
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyol synthesis, functionalisation, and biocompatibility studies of superparamagnetic iron oxide nanoparticles as potential MRI contrast agents†
Roxanne
Hachani
ab,
Mark
Lowdell
c,
Martin
Birchall
d,
Aziliz
Hervault
ab,
Damien
Mertz
e,
Sylvie
Begin-Colin
e and
Nguyễn Thị Kim
Thanh
*ab
aDepartment of Physics and Astronomy, University College London, Gower Street, London WC1E 6BT, UK
bUCL Healthcare and Biomagnetics and Nanomaterials Laboratory, 21 Albemarle Street, London W1S 4BS, UK. E-mail: ntk.thanh@ucl.ac.uk; Fax: +44 (0)207 670 2920; Tel: +44 (0)207 491 6509
cDepartment of Haematology, Royal Free Hospital, University College London, London NW3 2QG, UK
dUniversity College London Ear Institute, 332 Gray's Inn Road, London WC1X 8EE, UK
eICPMS, UMR CNRS-UdS-ECPM 7504, 23 rue du loess BP 43, 67034 Strasbourg, France
First published on 5th October 2015
Abstract
Iron oxide nanoparticles (IONPs) of low polydispersity were obtained through a simple polyol synthesis in high pressure and high temperature conditions. The control of the size and morphology of the nanoparticles was studied by varying the solvent used, the amount of iron precursor and the reaction time. Compared with conventional synthesis methods such as thermal decomposition or co-precipitation, this process yields nanoparticles with a narrow particle size distribution in a simple, reproducible and cost effective manner without the need for an inert atmosphere. For example, IONPs with a diameter of ca. 8 nm could be made in a reproducible manner and with good crystallinity as evidenced by X-ray diffraction analysis and high saturation magnetization value (84.5 emu g−1). The surface of the IONPs could be tailored post synthesis with two different ligands which provided functionality and stability in water and phosphate buffer saline (PBS). Their potential as a magnetic resonance imaging (MRI) contrast agent was confirmed as they exhibited high r1 and r2 relaxivities of 7.95 mM−1 s−1 and 185.58 mM−1 s−1 respectively at 1.4 T. Biocompatibility and viability of IONPs in primary human mesenchymal stem cells (hMSCs) was studied and confirmed.
Introduction
Magnetic iron oxide nanoparticles have been the subject of great interest in recent years due to their various potential biological applications such as magnetic hyperthermia,1 MRI2–4 drug delivery or cell tracking.5–8 Currently, iron oxide nanoparZticles such as magnetite (Fe3O4) or its oxidised form, maghemite (Fe2O3), are the most frequently investigated. This is due to their biocompatibility, non-toxicity and non-immunogenicity in biological systems. Furthermore, they were approved for clinical applications by the Federal Drug Administration (FDA) in the 1980s and have since then been commercially available.9 These were developed as contrast agents for magnetic resonance anatomic imaging of malignant diseases in organs associated with the reticuloendothelial system (e.g., liver, spleen)10–12 and lymph nodes.13 Numerous commercially available particles such as Feridex© or Resovist© have been used to label and track hMSCs.14–18 Indeed, in cellular therapies, they represent a valuable tool to monitor the displacement and functionality of transplanted hMSCs in a safe and effective manner.Superparamagnetic IONPs may also be used as a multimodal platform for other medical applications. The nanoparticle surface provides the stability for these nanoparticles as well as their functionality by which biological moieties (antibodies, peptides, drug encapsulation) may potentially be grafted.19–24 For instance, many studies have recently explored their therapeutic potential to kill cancer cells by hyperthermia (heat induced after the application of an alternating magnetic field) and simultaneous triggered drug delivery.25–27 Furthermore, when a biological moiety (e.g., antibody, peptide) is conjugated to the surface of the nanoparticles, the latter may be actively targeted towards cancer cells to minimise any secondary effects from medical treatment on neighbouring healthy cells.23,28,29 Their small size also allows sub cellular targeting which can overcome certain physical barriers that larger molecules such as pharmaceutical drugs may not.30 Another application for cellular therapy includes their ability to magnetically guide cells especially on tissue-engineered scaffolds or in vivo.31–34
Despite all these advantages, the in vitro and in vivo use of IONPs faces some challenges. Amongst which is the lack of reproducibility of synthesis that yield water-dispersible nanoparticles. Indeed, while the co-precipitation of Fe(II) and Fe(III) ions in the presence of a base is a widespread method in literature due to the high yields obtained,35,36 the reproducibility of this route remains questionable.37 The other main synthetic pathways involve organic solvents which allow a better control of the size and shape of nanoparticles obtained. However, the latter involve post synthesis steps to render the nanoparticles hydrophilic and biocompatible which are difficult to monitor and control precisely in a quantitative manner.
Recently, water-dispersible IONPs have been developed through a simple polyol method which involves the thermal decomposition of iron precursors in a polyol solvent. The polyol acts as the solvent, a surfactant, as well as the reducing agent. The polyols are known to reduce the metal salts to metal nuclei which then nucleate to form metal particles.38 In 2007, Wan et al. synthesised IONPs by this approach under inert atmosphere.39 The particles obtained were ca. 8 ± 1.1 nm and with a saturation magnetisation of 80 emu g−1. More recently, the morphological evolution of IONPs though the reduction of iron chloride hexahydrate FeCl3·6H2O in ethylene glycol or 1,2-propylene glycol was investigated and demonstrated the importance in the choice of solvent regarding the properties of the nanoparticles.40 This was confirmed with several recent studies with the use of diethylene glycol, tetraethylene glycol, poly(vinyl alcohol), or 2-pyrrolidone.41–44 Although the exact steps during this reaction have not yet been clearly identified, a thorough mechanistic study on the different variables during this pathway was investigated by Miguel-Sancho et al.42,45 The nanoparticles were then further modified by exchange of the non-covalently bound polyol molecules with meso-2,3-dimercaptosuccinic acid (DMSA) and functionalised with an antibody that was successfully recognised by a model anti-rabbit IgG HRP in an indirect ELISA assay. The biocompatibility and low cytotoxic effects of these types of particles on HepG2, U87MG, and HeLa cells was also recently demonstrated.46 However, the obtained IONPs had several disadvantages such as having a rather low MRI relaxivity value (r2 = 119 mM−1 s−1),41 low stability of tri(ethylene glycol) coated IONPs and aggregation of dimercaptosuccinic acid-coated IONPs.45 Therefore, obtaining water dispersible IONPs which have colloidal stability in high concentration of electrolytes and a wide range of pH, appropriate surface coatings, high magnetic moment, and biological compatibility remains a significant challenge.
In this work we have adapted the polyol synthesis at high pressure and high temperature conditions of an autoclave which allows us to obtain IONPs with enhanced magnetic properties and better control of their morphology. We attempted to contribute to a better comprehension of the polyol synthesis and to improve the properties of the IONPs by carrying out a systematic study of the process. For this, the conditions were finely tuned (reaction time, type of solvent and concentration of iron precursor) and their effects on the final size, morphology and magnetic properties of the obtained NPs were explored. We also tuned the surface of the IONPs with various ligands. The suitability of these SPIONs as an MRI contrast agent and for other biomedical applications was then investigated in vitro with primary hMSCs. The immediate application of this study is to magnetically label MSCs seeded on a trachea scaffold to detect and track them non-invasively by MRI.
Experimental
Chemicals
Iron(III) acetylacetonate (Fe(acac)3, 99.9%), triethylene glycol (TREG) (99%), diethylene glycol, DEG, (99%) and acetone were purchased from VWR, UK; tetraethylene glycol, TEG, (99%), L-tartaric acid (≥99.5%) and 3,4-dihydroxyhydrocinnamic acid (98%) were purchased from Sigma-Aldrich. All chemicals were used as received without further purification.Nanoparticle synthesis
The magnetic iron oxide nanoparticles were synthesized by a modified polyol synthesis procedure described by both Cai and Wan, as well as Maity et al.47,48In a typical reaction, we used the following reaction conditions: 0.7 g Fe(acac)3 were dissolved in 20 ml of various polyol solvents, then heated up to 250 °C and maintained at that temperature during 8 h.
In a typical synthesis, a desired amount of Fe(acac)3 was mixed with 20 ml of TREG and sonicated during 30 min in order to homogenise the red dispersion. The resulting mixture was then transferred into a 45 ml capacity Teflon liner and the latter was assembled with the autoclave jacket and placed into an oven (Memmert, model UFP400). The autoclave vessels have a flat PTFE gasket that is sealed with a screw cap. The screw caps and bodies of the vessel are made of alloy steel which allows experimental conditions of up to 300 °C and maximum working pressure of 1700 psi (115 bar). A pre-set program was run where the oven heated up to a desired temperature, which was maintained for a required period of time before ramping down to room temperature (RT) for 2 h. The resulting black dispersion was washed with acetone three times and by centrifugation in a Heraeus Biofuge Stratos centrifuge at 8500 rpm for 10 min. The precipitated material was dispersed in water. This procedure yielded IONPs coated with polyols. In previous literature, most polyol syntheses of IONPs are carried out in two heating stages: the first from room temperature to approximately 180 °C, then from 180 °C to the reflux temperature. This is to ensure the thermal decomposition of Fe(acac)3 which occurs at 180–190 °C,49 however we found this leads to a bimodal distribution of the nanoparticle size obtained so proceeded without it.
To explore the effect of the different parameters in this synthesis, a combination of experiments was designed in which the value of one of the parameters in Table 1 was changed at a time. It should be noted that all reaction conditions were carried out in triplicate for statistical analysis and it was shown the synthesis was reproducible.
| Parameter tested | Values |
|---|---|
| Reaction time (h) | 0.5, 1, 2, 4, 8, 12, 24 |
| Mass of Fe(acac)3 (g) | 0.35, 0.7, 1.4, 2.1, 3.5 |
| Solvent used | DEG, TREG, TEG |
Modification of the nanoparticle coating
While it is common for IONPs synthesised in organic solvents to have better control of the shape and size, this process needs to be followed by a ligand-exchange with a hydrophilic ligand to render them water dispersible and suitable for biomedical applications. In our work, the post-synthesis ligand exchange reaction ensures the stabilisation of the NPs obtained and eases their functionalization. The concentration of IONPs was estimated using a Quantum Design superconducting quantum interference device – vibrating sample magnetometer (SQUID-VSM) MPMS-3. A solution containing 100 mg MNPs was mixed with 50 mg of various ligands dissolved in 1 ml of deionized water: 3,4-dihydroxyhydrocinnamic acid (DHCA) and tartaric acid (TA). The reaction took place under continuous stirring for 72 h to ensure the complete displacement of the polyol by the ligand of choice. Once completed, the excess ligand was removed by dialysis for 5 days in 5 L of distilled water (changed daily) by using a 10 kDa molecular weight cut off cellulose membrane (SnakeSkin Dialysis Tubing, Fisher Scientific, UK). The resulting functionalized IONPs were dispersed in deionized water.Characterisation of the nanoparticles obtained
Transmission electron microscopic (TEM) images were recorded on a JEOL-1200 EX microscope at an accelerating voltage of 120 kV. The NP dispersions were diluted in deionized water, and dropped onto a carbon-coated copper grid, and dried at room temperature. The hysteresis loop (at 300 K and 5 K) was recorded on a SQUID-VSM with applied magnetic fields between −7 T and 7 T. The saturation magnetisation for each sample was normalised by taking into account the organic ligand content measured by thermogravimetric analysis. The dynamic light scattering and ζ-potential measurements were performed on a Malvern Nanosizer ZS instrument (Laser He–Ne 633 nm, Malvern Instruments Ltd, Worcestershire, UK). For hydrodynamic diameter measurements, 20 μl of an aqueous solution of IONPs were pipetted into a disposable micro-cuvette (ZEN0040). For ζ-potential measurements, the NPs were diluted and adjusted to the desired pH (from 3 to 10). ζ-Potential measurements were recorded at 25 °C within a disposable capillary cell (DTS1070). The ζ-potential was automatically calculated from electrophoretic mobility based on the Smoluchowski equation, ν = (εE/η)ζ, where ν is the measured electrophoretic velocity, η is the viscosity, ε is the electrical permittivity of the electrolytic solution and E is the electric field. The phase composition of the NPs was characterized with a PANalytical XRD using Co Kα (λ = 1.789 Å) radiation. Fourier transform infrared (FTIR) spectroscopy spectra were recorded on a PerkinElmer spectrometer to confirm the grafting of ligands on the surface of nanoparticles. The element analysis of Fe in the samples was measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES). Thermogravimetric analysis (TGA) was performed for freeze-dried NP samples with a TGA Seiko Exstar6000 TG/DTA6200 instrument in an inert atmosphere using a heating rate of 10 °C min−1.Cytotoxicity study
The cytotoxicity of the functionalized nanoparticles was assessed by an MTS assay with hMSCs. The culture medium used was αMEM supplemented with 10% FBS (fetal bovine serum). Cells were seeded in a 96-well plate at a density of 104 cells per well and incubated at 37 °C in a humidified atmosphere with 5% CO2 and allowed to adhere overnight. Cell loading with nanoparticles was carried out at various concentrations up to 1 mg ml−1 for 24 h. Then 20 μl of the sterile-filtered MTS solution was added to each well. After 4 h of incubation, the absorbance was read at 492 nm using BMG FluoStar Galaxy Optima Microplate Reader (Dynex Technologies). All experiments were performed in triplicate.Relaxivity studies
A series of IONP aqueous dispersions with different Fe concentrations (0.25, 0.5, 0.75 and 1 mM) determined by ICP-OES were prepared for relaxivity studies. All experiments were performed on a Bruker MQ60 NMR Analyzer working at a Larmor frequency of 60 MHz (1.41 T) and at 37 °C.Results and discussion
Effect of synthesis parameters
The TEM analysis of IONPs synthesised with different polyols revealed that IONPs obtained with DEG and TREG were within the superparamagnetic regime with an average size of 5.8 ± 0.8 nm (σ = 14.1%) and 9.1 ± 0.9 (σ = 9.8%) nm respectively (Fig. 1). On the other hand, when TEG was used, larger nanoparticles of 13.9 nm ± 3.4 nm (σ = 24.5%) were produced with an increased polydispersity. There is a correlation between the length of glycol and size of NPs, as the higher the length of the glycol, the larger the size of the synthesized NPs. We measured the magnetic properties of these TEG coated IONPs at 300 K, and showed that they exhibited a superparamagnetic behaviour with a saturation magnetisation σs = 79.1 Am2 kg−1. A small coercivity Hc = 4.6 Am2 kg−1 was observed (Fig. 2) since the particle size distribution increased, it is plausible that the larger IONPs above the superparamagnetic threshold of iron oxide (15–20 nm) become blocked at 300 K.50 | ||
| Fig. 1 TEM images and particle size distributions of iron oxide nanoparticles synthesized using different polyols (A) TEG, (B) TREG and (C) DEG. Size distributions were fitted with a normal function (solid line), d = mean diameter, δd = standard deviation and n = number of particles counted. | ||
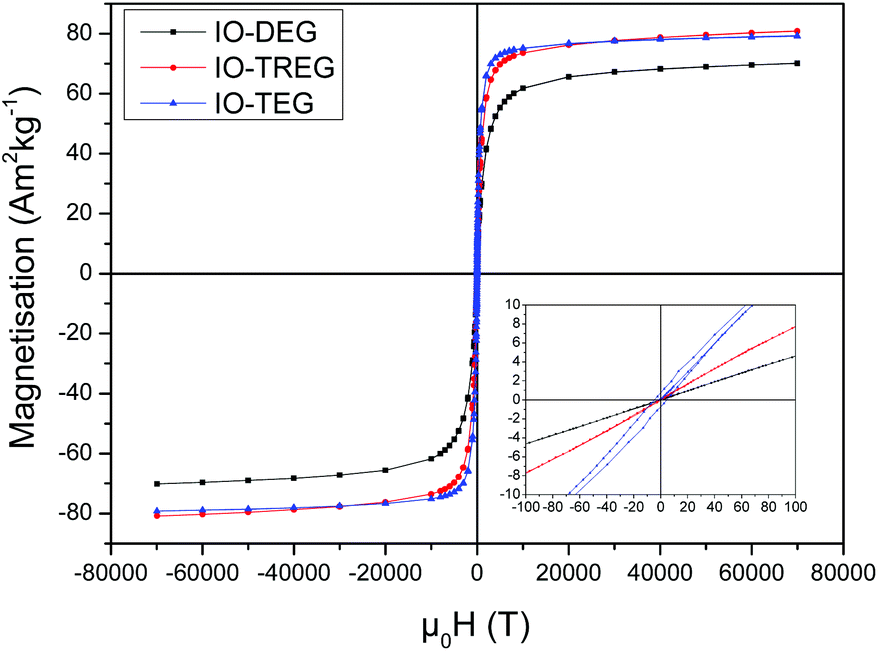 | ||
| Fig. 2 Magnetisation curves of IONPs obtained with different polyols (DEG, TREG and TEG). | ||
The presence of the different polyols on the surface of the NPs is confirmed by TGA measurements (Fig. S1 in ESI†). The weight loss occurs in two stages. The first slight weight loss from approximately 40 to 200 °C corresponds to the removal of the physically adsorbed water molecules. The second step from 200 to 570 °C, which represents the most significant loss, can be attributed to the removal of triethylene glycol in the sample (or other polyols such as diethylene glycol or tetraethylene glycol) as the boiling point is in that range.51,52
Different sizes of NPs were obtained (Fig. 1) in similar reaction conditions with different polyol ligands (Fig. 3) in high pressure and high temperature conditions. The exact nucleation/growth process for this reaction remains poorly understood to this day. As observed by Douglas et al., the mechanism here is not consistent with a classic LaMer mechanism.53
 | ||
| Fig. 3 Chemical structure and molecular weight of polyols used in the synthesis. | ||
It is plausible that the higher the length of the carbon chain of the polyol, the more effective they are as capping ligands to arrest the growth, hence bigger NPs are obtained as it was shown to be the case with different lengths of poly(ethylene glycol).54,55
This study confirms that the choice of the solvent is critical to obtaining high quality nanoparticles with a desired size and with a narrow size distribution.53,56,57
From these results, it was determined that IONPs produced in DEG and TREG were suitable for further investigations as they had a narrow size distribution, are superparamagnetic, and had a high magnetisation which are desired for biomedical applications.58
The reaction time also had an influence on the size of the nanoparticles obtained and therefore their magnetic properties. To study this, in a typical synthesis 1.4 g of Fe(acac)3 was dispersed in 20 ml of TREG. The reaction mixture was then transferred to a 45 ml capacity Teflon lined autoclave vessel before being heated up to 250 °C and maintained at that temperature for 1 h, 2 h, 4 h, 8 h, 12 h and 24 h. The increase in the reaction time from 1 h to 24 h led to an increase in the size of the NPs obtained from 7.2 nm ± 0.8 nm (σ = 11%) to 15 nm ± 1.9 nm (σ = 12.5%) (Fig. 4). This correlates to an increase of the saturation magnetization obtained from 61.2 Am2 kg−1 to 88 Am2 kg−1. Longer reaction times led to particle growth with narrow size distribution leading us to believe the reaction mechanism is through coalescence.59 This allows us to finely control the size of the nanoparticles which is crucial for their biomedical applications.
