
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProperties of the gold–sulphur interface: from self-assembled monolayers to clusters
Thomas
Bürgi
Department of Physical Chemistry, University of Geneva, Quai Ernest-Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: thomas.buergi@unige.ch
First published on 7th September 2015
Abstract
The gold–sulphur interface of self-assembled monolayers (SAMs) was extensively studied some time ago. More recently tremendous progress has been made in the preparation and characterization of thiolate-protected gold clusters. In this feature article we address different properties of the two systems such as their structure, the mobility of the thiolates on the surface and other dynamical aspects, the chirality of the structures and characteristics related to it and their vibrational properties. SAMs and clusters are in the focus of different communities that typically use different experimental approaches to study the respective systems. However, it seems that the nature of the Au–S interfaces in the two cases is quite similar. Recent single crystal X-ray structures of thiolate-protected gold clusters reveal staple motifs characterized by gold ad-atoms sandwiched between two sulphur atoms. This finding contradicts older work on SAMs. However, newer studies on SAMs also reveal ad-atoms. Whether this finding can be generalized remains to be shown. In any case, more and more studies highlight the dynamic nature of the Au–S interface, both on flat surfaces and in clusters. At temperatures slightly above ambient thiolates migrate on the gold surface and on clusters. Evidence for desorption of thiolates at room temperature, at least under certain conditions, has been demonstrated for both systems. The adsorbed thiolate can lead to chirality at different lengths scales, which has been shown both on surfaces and for clusters. Chirality emerges from the organization of the thiolates as well as locally at the molecular level. Chirality can also be transferred from a chiral surface to an adsorbate, as evidenced by vibrational spectroscopy.
 Thomas Bürgi | Thomas Bürgi studied chemistry and obtained hid PhD (1995) at the University of Berne (Switzerland). After a postdoc at MIT, he did his habilitation at ETH, Zürich. He became assistant professor at the University of Neuchâtel (Switzerland, 2005) and full professor at the University of Heidelberg (2008). In 2010 he moved to the University of Geneva, where he is professor of physical chemistry. His research focuses on chiral nanoparticles, plasmon-based metamaterials and in situ-vibrational spectroscopy. |
Introduction
Self-assembled monolayers (SAMs) of thiolates on gold surfaces1 have found applications in fields such as sensing,2 molecular electronics3 and surface patterning.4 The driving force for the anchoring of thiols on gold surfaces is the formation of a gold–sulphur bond.5 Thiols are also used to stabilize small metal particles from agglomeration.6 The use of thiols furthermore enables the preparation of very small gold particles or clusters.7,8 Recently direct structural information of small gold clusters could be obtained by X-ray crystallography,9–19 and by atomic resolution electron microscopy.20 The structure of the gold–thiolate interface that emerges from these studies is different from the model developed for SAMs in the 1980s and early 1990s. The latter is furthermore challenged based on these new findings.21,22 More recent X-ray diffraction studies and computational work on SAMs points towards a more dynamic nature of the Au–S interface.23 In this article we focus on the properties of the gold–sulphur interface. We compare recent findings gained from thiolate-protected gold clusters with properties of SAMs. Particularly we focus on the structure of the interface, on its flexibility, on chemical modifications by ligand exchange reactions, on its vibrational properties as well as on properties related to its chirality. We try to highlight differences and similarities between SAMs and particles/clusters. The com parison is sometimes obscured by the fact that the two systems are studied by different communities using different techniques.Structure of the gold–thiolate interface
Self-assembled monolayers (SAMs): original model
During the last 30 years the preparation and characterization of self-assembled monolayers witnessed large success.1,24 The self-assembly of thiols, above all alkanethiols, on gold has particularly attracted researchers.5,25,26 SAMs are attractive model systems to study the principles of self-assembly on surfaces but the interest in SAMs was manly fostered by the possibility to engineer surface properties like adhesion and wettability, to prevent corrosion processes and to structure surfaces. The structure of alkanethiolate SAMs on Au(111) and other single crystal faces was addressed in the late 80s and early 90s by electron diffraction studies.27,28 The conclusions drawn from such studies resulted in a model in which the sulphur atoms adopt a hexagonal structure with a sulphur–sulphur diastance of 4.97 Å. These studies as well as He diffraction29 and atomic force microscopy (AFM)30 indicate that the thiolates form a √3 × √3R 30° overlayer that is commensurate with the Au(111) surface underneath. Note that the samples for these studies were prepared by immersion of the gold surface into an ethanolic solution of the thiol. It was suggested that the sulphur atoms of the thiolates are bound to the threefold hollow sites of the (111) surface, in agreement with the observation that sulphur atoms bind to highly coordinated sites on transition metal surfaces. The early electron and He diffraction studies focused mainly on the symmetry of the diffraction pattern and provided information on the average symmetry of the overlayer. However, these studies did not give information on the lateral position of the anchoring sulphur atom of the thiolates. In order to obtain such information from scattering experiments the analysis of the intensities is required. Similarly, with scanning probe techniques, it is a priory difficult to learn about the exact location of the headgroup of (long) alkanethiolate SAMs because the respective Au–S interface is buried underneath the hydrocarbon chains. Further X-ray studies revealed a c(4 × 2) superlattice of the √3 × √3R 30° lattice. The diffraction patterns furthermore indicated that two of the four molecules of the unit mesh are distinct to a certain extent. An X-ray standing wave (XSW) study under ultrahigh vacuum (UHV) conditions revealed two different sulphur sites with distinct lateral and vertical positions with respect to the underlying Au(111) surface (see Fig. 1).31 Much less is known about the structure of SAMs on other crystal faces such as Au(100) although significant progress is being made more recently.32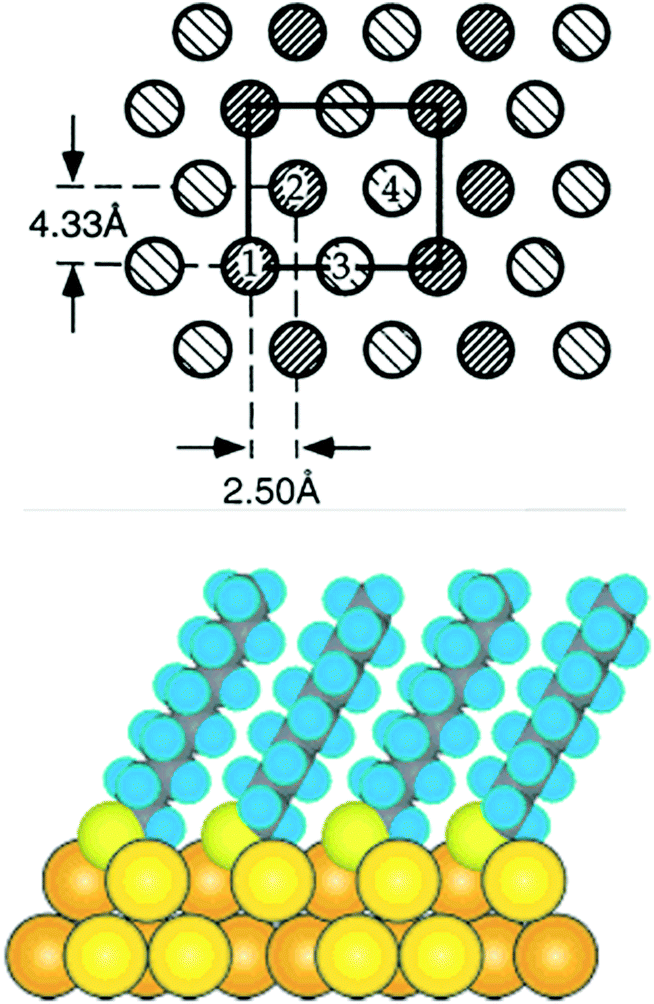 | ||
| Fig. 1 Top: model of the c(4 × 2) phase of a SAM in which each molecule is schematically drawn as circle. The two types of shading represent molecules that are distinct within the c(4 × 2) unit mesh (rectangle). Molecules 1 and 2 are equivalent but distinct from molecules 3 and 4. Reproduced from ref. 31 with permission from Elsevier. Bottom: cross section of a SAM formed from decanethiol. Reprinted with permission from ref. 24 Copyright (2005) American Chemical Society. | ||
Thiolate-protected clusters
In contrast to the model that was developed for SAMs as outlined above the structure of the gold–thiolate interface of thiolate-protected gold clusters is quite different.33 This was first evidenced through the determination of the crystal structure of Au102(pMBA)44 (pMBA = para mercaptobenzoic acid) by Kornberg and coworkers.10 This work revealed the existence of SR(–Au–SR)1,2 staple motifs, in which gold atoms are sandwiched between two sulphur atoms of the thiolates. Fig. 2 shows such staple motifs. Analogous staple motifs were also found for other clusters like Au25(SR)18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 and Au38(SR)24.11 The former cluster contains only dimeric SR(–Au–SR)2 staples, whereas the latter cluster contains a mixture of dimeric and monomeric SR–Au–SR staples. As can be deduced from Fig. 2 the sulphur atoms in the staple structures represent stereogenic centres. The sulphur is in fact bound to four different substituents, two different gold atoms, the organic part R of the thiolate and the electron lone-pair. In principle the organic part R of the thiolate can adopt two positions and the interconversion between the two is easily possible due to a low barrier.33
9 and Au38(SR)24.11 The former cluster contains only dimeric SR(–Au–SR)2 staples, whereas the latter cluster contains a mixture of dimeric and monomeric SR–Au–SR staples. As can be deduced from Fig. 2 the sulphur atoms in the staple structures represent stereogenic centres. The sulphur is in fact bound to four different substituents, two different gold atoms, the organic part R of the thiolate and the electron lone-pair. In principle the organic part R of the thiolate can adopt two positions and the interconversion between the two is easily possible due to a low barrier.33
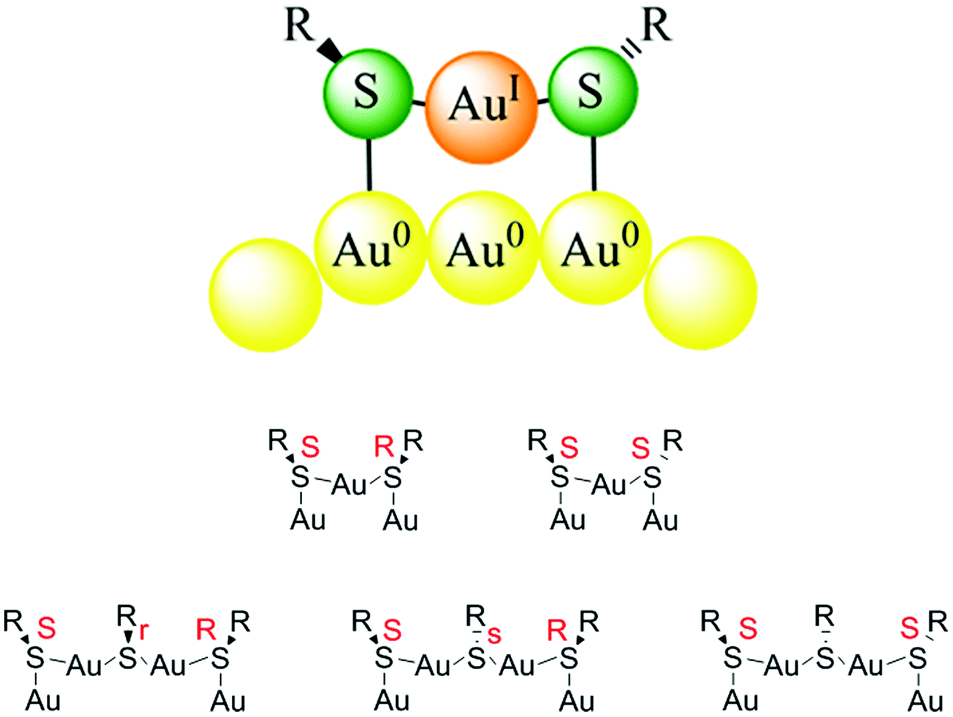 | ||
| Fig. 2 Top: schematic drawing of a monomeric Au–SR–Au staple motif. The organic substituent R of the thiolate can be oriented on the same side with respect to the staple plane (cis) or on opposite sides (trans). Bottom: stereochemistry of monomeric and dimeric staple motifs. Reprinted with permission from ref. 34 Copyright (2014) American Chemical Society. | ||
The structure of the staple motifs resembles the one of Au(I)–thiolate polymers that are formed when a Au(III) salt is mixed with a thiol during the preparation of thiolate-protected gold clusters according to the Brust synthesis.7 Such polymers are used as therapeutic agents against rheumatoid arthritis (myochrysine) for more than halve a century. X-ray structure determination of gold(I)–thiomalate single crystals revealed a gold–sulphur double helical structure.35 Each helix shows approximate four-fold symmetry. Left- and right-handed helices are found in the unit cell. The left-handed helix contains exclusively (S)-thiomalate, whereas the right-handed helix is built from (R)-thiomalate. In this structure the S–Au–S angle is 178.9° and 169.4° for the two distinguishable gold atoms in the unit cell, whereas the Au–S distances are 2.289 Å and 2.285 Å, respectively. It was furthermore suggested, based on NMR and wide angle X-ray scattering (WAXS)/differential anomalous scattering (DAS), that oligomers of similar structure exist in solution. The structural parameters given above are close to the ones determined for typical staple motifs of thiolate-protected gold clusters. For example for the Au38(PET)24 (PET = 2-phenylethylthiolate) cluster the average S–Au–S bond angle is 168.6° and the average Au–S bond length in the staple is 2.3 Å.11
The staple motifs described above are the most abundant structure elements found up to now in thiolate-protected gold clusters. More recent structure determinations indicate the presence of other binding motifs such as trimeric motifs SR(–Au–SR)3,13 bridging thiolates,12,16 cyclic –Au–SR– structures13 or even naked sulphur atoms.14 However, sometimes it is not straightforward to clearly distinguish between these binding motifs on the highly curved surface of a cluster.
