
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The rapid synthesis of oxazolines and their heterogeneous oxidation to oxazoles under flow conditions†
Steffen
Glöckner
,
Duc N.
Tran
,
Richard J.
Ingham
,
Sabine
Fenner
,
Zoe E.
Wilson
,
Claudio
Battilocchio
and
Steven V.
Ley
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: svl1000@cam.ac.uk; Web: http://www.leygroup.ch.cam.ac.uk/
First published on 28th October 2014
Abstract
A rapid flow synthesis of oxazolines and their oxidation to the corresponding oxazoles is reported. The oxazolines are prepared at room temperature in a stereospecific manner, with inversion of stereochemistry, from β-hydroxy amides using Deoxo-Fluor®. The corresponding oxazoles can then be obtained via a packed reactor containing commercial manganese dioxide.
Introduction
Oxazolines and oxazoles are important biological scaffolds present within many natural products (Fig. 1).1,2 Amongst the many synthetic approaches towards these structural motifs, one very useful strategy involves the cyclodehydration of β-hydroxy amides using reagents such as Burgess’ reagent,3 the Mitsunobu reagent,4 Martin sulfurane,5 Ph2-SO-Tf2O,6 PPh3-CCl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 or by treatment with fluorinating reagents such as diethylaminosulfur trifluoride (DAST) and Deoxo-Fluor®.8
7 or by treatment with fluorinating reagents such as diethylaminosulfur trifluoride (DAST) and Deoxo-Fluor®.8
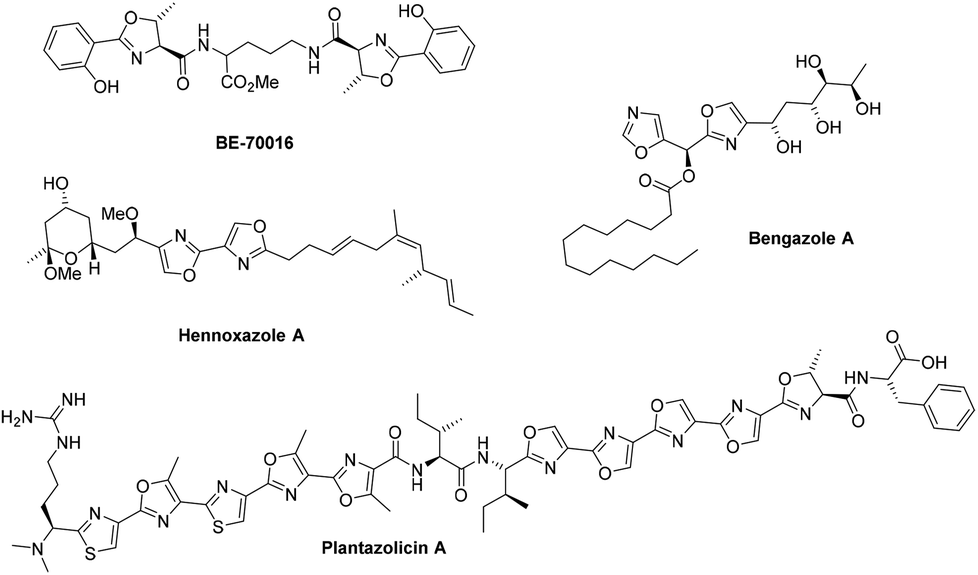 | ||
| Fig. 1 Oxazoline- and oxazole-containing natural products. | ||
The oxidative aromatisation of oxazolines to the corresponding oxazoles is typically achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and bromotrichloromethane.8,9 Other reagents such as NiO2,10 CuBr2/DBU,11 NBS/hv,12 CuBr/peroxide12 or O2 gas13 have also been successfully employed, although largely for the oxidation of C5-unsubstituted oxazoline rings. While manganese(IV) dioxide (MnO2) has been applied with some success to the preparation of thiazoles from thiazolidines,14 to date it has not been widely used for the synthesis of oxazoles and only a few examples exist in the literature.15,16
We have previously described the preparation of oxazolines in flow during the synthesis of o-methyl siphonazole,9 hennoxazole A17 and bisoxazoline PyBox chiral ligands.18 DAST was employed to effect the cyclisation of a β-hydroxy amide to the oxazoline at 70–90 °C, followed by in-line quenching of residual HF. This approach greatly improved the safety profile of the reaction compared to the batch synthesis, as well as providing pure products without need for additional purification. With these benefits in mind, it was decided to further investigate the scope of this flow transformation, employing the more stable Deoxo-Fluor® reagent.
Our experience has shown that manganese dioxide can be simply packed into an Omnifit® column19 and used successfully as a heterogeneous reagent in flow processes, without the problems usually associated with the use of this reagent in batch synthesis, i.e. its tendency to adhere to surfaces and cause blockages.20 Here we report the development of a flow protocol for the rapid cyclodehydration of a diverse range of β-hydroxy amides using Deoxo-Fluor® and the oxidative aromatisation of the resulting oxazolines to oxazoles using commercial activated and amorphous manganese dioxide.
Results and discussion
Our initial investigations focused on the flow synthesis of oxazolines, revealing that the use of a slight excess (1.35 eq.)21 of Deoxo-Fluor® resulted in >99% conversion of the β-hydroxy amides to the oxazolines. The use of higher quantities of fluorinating agent led to a large number of unwanted by-products. Unlike typical batch reactions using Deoxo-Fluor®, which usually require cooling to −20 °C,8 it was found that in flow optimal yields were obtained at room temperature, desirably removing the need for cooling.In a set of model experiments with amide 1a, where residence time was kept constant at 1.7 min, greater conversion was observed as flow rate increased, suggesting that mixing was a key factor for this reaction (Fig. 2).
 | ||
| Fig. 2 Effect of flow rate on cyclodehydration of 1a to 2a (residence time = 1.7 min). | ||
A flow rate of 6 mL min−1 gave full conversion to the desired oxazoline 2a.
