DOI:
10.1039/C5NR02506K
(Paper)
Nanoscale, 2015,
7, 16647-16657
Cellulose nanocrystals with tunable surface charge for nanomedicine†
Received
17th April 2015
, Accepted 18th June 2015
First published on 19th June 2015
Abstract
Crystalline nanoparticles of cellulose exhibit attractive properties as nanoscale carriers for bioactive molecules in nanobiotechnology and nanomedicine. For applications in imaging and drug delivery, surface charge is one of the most important factors affecting the performance of nanocarriers. However, current methods of preparation offer little flexibility for controlling the surface charge of cellulose nanocrystals, leading to compromised colloidal stability under physiological conditions. We report a synthesis method that results in nanocrystals with remarkably high carboxyl content (6.6 mmol g−1) and offers continuous control over surface charge without any adjustment to the reaction conditions. Six fractions of nanocrystals with various surface carboxyl contents were synthesized from a single sample of softwood pulp with carboxyl contents varying from 6.6 to 1.7 mmol g−1 and were fully characterized. The proposed method resulted in highly stable colloidal nanocrystals that did not aggregate when exposed to high salt concentrations or serum-containing media. Interactions of these fractions with four different tissue cell lines were investigated over a wide range of concentrations (50–300 μg mL−1). Darkfield hyperspectral imaging and confocal microscopy confirmed the uptake of nanocrystals by selected cell lines without any evidence of membrane damage or change in cell density; however a charge-dependent decrease in mitochondrial activity was observed for charge contents higher than 3.9 mmol g−1. A high surface carboxyl content allowed for facile conjugation of fluorophores to the nanocrystals without compromising colloidal stability. The cellular uptake of fluoresceinamine-conjugated nanocrystals exhibited a time–dose dependent relationship and increased significantly with doubling of the surface charge.
Introduction
Cellulose nanocrystals (CNCs) are the elementary building blocks of cellulosic materials. These rod-shaped nanocrystals exhibit remarkable properties, namely, high tensile strength, low density, low coefficients of thermal expansion, high aspect ratio, high surface area, as well as optical and electrical functionality.1 They have attracted a tremendous level of attention as indicated by the increasing number of scientific contributions1–3 and industrial investments in the field.4 CNCs provide a potentially green alternative to common functional nanoparticles since they degrade faster than macroscopic cellulose, whereas other important and widely used nanoparticles such as fullerenes and carbon nanotubes are not biodegradable.5 These properties, combined with the relative ease of surface modification (due to the reactive hydroxyl groups of cellulose), present CNCs as a potential game changer in many fields ranging from automotive, aerospace and electronics industries to medical devices, drug delivery and medical imaging.
CNCs exhibit attractive properties as nanoscale carriers for bioactive molecules in nanobiotechnology and nanomedicine, namely elongated morphology,6 high aspect ratio,7 large surface area, ease of bioconjugation,8 biocompatibility and biodegradability. For applications in imaging and drug delivery, surface charge is one of the most important factors affecting the performance of nanoparticles.9 Not only does surface charge affect the biocompatibility and uptake of the nanoparticles,10 it can also determine how fast the nanoparticles are cleared out by the host immune system.11 Moreover, controlling surface charge can increase colloidal stability in complex fluids since nanocarriers are prone to aggregation in the presence of salt, blood or plasma thereby drastically reducing their cellular uptake. CNCs synthesized by conventional acid hydrolysis have a maximum negative charge of 0.8 mmol g−1 and methods such as TEMPO oxidation lead to slightly higher charges.12 It is thus desirable to synthesize CNCs with adequate and tunable surface charge to guarantee their stability in complex media without adversely affecting their biocompatibility and cellular uptake.
Acid hydrolysis is the most common method to synthesize CNCs.13 Sulphuric acid is typically used to treat cellulosic biomass to remove the amorphous regions in cellulose fibers, leaving the highly ordered crystalline regions intact.14 This method results in negatively charged, sulfonated nanoparticles.15 The alternative is to oxidize the pulp using 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)–NaBr–NaClO,16 or ammonium persulfate,17 both of which produce negatively charged, carboxylated CNCs. Each method has important advantages for certain applications but neither offer any control over particle charge; slight modification of particle charge is only possible by varying the source of cellulosic biomass18 or significantly changing the reaction conditions.19 Furthermore, due to the relatively low surface charge, all the methods mentioned require ultrasonication to disperse and stabilize the CNC suspension. Colloidal CNCs prepared in this manner have a high tendency to aggregate in media with higher salt content or with a change of pH; for example, during bioconjugation reactions, or when subjected to complex fluids such as blood. Our group previously reported that using a two-step oxidation process, cellulosic fibres can be separated into their nano-sized crystalline building blocks without the necessity of intensive treatments such as mechanical shear or ultrasonication.20 Further investigations revealed that these nanocrystals were decorated with a surprisingly high number of carboxyl groups (more than 6 mmol g−1), with zeta potentials ranging from −40 mV at high and low pH to −100 mV at physiological pH.21 Our investigations provided evidence for the existence of a soft, porous layer of dicarboxylic cellulose (DCC) chains on the poles of the nanocrystals that host most of the carboxyl groups.21 These polymeric chains expand and collapse with ionic strength (Scheme S1†), electrosterically stabilizing the CNC dispersions at ionic strengths up to 2 M,22 whereas CNC suspensions prepared via conventional methods aggregate at 30 mM. Herein, we have developed a novel and easily scalable method to synthesize CNCs with tunable surface charge from a single sample of starting pulp without the need to modify the reaction conditions. To assess their applicability as nanocarriers, the interaction of nanocrystal fractions having six different surface charge contents was investigated with phagocytic and non-phagocytic cell lines to quantify their uptake and biocompatibility as a function of surface charge. To confirm that the obtained results are not biased by the specific physiology of a single cell line, three different immortalized non-phagocytic cell lines were utilized in this investigation.
Results and discussion
CNCs are rod-shaped nanocrystals that contain only a small number of defects resulting in an extraordinary axial Young's modulus, close to that derived from theoretical chemistry. Unlike most other nanocrystals, which require synthesis under highly controlled conditions, these nanocrystals are developed by nature and packed into trees and plants and thus only require isolation from natural resources. CNCs are of interest for biomedical applications due to their shape, surface chemistry and biocompatible/biodegradable nature. The motivation for this work was to develop CNCs that are stable under physiological conditions and to develop an approach to tune the surface charge of CNCs during synthesis as a means of controlling their interactions with tissue cells.