SAMs re-visited
Later STM and diffraction studies on SAMs of short thiols (methylthiol, Fig. 3) prepared under UHV conditions also revealed surface complexes where two sulphur atoms are joined by a gold adatom, both at low and high coverage.21,36,37 Interestingly, Vericat and coworkers studied SAMs of pMBA (the thiolate used to evidence ad-atoms on the Au102 cluster), on Au(111) in UHV and found no evidence for gold ad-atoms,38 which raises the question if the ad-atom structure is a general feature for flat surfaces as well.39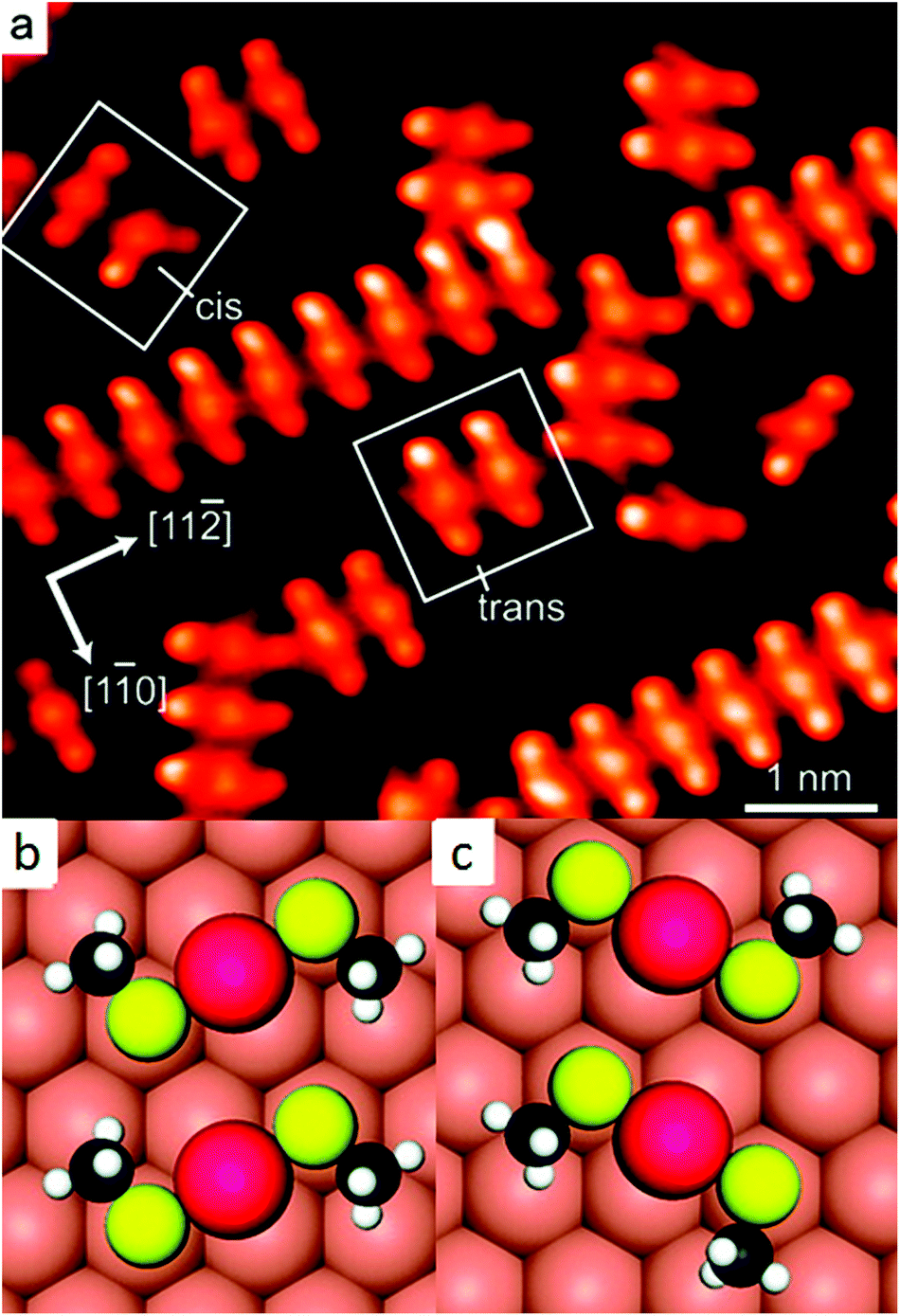 | ||
| Fig. 3 STM image of self-assembled structures of methylthiolate at low coverage on Au(111), produced by heating the gold crystal predosed with CH3SSCH3 above 200 K for ∼10 min (a). Highlighted are two trans-(CH3S)2Au complexes and adjacent cis- and trans-adatom complexes. Their schematic models are shown in (b) and (c), respectively. Reprinted with permission from ref. 21 Copyright (2009) American Chemical Society. | ||
The creation of gold adatoms is consistent with the observation of etch pits (vacancy islands) on gold surfaces exposed to thiols. For SAMs of long-chain alkanethiols the gold–sulphur interface is buried and precludes direct observation by scanning probe techniques. Structure determination remains therefore a real challenge and the same is true for calculations, since different structural models have similar energy22 and a key role may be played by intermolecular interactions between the alkyl chains. Such interactions represent a real challenge for density functional theory (DFT) calculations. Using grazing incidence X-ray diffraction (GIXRD) the structure of a hexanethiolate SAM prepared in UHV on Au(111) at high coverage was reinvestigated.23 Best-fit conditions were found for a model with adatoms and vacancies in the topmost Au layer. In this model both vacancies and adatoms are delocalized (partially occupied atomic sites), which reflects the dynamic character indicated by density functional theory based molecular dynamics simulations. The dynamic nature of the interface stems from the interconversion between two different thiol adsorption geometries as well as vacancy migration. The structure derived from the fit to the experiment is therefore an average structure consisting of one-dimensional zigzag chains –S–Au–S–Au–S– as well as thiolates adsorbed in a bridged adsorption mode. The chains resemble the staple motifs found on several thiolate-protected gold clusters10 and the structure of gold–thiolate polymers.35
The X-ray diffraction studies mentioned above provide averaged and static information and do not directly probe the dynamic aspect of the gold–thiolate interface. However, the appearance of partially occupied sites in the best fit model at least indicates some disorder in the Au–S interface and points towards the dynamic nature of the structure.
Dynamic aspects of the gold–thiolate interface
Mobility of thiolates on gold surfaces
Several experiments indicate that the gold–thiolate interface of thiolate SAMs on flat gold surfaces is not rigid.23,40–43 These observations include the movement of etch pits, the diffusion of thiolates on the gold surface and the exchange of one thiolate by another. Some of these experiments were reported before the existence of staple motifs was evidenced. The diffusion of thiolates on gold surfaces has important implications for applications of SAMs. For example the spreading of alkanethiol “ink” molecules represents one limitation of microcontact printing for obtaining high-resolution nano-patterns. STM work showed that alkanthiolate SAMs of mixed composition can undergo phase segregation into domains,41,42 which implies some mobility of the thiolates on the gold surface. Also, etch pits formed during thiol adsorption on Au(111) were shown to diffuse on the surface without damage of the monolayer.44 Note that the STM imaging was done in air but the SAMs were prepared in vacuum. Interestingly, the diffusion rate was independent of the alkanthiol chain length. STM work also showed that step edges of a Au(111) surface covered by an alkanethiolate SAM can move. The simplest explanation for this observation, as put forward by the authors, is that Au substrate atoms and the thiolate layer move at the same time, in a concerted fashion.43 It was proposed that a Au–thiolate complex (of unknown stoichiometry) at a step edge is moving. Interestingly, different defects were discriminated based on their size and morphology as well as their mobility. For example, point defects appear as 18–25 Å diameter, 1 Å deep depressions in the STM images. These were assigned to molecular voids (missing molecules), and were never observed to move at room temperature on terraces. It was therefore argued that thiolates are not able to move independently on terraces.43The diffusion of thiolates on gold was also studied by cyclic voltammetry.45 A phase-separated SAM of two different thiolates shows a different cyclic voltammogram compared to a homogeneously mixed SAM of the same thiolates. Starting from artificially separated domains of 1-undecanethiol (UDT) and 11-mercaptoundecanoic acid it took over 300 hours at 60 °C to convert into a homogeneously mixed SAM. The authors attributed the change in mixing state to the lateral diffusion of the thiolates on gold, since desorption of thiolates into water was considered unlikely. Similarly, a change in mixing state was also observed when annealing the SAM at 60 °C in air. From these experiments a diffusion coefficient D of 10−18 cm2 s−1 was roughly estimated at 60 °C. This is slow, on the order of 1 nm per hour lateral diffusion. Using STM other researchers estimated diffusion coefficients that are roughly one order of magnitude larger for the coalescence of holes in a dodecanethiol SAM at 90 °C or for the diffusion of a single pit in a dodecanethiol SAM.46,47 It is however a priori not clear if the diffusion processes mentioned above in the STM and cyclic voltammetry studies refer to the same physical processes, i.e. diffusion of thiolates, adsorbate complexes or surface gold atoms, which might explain the order of magnitude difference in the estimated diffusion constants for the different experiments.
To sum up, there is evidence for the mobility of thiolates on flat gold surfaces. Several cases have to be distinguished, including (i) the diffusion of thiolates on otherwise free gold surfaces, (ii) the diffusion of thiolates within a fully covered surface and (iii) the diffusion of gold atoms. Process (i) is important during the formation of SAMs (low coverage) and also for microcontact printing. Process (ii) has implications for phase separation processes of mixed SAMs driven by thermodynamic factors. Process (iii) is evident from the movement of etch pits on SAMs and can be used to heal such defects. The mechanism of these processes is largely unclear and especially for processes (ii) and (iii) it is unclear whether a gold–thiolate complex is moving and, if yes, what the stoichiometry of such a complex could be.
