
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel hollow all-carbon structures†
Dage
Sundholm
*a,
Lukas N.
Wirz
b and
Peter
Schwerdtfeger
*c
aUniversity of Helsinki, Department of Chemistry, P.O. Box 55 (A.I. Virtanens plats 1), FIN-00014 University of Helsinki, Finland. E-mail: Dage.Sundholm@helsinki.fi
bCentre of Theoretical Chemistry and Physics, The New Zealand Institute for Advanced Study, Massey University Auckland, Private Bag 102904, 0632 Auckland, New Zealand. E-mail: mail@lukas-wirz.de
cCentre of Theoretical Chemistry and Physics, The New Zealand Institute for Advanced Study, Massey University Auckland, Private Bag 102904, 0632 Auckland, New Zealand. E-mail: p.a.schwerdtfeger@massey.ac.nz
First published on 24th August 2015
Abstract
A new family of cavernous all-carbon structures is proposed. These molecular cage structures are constructed by edge subdivisions and leapfrog transformations from cubic polyhedra or their duals. The obtained structures were then optimized at the density functional level. These hollow carbon structures represent a new class of carbon allotropes which could lead to many interesting applications.
1. Introduction
Graphene is a hexagonal two-dimensional (2D) lattice connected by trivalent vertices representing sp2 carbon atoms. This simple network can be modified by introducing different atoms or more complex molecular units for the vertices, and/or by edge extensions and modifications such as adding extra atoms or replacing them by molecular units with incoming and outgoing bonds lying on straight lines.1–4 Alternatively, one could also work with its dual, i.e., a triangulated planar surface of hexagonal symmetry containing vertices of degree six, and modify its topology accordingly. There are many different combinations and alterations possible to form such 2D molecular networks, which remain mostly unexplored and could have many interesting electronic properties and applications.2,3Graphyne shown in Fig. 1 represents such a class of 2D all-carbon allotropes,4–6 that is either built from a graphene template or its dual, and some building blocks have already been synthesized by organic chemists.7–13 Graphynes are one carbon atom thick and consist of sp and sp2 hybridized carbons in contrast to graphene whose 2D structure consists of six-membered rings of sp2 hybridized carbons. The α-graphyne structure is formally obtained by inserting –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– units into every carbon–carbon bond of graphene leading to a 2D carbon structure consisting of large hexagonal rings with frustrated three-coordinated carbons in each corner of the 18-membered hexagonal carbon rings. This bond assignment reflects well the symmetry of the structure and approximate bond lengths; however, configurations with only paired electrons are likely to contribute significantly to the electronic structure. The β-graphyne structure consists of similar hexagonal rings as in α-graphyne. However, each of the hexagonal rings is surrounded by six hexadehydro[12]annulene rings yielding an infinite 2D structure with all carbons formally four-valent. The γ-graphyne structure also shown in Fig. 1 consists of an infinite 2D structure with the six-membered hexagonal carbon (benzoic) rings surrounded by six hexadehydro[12]annulene rings having all carbons four-valent as in β-graphyne. It can be easily seen that this structure can be obtained by replacing certain edges in graphene with acetylene units (in graph theoretical terms this is called an edge subdivision), or alternatively by replacing all vertices in the dual triangulated network with benzene units and edges between them with –CC– groups. Graphdiynes are derived from the corresponding graphyne structures by replacing the –CC– groups with –CC–CC– units. Graphdiyne films have been synthesized on copper surfaces.14 The graphyne and graphdiyne structures are shown in Fig. 1. Of course, there are many other graphyne structures possible, see for example Peng et al. for a recent review.4
C– units into every carbon–carbon bond of graphene leading to a 2D carbon structure consisting of large hexagonal rings with frustrated three-coordinated carbons in each corner of the 18-membered hexagonal carbon rings. This bond assignment reflects well the symmetry of the structure and approximate bond lengths; however, configurations with only paired electrons are likely to contribute significantly to the electronic structure. The β-graphyne structure consists of similar hexagonal rings as in α-graphyne. However, each of the hexagonal rings is surrounded by six hexadehydro[12]annulene rings yielding an infinite 2D structure with all carbons formally four-valent. The γ-graphyne structure also shown in Fig. 1 consists of an infinite 2D structure with the six-membered hexagonal carbon (benzoic) rings surrounded by six hexadehydro[12]annulene rings having all carbons four-valent as in β-graphyne. It can be easily seen that this structure can be obtained by replacing certain edges in graphene with acetylene units (in graph theoretical terms this is called an edge subdivision), or alternatively by replacing all vertices in the dual triangulated network with benzene units and edges between them with –CC– groups. Graphdiynes are derived from the corresponding graphyne structures by replacing the –CC– groups with –CC–CC– units. Graphdiyne films have been synthesized on copper surfaces.14 The graphyne and graphdiyne structures are shown in Fig. 1. Of course, there are many other graphyne structures possible, see for example Peng et al. for a recent review.4
 | ||
| Fig. 1 The molecular structure of (a) α-graphyne, (b) β-graphyne, (c) γ-graphyne and (d) γ-graphdiyne. | ||
The graphene structure consisting of an infinite 2D layer of six-membered rings can be wrapped onto a sphere to give cavernous fullerene-like structures,15 but with the condition that e.g., 12 six-membered rings are replaced by five-membered ones such that Euler's polyhedral formula is fulfilled.16 Carbon nanotubes are similarly obtained by rolling up graphene sheets onto a cylinder.17 Fullerenes, carbon nanotubes, and other carbon-based nanostructures such as graphyne, graphdiyne, graphone, and graphane have been proposed as basic building blocks for a variety of interesting nanotechnological applications.4,18–30 As for graphene, hollow graphyne and graphdiyne structures can be obtained by wrapping up graphyne sheets, with modifications of course so that Euler's polyhedral formula is fulfilled. Analogously, graphyne-based and graphdiyne-based carbon nanotubes can be obtained by rolling up the corresponding planar sheets. Fullerene like graphynes (fullerynes) were already introduced by Baughman et al. in 1993,29 and more generally, vertex insertions into cubic polyhedral graphs were already discussed by Fowler and Rogers in 1998.30 Here we also note that the insertion of dicarbon units into all bonds of a chemical structure leads to the concept of carbomers (see for example the study of Chauvin and co-workers31–33).