Characterization of CNCs
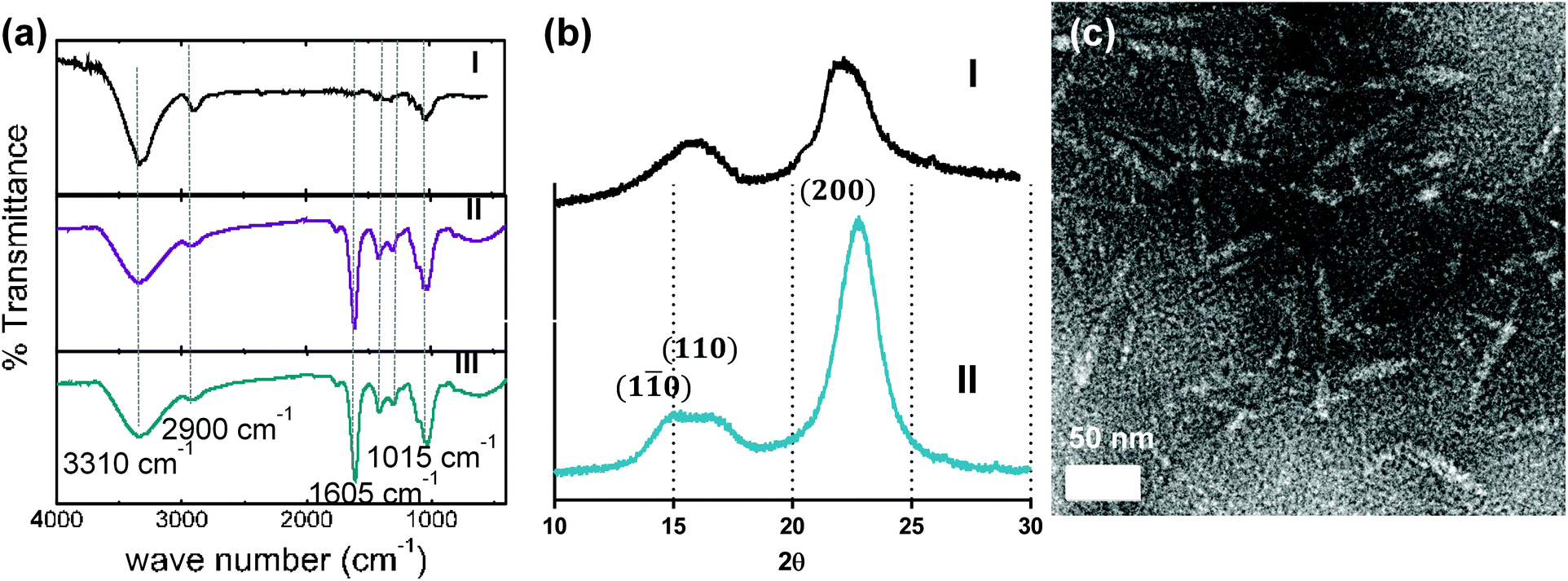
Softwood cellulose pulp was oxidized with sodium periodate to introduce aldehyde groups on the cellulose chains followed by sodium chlorite oxidation to convert the aldehyde groups to carboxyl groups. The carboxylated glucose chains were further cleaved with HCl to various degrees to achieve nanocrystals with different carboxyl contents. FTIR spectra for the treated pulp are shown in Fig. 1a. The band at 1200 cm−1 was used for normalization of FTIR spectra, as is common for cellulosic samples.22 Non-treated pulp (curve I) showed the characteristic peaks for cellulose, namely (1) a broad peak at 3310 cm−1 due to the stretching of hydroxyl groups, (2) the peak at 1298 cm−1 due to the hydroxyl bending vibration,23 (3) peaks at 2900, 1416 and 1015 cm−1 due to C–H stretching vibration, –CH2 scissoring and ˙CH2–O–CH2 stretching, respectively.24 Curves II (CNC before HCl treatment) and III (CNC after 3 h of HCl treatment) showed a distinctive peak at 1605 cm−1 for carboxyl vibration in the sodium form.25 The crystalline properties of each fraction were examined by XRD as shown in Fig. 1b. The 2θ angles of 15.3°, 16.5° and 22.6° in Fig. 1b correspond to the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), (110) and (200) peaks, which is typical of cellulose Iβ.12,26 Using eqn (1) in the Experimental section, the crystallinity index (C.I.) of the starting cellulose was calculated to be 79%, the C.I. of the fraction with the highest carboxyl content was 90%, which indicates the high crystallinity of this fraction. The C.I. of the fractions treated with HCl increased to 96%, indicating that the acid treatment cleaved the amorphous carboxylated cellulose chains, thus increasing overall crystallinity (Table 1).
0), (110) and (200) peaks, which is typical of cellulose Iβ.12,26 Using eqn (1) in the Experimental section, the crystallinity index (C.I.) of the starting cellulose was calculated to be 79%, the C.I. of the fraction with the highest carboxyl content was 90%, which indicates the high crystallinity of this fraction. The C.I. of the fractions treated with HCl increased to 96%, indicating that the acid treatment cleaved the amorphous carboxylated cellulose chains, thus increasing overall crystallinity (Table 1).
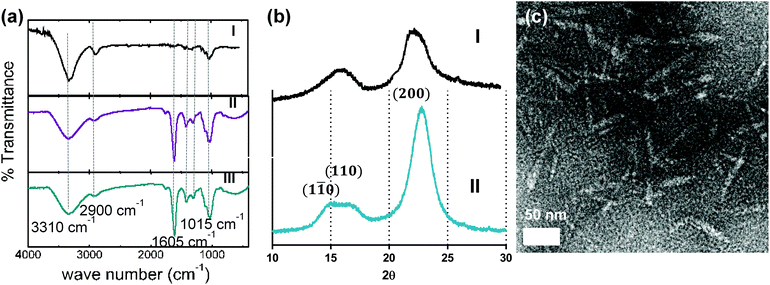 |
| | Fig. 1 (a) FTIR spectra of (I) original pulp, (II) non-hydrolysed CNC with high carboxyl content and (III) 3 h-hydrolysed CNC with low carboxyl content, (b) X-ray diffractogram of (I) starting pulp and (II) CNC oxidized for 3 h, (c) tunneling electron micrograph of CNC hydrolysed for 3 h. | |
Table 1 Characterization of CNC fractions with controlled surface charge
| Fraction # |
Hydrolysis time (h) |
–COOH content (mmol g−1) |
C.I. |
Hydrodynamic diameter (nm) |
TEM size (nm), length–width |
|
FL: fluoresceinamine.