Mobility of thiolates on nanoparticles and clusters
The mobility of thiolates on the surface of thiolate-protected nanoparticles and clusters is an important aspect particularly for applications of particles with mixed ligand shells. The latter can be obtained directly during the synthesis,48 by using a mixture of different thiols, or afterwards by ligand exchange49 or chemical reaction50 of one part of the thiolate ligands. Right after preparation the distribution of ligands in mixed ligand shells is likely random, to some extent, depending on the preparation method. Such a distribution might be far from the thermodynamic most stable structure. Mobility of the thiolates would then be required to reach this situation. Such mobility is crucial for the formation and/or the stability of Janus type nanoparticles that are covered by two different thiolates.51,52 Also, for obtaining liquid-crystalline gold nanoparticles by attaching mesogenes to preformed thiolate-protected gold nanoparticles the arrangement of the liquid-crystalline parts on the particle surface is crucial.53,54 Therefore the mobility of the thiolates is advantageous for the formation of the liquid-crystalline phase, which is usually observed after annealing the sample.The diffusion of thiolates on gold nanoparticles was proposed by Murray and coworkers to rationalize the kinetics of thiolate-for-thiolate exchange reactions.55 However, the rate of diffusion was proposed to be very low, resulting in typical reaction times of weeks at room temperature. Similarly, the extent of photochemical reactions, which depends on the positions of the reacting groups on the gold nanoparticle surface, were observed to be more pronounced for aged samples (after weeks), which was explained by slow lateral diffusion of ligands.56 In contrast, other work indirectly indicated fast diffusion of thiolates on gold nanoparticles.57,58 For example, the results of fluorescence titration experiments using pyrenyl chromophores to monitor the adsorption of ligands on gold nanoparticles were rationalized by equilibration of the ligands on the nanoparticle surface on the time scale of 1–2 hours.58
There is some experimental evidence of phase segregation of mixed thiolates on the surface of gold nanoparticles, driven by thermodynamic factors, which implies some mobility of the thiolates.59 Indication of phase segregation stems from MALDI (matrix-assisted laser desorption/ionization) mass spectrometry,51 scanning tunnelling microscopy (STM),59 and spectroscopy such as electron paramagnetic resonance (EPR),60 infrared (IR)61 and nuclear magnetic resonance (NMR).62 The interpretation of STM is challenging for this purpose but MALDI is an elegant way to study segregation processes on gold nanoparticles (see Fig. 4).51 In MALDI experiments of thiolate-protected gold nanoparticles fragments of composition Au4(SR)4 are often observed. These fragments are thought to be formed by the rearrangement of the staple motifs discussed above during the MALDI process. The ligands in these fragments likely stem from the same location on the nanoparticle. By analysing the distribution of fragments of mixed SR/SR′ thiolate-protected gold particles one can gain information on the distribution of the thiolates on the original cluster, which can in principle vary from completely phase separated (Janus type) to homogeneously (statistically) mixed. For phase segregated particles one would expect large abundance of Au4(SR)4 and Au4(SR′)4 fragments and low abundance of mixed fragments Au4(SR)x(SR′)4−x (x = 1…3). Using this method phase segregation was found for certain ligand combinations on 2–4 nm (core diameter) nanoparticles.51 However, the most pronounced phase separation effects were found for samples that were prepared via ligand exchange rather than direct synthesis. The lack of phase separation in nanoparticles that were prepared by the direct route, even after their heating to 55 °C for one hour indicated that mobility of the thiolates on the gold nanoparticle surface is limited at room temperature or slightly above. The pronounced phase separation via ligand exchange is in line with work on SAMs on Au(111) that showed domainwise exchange of hexadecanethiol with 12-mercaptododecanoic acid in ethanol.63
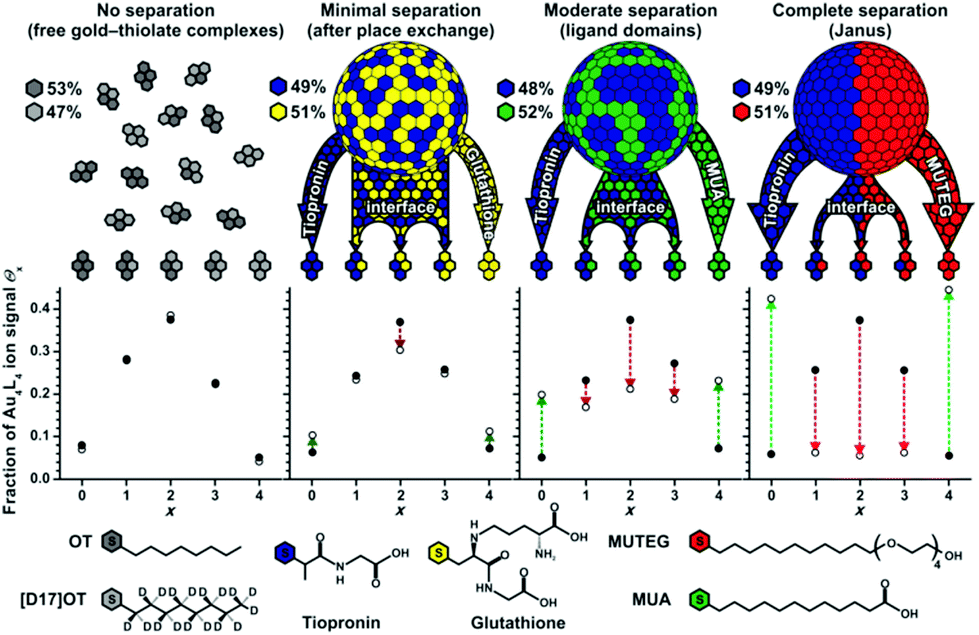 | ||
| Fig. 4 Comparison of experimental (open circles) and theoretical (filled circles) ligand distributions for free gold–thiolate complexes and three mixed-ligand AuNPs obtained by ligand-exchange reactions of tiopronin AuNPs with free glutathione, 11-mercaptoundecanoic acid (MUA), or mercaptoundecyltetra-ethylene glycol (MUTEG). Deviation from the theoretical model (indicated by arrows) reveals phase-segregated gold–thiolate monolayers on AuNPs. Various ligand mixtures yield different degrees of nanophase separation. Reprinted with permission from ref. 51 Copyright (2011) John Wiley and Sons. | ||
Electron paramagnetic resonance (EPR) spectroscopy was also used to study lateral diffusion of thiolates on gold nanoparticles.65,66 Chechik and coworkers designed a disulphide ligand with two spin labels connected by a cleavable bridge.65 After adsorption on gold nanoparticles the bridge was broken and the initially close spin labels could diffuse away from each other, which was probed by EPR spectroscopy. The results suggested that at room temperature there is virtually no lateral mobility, whereas at 90 °C redistribution was observed in the course of several hours. The authors suggested that this redistribution is at least partially due to desorption of ligands and re-adsorption on different nanoparticles.
The studies mentioned above were performed with small nanoparticles (2–4 nm core diameter). Such samples are typically not monodisperse and may contain particles and clusters that are not infinitely stable particularly at elevated temperatures, which may interfere with the interpretation of the obtained results. A way to circumvent this drawback is the preparation of well-defined clusters, which can be obtained by synthesis and subsequent separation for example by size exclusion chromatography.67 In such samples decomposition and size evolution can readily be monitored. Direct evidence for the movement of thiolates on the surface of a gold cluster was obtained from racemization studies of a chiral Au38(PET)24 cluster. The structure of this cluster is known (see Fig. 5)11 and its enantiomers could be separated using chiral chromatography (HPLC, Fig. 5).64
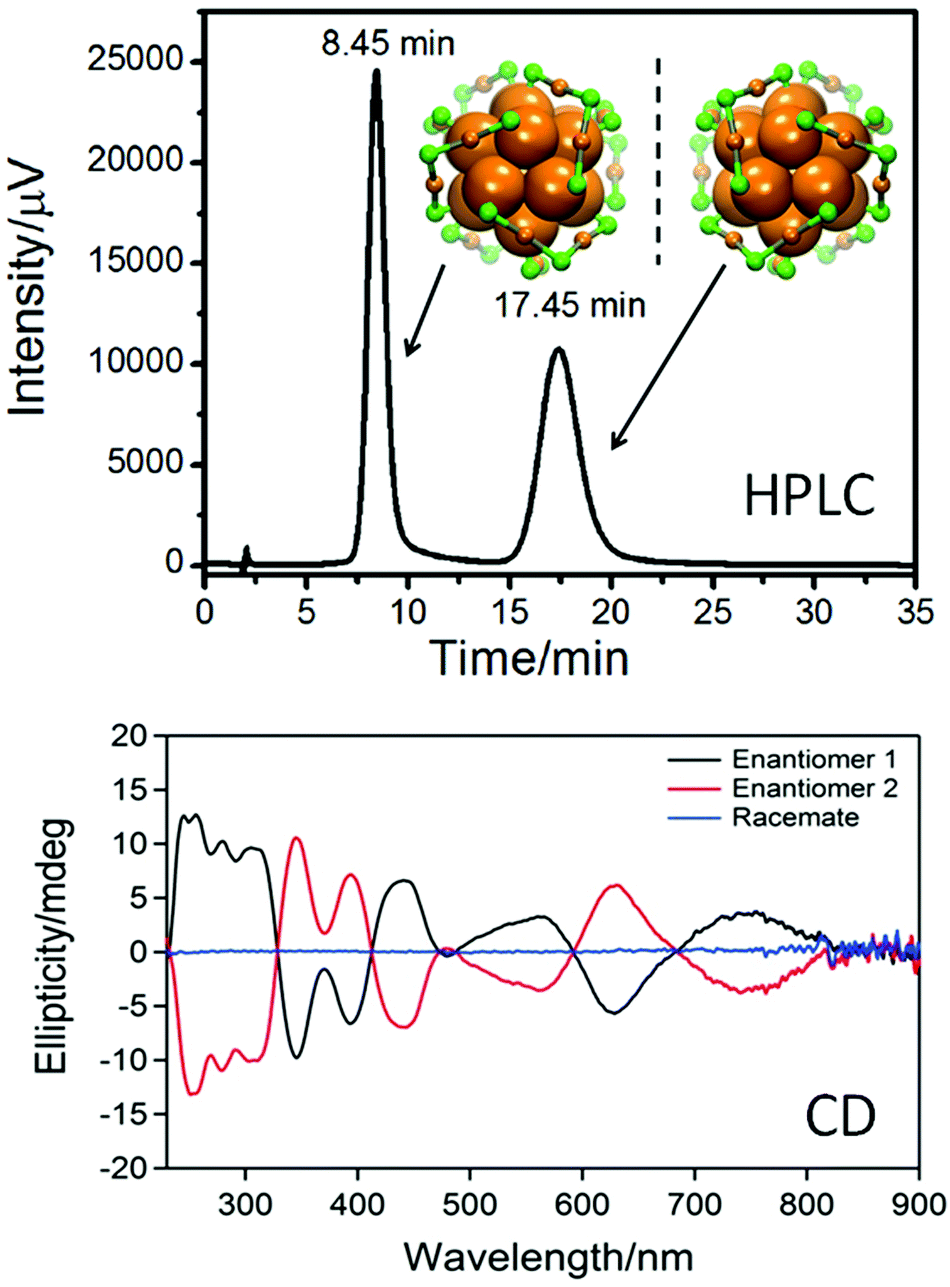 | ||
| Fig. 5 Top: HPLC chromatogram of the enantioseparation of racemic Au38(PET)24. The structure of the left- and right-handed cluster enantiomers is also shown viewed along the long axis of the cluster. Au core atoms: bold orange, Au atoms of the staples: orange, sulphur in the staples: green. The PET ligand is omitted for clarity. Bottom: circular dichroism (CD) spectra of the two enantiomers. Reproduced from ref. 64. | ||
Racemization of the cluster was evidenced both by circular dichroism (CD) spectroscopy and HPLC and the kinetics of this process at different temperatures was followed by CD spectroscopy.68 Racemization takes about half an hour at 80 °C, whereas it is very slow at room temperature. Furthermore, it is not affected by the addition of free thiol. The racemization proves the mobility of the staple motifs on the cluster surface. In the racemization process all six dimeric staple motifs (SR–Au–SR–Au–SR) of the cluster are involved. These are arranged in two chiral three-blade fans of the same handedness at the poles of the cluster (Fig. 5). During racemization both fans invert their handedness. No direct information on the mechanism of this reaction is available. Since there is some evidence for thiolate desorption from gold particles into the solution at elevated temperature (see below) a mechanism involving desorption of thiolates has to be considered. There are two arguments against such a mechanism in the discussed case: (i) addition of thiolates to the solution does not affect the kinetics, and (ii) the activation energy for the process was determined to be less than 30 kcal mol−1,68 whereas a Au–S bond is worth 40 kcal mol−1.1,69 Two possible mechanisms without complete Au–S bond breaking have been proposed, one including the sliding of thiolates of the staple motif over the cluster core surface and the other involving a series of inter-staple SN2-type reactions, where old bonds are broken and new bonds are formed simultaneously. Note that in the first mechanism (sliding) the staple motifs stay intact, whereas in the SN2 mechanism staple motifs are broken and new ones are formed, whereby thiolates exchange between different staples. A similar “crossover” mechanism, with concerted Ag–S bond breaking and formation, was proposed to explain the transformation of polymeric –Ag–SR–Ag–SR– chains in the solid of (3-methylpentane-3-thiolato)silver into (AgSR)8 molecules in solution.70 Similarly, for ligand exchange reactions calculations indicate that concerted Au–S bond formation and bond breaking could play a role.71
Racemization was also studied for Au40(PET)24.72 The enantiomers of this chiral cluster could be separated using chiral HPLC,73 but its structure has not been resolved yet by X-ray crystallography. A structural model has been proposed, which is supported by the comparison of calculated and experimental circular dichroism (CD) spectra.74 Racemization of Au40(PET)24 proceeds at about 50 °C higher temperatures compared to Au38(PET)24, although the activation energies are quite similar for the two cases. For the Au38(PET)24 cluster it was furthermore demonstrated that the incorporation of one rigid dithiol, 1,1′-binaphthyl-2,2′-dithiol (BINAS), into the ligand shell significantly affects the racemization.75 Species with composition Au38(PET)22(BINAS)1 were obtained by ligand exchange, followed by HPLC purification.76 The BINAS ligand is thought to bridge two dimeric staple units.77,78 The effect on racemization is quite drastic. Compared to the parent Au38(PET)24 cluster the incorporation of only one dithiolate increases the temperature of racemization by 40–50 °C. The effect of doping on racemization was also studied. Doping of the Au38(PET)24 cluster with two Pd atoms leads to a drastic lowering of the racemization temperature.79 The doped Pd2Au36(PET)24 cluster is thought to have a similar structure as the parent Au38(PET)24 cluster. The effect of doping on the racemization may therefore indicate the importance of electronic effects.
Fig. 6 summarizes the racemization experiments described above. The activation entropies are distinctly different for the investigated clusters. Note that the two enantiomers of the Au38 cluster, exchanged with enantiopure R-BINAS, show slightly different behaviour. This indicates that diastereomeric interactions play a role in the activation step.