One of the smallest members of this class of molecules is the recently proposed gaudiene molecule, which is an all-carbon molecule consisting of 72 carbon atoms forming a hollow structure of Oh symmetry (Fig. 2). Gaudiene can be constructed from a truncated octahedron with two thirds of the edges replaced by –CC– units.34 Quantum chemical calculations showed that β-C72 is an aromatic molecule with a rather large optical gap. Here, a novel class of hollow carbon structures is proposed. We call this class of polyhedral molecules gaudienes, because the initial polyhedron was inspired by the work of the Spanish architect Antoni Gaudí.34 Gaudienes are more general than carbomers as the insertion of dicarbon units may not take place in every chemical bond.
 | ||
| Fig. 2 The molecular structure of β-C72 of Oh symmetry, the smallest β-gaudiene investigated. | ||
2. Graph theoretical considerations
Before we start with the discussion of the different gaudienes we briefly analyze their topology and introduce a classification scheme. For the moment we do not distinguish between single, double or triple bonding in the polyhedral graph. Formally, gaudienes belong to the class of convex polyhedra (although a quantum theoretical treatment might end up with a locally non-convex structure).35 The classification of (non-regular) polytopes is currently an open problem.36 For the special class of fullerenes a face-spiral classification scheme has been developed by Manolopoulos et al.,37,38 which can be generalized for cubic polyhedra.35,39 Here, we use the fact that simple insertions (deletions) of divalent vertices into edges (edge subdivisions) of a graph G results in a graph G′ (which is homeomorphic to G) with a larger vertex set. Note that even though we have a homeomorphism between G and G′, G is not a subgraph of G′ as the vertex set E is not a subset of E′.A polyhedral graph G satisfies Euler's polyhedral formula, and by using the handshaking lemma we obtain
 | (1) |
 | (2) |
Thus, we conclude that one cannot tile a sphere with hexagons only, i.e., one has to introduce extra n-gons (for example 12 pentagons for fullerene graphs; this however does not guarantee that a certain polyhedron exists as for example the fullerene C22 does not exist). Introducing extra n-gons can be done in many different ways as we shall see when we discuss the gaudienes in detail.
For the characterization of the various gaudienes we have to describe the cubic graph G or, if G is obtained by some other graph transformation T of the original graph G0, i.e., G = T(G0). We list the vertices and faces according to Pedersen36 and describe the subsequent list of vertex insertions into the edges of each n-gon separately. For G, or G0 with no divalent vertices, we use the notation
| PG[N3, N4, N5, N6,…; F3, F4, F5, F6,…] | (3) |
| Ih[60;0, 0, 12, 20] | (4) |
This scheme does not uniquely classify the graph, as there are, for example, many non-isomorphic structures for a fullerene with a certain vertex count and symmetry,35 but becomes useful for the gaudienes introduced here as we shall see.
α-Graphyne based polyhedral structures (in their most general form) can be derived from any polyhedral graph by inserting linear –CC– units into every bond (see Fig. 1). It is thus identical to the carbomer concept. This insertion is performed irrespective of the assignment of double bonds in the original structure. For cubic graphs this scheme was already introduced by Fowler and Rogers.30 We denote the transformed version of a graph G0 according to this scheme as G = α(G0). For example, if we chose as the graph G0 of an α-gaudiene a fullerene with N0 vertices, denoted as PG[N0; 0, 0, 12, (N0/2–10)] and we insert two divalent vertices into all 3N0/2 edges, we get a total vertex count of NG = 4N0, where NG is the number of vertices in the α-gaudiene. Such an edge subdivision conserves the point group of G0.
It follows from Thurston's proof, which considers the number of non-isomorphic triangulations of a sphere with all N vertices of valency six or smaller, that the number of nonisomorphic cubic graphs with faces up to hexagons grow as  .40 As every polyhedral graph can be transformed into an α-gaudiene, we can estimate the number of derived α-gaudienes.
.40 As every polyhedral graph can be transformed into an α-gaudiene, we can estimate the number of derived α-gaudienes.
β-Graphyne analogue carbon cages can be generated starting with any polyhedral (i.e., three connected and planar) graph G0. First we perform a leapfrog transformation (LF), which is a (1,1) Goldberg–Coxeter transformation.35,41 The Goldberg–Coxeter transformation of a polyhedral graph is a polyhedral graph itself, implying that the initial graph can be subjected to any number of consecutive leapfrog transformations. In the obtained graph all faces with sizes ≠6 and a subset of all hexagons are selected, such that each vertex is adjacent to exactly one selected face. Such a selection is guaranteed to exist and to be unique for every polyhedral graph that is obtained through a leapfrog transformation. The edges of all selected faces (i.e., two thirds of all edges) are then replaced by a linear segment with two vertices. The leapfrog transformation triples the number N0 of vertices of a graph and introduces N0 extra hexagons,38 while the described replacement triples the number of vertices again. Therefore, β-graphyne cages with NG = (3 × 3)N0 vertices are accessible from polyhedra with N0 vertices. We denote this sequence of one leapfrog transformation and the following edge subdivision of G0 as β(LF(G0)). We denote k consecutive leapfrog transformations of a graph G0 as LFk(G0).