 | ||
| Fig. 4 TEM images of iron oxide nanoparticles synthesized using different reaction times in tri(ethylene glycol): (A) 1 h, (B) 2 h, (C) 4 h, (D) 8 h, (E) 12 h, and (F) 24 h. | ||
This method, in comparison to previous conventional polyol methods, has proved to yield water-dispersible IONPs of which the size and monodispersity can be carefully controlled with the reaction conditions. In addition, while Wan et al. synthesised IONPs with the classic polyol method using the Schlenk line,39 our NPs obtained in this work at high pressure and high temperature conditions lead to higher saturation magnetisation. When replicating the same chemical reaction using a conventional set-up consisting of a round bottom flask, condenser and magnetic stirring we found the IONPs obtained different by their polydispersity and magnetic properties (Fig. S3†). We measured the magnetic properties of these TREG coated IONPs at 300 K, and showed that they exhibited a superparamagnetic behaviour with a saturation magnetisation σs = 81.8 Am2 kg−1. While the magnetic properties are similar with high pressure and high temperature conditions, the size distribution is much larger with a polydispersity of σ = 18%. We suppose that the high pressure conditions allows a better control of the morphology of the IONPs obtained hence leading to the lower dispersity. The next step was to prove how robust this method was in producing reproducible nanoparticles from one batch to another.
Reproducibility studies
While it was found that the IONPs obtained were consistent under the same experimental conditions, we decided to investigate the reproducibility of the synthetic conditions in order to evaluate the suitability of this method for mass-production of IONPs. Each reaction, using TREG as the polyol capping agent, was repeated three times and the properties of the NPs obtained were assessed. We chose a standard condition for the reaction in which 1.4 g of Fe(acac)3 and 20 mL of TREG were heated up to 250 °C and maintained at that temperature during 8 h before cooling down to RT over 2 h. For simplicity, these reactions are designed by IONP-A, IONP-B and IONP-C. The results are illustrated in Table 2.| Sample | D TEM ± σ TEM (nm) | D XRD (nm) | Ms (Am2 kg−1) |
|---|---|---|---|
| IONP-A | 9.9 ± 1.1 | 9.4 | 70.5 |
| IONP-B | 10.9 ± 1.1 | 8.9 | 77.8 |
| IONP-C | 10.5 ± 1.1 | 9.3 | 81.1 |
| Overall average | 10.2 ± 1.2 | 9.2 ± 0.3 | 76.5 ± 5.4 |
The core size determined by TEM images of three reactions realised in the same experimental conditions showed a well-controlled synthesis route with narrow size distribution. Similarly, the crystallite size determined by XRD only presented a standard deviation of 0.3 nm (Fig. 5).
 | ||
| Fig. 5 XRD pattern of IONPs synthesized in different polyol solvents (DEG, TREG and TEG). Peaks have been indexed according to the reference pattern for magnetite (pdf ref. 01-088-0315). Diffraction patterns have been normalised and offset along the y-axis for better comparison. | ||
XRD does not allow distinction between maghemite (γ-Fe2O3) and magnetite (Fe3O4) phase. Room temperature 57Fe Mossbauer spectra were recorded on selected samples. These spectra (not shown) exhibited absorption out to ±8 mm s−1, indicative of magnetic hyperfine splitting, but were dominated by a very broad central absorption peak, as is typical of disordered and/or fine particle materials.60 The mean isomer shift of the spectra, relative to alpha-iron, was determined to be in the range delta = 0.38 to 0.42 mm s−1. These values lie between those of pure maghemite (delta = 0.32 mm s−1) and pure magnetite (delta = 0.53 mm s−1), implying that the samples were either non-stoichiometric magnetites, or magnetite/maghemite composites, or some combination of the two.61
These NPs were well dispersed in water; however we found that the hydrodynamic diameters obtained were quite large (above 200 nm) in comparison to the core size determined by TEM or XRD. Furthermore, sedimentation appeared in the centrifuge tubes after several days. This led us to believe that the polyol coating was unstable. Because, even in water or when salt is added the NPs precipitated out of solution in less than 30 min. While the exact binding mechanism of polyols to the surface of iron oxide nanoparticles has not been established, recent literature suggests that the polyol, such as DEG, chelates iron. However, the exact structure of this chelate remains undetermined. Recently, TREG coated IONPs synthesized by the polyol method in an autoclave or in standard conditions were found to be unstable and aggregate, as it is believed that the polyol coating is labile and is progressively lost, thus leading to the aggregation of the nanoparticles.45,62
Nanoparticle functionalization
The stabilisation of the NPs obtained is critical for any biomedical applications, as IONPs aggregates at physiological pH. This issue has been addressed by many groups, and a number of ligands have shown to interact with IONPs and to stabilise them, these include phosphonates,63,64 poly(ethylene glycol),65–67 siloxanes,68,69 or various starch molecules such as dextran.70,71 Another group of interest are carboxylic ligands such as citric acid,1,6 dimercaptosuccinic acid (DMSA)72–74 or folic acid75 which have been used recently on the surface of IONPs as they can provide biocompatibility, stability and targeting properties.76–78 In addition to the previously stated ones, we tested a variety of ligands to optimise the stabilisation process by charge or steric hindrance. Two ligands were shown to be successful: 3,4-dihydroxyhydrocinnamic acid (IONP-DHCA) and tartaric acid (IONP-TA). As mentioned previously, 100 mg of IONP-TREG was mixed with 50 mg of ligand and left under continuous stirring during 72 h. Once completed, the excess ligand was removed by dialysis and the resulting functionalized IONPs were dispersed in deionized water.After the ligand exchange step, the hydrodynamic size of the IONPs decreased: 154 nm and 193 nm in water for the IONP-DHCA and IONP-TA respectively (Fig. S2 and S3†), in comparison to the 270 nm for TREG coated IONPs. This confirms that the ligands help stabilise the particles and prevent aggregation.79 In PBS, the hydrodynamic diameter of IONP-DHCA decreased to 140 nm, and no aggregation was observed (Fig. S2†). The hydrodynamic diameter in serum containing media, Roswell Park Memorial Institute (RPMI) medium containing 10% Fetal Bovine Serum) was also measured (Fig. S8†). We were able to decrease the hydrodynamic diameter down to approximately 85 nm in water and 104 nm in PBS when increasing the dialysis time up to 7 days (Fig. S9†). Furthermore, IONP-TA did show slight aggregation in water which became more significant in PBS with a hydrodynamic diameter that increased to 243 nm (Fig. S3†). Due to the smaller hydrodynamic diameter obtained with DHCA, we decided to study the long term stability of IONP-DHCA. 72 days later, the IONP-DHCA in water remained stable with a measured diameter of 140 nm. Moreover, the zeta potential is strongly negative (Fig. 6): −46.8 mV for IONP-DHCA and −35.5 mV for IONP-TA, which confirms the stability of these IONPs in moderate electrolyte conditions (100 mM NaCl was tested). In the case of IONP-DHCA, the catechol functional groups are attached to the nanoparticles, and therefore the carboxylic groups remain unbound, which provide the negative surface charge and are ideal for any further surface functionalization. In the case of IONP-TA, one of the carboxylic groups is an anchor to the nanoparticle and the other one provides the negative surface charge and functionality as it remains unbound.
 | ||
| Fig. 6 Evolution of zeta potential in NaCl 100 mM of the functionalized iron oxide nanoparticles after coating with DHCA and TA, as a function of the pH. | ||
The ligand exchange of IONP-TREG with DHCA was confirmed by FT-IR measurements. The corresponding FT-IR spectra are displayed in Fig. 7. For polyol coated NPs, bands at 1116–1050 cm−1 are characteristic of C–O stretching and confirm the attachment of TREG on the surface (Fig. S11†).
 | ||
| Fig. 7 ATR-FTIR spectrum of IONPs coated with 2,3-dihydroxyhydrocinnamic acid (DHCA) and tartaric acid (TA). The 1720–4000 cm−1 range has been excluded as no significant information was found. | ||
The broad band (3600–2500 cm−1) centred around 3400 cm−1 is due to hydrogen bonded O–H stretching vibration from surface hydroxyl groups on nanoparticles and adsorbed TREG and water.80 The characteristic Fe–O stretching vibration band is also present at 540 and 545 cm−1.81,82
In the case of DHCA functionalized NPs, the broad band at approximately 1490 cm−1 and peaks at 1247 cm−1 are present when catechol binds covalently to various metal oxides as catechol anions.83 They can be attributed to the benzene ring vibration and a C–O stretch, respectively. The band at 1403 cm−1 was assigned to symmetric vibrations of carboxyl groups COO− and COOH.
Tartaric acid functionalized NPs also exhibited specific peaks. At 1613 cm−1 a sharp peak corresponds to the asymmetric stretching of carboxylic groups, and is shifted towards lower wavenumber as it is bound to the surface of the NPs.84,85 The band observed at 1089 cm−1 is due to the C–O stretch in hydroxyl groups, and the band observed at 1370 cm−1 is characteristic of the bending of alkanes –C–H. Both bands can be attributed to the bound tartaric acid on the surface of NPs. The broad band (3600–2500 cm−1) centred around 3400 cm−1 is due to hydrogen bonded O–H stretching vibration from surface hydroxyl groups on nanoparticles and adsorbed TREG and water, therefore it cannot be attributed specifically to the hydroxyl groups of tartaric acid.80
In vitro characterization
We next investigated the ability of the functionalized nanoparticles as MRI contrast agent by performing the relaxivity measurements at 1.4 T. We prepared samples of four different concentrations of Fe up to 1 mM (determined by ICP-AES). We measured the r1 and r2 values according to the linear relationship of longitudinal and transverse relaxation rates versus the magnetic metal concentrations of Fe2O3 (Fig. 8). The IONP-DHCA nanoparticles exhibited the highest relaxation enhancement, with an r2 value of 185.58 mM−1 s−1 and r1 of 7.95 mM−1 s−1 at 1.4 T. For tartaric acid functionalized nanoparticles, the r2 decreases to 149.68 mM−1 s−1 and r1 increases to 8.63 mM−1 s−1. The relatively high r2/r1 ratio (Table 3) suggests these nanoparticles are T2 weighted contrast agents. | ||
| Fig. 8 Top plot of relaxation rate 1/T1 over Fe2O3 concentration of the IO-DHCA nanoparticles. The slope indicates the specific relaxivity (r1); bottom plot of 1/T2 over Fe2O3 concentration of the IO-DHCA nanoparticles. The slope indicates the specific relaxivity (r2). ○ IONP-TA and Δ IONP-DHCA. | ||
| r 2 (mM−1 s−1) | r 1 (mM−1 s−1) | r 2/r1 | |
|---|---|---|---|
| IONP-DHCA 8 h | 185.58 | 7.95 | 23.3 |
| IONP-TA 8 h | 149.68 | 8.63 | 17.3 |
| IONP-DHCA 12 h | 160.6 | 5.16 | 31.1 |
| IONP-DHCA 24 h | 259.1 | 9.9 | 26.2 |
| Endorem® | 41 | 4.7 | 8.72 |
| Resovist® | 61 | 8.7 | 7 |
At the same frequency, in comparison to FDA-approved nanoparticles for MRI Resovist® and Endorem®, the NPs obtained had an improved r2/r1 ratio by a factor of more than 3.86 Furthermore, the promising features of these IONPs as MRI contrast agents was confirmed with relaxivity measurements of IONPs obtained in the same manner as the previous IONP-DHCA sample but being maintained at 250 °C during 12 h and 24 h (Table 3), thus having higher saturation magnetisation of 84.1 and 85.4 emu g−1 respectively (Fig. S12†).
Biocompatibility is the bottleneck of many of today's current applications of nanomaterials in biomedical applications. To verify the suitability of the functionalized NPs, cell proliferation was measured by an MTS assay. Fig. 9 shows the effect of IONPs on cell proliferation after incubation at various concentrations for 24 h.
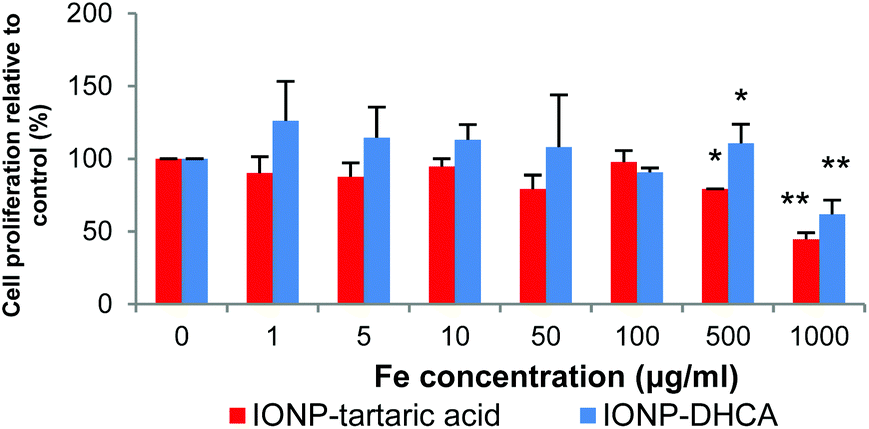 | ||
| Fig. 9 Cell proliferation of hMSCs after being incubated with IONP-DHCA and IONP-TA during 24 h at various concentrations. Data indicated as the mean ± standard deviation of 3 independent experiments. *p < 0.05 and **p < 0.01. | ||
There is no significant difference in cell viability for both types of nanoparticles up to 500 μg ml−1. However, there is significant decrease in viability at concentrations of 1 mg ml−1, at 60% for IONP-DHCA and 45% for IONP-TA relative to the control.
These results demonstrate that the functionalized iron oxide nanoparticles have excellent biocompatibility and little toxicity is observed for the nanoparticles that could be used as MRI T2 contrast agents in hMSCs.
Conclusions
In summary, water-dispersible IONPs have been successfully obtained via a simple and reproducible polyol synthesis modified by using high pressure and high temperature conditions. Their size and magnetic properties could be finely tuned by modifying the solvent, reaction time and concentration of iron precursor Fe(acac)3. The as-synthesised nanoparticles had a high saturation magnetization (ca. 80 emu g−1) but were not stable as indicated by the hydrodynamic diameter measurements and their precipitation in solution. The modification of the TREG coating by a ligand exchange process yielded stable nanoparticles with carboxylic ligands: 3,4-dihydroxyhydrocinnamic acid (IONP-DHCA) and tartaric acid (IONP-TA). The best results were obtained with IONP-DHCA which demonstrated long term stability, even when subjected to various salt concentrations (up to 1 M NaCl). The potential of these novel particles as MRI contrast agents is demonstrated with better relaxivity values when compared to commercial contrast agents: r2 = 185.58 mM−1 s−1 and r1 = 7.95 mM−1 s−1 for IONP-DHCA while r2 = 149.68 mM−1 s−1 and r1 = 8.63 mM−1 s−1 for IONP-TA. We believe that such highly magnetic and stable iron oxide nanoparticles hold great promise in serving as novel and effective MRI contrast agents for applications such as stem cell tracking or cancer cell targeted imaging for example.Acknowledgements
N. T. K. Thanh thanks the Royal Society for her university research fellowship and EPSRC (Grant no. EPSRC, EP/M015157/1) for the financial support. R. Hachani thanks UCL for her SLMS PhD studentship. We would like to acknowledge Dr Lara K. Bogart, for the 57Fe Mossbauer measurements and analysis performed; Miss Carla Carvalho, for her assistance with the hMSC cultures; and Dr Cristina Blanco-Andujar for relaxivity measurements.Notes and references
- C. Blanco-Andujar, D. Ortega, P. Southern, Q. A. Pankhurst and N. T. K. Thanh, Nanoscale, 2015, 7, 1768–1775 RSC.
- K. Niemirowicz, K. Markiewicz, A. Wilczewska and H. Car, Adv. Med. Sci., 2012, 1–12 Search PubMed.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS PubMed.
- R. Hachani, M. Lowdell, M. Birchall and N. T. K. Thanh, Nanoscale, 2013, 5, 11362–11373 RSC.
- S. M. C. Berman, P. Walczak and J. W. M. Bulte, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 343–355 CrossRef CAS PubMed.
- K. Andreas, R. Georgieva, M. Ladwig, S. Mueller, M. Notter, M. Sittinger and J. Ringe, Biomaterials, 2012, 33, 4515–4525 CrossRef CAS PubMed.
- M. Mahmoudi, H. Hosseinkhani, M. Hosseinkhani, S. Boutry, A. Simchi, W. S. Journeay, K. Subramani and S. Laurent, Chem. Rev., 2011, 111, 253–280 CrossRef CAS PubMed.
- H. Markides, R. Morris, S. Roberts and J. El Haj, J. Tissue Eng. Regener. Med., 2012, 6, 53–53 Search PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- J. T. Ferrucci and D. D. Stark, Am. J. Roentgenol., 1990, 155, 943–950 CrossRef CAS PubMed.
- C. Poeckler–Schoeniger, J. Koepke, F. Gueckel, J. Sturm and M. Georgi, Magn. Reson. Imaging, 1999, 17, 383–392 CrossRef.
- P. Reimer and T. Balzer, Eur. J. Radiol., 2003, 13, 1266–1276 Search PubMed.
- Y.-X. Wang, S. Hussain and G. Krestin, Eur. J. Radiol., 2001, 11, 2319–2331 CrossRef CAS PubMed.
- L. Kostura, D. L. Kraitchman, A. M. Mackay, M. F. Pittenger and J. W. M. Bulte, NMR Biomed., 2004, 17, 513–517 CrossRef PubMed.
- I. Kassis, A. Vaknin-Dembinsky, J. Bulte and D. Karussis, Int. J. Stem Cells, 2010, 3, 144–153 CrossRef CAS PubMed.
- H. S. Kim, S. Y. Oh, H. J. Joo, K.-R. Son, I.-C. Song and W. K. Moon, NMR Biomed., 2010, 23, 514–522 CrossRef CAS PubMed.
- T. D. Henning, E. J. Sutton, A. Kim, D. Golovko, A. Horvai, L. Ackerman, B. Sennino, D. McDonald, J. Lotz and H. E. Daldrup-Link, Contrast Media Mol. Imaging, 2009, 4, 165–173 CrossRef CAS PubMed.
- Y. Wang, C. Xu and H. Ow, Theranostics, 2013, 3, 544 CrossRef PubMed.
- T. Q. Huy, P. Van Chung, N. T. Thuy, C. Blanco-Andujar and N. T. K. Thanh, Faraday Discuss., 2014, 175, 73–82 CAS.
- N. T. K. Thanh and L. A. W. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- A. Quarta, D. Bernareggi, F. Benigni, E. Luison, G. Nano, S. Nitti, M. C. Cesta, L. Di Ciccio, S. Canevari, T. Pellegrino and M. Figini, Nanoscale, 2015, 7, 2336–2351 RSC.
- J.-M. Montenegro, V. Grazu, A. Sukhanova, S. Agarwal, J. M. de la Fuente, I. Nabiev, A. Greiner and W. J. Parak, Adv. Drug Delivery Rev., 2013, 65, 677–688 CrossRef CAS PubMed.
- K. L. Vigor, P. G. Kyrtatos, S. Minogue, K. T. Al-Jamal, H. Kogelberg, B. Tolner, K. Kostarelos, R. H. Begent, Q. A. Pankhurst, M. F. Lythgoe and K. A. Chester, Biomaterials, 2010, 31, 1307–1315 CrossRef CAS PubMed.
- B. Kozissnik, L. A. W. Green, K. A. Chester and N. T. K. Thanh, Strategy for Functionalisation of Magnetic Nanoparticles for Biological Targets, in Magnetic Nanoparticles: From Fabrication to Clinical Applications, ed. N. T. K. Thanh, CRC Press, Taylor & Francis, Boca Raton, London, New York, 2012, pp. 129–150 Search PubMed.
- G. Bealle, R. Di Corato, J. Kolosnjaj-Tabi, V. Dupuis, O. Clement, F. Gazeau, C. Wilhelm and C. Menager, Langmuir, 2012, 28, 11843–11851 CrossRef PubMed.
- S. Laurent, S. Dutz, U. O. Häfeli and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8–23 CrossRef CAS PubMed.
- A. Hervault and N. n. T. K. Thanh, Nanoscale, 2014, 6, 11553–11573 RSC.
- L. Fiandra, S. Mazzucchelli, C. De Palma, M. Colombo, R. Allevi, S. Sommaruga, E. Clementi, M. Bellini, D. Prosperi and F. Corsi, ACS Nano, 2013, 7, 6092–6102 CrossRef CAS PubMed.
- K. Hayashi, K. Ono, H. Suzuki, M. Sawada, M. Moriya, W. Sakamoto and T. Yogo, ACS Appl. Mater. Interfaces, 2010, 2, 1903–1911 CAS.
- L. Zhu and V. P. Torchilin, Integr. Biol., 2013, 5(1), 96–107 RSC.
- J. Riegler, A. Liew, S. O. Hynes, D. Ortega, T. O'Brien, R. M. Day, T. Richards, F. Sharif, Q. A. Pankhurst and M. F. Lythgoe, Biomaterials, 2013, 34, 1987–1994 CrossRef CAS PubMed.
- N. Landázuri, S. Tong, J. Suo, G. Joseph, D. Weiss, D. J. Sutcliffe, D. P. Giddens, G. Bao and W. R. Taylor, Small, 2013, 9, 4017–4026 CrossRef PubMed.
- D. Tukmachev, O. Lunov, V. Zablotskii, A. Dejneka, M. Babic, E. Sykova and S. Kubinova, Nanoscale, 2015, 7, 3954–3958 RSC.
- F. Schulze, A. Dienelt, S. Geissler, P. Zaslansky, J. Schoon, K. Henzler, P. Guttmann, A. Gramoun, L. A. Crowe, L. Maurizi, J.-P. Vallée, H. Hofmann, G. N. Duda and A. Ode, Small, 2014, 10, 4340–4351 CAS.
- L. Babes, B. Denizot, G. Tanguy, J. J. Le Jeune and P. Jallet, J. Colloid Interface Sci., 1999, 212, 474–482 CrossRef CAS PubMed.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247–1248 CrossRef.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- H. Dong, Y. C. Chen and C. Feldmann, Green Chem., 2015, 17, 4107–4132 RSC.
- J. Wan, W. Cai, X. Meng and E. Liu, Chem. Commun., 2007, 5004–5006 RSC.
- C. M. Cheng, F. J. Xu and H. C. Gu, New J. Chem., 2011, 35, 1072–1079 RSC.
- F. Hu, K. W. MacRenaris, E. A. Waters, T. Liang, E. A. Schultz-Sikma, A. L. Eckermann and T. J. Meade, J. Phys. Chem. C, 2009, 113, 20855–20860 CAS.
- N. Miguel-Sancho, O. Bomati-Miguel, A. G. Roca, G. Martinez, M. Arruebo and J. Santamaria, Ind. Eng. Chem. Res., 2012, 51, 8348–8357 CrossRef CAS.
- D. Arndt, V. Zielasek, W. Dreher and M. Baumer, J. Colloid Interface Sci., 2014, 417, 188–198 CrossRef CAS PubMed.
- Z. Li, H. Chen, H. Bao and M. Gao, Chem. Mater., 2004, 16, 1391–1393 CrossRef CAS.
- N. Miguel-Sancho, O. Bomati-Miguel, G. Colom, J. P. Salvador, M. P. Marco and J. Santamaria, Chem. Mater., 2011, 23, 2795–2802 CrossRef CAS.
- O. Bomati-Miguel, N. Miguel-Sancho, I. Abasolo, A. Candiota, A. Roca, M. Acosta, S. Schwartz Jr., C. Arus, C. Marquina, G. Martinez and J. Santamaria, J. Nanopart. Res., 2014, 16, 1–13 CrossRef.
- W. Cai and J. Wan, J. Colloid Interface Sci., 2007, 305, 366–370 CrossRef CAS PubMed.
- D. Maity, S. N. Kale, R. Kaul-Ghanekar, J.-M. Xue and J. Ding, J. Magn. Magn. Mater., 2009, 321, 3093–3098 CrossRef CAS.
- Q. Song, Y. Ding, Z. L. Wang and Z. J. Zhang, Chem. Mater., 2007, 19, 4633–4638 CrossRef CAS.
- K. M. Krishnan, IEEE Trans. Magn., 2010, 46, 2523–2558 CrossRef CAS PubMed.
- S. Kazan, E. E. Tanrıverdi, R. Topkaya, Ş. Demirci, Ö. Akman, A. Baykal and B. Aktaş, Arabian J. Chem., 2012 DOI:10.1016/j.arabjc.2011.12.005.
- T. G. Altincekic, İ. Boz, A. Baykal, S. Kazan, R. Topkaya and M. S. Toprak, J. Alloys Compd., 2010, 493, 493–498 CrossRef CAS.
- F. J. Douglas, D. A. MacLaren and M. Murrie, RSC Adv., 2012, 2, 8027–8035 RSC.
- G. Fontana, M. Licciardi, S. Mansueto, D. Schillaci and G. Giammona, Biomaterials, 2001, 22, 2857–2865 CrossRef CAS PubMed.
- P. G. Koutsoukos, Trends in Colloid and Interface Science XV, Springer, Berlin Heidelberg, 2003 Search PubMed.
- Y. Zhu, T. Mei, Y. Wang and Y. Qian, J. Mater. Chem., 2011, 21, 11457–11463 RSC.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805–1813 CrossRef CAS.
- N. T. K. Thanh, V. F. Puntes, L. D. Tung and D. G. Fernig, J. Phys.: Conf. Ser., 2005, 17, 70–76 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- H. Y. Hah, S. Gray, C. E. Johnson, J. A. Johnson, V. Kolesnichenko, P. Kucheryavy and G. Goloverda, J. Phys.: Conf. Ser., 2014, 548 Search PubMed.
- G. M. Da Costa, C. Blanco-Andujar, E. De Grave and Q. A. Pankhurst, J. Phys. Chem. B, 2014, 118, 11738–11746 CrossRef CAS PubMed.
- I.-M. Grabs, C. Bradtmöller, D. Menzel and G. Garnweitner, Cryst. Growth Des., 2012, 12, 1469–1475 CAS.
- M. Das, D. Mishra, P. Dhak, S. Gupta, T. K. Maiti, A. Basak and P. Pramanik, Small, 2009, 5, 2883–2893 CrossRef CAS PubMed.
- Y. Lalatonne, C. Paris, J. M. Serfaty, P. Weinmann, M. Lecouvey and L. Motte, Chem. Commun., 2008, 2553–2555 RSC.
- F. Hu, Q. Jia, Y. Li and M. Gao, Nanotechnology, 2011, 22, 245604 CrossRef PubMed.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- C. Barrera, A. Herrera, Y. Zayas and C. Rinaldi, J. Magn. Magn. Mater., 2009, 321, 1397–1399 CrossRef CAS.
- C. W. Lu, Y. Hung, J. K. Hsiao, M. Yao, T. H. Chung, Y. S. Lin, S. H. Wu, S. C. Hsu, H. M. Liu, C. Y. Mou, C. S. Yang, D. M. Huang and Y. C. Chen, Nano Lett., 2007, 7, 149–154 CrossRef CAS PubMed.
- Y. X. J. Wang, T. Quercy-Jouvet, H. H. Wang, A. W. Li, C. P. Chak, S. H. Xuan, L. Shi, D. F. Wang, S. F. Lee, P. C. Leung, C. B. S. Lau, K. P. Fung and K. C. F. Leung, Materials, 2011, 4, 703–715 CrossRef CAS.
- S. L. Easo and P. V. Mohanan, Carbohydr. Polym., 2013, 92, 726–732 CrossRef CAS PubMed.
- T. D. Henning, S. Boddington and H. E. Daldrup-Link, J. Visualized Exp., 2008, 685 Search PubMed.
- L. Maurizi, H. Bisht, F. Bouyer and N. Millot, Langmuir, 2009, 25, 8857–8859 CrossRef CAS PubMed.
- A. Ruiz, P. Morais, R. Bentes de Azevedo, Z. M. Lacava, A. Villanueva and M. del Puerto Morales, J. Nanopart. Res., 2014, 16, 1–20 CrossRef CAS.
- M. Geppert, M. C. Hohnholt, K. Thiel, S. Nürnberger, I. Grunwald, K. Rezwan and R. Dringen, Nanotechnology, 2011, 22 Search PubMed.
- Y. L. Liu, J. L. Zhang, G. Cheng, G. Y. Hong and J. Z. Ni, Nanotechnology, 2012, 23 Search PubMed.
- Q. L. Jiang, S. W. Zheng, R. Y. Hong, S. M. Deng, L. Guo, R. L. Hu, B. Gao, M. Huang, L. F. Cheng, G. H. Liu and Y. Q. Wang, Appl. Surf. Sci., 2014, 307, 224–233 CrossRef CAS.
- M. Kumar, G. Singh, V. Arora, S. Mewar, U. Sharma, N. R. Jagannathan, S. Sapra, A. K. Dinda, S. Kharbanda and H. Singh, Int. J. Nanomed., 2012, 7, 3503–3516 CAS.
- S. Mohapatra, S. K. Mallick, T. K. Maiti, S. K. Ghosh and P. Pramanik, Nanotechnology, 2007, 18, 385102 CrossRef.
- J. Lim, S. P. Yeap, H. X. Che and S. C. Low, Nanoscale Res. Lett., 2013, 8, 381–381 CrossRef PubMed.
- G. Busca, V. Lorenzelli, G. Ramis and R. J. Willey, Langmuir, 1993, 9, 1492–1499 CrossRef CAS.
- M. Mahdavi, M. B. Ahmad, M. J. Haron, Y. Gharayebi, K. Shameli and B. Nadi, J. Inorg. Organomet. Polym. Mater., 2013, 23, 599–607 CrossRef CAS.
- L. Levy, Y. Sahoo, K.-S. Kim, E. J. Bergey and P. N. Prasad, Chem. Mater., 2002, 14, 3715–3721 CrossRef CAS.
- W. Huang, P. Jiang, C. Wei, D. Zhuang and J. Shi, J. Mater. Res., 2008, 23, 1946–1952 CrossRef CAS.
- S. Nigam, K. C. Barick and D. Bahadur, J. Magn. Magn. Mater., 2011, 323, 237–243 CrossRef CAS.
- M. Răcuciu, D. E. Creangă and A. Airinei, Eur. Phys. J. E, 2006, 21, 117–121 CrossRef PubMed.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H. J. Weinmann, Invest. Radiol., 2005, 40, 715–724 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr03867g |
| This journal is © The Royal Society of Chemistry 2016 |