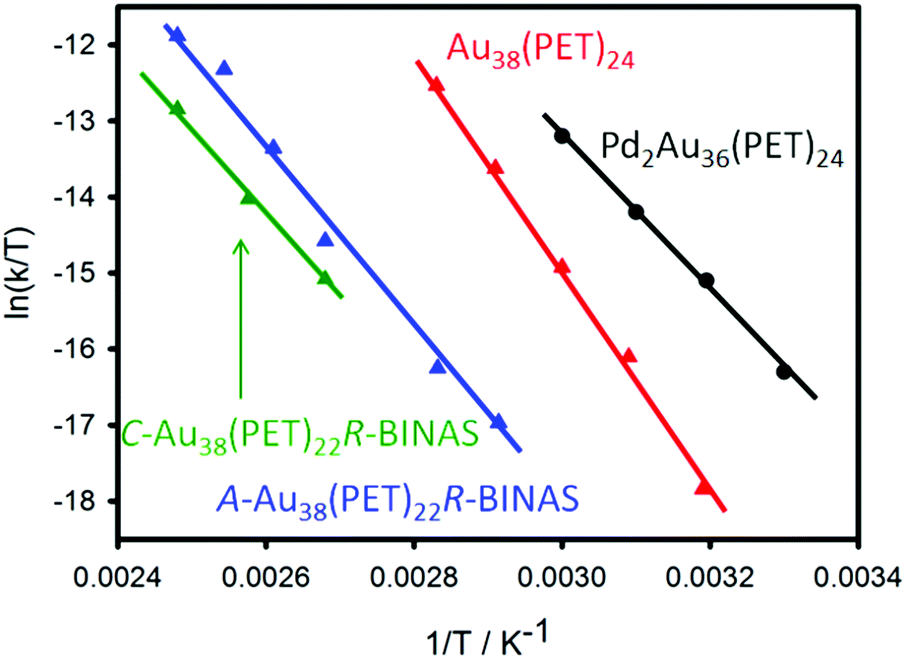 | ||
| Fig. 6 Comparative Eyring plot of different clusters.72,79 The C-Au38(PET)22R-BINAS (A-Au38(PET)22R-BINAS) is the clockwise (anticlockwise) Au38 cluster containing 22 2-PET and one R-BINAS ligand. | ||
Some more information on the mobility of thiolates on the cluster surface was obtained from ligand exchange experiments with a monothiol ([2.2]paracyclophane-4-thiol).80Fig. 7 shows that there are only four different symmetry-unique thiolates on the Au38(PET)24 cluster. Put in other words, ligand exchange can lead, in principle, to only four isomers (regioisomers) of composition Au38(PET)23(SR′)1, where SR′ is the incoming thiolate. Note that the number of isomers increases drastically with the number of exchanged ligands. When ligand exchange was stopped at short times, a mixture of un-exchanged and singly exchanged species was obtained, as verified by MALDI mass spectrometry. The same sample showed at least three additional weak peaks in the HPLC chromatogram, which were assigned to regioisomers of Au38(PET)23(SR′)1. A single regioisomer was then isolated and HPLC experiments showed that such a species is stable at room temperature and slightly elevated temperatures. This proves that the incoming ligand does not move to different symmetry-unique sites. However, at 80 °C migration of thiolates to different symmetry-unique sites was clearly observed (Fig. 7).80
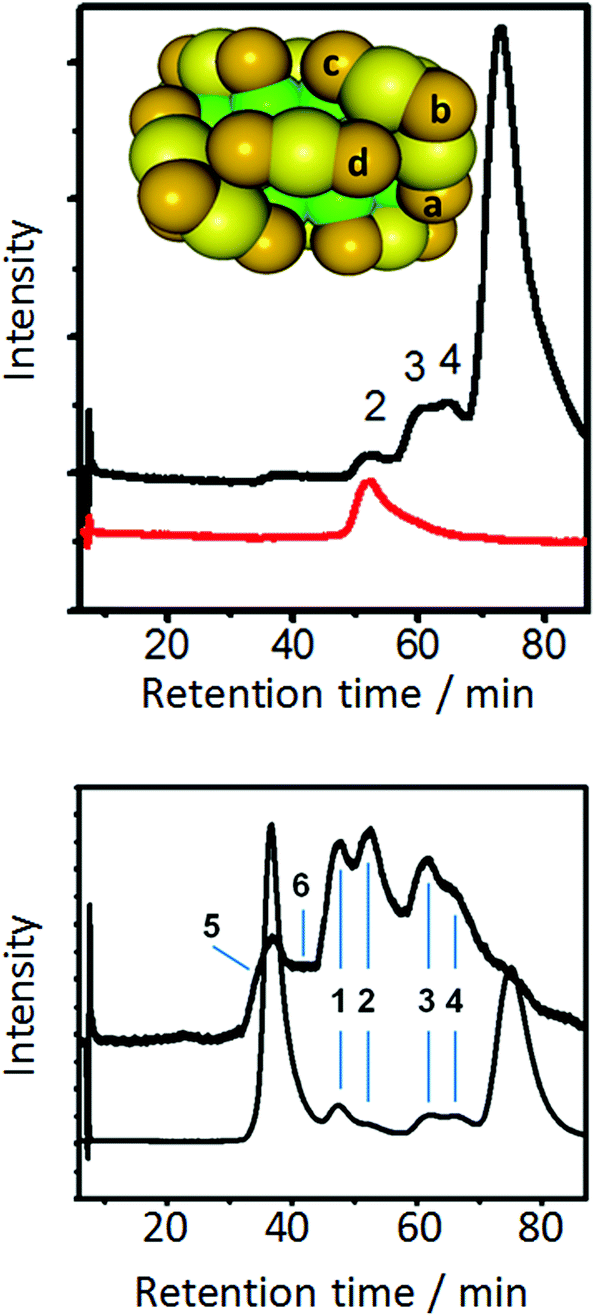 | ||
| Fig. 7 Top: model of Au38(PET)24 cluster. Green: Au atoms in the core, yellow: surface Au atoms, orange: sulphur atoms. The PET ligand is omitted for clarity. The letters a–d indicate the four symmetry unique sites of the cluster. The black HPLC chromatogram was obtained by performing ligand exchange with [2.2]paracyclophane-4-thiol (PCP4). The dominant peak is due to unexchanged Au38(PET)24 (enantiomer 2). The smaller peaks are due regioisomers of singly exchanged cluster Au38(PET)23(PCP4)1. Peak 2 was collected and re-injected (red trace). Bottom: HPLC chromatogram (top trace) of the collected peak 2 corresponding to a specific regioisomer of Au38(PET)23(PCP4)1 after heating to 80° C for 90 min. For comparison the HPLC chromatogram of a ligand exchange reaction of racemic Au38(PET)24 with PCP4 is shown. Reprinted with permission from ref. 80. Copyright (2013) American Chemical Society. | ||
The study mentioned above furthermore shows that ligand exchange at the four symmetry-unique sites is not equally likely because the bands in the HPLC associated with the formed regioisomers have different intensity (see Fig. 7, top).80 NMR experiments showed this also for Au25(PET)18 clusters.81 In the latter cluster only two different positions are available for exchange, namely in the centre of a dimeric staple (called outer ligand by the authors) or at the end of the dimeric staple (called inner ligand by the authors). The experiments showed that exchange is favoured for the outer ligand (middle position, SR–Au–SR–Au–SR) and that the exchange rate at the outer ligand is up to 3.5 times higher than for the inner ligand, depending on the nature of the incoming ligand. Interestingly, a single crystal X-ray study revealed ligand exchange only at the most solvent-exposed site, at the end of the dimeric staple,82 which seems to be in contradiction with the aforementioned study. Possibly crystallization leads to a selection of a subset of ligand-exchange products.
The stability of thiolate-protected gold clusters and nanoparticles indicates that desorption of thiolates or of gold–thiolate complexes from the gold cluster/nanoparticle surface is not an important process, despite the fact that the thiolates are mobile to a certain extent on the gold cluster surface. However, some experimental observations show that thiolates can be exchanged between different clusters. Negishi and coworkers prepared Au25(SC12H25)18 and Au25(SC10H21)18 clusters.83 Mixing of the two clusters in CH2Cl2 solution for 10 minutes at room temperature lead to a mixed thiolate layer as evidence by mass spectrometry, which showed that up to four thiolates exchanged between the particles, as can be seen in Fig. 8. The effect was less pronounced for a Pd doped cluster.
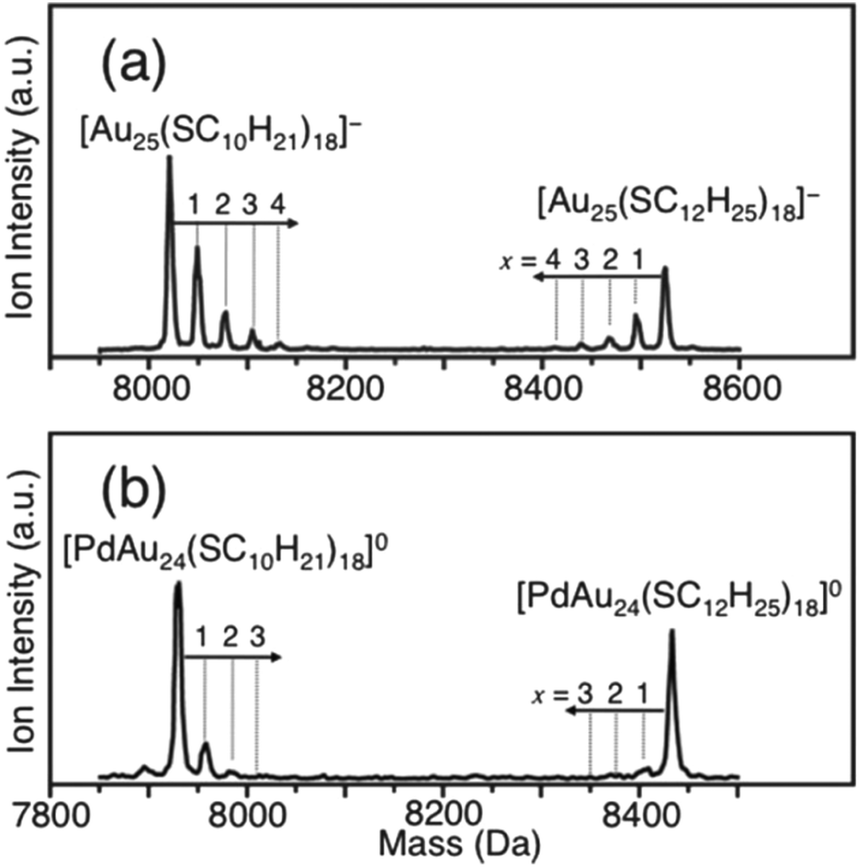 | ||
| Fig. 8 Negative-ion MALDI mass spectra of a mixture of [Au25(SC12H25)18]− and [Au25(SC10H21)18]− after 10 min in CH2Cl2 (a) and of [PdAu24(SC12H25)18]0 and [PdAu24(SC10H21)18]0 after 10 min in CH2Cl2 (b). The dominant peak corresponds to the parent clusters, whereas the small peaks are due to exchange clusters with mixed ligand shell. Reproduced from ref. 83 with permission from The Royal Society of Chemistry. | ||
EPR was also used to study exchange of thiolates between different particles.84 Two types of particles, initially covered by different thiolates each, were mixed. Interparticle exchange of ligands then leads to a change in interaction between adjacent ligands, which is sensitively monitored by EPR. Changes were very slow at room temperature, but at 70 °C alterations in EPR line-shapes were observed within minutes, indicating the interparticle exchange of ligands. Interestingly, even after prolonged heating the presence of free ligands in solution was minimal, showing that the steady state concentration of free ligand is low. Based on the kinetics of this reaction the authors proposed a mechanism where the rate-determining step is the desorption of a thiolate species (of unknown nature and stoichiometry), which creates a vacancy for the readsorption of (another) thiolate.
These clusters studies seem to be in agreement with studies performed 20 years ago on SAMs on gold of octadecanethiol containing radiolabeled head groups (35S).85 These studies showed that thiols can desorb into solution on a timescale of a day. The desorption process was not complete, followed pseudo first order kinetics and depended on the solvent. The finding is consistent with earlier studies, which indicated the transfer of thiolates (4-carboxythiophenol) between gold and silver colloids.86
In summary, the gold–thiolate interface is not rigid, both in clusters and in SAMs. Thiolates can move between different sites on clusters. Several studies indicate that this is a slow process at room temperature both for clusters and nanoparticles. At slightly elevated temperatures (70–90° C) migration is evidenced within tens of minutes. Also, entire staple motifs can move on the cluster surface, as is shown by racemization studies. Again, this process is observed above room temperature. In addition, there is evidence both for clusters and for flat gold surfaces that thiolates can desorb into solution even at room temperature within tens of minutes. The stoichiometry of the desorbing species is unknown. However, work by Murray and coworkers indicated that even metal atoms can be exchanged between gold and silver clusters protected by thiolate monolayers87 indicating that even metal atoms/ions can desorb. Despite the large body of experiments described above there is not yet a clear picture emerging, for example as concerns the relative importance of ligand desorption/re-adsorption and direct migration on the surface. Some findings remain somehow contradictory. Possibly there is a particle size effect of these properties, as the work of Stellacci and Murry seems to indicate by showing that the formation of striped (phase segregated) nanoparticles is size dependent.88 Open questions to be addressed in the future concern the stoichiometry of the species desorbing from or migrating on the cluster surface, the role of Au(I) in the staples for these processes and the migration/desorption of dithiolates.
