
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of 1,2-diamine containing indolines by a conjugate addition nitro-mannich reaction†
James C.
Anderson
*a,
Ian B.
Campbell
b,
Sebastien
Campos
b,
Jonathan
Shannon
a and
Derek A.
Tocher‡
a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: j.c.anderson@ucl.ac.uk
bGlaxoSmithKline, Medicines Research Centre, SG1 2NY Stevenage, UK
First published on 20th October 2014
Abstract
A conjugate addition nitro-Mannich reaction followed by nitro reduction and intramolecular N-arylation gives diastereomerically pure substituted 1,2-diamine containing indolines. Placing the N-arylation cyclisation handle on the imine precursor derived from an ortho-bromine substituted aromatic aldehyde gave the corresponding β-nitroamines in 55–72% yields as single diastereoisomers. Nitro reduction was effected with modified quantities of Zn/HCl and a subsequent Pd(0) catalysed Buchwald Hartwig cyclisation gave indoline products in 40–70% yields as single diastereoisomers.
Introduction
The nitro-Mannich (or aza-Henry) reaction has emerged as a reliable and predictable reaction for the synthesis of β-nitroamines in stereochemically pure form.1,2 These products have proven useful in the synthesis of many nitrogen containing functional groups including 1,2-diamines,3–10 α-amino carbonyls,11–13 peptidomimetics,14 natural products15–20 and many heterocyclic small molecules21–32 of importance to drug discovery. The anti-diastereoisomer dominates with higher homologues of nitromethane, with there being only a few methods for syn-selective nitro-Mannich reactions.26,33–35 We reported the enantioselective alkyl conjugate addition nitro-Mannich reaction of dialkyl zincs to nitroalkenes to synthesise complex β-nitroamines containing three contiguous stereocentres (Scheme 1).36 The judicious choice of solvent determined whether the syn,anti- or syn,syn-diastereoisomer was formed and provided another method for the synthesis of syn-β-nitroamines. As part of an investigation into using the nitro-Mannich reaction in diversity-oriented array synthesis, we were interested in the synthesis of arrays of stereochemically diverse fused heterocyclic ring systems. We have recently shown that the reductive nitro-Mannich reaction with arylbromide nitrostyrenes can deliver stereodefined functionalised diamine building blocks that are precursors to either 3-aminotetrahydroquinolines or 2-aminomethylene indolines via N-arylation (Scheme 1).37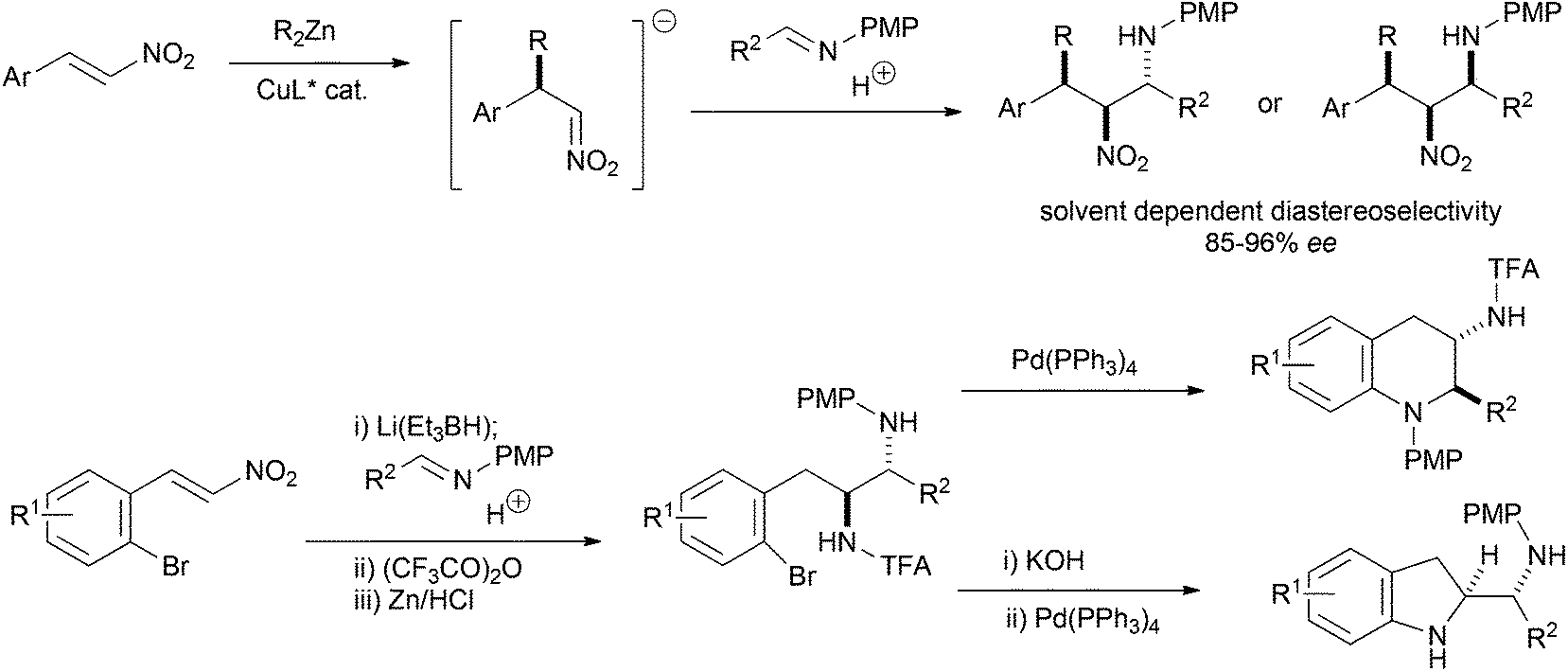 | ||
| Scheme 1 Previous work. | ||
We detail here our investigation into the combination of the alkyl conjugate addition nitro-Mannich reaction with suitable aryl bromide containing coupling partners and their subsequent intramolecular cyclisation by palladium catalysed N-arylation to yield novel drug like heterocyclic ring systems (Scheme 2).