Equivalently, β-graphynes can be created from the same original graph G0 by first taking its dual G*0. Then, each n-valent vertex in the triangulation is replaced by an n-gon in which into each edge a –CC– unit has been inserted. For every pair of vertices connected by an edge in the original graph, the resulting two expanded polygons are connected by an edge.
In a similar fashion, γ-graphyne analogue carbon cages are generated starting with any polyhedral graph G0. Transformation and selection of faces are performed in the same fashion as above. Then, all edges that are not adjacent to any selected face (one third of the edge set), i.e., the complement of the previously picked edges, are replaced by two additional vertices. This replacement scheme doubles the number of vertices, transforming the polyhedral graphs with N0 vertices into NG = (3 × 2)N0 vertex cages. Analogously to the previous transformation this is denoted as γ(LF(G0)).
Alternatively, we can start with the dual graph G*0, and replace every n-valent vertex by an n-gon. These n-gons are then connected – according to the connectivity of the vertices they originated from – by bonds into which –CC– units have been inserted.
The α-, β-, and γ-graphdiyne like cages are generated in the same way as the respective graphynes but by replacing the edges with linear four-vertex segments instead of two-vertex segments. N0 vertex polyhedral graphs are then transformed into NG = 7N0, NG = (3 × 5)N0, and NG = (3 × 3)N0 vertex cages respectively. Transformations of graph G0 to graphdiynes are written with a prepended ‘C4’, e.g., C4-α(G0).
3. Computational methods
Computationally, all structures were generated with a modified version of the program Fullerene.42 Selected small polyhedral graphs were hard coded. These graphs were subjected to leapfrog transformations as detailed in the previous section. The generation of all the gaudienes considered here is summarized in Table 1.| N | T | G 0 | Polyhedron (G0) |
|---|---|---|---|
| 80 | α | I h [20; 0, 0, 12] | Dodecahedron |
| 72 | β (LF) | O h [8; 0, 6 ] | Cube |
| 216 | β (LF2) | O h [8; 0, 6 ] | Cube |
| 648 | β (LF3) | O h [8; 0, 6] | Cube |
| 120 | C4-β (LF) | O h [8; 0, 6] | Cube |
| 360 | C4-β (LF2) | O h [8; 0, 6] | Cube |
| 162 | β (LF) | C 2v [18; 0, 2, 8, 1] | FS (45556555545) |
| 288 | β (LF) | C2 [32; 0, 0, 12, 6] | Fullerene C32 (1) |
| 288 | β (LF) | D 2 [32; 0, 0, 12, 6] | Fullerene C32 (2) |
| 288 | β (LF) | D 3d [32; 0, 0, 12, 6] | Fullerene C32 (3) |
| 288 | β (LF) | C2 [32; 0, 0, 12, 6] | Fullerene C32 (4) |
| 288 | β (LF) | D 3h [32; 0, 0, 12, 6] | Fullerene C32 (5) |
| 288 | β (LF) | D 3 [32; 0, 0, 12, 6] | Fullerene C32 (6) |
| 72 | γ (LF) | D 6h [12; 0, 6, 0, 2] | Hexagonal prism |
| 648 | γ (LF3) | D 6h [12; 0, 6, 0, 2] | Hexagonal prism |
| 108 | C4-γ (LF) | D 6h [12; 0, 6, 0, 2] | Hexagonal prism |
| 432 | γ (LF2) | D 6d [24; 0, 0, 12, 2] | Fullerene C24 (1) |
| 288 | γ (LF) | D 2h [48; 0, 0, 12, 14] | Fullerene C48 (15) |
| 288 | γ (LF) | C 2v [48; 0, 0, 12, 14] | Fullerene C48 (17) |
| 288 | γ (LF) | D 2h [48; 0, 0, 12, 14] | Fullerene C48 (41) |
| 288 | γ (LF) | C 2v [48; 0, 0, 12, 14] | Fullerene C48 (56) |
| 288 | γ (LF) | C 2h [48; 0, 0, 12, 14] | Fullerene C48 (80) |
| 288 | γ (LF) | C 2v [48; 0, 0, 12, 14] | Fullerene C48 (138) |
| 288 | γ (LF) | D 6d [48; 0, 0, 12, 14] | Fullerene C48 (186) |
| 288 | γ (LF) | D 6d [48; 0, 0, 12, 14] | Fullerene C48 (189) |
As the force field in program Fullerene can only operate on cubic graphs and triangulations, polyhedra were generated at this stage and optimized to bond lengths that correspond to the polyhedron after replacing a subset of bonds with elongated linear segments. After the optimization of the Cartesian coordinates, atoms were inserted and placed in 3D by linear interpolation.
The molecular structures were then further optimized at the density functional theory (DFT) level using the Becke–Perdew generalized gradient approximation (GGA) functional (BP86) in combination with the Karlsruhe split-valence polarization (SVP) basis sets.43–46 Since the calculations at the BP86 level slightly underestimate the gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), single-point calculations were performed at the DFT level using Becke's three-parameter functional (B3LYP) together with a SVP basis set.46–48 The semi-empirical dispersion correction of Grimme has been employed in the molecular structure optimization.49,50 The DFT calculations were performed with Turbomole version 6.5.51–53 The Cartesian coordinates of the atomic positions of all studied structures are given in the ESI.† Improving the basis set to a triple-zeta split-valence polarization set for β-C72 leads only to a small change in geometry (change in bond distances |Δre| < 0.012 Å and bond angles |Δαe| < 0.04 degrees). These errors are within the accuracy of DFT and therefore do not warrant more extensive computer calculations for the discussion of the structures presented here.