Chirality
Chiral SAMs
Chiral metal surfaces have attracted considerable interest due to potential applications in chiral catalysis89,90 and electrocatalysis91 but also due to fundamental aspects that are linked to 2-dimensional supramolecular phenomena, chiral recognition and symmetry breaking.92–95 There are also some reports on chiral SAMs of thiolates on gold.96–102 As mentioned above, STM work revealed the existence of monomeric staples of methyl thiolates on Au(111).21 Locally this produces a chiral structure because the sulphur atoms represent stereogenic centres similar as a carbon atom with four different substituents (see Fig. 2 and 3).The structure of SAMs of BINAS on Au(111) was also studied by STM, which revealed a 2D chiral arrangement.103 These SAMs were prepared from solution and investigated in air. A honeycomb structure was found, consisting of screw-like entities with 3-fold rotational symmetry, each of which is composed of three BINAS molecules. The formation of orderd structures implies some degree of mobility of the thiolates on the surface. Such a SAM of BINAS on gold was shown later by quartz crystal microbalance (QCM) to discriminate between the enantiomers of thalidomide.104R-thalidomide adsorbed on R-BINAS SAMs but not or hardly on S-BINAS SAMs.
An interesting STM study revealed the assembly of an achiral adamantane tripod (trithiol) on Au(111) into a chiral framework.105 The sample was prepared and examined in UHV. The rigid molecule forms three Au–S bonds with the surface. The adsorbed molecules develop chiral trimers, which act as building block for even larger hexagonal chiral structures. This shows that symmetry can be broken by achiral molecules leading to locally chiral structures on a globally racemic surface. The formation of such highly ordered hierarchical structures again indicates some degree of mobility of the individual molecules on the surface after their adsorption. In these experiments the molecules were evaporated onto the surface at around 330 K. Similarly, supramolecular chiral structures were found for SAMs of a dithiocarbamate on Au(111).106
Waldeck and coworkers prepared SAMs on gold from thiol-terminated chiral scaffold molecules containing a porphyrine chromophore at its end.107 The SAMs were prepared from solution. Illumination by visible light within an electrochemical cell generated a cathodic photocurrent. Interestingly, the authors observed a strong difference of this photocurrent depending on the circularity of the light used (left- or right-circular polarized). This asymmetry in the photocurrent was explained with a symmetry constraint on the electronic coupling between the porphyrine moiety and the chiral scaffold.
Nakanishi and coworkers prepared SAMs containing leucine molecules and studied the crystallization of leucine on top of these SAMs.108 It was found that the crystallization was enantioselective. When immersing a D-leucine SAM in a D-leucine solution, crystals of D-leucine were grown. However, when the D-leucine SAM was immersed in a L-leucine solution, no crystals were formed (and vice versa). Similarly, Amabilino and coworkers prepared SAMs from a chiral resolving agent containing a cyclic disubstituted phosphate group.109 These SAMs were used as templates for the growth of crystals of organic molecules with a similar structure as the ones used for SAM formation.
Using UHV STM Besenbacher and coworkers showed that cysteine on Au(110) forms dimers and that the molecular pairs formed from a racemic mixture are exclusively homochiral, which is evidence for strong chiral recognition.97 Chiral SAMs prepared from homocysteine on Au(111) were used as electrodes to study the redox behaviour of catechins using cyclic voltammetry.110 The formation of the SAM blocks, to some extent, the redox reaction. However, the extent of blocking was different for the two enantiomers of the catechins. The preference for one enantiomer was inverted in acidic solution. These experiments showed that the homocysteine SAM can distinguish enantiomers of catechin. By comparison between catechin and epicatechin the authors concluded that the chiral SAM is able to discriminate the absolute configuration at one of the two chiral centres. Homocysteine SAMs were also integrated into the gate of a field effect transistor, which allowed the discrimination between the enantiomers of alanine.111
SAMs of chiral N-acetyl-L-cysteine112 and glutathione113 on gold were also prepared directly on internal reflection elements, which enabled in situ studies by attenuated total reflection infrared spectroscopy (ATR-IR). The ability of these two SAMs to discriminate between proline enantiomers was then studied using modulation excitation spectroscopy combined with a phase-sensitive detection.114–117 For this, solutions of the two enantiomers of the probe molecule (proline) were flown alternatingly over the SAM and the response of the system was recorded in situ. For the glutathione SAM it was shown that the adsorption and desorption kinetics (but particularly the desorption) are different for the two enantiomers of proline and therefore the chiral SAM can discriminate between them. All these examples show that chiral SAMs have the ability to discriminate between enantiomers of molecules that interact with the SAMs.
Chiral thiolate-protected gold clusters and particles
The interest in chiral gold clusters is increasing due to possible applications in chiral technology.34,118 Evidencing chiral properties in this case is different compared to flat gold surfaces and often easier. Chiroptical methods proved particularly useful for this purpose as they are a selective probe of chirality. Also, crystallography and theory helped a lot for the understanding of chirality of thiolate-protected gold clusters.The first experimental manifestation of chirality in thiolate-protected clusters stems from circular dichroism (CD) measurements on gold clusters protected by (chiral) glutathione.119 Strong CD signals were observed in transitions that are mainly located in the metal core. Similar observations were made for other gold clusters protected by chiral thiolates.120–126 Until direct structural information was available the origin of the optical activity of was not clear. Particularly it was not clear if the structure of the cluster is intrinsically chiral127 or if the optical activity is “just” induced by the chiral ligands around a symmetric particle core.118,128 The first crystal structure of a thiolate-protected Au102(pMBA)44 cluster then revealed a symmetric core protected by gold–thiolate staples (see Fig. 2 and 3). The staples are arranged in a chiral fashion even though the thiol itself is not chiral. Furthermore, each sulphur atom represents a stereogenic centre. The unit cell of the crystal contains both enantiomers of the cluster and therefore such a sample is racemic and does not show optical activity. A similar situation was later found for the chiral Au38(PET)24 cluster (see Fig. 7).11 The enantiomers of the cluster could be separated into its enantiomers using chiral chromatography.64 This allowed one to measure the CD spectrum of the cluster, which was quite pronounced despite the fact that the cluster is protected by achiral ligands. The enantiomers of other chiral gold clusters were later also separated.16,73,79,129
An interesting case represents the Au25(SR)18 cluster.9 Its Au–S framework is achiral consisting of an Au13 icosahedral core protected by six long staple motifs (three orthogonal pairs). The cluster shows optical activity when covered by a chiral thiolate.130 However, the anisotropy factors are much weaker compared to the Au38 case, which indicates that the chiral arrangement of thiolates gives rise to stronger optical activity compared to the optical activity induced by chiral thiolates.
Chiral Au38(PET)24 clusters were shown to be able to discriminate between enantiomers of a chiral thiols in ligand exchange reactions (Fig. 9). Specifically, a racemic mixture of the cluster was exposed to R-BINAS and the ligand exchange was monitored in situ by chiral HPLC.76 Analysis of the kinetics revealed that the left-handed cluster reacted about four times faster with R-BINAS than the right-handed cluster. Note that the thiolates initially adsorbed on the cluster are not chiral.
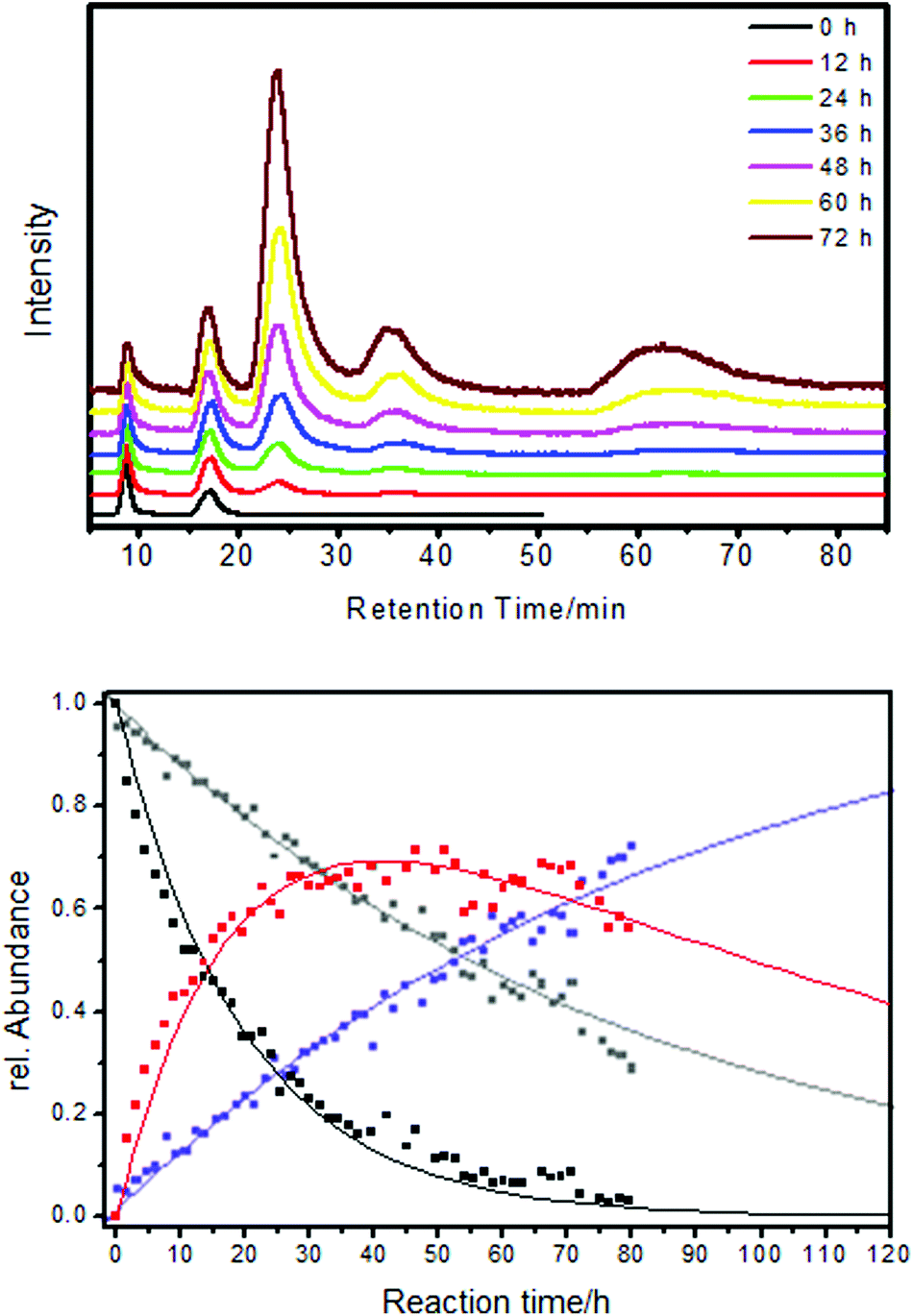 | ||
| Fig. 9 The ligand exchange between Au38(PET)24 and BINAS is diastereomeric. Top: HPLC traces recorded in situ during the ligand exchange between racemic Au38(PET)24 and R-BINAS. The first two peaks belong to the two enantiomer of Au38(PET)24 (A-Au38(PET)24 and C-Au38(PET)24) The next two peaks belong to the corresponding cluster with one R-BINAS incorporated (A-Au38(PET)22(R-BINAS)1 and C-Au38(PET)22(R-BINAS)1). The broad peak at higher retention times corresponds to higher exchange products. All chromatograms were scaled to the intensity of the first peak. Bottom: kinetic analysis of the ligand exchange. Dots are measurements (HPLC) and solid lines are derived from a fit to a kinetic model. Black: A-Au38(PET)24, gray: C-Au38(PET)24, red: A-Au38(PET)22(R-BINAS)1, blue: C-Au38(PET)22(R-BINAS)1. Reprinted with permission from ref. 76 Copyright (2012) American Chemical Society. | ||
Gellman and coworkers also demonstrated that gold nanoparticles of about 5 nm in size and covered by enantiomers of cysteine show enantioselectivity in adsorption of propylene oxide on their surface.131 The authors used optical rotation to demonstrate enantiospecific adsorption and used the fact that the specific optical rotation is enhanced for molecules interacting with the gold nanoparticles. The analysis can be made in a quantitative manner in order to extract enantiospecific adsorption equilibrium constants.132 In summary, not much has been done to demonstrate and apply the enantiospecific properties of gold nanoparticles and clusters although their potential has been shown. With the recent progress in the preparation of chiral clusters and particles more work will certainly be done in this field.
