
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive nanoparticles from ionic cellulose derivatives†
Yonggui
Wang
a,
Thomas
Heinze
b and
Kai
Zhang
*a
aWood Technology and Wood Chemistry, Georg-August-Universität Göttingen, Büsgenweg 4, D-37077 Göttingen, Germany. E-mail: kzhang1@uni-goettingen.de
bCenter of Excellence for Polysaccharide Research, Institute of Organic Chemistry and Macromolecular Chemistry, Friedrich Schiller University of Jena, Humboldtstr. 10, D-07743 Jena, Germany
First published on 19th November 2015
Abstract
Stimuli-responsive nanoparticles (NPs) based on sustainable polymeric feedstock still need more exploration in comparison with NPs based on synthetic polymers. In this report, stimuli-responsive NPs from novel ionic cellulose derivatives were prepared via a facile nanoprecipitation. Cellulose 10-undecenoyl ester (CUE) with a degree of substitution (DS) of 3 was synthesized by esterification of cellulose with 10-undecenoyl chloride. Then, CUE was modified by photo-induced thiol–ene reactions, in order to obtain organo-soluble ionic cellulose derivatives with DSs of ∼3, namely cellulose 11-((3-carboxyl)ethylthio)undecanoate (CUE–MPA), cellulose 11-((2-aminoethyl)thio)undecanoate (CUE–CA), cellulose 11-(2-(2-(diethylamino)ethyl)thio)undecanoate (CUE–DEAET) and cellulose 11-(2-(2-(dimethylamino)ethyl)thio)undecanoate (CUE–DMAET). CUE–MPA could be transformed into NPs with average diameters in the range of 80–330 nm, but these NPs did not show particular stimuli-responsive properties. Moreover, the dropping technique resulted in smaller NPs than a dialysis technique. Stable NPs with average diameters in the range of 90–180 nm showing pH-responsive and switchable sizes were obtained from CUE–DEAET and CUE–DMAET possessing tertiary amines using nanoprecipitation. Thus, altering the terminal functional groups will be a new approach to prepare stimuli-responsive cellulose-derived polymeric NPs.
Introduction
Over the past few decades, functional polymeric nanoparticles (NPs) have been of special interest due to the variable chemical structures of constructing polymers and a wide range of applications from controlled drug delivery to biosensors.1–4 Polymeric NPs can either be prepared during polymerization, e.g., emulsion polymerization, or by post-shaping methods including nanoprecipitation or polyelectrolyte complexation.5–7 Among various NPs, the complexes in the form of nanocapsules from oppositely charged ionic polymers due to cooperative electrostatic interactions have shown huge potential in various applications.8,9 The change of ionic strengths or pH values can lead to a screening of the charges and to a disassociation of the complexes.10,11 This allows nanocapsules from ionic polymers to be used as delivery systems for biological applications.12–15 The capsule formation using oppositely charged ionic polymers has been extensively investigated from both theoretical and experimental aspects.8,16,17 However, due to the excellent water-solubility of most ionic polymers, studies on the formation of polymeric NPs from a single ionic polymer are still very rare. Among the functional groups of ionic polymers including polyelectrolytes, moieties containing carboxyl and amino groups are promising candidates, because they could undergo a pH-triggered solubility change, structure degradation, or destruction of cross-linkings.18–21On the other hand, more research interest is evoked towards the use of sustainable materials, such as polysaccharides.22–26 Polysaccharides as biopolymers have several advantageous features, such as abundance, renewability, nontoxicity, biocompatibility and biodegradability.14,27 Among them, cellulose is the most abundant natural polysaccharide, which consists of β-(1 → 4)-linked anhydroglucose units (AGUs). Three hydroxyl groups are present in each AGU and they can be modified by diverse functional groups, e.g. sulfate, carboxymethyl and methyl groups.28–30
Diverse ionic cellulose derivatives can be synthesized by the introduction of ionic groups into the cellulose backbone. Cellulose derivatives containing carboxyl groups with a pKa value in the range of 4–5 are protonated in the strongly acidic environment of the stomach and are ionized in the near-neutral small intestinal milieu.31 Amino group-containing cellulose derivatives show remarkable self-association behavior32 and may find numerous applications in a variety of fields including food, chemical, pharmaceutical and paper industries.33 A high degree of substitution (DS) is often desired for distinct applications. As a typical example, cellulose sulfates with high DSs showed higher biological activities towards enhancing the cell activities.34,35 However, ionic cellulose derivatives containing high amounts of carboxyl or amino groups, in particular with a DS of 3, are still rare. A major reason is limited synthesis routes for the efficient derivatization of all hydroxyl groups at the cellulose backbone. Esterification of cellulose using acid chlorides is among few methods an effective one leading to derivatives with a DS of 3.36 Moreover, stimuli-responsive NPs from cellulose derivatives, in particular ionic cellulose derivatives, also need more exploration in comparison to NPs from synthetic polymers exhibiting versatile chemical components.37,38
Thus, we report in the present study the formation of pH-responsive NPs from ionic cellulose derivatives with high DSs (of ∼3). At first, novel ionic cellulose derivatives containing carboxyl or primary/tertiary amino groups were synthesized by UV-induced thiol–ene reactions on cellulose 10-undecenoyl ester (CUE) with a DS of 3 without a photo-initiator. By using thiols containing carboxyl- or amine groups, namely 3-mercaptopropionic acid (MPA), cysteamine hydrochloride (CA), 2-(diethylamino)ethanethiol hydrochloride (DEAET) and 2-(dimethylamino)ethanethiol hydrochloride (DMAET), four ionic cellulose derivatives with a DS of ∼3 for corresponding functional groups were obtained. The organo-soluble ionic cellulose derivatives including cellulose 11-((3-carboxyl)ethylthio)undecanoate (CUE–MPA), cellulose 11-(2-(2-(diethylamino)ethyl)thio)undecanoate (CUE–DEAET) and cellulose 11-(2-(2-(dimethylamino)ethyl)thio)undecanoate (CUE–DMAET) were further transformed into NPs via a facile nanoprecipitation. The properties of obtained NPs regarding their sizes, morphologies and switchable properties were further analyzed.