4. Molecular structures
4.1 α-Gaudienes
The optimized structure of the smallest fullerene-like carbomer based on α-graphyne, the gaudiene α-C80, is shown in Fig. 3. The structure belongs to the icosahedral (Ih) point group as does its origin, the dodecahedron represented by the smallest fullerene Ih-C20. Hence, this gaudiene consists of 12 pentagonal 15-membered carbon rings forming a cage. The atoms shared by the three rings are formally sp3 hybridized carbons with only three neighbor carbons leading to a relatively small HOMO–LUMO gap of 0.740 eV at the BP86 level, which can be compared with the HOMO–LUMO gaps of 1.66 eV, 1.45 eV and 1.23 eV for the C60, C180 and C240 fullerenes calculated at the GGA level.54 For comparison, DFT calculations using the BP86 functional yield HOMO–LUMO gaps of 1.24 eV and 0.21 eV for β-C72 and γ-C72, respectively. At the B3LYP level the HOMO–LUMO gap for α-C80 is 1.22 eV. The adjacency spectrum of fullerene-like carbomers and their open/closed shell character have been analyzed in detail by Fowler and Rogers.30 | ||
| Fig. 3 The molecular structure of α-C80 of Ih symmetry, which is the smallest α-gaudiene investigated. | ||
A very stable molecule with a HOMO–LUMO gap of 5.17 eV is obtained by adding 20 hydrogens to the corners of the α-C80 icosahedron. Larger α-gaudienes can easily be constructed by using fullerenes as templates and replacing each carbon–carbon bond with a –CC– moiety. All α-gaudienes have three-coordinated carbons in the intersections between three carbon rings. More stable molecules can be obtained by adding substituents to these three-coordinated carbons.
We should mention that eqn (2) also allows one to use other cubic polyhedra. For example, if we chose the truncated octahedron belonging to the Archimedean solids, Oh[24; 0, 8, 0, 6], containing six squares according to eqn (2), we can expand this graph for all edges in a similar way and we arrive at Oh-C96, another type of α-gaudiene. Hence the playing field is huge for the construction of new 3D polyhedral molecular structures.
4.2 β-Gaudienes
The smallest member of the β-gaudiene class of molecules with four- and six-membered rings involved is the previously proposed β-C72 shown in Fig. 2.34 It belongs to the Oh point group and can be generated from the simple cube G0 = Oh[8; 0, 6] or its dual, the octahedron G*0 = Oh[0, 6; 8]. When folding β-graphyne to the hollow C72 structure, the edges of eight hexadehydro[12]annulene rings of β-graphyne form six four-sided rings having four carbons on each side with alternating triple and single bonds. Even though hexadehydro[12]annulene is formally antiaromatic,55–57 β-C72 is aromatic according to the ring-current criterion sustaining a ring current strength of 44.3 nA T−1 around the molecule, which can be compared to the ring-current strength of benzene of 11.8 nA T−1.34,58 Since all carbons have formally four bonds, β-C72 is a stable molecule with a HOMO–LUMO gap of 1.82 eV at the B3LYP/SVP level.A number of β-gaudiene structures were constructed using small polyhedral graphs as starting points. The smallest one constructed using that procedure was β-C162 (C2v), which was obtained using an 18-vertex graph as the initial structure, C2v[18; 0, 2, 8, 1], belonging to a non-regular polyhedron (see Fig. 4). This polyhedron does not belong to the common solids and we therefore give the face spiral indices in Table 1. The optimized molecular structure of β-C162 (C2v) is shown in Fig. 5a. It has a rather small HOMO–LUMO gap of 0.48 eV as obtained at the B3LYP level.
 | ||
| Fig. 4 Graph of the non-regular C2v [18; 0, 2, 8, 1] polyhedron. | ||
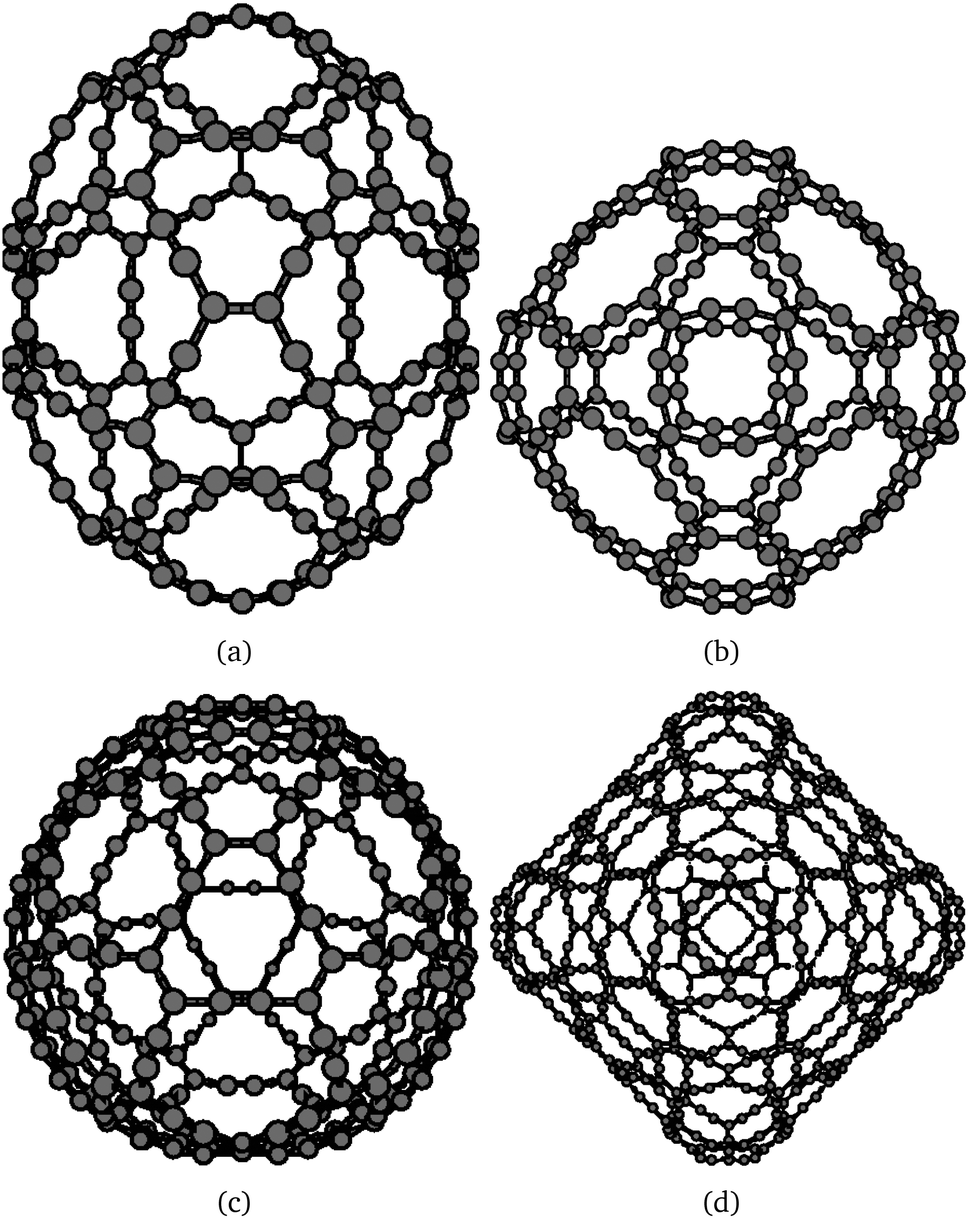 | ||