Vibrational properties
Vibrational properties of SAMs
Vibrational spectra are a sensitive probe of structure although the information is not immediately evident from the spectrum. However, it is clear that different structures of adsorbate layers, e.g. adsorbate layers of thiolates with or without ad-atoms, are characterized by different vibrational spectra. In addition, the vibrational properties are usually quite local, in contrast for example to the electronic properties and it is therefore anticipated that comparison between vibrational spectra of SAMs of thiolates on flat surfaces and thiolate-protected gold clusters can directly be compared. Vibrational spectroscopy could therefore be a key tool to address similarities and differences of the two systems, SAMs and clusters, as concerns their structure.Vibrational spectra of adsorbed thiolates on gold surfaces provide useful information on interactions between molecules in the SAM, on the orientation of molecules with respect to the surface112,133 and the order within the layer. Information on the order within an alkanethiol SAM is provided by the frequency and width of the C–H stretching bands.134 Vibrational spectra can be obtained by several techniques including infrared and Raman spectroscopy, high-resolution electron energy loss spectroscopy (HREELS) and non-linear optical techniques such as sum-frequency generation. Of particular interest here are the vibrational properties of the Au–S interface, which are quite low in frequency. Although vibrational spectroscopy was frequently used to characterize SAMs of thiolates not much attention was paid to the low frequency region of the spectrum, a fact related with experimental challenges. Raman signals of monolayers on flat surfaces are very small and far infrared (FIR) spectroscopy requires special equipment. Typical Au–S stretching vibrations give rise to bands around 220–240 cm−1 in the spectrum, as evidenced by Raman and HREELS measurements.135–137 A systematic study of the HREELS spectra of SAMs formed by alkanthiolates of different lengths on Au(111) revealed multiple Au–S stretching bands, which were assigned to multiple adsorption sites of the thiolate on the gold surface.138 The authors also observed a dependence on the chain lengths. At the time of the study the staple structure was not yet discovered. It is clear that such a staple structure should give rise to multiple Au–S stretching modes at different frequencies due to the different nature of the Au–S bonds involved (Au–S within the staple and Au–S involving a core gold atom).
Vibrational properties of thiolate-protected gold nanoparticles and clusters
Not too much attention has been paid up to now to the vibrational properties of thiolate-protected gold clusters. The mid infrared region of the spectrum mainly contains information about the ligand themselves and possibly on their electronic interaction with the metal core.139 The vibrations of the Au–S interface are expected below about 350 cm−1 and the vibrations within the metal core (Au–Au vibrations) are even lower in energy (≪200 cm−1).140,141 The latter vibrations are expected to be weak in the IR spectrum due to the low polarity but should be prominent in the Raman spectrum.In principle a vibrational spectrum contains information about the structure (conformation) of a molecule. In many cases it is however not trivial to extract such information from the spectra.
Creutz and co-workers reported far-IR spectra of alkanethiol capped gold nanoparticles (about 2 nm).143 The spectra are characterized by quite broad bands that change with the length of the alkanethiol. Bands at 260–270 cm−1 and around 180 cm−1 were assigned to Au–S vibrations. Murray and coworkers reported the Raman spectrum of Au25(PET)18. A band at around 290 cm−1 was assigned to Au–S vibrations.144
Halas and coworkers used surface enhanced Raman spectroscopy to study the low-energy region of SAMs of alkanethiols of different lengths (C10, C11…C16) on plasmonic gold particles.145 The low-energy vibrational spectrum (below 400 cm−1), where the Au–S vibrations are expected, showed several bands and the spectra were quite different from the Raman spectra of the alkanethiol liquids. In particular the spectra were characterized by a sharp band that shifted drastically with chain lengths from 367 cm−1 (C10) to 278 cm−1 (C16) and a second band at lower frequency that shifted as well.
These observations were explained by a coupling between the Au–S stretching and longitudinal acoustic modes (LAM) of the alky chain. The latter modes were also observed for the alkanethiol liquids although less sharp and with a smaller shift as a function of chain length. It is likely that the coupling and therefore the spectrum is sensitive to the geometry and binding of the thiolate head-group. However, the coupling also complicates a more direct analysis of the low-frequency vibrational spectra.
In an attempt to relate vibrational signatures to structural information of the Au–S interface Raman and far-IR spectra of well-defined clusters were measured.142,146 By comparison with calculations one can assign different types of Au–S vibrations notably Au–S–C bending modes around 180 cm−1, radial Au–S stretching modes around 220–280 cm−1 and tangential Au–S stretching modes around 320 cm−1 and above. (see Fig. 10).
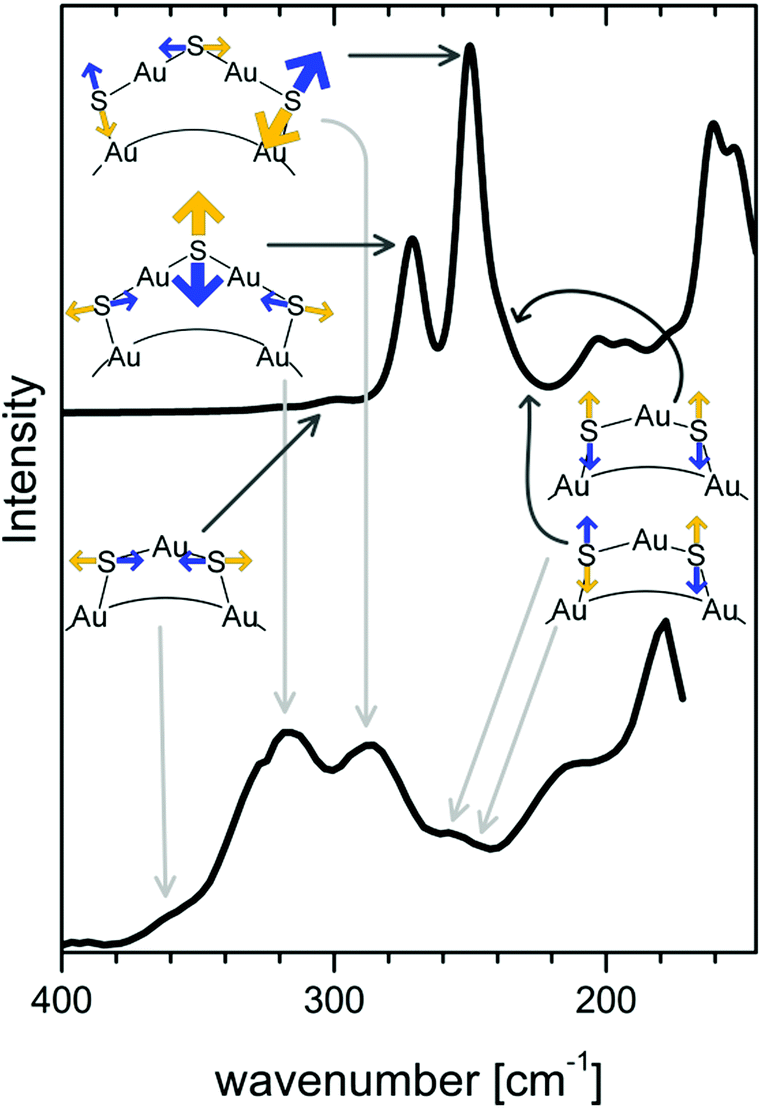 | ||
| Fig. 10 Calculated (top) and experimental (bottom) Raman spectrum of the Au38(SCH3)24 and Au38(PET)24 cluster, respectively. Radial and tangential Au–S modes of the staples are schematically represented. Radial vibrations of the long staples are responsible for bands with high intensity. Modes associated with the short staples (symmetric and antisymmetric stretching and tangential vibrations) have lower Raman intensity. Reprinted with permission from ref. 142 Copyright (2014) American Chemical Society. | ||
Radial modes are lower in frequency and involve the stretching between sulphur atoms of the staple and gold core atoms. Tangential modes involve stretching motions between sulphur and gold within staples. As mentioned above good quality HREELS spectra of alkanethiol SAMs evidence multiple peaks in the 200 cm−1–350 cm−1 spectral region,138 which might indeed be attributed to staple-like structures instead of different adsorption sites of the thiolate. Vibrational spectroscopy has therefore the potential to shed more light on the nature of the Au–S interface on flat SAMs but for this more systematic studies are needed with high quality spectra of the low frequency vibrations.
A special technique that is particularly sensitive to conformation is vibrational circular dichroism (VCD), i.e. the differential absorption of left- and right-circular polarized light by a chiral sample.147,148 This technique has been applied to gold clusters in order to extract information on the conformation of the adsorbed chiral thiolates.122,149–151 VCD signals are in general very weak and the technique cannot be applied to surfaces. However, VCD on clusters is a powerful technique to study the structure of adsorbed molecules.
Achiral molecules and racemic mixtures do not show VCD signals. However, it has been demonstrated that the achiral 2-phenylethylthiol gives rise to strong VCD signals when adsorbed on the chiral Au38 cluster (Fig. 11).152 For the VCD measurements the enantiomers of the cluster were separated. The reason for this is a preferred chiral conformation of the molecule adsorbed on the cluster. More precisely the molecule can exist in (achiral) anti and (transiently chiral) gauche conformation, referring to the relative orientation of the phenyl group and the sulphur atom. The infrared absorption spectra indicated a pronounced abundance of gauche molecules. For the gauche conformation two enantiomeric forms exist and in solution the two forms are equally abundant. Due to the chirality of the cluster one gauche enantiomer becomes more stable, which gives rise to the observed VCD signals. This shows that the chiral cluster transfers its chirality to the achiral molecule. The study furthermore indicates that the conformation of the thiolates on the clusters is different in solution and in the solid state (crystal).
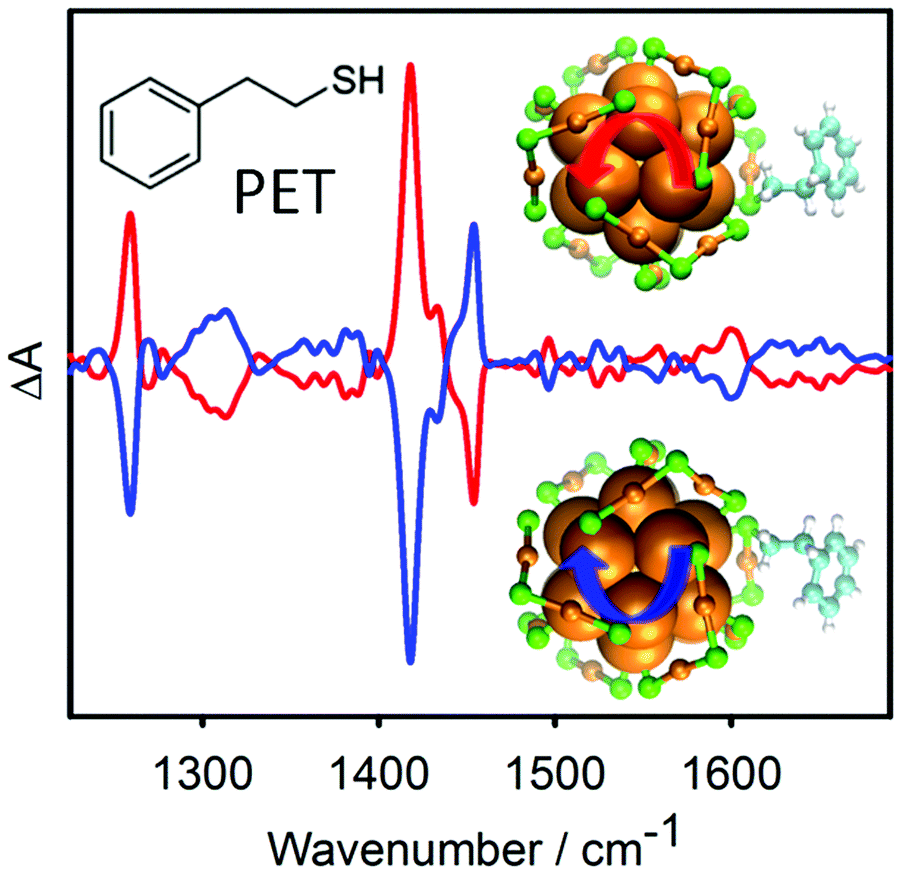 | ||
| Fig. 11 VCD spectra of the two enantiomers of Au38(PET)24. The bands in the spectra are due to the 2-phenylethylthiolate (PET) adsorbed on the cluster. PET is an achiral molecule, but the chiral cluster stabilizes one enantiomeric form of a transiently chiral gauche conformation, which leads to the VCD signals. The models show the chiral Au–S framework of the cluster and one PET ligand adopting a chiral conformation. Reproduced from ref. 152. | ||
Conclusions
In this article we have compared properties of thiolate SAMs on gold and thiolate-protected gold clusters focusing on their structure and chirality, on their dynamic nature and vibrational properties. The Au–S interface proved to be dynamic in both cases. Due to the highly curved surface of small gold clusters different properties can be expected compared to SAMs on flat gold surfaces. It seems however, that the two systems have a lot of similarities, which may, at the end, be due to similar structure elements of the Au–S interface in the two cases.There is strong evidence for gold ad-atoms or staple-like structures on flat gold surfaces (at least for Au(111)). However, there are also recent studies, which do not give direct evidence for gold ad-atoms. On the other hand, there is now also evidence from single crystal X-ray crystallography for other structural motifs, besides staple motifs, such as bridged adsorption sites on clusters. If a unified model of the Au–S interface will emerge for both the cluster and SAM field remains to be seen. What is clear is that the Au–S interface is more complex than it was thought one decade ago.