Under these conditions, a solution of β-hydroxy amide (0.25 M) and a solution of Deoxo-Fluor®, each delivered at a flow rate of 3.00 mL min−1, were combined at a T-piece before passing into a reactor coil (1/16′′ o.d., 1 mm i.d., 10 mL, PFA) at 25 °C. The exiting stream was combined with an aqueous sodium bicarbonate solution at a T-mixer (flow rate of 9 mL min−1) to quench any residual HF and then directed into a Zaiput liquid–liquid membrane separator.22,23 This device enabled separation of the two phases at a high combined flow rate (15 mL min−1), with minimal breakthrough of the aqueous phase into the organic phase (Scheme 1).
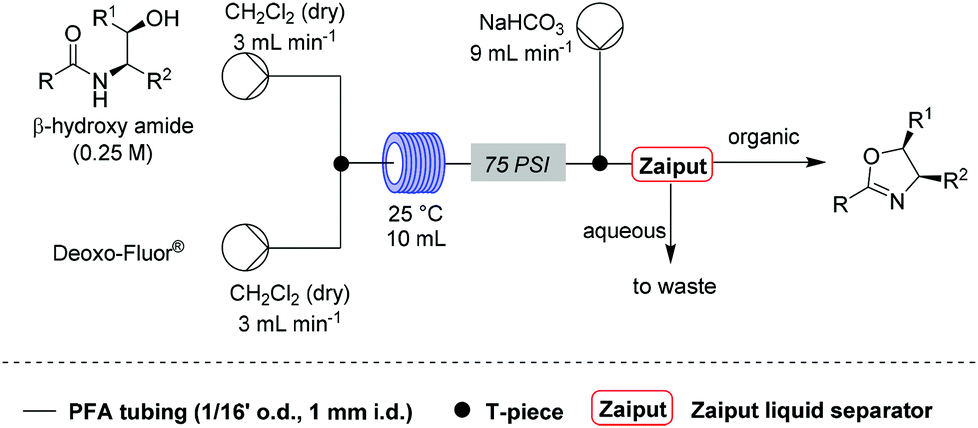 | ||
| Scheme 1 Optimised conditions for the flow synthesis of oxazolines. | ||
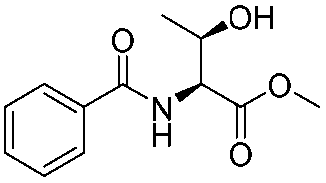
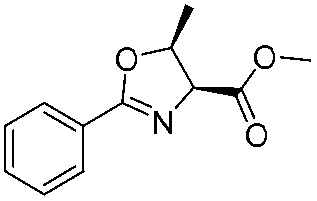
The optimised conditions were successfully applied on a 2 mmol scale to a selection of β-hydroxy amides to obtain the desired oxazolines in moderate to excellent yields with no need for further purification (Table 1). Aryl oxazolines 2a–f were obtained in excellent yields and diastereoselectivities. The dipeptide derivatives subjected to these conditions (1g, 1h and 1i) showed excellent reactivities, yielding 92–98% of the desired oxazolines. Moreover, dicyclisation processes were possible (2k and 2l) with good yield, although higher flow rates (10 mL min−1) were required. It was found that the yields under flow conditions were typically 10% higher than the corresponding batch system, with the efficient heat transfer and rapid mixing greatly improving the safety profile when compared to the batch reaction.8
| Entrya | Substrate | Product | Isolated yieldb | ||
|---|---|---|---|---|---|
| a Reactions were run on a 2 mmol scale. b Compounds were isolated without purification. c The crude material was passed through a plug of calcium carbonate/silica in place of an aqueous work up. d Total flow rate = 10 mL min−1 with 2.6 eq. of Deoxo-Fluor®. | |||||
| 1 |
|
1a |
|
2a | 98% |
| 2 |
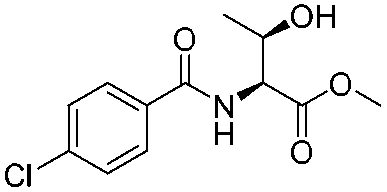
|
1b |
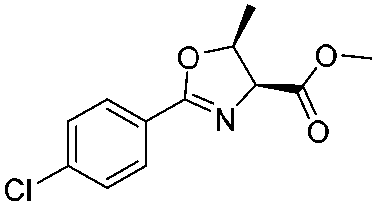
|
2b | 98% |
| 3 |
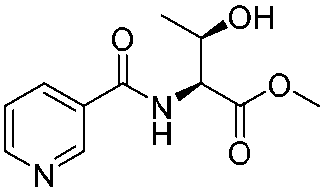
|
1c |
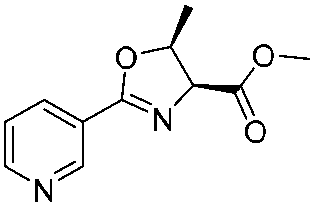
|
2c | 79%c |
| 4 |
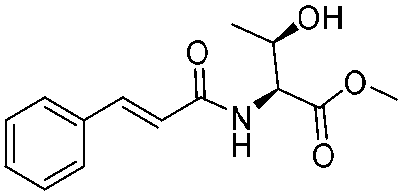
|
1d |
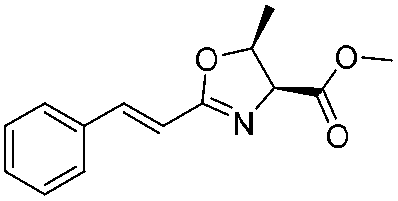