|
| 1 |
0 |
6.6 |
90.0% |
234 ± 4 |
97 ± 8–4 ± 2.1 |
| 2 |
0.5 |
4.7 |
90.0% |
227 ± 2 |
107 ± 3–5 ± 2 |
| 3 |
1 |
3.8 |
— |
219 ± 4 |
103 ± 2–6 ± 1 |
| 4 |
1.5 |
2.9 |
— |
197 ± 2 |
105 ± 8–4 ± 2 |
| 5 |
2 |
2.2 |
95.3% |
151 ± 6 |
110 ± 3–7 ± 1 |
| 6 |
3 |
1.7 |
96.1% |
125 ± 7 |
107 ± 1–7 ± 2 |
| 1 + FLa |
— |
2.8 |
— |
233 ± 8 |
107 ± 3–5 ± 1 |
| 2 + FLa |
— |
1.6 |
— |
228 ± 3 |
98 ± 6–4 ± 3 |
The amount of carboxyl groups on each CNC fraction was determined using conductometric titrations. The data in Table 1 show a decrease in the concentration of carboxyl groups with increasing HCl treatment time. It is important to note that based on our previously published surface charge model,21 most of the carboxyl groups are located on amorphous carboxylated cellulose chains protruding from the poles of the nanocrystals, leading to a zeta potential of −90 to −100 mV under physiological conditions. Acid hydrolysis cleaves these chains, thus decreasing the number of carboxyl groups. The number of carboxylic groups for fractions 1 and 2 also decreased after conjugation with fluoresceinamine as approximately half of the carboxyl groups were covalently bound to the amine group on the fluorophore. The DLS hydrodynamic diameter of all fractions measured in DI is presented in Table 1. Hydrodynamic diameters for the nanocrystals dispersed in PBS and cell culture medium are presented in Table S1.† All the fractions displayed stable colloidal dispersions in DI, PBS and cell culture medium during the period of DLS experiments. CNCs exhibit smaller hydrodynamic diameters in PBS and cell culture medium, which can be explained by the expanding and coiling of the amorphous DCC chain with a change in ionic strength.21,22 Accordingly, fractions 5 and 6, that have been treated with acid for a longer time, and are thus expected to have shorter DCC chains, exhibit a relatively smaller change in diameter with increasing ionic strengths. Fluoresceinamine-conjugated CNCs exhibited comparable hydrodynamic diameters to the bare CNCs under the same physicochemical conditions. This demonstrates one of the key advantages of the proposed CNC synthesis approach; namely, decorating the CNCs with a high number of carboxyl groups provides a large experimental window for functionalizing the particles without destabilizing the suspension and aggregating the particles due to complete masking of surface charge. This is of particular interest for nanobiotechnology and nanomedicine applications since the nanocrystals must be stable under physiological pH and ionic strengths after they are loaded with drug molecules, nucleic acid fragments or fluorophores. TEM imaging confirmed the sizes obtained with DLS and showed that the dimensions (length and diameter) of the nanoparticles did not change significantly for the different fractions (Fig. 1c and Table 1).