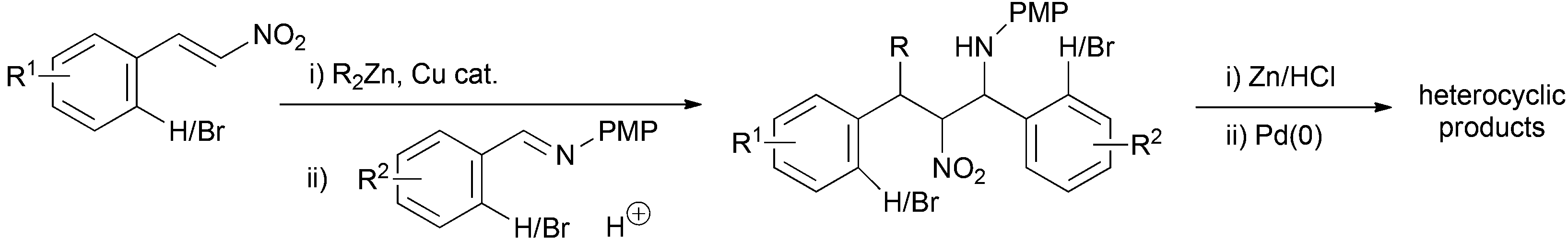 | ||
| Scheme 2 Proposed array synthesis of complex heterocycles. | ||
Results and discussion
Using conditions previously developed by us, the simplest 2-bromonitrostyrene (1a) underwent smooth Cu(OTf)2 catalysed (5 mol%) conjugate addition of ZnEt2 (1.1 equiv.) as judged by TLC. The resultant nitronate species was then reacted with PMP protected imine 2a (2.2 equiv.) and TFA (2.6 equiv.) with quenching at −78 °C. Crude 3aa was isolated in a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 ratio of two of the possible four diastereoisomers, determined by 1H NMR (Scheme 3). Preliminary investigation of the reaction conditions varied the temperature after TFA addition and the time before quenching. The selectivity tended towards 1:1 and the crude product was unstable over time, reverting to starting materials.38 Usually formation of the corresponding trifluoroacetamide leads to an isolable product, but in this instance the instability of 3aa and the reluctance of similar syn,syn-diastereoisomers to be protected, led to the isolation of only 34% of the syn,anti-4aa diastereoisomer as a single diastereoisomer.36,§ This product was most likely the minor diastereoisomer in the mixture of 3aa.
:40 ratio of two of the possible four diastereoisomers, determined by 1H NMR (Scheme 3). Preliminary investigation of the reaction conditions varied the temperature after TFA addition and the time before quenching. The selectivity tended towards 1:1 and the crude product was unstable over time, reverting to starting materials.38 Usually formation of the corresponding trifluoroacetamide leads to an isolable product, but in this instance the instability of 3aa and the reluctance of similar syn,syn-diastereoisomers to be protected, led to the isolation of only 34% of the syn,anti-4aa diastereoisomer as a single diastereoisomer.36,§ This product was most likely the minor diastereoisomer in the mixture of 3aa.
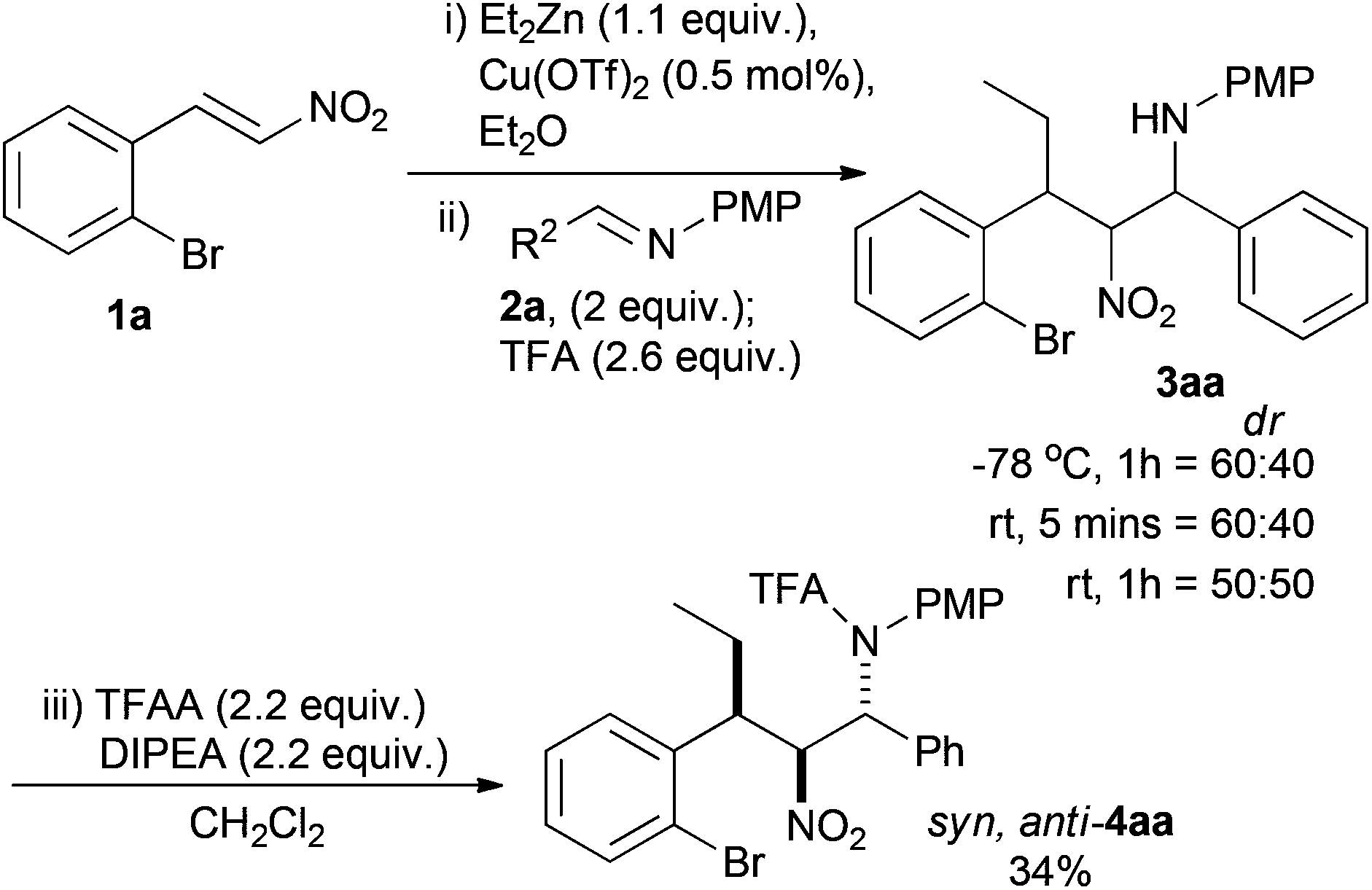 | ||
| Scheme 3 Preliminary conjugate addition nitro-Mannich reaction. | ||
A screen of different protecting groups of the imine partner 2 (COPh, CO2Bn, POPh2) along with various Lewis and Bronsted acid promoters in place of TFA (AcOH, CF3SO3H, MeSO3H, Yb(OTf)3, AlCl3, Zn(OTf)2, BF3·Et2O) was undertaken. Disappointingly no conditions were found that gave either high conversion or a good level of diastereoselectivity with the 2-bromonitrostyrene (1a). These results were similar to what we had found in our earlier work with other ortho-substituted nitrostyrenes.36
An alternative cyclisation precursor was envisaged where the halide coupling partner was part of the aldehyde that the imine was derived from. This small change had a dramatic effect on the extent of diastereoselectivity, and stability of the nitro-Mannich product (Scheme 4). Complete consumption of 1b was verified by TLC and the reaction went to complete conversion as judged by 1H NMR. The diastereoselectivity of the crude product did not change over reaction time or temperature after addition of TFA, indicating that the major stereoisomer was thermodynamically stable. We tentatively assigned the stereochemistry as the syn,syn-diastereoisomer by analogy to previous work where we found that this particular diastereoisomer was relatively stable and could be purified by column chromatography to give diastereomerically pure material.36 With 2 equivalents of imine 2b the major diastereoisomer syn,syn-3bb could be isolated in 72% yield¶ and the relative stereochemistry was confirmed by single crystal X-ray analysis (see ESI†). Reducing the quantity of imine 2b to 1.1 equivalents led to no change in crude diastereoselectivity (85:15), but the major compound was significantly easier to purify and was isolated in 68% yield.