|
2d | 98% |
| 5 |
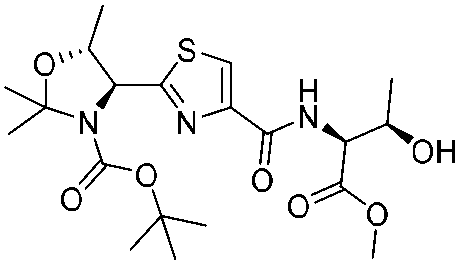
|
1e |
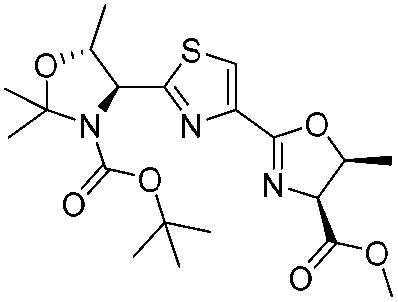
|
2e | 99% |
| 6 |
|
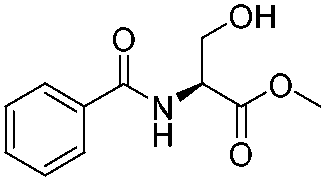
1f |
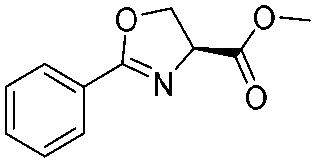
|
2f | 95% |
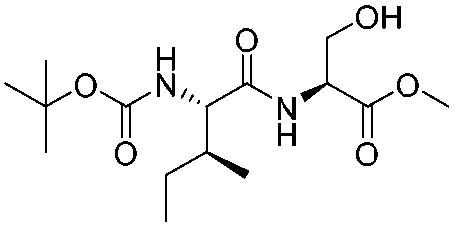
| 7 |
|
1g |
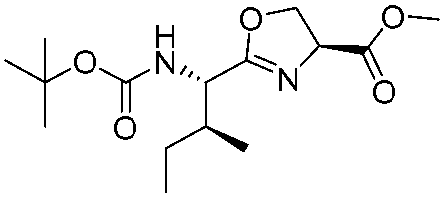
|
2g | 98% |
| 8 |
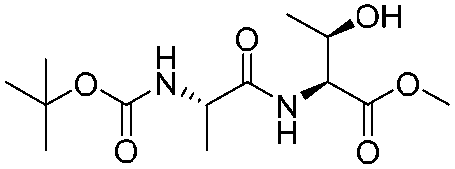
|
1h |
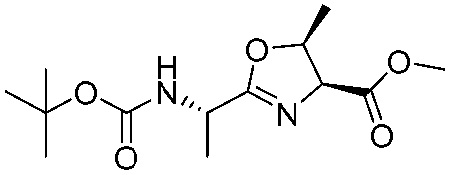
|
2h | 92% |
| 9 |
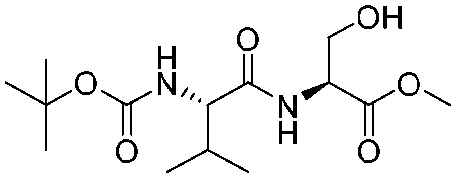
|
1i |
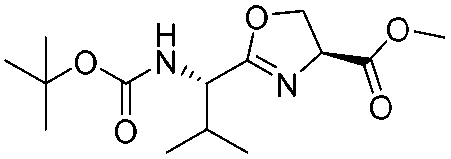
|
2i | 95% |
| 10 |
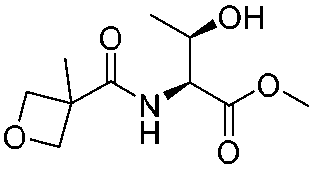
|
1j |
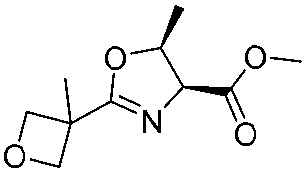
|
2j | 60% |
| 11 |
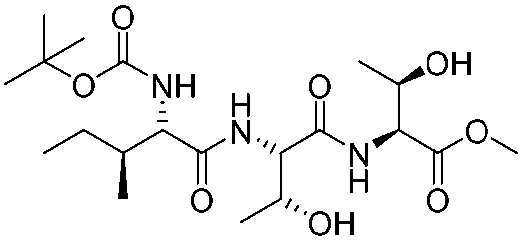
|
1k |
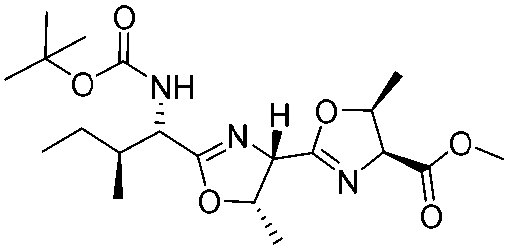
|
2k | 85%d |
| 12 |
|
1l |
|
2l | 92%d |
Such a dependence on the flow rate suggested that the mixing efficiency was a significant factor affecting the rate of cyclisation. Microfluidics are known to provide a highly efficient static mixing23–26 and therefore the cyclodehydration of β-hydroxy amides 1b and 1i was attempted on a 1 μL (Labtrix Start) microchip (Scheme 2).27 It was found that both substrates could be cyclodehydrated in excellent yield at room temperature with residence times around 1 s, further supporting the importance of mixing for this process.28
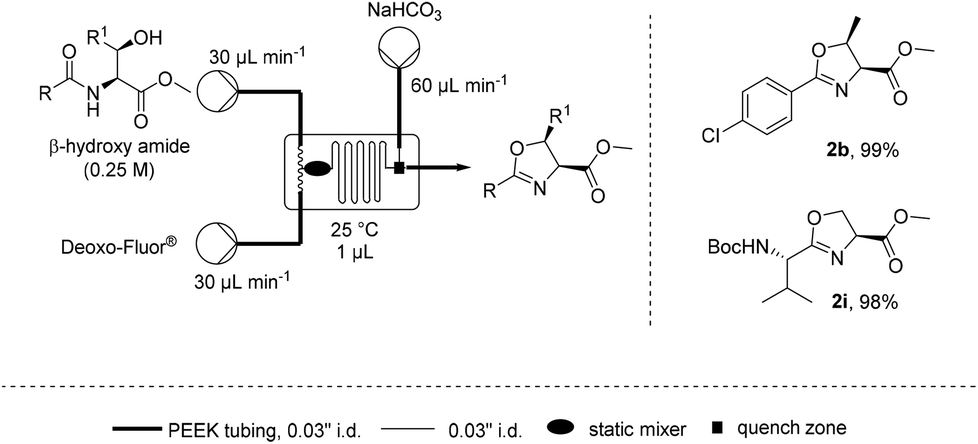 | ||
| Scheme 2 Microchip reaction for the preparation of oxazolines. | ||
Additionally, we wanted to investigate whether this flow protocol could be run continuously to afford larger amounts of material, which is frequently a requirement in synthesis. A microchip reactor would not be appropriate because whilst it generates an improved reaction rate, it cannot provide the same potential throughput as a mesofluidic system. Preliminary investigations into this were carried out using 30 g of β-hydroxy amide 1a. A magnetic mixer was introduced to ensure complete quenching over an extended period as without this device, residual HF was observed in the organic stream. We also took the opportunity to test a simple large-scale gravity separation device (see SI) to recover the organic phase. Using this specific system, 10.2 g h−1 of 2a could be generated in a single continuous run over 2 h and 40 min, with 27.2 g of 2a being produced (98% yield) (Scheme 3).
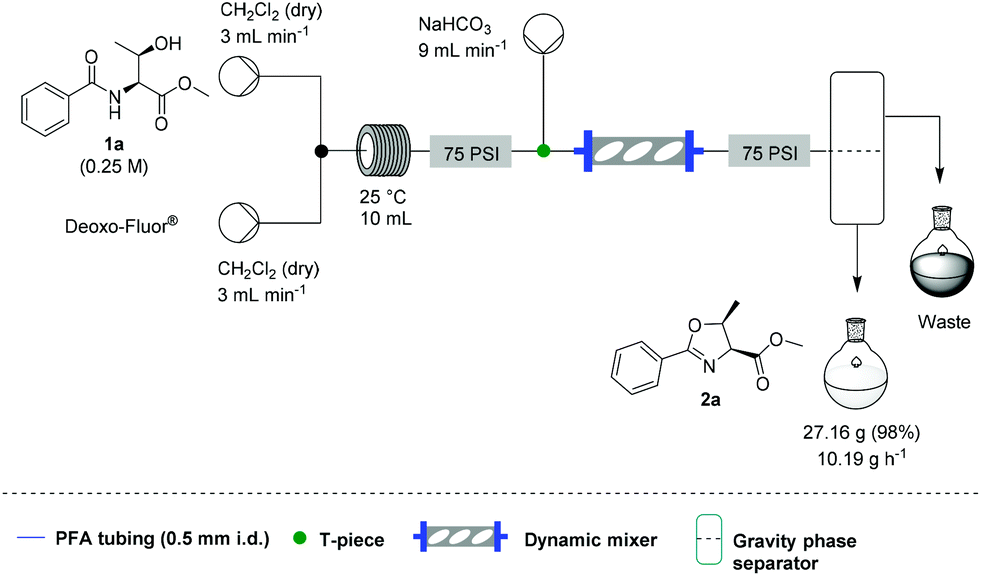 | ||
| Scheme 3 Platform set up for the scale up experiment. | ||
With flow conditions established for the cyclodehydration, we turned our attention to the MnO2-mediated oxidative aromatisation reaction, using oxazoline 2a as a model substrate. Previous studies had demonstrated that the solvent and temperature have a strong influence on the general oxidative reactivity of MnO2.29–31 Oxazoline 2a (0.5 mmol) in 8 mL of a range of solvents was passed through a column reactor packed with activated MnO232 (3 g) with an initial flow rate of 100 μL min−1, which was reduced to 60 μL min−1 after 50 min. The flow output was directly collected and evaporated (Scheme 4). Dichloromethane, trifluorotoluene and isopropyl acetate afforded pure desired product at 40 °C, although in low isolated yield (40%, 20% and 32% respectively). We speculate that the remaining material has decomposed and is retained in the MnO2 column. When isopropyl acetate was used as solvent at lower temperature (0 °C), although less material was lost, the conversion was poorer and the yield significantly reduced (31%). Neither acetone or dimethoxy ethane (DME) afforded full conversion at 40 °C, but after purification afforded higher yields of the desired product (59% and 63% respectively). Pleasingly, it was found that by increasing the reaction temperature to 60 °C with DME as solvent, pure 3a could be isolated in 79% by simply removing the solvent in vacuo (Table 2).
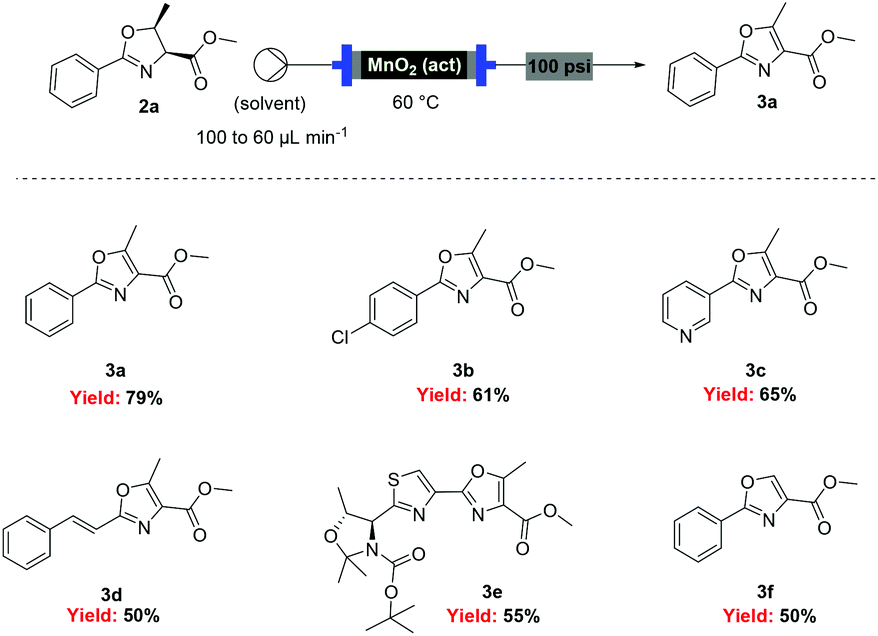 | ||
| Scheme 4 Flow oxidation of aryl-oxazolines using activated MnO2. | ||
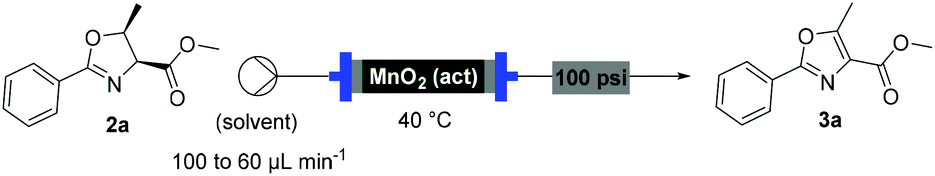
Under these optimised conditions we were able to process several 2-aryl-substituted oxazolines to oxazoles with isolated yields of 50–79% (Scheme 4). Notably, pyridyl (3c) and styryl (3d) oxazolines were compatible with the process giving 65% and 50% yields respectively. The mild reaction conditions allowed oxidation in the presence of an acid labile protecting group (3e). Desirably, all these oxazoles were obtained in analytically pure form without any further purification, with inductively coupled plasma-mass spectrometry (ICP-MS) analyses showing negligible leaching of MnO2 (less than 10 ppb).
However, when these optimised conditions were applied to 2-alkyl-substituted oxazolines, extensive decomposition of these substrates to unidentified by-products was observed. It was suspected that the high reactivity of activated MnO2 could be responsible for this decomposition, so the reaction was attempted using the less active amorphous MnO233 under otherwise identical conditions. At 60 °C only partial conversion to the desired oxazoles was detected whereas increasing the temperature at 100 °C led to complete conversion of the starting material. This allowed access to a range of alkyl substituted oxazoles in moderate to good yields (Scheme 5), including sensitive substrates such as oxetane containing 3j. Notably, the double oxidation of 4 could be carried out in 83% yield to afford 5, an intermediate in our recent total synthesis of plantazolicin A and B.34 When the same reaction was attempted in batch the yield could not be optimised beyond 20%, necessitating a stepwise synthesis of 5.
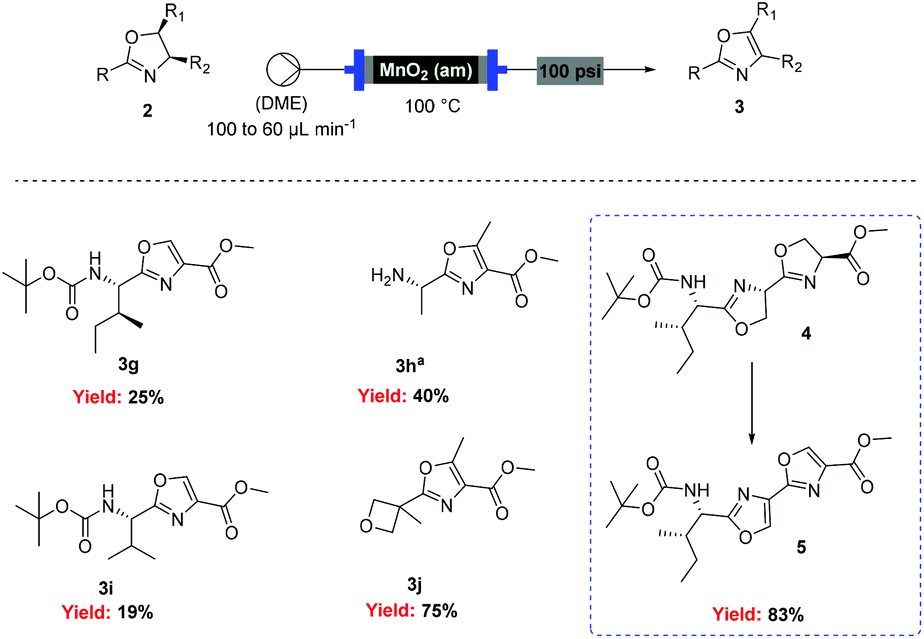 | ||
| Scheme 5 Flow oxidation of 2-alkyl-oxazolines using amorphous MnO2. aDeprotection was observed. | ||
When using a flow system, there is the possibility to take advantage of the ability to perform reactions and processes under computer control.35 In this case, the addition of an automation layer enabled up to four reactions using different substrates to be performed in sequence, with no manual intervention required apart from loading the reagents before starting the procedure. A reactor configuration (Scheme 6) was devised that incorporated a multi-port switching valve (V1) such that four separate substrates could be passed through four separate columns to prevent any cross-contamination between samples. A secondary valve (V2) enabled the flow stream to be thoroughly purged between each sample, and prevented cross-contamination as V1 moved between the different positions. The pre-prepared reagent solutions were drawn from four separate bottles (selected by V1) and passed through the heated MnO2 columns (1–4). A software protocol created in the Python programming language35,36 and running on a Raspberry Pi® microcomputer37 controlled the individual reactor components according to a pre-programmed sequence of timed actions, to co-ordinate the pumps and valves to perform the reactions. Using this system, a batch of four substrates was processed overnight (12 h). This demonstrates the ability for automated machine-assisted systems to save skilled researcher time in the laboratory.