The Au–S interface is far from being rigid and static both on flat surfaces and on clusters. Different processes including migration of thiolates and desorption of thiolates (and re-adsorption) contribute to the flexibility of the interface. Migration of thiolates was evidenced for clusters and surfaces at temperatures above ambient whereas there is evidence for desorption even at room temperature both from flat surfaces and from clusters. On flat surfaces the migration of etch pits is also observed and it is not clear if there is an analogous process for clusters. The flexibility of the Au–S interface is an important property for applications of clusters (and SAMs) and it will be important in the future to clarify the composition of the migrating and of the desorbing species.
Chirality of SAMs and of thiolate-protected clusters is an interesting property with possible applications in chiral technology. Chirality in both cases arises at different levels that is at the level of arrangement of the thiolates (organization level) and at the molecular level (local level). In both cases achiral molecules can become chiral upon adsorption which has to do with symmetry breaking imposed by the surface. Chiral recognition and chirality transfer effects have been demonstrated and it remains to be shown if chiral gold clusters may be used for chirality sensing or enantioselective catalysis.
Comparison of properties of the two systems considered here (thiolate SAMs on flat surfaces and thiolate-protected clusters) is sometimes difficult due to the different experimental techniques that have to be applied. In this sense vibrational spectroscopy stands out because it can be equally applied to both systems and the vibrational spectrum is a local property, i.e. whether the surface is flat or curved does not matter much as concerns the vibrational spectrum of the adsorbate. Indeed, spectroscopy of the Au–S vibrations could play an important role to characterize the structure of various Au–S interfaces. Unfortunately this is hampered by the relative complexity of the vibrational signatures. Future theoretical studies are needed here. In addition, there are only few vibrational spectra available of clusters and good quality low-frequency vibrational spectra of thiolated SAMs are surprisingly scarce.
Acknowledgements
Financial support from the Swiss National Science Foundation (grant number 200020_152596) and the University of Geneva are kindly acknowledged. The author thanks all PhD students and PostDocs involved in this project for their work during the last decade, notably Cyrille Gautier, Stefan Knoppe, Julien Boudon, Birte Varnholt, Bei Zhang, Annelies Sels, Ani Baghdasaryan, Satyabrata Si, Lule Beqa, Noelia Barrabes and Igor Dolamic.Notes and references
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS.
- N. K. Chaki and K. Vijayamohanan, Biosens. Bioelectron., 2002, 17, 1–12 CrossRef CAS.
- G. Heimel, L. Romaner, E. Zojer and J. L. Bredas, Acc. Chem. Res., 2008, 41, 721–729 CrossRef CAS PubMed.
- Y. N. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- A. Badia, S. Singh, L. Demers, L. Cuccia, G. R. Brown and R. B. Lennox, Chem. – Eur. J., 1996, 2, 359–363 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802, 10.1039/c39940000801.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- H. F. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. C. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- C. J. Zeng, H. F. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. C. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114–13118 CrossRef CAS PubMed.
- C. J. Zeng, C. Liu, Y. X. Chen, N. L. Rosi and R. C. Jin, J. Am. Chem. Soc., 2014, 136, 11922–11925 CrossRef CAS PubMed.
- D. Crasto, S. Malola, G. Brosofsky, A. Dass and H. Hakkinen, J. Am. Chem. Soc., 2014, 136, 5000–5005 CrossRef CAS PubMed.
- A. Das, T. Li, K. Nobusada, C. J. Zeng, N. L. Rosi and R. C. Jin, J. Am. Chem. Soc., 2013, 135, 18264–18267 CrossRef CAS PubMed.
- C. J. Zeng, T. Li, A. Das, N. L. Rosi and R. C. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- P. R. Nimmala, S. Knoppe, V. R. Jupally, J. H. Delcamp, C. M. Aikens and A. Dass, J. Phys. Chem. B, 2014, 118, 14157–14167 CrossRef.
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Y. Sfeir and R. Jin, Sci. Adv., 2015, 1, 1:e150004 Search PubMed.
- A. Dass, S. Theivendran, P. R. Nimmala, C. Kumara, V. R. Jupally, A. Fortunelli, L. Sementa, G. Barcaro, X. Zuo and B. C. Noll, J. Am. Chem. Soc., 2015, 137, 4610–4613 CrossRef CAS PubMed.
- M. Azubel, J. Koivisto, S. Malola, D. Bushnell, G. L. Hura, A. L. Koh, H. Tsunoyama, T. Tsukuda, M. Pettersson, H. Hakkinen and R. D. Kornberg, Science, 2014, 345, 909–912 CrossRef CAS PubMed.
- O. Voznyy, J. J. Dubowski, J. T. Yates and P. Maksymovych, J. Am. Chem. Soc., 2009, 131, 12989–12993 CrossRef CAS PubMed.
- H. Gronbeck, H. Hakkinen and R. L. Whetten, J. Phys. Chem. C, 2008, 112, 15940–15942 Search PubMed.
- A. Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, Science, 2008, 321, 943–946 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152–7167 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Corthey, E. Pensa, E. Cortes, M. H. Fonticelli, F. Ibanez, G. E. Benitez, P. Carro and R. C. Salvarezza, RSC Adv., 2014, 4, 27730–27754 RSC.
- L. Strong and G. M. Whitesides, Langmuir, 1988, 4, 546–558 CrossRef CAS.
- L. H. Dubois, B. R. Zegarski and R. G. Nuzzo, J. Chem. Phys., 1993, 98, 678–688 CrossRef CAS PubMed.
- C. E. D. Chidsey, G. Y. Liu, P. Rowntree and G. Scoles, J. Chem. Phys., 1989, 91, 4421–4423 CrossRef CAS PubMed.
- C. A. Alves, E. L. Smith and M. D. Porter, J. Am. Chem. Soc., 1992, 114, 1222–1227 CrossRef CAS.
- P. Fenter, F. Schreiber, L. Berman, G. Scoles, P. Eisenberger and M. J. Bedzyk, Surf. Sci., 1998, 412–13, 213–235 CrossRef.
- D. Grumelli, L. J. Cristina, F. L. Maza, P. Carro, J. Ferron, K. Kern and R. C. Salvarezza, J. Phys. Chem. C, 2015, 119, 14248–14254 CAS.
- H. Hakkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- S. Knoppe and T. Burgi, Acc. Chem. Res., 2014, 47, 1318–1326 CrossRef CAS PubMed.
- R. Bau, J. Am. Chem. Soc., 1998, 120, 9380–9381 CrossRef CAS.
- R. Mazzarello, A. Cossaro, A. Verdini, R. Rousseau, L. Casalis, M. F. Danisman, L. Floreano, S. Scandolo, A. Morgante and G. Scoles, Phys. Rev. Lett., 2007, 98, 4 CrossRef.
- P. Maksymovych, D. C. Sorescu and J. T. Yates, Phys. Rev. Lett., 2006, 97, 4 CrossRef.
- E. Pensa, A. A. Rubert, G. Benitez, P. Carro, A. G. Orive, A. H. Creus, R. C. Salvarezza and C. Vericat, J. Phys. Chem. C, 2012, 116, 25765–25771 CAS.
- E. Pensa, E. Cortes, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benitez, A. A. Rubert and R. C. Salvarezza, Acc. Chem. Res., 2012, 45, 1183–1192 CrossRef CAS PubMed.
- S. Seo and H. Lee, J. Phys. Chem. C, 2011, 115, 15480–15486 CAS.
- S. J. Stranick, A. N. Parikh, Y. T. Tao, D. L. Allara and P. S. Weiss, J. Phys. Chem., 1994, 98, 7636–7646 CrossRef CAS.
- R. K. Smith, S. M. Reed, P. A. Lewis, J. D. Monnell, R. S. Clegg, K. F. Kelly, L. A. Bumm, J. E. Hutchison and P. S. Weiss, J. Phys. Chem. B, 2001, 105, 1119–1122 CrossRef CAS.
- S. J. Stranick, A. N. Parikh, D. L. Allara and P. S. Weiss, J. Phys. Chem., 1994, 98, 11136–11142 CrossRef CAS.
- R. L. McCarley, D. J. Dunaway and R. J. Willicut, Langmuir, 1993, 9, 2775–2777 CrossRef CAS.
- S. Imabayashi, D. Hobara and T. Kakiuchi, Langmuir, 2001, 17, 2560–2563 CrossRef CAS.
- F. T. Arce, M. E. Vela, R. C. Salvarezza and A. J. Arvia, Electrochim. Acta, 1998, 44, 1053–1067 CrossRef.
- C. Schonenberger, J. Jorritsma, J. A. M. Sondaghuethorst and L. G. J. Fokkink, J. Phys. Chem., 1995, 99, 3259–3271 CrossRef.
- S. W. Chen and R. W. Murray, J. Phys. Chem. B, 1999, 103, 9996–10000 CrossRef CAS.
- R. S. Ingram, M. J. Hostetler and R. W. Murray, J. Am. Chem. Soc., 1997, 119, 9175–9178 CrossRef CAS.
- S. Mischler, S. Guerra and R. Deschenaux, J. Chem. Soc., Chem. Commun., 2012, 2183–2185 RSC.
- K. M. Harkness, A. Balinski, J. A. McLean and D. E. Cliffel, Angew. Chem., Int. Ed., 2011, 50, 10554–10559 CrossRef CAS PubMed.
- S. Pradhan, L. P. Xu and S. W. Chen, Adv. Funct. Mater., 2007, 17, 2385–2392 CrossRef CAS PubMed.
- S. Frein, J. Boudon, M. Vonlanthen, T. Scharf, J. Barbera, G. Suss-Fink, T. Burgi and R. Deschenaux, Helv. Chim. Acta, 2008, 91, 2321–2337 CrossRef CAS PubMed.
- L. Cseh and G. H. Mehl, J. Am. Chem. Soc., 2006, 128, 13376–13377 CrossRef CAS PubMed.
- M. J. Hostetler, A. C. Templeton and R. W. Murray, Langmuir, 1999, 15, 3782–3789 CrossRef CAS.
- A. J. Kell, R. L. Donkers and M. S. Workentin, Langmuir, 2005, 21, 735–742 CrossRef CAS PubMed.
- K. Norgaard, M. J. Weygand, K. Kjaer, M. Brust and T. Bjornholm, Faraday Discuss., 2004, 125, 221–233 RSC.
- M. H. V. Werts, H. Zaim and M. Blanchard-Desce, Photochem. Photobiol. Sci., 2004, 3, 29–32 CAS.
- A. M. Jackson, J. W. Myerson and F. Stellacci, Nat. Mater., 2004, 3, 330–336 CrossRef CAS PubMed.
- C. Gentilini, P. Franchi, E. Mileo, S. Polizzi, M. Lucarini and L. Pasquato, Angew. Chem., Int. Ed., 2009, 48, 3060–3064 CrossRef CAS PubMed.
- A. Centrone, Y. Hu, A. M. Jackson, G. Zerbi and F. Stellacci, Small, 2007, 3, 814–817 CrossRef CAS PubMed.
- G. Guarino, F. Rastrelli, P. Scrimin and F. Mancin, J. Am. Chem. Soc., 2012, 134, 7200–7203 CrossRef CAS PubMed.
- T. Kakiuchi, K. Sato, M. Iida, D. Hobara, S. Imabayashi and K. Niki, Langmuir, 2000, 16, 7238–7244 CrossRef CAS.
- I. Dolamic, S. Knoppe, A. Dass and T. Buergi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- P. Ionita, A. Volkov, G. Jeschke and V. Chechik, Anal. Chem., 2008, 80, 95–106 CrossRef CAS PubMed.
- M. Yulikov, P. Lueders, M. F. Warsi, V. Chechik and G. Jeschke, Phys. Chem. Chem. Phys., 2012, 14, 10732–10746 RSC.
- S. Knoppe, J. Boudon, I. Dolamic, A. Dass and T. Buergi, Anal. Chem., 2011, 83, 5056–5061 CrossRef CAS PubMed.
- S. Knoppe, I. Dolamic and T. Buergi, J. Am. Chem. Soc., 2012, 134, 13114–13120 CrossRef CAS PubMed.
- L. H. Dubois and R. G. Nuzzo, Annu. Rev. Phys. Chem., 1992, 43, 437–463 CrossRef CAS.
- I. G. Dance, L. J. Fitzpatrick, A. D. Rae and M. L. Scudder, Inorg. Chem., 1983, 22, 3785–3788 CrossRef CAS.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Makinen, O. A. Wong, H. Hakkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 CrossRef CAS PubMed.