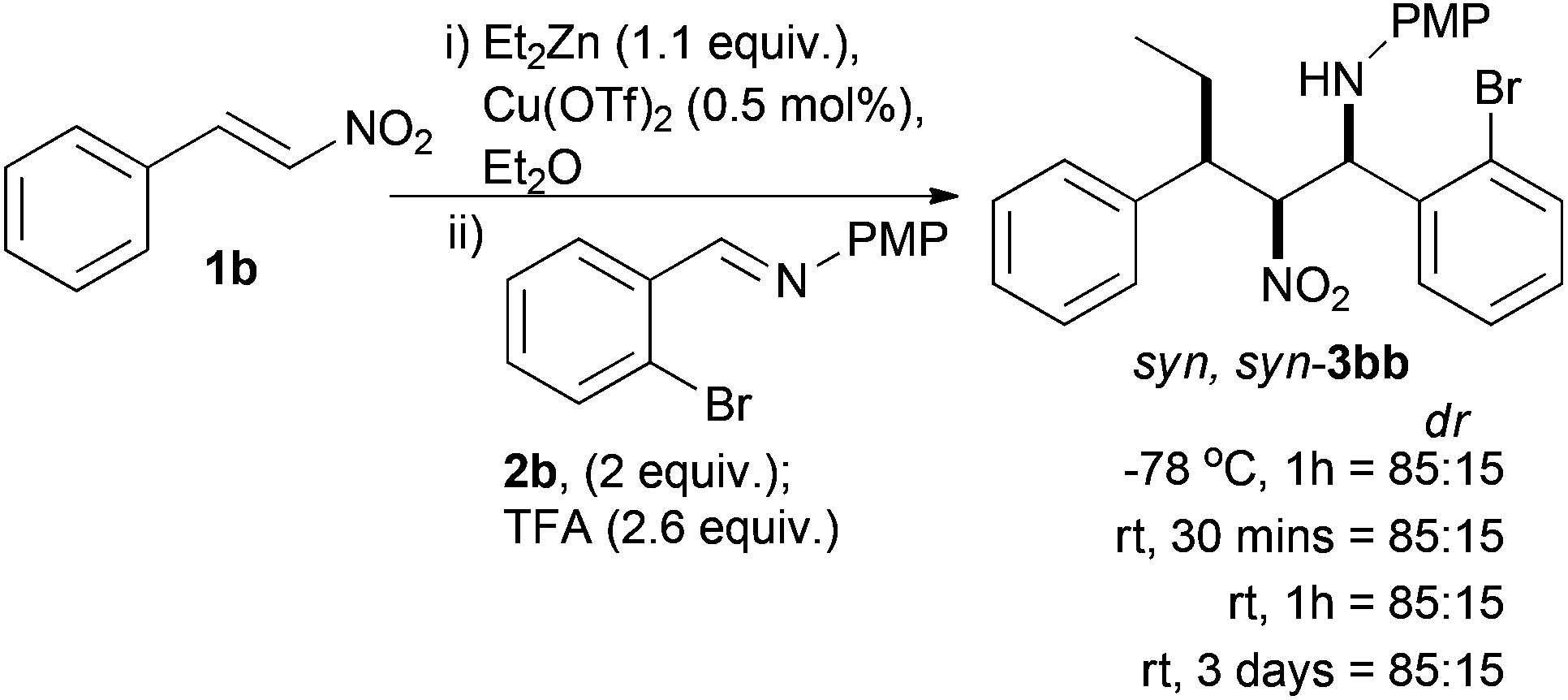 | ||
| Scheme 4 Stable cyclisation precursor. | ||
A solvent screen (see ESI†) showed that Et2O gave the highest diastereoselectivity followed by toluene (80:20), which contained more side products and unlike Et2O was a homogenous solution. Other solvents which gave homogeneous reaction mixtures did not give as the major diastereoisomer the syn,anti-product which had been previously observed with very similar substrates.36 In most cases the syn,syn-diastereoisomer was the major compound to varying degrees and all attempts at protection of the amine as a trifluoroacetamide were unsuccessful, which is in agreement with our previous observations for this particular diastereoisomer.36
Intramolecular N-arylation required the reduction of the nitro function to an amine. Investigations commenced with 3bb and initial conditions we have developed for the reduction of β-nitroamines into 1,2-diamines using Zn/HCl (Scheme 5, conditions A).36–38 Although the nitro group was completely reduced, a 1:1 mixture of the desired amine 5bb and the debrominated material 6 were obtained. Optimisation studies found that reducing the amount of reductant zinc, but increasing the relative amount of HCl lead cleanly to the desired diamine. Normally β-nitroamines (like 3bb) would be isolated as their corresponding trifluoroacetamides and subsequent reduction is accompanied by migration of the trifluoroacetyl group to the primary nitrogen, giving a stable isolable product.36–38 However, as the syn,syn-diastereoisomers like 3bb are in general inert to trifluoroacetylation,36 the resultant 1,2-diamine reduction product 5bb was found to become less pure upon column chromatography. As a consequence intramolecular cyclisation using palladium catalysed N-arylation was attempted on the crude reduction material.