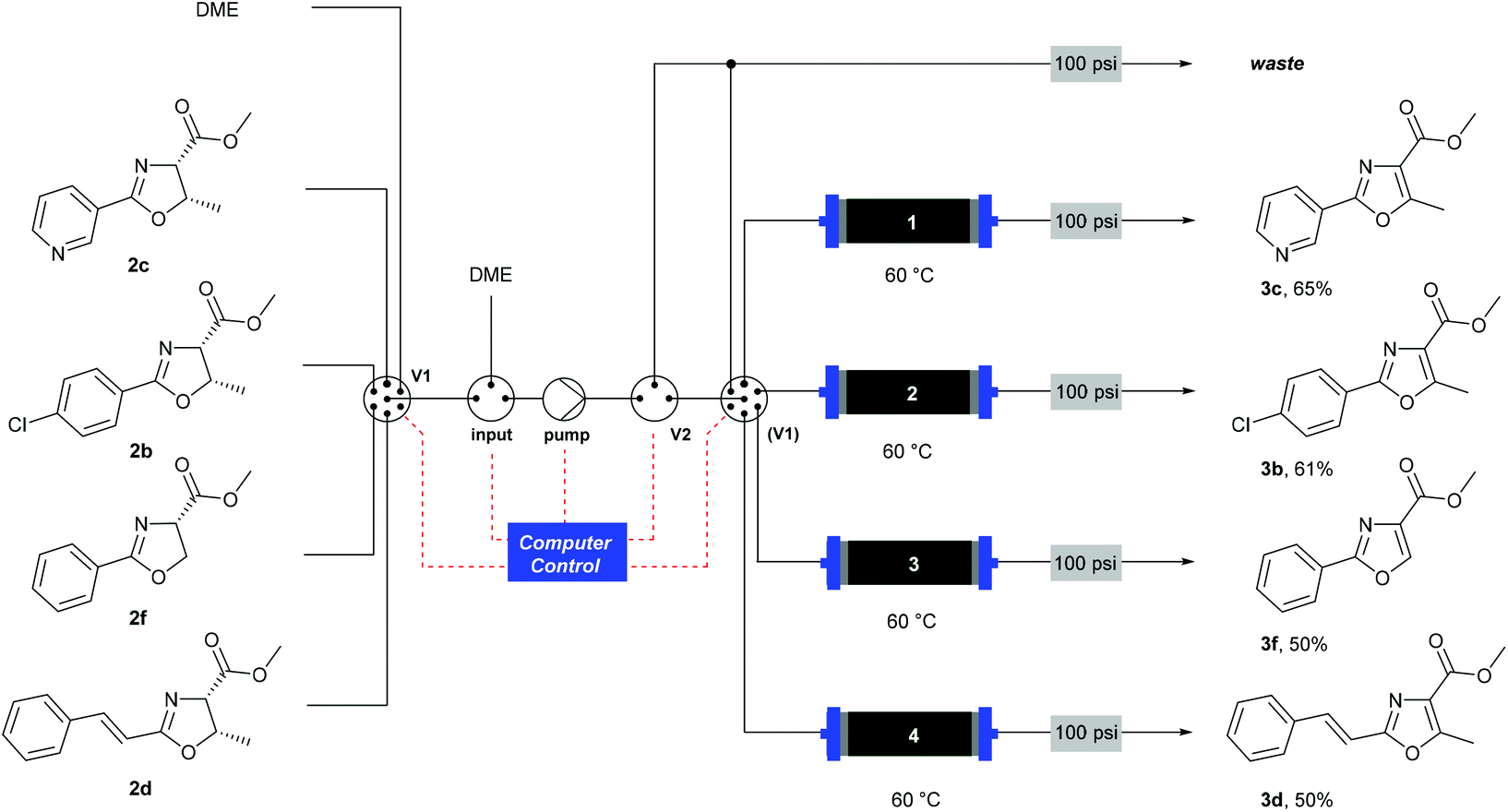 | ||
| Scheme 6 Automated oxidation of oxazolines using a Raspberry Pi® computer and a multiple position valve. | ||
Conclusions
In conclusion, an improved rapid method for the flow synthesis of oxazolines at room temperature has been developed. A multi-hour continuous flow run illustrates the potential of this method for the safe large scale production of oxazolines. It was found that aryl-substituted oxazolines undergo smooth oxidative aromatisation when simply flowed through a column of activated MnO2 at 60 °C, with isolated yields between 50–79%. Advantageously, unlike batch protocols, no downstream workup or purification was found to be required. The oxidation step has also been demonstrated to be amenable to automation which allows the sequential processing of 4 different substrates on a single flow platform, with minimal operator intervention. For the more sensitive 2-alkyl-substituted oxazolines, it was found that good results could be achieved using amorphous manganese dioxide at increased temperature, increasing the scope of this process.Experimental section
General experimental section
1H-NMR spectra were recorded on a Bruker Avance DPX-400 spectrometer with the residual solvent peak as the internal reference (CDCl3 = 7.26 ppm, d6-DMSO = 2.50 ppm). 1H resonances are reported to the nearest 0.01 ppm. 13C-NMR spectra were recorded on the same spectrometers with the central resonance of the solvent peak as the internal reference (CDCl3 = 77.16 ppm, d6-DMSO = 39.52 ppm). All 13C resonances are reported to the nearest 0.1 ppm. DEPT 135, COSY, HMQC, and HMBC experiments were used to aid structural determination and spectral assignment. The multiplicity of 1H signals are indicated as: s = singlet, d = doublet, t = triplet, m = multiplet, br. = broad, or combinations of thereof. Coupling constants (J) are quoted in Hz and reported to the nearest 0.1 Hz. Where appropriate, averages of the signals from peaks displaying multiplicity were used to calculate the value of the coupling constant. Infrared spectra were recorded neat on a PerkinElmer Spectrum One FT-IR spectrometer using Universal ATR sampling accessories. Unless stated otherwise, reagents were obtained from commercial sources and used without purification. The removal of solvent under reduced pressure was carried out on a standard rotary evaporator. Melting points were performed on either a Stanford Research Systems MPA100 (OptiMelt) automated melting point system and are uncorrected. High resolution mass spectrometry (HRMS) was performed using a Waters Micromass LCT Premier™ spectrometer using time of flight with positive ESI, or conducted by Mr Paul Skelton on a Bruker BioApex 47e FTICR spectrometer using (positive or negative) ESI or EI at 70 eV to within a tolerance of 5 ppm of the theoretically calculated value. LC-MS analysis was performed on an Agilent HP 1100 series chromatography (Mercury Luna 3u C18 (2) column) attached to a Waters ZQ2000 mass spectrometer with ESCi ionization source in ESI mode. Elution was carried out at a flow rate of 0.6 mL min−1 using a reverse phase gradient of acetonitrile and water containing 0.1% formic acid. Retention time (Rt) is given in min to the nearest 0.1 min and the m/z value is reported to the nearest mass unit (m.u.). Specific optical rotation was recorded on a Perkin-Elmer Model 343 digital polarimeter, using a Na/Hal lamp set at 589 nm and with a path length of 100 mm. All [α]D values were measured using spectroscopy grade solvent at the specified concentration and temperature. Unless otherwise specified all the flow reactions were performed using a Vapourtec R2/R4+ flow platform.38General protocol for the preparation of oxazoline in flow
A solution of Deoxo-Fluor® (1 mL, 50% in toluene) in CH2Cl2 (7.0 mL) and a solution of β-hydroxy amide (2 mmol) in CH2Cl2 (8 mL) were combined at a T-piece (each stream run at 3.0 mL min−1) and reacted at rt in a 10 mL PFA reactor coil. The combined stream was then directed to an aqueous quenching stream (9 mL min−1) and the solution directed to a liquid/liquid separator.22![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 2953, 1736, 1645, 1603, 1580, 1496, 1450, 1384, 1349, 1244, 1197, 1174, 1067, 1045, 1001, 973, 934, 904, 886, 851, 778, 695; specific rotation: [α]24.1D = +58.58° cm3 g−1 dm−1 (c = 8.5 in ethanol). Lit.: [α]20D = +69.4° cm3 g−1 dm−1 (c = 8.5 in EtOH).39
(cm−1) = 2953, 1737, 1646, 1599, 1491, 1437, 1404, 1384, 1355, 1244, 1197, 1172, 1091, 1075, 1045, 1015, 935, 904, 887, 840, 780, 731, 680; specific rotation: [α]24.0D = +48.2° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2953, 1743, 1650, 1593, 1432, 1417, 1353, 1246, 1198, 1176, 1082, 1044, 1023, 936, 905, 886, 821, 781, 708, 620; specific rotation: [α]24.0D = +53.0° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2951, 1735, 1652, 1607, 1496, 1449, 1358, 1242, 1197, 1174, 1045, 972, 948, 930, 840, 758, 732, 692; specific rotation: [α]24.0D = +27.9° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2977, 1753, 1740, 1704, 1649, 1538, 1453, 1361, 1337, 1303, 1261, 1213, 1177, 1132, 1088, 1066, 1047, 982, 957, 918, 897, 878, 855, 835, 776, 755, 725; specific rotation: [α]23.9D = −7.4° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2926, 1738, 1658, 1450, 1361, 1255, 1198, 1175, 1133, 1044, 982, 945, 844, 781, 732; specific rotation: [α]23.9D = +47.2° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 3342, 3256, 2967, 1740, 1687, 1658, 1626, 1521, 1454, 1390, 1365, 1288, 1240, 1200, 1170, 1042, 1019, 870, 777; specific rotation: [α]26.0D = +19.7° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2953, 1736, 1645, 1603, 1580, 1496, 1450, 1384, 1349, 1244, 1197, 1174, 1067, 1045, 1001, 973, 934, 904, 886, 851, 778, 695; specific rotation: [α]24.1D = +58.58° cm3 g−1 dm−1 (c = 8.5 in ethanol). Lit.: [α]20D = +69.4° cm3 g−1 dm−1 (c = 8.5 in EtOH).39
(cm−1) = 2953, 1737, 1646, 1599, 1491, 1437, 1404, 1384, 1355, 1244, 1197, 1172, 1091, 1075, 1045, 1015, 935, 904, 887, 840, 780, 731, 680; specific rotation: [α]24.0D = +48.2° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2953, 1743, 1650, 1593, 1432, 1417, 1353, 1246, 1198, 1176, 1082, 1044, 1023, 936, 905, 886, 821, 781, 708, 620; specific rotation: [α]24.0D = +53.0° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2951, 1735, 1652, 1607, 1496, 1449, 1358, 1242, 1197, 1174, 1045, 972, 948, 930, 840, 758, 732, 692; specific rotation: [α]24.0D = +27.9° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2977, 1753, 1740, 1704, 1649, 1538, 1453, 1361, 1337, 1303, 1261, 1213, 1177, 1132, 1088, 1066, 1047, 982, 957, 918, 897, 878, 855, 835, 776, 755, 725; specific rotation: [α]23.9D = −7.4° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 2926, 1738, 1658, 1450, 1361, 1255, 1198, 1175, 1133, 1044, 982, 945, 844, 781, 732; specific rotation: [α]23.9D = +47.2° cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) = 3342, 3256, 2967, 1740, 1687, 1658, 1626, 1521, 1454, 1390, 1365, 1288, 1240, 1200, 1170, 1042, 1019, 870, 777; specific rotation: [α]26.0D = +19.7° cm3 g−1 dm−1 (c = 1 in CHCl3).
General protocol for the preparation of 2-aryl-oxazoles in flow
A solution of oxazoline (0.5 mmol) in DME (8 mL) was passed through a column reactor packed with activated MnO2 at 60 °C (flow rate 100 μL min−1 for 50 min then 60 μL min−1). The machine output was collected and concentrated under vacuum to give the corresponding oxazoles in good yield, without the need of further purification. (cm−1) = 3001, 2950, 2857, 1717, 1614, 1585, 1559, 1485, 1450, 1346, 1278, 1235, 1109, 1090, 1056, 1011, 827, 726, 692.
(cm−1) = 3011, 2953, 1713, 1614, 1577, 1483, 1431, 1418, 1386, 1347, 1235, 1197, 1109, 1071, 1018, 979, 820, 783, 719, 703.
(cm−1) 2954, 1715, 1644, 1544, 1438, 1388, 1367, 1220, 1180, 1102, 988, 965, 757.
(cm−1) 2977, 2364, 1706, 1616, 1443, 1365, 1262, 1175, 1106, 984, 855, 731.
General protocol for the preparation of 2-alkyl-oxazoles in flow
A solution of oxazoline (0.125 mmol) in DME (2 mL) was loaded into a 2 mL loop and then passed through a column reactor packed with activated MnO2 at 100 °C (flow rate 100 μL min−1 for 50 min then 60 μL min−1). Unless otherwise stated, the machine output was collected and concentrated under vacuum to give the corresponding oxazoles. (cm−1) 3323, 2972, 1728, 1698, 1530, 1328, 1232, 1152, 1105; specific rotation: [α]28.3D = −26.2 cm3 g−1 dm−1 (c = 1 in CHCl3).
(cm−1) 2956, 2880, 1719, 1668, 1621, 1585, 1441, 1350, 1250, 1180, 1132, 1090, 980, 787.
(cm−1) 3141, 2959, 1721, 1690, 1509, 1247, 1147, 1001, 870, 729; specific rotation: [α]D25.0 = −34.0 cm3 g−1 dm−1 (c = 1 in CHCl3).
Acknowledgements
We are grateful to the Swiss National Science Foundation (DNT), the Ralph Raphael fellowship (RJI), the German Academic Exchange Service DAAD (SF), the Royal Society Newton International Fellowship (ZEW), Pfizer Worldwide Research and Development (CB) and the EPSRC (SVL, SF and ZEW, grant n° EP/K0099494/1) for financial support. The Labtrix Start was kindly loaned by Chemtrix BV and the liquid-liquid phase separator by Zaiput Flow Technologies (we are grateful to Dr Andrea Adamo for technical support with this device).Notes and references
- L. T. Tan, R. T. Williamson, W. H. Gerwick, K. S. Watts, K. McGough and R. Jacobs, J. Org. Chem., 2000, 65, 419–425 CrossRef CAS PubMed.
- J. A. McIntosh, M. S. Donia and E. W. Schmidt, Nat. Prod. Rep., 2009, 26, 537–559 RSC.
- P. Wipf and C. P. Miller, Tetrahedron Lett., 1992, 907–910 CrossRef CAS.
- P. Wipf and C. P. Miller, Tetrahedron Lett., 1992, 33, 6267–6270 CrossRef CAS.
- F. Yokokawa and T. Shioiri, Tetrahedron Lett., 2002, 43, 8679–8682 CrossRef CAS.
- F. Yokokawa, Y. Hamada and T. Shioiri, Synlett, 1992, 153–155 CrossRef CAS PubMed.
- A. I. Meyers and D. Hoyer, Tetrahedron Lett., 1985, 26, 4687–4690 CrossRef CAS.
- A. J. Phillips, Y. Uto, P. Wipf, M. J. Reno and D. R. Williams, Org. Lett., 2000, 2, 1165–1168 CrossRef CAS.
- M. Baumann, I. R. Baxendale, M. Brasholz, J. J. Hayward, S. V. Ley and N. Nikbin, Synlett, 2011, 1375–1380 CAS.
- D. W. Knight, G. Pattenden and D. E. Rippon, Synlett, 1990, 36–37 CrossRef CAS.
- J. A. Lafontaine, D. P. Provencal, C. Gardelli and J. W. Leahy, J. Org. Chem., 2003, 68, 4215–4234 CrossRef CAS PubMed.
- A. I. Meyers and F. Tavares, Tetrahedron Lett., 1994, 35, 2481–2484 CrossRef CAS.
- Y. Huang, L. Ni, H. Gan, Y. He, J. Xu, X. Wu and H. Yao, Tetrahedron, 2011, 67, 2066–2071 CrossRef CAS PubMed.
- Y.-B. Yu, H.-L. Chen, L.-Y. Wang, X.-Z. Chen and B. Fu, Molecules, 2009, 14, 4858–4865 CrossRef CAS PubMed.
- K. Meguro, H. Tawada, Y. Sugiyama, T. Fujita and Y. Kawamatsu Awamatsu, Chem. Pharm. Bull., 1986, 34, 2840–2851 CrossRef CAS.
- C. Shin, Y. Nakamura and K. Okumura, Chem. Lett., 1993, 1405–1408 CrossRef CAS.
- For information about Omnifit® glass columns, see: http://www.omnifit.com/.
- C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2014, 16, 1060–1063 CrossRef CAS PubMed.
- A. Fernández, Z. C. Levine, M. Baumann, S. Sulzer-Mossé, C. Sparr, S. Schläger, A. Metzger, I. R. Baxendale and S. V. Ley, Synlett, 2013, 514–518 Search PubMed.
- C. Battilocchio, M. Baumann, I. R. Baxendale, M. Biava, M. O. Kitching, S. V. Ley, R. E. Martin, S. A. Ohnmacht and N. D. C. Tappin, Synthesis, 2012, 635–647 CAS.
- (a) Due to the approximate concentration of Deoxo-Fluor® solutions (≈50% in toluene) in commercial samples, we could only obtain reproducible results using 1.35 equiv. of reagent; (b) Deoxo-Fluor® (≈50% in toluene) was purchased from Sigma Aldrich (94324-50ML-F) and used as supplied.
- (a) For information about the Zaiput liquid-liquid separator, see https://www.zaiput.com; for a description of the device, see also: (b) A. Adamo, P. L. Heider, N. Weeranoppanant and K. F. Jensen, Ind. Eng. Chem. Res., 2013, 52, 10802–10808 CrossRef CAS; (c) T. A. Hamlin, G. M. L. Lazarus, C. B. Kelly and N. E. Leadbeater, Org. Process Res. Dev., 2014, 18, 1253–1258 CrossRef CAS.
- J.-i. Yoshida, Flash Chemistry: Fast Organic Synthesis in Microsystems, Wiley-VCH, 2008 Search PubMed.
- V. Hessel, H. Löwe and F. Schönfeld, Chem. Eng. Sci., 2005, 60, 2479–2501 CrossRef CAS PubMed.
- A. D. Stroockl, S. K. W. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647–651 CrossRef PubMed.
- X. Fu, S. Liu, X. Ruan and H. Yang, Sens. Actuators, B, 2006, 114, 618–624 CrossRef CAS PubMed.
- For information about the Labtrix® Start system, see: http://www.chemtrix.com.
- For a recent review on mixing effect under flow conditions, see: R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- R. J. Gritter, G. D. Dupre and T. J. Wallace, Nature, 1964, 202, 179–181 CrossRef CAS.
- A. J. Fatiadi, Synthesis, 1976, 65–104 CrossRef CAS.
- R. J. K. Taylor, M. Reid, J. Foot and S. A. Raw, Acc. Chem. Res., 2005, 38, 851–869 CrossRef CAS PubMed.
- Activated MnO2 was purchased from Sigma-Aldrich (217646-100G) and used as supplied.
- Amorphous MnO2 was purchased from Sigma-Aldrich (310700-500G) and used as supplied.
- Z. E. Wilson, S. Fenner and S. V. Ley, submitted manuscript.
- R. J. Ingham, C. Battilocchio, J. M. Hawkins and S. V. Ley, Beilstein J. Org. Chem., 2014, 10, 641–652 CrossRef CAS PubMed.
- For information about the Python programming language, see: http://www.python.org.
- For information about the Raspberry Pi® microcomputer, see: http://www.raspberrypi.org.
- For information about the Vapourtec R2+/R4 reactor, see: http://www.vapourtec.co.uk/.
- H. Aït-Haddou, O. Hoarau, D. Cramailére, F. Pezet, J.-C. Daran and G. G. A. Balavoine, Chem. – Eur. J., 2004, 10, 699–707 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical characterisation of compounds and spectra. See DOI: 10.1039/c4ob02105c |
| This journal is © The Royal Society of Chemistry 2015 |