Results and discussion
Cellulose 10-undecenoyl ester (CUE) and ionic cellulose derivatives
Cellulose 10-undecenoyl ester (CUE) was synthesized in pyridine under heterogeneous conditions (Scheme 1). FTIR and NMR spectroscopy showed the presence of 10-undecenoyl groups at the cellulose backbone (Fig. 1, S1 and S2†). In the 13C NMR spectrum of CUE, the signals at 114 ppm (C-17) and 139 ppm (C-16) occur from carbons in terminal olefin groups, while signals between 40 and 10 ppm are attributed to the other carbons of 10-undecenoyl moieties (Fig. 1a).31 Based on 1H NMR spectroscopy, the hydroxyl groups within the AGUs of cellulose were totally derivatized. Thus, the CUE has a degree of substitution (DS) of 3 (Fig. S3†). The CUE has a lower weight-averaged degree of polymerization (DPW) of 62 ± 3 than the starting MCC (with a DP of ∼270), which indicates a severe degradation of cellulose chains during esterification.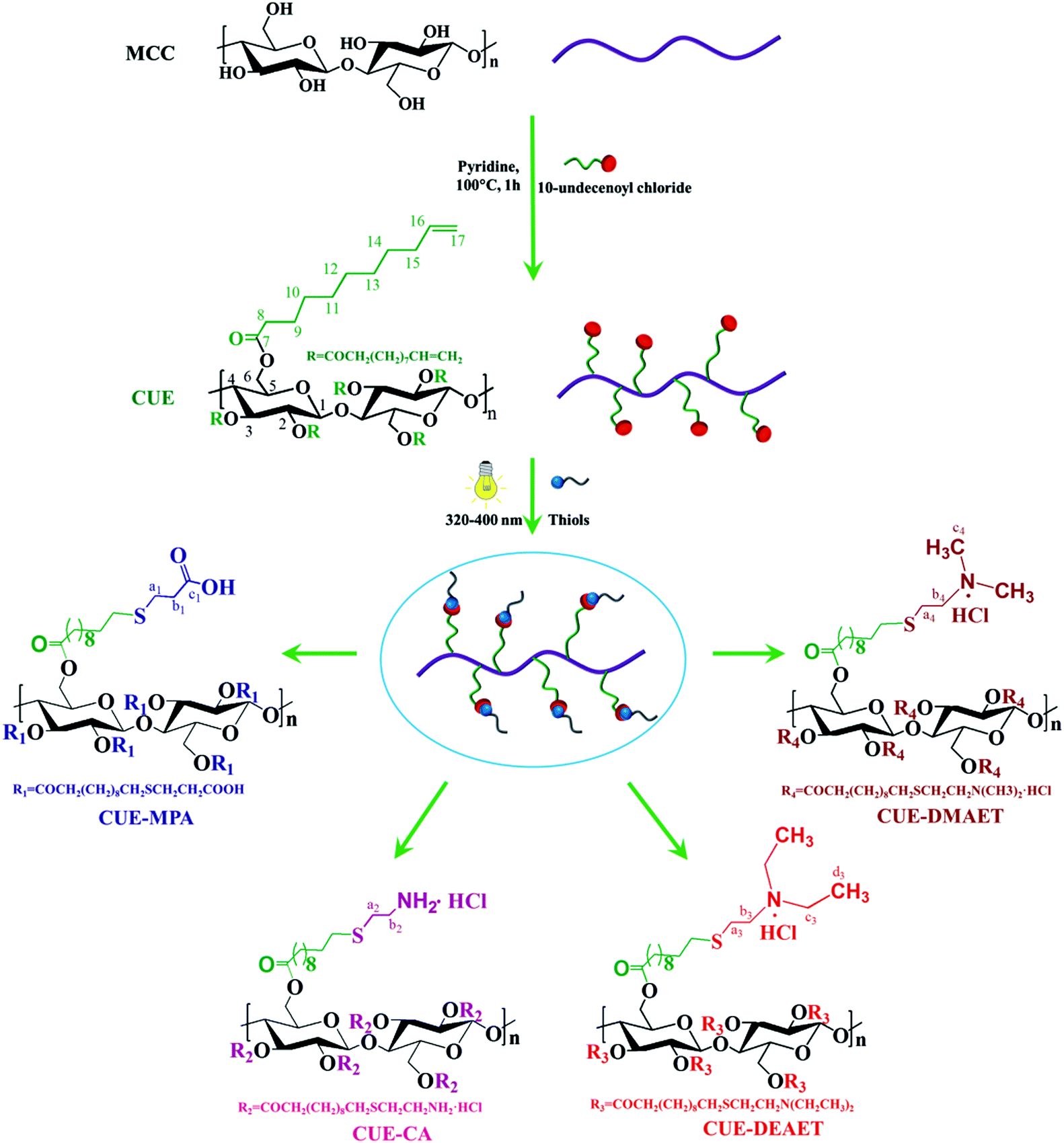 | ||
| Scheme 1 Schematic illustration for the synthesis of cellulose 10-undecenoyl ester (CUE), cellulose 11-((3-carboxyl)ethylthio)undecanoate (CUE–MPA), cellulose 11-((2-aminoethyl)thio)undecanoate (CUE–CA), cellulose 11-(2-(2-(diethylamino)ethyl)thio)undecanoate (CUE–DEAET) and cellulose 11-(2-(2-(dimethylamino)ethyl)thio)undecanoate (CUE–DMAET). | ||
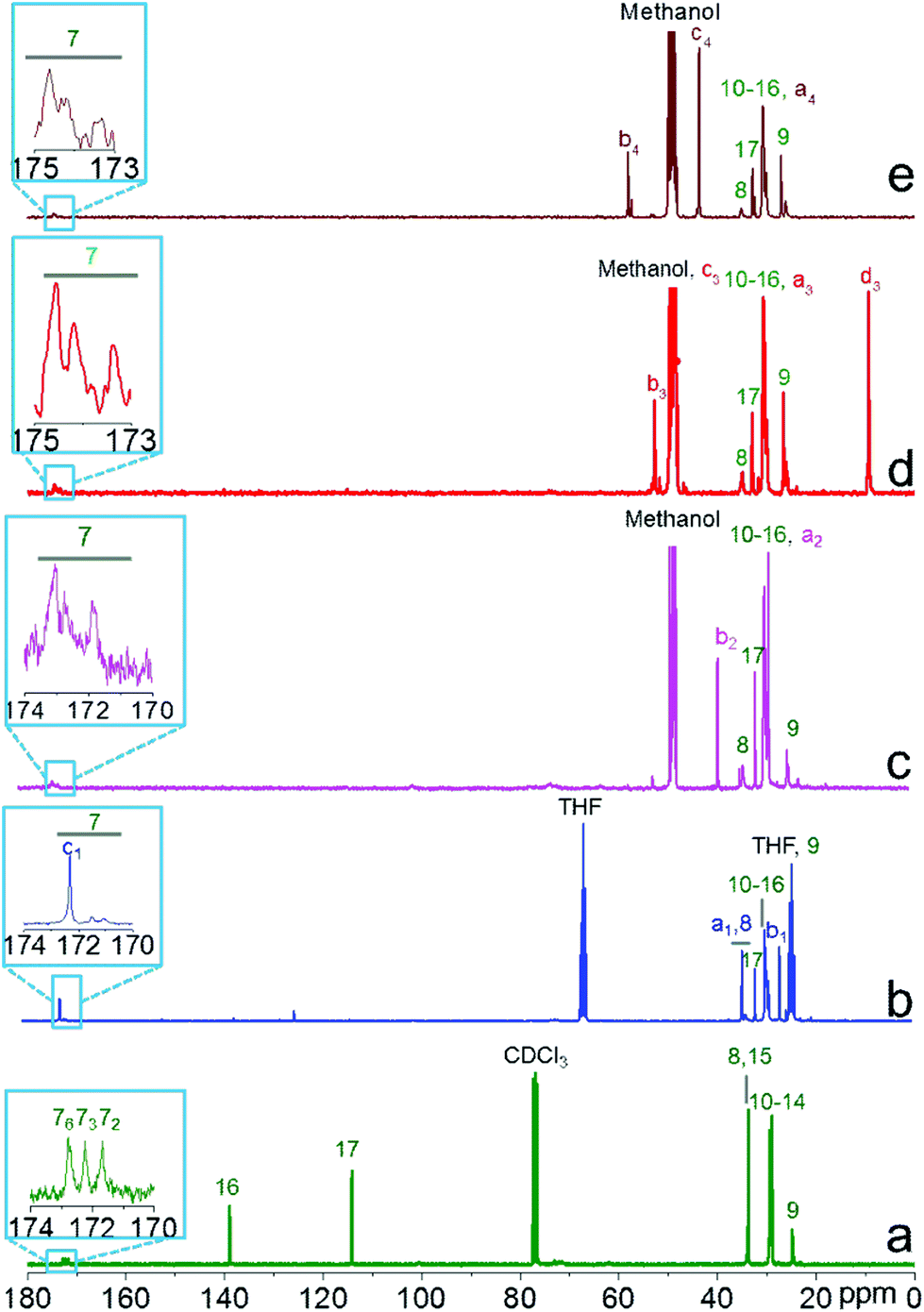 | ||
| Fig. 1 13C NMR spectra of (a) cellulose 10-undecenoyl ester (CUE), (b) cellulose 11-((3-carboxyl)ethylthio)undecanoate (CUE–MPA), (c) cellulose 11-((2-aminoethyl)thio)undecanoate (CUE–CA), (d) cellulose 11-(2-(2-(diethylamino)ethyl)thio)undecanoate (CUE–DEAET), (e) cellulose 11-(2-(2-(dimethylamino)ethyl)thio)undecanoate (CUE–DMAET). | ||
Due to the presence of terminal vinyl groups in CUE, the introduction of further functional groups leading to ionic cellulose derivatives by means of thiol–ene chemistry is possible. The thiol–ene reaction, which has been known for over 100 years,7 has recently attracted great interest for the synthesis of functional materials. For example, it has been used for the synthesis of silicon NPs,39,40 responsive hybrid microcapsules41 and carbosilane-thioether dendrimers.42–44 This fact is due to several features of thiol–ene reactions, such as the absence of metal catalysts, mild reaction conditions and insensitiveness of thiol–ene reactions to water or oxygen.45,46 In the present study, CUE was modified through the reaction of the terminal vinyl groups with MPA, CA, DEAET and DMAET. The thiol–ene reactions were photo-induced by UV light of the wavelength 320–400 nm without the presence of any photo-initiator (Scheme 1). After the reaction, CUE–MPA, CUE–CA, CUE–DEAET and CUE–DMAET were obtained according to FTIR, 1D and 2D NMR spectroscopy (Fig. 1, S1 and S4–S9†). In the 13C NMR spectrum of CUE–MPA (Fig. 1b), the signals at 26.8, 34.4 and 172.3 ppm are derived from carbons in CH2 next to carboxyl groups (C-b1), carbons in CH2 next to thioether bonds (C-a1) and carbons in carboxyl groups (C-c1), respectively, confirming the introduction of 3-mercaptopropionic groups. Within the 13C NMR spectrum of CUE–CA, the signals at 28.3 and 38.6 ppm are assigned to the carbons of the CH2 of (2-aminoethyl)thiol groups (C-a2 and C-b2) (Fig. 1c). Within the 13C NMR spectrum of CUE–DEAET, the signal at 9.4 ppm is attributed to the carbons in terminal CH3 groups (C-d3) (Fig. 1d). Moreover, the signals at 43.7 and 58.1 ppm within the 13C NMR spectrum of CUE–DMAET are assigned to the carbons of the terminal CH3 (C-c4) and CH2 next to amino groups (C-b4) (Fig. 1e). Furthermore, within the 13C and 1H NMR spectra (Fig. 1, S4, S6, S8 and S9†), no signal of carbons and protons in terminal alkenes is notable. Moreover, the crosslinking between vinyl groups should be possible during the thiol–ene reaction.47 But the signal caused by crosslinking was not detected in the samples by means of NMP spectroscopy. Thus, almost all the terminal vinyl groups were modified during the thiol–ene reactions, leading to four ionic cellulose derivatives with a DS of ∼3 for corresponding functional groups.
Nanoprecipitation using solutions of CUE–MPA
The ionic cellulose derivatives were further transformed into NPs. Polymeric NPs, in particular stimuli-responsive NPs, can be prospective candidates in diverse application fields, such as for functional surface coating, targeted drug delivery or sensors.14,34,48–50 The CUE–MPA was converted into NPs via nanoprecipitation through a solvent exchange process, which has also been named as a polymeric ‘Ouzo effect’.51,52 Two techniques as dropping and dialysis were used (Scheme 2). After nanoprecipitation, NPs with average diameters in the range of 80–330 nm were obtained using the solutions of CUE–MPA in THF with diverse concentrations under varied conditions (Fig. 2, Table S1†).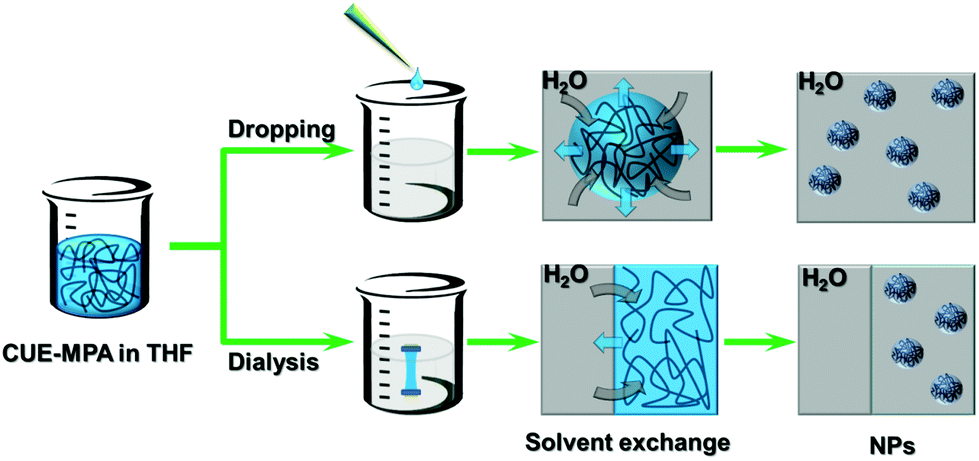 | ||
| Scheme 2 Schematic illustration for the nanoprecipitation of CUE–MPA solutions using dropping and dialysis techniques. Blue areas: THF, black curves: CUE–MPA chains. The scheme is not in real scale. | ||
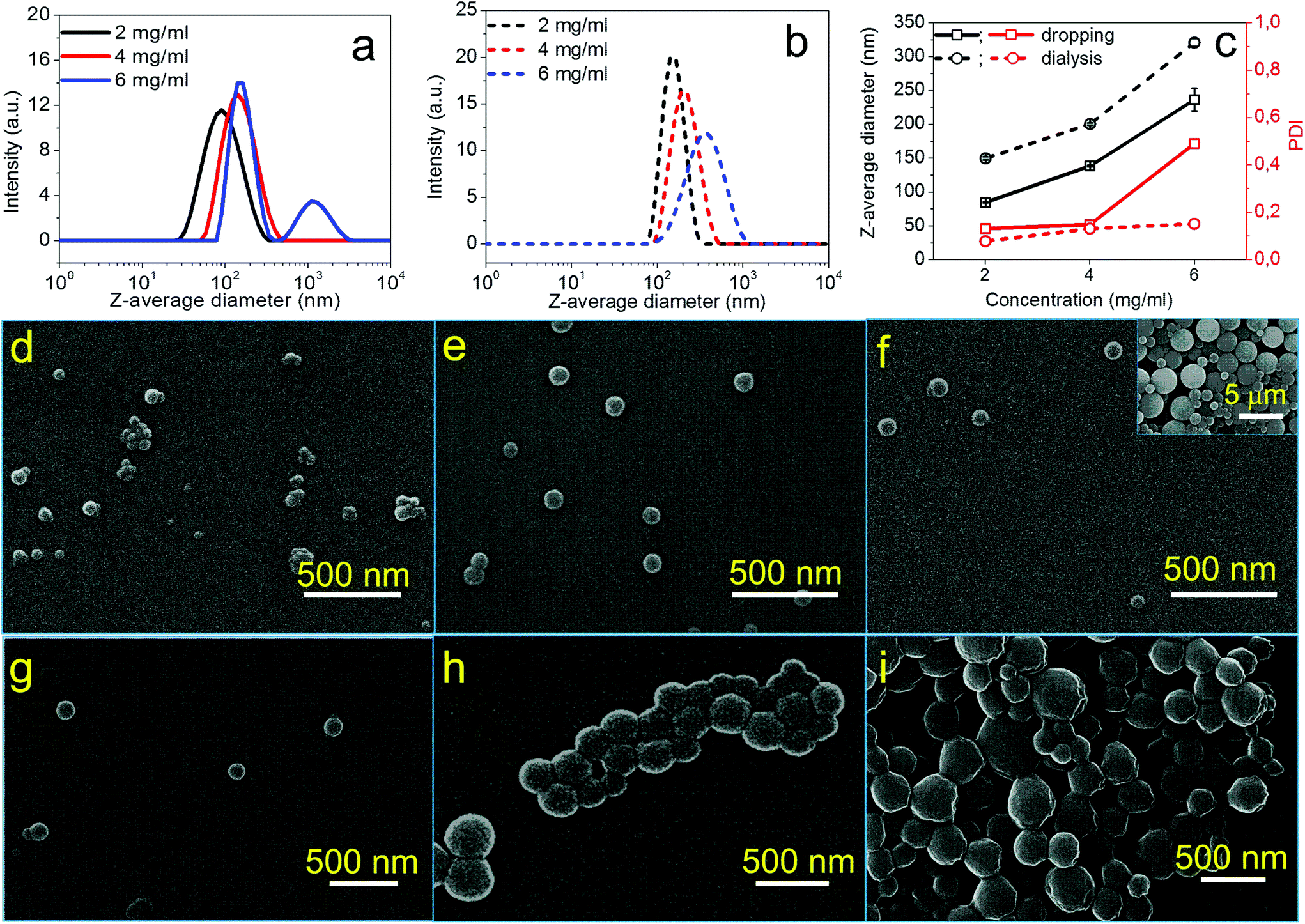 | ||
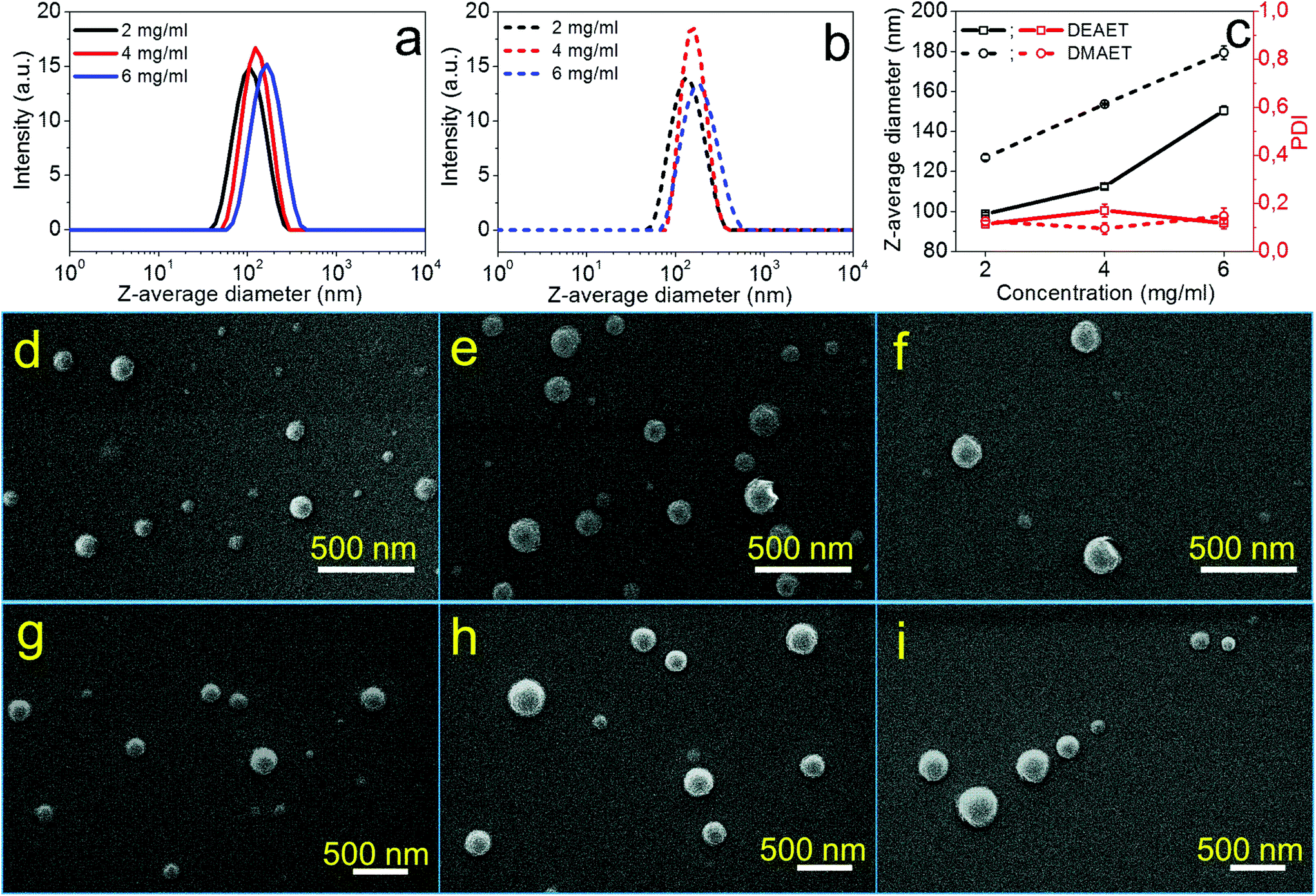
| Fig. 2 DLS curves of NPs from CUE–MPA solutions with diverse concentrations prepared via (a) dropping and (b) dialysis technique. (c) Z-average diameters and PDI of obtained NPs in correlation with the concentrations of starting solutions. (d–f) SEM images of obtained NPs via a dropping technique using solutions of CUE–MPA with diverse concentrations: (d) 2 mg ml−1, (e) 4 mg ml−1 and (f) 6 mg ml−1. (g–i) SEM images of obtained NPs via a dialysis technique using solutions of CUE–MPA with diverse concentrations: (g) 2 mg ml−1, (h) 4 mg ml−1 and (i) 6 mg ml−1. Scale bar in (d–i): 500 nm. The inset in (f) with a scale bar of 5 μm shows the presence of microparticles. | ||
The formation process of NPs using CUE–MPA solutions was analyzed in detail by alternating the parameters of the nanoprecipitation, such as the concentrations of solutions, and the dropping or dialysis technique. Fig. 2 shows the dependence of the average sizes and PDI (i.e. the size distribution) of NPs on the concentrations of initial polymer solutions. Using the dropping technique, the average diameters of NPs increased from 85 ± 1 to 139 ± 1 nm with a rising concentration from 2 to 4 mg ml−1. However, CUE–MPA solution of 6 mg ml−1 resulted in NPs with bi-modally distributed sizes (Fig. 2a). Also a small amount of microparticles (∼10% based on signal intensities of DLS measurements ascribed to NPs and microparticles) is present among NPs (Fig. 2f). Because the nanoprecipitation using CUE–MPA solutions was carried out under comparable conditions except for the concentration, a concentration of 6 mg ml−1 was too high for the fabrication of NPs with uniform sizes.
Using the dialysis technique, the average diameters of NPs increased from 150 ± 2 to 321 ± 6 nm with an increasing concentration from 2 to 6 mg ml−1. PDI increases simultaneously from 0.078 to 0.151. The positive increase of the average diameters of NPs with higher polymer concentrations is related to the viscosity of the solutions and has been observed for NPs from other polymers, such as cellulose stearoyl esters, hydrophobic esters of dextran, pullulan and starch.51,53,54
In addition, NPs prepared using the dialysis technique have larger average diameters than NPs prepared using the dropping technique (Fig. 2). This difference is attributed to distinct formation processes of NPs via dropping and dialysis. Using the dropping technique, the formation of primary NPs or nuclei is based on the aggregation of single polymer chains during the fast diffusion-out of solvent from the drops of polymer solutions. The fast diffusion-out of THF from drops of the polymer solution leads to nascent clusters of polymer chains and then NPs.54 This process takes place very quickly and is assumed at a time scale of milliseconds.54,55 In comparison, the exchange of organic solvents in the polymer solution with water during the dialysis is based on the passive transport of solvents, which is comparatively very slow (Fig. 2). The aggregation of polymer chains is primarily driven by an increasing interfacial tension during the solvent exchange.56,57
Modified nanoprecipitation using ionic cellulose derivatives containing amino groups
The ionic cellulose derivatives containing amino groups were also used for the preparation of NPs by a modified dropping technique. After the addition of solutions of CUE–CA, CUE–DEAET and CUE–DMAET into water, transparent suspensions with pH values around 4.5 were obtained (Fig. S10 and S11†). These initial suspensions contained multi-modally distributed polymer colloids as shown by DLS measurements (Fig. 3 and S10†). The presence of these colloids is probably due to the strong interaction between the protonated amino groups at polymer chains and water, such as ion-dipole bonding interactions,58 leading to swollen clusters of polymer chains of various sizes. Cationic polymers containing amino groups generally exhibit pH-responsive behaviors.59,60 Their charge density depends on the pH values of the solutions or dispersion solvents, which affects the interaction of polymer chains with solvent molecules and in turn influences the polymer conformation.60 It is also known that ionized amino groups with a cationic nitrogen center caused by an acid, such as HCl, can participate in ion-dipole bonding interactions with water and thereby show enhanced solubility in water compared to the non-ionized counterpart. Thus, deionization treatment was adopted by adding a NaOH solution (0.01 M), in order to form NPs by decreasing the interaction of polymer chains with water. With increasing pH values of CUE–DEAET and CUE–DMEAET suspensions from pH 4.5 to pH 7, the interaction between the polymers and water became weaker due to the deprotonation of the ionized amino groups. Subsequently, swollen colloids were converted into shrunken and stable NPs from CUE–DEAET and CUE–DMAET at pH 7 with the average diameter of 130 ± 4 and 177 ± 1 nm, respectively (Fig. 3). When the pH values of NP suspensions were further increased from pH 7 to pH 8, only a slight increase of the average diameter of NPs is observed. It should be caused by the slight aggregation of NPs due to lower zeta potentials (Fig. 3). When the pH values of the suspensions were further elevated to pH 9, the zeta potentials of CUE–DEAET and CUE–DMAET suspensions decreased to 0.3 ± 6.0 mV and 15.1 ± 6.7 mV respectively. The suspensions with these low zeta potentials are unstable according to the electrostatic stabilization theory,61 and severe aggregations are notable (Fig. 3 and S11†). Thus, CUE–DEAET and CUE–DMAET can be converted into NPs by a modified nanoprecipitation route, which includes a conventional dropping technique with solvent exchange and a further instant formation of NPs induced by pH-change (Scheme 3). There are only tiny differences between CUE–DEAET and CUE–DMAET regarding the average sizes and pH values of the suspensions right after the nanoprecipitation, which is due to the different substituents at tertiary amino groups as diethyl or dimethyl groups. In comparison, CUE–CA could not form stable NPs using this modified nanoprecipitation route, but only floppy aggregates (Fig. S10†).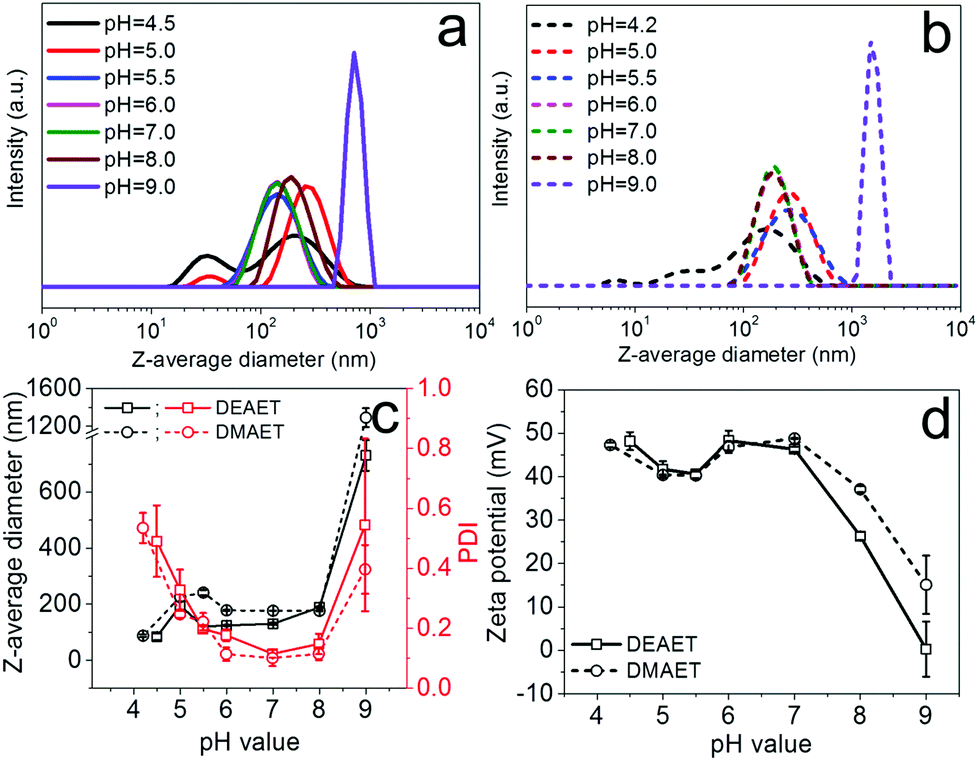 | ||
| Fig. 3 DLS curves of colloids or NPs from (a) CUE–DEAET and (b) CUE–DMAET immediately after dropping the polymer solutions (4 mg ml−1) into water and the subsequent adjustment of pH values to different pH values using NaOH solution (0.01 M); (c) Z-average diameters and PDI of obtained colloids or NPs; (d) zeta potentials of obtained colloids or NPs. | ||
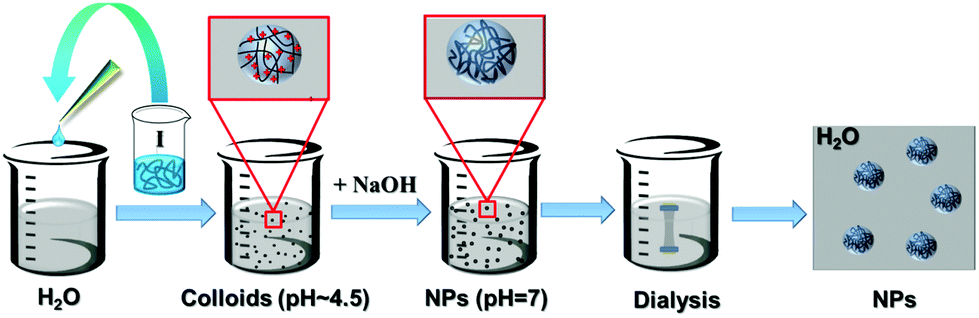 | ||
| Scheme 3 Schematic illustration for the modified nanoprecipitation of CUE–DEAET and CUE–DMAET solutions. Black curves: CUE–DEAET or CUE–DMAET chains; red crosses: H+ ions; I: polymer solutions in methanol. The scheme is not in real scale. | ||
After further dialysis of resulting NP suspensions at pH 7, the NPs with a spherical morphology were obtained according to the SEM images (Fig. 4d–i). For both ionic cellulose derivatives, the average diameters of NPs increased with higher concentrations of starting solutions. To be specific, with a rising concentration from 2 to 6 mg ml−1, the average diameter of NPs from CUE–DEAET solutions increased from 99 ± 1 to 151 ± 2 nm, while the average diameter of NPs from CUE–DMAET solutions increased from 127 ± 1 to 179 ± 3 nm (Fig. 4 and Table S2†). At the same time, all NPs maintained low PDI between 0.096 and 0.171, indicating a narrow size distribution of NPs (Table S2†). The increase of NP sizes is assumed to be positively related to the viscosity of the polymer solutions as shown by other solvent exchange nanoprecipitation processes.51,53,54 Moreover, the dialyzed NPs are smaller than their precursors before the dialysis (Fig. 3 and 4). This slight decrease is due to the removal of encapsulated methanol residue inside the precursors.
 | ||
| Fig. 4 DLS curves of NPs from (a) CUE–DEAET and (b) CUE–DMAET solutions with distinct concentrations after the adjustment of pH value to 7 and dialysis in water. (c) Z-average diameters and PDI of obtained NPs in correlation with the concentrations of starting solutions. (d–f) SEM images of obtained NPs using solution of CUE–DEAET with diverse concentrations: (d) 2 mg ml−1, (e) 4 mg ml−1 and (f) 6 mg ml−1. (g–i) SEM images of obtained NPs using solutions of CUE–DMAET with diverse concentrations: (g) 2 mg ml−1, (h) 4 mg ml−1 and (i) 6 mg ml−1. Scale bar in (d–i): 500 nm. | ||
PH-responsive NPs with switchable sizes
Reversible, pH-responsive swelling and deswelling capacities of NPs have drawn widespread attention in various fields, such as drug delivery systems.62,63 Thus, pH-sensitivity of obtained NPs and their swelling behaviors at distinct pH values were further investigated. In comparison to NPs of CUE–MPA with a stable size at various pH values (pH 3-10), NPs of CUE–DEAET and CUE–DMAET showed excellent pH-responsiveness (Fig. 5 and S12†). With a lower pH value from 7 to 4, the average diameter of NPs of CUE–DEAET increased from 113 ± 1 to 144 ± 2 nm with an accompanying volume increase of 106%, which is ascribed to the significant swelling of NPs due to the ionization of tertiary amino groups (Fig. 5a). With a decreasing pH value from 4 to 3, the average diameter of the NPs slightly decreased. This fact is due to the stronger ionization of amino groups at pH 3 and therefore a partial disassociation of NPs into swollen nanocolloids. Similar to CUE–DEAET, the average diameter of NPs from CUE–DMAET increased from 154 ± 1 nm at pH 7 to 185 ± 3 nm at pH 4, i.e., a volume increase of 73%, and NPs with a higher volume increase of 119% were obtained at pH 3 with the average diameter of 200 ± 1 nm (Fig. 5a).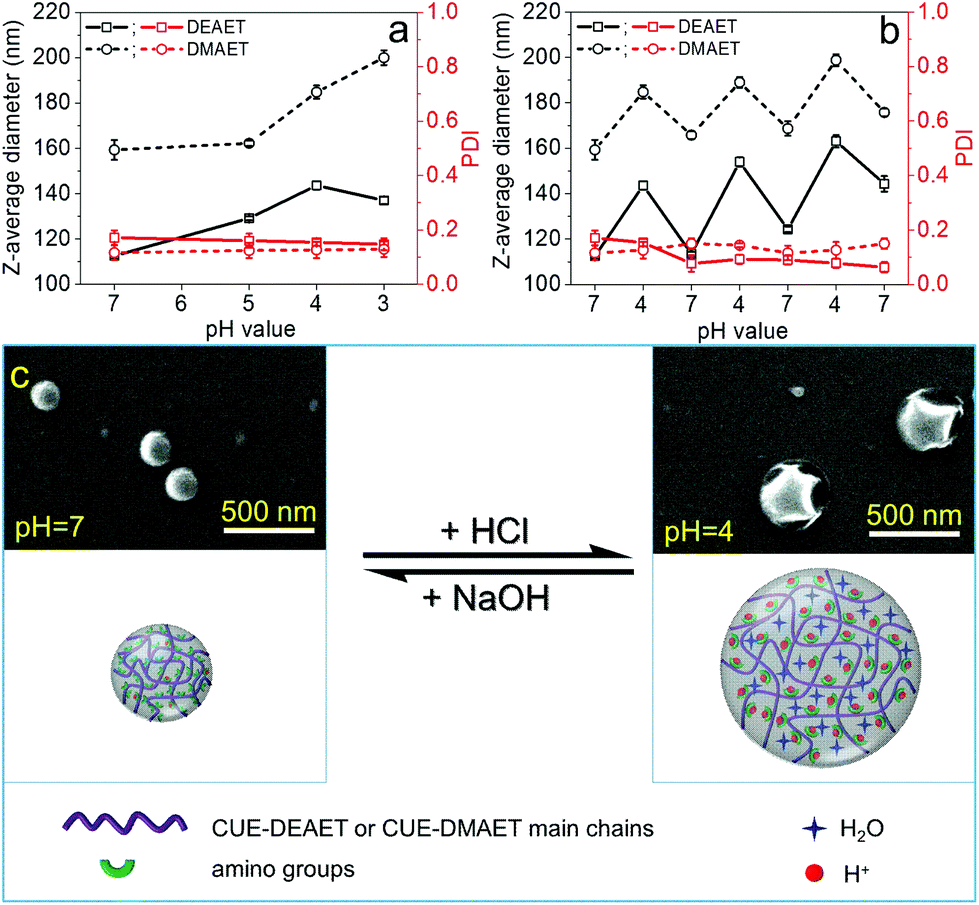 | ||
| Fig. 5 (a) Z-average diameters and PDI of NPs of CUE–DEAET and CUE–DMAET showing swelling behaviors during the decrease of the pH value from 7 to 3 using aqueous HCl solution (0.01 M); (b) Z-average diameters and PDI of NPs of CUE–DEAET and CUE–DMAET showing switchable sizes at the pH values of 7 and 4; (c) SEM images and schematic illustration of NPs from CUE–DEAET with switchable sizes at pH values of 7 and 4. | ||
This switchable swelling behavior of NPs was also visualized by SEM images. Dried NPs of CUE–DEAET from their dispersions at pH 7 exhibit a solid and spherical morphology according to their SEM images (Fig. 5c). In comparison, a shrunk and collapsed morphology was visible for the dried NPs of CUE–DEAET from their dispersion at pH 4 as shown by their SEM images. The shrinkage of the NPs during drying confirms the swelling behavior of the NPs at pH 4 (Fig. 5c and S13†).
Furthermore, after the repetitive swelling and deswelling, the average sizes of NPs increased at both pH values of 4 and 7. This size increase is assumed to be related to the accumulation of sodium chloride during the adjustment of pH values using aqueous NaOH or HCl solution (Fig. 5b). The presence of salt can influence the osmotic pressure between the internal network formed by some ionic polymers (e.g.: weak polyelectrolytes) and external solutions, and thereby change the swelling ability of NPs.64,65 In addition, the transformation from the swollen state to the non-swollen state by adding NaOH solution starts from the outer layer of the NPs, which impedes the deprotonation of amino groups within NPs and further decreases the deswelling degree, leading to bigger NPs compared to the previous cycle.
The triggering pH values for the swelling and deswelling of NPs lie generally in the range of 4–7, which matches the environment of the small intestinal and stomach milieu.63,66 In addition, the NPs from both ionic cellulose derivatives still maintained pH-regulated, size-switchable ability after three swelling–deswelling cycles (Fig. 5b). Thus, the pH-responsive properties of these NPs from CUE–DEAET and CUE–DMAET allow them to be promising potential candidates for diverse applications, such as colloid systems for food, medicine and pharmacy.
Conclusion
On the whole, pH-responsive NPs with switchable sizes were prepared from ionic cellulose derivatives. The synthesis of novel organo-soluble ionic cellulose derivatives containing carboxyl or amino groups with a degree of substitution (DS) of ∼3 was described for the first time. First, cellulose 10-undecenoyl ester (CUE) with a DS of 3 according to 1H NMR spectroscopy was obtained after the esterification of cellulose by 10-undecenoyl chloride. Then, CUE was successfully modified by grafting the terminal vinyl groups with MPA, CA, DEAET or DMAET via photo-induced thiol–ene reactions under UV irradiation without any photo-initiator. The ionic cellulose derivatives, CUE–MPA, CUE–CA, CUE–DEAET and CUE–DMAET with DSs of ∼3 were synthesized. CUE–MPA can be further transformed into stable nanoparticles (NPs) with average diameters in the range of 80–330 nm via dropping and dialysis techniques under diverse conditions. However, these NPs did not show responsive properties. In comparison, NPs from CUE–DEAET and CUE–DMAET via the modified dropping technique exhibit pH-responsive, size-switchable properties as shown by alternately changing the pH value between 7 and 4. The NPs became more expanded at pH 4, whereas shrinkage occurred when the pH increased to 7 while no stable NPs were formed from CUE–CA via the same process. Based on the sustainable character of cellulose and reversible pH-sensitivity, we foresee versatile applications for the NPs of organo-soluble, ionic cellulose derivatives, such as non-adhesive coatings and targeted drug delivery.Experimental
Materials
Microcrystalline cellulose (MCC) with a granule size of 50 μm and a DP of ∼270 was purchased from Sigma-Aldrich (Steinheim, Germany). 10-Undecenoyl chloride was received from Merck Schuchardt OHG (Hohenbrunn, Germany). 3-Mercaptopropionic acid (MPA) (≥99%) was purchased from Merck KGaA (Darmstadt, Germany). Cysteamine hydrochloride (CA) (≥98%) was purchased from AppliChem GmbH (Darmstadt, Germany). 2-(Diethylamino)ethanethiol hydrochloride (DEAET) and 2-(dimethylamino)ethanethiol hydrochloride (DMAET) were purchased from Sigma-Aldrich (Steinheim, Germany). Dry pyridine and tetrahydrofuran (THF) were received from VWR International GmbH (Darmstadt, Germany). Deionized water (DI water) was used in all experiments. Other chemicals are all of analytical grade and used as received.Synthesis of cellulose 10-undecenoyl ester (CUE)
CUE was prepared according to ref. 50 with a few modifications. In a typical case, 1 g dried MCC was washed with methanol and pyridine to remove traces of moisture before it was suspended in 30 ml pyridine. Then, the cellulose suspension was heated up to 100 °C and 8.4 ml 10-undecenoyl acid chloride (6 mol 10-undecenoyl chloride per mol AGUs of cellulose) was dropped into the hot cellulose suspension, while it was purged with nitrogen gas. After 1 h stirring at 100 °C, the mixture was added into 200 ml methanol. The precipitate was separated by centrifugation at 4500 rpm for 10 min. Thereafter, the product was purified through repeated dissolution in THF and precipitation in methanol. Finally, the product was dissolved in THF and stored in the dark for further use. Yield: 95.6%.Synthesis of cellulose 11-((3-carboxyl)ethylthio)undecanoate (CUE–MPA) via thiol–ene reaction of CUE with MPA
MPA (2 mol per mol C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds) was added into the THF-solution of CUE (15 mg ml−1). The mixture was exposed to UV light (320–400 nm with the intensity of ∼100 mW cm−2) at room temperature (RT). After 2 h stirring, the reaction mixture was added into 5 volumes of water. The precipitate was separated by centrifugation. Thereafter, the product was purified through repeated dissolution in THF and precipitation in water. Finally, the product was dissolved in THF for further use. Yield: 94.3%.
C double bonds) was added into the THF-solution of CUE (15 mg ml−1). The mixture was exposed to UV light (320–400 nm with the intensity of ∼100 mW cm−2) at room temperature (RT). After 2 h stirring, the reaction mixture was added into 5 volumes of water. The precipitate was separated by centrifugation. Thereafter, the product was purified through repeated dissolution in THF and precipitation in water. Finally, the product was dissolved in THF for further use. Yield: 94.3%.
Synthesis of cellulose 11-((2-aminoethyl)thio)undecanoate (CUE–CA), cellulose 11-(2-(2-(diethylamino)ethyl)thio)undecanoate (CUE–DEAET) and cellulose 11-(2-(2-(dimethylamino)ethyl)thio)undecanoate (CUE–DMAET) via thiol–ene reaction of CUE with CA, DEAET or DMAET
The CA, DEAET or DMAET (2 mol per mol CC double bonds) was added to the THF-solution of CUE (15 mg ml−1). Because the three thiol compounds cannot be dissolved in THF, a two-step reaction was applied. After the addition of thiol compounds, the mixture was exposed to UV light (320–400 nm, the intensity ∼100 mW cm−2) under stirring at RT. After 1 h reaction, products precipitated out and were separated via centrifugation. For the next step, the precipitates were dissolved in methanol. Then, the mixture was irradiated using UV light under stirring at RT for another 1 h. After that, the solutions were dialyzed in methanol. The purified products were dissolved in methanol for further use. Yields: 90.7% (CUE–CA), 94.1% (CUE–DEAET) and 95.7% (CUE–DMAET).
Nanoprecipitation using the solution of CUE–MPA
Nanoprecipitation using the solutions of CUE–CA, CUE–DEAET and CUE–DMAET
CUE–CA, CUE–DEAET and CUE–DMAET were turned into NPs via a modified dropping technique (Scheme 3). Briefly, CUE–CA, CUE–DEAET or CUE–DMAET was dissolved in methanol at a concentration of 2–6 mg ml−1. Then, 1 ml solution was added drop by drop into 10 ml water (pH of ∼7) under stirring of 500 rpm at RT, leading to transparent suspensions with the pH values of 4.2–4.5. The pH values of the transparent suspensions were adjusted using aqueous NaOH solution (0.01 M) to various pH values (pH = 5, 5.5, 6, 7, 8 and 9) to form NPs. The dispersions with the pH of 7 were dialysed in water to remove salt and methanol residue before further characterization.Examination of pH-responsive properties
The pH values of obtained NP suspensions from CUE–MPA, CUE–DEAET and CUE-DMAET with the initial concentration of 4 mg ml−1 were adjusted using aqueous NaOH solution (0.01 M) and HCl solution (0.01 M) to investigate the pH-responsive, size-switchable properties.Size exclusion chromatography (SEC)
The average molecular weight of CUE was measured in THF solution on a SECurity GPC system consisting of a pump, an autosampler, and a RI-detector of Agilent 1200 Series (Waldbronn, Germany) with a set of columns consisting of PSS Gram 5 and PSS Gram 1000 columns. An injection volume of 100 μl of CUE solution in THF at a concentration of 3 g l−1 was measured with the column temperature of 25 °C and the solvent flow of 1 ml min−1.One-dimensional (1D) and two-dimensional (2D) NMR spectroscopy
The liquid-state 1H and 13C NMR spectra of CUE in deuterated chloroform, CUE–MPA in deuterated THF and CUE–CA, CUE–DEAET as well as CUE–DMAET in deuterated methanol were recorded at RT on a Bruker DRX 500 spectrometer (Bruker, Biospin GmbH, Ettlingen, Germany) with a frequency of 300 MHz. A total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans for 13C NMR and 180 scans for 1H NMR were accumulated. 1H–1H HSQC spectra and 1H–13C COSY spectra of CUE–MPA and CUE–CA were recorded for the assignment of the signals within 1H and 13C NMR spectra (Fig. S5 and S7†).
000 scans for 13C NMR and 180 scans for 1H NMR were accumulated. 1H–1H HSQC spectra and 1H–13C COSY spectra of CUE–MPA and CUE–CA were recorded for the assignment of the signals within 1H and 13C NMR spectra (Fig. S5 and S7†).
Scanning electron microscopy (SEM)
SEM images were obtained on a Philips XL30 FEG high-resolution scanning electron microscope (HR-SEM) (FEI Deutschland GmbH, Frankfurt/Main, Germany). A layer from platinum/palladium (80/20) of 10 nm was coated on the surface of samples before SEM measurements.Dynamic light scattering (DLS)
The DLS measurements were performed on a Zetasizer Nano ZS (Malvern Instruments Ltd, UK) using a 5 mW laser with the incident beam of 633 nm (He–Ne laser). 1 ml of NP suspensions (0.1–0.4 mg ml−1 water) were used for the size measurement (Z-average diameter) in a quartz cuvette (Starna, Pfungstadt, Germany), and were used for zeta potential measurement in a disposable zeta cuvette (DTS1060C from Malvern Instruments Ltd). The sizes and zeta potentials (mV) of NPs were measured three times with 10 runs and 20 runs for each measurement, respectively.Acknowledgements
K.Z. thanks Georg-August-Universität Göttingen for the starting grant and Fonds der Chemischen Industrie (FCI) for financial support. Y.W. thanks the China Scholarship Council (CSC) for financial support.References
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKHzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- H. J. Chung, C. M. Castro, H. Im, H. Lee and R. Weissleder, Nat. Nanotechnol., 2013, 8, 369–375 CrossRef CAS PubMed.
- F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef CAS.
- A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 2140–2154 CrossRef CAS PubMed.
- E. Akiyama, N. Morimoto, P. Kujawa, Y. Ozawa, F. M. Winnik and K. Akiyoshi, Biomacromolecules, 2007, 8, 2366–2373 CrossRef CAS PubMed.
- A. Geissler, D. Scheid, W. Li, M. Gallei and K. Zhang, Cellulose, 2014, 21, 4181–4194 CrossRef CAS.
- F. Ganachaud and J. L. Katz, ChemPhysChem, 2005, 6, 209–216 CrossRef CAS PubMed.
- W. B. Wu, F. Huang, S. B. Pan, W. Mu, X. Z. Meng, H. T. Yang, Z. Y. Xu, A. J. Ragauskas and Y. L. Deng, J. Mater. Chem. A, 2015, 3, 1995–2005 CAS.
- D. V. Pergushov, A. H. E. Muller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
- C. V. Synatschke, F. H. Schacher, M. Fortsch, M. Drechsler and A. H. E. Muller, Soft Matter, 2011, 7, 1714–1725 RSC.
- D. V. Pergushov, E. V. Remizova, J. Feldthusen, A. B. Zezin, A. H. E. Muller and V. A. Kabanov, J. Phys. Chem. B, 2003, 107, 8093–8096 CrossRef CAS.
- S. Dumitriu and E. Chornet, Adv. Drug Delivery Rev., 1998, 31, 223–246 CrossRef CAS.
- X. T. Meng, J. B. Matson and K. J. Edgar, Biomacromolecules, 2014, 15, 177–187 CrossRef CAS PubMed.
- Z. H. Liu, Y. P. Jiao, Y. F. Wang, C. R. Zhou and Z. Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- Y. Lee and K. Kataoka, Soft Matter, 2009, 5, 3810–3817 RSC.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed.
- K. Sonaje, Y. H. Lin, J. H. Juang, S. P. Wey, C. T. Chen and H. W. Sung, Biomaterials, 2009, 30, 2329–2339 CrossRef CAS PubMed.
- J. Q. Zhao, H. Y. Wang, J. J. Liu, L. D. Deng, J. F. Liu, A. J. Dong and J. H. Zhang, Biomacromolecules, 2013, 14, 3973–3984 CrossRef CAS PubMed.
- L. L. Chang, J. J. Liu, J. H. Zhang, L. D. Deng and A. J. Dong, Polym. Chem., 2013, 4, 1430–1438 RSC.
- M. J. Barthel, A. C. Rinkenauer, M. Wagner, U. Mansfeld, S. Hoeppener, J. A. Czaplewska, M. Gottschaldt, A. Trager, F. H. Schacher and U. S. Schubert, Biomacromolecules, 2014, 15, 2426–2439 CrossRef CAS PubMed.
- H. Sehaqui, M. E. Galvez, V. Becatinni, Y. C. Ng, A. Steinfeld, T. Zimmermann and P. Tingautt, Environ. Sci. Technol., 2015, 49, 3167–3174 CrossRef CAS PubMed.
- A. Ciferri and S. Kudaibergenov, Macromol. Rapid Commun., 2007, 28, 1953–1968 CrossRef CAS.
- M. N. Kumar, R. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017–6084 CrossRef PubMed.
- K. Junka, O. Sundman, J. Salmi, M. Osterberg and J. Laine, Carbohydr. Polym., 2014, 108, 34–40 CrossRef CAS PubMed.
- H. F. Pan, L. Song, L. Y. Ma, Y. Pan, K. M. Liew and Y. Hu, Cellulose, 2014, 21, 2995–3006 CrossRef CAS.
- S. N. Pawar and K. J. Edgar, Biomaterials, 2012, 33, 3279–3305 CrossRef CAS PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- T. Heinze, T. F. Liebert, K. S. Pfeiffer and M. A. Hussain, Cellulose, 2003, 10, 283–296 CrossRef CAS.
- D. Klemm, B. Philipp, T. Heinze, U. Heinze and W. Wagenknecht, Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose, Wiley-VCH Verlag GmbH, Weinheim, 1998 Search PubMed.
- P. Zugenmaier, Crystalline Cellulose and Derivatives: Characterization and Structures, Springer, Heidelberg, 2008 Search PubMed.
- J. P. Reddy and J. W. Rhim, Mater. Lett., 2014, 129, 20–23 CrossRef.
- T. Heinze, M. Nikolajski, S. Daus, T. M. D. Besong, N. Michaelis, P. Berlin, G. A. Morris, A. J. Rowe and S. E. Harding, Angew. Chem., Int. Ed., 2011, 50, 8602–8604 CrossRef CAS PubMed.
- W. X. Wang, M. D. Mozuch, R. C. Sabo, P. Kersten, J. Y. Zhu and Y. C. Jin, Cellulose, 2015, 22, 351–361 CrossRef CAS.
- N. Aggarwal, N. Altgarde, S. Svedhem, K. Zhang, S. Fischer and T. Groth, Colloids Surf., B, 2014, 116, 93–103 CrossRef CAS PubMed.
- L. H. Fan, X. Y. Zhou, P. H. Wu, W. G. Xie, H. Zheng, W. Tan, S. H. Liu and Q. Y. Li, Int. J. Biol. Macromol., 2014, 66, 245–253 CrossRef CAS PubMed.
- K. Zhang, A. Geissler and T. Heinze, Part. Part. Syst. Charact., 2015, 32, 258–266 CrossRef CAS.
- K. T. Kim, S. A. Meeuwissen, R. J. Nolte and J. C. van Hest, Nanoscale, 2010, 2, 844–858 RSC.
- A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 2140–2154 CrossRef CAS PubMed.
- L. Ruizendaal, S. P. Pujari, V. Gevaerts, J. M. J. Paulusse and H. Zuilhof, Chem –Asian J., 2011, 6, 2776–2786 CrossRef CAS PubMed.
- Y. Kotsuchibashi, M. Ebara, T. Aoyagi and R. Narain, Polym. Chem., 2012, 3, 2545–2550 RSC.
- S. Boufi and A. Gandini, RSC Adv., 2015, 5, 3141–3151 RSC.
- J. Justynska, Z. Hordyjewicz and H. Schlaad, Polymer, 2005, 46, 12057–12064 CrossRef CAS.
- N. ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef CAS.
- C. Rissing and D. Y. Son, Organometallics, 2009, 28, 3167–3172 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- O. Okay, M. Kurz, K. Lutz and W. Funke, Macromolecules, 1995, 28, 2728–2737 CrossRef CAS.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- A. Geissler, L. Q. Chen, K. Zhang, E. Bonaccurso and M. Biesalski, Chem. Commun., 2013, 49, 4962–4964 RSC.
- E. Aschenbrenner, K. Bley, K. Koynov, M. Makowski, M. Kappl, K. Landfester and C. K. Weiss, Langmuir, 2013, 29, 8845–8855 CrossRef CAS PubMed.
- P. Lucas, M. Vaysse, J. Aubry, D. Mariot, R. Sonnier and F. Ganachaud, Soft Matter, 2011, 7, 5528 RSC.
- R. Merindol, S. Diabang, O. Felix, T. Roland, C. Gauthier and G. Decher, ACS Nano, 2015, 9, 1127–1136 CrossRef CAS PubMed.
- A. Geissler, M. Biesalski, T. Heinze and K. Zhang, J. Mater. Chem. A, 2014, 2, 1107–1116 CAS.
- L. Wagberg, G. Decher, M. Norgren, T. Lindstrom, M. Ankerfors and K. Axnas, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- M. Li, L. J. Wang, D. Li, Y. L. Cheng and B. Adhikari, Carbohydr. Polym., 2014, 102, 136–143 CrossRef CAS PubMed.
- M. Hamedi, E. Karabulut, A. Marais, A. Herland, G. Nystrom and L. Wagberg, Angew. Chem., Int. Ed., 2013, 52, 12038–12042 CrossRef CAS PubMed.
- A. J. L. Jesus and J. S. Redinha, Comput. Theor. Chem., 2013, 1023, 74–82 CrossRef.
- J. J. Harris and M. L. Bruening, Langmuir, 2000, 16, 2006–2013 CrossRef CAS.
- S. E. Burke and C. J. Barrett, Pure Appl. Chem., 2004, 76, 1387–1398 CrossRef CAS.
- N. Mandzy, E. Grulke and T. Druffel, Powder Technol., 2005, 160, 121–126 CrossRef CAS.
- H. Gao, Y. N. Ma, X. Y. Lu, Y. R. Liang, B. Q. Chen and J. B. Ma, Eur. Polym. J., 2011, 47, 1232–1239 CrossRef.
- W. W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharm., 2010, 7, 1913–1920 CrossRef CAS PubMed.
- J. H. Holtz and S. A. Asher, Nature, 1997, 389, 829–832 CrossRef CAS PubMed.
- H. N. Zhang and J. Ruhe, Macromolecules, 2005, 38, 4855–4860 CrossRef CAS.
- F. K. Tanno, S. Sakuma, Y. Masaoka, M. Kataoka, T. Kozaki, R. Kamaguchi, Y. Ikeda, H. Kokubo and S. Yamashita, J. Pharm. Sci., 2008, 97, 2665–2679 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr05862g |
| This journal is © The Royal Society of Chemistry 2016 |