| Fig. 5 The molecular structure of the energetically lowest isomers of β-gaudienes: (a) β-C162 (C2v), (b) β-C216 (Oh), (c) β-C288 (D3d), and (d) β-C648 (Oh). | ||
Since the topological method ensures that the symmetry of the seed structure is conserved, many different isomers can be constructed when the corresponding fullerene structures are available. The next larger β-gaudiene structures of Oh symmetry are β-C216 and β-C648, which have HOMO–LUMO gaps of 0.415 eV and 0.225 eV at the B3LYP level, respectively. These are also shown in Fig. 5.
A large number of β-gaudienes consisting of 288 carbons can be constructed by using different fullerene isomers such as those of C32 as starting structures. The energetically lowest β-C288 structure of all investigated isomers (see Table 1) belongs to the D3d point group. The energetically lowest β-gaudiene structures obtained in this work are shown in Fig. 5. The rest of the studied β-C288 structures are shown in the ESI.† The energetically lowest β-C288 isomers are 48.8 kJ mol−1 (D3), 51.9 kJ mol−1 (D2), 53.2 kJ mol−1 (C2), 65.6 kJ mol−1 (C2), and 146.6 kJ mol−1 (D3h) above the D3d structure. The β-C288 (Oh) isomer consisting of a β-C72 molecule inside β-C216 (β-C72@β-C216) is 683 kJ mol−1 above the lowest β-C288 isomer of D3d symmetry. The HOMO–LUMO gaps of the β-C288 structures are in the range of 0.27–0.42 eV, with the largest gap for the most stable isomer. The two β-C288 structures of C2 symmetry have negative HOMO–LUMO gaps at the BP86 level.
The largest molecule considered in this study was an onion-like β-C72@β-C216@β-C648 cluster of Oh symmetry. The structure is shown in the ESI.†
4.3 γ-Gaudienes
The smallest γ-gaudiene (γ-C72) studied belongs to the D6h point group and has a HOMO–LUMO gap of 0.519 eV. The optimized molecular structure is shown in Fig. 6. The benzoic six-membered carbon ring is surrounded by six hexadehydro[12]annulene rings that are fused at the molecular edge. The correct topology of the closed surface is ensured by the six four-membered carbon rings between the hexadehydro[12]annulenes at the edge of the molecule. The next larger γ-gaudiene structure of Oh symmetry is γ-C432 (Oh) with a HOMO–LUMO gap of 0.491 eV. The largest γ-gaudiene studied in this work is γ-C648, which has D6h symmetry and a small HOMO–LUMO gap of 0.253 eV at the B3LYP level. | ||
| Fig. 6 The molecular structure of γ-C72 of D6h symmetry, which is the smallest γ-gaudiene investigated. | ||
A large number of γ-C288 structures were constructed with different fullerene isomers of C48 as the origin graph. The energetically lowest γ-C288 belongs to the C2v point group. The structure of the energetically lowest γ-C288 (C2v) isomer is shown in Fig. 7. The structures and atomic coordinates of the remaining studied γ-C288 clusters are shown in the ESI.† The energetically lowest γ-C288 isomers are 44.3 kJ mol−1 (D2h), 47.8 kJ mol−1 (C2h), 160.0 kJ mol−1 (D6d), 167.5 kJ mol−1 (C2v), 287.2 kJ mol−1 (C2v), 565.5 kJ mol−1 (D2h), and 873.1 kJ mol−1 (D6d) above the lowest C2v structure. The calculations show that there are only three low-lying γ-C288 isomers among the studied ones.
 | ||
| Fig. 7 The molecular structure of the energetically lowest isomers of (a) γ-C288 (C2v), (b) γ-C432 (Oh), and (c) γ-C648 (D6h) gaudienes. | ||
The HOMO–LUMO gap of the lowest γ-C288 structure is 0.36 eV at the B3LYP level, whereas the HOMO–LUMO gap of the γ-C288 is in the range of 0.18–0.47 eV, with the largest gap for γ-C288 (D2h). The energetically lower isomer of D6d symmetry has a negative HOMO–LUMO gap at the BP86 level. The molecular structure of the energetically lowest γ-gaudienes obtained in this work is shown in Fig. 7.
4.4 Graphdiyne-based gaudienes
Graphdiyne-based gaudienes are obtained by replacing the –CC– units of gaudienes with the twice longer –CC–CC– (C4) group.
Even though the obtained molecules have a larger number of triple bonds, the HOMO–LUMO gap is wider than the corresponding graphyne-based molecules. The HOMO–LUMO gaps calculated at the B3LYP level are 0.877 eV, 1.356 eV, and 1.168 eV for C4-γ-C108, C4-β-C120, and C4-β-C360, respectively. The larger stabilization of the graphdiyne-based gaudienes can be traced back to the molecular properties of dodecadehydro[18]annulene, which is an aromatic molecule.56 The HOMO–LUMO gap of C4-γ-C108 is smaller than the other two studied C4-gaudienes due to the significantly larger bond strain of the small four-membered carbon rings at the edge of the molecule. For the C4-β-gaudienes, the correct topology is obtained by the six 20-membered carbon rings. The molecular structures of the studied C4-gaudienes are shown in Fig. 8. The Cartesian coordinates of the atomic positions are given in the ESI.†
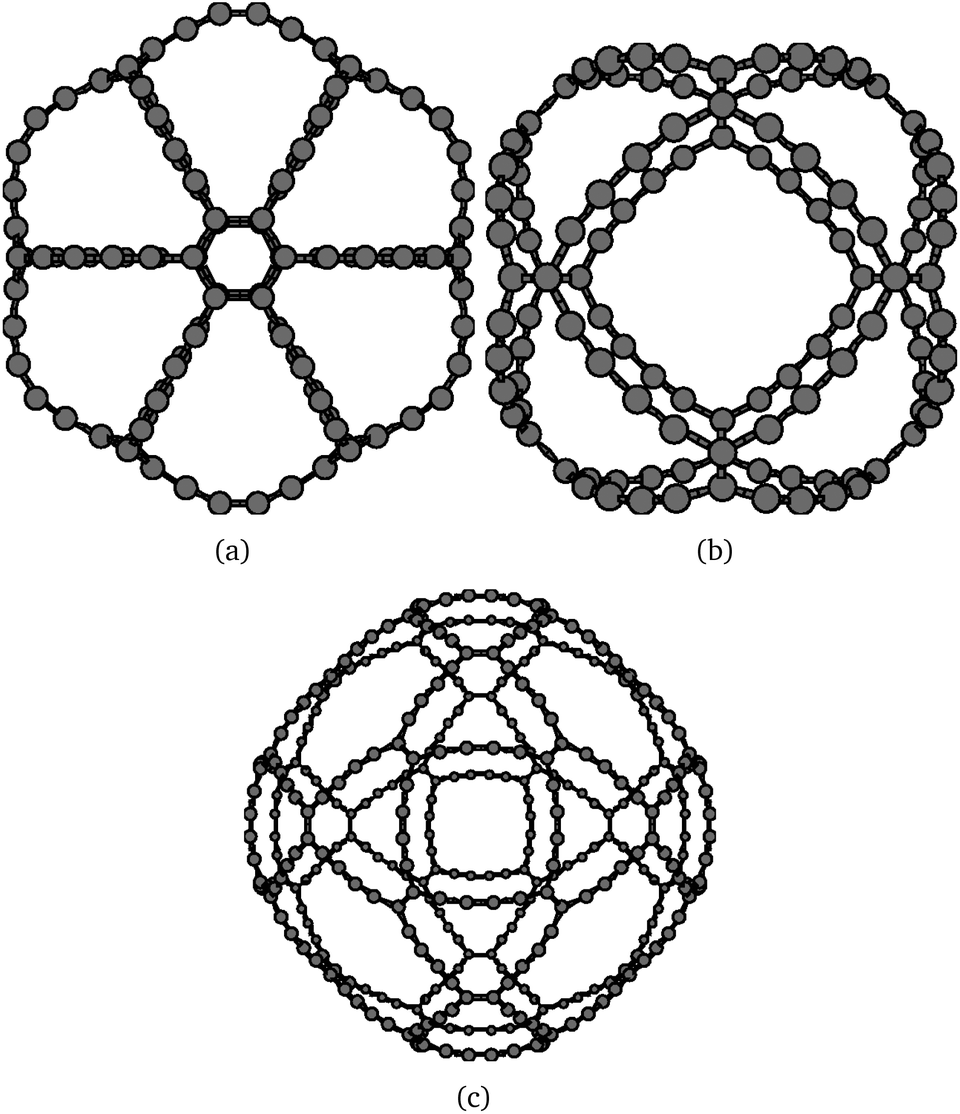 | ||
| Fig. 8 The molecular structure of the investigated graphdiyne-based (C4) gaudienes: (a) C4-γ-C108 (D6h), (b) C4-β-C120 (Oh), and (c) C4-β-C360 (Oh). | ||
5. Discussion and conclusions
The γ-gaudienes are generally significantly lower in energy than the β-gaudienes for the same number of carbon atoms. The most important exception is β-C72, which is 128.9 kJ mol−1 below γ-C72 due to the ring strain of the four-membered carbon rings of γ-C72 and maybe also due to the aromatic stabilization of β-C72.34 The HOMO–LUMO gap of β-C72 of 1.82 eV calculated at the B3LYP level is also much larger than the HOMO–LUMO gap of 0.52 eV for γ-C72. For the small γ-C72 cage, the bond strain of the four-membered carbon rings is not compensated by the energetically favorable structure of γ-graphyne. For larger gaudienes, the γ-graphyne structure has significantly lower energies than the corresponding β-gaudiene structures. The lowest isomer of γ-C288 is more than 5000 kJ mol−1 (!) below the energy of the lowest β-C288 structure. For larger gaudienes, the energy difference between the γ-gaudiene and β-gaudiene is even bigger. For the largest studied gaudienes, γ-C648 is about 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kJ mol−1 below β-C648.
000 kJ mol−1 below β-C648.
The relative energy per carbon atom is given as a function of 1/N in Fig. 9, where N is the number of carbon atoms. The relative energy depends roughly linearly on 1/N leading to the three straight lines in Fig. 9 showing that γ-gaudiene is the most stable structure and that C4-gaudienes are the energetically highest ones. Extrapolation to 1/N → 0 yields the relative energy of β-graphyne, γ-graphyne, and β-graphdiyne. The calculations show that β-graphyne is 17 kJ per mol per carbon higher in energy than γ-graphyne and that β-graphdiyne is only 7 kJ per mol per carbon above β-graphyne.
 | ||
| Fig. 9 The relative energy (in kJ mol−1) as a function of 1/N of the studied molecular classes. N is the number of atoms. The relative energies are fitted to a line. The outlier at 37 kJ mol−1 for γ-C72 is not considered in the fit. | ||
The HOMO–LUMO gaps calculated at the B3LYP level are plotted as a function of 1/N in Fig. 10 yielding three straight lines for three gaudiene classes. Extrapolation of the HOMO–LUMO gap to the limit of 1/N → 0 yields the estimated HOMO–LUMO gaps of the β-graphyne, γ-graphyne and β-graphdiyne sheets. The three lines suggest that the HOMO–LUMO gaps for the infinite planar structures are larger than zero. The extrapolated HOMO–LUMO gap of β-graphyne is 0.17 eV. For γ-graphyne we obtained an extrapolated HOMO–LUMO gap of 0.33 eV. The extrapolated HOMO–LUMO gap of 1.25 eV for β-graphdiyne agrees well with the previously reported value of 1.22 eV, which was calculated at the DFT level using a hybrid functional59 and it is significantly larger than the HOMO–LUMO gap of 0.46 eV calculated at the DFT level using the generalized gradient approximation.60 Even though the error bars of the present extrapolated values are large due to the few number of points used in the fit, the present calculations suggest that the HOMO–LUMO gap of graphdiyne is larger than 1 eV.
 | ||
| Fig. 10 The HOMO–LUMO gap (in eV) as a function of 1/N of the studied molecular classes. N is the number of atoms. The relative energies are fitted to a line. The outlier at 1.82 eV for β-C72 is not considered in the fit. | ||
The hollow gaudiene structures might open the avenue to novel materials with interesting properties. For example, triple bonds provide possibilities for functionalizing the molecules with different kinds of substituents that might even couple several gaudiene molecules with polymers or solid-state materials. The gaudiene structures can also be fully or partially saturated with hydrogens without destroying their cage structures. Such molecules are ordinary saturated hydrocarbons with extraordinary hollow structures. By attaching substituents to the corners of α-gaudienes, molecules with a very large HOMO–LUMO gap are obtained. The optical gap of the C4-gaudienes is significantly larger than the two other classes of molecules. Extrapolation to infinitely large clusters suggests that planar graphdiyne has indeed a HOMO–LUMO gap that is significantly larger than zero, which might provide novel possibilities for carbon-based mono-layered structures.4
Finally, we point out that there are also other graph theoretical procedures for obtaining cavernous all-carbon structures that have not been discussed here.61,62 For example, one can insert –CC– units into the edges running parallel to the main axis of a fullerene nanotube. One can also use other building blocks besides carbon which fit the required topologies, e.g., building units which are used in metal–organic frameworks. This opens up a whole new area of interesting cage structures and materials which could be useful in many interesting applications.
Acknowledgements
This work has been supported by the Academy of Finland through projects (266227 and 275845) and its Computational Science Research Programme (LASTU/258258). DS acknowledges Magnus Ehrnrooth Foundation, the Swedish Cultural Foundation in Finland, and the Alexander von Humboldt Stiftung for financial support. DS also thanks the New Zealand Institute for Advanced Study at the Massey University for the kind hospitality. CSC – the Finnish IT Center for Science – is acknowledged for computer time. We would like to thank James Avery (University of Copenhagen) for providing the generalized Goldberg–Coxeter program.References
- H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 122, 4153–4156 CrossRef PubMed
.
- A. L. Ivanovskii, Russ. Chem. Rev., 2012, 81, 571–605 CrossRef PubMed
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed
- Q. Peng, A. K. Dearden, J. Crean, L. Han, S. Liu, X. Wen and S. De, Nanotechnol. Sci. Appl., 2014, 7, 1–29 CrossRef CAS PubMed
- R. H. Baughman, H. Eckhardt and M. Kertesz, J. Chem. Phys., 1987, 87, 6687–6699 CrossRef CAS PubMed
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef CAS PubMed
- F. Diederich, Nature, 1994, 369, 199–207 CrossRef CAS PubMed
- M. M. Haley, S. C. Brand and J. J. Pak, Angew. Chem., Int. Ed., 1997, 36, 836–838 CrossRef CAS PubMed
- U. H. F. Bunz, Y. Rubin and Y. Tobe, Chem. Soc. Rev., 1999, 28, 107–119 RSC
- E. L. Spitler, C. A. Johnson and M. M. Haley, Chem. Rev., 2006, 106, 5344–5386 CrossRef CAS PubMed
- M. M. Haley, Pure Appl. Chem., 2008, 80, 519–532 CrossRef CAS
- F. Diederich and M. Kivala, Adv. Mater., 2010, 22, 803–812 CrossRef CAS PubMed
- K. Tahara, Y. Yamamoto, D. E. Gross, H. Kozuma, Y. Arikuma, K. Ohta, Y. Koizumi, Y. Gao, Y. Shimizu, S. Seki, K. Kamada, J. S. Moore and Y. Tobe, Chem. – Eur. J., 2013, 19, 11251–11260 CrossRef CAS PubMed
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS PubMed
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS PubMed
- S. Ihara and S. Itoh, Carbon, 1995, 33, 931–939 CrossRef CAS
-
M. Meyyappan and D. Srivastava, Handbook of Nanoscience, Engineering, and Technology, CRC Press, Boca Raton FL, 2003, ch. 18, pp. 1–26 Search PubMed
- T. C. Dinadayalane and J. Leszczynski, Struct. Chem., 2010, 21, 1155–1169 CrossRef CAS
- A. N. Enyashin and A. L. Ivanovskii, Phys. Status Solidi B, 2011, 248, 1879–1883 CrossRef CAS PubMed
- R. Mas-Ballesté, C. Gómez-Navarro, J. Gómez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC
- D. Malko, C. Neiss, F. Viñes and A. Görling, Phys. Rev. Lett., 2012, 108, 086804 CrossRef
- S. Zhang, Y. Zhang, S. Huang and C. Wang, Nanoscale, 2012, 4, 2839–2842 RSC
- L. Zoppi, J. S. Siegel and K. K. Baldridge, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 1–12 CrossRef CAS PubMed
- J.-J. Zheng, X. Zhao, Y. Zhao and X. Gao, Sci. Rep., 2013, 3, 1271 Search PubMed
- J. Xi, D. Wang and Z. Shuai, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 215–227 CrossRef CAS PubMed
- Z. Li, M. Smeu, A. Rives, V. Maraval, R. Chauvin, M. A. Ratner and E. Borguet, Nat. Commun., 2015, 6, 6321 CrossRef CAS PubMed
- R. H. Baughman, D. S. Galvão, C. Cui, Y. Wang and D. Tománek, Chem. Phys. Lett., 1993, 204, 8–14 CrossRef CAS
- P. W. Fowler and K. M. Rogers, J. Chem. Soc., Faraday Trans., 1998, 94, 1019–1027 RSC
- R. Chauvin, Tetrahedron Lett., 1995, 36, 401–404 CrossRef CAS
- R. Chauvin, Tetrahedron Lett., 1995, 36, 397–400 CrossRef CAS
- V. Maraval and R. Chauvin, New J. Chem., 2007, 31, 1853–1873 RSC
- D. Sundholm, Phys. Chem. Chem. Phys., 2013, 15, 9025–9028 RSC
- P. Schwerdtfeger, L. N. Wirz and J. Avery, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 96–145 CrossRef CAS PubMed
- J. J. Pedersen, Calif. Math., 1977, 2, 21–28 Search PubMed
- D. E. Manolopoulos, J. C. May and S. E. Down, Chem. Phys. Lett., 1991, 181, 105–111 CrossRef CAS
-
P. W. Fowler and D. E. Manolopoulos, An Atlas of Fullerenes, Dover Publications Inc., Mineola, New York, 2nd edn, 2006 Search PubMed
- L. N. Wirz, R. Tonner, J. Avery and P. Schwerdtfeger, J. Chem. Inf. Model., 2014, 54, 121–130 CrossRef CAS PubMed
- W. P. Thurston, Geom. Topol. Monogr., 1998, 1, 511–549 Search PubMed
- J. Avery, to be submitted, 2015.
- P. Schwerdtfeger, L. Wirz and J. Avery, J. Comput. Chem., 2013, 34, 1508–1526 CrossRef CAS PubMed
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS
- TURBOMOLE V6.5 2013, a development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip.
Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS
- G. Casella, A. Bagno and G. Saielli, Phys. Chem. Chem. Phys., 2013, 15, 18030–18038 RSC
- J. A. Pople and K. G. Untch, J. Am. Chem. Soc., 1966, 88, 4811–4815 CrossRef CAS
- J. Jusélius and D. Sundholm, Phys. Chem. Chem. Phys., 2001, 3, 2433–2437 RSC
- J. Jusélius and D. Sundholm, Phys. Chem. Chem. Phys., 2008, 10, 6630–6634 RSC
- H. Fliegl, D. Sundholm, S. Taubert, J. Jusélius and W. Klopper, J. Phys. Chem. A, 2009, 113, 8668–8676 CrossRef CAS PubMed
- Y. Jiao, A. Du, M. Hankel, Z. Zhu, V. Rudolph and S. C. Smith, Chem. Commun., 2011, 47, 11843–11845 RSC
- M. Long, L. Tang, D. Wang, Y. Li and Z. Shuai, ACS Nano, 2011, 5, 2593–2600 CrossRef CAS PubMed
- D. Sebastiani and M. A. Parker, Symmetry, 2009, 1, 226–239 CrossRef CAS PubMed
- A. Ivanovskii, Prog. Solid State Chem., 2013, 41, 1–19 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of the studied molecules. See DOI: 10.1039/c5nr04370k |
| This journal is © The Royal Society of Chemistry 2015 |