- B. Varnholt, I. Dolamic, S. Knoppe and T. Buergi, Nanoscale, 2013, 5, 9568–9571 RSC.
- S. Knoppe, I. Dolamic, A. Dass and T. Buergi, Angew. Chem., Int. Ed., 2012, 51, 7589–7591 CrossRef CAS PubMed.
- S. Malola, L. Lehtovaara, S. Knoppe, K.-J. Hu, R. E. Palmer, T. Buergi and H. Hakkinen, J. Am. Chem. Soc., 2012, 134, 19560–19563 CrossRef CAS PubMed.
- S. Knoppe, S. Michalet and T. Buergi, J. Phys. Chem. C, 2013, 117, 15354–15361 CAS.
- S. Knoppe, R. Azoulay, A. Dass and T. Buergi, J. Am. Chem. Soc., 2012, 134, 20302–20305 CrossRef CAS PubMed.
- B. Molina, A. Sanchez-Castillo, S. Knoppe, I. L. Garzon, T. Buergi and A. Tlahuice-Flores, Nanoscale, 2013, 5, 10956–10962 RSC.
- S. Knoppe and T. Buergi, Phys. Chem. Chem. Phys., 2013, 15, 15816–15820 RSC.
- N. Barrabes, B. Zhang and T. Burgi, J. Am. Chem. Soc., 2014, 136, 14361–14364 CrossRef CAS PubMed.
- L. Beqa, D. Deschamps, S. Perrio, A.-C. Gaumont, S. Knoppe and T. Buergi, J. Phys. Chem. C, 2013, 117, 21619–21625 CAS.
- P. Pengo, C. Bazzo, M. Boccalon and L. Pasquato, J. Chem. Soc., Chem. Commun., 2015, 3204–3207 RSC.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 CrossRef CAS PubMed.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 RSC.
- M. Zachary and V. Chechik, Angew. Chem., Int. Ed., 2007, 46, 3304–3307 CrossRef CAS PubMed.
- J. B. Schlenoff, M. Li and H. Ly, J. Am. Chem. Soc., 1995, 117, 12528–12536 CrossRef CAS.
- K. S. Mayya and M. Sastry, Langmuir, 1998, 14, 6344–6346 CrossRef CAS.
- Y. Song, T. Huang and R. W. Murray, J. Am. Chem. Soc., 2003, 125, 11694–11701 CrossRef CAS PubMed.
- R. P. Carney, G. A. DeVries, C. Dubois, H. Kim, J. Y. Kim, C. Singh, P. K. Ghorai, J. B. Tracy, R. L. Stiles, R. W. Murray, S. C. Glotzer and F. Stellacci, J. Am. Chem. Soc., 2008, 130, 798–799 CrossRef CAS PubMed.
- T. Burgi and A. Baiker, Acc. Chem. Res., 2004, 37, 909–917 CrossRef PubMed.
- D. Ferri and T. Burgi, J. Am. Chem. Soc., 2001, 123, 12074–12084 CrossRef CAS PubMed.
- G. A. Attard, J. Phys. Chem. B, 2001, 105, 3158–3167 CrossRef CAS.
- A. J. Gellman, Y. Huang, X. Feng, V. V. Pushkarev, B. Holsclaw and B. S. Mhatre, J. Am. Chem. Soc., 2013, 135, 19208–19214 CrossRef CAS PubMed.
- C. F. McFadden, P. S. Cremer and A. J. Gellman, Langmuir, 1996, 12, 2483–2487 CrossRef CAS.
- R. Fasel, M. Parschau and K. H. Ernst, Nature, 2006, 439, 449–452 CrossRef CAS PubMed.
- M. O. Lorenzo, C. J. Baddeley, C. Muryn and R. Raval, Nature, 2000, 404, 376–379 CrossRef CAS PubMed.
- A. Cossaro, S. Terreni, O. Cavalleri, M. Prato, D. Cvetko, A. Morgante, L. Floreano and M. Canepa, Langmuir, 2006, 22, 11193–11198 CrossRef CAS PubMed.
- A. Kuhnle, T. R. Linderoth, B. Hammer and F. Besenbacher, Nature, 2002, 415, 891–893 CrossRef CAS PubMed.
- T. Greber, Z. Sljivancanin, R. Schillinger, J. Wider and B. Hammer, Phys. Rev. Lett., 2006, 96, 056103 CrossRef CAS.
- G. Gonella, S. Terreni, D. Cvetko, A. Cossaro, L. Mattera, O. Cavalleri, R. Rolandi, A. Morgante, L. Floreano and M. Canepa, J. Phys. Chem. B, 2005, 109, 18003–18009 CrossRef CAS PubMed.
- G. Dodero, L. De Michieli, O. Cavalleri, R. Rolandi, L. Oliveri, A. Dacca and R. Parodi, Colloids Surf., A, 2000, 175, 121–128 CrossRef CAS.
- O. Cavalleri, G. Gonella, S. Terreni, M. Vignolo, L. Floreano, A. Morgante, M. Canepa and R. Rolandi, Phys. Chem. Chem. Phys., 2004, 6, 4042–4046 RSC.
- M. M. Beerbom, R. Gargagliano and R. Schlaf, Langmuir, 2005, 21, 3551–3558 CrossRef CAS PubMed.
- B. Ohtani, A. Shintani and K. Uosaki, J. Am. Chem. Soc., 1999, 121, 6515–6516 CrossRef CAS.
- T. Nakanishi, N. Yamakawa, T. Asahi, N. Shibata, B. Ohtani and T. Osaka, Chirality, 2004, 16, S36–S39 CrossRef CAS PubMed.
- S. Katano, Y. Kim, H. Matsubara, T. Kitagawa and M. Kawai, J. Am. Chem. Soc., 2007, 129, 2511–2515 CrossRef CAS PubMed.
- P. Morf, N. Ballav, M. Putero, F. von Wrochem, J. M. Wessels and T. A. Jung, J. Phys. Chem. Lett., 2010, 1, 813–816 CrossRef CAS.
- J. J. Wei, C. Schafmeister, G. Bird, A. Paul, R. Naaman and D. H. Waldeck, J. Phys. Chem. B, 2006, 110, 1301–1308 CrossRef CAS PubMed.
- T. Nakanishi, N. Banno, M. Matsunaga, T. Asahi and T. Osaka, Colloids Surf., A, 2006, 284, 270–275 CrossRef PubMed.
- A. Bejarano-Villafuerte, M. W. van der Meijden, M. Lingenfelder, K. Wurst, R. M. Kellogg and D. B. Amabilino, Chem. – Eur. J., 2012, 18, 15984–15993 CrossRef PubMed.
- T. Nakanishi, M. Matsunaga, M. Nagasaka, T. Ueno and T. Osaka, Electrochim. Acta, 2008, 53, 6209–6214 CrossRef CAS PubMed.
- M. Matsunaga, D. Yamamoto, T. Nakanishi and T. Osaka, Electrochim. Acta, 2010, 55, 4501–4505 CrossRef CAS PubMed.
- M. Bieri and T. Burgi, J. Phys. Chem. B, 2005, 109, 22476–22485 CrossRef CAS PubMed.
- M. Bieri and T. Burgi, Langmuir, 2005, 21, 1354–1363 CrossRef CAS PubMed.
- M. Bieri and T. Burgi, ChemPhysChem, 2006, 7, 514–523 CrossRef CAS PubMed.
- M. Bieri and T. Burgi, J. Phys. Chem. B, 2005, 109, 10243–10250 CrossRef CAS PubMed.
- R. Wirz, T. Burgi, W. Lindner and A. Baiker, Anal. Chem., 2004, 76, 5319–5330 CrossRef CAS PubMed.
- R. Wirz, T. Burgi and A. Baiker, Langmuir, 2003, 19, 785–792 CrossRef CAS.
- C. Gautier and T. Burgi, ChemPhysChem, 2009, 10, 483–492 CrossRef CAS PubMed.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2000, 104, 2630–2641 CrossRef CAS.
- H. Yao, T. Fukui and K. Kimura, J. Phys. Chem. C, 2007, 111, 14968–14976 CAS.
- H. Yao, K. Miki, N. Nishida, A. Sasaki and K. Kimura, J. Am. Chem. Soc., 2005, 127, 15536–15543 CrossRef CAS PubMed.
- C. Gautier and T. Burgi, J. Am. Chem. Soc., 2006, 128, 11079–11087 CrossRef CAS PubMed.
- C. Gautier, R. Taras, S. Gladiali and T. Burgi, Chirality, 2008, 20, 486–493 CrossRef CAS PubMed.
- C. Gautier and T. Burgi, J. Am. Chem. Soc., 2008, 130, 7077–7084 CrossRef CAS PubMed.
- S. Knoppe, A. C. Dharmaratne, E. Schreiner, A. Dass and T. Buergi, J. Am. Chem. Soc., 2010, 132, 16783–16789 CrossRef CAS PubMed.
- S. Knoppe, A. Dass and T. Buergi, Nanoscale, 2012, 4, 4211–4216 RSC.
- I. L. Garzon, J. A. Reyes-Nava, J. I. Rodriguez-Hernandez, I. Sigal, M. R. Beltran and K. Michaelian, Phys. Rev. B: Condens. Matter, 2002, 66, 4 CrossRef.
- M. R. Goldsmith, C. B. George, G. Zuber, R. Naaman, D. H. Waldeck, P. Wipf and D. N. Beratan, Phys. Chem. Chem. Phys., 2006, 8, 63–67 RSC.
- S. Knoppe, O. A. Wong, S. Malola, H. Hakkinen, T. Burgi, T. Verbiest and C. J. Ackerson, J. Am. Chem. Soc., 2014, 136, 4129–4132 CrossRef CAS PubMed.
- S. Knoppe, N. Kothalawala, V. R. Jupally, A. Dass and T. Burgi, J. Chem. Soc., Chem. Commun., 2012, 4630–4632 RSC.
- N. Shukla, M. A. Bartel and A. J. Gellman, J. Am. Chem. Soc., 2010, 132, 8575–8580 CrossRef CAS PubMed.
- N. Shukla, N. Ondeck and A. J. Gellman, Surf. Sci., 2014, 629, 15–19 CrossRef CAS PubMed.
- R. Arnold, A. Terfort and C. Woll, Langmuir, 2001, 17, 4980–4989 CrossRef CAS.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559–3568 CrossRef CAS.
- A. Kudelski, Vib. Spectrosc., 2005, 39, 200–213 CrossRef CAS PubMed.
- V. De Renzi, L. Lavagnino, V. Corradini, R. Biagi, M. Canepa and U. del Pennino, J. Phys. Chem. C, 2008, 112, 14439–14445 CAS.
- T. Hayashi, Y. Morikawa and H. Nozoye, J. Chem. Phys., 2001, 114, 7615–7621 CrossRef CAS PubMed.
- H. S. Kato, J. Noh, M. Hara and M. Kawai, J. Phys. Chem. B, 2002, 106, 9655–9658 CrossRef CAS.
- M. Farrag, M. Tschurl, A. Dass and U. Heiz, Phys. Chem. Chem. Phys., 2013, 15, 12539–12542 RSC.
- A. Tlahuice-Flores, Mol. Simul., 2013, 39, 428–431 CrossRef CAS PubMed.
- A. Tlahuice-Flores, R. L. Whetten and M. Jose-Yacaman, J. Phys. Chem. C, 2013, 117, 12191–12198 CAS.
- B. Varnholt, P. Oulevey, S. Luber, C. Kumara, A. Dass and T. Burgi, J. Phys. Chem. C, 2014, 118, 9604–9611 CAS.
- J. Petroski, M. Chou and C. Creutz, J. Organomet. Chem., 2009, 694, 1138–1143 CrossRef CAS PubMed.
- J. F. Parker, J. P. Choi, W. Wang and R. W. Murray, J. Phys. Chem. C, 2008, 112, 13976–13981 CAS.
- C. S. Levin, B. G. Janesko, R. Bardhan, G. E. Scuseria, J. D. Hartgerink and N. J. Halas, Nano Lett., 2006, 6, 2617–2621 CrossRef CAS PubMed.
- I. Dolamic, B. Varnholt and T. Burgi, Phys. Chem. Chem. Phys., 2013, 15, 19561–19565 RSC.
- L. A. Nafie, J. C. Cheng and P. J. Stephens, J. Am. Chem. Soc., 1975, 97, 3842–3843 CrossRef CAS.
- T. Burgi, A. Urakawa, B. Behzadi, K. H. Ernst and A. Baiker, New J. Chem., 2004, 28, 332–334 RSC.
- C. Gautier and T. Burgi, J. Chem. Soc., Chem. Commun., 2005, 5393–5395, 10.1039/b509346c.
- C. Gautier and T. Burgi, J. Phys. Chem. C, 2010, 114, 15897–15902 CAS.
- H. Yao, N. Nishida and K. Kimura, Chem. Phys., 2010, 368, 28–37 CrossRef CAS PubMed.
- I. Dolamic, B. Varnholt and T. Burgi, Nat. Commun., 2015, 6, 7117 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |