Dd2b | NFFc | 3D7 | Dd2 | ||
| a 3D7 = P. falciparum chloroquine-sensitive line. b Dd2 = P. falciparum multi-drug-resistant line. c NFF = neonatal foreskin fibroblast cells. d SI = selectivity index = NFF cell-line IC50/P. falciparum IC50. e CQ = chloroquine (positive control). | ||||||
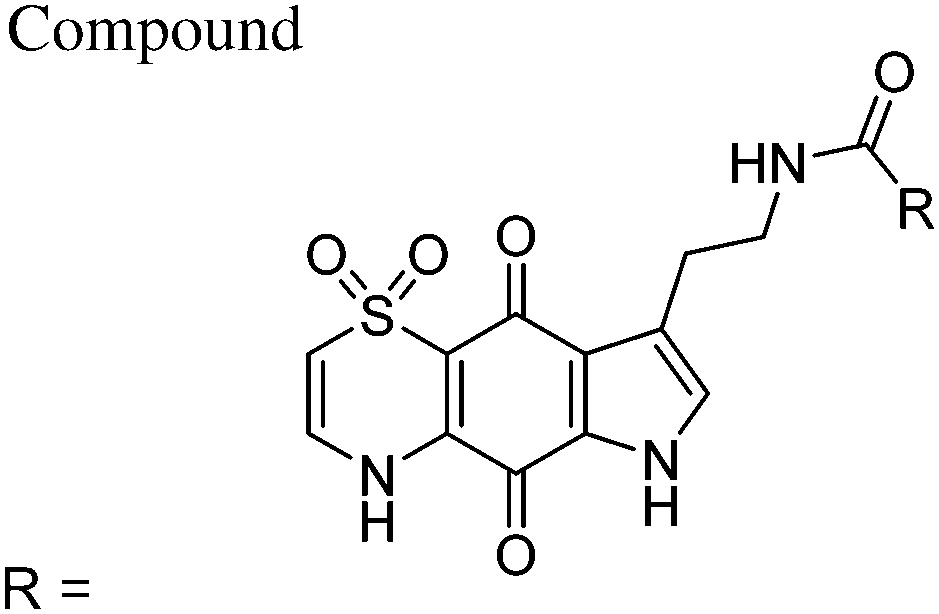
| 33 | N(H)n-Bu | 709 ± 159 | 495 ± 119 | 73000 ± 21000 |
103 | 147 |
| 34 | N(H)t-Bu | 685 ± 101 | 787 ± 97 | >100000 |
>145 | >127 |
| 35 | Ot-Bu | 753 ± 105 | 418 ± 88 | 17800 ± 2000 |
24 | 46 |
| 36 | N(H)-3,4-dimethoxybenzyl | 1110 ± 146 | 1100 ± 58 | >100000 |
>90 | >91 |
| 37 | N(Me)OMe | 741 ± 127 | 618 ± 16 | 57900 ± 21800 |
78 | 94 |
| 38 | N(H)CH2CO2Et | 663 ± 98 | 540 ± 85 | >100000 |
>150 | >185 |
| 39 | N(H)-adamant-2-yl | 631 ± 328 | 529 ± 194 | 63000 ± 8000 |
100 | 119 |
| CQ | 12 ± 4 | 109 ± 60 | 46000 ± 14000 |
3833 | 422 | |
|
|
Mean IC50 ± SD (nM) | SId | ||||
|---|---|---|---|---|---|---|
| 3D7a | Dd2b | NFFc | 3D7 | Dd2 | ||
| a 3D7 = P. falciparum chloroquine-sensitive line. b Dd2 = P. falciparum multi-drug-resistant line. c NFF = neonatal foreskin fibroblast cells. d SI = selectivity index = NFF cell-line IC50/P. falciparum IC50. e CQ = chloroquine (positive control). | ||||||
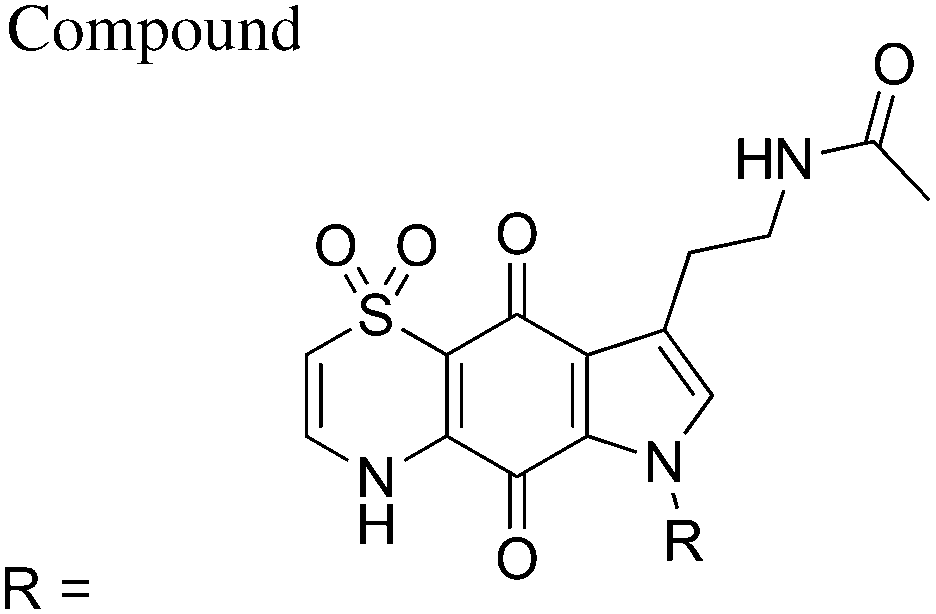
| 40 | Methyl | 684 ± 104 | 706 ± 146 | >100000 |
>146 | >142 |
| 41 | Isopropyl | 937 ± 160 | 845 ± 329 | >100000 |
>107 | >118 |
| 42 | Cyclopentyl | 933 ± 149 | 666 ± 86 | >100000 |
>107 | >150 |
| 43 | (3-Methyloxetan-3-yl)methyl | 822 ± 56 | 770 ± 211 | >100000 |
>122 | >130 |
| 44 | Benzyl | 441 ± 69 | 316 ± 55 | 78000 ± 10000 |
178 | 246 |
| 45 | 4-Chlorobenzyl | 635 ± 99 | 564 ± 136 | >100000 |
>158 | >178 |
| CQ | 12 ± 4 | 109 ± 60 | 46000 ± 14000 |
3833 | 422 | |
Structure–activity relationships
In regards to the amide analogues (8–32), the series of aliphatic amides showed minimal differences in potency and selectivity as the aliphatic chain increased in length or branching. For instance, replacing the acetamide side chain in 8 with an n-pentylamide unit in 13 resulted in a 1.3-fold and 2.8-fold decrease in potency against the 3D7 and Dd2 lines, respectively. The SI for the straight chain amides (8–10, 13) clearly showed a decreasing trend as the chain was extended from C2 to C5; for example the C2 amide 8 displayed 3.6-fold (Dd2 line) and 1.7-fold (3D7 line) better SI values compared to the C5 amide 13. While substitution with cyclic sidechains, as in the cyclopropyl and cyclobutyl derivatives 11 and 15, respectively, showed no improvement to the potency or selectivity, the isopropyl and isobutyl amides, 12 and 14, appeared to improve selectivity. Substituting the aliphatic amide motif with benzamide structures, such as those found in 16–28, typically resulted in less active antimalarial compounds and lower SI values. The urea/carbamate series (33–39), whilst still displaying in vitro P. falciparum growth inhibition activity, and showing SI values comparable to some of the better amide analogues, did not result in the identification of any significantly potent compounds. Several indole-N-alkylacetamide analogues (40–45) were synthesised in order to determine if N-substitution of the indoloquinone improved the parasite growth inhibition activity and/or selectivity. Analogues 40–45 were based on the acetamide scaffold 8, which had been shown to display the best antimalarial activity and selectivity of the amide series. The indole-N-benzyl analogue 44 was shown to have similar or slightly better antimalarial activity and selectivity profiles towards the 3D7 and Dd2 lines compared to the acetamide analogue 8, however the additional synthetic steps and lower yields associated with 44 precluded this compound from further evaluation.In vitro profiling of several analogues towards an atovaquone-resistant Plasmodium falciparum line (C2B)
Atovaquone is a synthetic quinone-based antimalarial drug, which is typically co-administered with proguanil for the treatment of P. falciparum infection. The mechanism of action for atovaquone involves the inhibition of the cytochrome bc1 complex in Plasmodium parasites.17–19 As the thiaplakortone scaffold also contains a quinone motif we examined the activity of several of our more potent compounds, which included the synthesised natural products (1 and 2), along with the amide (8 and 16) and urea analogues (33 and 38) against an atovaquone-resistant P. falciparum line (C2B).20–22In vitro screening results using the C2B line showed that 1, 2, 8, 16, 33 and 38 had IC50 values of 52 ± 9.8, 377 ± 49, 324 ± 70, 286 ± 26, 383 ± 108, and 331 ± 124 nM, respectively. These C2B data when compared to the 3D7 strain (Tables 1 and 2) showed only minimal differences, with most compounds displaying slightly more activity towards the C2B line (cf. to 3D7). Compound 39 showed the largest IC50 difference, however this equated to only a 2.0-fold change in IC50 value. These data indicate that the thiaplakortone structure class does not affect Plasmodium parasite growth via inhibition of the cytochrome bc1 complex.ADME and pharmacokinetic profiling
A series of eight compounds was assessed for physicochemical (Table 4) and metabolic stability characteristics (Table 5). All compounds exhibited high total polar surface area values in excess of 120 Å2 and a high number of hydrogen bond accepting groups, suggesting that absorption following oral administration would likely be compromised. With the exception of 32 and 39, all compounds had relatively low logD7.4 values (0.3–1.3) and the majority had good aqueous solubility (50–100 μg mL−1 or higher) at pH values representative of the gastric (pH 2.0) and intestinal (pH 6.5) environments. For 32 and 39, logD7.4 values were higher (1.9 and 2.3, respectively) and aqueous solubility values were poor at both pH 2 and 6.5 (<12.5 μg mL−1).
| Compd | PSAa (Å2) | FRBb | HBD/HBAc | LogD7.4 |
Solubility (μg mL−1) pH 2.0/6.5 |
|---|---|---|---|---|---|
| a Total polar surface area. b Freely rotatable bonds. c Hydrogen bond donors/hydrogen bond acceptors. | |||||
| 8 | 125 | 3 | 3/8 | 0.3 | >100/>100 |
| 16 | 125 | 4 | 3/8 | 1.3 | 25–50/25–50 |
| 32 | 141 | 4 | 4/9 | 1.9 | 12.5–25/6.3–12.5 |
| 33 | 137 | 6 | 4/9 | 1.1 | >100/>100 |
| 34 | 137 | 4 | 4/9 | 1.1 | >100/>100 |
| 37 | 138 | 4 | 3/10 | 0.7 | >100/50–100 |
| 38 | 163 | 7 | 4/11 | 0.7 | >100/>100 |
| 39 | 137 | 4 | 4/9 | 2.3 | 6.3–12.5/6.3–12.5 |
| Compd | Species | Half-life (min) | In vitro CLint (μL min−1 mg−1 protein) |
|---|---|---|---|
| a CQ = chloroquine. | |||
| 8 | Mouse | >240 | <7 |
| 16 | Mouse | 185 | 9 |
| 32 | Mouse | 17 | 104 |
| 33 | Mouse | 167 | 10 |
| 34 | Mouse | 211 | 8 |
| 37 | Mouse | >240 | <7 |
| 38 | Mouse | >240 | <7 |
| 39 | Mouse | 1 | 1413 |
| CQ | Mouse | 177 | 10.2 |
The metabolic stability of the same eight compounds was then assessed in vitro using NADPH supplemented mouse liver microsomes.23 All of the compounds exhibited good metabolic stability (in vitro intrinsic clearance (CLint) of ≤10 μL min−1 mg−1 protein) with the exception of 32 and 39, which both exhibited a high degree of metabolic lability (CLint > 100 μL min−1 mg−1 protein). Two compounds, the amide analogue 8 and the urea analogue 33, were progressed to an in vivo study where they were administered to mice at a dose of 32 mg kg−1via either the oral or subcutaneous route (Fig. 3). The apparent half-lives were approximately 2 h, and as expected given the physicochemical properties, the exposure [area under the curve (AUC) over 8 h and maximum plasma concentration (Cmax)] following oral administration was more than 10-fold lower than that after subcutaneous dosing. Following subcutaneous administration, plasma concentrations for each compound remained above 0.2 μM for at least 8 h.
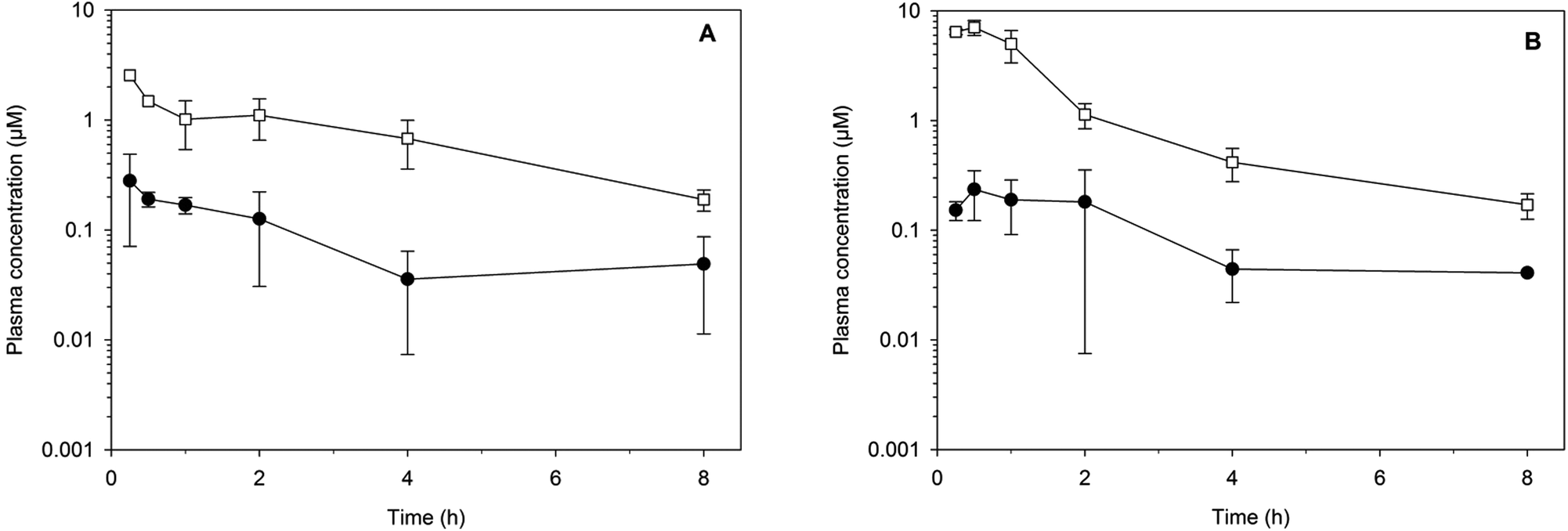 | ||
| Fig. 3 Plasma concentration versus time profiles for (A) 8 and (B) 33. Symbols represent concentrations after oral (filled circles) or subcutaneous (open squares) administration of 32 mg kg−1 (mean ± SD, n = 3). | ||
In vivo tolerability studies in mice
The amide (8, 16, 32) and urea (33, 34, 37, 38) analogues were evaluated for tolerability in mice at 8 mg kg−1, 16 mg kg−1 and 32 mg kg−1 given twice daily for 4 days by subcutaneous administration. At these doses the analogues were found to be well tolerated in healthy mice with no observable adverse events such as tremors, panting, loss of weight (>20%) and lack of natural movement. These findings are in contrast to the poor tolerance seen with the methyl derivatives of thiaplakortone A reported elsewhere.12In vivo efficacy studies in the P. berghei malaria mouse model
Based on the favorable tolerability findings of the thiaplakortone A amide derivatives (8, 16, 32) and urea (33, 34, 37, 38) analogues, the efficacy of these compounds was assessed in the rodent-P. berghei model using the highest tested dose of 32 mg kg−1. Compounds were administered subcutaneously twice daily for 4 days. Of the amide analogues tested, 8 was the most active with a mean suppression of blood stage parasitemia of 52% on D + 4 post inoculation of P. berghei to mice. The next most active amide analogue was 32 with 11% suppression on D + 4. Fig. 4 compares the mean parasitemia profile of P. berghei infected mice treated with either 8, chloroquine (positive control) or drug-free vehicle (control). Chloroquine suppression at D + 4 was 65% at 1 mg kg−1 day−1 given subcutaneously, which is comparable to the 50% suppression at 1.5 mg kg−1 day−1 described by Ridley et al.24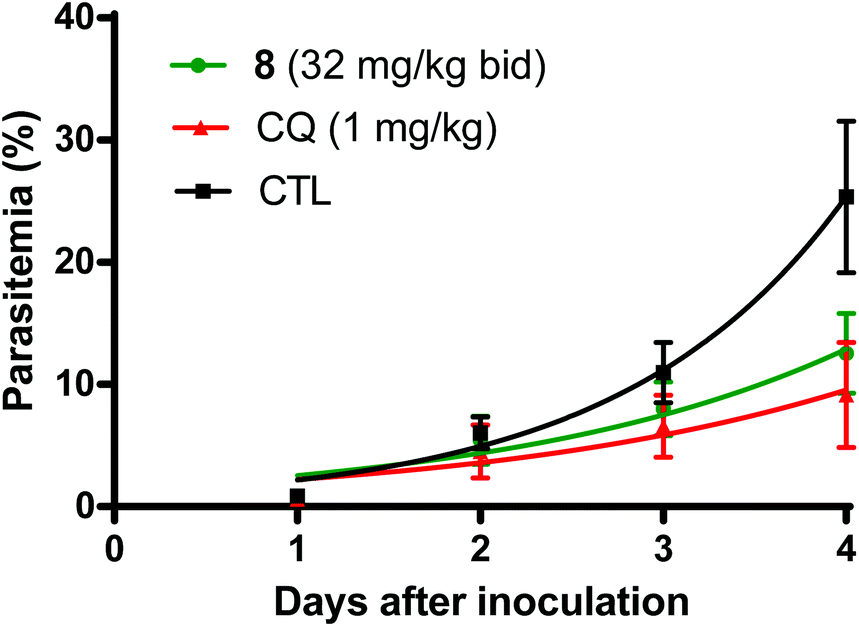 | ||
| Fig. 4 Mean (±SD, n = 6) parasitemia profile of P. berghei infected mice treated with either compound 8 (32 mg kg−1 twice daily for 4 days), the positive control chloroquine (CQ, 1 mg kg−1 daily for 4 days) or drug-free vehicle (CTL). | ||
Of the urea analogues tested, 33 was the most active with 26% suppression of parasitemia on D + 4 following subcutaneous administration. Analogues 37 and 38 had similar suppression values of 16% and 14%, respectively, on D + 4. When orally administered at 32 mg kg−1 twice daily for 4 days both compounds 8 and 33 did not significantly inhibit parasitemia (<3%) on D + 4.
Conclusions
In summary, 31 amide and 7 urea derivatives based on the thiaplakortone A tricyclic scaffold were synthesised and evaluated for their in vitro antimalarial activity and mammalian cell toxicity. Of these compounds, eight were chosen for ADME profiling after which several analogues were prioritised for in vivo pharmacokinetic analysis, followed by tolerability and antimalarial efficacy in a murine malaria model. Compounds 8 and 33 were both well tolerated in mice when administered subcutaneously at 32 mg kg−1 twice daily for 4 days. Furthermore, using this regimen blood stage P. berghei was suppressed by 52% for 8 and 26% for 33, relative to the vehicle control. Unfortunately, oral administration of these compounds at 32 mg kg−1 twice daily for 4 days did not suppress parasitemia, most likely due to poor oral bioavailability and limited exposure. While these data highlight the potential of the natural product derived thiazino-quinone scaffold as an anti-malarial starting point, the lack of oral bioavailability currently impinges on the further development of this series.Experimental
General procedures
Melting points were recorded on a capillary melting point apparatus and are uncorrected. Unless otherwise specified, 1H and 13C NMR spectra were recorded at 30 °C in DMSO-d6 on a Varian INOVA 500 NMR spectrometer. The 1H and 13C NMR chemical shifts were referenced to the solvent peak for DMSO-d6 at δH 2.50 and δC 39.5. LRESIMS was obtained from LC-MS data generated using a Waters Alliance 2790 HPLC equipped with a Waters 996 photodiode array detector and an Alltech evaporative light scattering detector that was attached to a Water ZQ mass spectrometer. HRESIMS were recorded on a Bruker MicrOTof-Q spectrometer (Dionex UltiMate 3000 micro LC system, ESI mode). Analytical thin layer chromatography (TLC) was performed on aluminum-backed 0.2 mm thick silica gel 60 F254 plates as supplied by Merck. Eluted plates were visualised using a 254 nm UV lamp and/or by treatment with a suitable dip followed by heating. These dips included phosphomolybdic acid–Ce(SO4)2–H2SO4 (conc.)–H2O (37.5 g:7.5 g:37.5 g:720 mL) or KMnO4–K2CO3–5% NaOH aqueous solution–H2O (3 g:20 g:5 mL:300 mL). Flash chromatographic separations were carried out following protocols defined by Still et al.25 with silica gel 60 (40–63 mm, supplied by GRACE) or amino bonded silica gel (Davisil®) as the stationary phase and using the AR- or HPLC-grade solvents indicated. Semi-preparative HPLC work was performed using a Waters 600 pump and 966 PDA detector, a Gilson 715 liquid handler and a C18-bonded silica Betasil 5 μm 143 Å column (21.2 × 150 mm). Alltech sample preparative C18-bonded silica (35–75 μm, 150 Å) and an Alltech stainless steel guard cartridge (10 × 30 mm) were used for preadsorption and HPLC work. A Phenomenex C18-bonded silica Luna 3 μm 100 Å (4.6 × 50 mm) column was used for LC-MS studies. All compounds were analysed for purity using LC-MS and shown to be >95% pure, unless otherwise stated. Starting materials and reagents were generally available from the Sigma-Aldrich, Merck, AK Scientific Inc., Matrix Scientific Chemical Companies and were used as supplied. THF, MeOH and DCM were dried using a Glass Contour solvent purification system that is based upon a technology originally described by Grubbs et al.26 Triethylamine (TEA) was freshly distilled over calcium hydride before use. Where necessary, reactions were performed under a nitrogen atmosphere and glassware was heated in an oven at 140 °C then dried under vacuum prior to use. Temperatures quoted as 0 °C and −78 °C were obtained by cooling the reaction vessel in baths of ice/H2O and CO2(s)/acetone, respectively. Compounds for biological studies were place under high vacuum (0.05 mmHg) for several hours before testing to remove trace, residual solvents.
:3, 3 × 1 mL). The remaining solid was purified by flash chromatography (silica, 1:5 v/v MeOH–DCM elution) to afford the amide.
:10 v/v MeOH–DCM elution) to afford the amide.
:10 v/v MeOH–DCM elution) to afford the urea.
:10 v/v MeOH–DCM elution) to afford the amide.
:10 v/v MeOH–DCM elution) to afford the N-alkylated analogues.
:5 v/v MeOH–DCM elution) to afford compound 11 (1.5 mg, 14%) as a brown powder. 1H NMR (500 MHz, DMSO-d6) thiazine and indole NH not observed. δ 8.08 (t, J = 5.6 Hz, 1H), 7.27 (s, 1H), 7.05 (d, J = 8.8 Hz, 1H), 6.40 (dd, J = 8.8, 1.4 Hz, 1H), 3.32–3.28 (m, 2H, CH2N– obscured by H2O) 2.84 (t, J = 7.2 Hz, 2H), 1.54–1.45 (m, 1H), 0.68–0.57 (m, 4H); (−)-LRESIMS m/z (rel. int.) 360 (100) [M − H]−; (+)-HRESIMS calcd for C16H15N3NaO5S [M + Na]+ 384.0625, found 384.0622.
:10 v/v MeOH–DCM elution) to afford the compound 16 (54 mg, 45%) as a yellow powder. 1H NMR (500 MHz, DMSO-d6) δ 12.91 (br s, 1H), 11.00 (br s, 1H), 8.48 (t, J = 5.6 Hz, 1H), 7.81 (d, J = 7.3 Hz, 2H), 7.52–7.48 (m, 1H), 7.46–7.42 (m, 2H), 7.31 (s, 1H), 7.05 (d, J = 8.8 Hz, 1H), 6.42 (d, J = 8.7 Hz, 1H), 3.53 (app. q, J = 6.8 Hz, 2H), 2.98 (t, J = 7.0 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 178.5, 167.5, 166.1, 139.9, 134.5, 130.9, 130.1, 128.8, 128.2 (2C), 127.7, 127.1 (2C), 124.6, 123.3, 114.2, 111.6, 39.1, 25.2. (+)-LRESIMS m/z (rel. int.) 398 (100) [M + H]+; (+)-HRESIMS calcd for C19H15N3NaO5S [M + Na]+ 420.0625, found 420.0623.
:10 v/v MeOH–DCM elution) to afford urea 37 (67 mg, 26%) as an orange powder; 1H NMR (500 MHz, DMSO-d6) thiazine and indole NH not observed δ 7.28 (s, 1H), 7.11 (app. t, J = 5.5 Hz, 1H), 7.05 (d, J = 8.8 Hz, 1H), 6.40 (d, J = 8.8 Hz, 1H), 3.53 (s, 3H), 3.31–3.28 (m, 2H), 2.90 (s, 3H), 2.88 (app. t, J = 6.9 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 178.5, 167.5, 159.9, 139.9, 130.1, 128.8, 127.7, 124.7, 123.4, 114.2, 111.5, 61.1, 39.4, 35.4, 25.5. (−)-LRESIMS m/z (rel. int.) 379 (100) [M − H]−; (+)-HRESIMS calcd for C15H16N4NaO6S [M + Na]+ 403.0683, found 403.0679.
:10 v/v MeOH–DCM elution) to afford compound 39 (16 mg, 37%) as an orange powder. 1H NMR (500 MHz, DMSO-d6) δ 12.96 (br s, 1H), 10.99 (br s, 1H), 7.28 (s, 1H), 7.05 (d, J = 8.8 Hz, 1H), 6.42 (d, J = 8.8 Hz, 1H), 6.03 (d, J = 8.1 Hz, 1H), 5.85 (t, J = 5.8 Hz, 1H), 3.65 (d, J = 8.1 Hz, 1H), 3.28–3.21 (m, 2H), 2.80 (t, J = 7.0 Hz, 2H), 1.86–1.61 (m, 12H), 1.49 (d, J = 12.5 Hz, 2H); (+)-LRESIMS m/z (rel. int.) 471 (100) [M + H]+; (+)-HRESIMS calcd for C23H26N4NaO5S [M + Na]+ 493.1516, found 493.1519.
:10 v/v MeOH–DCM elution) to afford acetamide 41 (7 mg, 43%) as an orange powder; 1H NMR (500 MHz, DMSO-d6) δ 10.93 (br s, 1H), 7.88 (app. t, J = 5.7 Hz, 1H), 7.59 (s, 1H), 7.07 (dd, J = 8.7, 1.0 Hz, 1H), 6.42 (d, J = 8.7 Hz, 1H), 5.20 (m, 1H), 3.28–3.24 (m, 2H), 2.83 (app. t, J = 7.3 Hz, 2H), 1.78 (s, 3H), 1.43 (d, J = 6.7 Hz, 6H); 13C NMR (125 MHz, DMSO-d6) δ 178.1, 169.0, 167.5, 140.4, 130.3, 129.0, 125.4, 124.0, 123.8, 113.6, 111.4, 49.9, 38.3, 25.5, 22.6, 22.4; (+)-LRESIMS m/z (rel. int.) 378 (100) [M + H]+; (+)-HRESIMS calcd for C17H19N3NaO5S [M + Na]+ 400.0938, found 400.0935.
:10 v/v MeOH–DCM elution) to afford acetamide 44 (9 mg, 47%) as an orange powder; 1H NMR (500 MHz, DMSO-d6) δ 10.98 (s, 1H), 7.86 (t, J = 5.4 Hz, 1H), 7.52 (s, 1H), 7.33 (t, J = 7.3 Hz, 2H), 7.28 (t, J = 7.2 Hz, 1H), 7.24 (d, J = 7.2 Hz, 2H), 7.04 (d, J = 8.8 Hz, 1H), 6.42 (d, J = 8.8 Hz, 1H), 5.54 (s, 2H), 3.27 (dd, J = 13.0, 6.8 Hz, 2H), 2.83 (t, J = 7.1 Hz, 2H), 1.75 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 187.9, 179.0, 177.6, 145.0, 146.8, 143.5, 140.1, 138.6 (2C), 137.7, 137.0, 135.7 (2C), 134.1, 134.0, 124.0, 121.5, 61.5, 48.2, 35.2, 32.5. (+)-LRESIMS m/z (rel. int.) 426 (100) [M + H]+; (+)-HRESIMS calcd for C21H19N3NaO5S [M + Na]+ 448.0968, found 448.0933.
X-ray structure determination of thiaplakortone A (1) and n-propyl amide analogue (9)
Unique data sets for compounds 1 as the dihydrate and 9 with solvated DMSO were measured at 223 and 200 K respectively on an Oxford-Diffraction GEMINI S Ultra CCD diffractometer (Mo-Kα radiation, graphite monochromation) utilising CrysAlis software.27 The structures were solved by direct methods and refined by full matrix least squares refinement on F2. Anisotropic thermal parameters were refined for non-hydrogen atoms; (x, y, z, Uiso)H were included and constrained at estimated values. Conventional residuals at convergence are quoted; statistical weights were employed. Computation used, SIR-97,28 SHELX97,29 TeXsan,30 ORTEP-331 and PLATON32 programs and software systems.Full .cif depositions reside with the Cambridge Crystallographic Data Centre, CCDC Nos. 998930 (1) and 998931 (9).
In vitro P. falciparum growth inhibition assays
The in vitro growth inhibitory activity of compounds was tested against P. falciparum lines 3D7, Dd2, and C2B using the [3H]-hypoxanthine incorporation assay. Chloroquine (positive control) was prepared as a 10 mM stock solution in PBS. All other compounds were prepared as 20 mM stock solutions in DMSO. Serial dilutions of compounds/controls were prepared in hypoxanthine-free culture media, followed by addition of [3H]-hypoxanthine (0.5 μCi per well) and asynchronous cultures at 1% parasitemia and 1% final haematocrit. The antimalarial drugs chloroquine or atovaquone were included in assays as internal controls. Following incubation for 48 h, the amount of [3H]-hypoxanthine incorporated into parasites was determined by harvesting cultures onto glass fiber filter mats and counting using a Perkin Elmer/Wallac Trilux 1450 MicroBeta scintillation counter. Percentage inhibition of growth compared to matched DMSO controls (0.5%) was determined. IC50 values were determined using log-linear interpolation of inhibition curves and are presented as mean (± SD) of three independent assays, each carried out in triplicate wells. Chloroquine [IC50 = 0.109 ± 0.060 μM (Dd2); IC50 = 0.012 ± 0.004 μM (3D7)] and atovaquone [IC50 = 0.0002 ± 0.0001 μM (3D7) IC50 = 5.750 ± 0.778 μM (C2B)] served as positive controls.In vitro cytotoxicity assay
Cytotoxicity against a mammalian cell line (neonatal foreskin fibroblast cells; NFF) was assessed by culturing cells in RPMI media (Life Technologies) supplemented with 10% heat inactivated foetal calf serum (CSL Biosciences) and 1% streptomycin (Life Technologies). Chloroquine was prepared as a 10 mM stock solution in PBS. All other compounds were prepared as 20 mM stock solutions in DMSO. Cells were seeded into wells of 96-well tissue culture plates (3000 cells per well) and cultured for 24 h at 37 °C in 5% CO2 before being treated with a dilution series of each compound/control. After 72 h, medium was removed and plates washed in phosphate buffered saline pH 7.4 (PBS), before fixing with denatured alcohol. Fixative was removed and washed from cells before the addition of sulforhodamine B (0.4%; Sigma 50 μL). After staining for 1 h, plates were washed three times with 1% acetic acid, then 100 μL of 10 mM Tris base (unbuffered, pH > 9) was added to each well. Plates were read at 564 nm in an ELISA micro-plate reader. Percentage inhibition of growth as compared to matched DMSO controls (0.5%) was determined and IC50 values calculated using log-linear interpolation of inhibition curves. Data are presented as mean ± SD of three independent assays, each carried out in triplicate wells. The antimalarial drug chloroquine was included as an internal control in each assay, and was shown to display an IC50 of 46 ± 14 μM.Physicochemical and in vitro metabolic stability assays
In silico parameters were calculated using JChem for excel (ChemAxon, Budapest). Kinetic solubility in 0.01 M HCl (pH 2) and phosphate buffer (pH 6.5) was determined by serial dilution of a concentrated stock solution prepared in DMSO and the solubility range was determined by nephelometry. LogD was estimated at pH 7.4 using a chromatographic method as described previously.23
Metabolic stability was assessed in vitro by incubating with mouse liver microsomes (Xenotech, Lenexa, KS) at 37 °C using substrate concentrations of 1 μM and a microsomal protein concentration of 0.4 mg mL−1. Addition of an NADPH-regenerating buffer system (containing 1 mg mL−1 NADP, 1 mg mL−1 glucose-6-phosphate, 1 U mL−1 glucose-6-phosphate dehydrogenase and 0.67 mg mL−1 MgCl2) was used to initiate the metabolic reactions, and samples were quenched at various time points over 60 min by the addition of chilled MeCN. Control samples (containing no NADPH) were included to monitor for potential degradation in the absence of cofactor. Samples were analysed by LC-MS on a Waters/Micromass Xevo G2 QTOF or a Waters/Micromass Quattro Ultima PT Triple Quadrupole MS, each coupled to a Waters Acquity uPLC (Milford, MA) under positive electrospray ionisation. Chromatography was conducted using a Supelco Ascentis Express reverse phase C8 or C18 column (50 × 2.1 mm, 2.7 μm) (Sigma-Aldrich, St Louis, MO), equipped with a Phenomenex SecurityGuard column hosting a Luna C8 cartridge (Torrance, CA) and both were maintained at a temperature of 40 °C. The mobile phase consisted of MeCN and H2O (containing 0.05% formic acid) mixed using a binary gradient at a flow rate of 0.4 mL min−1. The first-order rate constant for substrate depletion was used to calculate the in vitro intrinsic clearance.
In vivo amide and urea analogue exposure in mice
All animal studies were conducted using established procedures in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and the study protocols were reviewed and approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee. The systemic exposure of the amide 8 and the urea 33 analogues was assessed in non-fasted, noninfected female Swiss Outbred mice following administration of 32 mg kg−1 by oral gavage (0.2 mL per mouse) or subcutaneous injection (0.2 mL per mouse) under the skin in the abdomen. Formulations for both routes comprised 10% v/v EtOH, 10% v/v Tween 80 (Sigma Chemical Co., St Louis, MO), and 80% v/v MilliQ H2O. Blood samples were taken over 8 h with two samples per mouse; one via submandibular bleed (∼120 μL, conscious sampling) and the other via terminal cardiac puncture (0.6 mL under isofluorane anesthesia) after which animals were euthanised by cervical dislocation. Blood samples were transferred to tubes containing heparin and a stabilisation cocktail (complete protease inhibitor cocktail (Roche), potassium fluoride, and EDTA) to minimise the potential for ex vivo degradation.Following collection, blood samples were centrifuged and plasma stored at −20 °C prior to analysis. Plasma samples were assayed by first thawing, spiking with internal standard (diazepam) and precipitating plasma proteins with the addition of MeCN (2-fold volume ratio), centrifuging and injecting the supernatant onto an LCMS system (Micromass Xevo TQ triple quadrupole mass spectrometer coupled to a Waters Acquity UPLC). The column was a Supelco Ascentis Express RP Amide column (2.7 μm, 50 mm × 2.1 mm i.d.) equipped with a Phenomenex Security Guard column with Synergy Polar packing material, and both columns were maintained at 40 °C. The mobile phase consisted of MeCN and H2O, both containing 0.05% v/v formic acid, and delivered using a linear gradient over 3.3 min followed by re-equilibration to the starting conditions. The flow rate was 0.4 mL min−1, and the injection volume was 3 μL. LCMS analysis was conducted in positive mode electrospray ionisation and elution of the analytes monitored in MRM mode. Concentrations were determined by comparison to a set of calibration standards prepared in blank mouse plasma. Accuracy was within ± 10%, and precision (% RSD) was less than 11%. The limit of quantitation was 0.5 ng mL−1 for 8 and 1 ng mL−1 for 33.
Pharmacokinetic parameters were determined using non-compartmental analysis (WinNonlin version 5.2, Pharsight, Mountain View, CA) and included the maximum plasma concentration (Cmax), the time to reach the maximum concentration (Tmax), the apparent terminal elimination half-life (where it could be determined) and the area under the plasma concentration versus time profile from 0 to 8 h (AUC0–8h).
In vivo tolerability assessment in mice
Animal studies were approved by the Army Malaria Institute Animal Ethics Committee (AEC no. 05/13) in accord with the Australian Code of Practice for the care and Use of Animals for Scientific Purposes. Tolerability assessment of amide and urea analogues were carried out in groups of three healthy mice administered different doses of compound. The mice were male and female Outbred ARC Swiss mice (Animal Resource Centre, Western Australia) aged between 6 and 7 weeks, with a mean weight of 28 ± 3 g. The animals were observed for physical distress twice daily. Physical adverse events that were monitored included reduced activity and movement, tremour, panting, behaviour changes, reduced appetite, extreme pallor, ruffled coat and weight loss. If any animal exhibited physical adverse events that appeared highly stressful they were euthanised. All compounds were dissolved in 10% EtOH/10% Tween 80/80% distilled H2O and administered subcutaneously to mice twice daily at about 6 h apart for 4 consecutive days.In vivo efficacy assessment in the rodent-P. berghei model
The in vivo efficacy of amide and urea analogues were determined using a modified Peters 4 day test.33 This test measures the suppressive activity of blood schizontocides over 4 days at a high-tolerated dose that does not cause physical stress in healthy mice. Briefly, female ARC mice (groups of six mice, age 6–7 weeks with a mean weight of 28 ± 3 g) were inoculated intraperitoneally with 20 × 106P. berghei (ANKA strain) infected erythrocytes. The mice were then treated subcutaneously or orally at about 1 h and 6 h after parasite inoculation (D0) and then twice daily at about 6 h apart for 3 consecutive days with either the analogue and drug-free vehicle (control). Chloroquine (positive control) was given daily for 4 days. The analogues were dissolved in 10% EtOH/10% Tween 80/80% distilled H2O and chloroquine was dissolved in H2O. The drug-free vehicle was 10% EtOH/10% Tween 80/80% distilled H2O. Thin blood smears were made on D + 1, D + 2, D + 3, and D + 4 and stained with Giemsa. The degree of infection (parasitemia expressed as percentage of infected erythrocytes) was determined microscopically. Blood smears were read independently by two microscopists with results averaged.Conflict of Interest
The authors declare no competing financial interest. The opinions expressed herein are those of the authors’ and do not necessarily reflect those of the Australian Defence Force, Joint Health Command or any extant policy.Acknowledgements
The authors acknowledge the National Health and Medical Research Council for financial support (grant APP1024314) and the Australian Research Council (ARC) for fellowship support (grant FT0991213 to KTA.). We thank the ARC for support toward NMR and MS equipment (grant LE0668477 and LE0237908). We thank the Australian Red Cross Blood Service for the provision of human blood and sera. We are grateful to Stephen McLeod-Robertson and Thomas Travers for the in vivo tolerability and efficacy mice studies.Notes and references
- C. J. Murray, L. C. Rosenfeld, S. S. Lim, K. G. Andrews, K. J. Foreman, D. Haring, N. Fullman, M. Naghavi, R. Lozano and A. D. Lopez, Lancet, 2012, 379, 413 CrossRef.
- WHO, World Malaria Report, World Health Organization, Geneva, 2013.
- S. T. Agnandji, B. Lell, S. S. Soulanoudjingar, J. F. Fernandes, B. P. Abossolo, C. Conzelmann, B. G. Methogo, Y. Doucka, A. Flamen, B. Mordmuller, S. Issifou, P. G. Kremsner, J. Sacarlal, P. Aide, M. Lanaspa, J. J. Aponte, A. Nhamuave, D. Quelhas, Q. Bassat, S. Mandjate, E. Macete, P. Alonso, S. Abdulla, N. Salim, O. Juma, M. Shomari, K. Shubis, F. Machera, A. S. Hamad, R. Minja, A. Mtoro, A. Sykes, S. Ahmed, A. M. Urassa, A. M. Ali, G. Mwangoka, M. Tanner, H. Tinto, U. D'Alessandro, H. Sorgho, I. Valea, M. C. Tahita, W. Kabore, S. Ouedraogo, Y. Sandrine, R. T. Guiguemde, J. B. Ouedraogo, M. J. Hamel, S. Kariuki, C. Odero, M. Oneko, K. Otieno, N. Awino, J. Omoto, J. Williamson, V. Muturi-Kioi, K. F. Laserson, L. Slutsker, W. Otieno, L. Otieno, O. Nekoye, S. Gondi, A. Otieno, B. Ogutu, R. Wasuna, V. Owira, D. Jones, A. A. Onyango, P. Njuguna, R. Chilengi, P. Akoo, C. Kerubo, J. Gitaka, C. Maingi, T. Lang, A. Olotu, B. Tsofa, P. Bejon, N. Peshu, K. Marsh, S. Owusu-Agyei, K. P. Asante, K. Osei-Kwakye, O. Boahen, S. Ayamba, K. Kayan, R. Owusu-Ofori, D. Dosoo, I. Asante, G. Adjei, D. Chandramohan, B. Greenwood, J. Lusingu, S. Gesase, A. Malabeja, O. Abdul, H. Kilavo, C. Mahende, E. Liheluka, M. Lemnge, T. Theander, C. Drakeley, D. Ansong, T. Agbenyega, S. Adjei, H. O. Boateng, T. Rettig, J. Bawa, J. Sylverken, D. Sambian, A. Agyekum, L. Owusu, F. Martinson, I. Hoffman, T. Mvalo, P. Kamthunzi, R. Nkomo, A. Msika, A. Jumbe, N. Chome, D. Nyakuipa, J. Chintedza, W. R. Ballou, M. Bruls, J. Cohen, Y. Guerra, E. Jongert, D. Lapierre, A. Leach, M. Lievens, O. Ofori-Anyinam, J. Vekemans, T. Carter, D. Leboulleux, C. Loucq, A. Radford, B. Savarese, D. Schellenberg, M. Sillman and P. Vansadia, N. Engl. J. Med., 2011, 365, 1863 CrossRef PubMed.
- S. Abdulla, N. Salim, F. Machera, R. Kamata, O. Juma, M. Shomari, S. Kubhoja, A. Mohammed, G. Mwangoka, T. Aebi, H. Mshinda, D. Schellenberg, T. Carter, T. Villafana, M.-C. Dubois, A. Leach, M. Lievens, J. Vekemans, J. Cohen, W. R. Ballou and M. Tanner, Malar. J., 2013, 12, 11 CrossRef PubMed.
- A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455 CrossRef CAS PubMed.
- A. M. Dondorp and P. Ringwald, Trends Parasitol., 2013, 29, 359 CrossRef CAS PubMed.
- C. Wongsrichanalai, Lancet Infect. Dis., 2013, 13, 114 CrossRef CAS PubMed.
- C. H. Sibley, Lancet Infect. Dis., 2013, 13, 999 CrossRef CAS PubMed.
- D. Camp, R. A. Davis, M. Campitelli, J. Ebdon and R. J. Quinn, J. Nat. Prod., 2012, 75, 72 CrossRef CAS PubMed.
- S. Duffy and V. M. Avery, Am. J. Trop. Med. Hyg., 2012, 86, 84–92 CrossRef CAS PubMed.
- R. A. Davis, S. Duffy, S. Fletcher, V. M. Avery and R. J. Quinn, J. Org. Chem., 2013, 78, 9608 CrossRef CAS PubMed.
- R. H. Pouwer, S. M. Deydier, P. V. Le, B. D. Schwartz, N. C. Franken, R. A. Davis, M. J. Coster, S. A. Charman, M. D. Edstein, T. S. Skinner-Adams, K. T. Andrews, I. D. Jenkins and R. J. Quinn, ACS Med. Chem. Lett., 2014, 5, 178 CrossRef CAS PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- H. A. Staab, Angew. Chem., Int. Ed. Engl., 1962, 1, 351 CrossRef.
- S. T. Heller and R. Sarpong, Org. Lett., 2010, 12, 4572 CrossRef CAS PubMed.
- D. Carniato, C. Charon, J. Gleitz, D. Roche and B. Hock, International Patent ApplicationWO 2007/068330 A068331, 2007 Search PubMed.
- A. T. Hudson, Parasitol. Today, 1993, 9, 66 CrossRef CAS PubMed.
- M. W. Mather, K. W. Henry and A. B. Vaidya, Curr. Drug Targets, 2007, 8, 49 CrossRef CAS PubMed.
- P. Olliaro, Pharmacol. Ther., 2001, 89, 207 CrossRef CAS PubMed.
- S. Schwikkard and F. R. Heerden, Nat. Prod. Rep., 2002, 19, 675 RSC.
- K. Kaur, M. Jain, T. Kaur and R. Jain, Bioorg. Med. Chem., 2009, 17, 3229 CrossRef CAS PubMed.
- J.-R. Ioset, Curr. Org. Chem., 2008, 12, 643 CrossRef CAS.
- J. M. Coteron, M. Marco, J. Esquivias, X. Deng, K. L. White, J. White, M. Koltun, F. El Mazouni, S. Kokkonda, K. Katneni, R. Bhamidipati, D. M. Shackleford, I. Angulo-Barturen, S. B. Ferrer, M. B. Jimenez-Diaz, F.-J. Gamo, E. J. Goldsmith, W. N. Charman, I. Bathurst, D. Floyd, D. Matthews, J. N. Burrows, P. K. Rathod, S. A. Charman and M. A. Phillips, J. Med. Chem., 2011, 54, 5540 CrossRef CAS PubMed.
- R. G. Ridley, H. Matile, C. Jaquet, A. Dorn, W. Hofheinz, W. Leupin, R. Masciadri, F.-P. Theil, W. F. Richter, M.-A. Girometta, A. Guenzi, H. Urwyler, E. Gocke, J.-M. Potthast, M. Csato, A. Thomas and W. Peters, Antimicrob. Agents Chemother., 1997, 41, 677 CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- S. C. C. Agilent Technologies Inc, USA, 2012.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spaagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- TeXsan for Windows, Version 1.06. MSC 9009 New Trails Drive, The Woodlands, TX 77381, USA, 1997–2001 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- W. Peters, J. H. Portus and B. L. Robinson, Ann. Trop. Med. Parasitol., 1975, 69, 155 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and representative 13C NMR spectra of amide (8–32, 40–45) and urea (33, 34, 36–39) analogues. CCDC 998930 and 998931. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01849d |
| This journal is © The Royal Society of Chemistry 2015 |