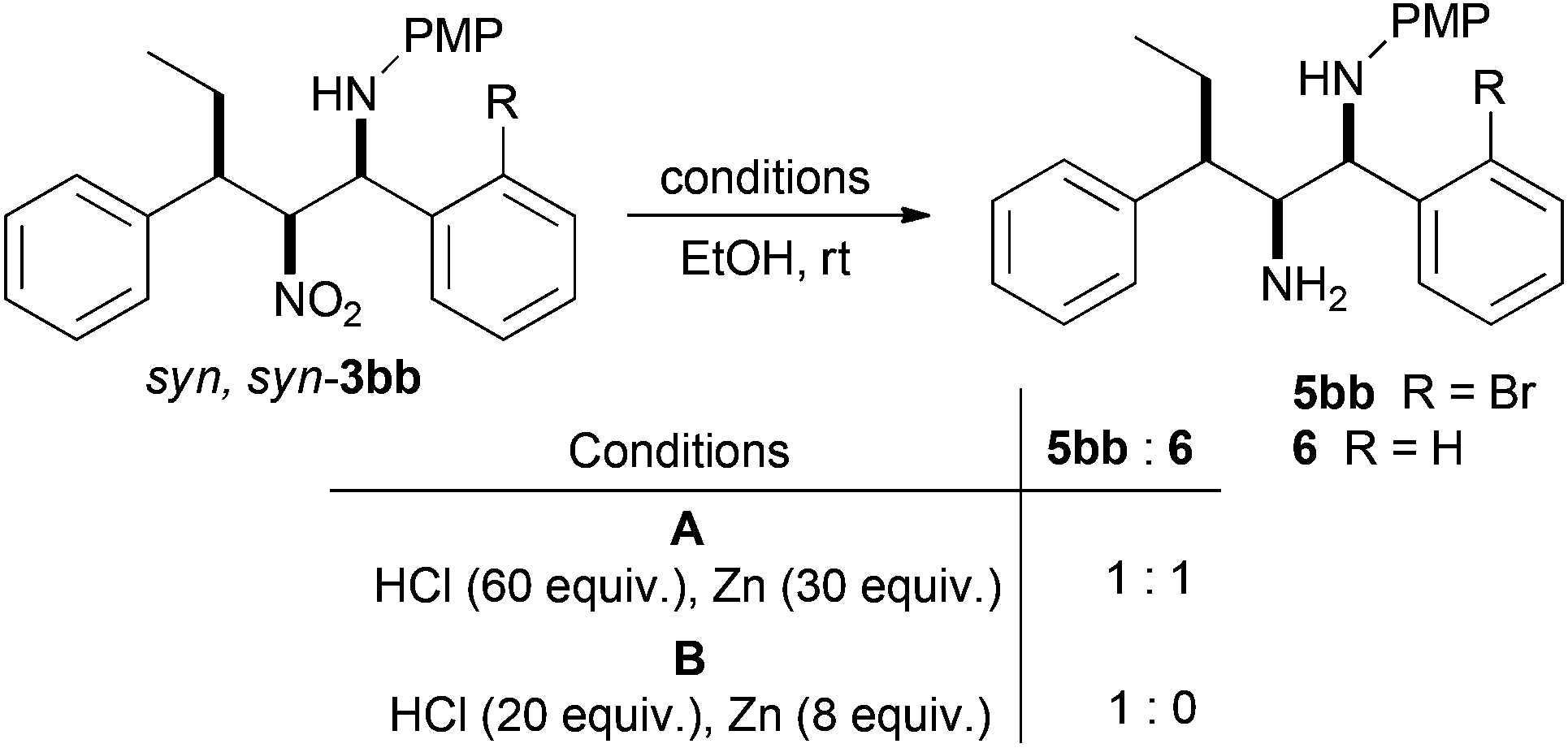 | ||
| Scheme 5 Nitro reduction. | ||
Treatment of 5bb under standard Pd(PPh3)4 Buchwald–Hartwig conditions that we have used to perform analogous cyclisations (Scheme 1),37 led cleanly to the indoline 7bb by 1H NMR of the crude cyclisation product (Scheme 6). Initial attempts at purification using silica gel chromatography led to degradation and isolation of the corresponding indole 8 (∼25%) as a mixture with p-anisidine. Indoline 7bb is unstable to acidic conditions, but using basic alumina chromatography we were able to isolate 7bb in a good 60% yield over two steps as a single diastereoisomer. Use solely of NaOtBu as base led to 64% yield of indoline 7bb (Scheme 6).
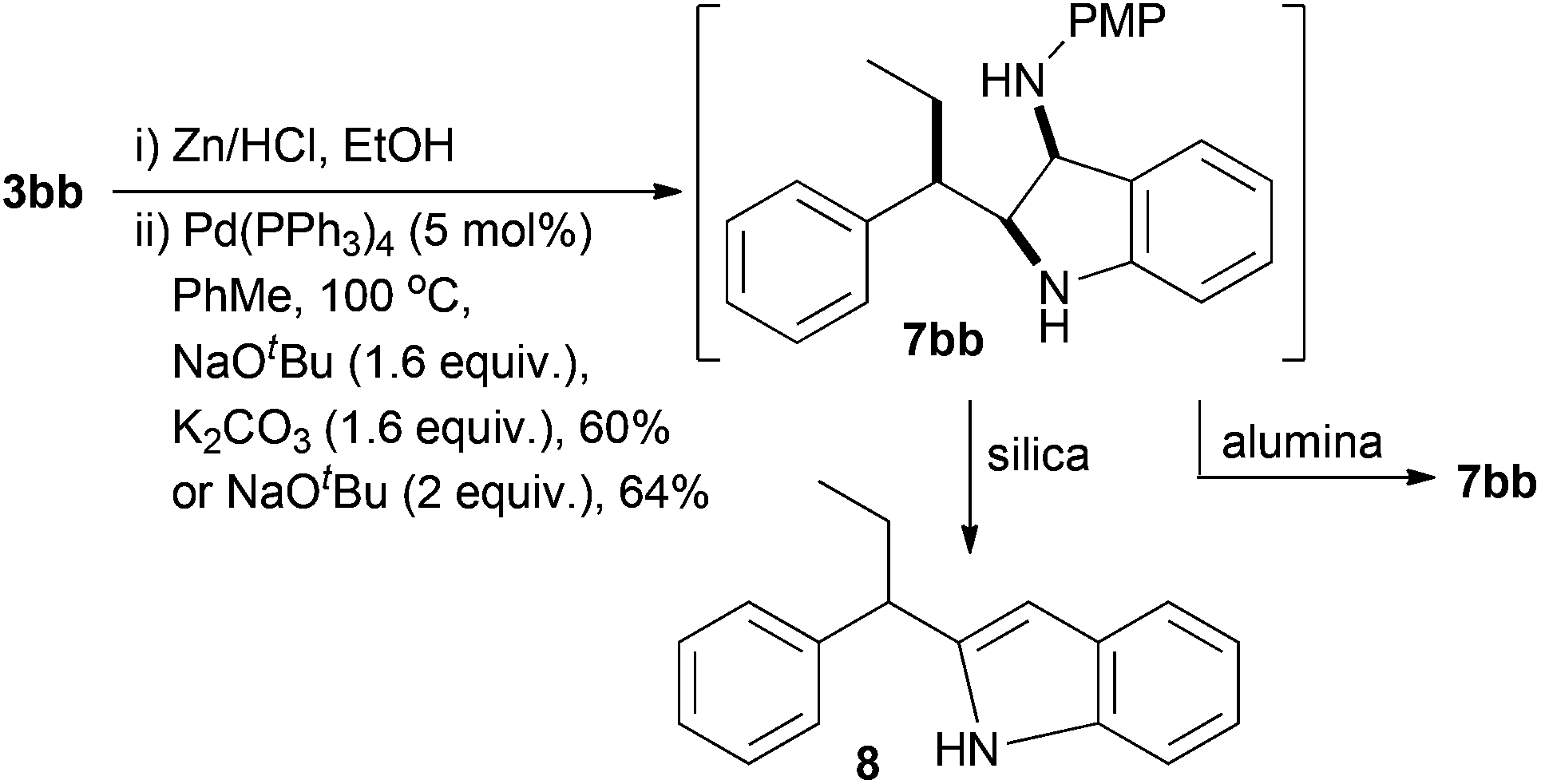 | ||
| Scheme 6 Indoline formation. | ||
The scope of this reaction sequence for the synthesis of the 1,2-diamine indolines was then investigated with a series of aldimines containing an ortho-bromine susbstituent and nitro styrenes under the optimised conditions described above (Scheme 7 and Table 1).
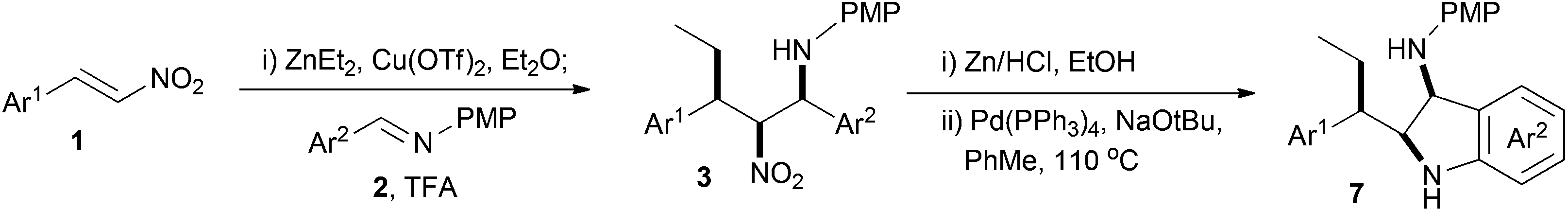 | ||
| Scheme 7 Scope of indoline formation. | ||
| Entry | Ar1 | Ar2 | 3 dr crudeb | Yield 3c (%) | Yield 7c (%) |
|---|---|---|---|---|---|
| a Nitro-Mannich: 1 (3 mmol), Cu(OTf)2 (5 mol%), ZnEt2 (1.1 equiv.), Et2O, −78 °C, 5 min, then RT 2 h; 2 (1.1 equiv.), Et2O, −78 C, 10 min; TFA (2.6 equiv.), Et2O, −78 °C, 1 h then to RT 1 h. Nitro reduction: conc. HCl (10 mmol) added to 3 (0.50 mmol), Zinc (4.00 mmol), in EtOH (7.5 mL) at 0 °C, 5 min then RT. N-arylation: crude 1,2-diamine in PhMe (1.5 ml) added to NaOtBu (1.00 mmol), Pd(PPh3)4 (0.025 mmol) in PhMe (1 ml) at RT then 90 °C, 16–24 h. b Diastereoselectivities were calculated by comparison of the 1H NMR signals for the CHCHNO2 protons (δ 3.1–3.6 ppm) of the crude reaction mixture. c Isolated yield of pure syn,syn-diastereoisomer. d Reaction failed at cyclisation. e Reaction failed at reduction. | |||||
| 1 | Ph | 2-BrPh | 85:15 |
68 | 64 |
| 2 | Ph | 2-Br-5-FPh | 90:10 |
72 | 53 |
| 3 | Ph | 2-Br-5-MeOPh | 80:20 |
63 | 40 |
| 4 | Ph | 2-Br-4,5-(MeO)2Ph | 80:20 |
57 | 0d |
| 5 | Ph | 2-Br-4-MePh | 85:15 |
63 | 64 |
| 6 | Ph | 2-Br-3-pyridinyl | 80:20 |
55 | 0e |
| 7 | 4-MePh | 2-BrPh | 80:20 |
64 | 64 |
| 8 | 2-MePh | 2-BrPh | 80:20 |
62 | 66 |
| 9 | 4-FPh | 2-BrPh | 75:25 |
62 | 70 |
The conjugate addition nitro-Mannich reaction proceeds well with a range of imines 1 and nitrostyrenes 2 to give good diastereoslectivites of syn,syn-products and good isolated yields of diastereomerically pure β-nitroamines 3. The reductions proceeded smoothly except for the β-nitroamine derived from 2-bromo-3-pyridinylaldehyde (entry 6), which led to an inseparable mixture of unidentified products. Use of Sn/HCl gave similar results and Al/Hg amalgam, which we have used for the reduction of sensitive β-nitroamines,39 gave a complex mixture of products. No doubt the sensitivity of the pyridine function and the C–Br bond are both compounding this reduction. Cyclisations to give the indoline nucleus by standard Pd(PPh3)4 Buchwald–Hartwig conditions gave good yields in most cases. The β-nitroamine derived from 2-bromo-5-methoxy benzaldehyde (entry 3) cyclised using Pd(PPh3)4 albeit with a moderate yield of 40%. The congener derived from 2-bromo-4,5-dimethoxy benzaldehyde (entry 4) failed to cyclise and gradually led to decomposition of the β-nitroamine over longer reaction times. Changing to a Binap or X-Phos ligand system, which is known to coupling electron rich aryl bromides,40 also led to gradual decomposition.
With respect to the conjugate addition of other carbon nucleophiles, we have already shown that methyl and phenyl dialkylzinc species work in the conjugate addition nitro-Mannich reaction and led to syn,syn-β-nitroamines in high diastereoselectivity.36 There are a number of chiral metal catalysed systems that control the enantioselectivity of dialkyl zinc addition to nitro-alkenes and we have already shown that this is a good way of making the conjugate addition nitro-Mannich products enantioselectively,36 which would in turn lead to the heterocyclic products described here in enantiomerically pure form. This methodology is limited by the availability of dialkylzincs and although it is possible to add functionalised dialkyl zincs that can be derived from the tandem hydroboration/boron-zinc exchange method developed by Knochel,41 it would be better if more readily available carbon nucleophiles could be found. In an attempt to widen the scope of the reaction to include organozinc halides||,42,43 test reactions revealed that benzylzinc bromide and allylzinc bromide both gave low diastereoselectivities and trace conversions (<10%) with 1b and 2b in THF and Et2O. By TLC the reactions seem to falter at the conjugate addition stage. Exploration of Grignard reagents was not successful with vinyl magnesium bromide, but changing to ethylmagnesium bromide gave an optimised yield of 70% (Scheme 8), but unfortunately a significant drop in diastereoselectivity (60:40).
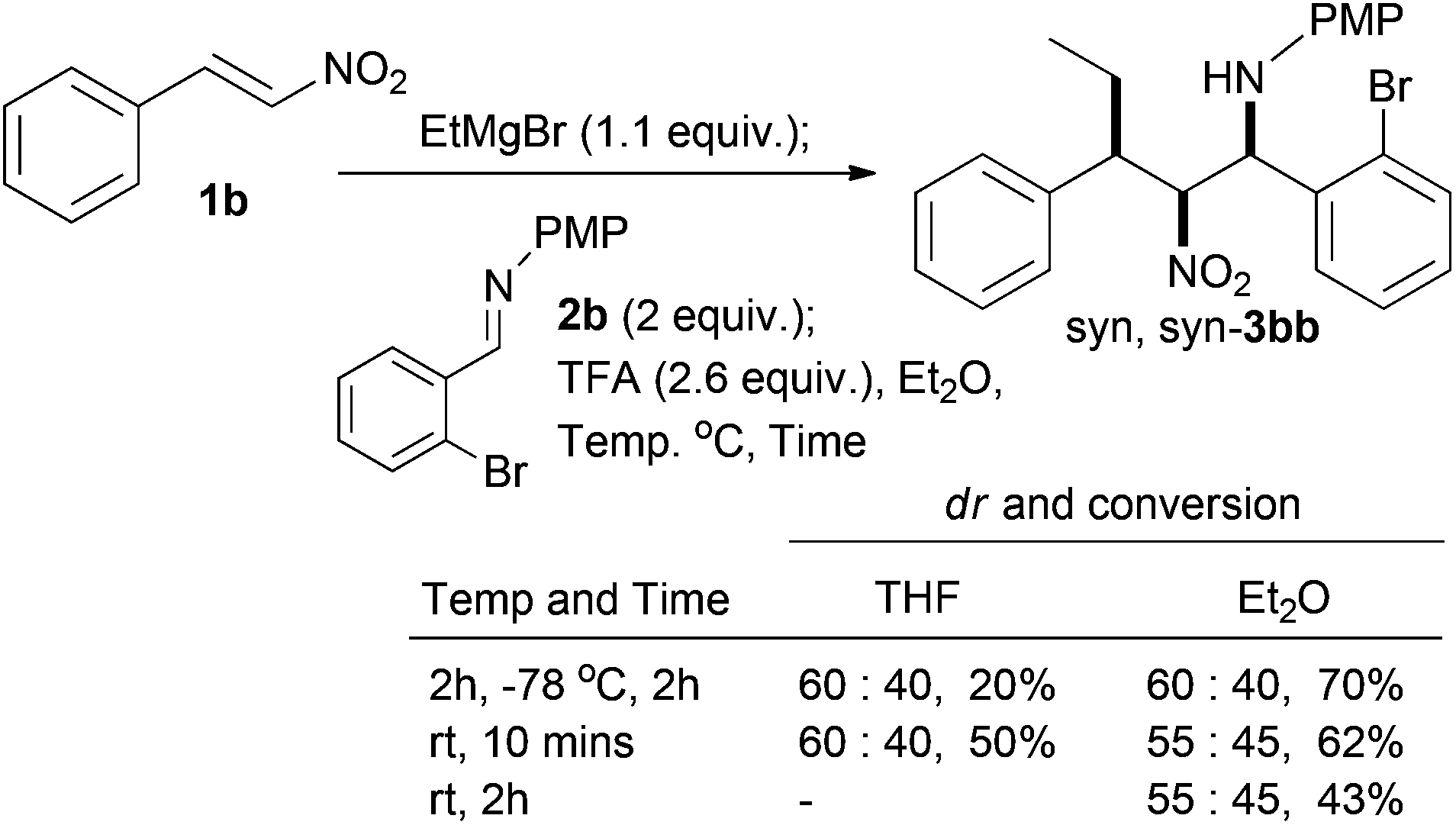 | ||
| Scheme 8 Use of Grignard nucleophile. | ||
We have found nucleophilic hydride useful in the conjugate addition nitro-Mannich reaction38,44 and with these particular substrates use of super hydride® led to anti-β-nitroamine 9, that could be smoothly reduced and cyclised to give the indoline 10 in 75% yield from 9 (Scheme 9). The sense of diastereoselection was anti- as previously described for other ortho-substituted aldimines.38
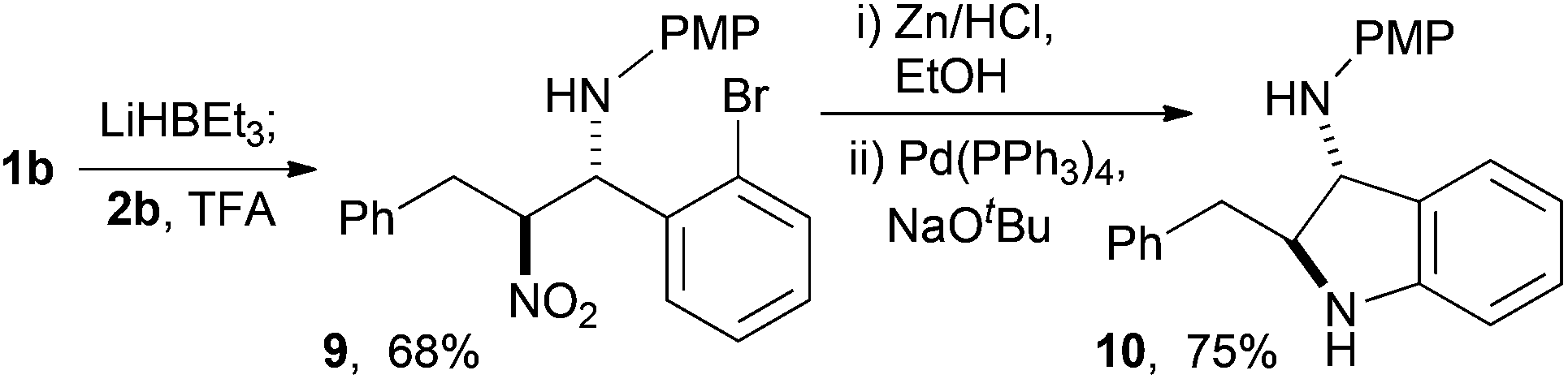 | ||
| Scheme 9 Sequence using the reductive nitro-Mannich reaction. | ||
Conclusion
We have developed a diastereoselective synthesis of substituted 1,2-diamine containing indolines, that represent novel drug like heterocyclic ring systems, from a conjugate addition nitro-Mannich reaction followed by nitro reduction and intramolecular N-arylation. Attempts to make indoline precursors from nitro styrenes containing an ortho-bromine substituent were low yielding and poorly diastereoselective (Scheme 3). Placing the N-arylation cyclisation handle on the imine precursor derived from an ortho-bromine substituted aromatic aldehyde was much more successful giving the corresponding β-nitroamine in good yield and as a single diastereoisomer. Nitro reduction was effected with modified quantities of Zn/HCl and a subsequent Pd(0) catalysed Buchwald Hartwig cyclisation gave indoline products in good yields as single diastereoisomers (Table 1). Electron rich aryl bromides were found to be reluctant to cyclise. Despite exploring other more readily available organometallic carbon nucleophiles, especially organozinc halides, a limitation to the current methodology is that dialkyl zinc species are the most efficient for the initial conjugate addition to nitro styrene. We have previously modified literature protocols to enable the enantioselective synthesis of suitably functionalised β-nitroamines for cyclisation to functionalised heterocycles36,37,44 and this methodology could also be used in this case. We are investigating the use of other nucleophiles to trigger this reaction and alternative cyclisation modes are being investigated to prepare alternate ring systems and will be reported in due course.Experimental section
General procedure for the synthesis of syn,syn-β-nitroamines 3 (Table 1)
To a stirred mixture of nitroalkene (3.00 mmol) and Cu(OTf)2 (0.05 mmol) in Et2O (5 mL) at −78 °C was added Et2Zn (3.3 mmol, 1 M in hexanes) dropwise. The mixture was stirred at this temperature for 5 min then at RT until the reaction was complete by TLC analysis (approximately 2 h). The reaction mixture was cooled to −78 °C and a solution of imine (3.3 mmol) in dry Et2O (5 mL) was added and the mixture stirred for 10 min. Then a solution of TFA (7.8 mmol) in Et2O (0.2 mL) was added dropwise over 20 s and the reaction stirred for 1 h. The reaction was warmed to room temperature over 1 h to provide a suspension of white solid in a vivid yellow supernatant. The reaction was quenched by the addition of Et2O and saturated aq. NaHCO3. The layers were separated, and the aqueous phase was extracted with Et2O. The organic layers were combined, and the solvent was removed in vacuo to provide crude β-nitroamine. Diastereoselectivities were calculated by comparison of the 1H NMR signals for the CHCHNO2 protons (δ 3.1–3.6 ppm). Purification by flash chromatography yielded diastereomerically pure syn,syn-β-nitroamines 3.General procedure for the synthesis of indolines 7 (Table 1)
To a stirred mixture of syn,syn-β-nitroamine 3 (0.50 mmol) and Zinc (4.00 mmol) in EtOH (7.5 mL) at 0 °C was added conc. hydrochloric acid (10 mmol). The mixture was stirred at this temperature for 5 min then at RT until the reaction was complete by TLC analysis (approximately 1 h). The reaction was quenched by the addition of EtOAc and saturated aq. NaHCO3. The layers were separated, and the aqueous phase was extracted with EtOAc. The organic layers were combined, and the solvent was removed in vacuo to provide crude 1,2-diamine.To a stirred mixture of sodium tert-butoxide (1.00 mmol), palladium tetrakistriphenylphosphine (0.025 mmol) in toluene (1 ml) under nitrogen at RT was added a solution of crude 1,2-diamine in toluene (1.5 ml). The reaction vessel was heated at 90 °C until the reaction was complete by TLC analysis (approximately 16 to 24 h). Reaction cooled to RT and filtered thought celite and concentrated to give a brown oil. Purification by flash chromatography using basic Alumina or aminopropyl (NH2) silica yielded the indoline 7 (Table 1).
Acknowledgements
We thank the EPSRC and GSK for funding (EP/F068344/2), Dr L. Harris for mass spectra and Ms J. Maxwell for microanalytical data.References
- For a comprehensive review of the nitro-Mannich reaction see: A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887 CrossRef CAS PubMed
.
- For a review specific to asymmetric methods see: E. Marquéz-López, P. Merino, T. Tejero and R. P. Herrera, Eur. J. Org. Chem., 2009, 2401 CrossRef
- D. H. Lloyd and D. E. Nichols, J. Org. Chem., 1986, 51, 4294 CrossRef CAS
- M. A. Sturgess and D. J. Yarberry, Tetrahedron Lett., 1993, 34, 4743 CrossRef CAS
- A. G. M. Barrett and C. D. Spilling, Tetrahedron Lett., 1998, 29, 5733 CrossRef
- S. D. Rychnovsky, T. Beauchamp, R. Vaidyanathan and T. Kwan, J. Org. Chem., 1998, 63, 6363 CrossRef CAS PubMed
- H. Adams, J. C. Anderson, S. Peace and A. M. K. Pennell, J. Org. Chem., 1998, 63, 9932 CrossRef CAS
- K.-I. Yamada, S. J. Harwood, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 3504 CrossRef
- L. Bernadi, B. F. Bonini, I. E. Capito, G. Dessole, M. Comes-Franchimi, M. Fochi and A. Ricci, J. Org. Chem., 2004, 69, 8168 CrossRef PubMed
- M. G. Núñez, A. J. M. Farley and D. J. Dixon, J. Am. Chem. Soc., 2013, 135, 16348 CrossRef PubMed
- K. R. Knudsen, T. Risgaard, N. Nishiwaki, K. V. Gothelf and K. A. Jørgensen, J. Am. Chem. Soc., 2001, 123, 5843 CrossRef CAS
- S. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017 CrossRef PubMed
- C. Palomo, M. Oiarbibe, R. Halder, A. Laso and R. López, Angew. Chem., Int. Ed., 2006, 45, 117 CrossRef CAS PubMed
- M. G. Bursavich and D. H. Rich, J. Med. Chem., 2002, 45, 541 CrossRef CAS
- B. Shen and J. N. Johnston, Org. Lett., 2008, 10, 4397 CrossRef CAS PubMed
- P. Jakubec, D. M. Cockfield and D. Dixon, J. Am. Chem. Soc., 2009, 131, 16632 CrossRef CAS PubMed
- P. Jakubec, A. F. Kyle, J. Calleja and D. J. Dixon, Tetrahedron Lett., 2011, 52, 6094 CrossRef CAS PubMed
- A. F. Kyle, P. Jakubec, D. M. Cockfield, E. Cleator, J. Skidmore and D. J. Dixon, Chem. Commun., 2011, 47, 10037 RSC
- G. Kumaraswarmy and A. Pitchaiah, Helv. Chim. Acta, 2011, 94, 1543 CrossRef
- P. Jakubec, A. Hawkins, W. Felzmann and D. J. Dixon, J. Am. Chem. Soc., 2012, 134, 17482 CrossRef CAS PubMed
- N. Tsuritani, K.-I. Yamada, N. Yoshikawa and M. Shibasaki, Chem. Lett., 2002, 276 CrossRef CAS
- X. Xu, T. Furukawa, T. Okino, H. Miyabe and Y. Takemoto, Chem. – Eur. J., 2006, 12, 466 CrossRef PubMed
- J. C. Anderson and H. A. Chapman, Org. Biomol. Chem., 2007, 5, 2413 CAS
- P. S. Hynes, P. A. Stupple and D. J. Dixon, Org. Lett., 2008, 10, 1389 CrossRef CAS PubMed
- K. L. Jensen, P. H. Poulsen, B. S. Donslund, F. Morana and K. A. Jørgensen, Org. Lett., 2012, 14, 1516 CrossRef CAS PubMed
- S. Handa, V. Gnanadesikan, S. Matsunga and M. J. Shibasaki, J. Am. Chem. Soc., 2010, 132, 4925 CrossRef CAS PubMed
- J. Weng, Y.-B. Li, R.-B. Wang, F.-Q. Li, C. Liu, A. S. C. Chan and G. Lu, J. Org. Chem., 2010, 75, 3125 CrossRef CAS PubMed
- G. Kumaraswarmy and A. Pitchaiah, Tetrahedron, 2011, 67, 2536 CrossRef PubMed
- T. A. Davis and J. N. Johnston, Chem. Sci., 2011, 2, 1076 RSC
- H. Xie, Y. Zhang, S. Zhang, X. Chen and W. Wang, Angew. Chem., Int. Ed., 2011, 50, 11773 CrossRef CAS PubMed
- T. A. Davis, M. W. Danneman and J. N. Johnston, Chem. Commun., 2012, 48, 5578 RSC
- J. C. Anderson, L. R. Horsfall, A. S. Kalogirou, M. R. Mills, G. J. Stepney and G. J. Tizzard, J. Org. Chem., 2012, 77, 6186 CrossRef CAS PubMed
- S. Handa, V. Gnanadesikan, S. Matsungaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 4900 CrossRef CAS PubMed
- D. Kundu, R. K. Debnath, A. Majee and A. Hajra, Tetrahedron Lett., 2009, 50, 6998 CrossRef CAS PubMed
- X. Wang, Y.-F. Chen, L.-F. Niu and P.-F. Xu, Org. Lett., 2009, 11, 3310 CrossRef CAS PubMed
- J. C. Anderson, G. J. Stepney, M. R. Mills, L. R. Horsfall, A. J. Blake and W. Lewis, J. Org. Chem., 2011, 76, 1961 CrossRef CAS PubMed
- J. C. Anderson, A. Noble and D. A. Tocher, J. Org. Chem., 2012, 77, 6703 CrossRef CAS PubMed
- J. C. Anderson, A. J. Blake, P. J. Koovits and G. J. Stepney, J. Org. Chem., 2012, 77, 4711 CrossRef CAS PubMed
- J. C. Anderson and H. A. Chapman, Synthesis, 2006, 3309 CrossRef CAS PubMed
- A. Neogi, T. P. Majhi, R. Mukhopadhyay and P. Chattopadhyay, J. Org. Chem., 2006, 71, 3291 CrossRef CAS PubMed
- F. Langer, L. Schwink, A. Devasagayaraj, P.-Y. Chavant and P. Knochel, J. Org. Chem., 1996, 61, 1841 CrossRef PubMed
- J. Shannon, D. Bernier, D. Rawson and S. Woodward, Chem. Commun., 2007, 3945 RSC
- A. J. Blake, D. Giunta, J. Shannon, M. Solinas, F. Walzer and S. Woodward, Collect. Czech. Chem. Commun., 2007, 72, 1107 CrossRef CAS
- J. C. Anderson and P. J. Koovits, Chem. Sci., 2013, 4, 2897 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental details, X-ray representations and copies of 1H and 13C NMR spectra. CCDC 1020679. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob01793e |
| ‡ Corresponding author for crystallographic results. |
| § β-Nitroamines without electron withdrawing protecting groups on nitrogen are known to be unstable to standard purification techniques and undergo retro-addition. See ref. 7 and 36. |
| ¶ A minor diastereoisomer was also isolated in 10% yield and is tentatively assigned anti,syn-3bb, by analogy to other work (ref. 36), which has been more rigorously corroborated with single crystal X-ray crystallography. This is based upon the diastereosiomer being stable to purification, so most probably has the syn-relative stereochemistry across the nitro-amine bond. |
| || Alkyl/aryl zinc halides have been shown to be useful in certain addition and conjugate addition reactions. |
| This journal is © The Royal Society of Chemistry 2015 |