Effect of CNCs on cell viability
Effect of CNCs on metabolic activity of cells.
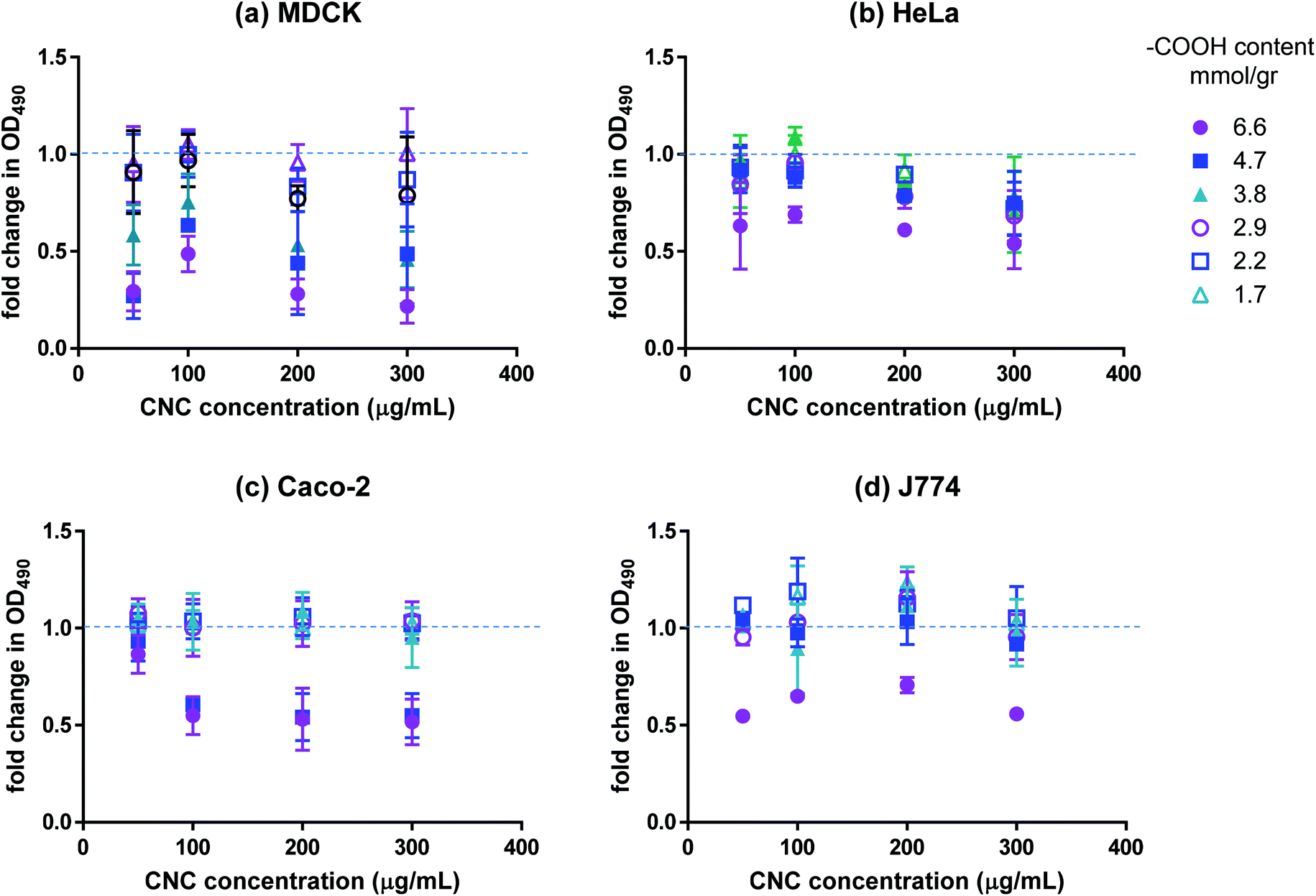
The effect of CNCs on the viability of cultured mammalian cell lines (listed in Table 2) was investigated using various in vitro assays. We used four different cell lines for our work; namely, cells from the human colon, kidney and cervix as well as macrophage cell lines. Macrophage cells are a part of the body's immune system and act to clear out nanocarriers (and other external agents) from the body. Kidney is one of the key points of accumulation of nanoparticles after they have been cleared by the body. Human colon cell lines were used due to the relatively higher feasibility of drug delivery to the gastrointestinal tract. There are concerns that injecting nanoparticles into the blood stream may cause severe stimulation of the immune system. However, nanodrug delivery through the GI tract is less risky and more feasible, making the cell lines from the GI tract a useful model for uptake studies. Finally, HeLa cells (cervix) are used as a general model for uptake. Cancer cells were used in all cases due to the relevance of nanodrug delivery to cancer therapy. The MTS assay was used to assess the metabolic activity of cells in contact with CNCs. The reduction of MTS into a soluble formazan by metabolically active cells was used to quantify the respiration level of the cells. The results of the MTS assay are presented in Fig. 2. Although the magnitude of the effect varied for the different cell lines, a clear charge-dependent loss of mitochondrial activity was observed. The MDCK cell line exhibited the most marked decrease, with the metabolic activity decreasing by more than 75% compared to the control for fraction 1 and by ∼50% for fractions 2 and 3. For the other cell lines, fraction 1 resulted in ∼50% reduction in metabolic activity with the effect diminishing sharply for the other fractions; for carboxyl contents below 3.8 mmol g−1 (fraction 4) the effect was negligible. Interestingly, the metabolic activity exhibited a weak dependence on CNC dosage in the range of 100–300 μg mL−1. It can be inferred from the data that CNCs with surface carboxyl contents above a certain threshold are not fully biocompatible. To investigate whether the decrease in metabolic activity observed in our work was a result of negative charge or the presence of carboxylated cellulose on the surface of the nanocrystals, cells were treated with dicarboxylic cellulose (DCC). No change in their metabolic activity was detected with the MTS assay (data not shown). Although there is no evidence in the literature for mitochondrial cytotoxicity of carboxyl groups (carboxyl polystyrene particles are believed to be benign towards mitochondria,27 whereas amine-functionalized polystyrene particles damage mitochondria and lysosomes28), high charge density of either polarity has been reported to result in increased in vitro and in vivo cytotoxicity.9 Most studies have reported negligible cytotoxicity13,18,29–31 and inflammatory response30,32 associated with CNCs. However, when CNCs prepared from different biomass sources were compared, a slight decrease in cell spreading was reported for cells with a higher carboxyl content (0.2 mmol g−1).33 In another study, fluorophore-conjugated CNCs with a net negative zeta potential were reported to result in a decrease in cell metabolic activity compared to the same nanocrystals conjugated to a different fluorophore which resulted in nanocrystals with a positive zeta potential.34 The latter report must be interpreted with caution, since the charge density on the negative CNC was at least 10 times higher than the positive CNC, and thus, mitochondrial toxicity cannot be attributed solely to the polarity of CNC charge, especially since there are numerous reports in the literature on the cytotoxicity of positively charged particles.9 It is noteworthy that all of the aforementioned studies used CNCs with a significantly lower surface carboxyl content (5–8 times lower) than our study and that we did not observe a significant loss in metabolic activity for carboxyl contents up to 3.8 mmol g−1. Once loaded with the desired cargo (fluorophores, drugs, DNA strands, etc.), CNCs with higher carboxyl contents are unlikely to have adverse effects on cell metabolic activity if the loading results in the decrease of their net negative charge as indicated by the results for fluoresceinamine-conjugated CNCs (Fig. S1†).
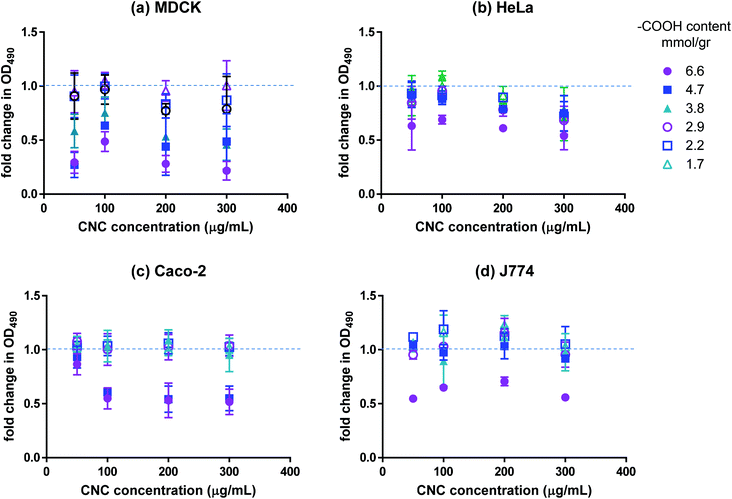 |
| | Fig. 2 Fold change in mitochondrial activity of (a) MDCK, (b) HeLa, (c) Caco-2, and (d) J774 as indicated by OD490 of CNC-treated cells incubated with MTS. Values present the average OD490 for the samples divided by the average OD490 of the negative control; n = 3, error bars represent 95% confidence intervals. | |
Table 2 Cell lines used in this study
| Cell line |
Tissue type |
Tissue |
Cell type |
Source |
| Caco-2 |
Human |
Colon |
Epithelial |
ATCC HTB-37 |
| HeLa |
Human |
Cervix |
Epithelial |
ATCC CCL-2 |
| MDCK |
Dog |
Kidney |
Epithelial |
ATCC CCL-34 |
| J774 |
Mouse |
Ascites |
Macrophage |
ATCC TIB-67 |
Effect of CNCs on cell membrane integrity
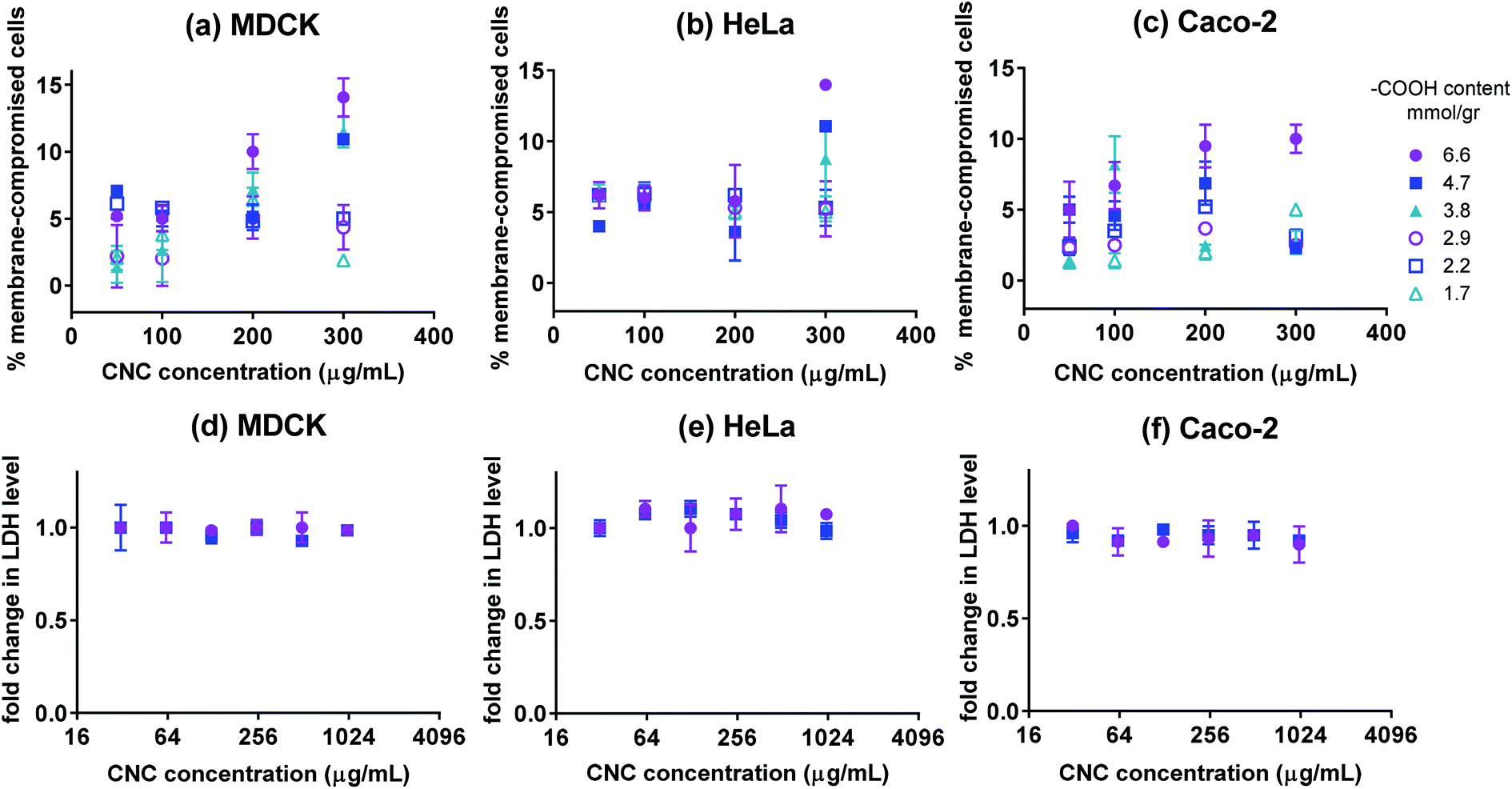
The Live/Dead fluorescence assay was used to quantify the percentage of live and dead cells after 24 h of incubation with CNCs. The Live/Dead assay distinguishes live cells by the presence of ubiquitous intracellular esterase activity. The non-fluorescent calcein AM is enzymatically converted to fluorescent calcein in live cells. Cells with damaged membranes are quantified by ethidium homodimer-1 (EthD-1) which only permeates cells with compromised membranes. The percentage of membrane-compromised cells is presented in Fig. 3(a–c). After 24 h of incubation with CNCs, all samples contained more than 90% cells with uncompromised membranes except for cells incubated with high concentrations of fraction 1. The negative control contained 5–8% membrane compromised cells, thus only fraction 1 at high concentrations appeared to have a pronounced effect on cell membrane integrity. To further confirm the results of the Live/Dead assay, LDH assay was used to verify possible release of cellular biomolecules into the medium by membrane-compromised cells. Lactate dehydrogenase (LDH) is a cytosolic enzyme present in different cell types. Plasma membrane damage releases LDH into the cell culture medium which can be quantified by a coupled enzymatic reaction. Fig. 3(d–f) show LDH levels for cells incubated with various concentrations of CNCs having 6.6 or 4.7 mmol g−1 of charge. The change in the level of LDH in the cell culture medium appeared to be lower than the resolution of the assay. This was expected since based on the Live/Dead assay, less than 10% of cells exhibited compromised membranes. The results of these two assays indicate that the CNCs used in this investigation have a negligible effect on cell membrane integrity; except when the fraction with the highest charge density was used at high concentrations. Previous reports indicate that carboxyl-functionalized nanoparticles did not cause membrane disruption in non-phagocytic cells as opposed to amine-functionalized nanoparticles which generally permeate the cell membrane.35
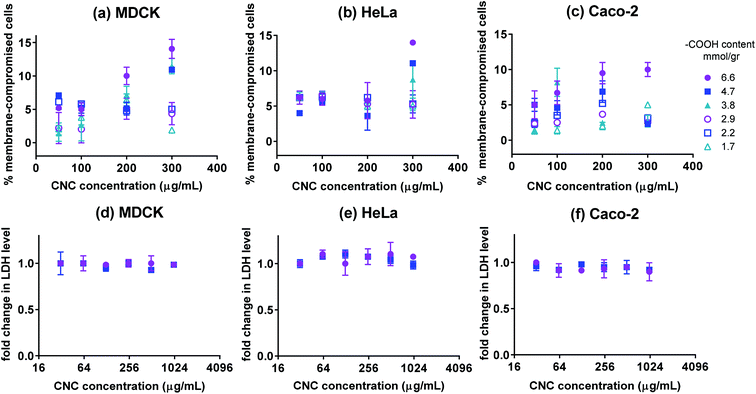 |
| | Fig. 3 The percentage of membrane-compromised cells after 24 h incubation with CNCs having various surface –COOH contents for cultures of (a) MDCK, (b) HeLa, and (c) Caco-2 cells. Fold change in LDH level for (d) MDCK, (e) HeLa, and (f) Caco-2 cells incubated for 24 h with CNCs (only fractions 1 and 2 with the highest charge); n = 3 and error bars represent 95% confidence intervals. | |
Cellular uptake of CNCs
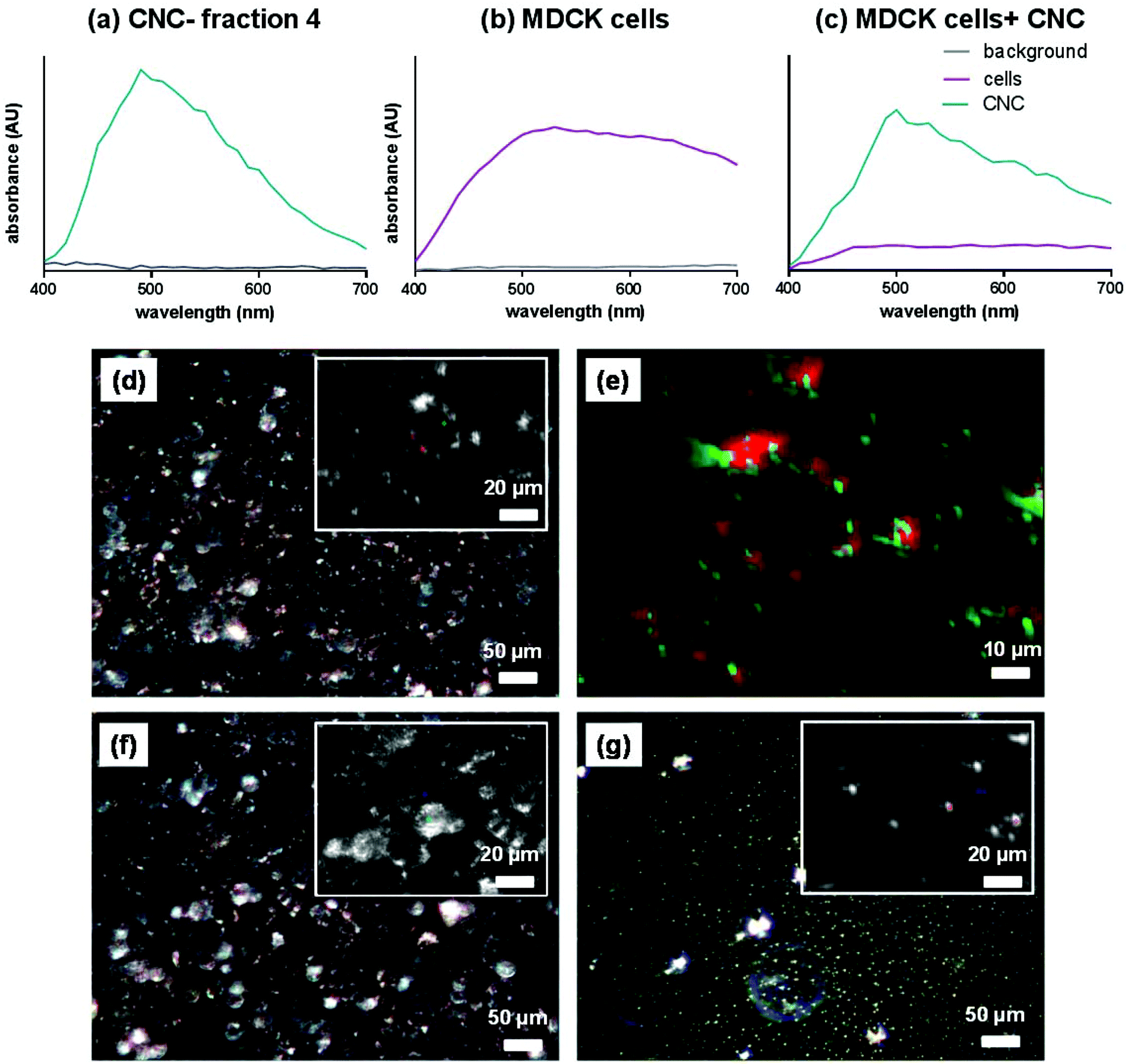
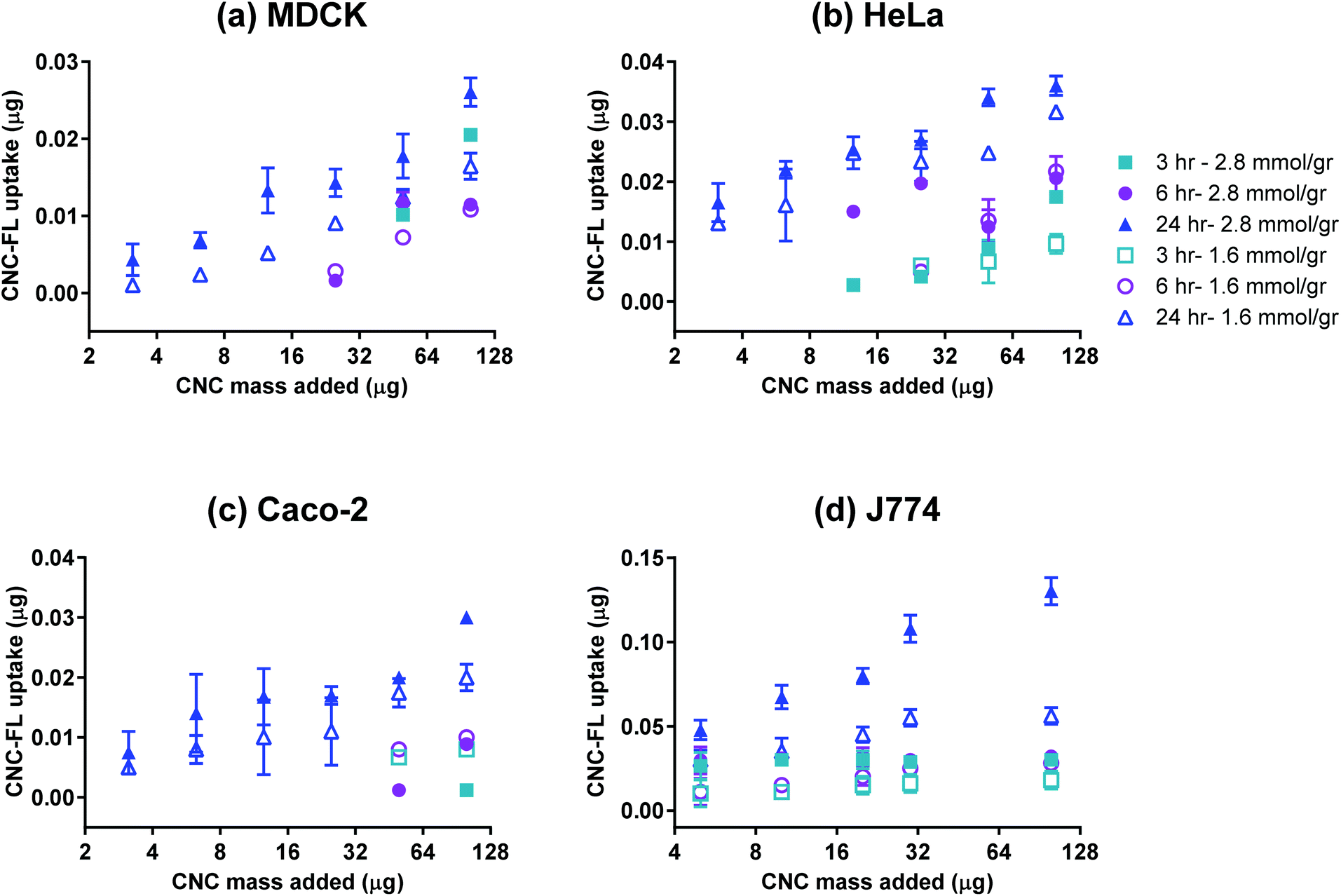
The effective cellular uptake of CNCs was qualitatively confirmed using darkfield hyperspectral imaging (Fig. 4) for bare CNCs (fraction 4) and confocal microscopy for fluoresceinamine-conjugated CNCs (Fig. 5). Darkfield hyperspectral microscopy provides the possibility of imaging nanoscale structures within live or fixed cells without the need for nanoparticle labeling. The spectral data in Fig. 4a–c show that the CNCs exhibit a strong spectral profile which is clearly distinct from that of the HeLa cells. Thus, the CNCs are readily identified in tissue cultures that were incubated with the nanocrystals once the free nanocrystals are washed away (Fig. 4a and b). Confocal microscopy provided further confirmation of the presence of CNCs (fluorophore conjugated) inside the cells (Fig. 5). Since membrane integrity of the cells was shown to be intact, the presence of CNCs inside the cells indicates a mechanism of active or passive uptake. The uptake of fluoresceinamine-conjugated CNCs was quantified in terms of fluorescence intensity for cells treated with these nanocrystals. This fluorescence intensity was converted to the mass of nanoparticles uptaken using a calibration curve (a representative calibration curve is shown in Fig. S2†). CNC uptake exhibited clear time- and dose-dependent relationships (Fig. 6), although the mass of uptaken particles was generally a low percentage of the particles added to the cells. Nonetheless, even this low percentage was enough to be clearly imaged via fluorescence and hyperspectral microscopy (Fig. 4). The macrophage cells (J774) exhibited particle uptake 4 times higher than that of the non-phagocytic cells (Fig. 6) which was expected due to their nature. The uptake was minimal after 3 h but significantly increased after incubation for 24 h. Interestingly, uptake increased with increasing particle charge density. Although there is no consensus in the literature regarding the effect of surface charge on the uptake of nanoparticles, there have been reports of carboxylated nanoparticles exhibiting higher cellular uptake.36 It was previously shown that oblong particles are uptaken by the cells starting from their poles,6,37 thus the chemistry on the poles of the nanocrystal could be a determining factor in particle uptake. The CNCs synthesized in the current investigation possess soft, amorphous, highly carboxylated DCC chains on their poles.21,22 It can be hypothesized that the increase in uptake for CNCs with a higher charge is due to the interaction of these carboxyl-rich chains with the cell membrane. This would explain the observed decrease in particle uptake when the length and/or density of these chains are decreased. Confirmation of this hypothesis requires a more in-depth investigation. It is noteworthy that the uptake experiments were conducted with nanocrystals having charge densities below the threshold limit where the CNCs exhibit mitochondrial toxicity and a (mild) effect on membrane integrity. Obtaining results for higher CNC charges was not possible due to the inevitable masking of CNC charge during the process of conjugation with fluorophores. The uptake behavior may change for CNC charges closer to the threshold surface charge.
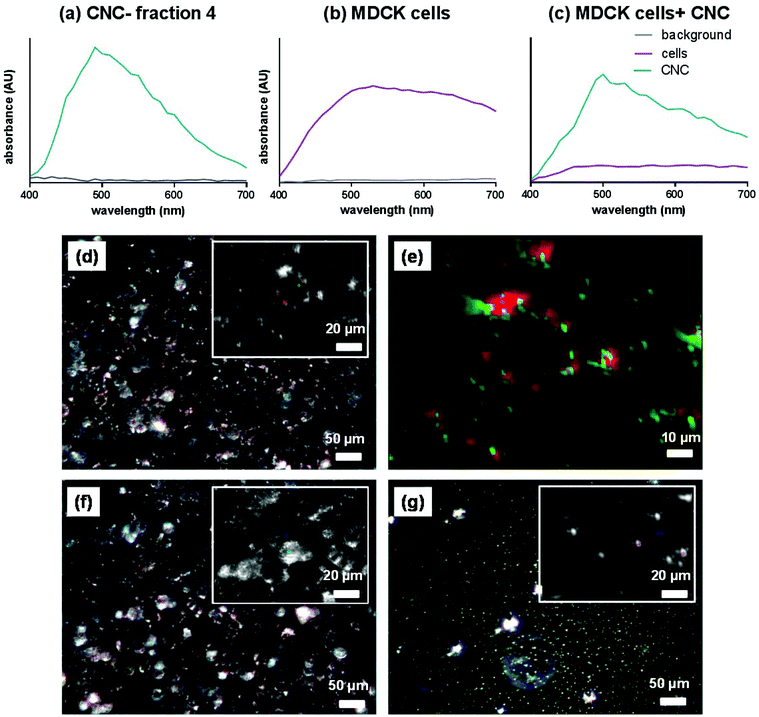 |
| | Fig. 4 Intensity of spectra for (a) CNCs only (fraction 4), (b) cells only and (c) cells and CNCs (fraction 4). Representative darkfield spectral images for (d) HeLa cells with CNCs (fraction 4), (e) the digitally magnified version of the inset from panel (d) with CNCs digitally colored in green and the cells digitally colored in red, (f) HeLa cells without CNCs, and (g) CNCs only. The insets in panels (d), (f) and (g) show optically magnified sections of each image. | |
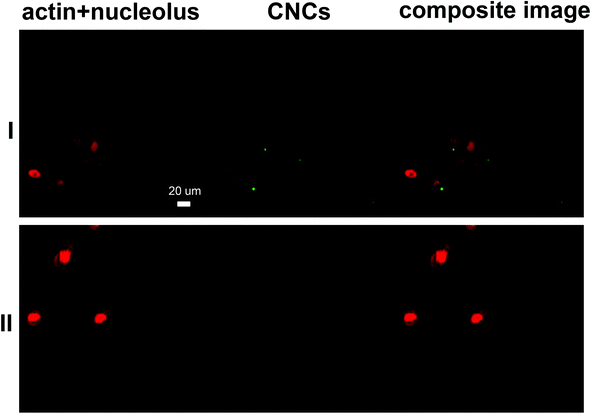 |
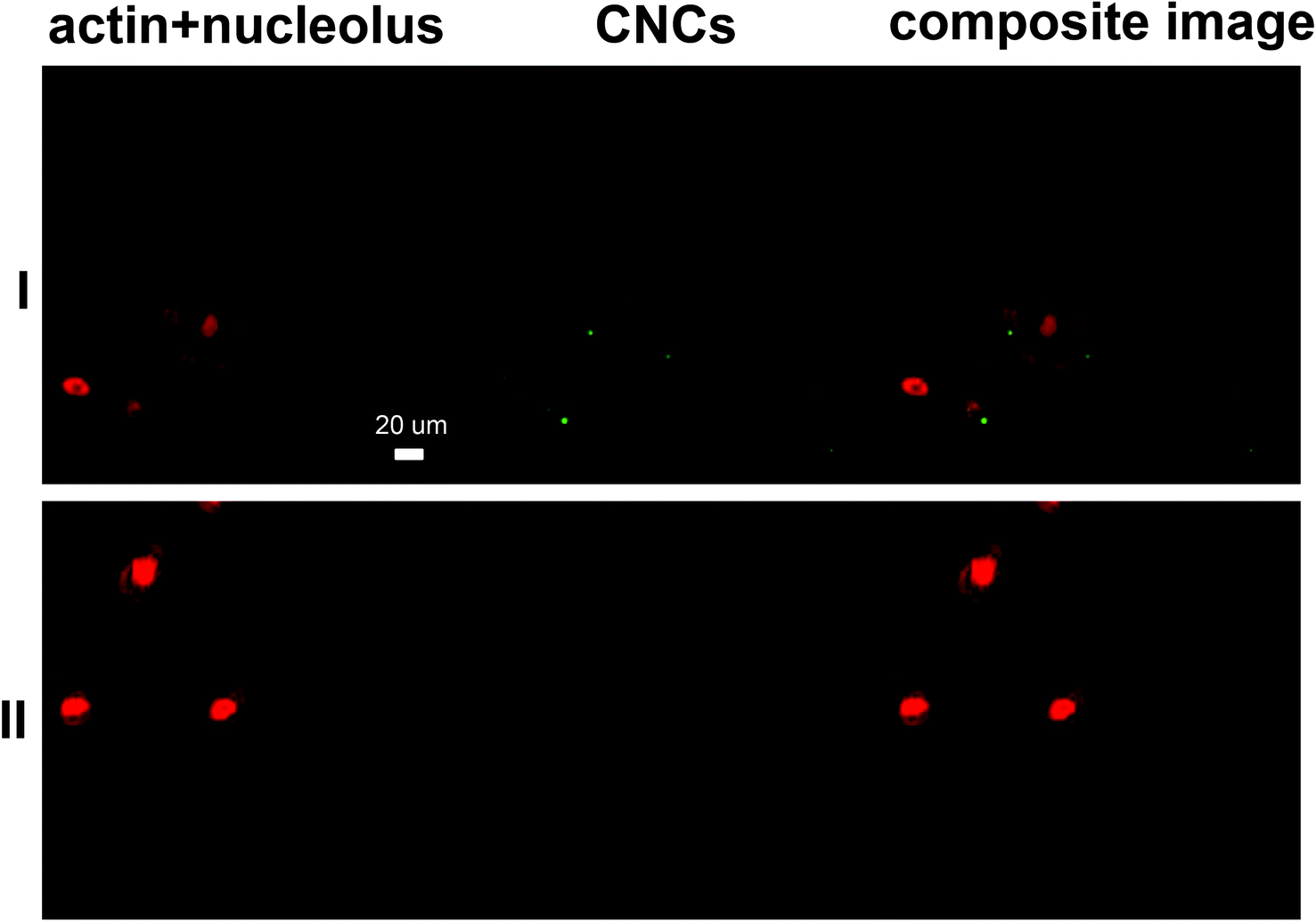
| | Fig. 5 Representative CLSM images of MDCK cells incubated with (I) CNCs and (II) the negative control. Channel 1 presents cells stained with Eth-1 and Alexa Fluor® 633 (both in red) and channel 2 represents fluoresceinamine-conjugated CNCs (green dots). | |
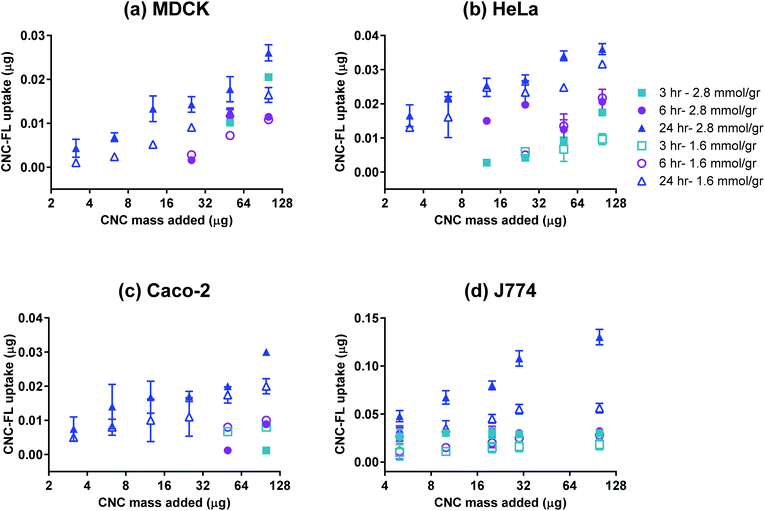 |
| | Fig. 6 Uptake of CNC-FL by (a) MDCK, (b) HeLa, (c) Caco-2, and (d) J774 cell lines; n = 3, error bars represent 95% confidence intervals. | |
Experimental
Preparation of cellulose nanocrystals
CNCs were prepared based on the method of Yang et al.22 with key modifications to allow for tunable control of surface charge. Q-90 softwood pulp sheets (10 g) were soaked in DI water overnight. The wet pulp was thoroughly dispersed by using a disintegrator (Noram), filtered through a nylon mesh and strained to remove excess water and added to a solution of 0.093 M NaIO4 and 1 M NaCl. The periodate oxidation reaction was performed at room temperature in a continuously stirred reaction vessel protected from light (96 h, 105 rpm). The reaction was stopped by adding 4% ethylene glycol to quench the residual periodates; the aldehyde-derivatized pulp was subsequently washed thoroughly and resuspended in deionized water (DI) (1 g of pulp in 50 mL DI). To convert the aldehyde groups to carboxyl, NaCl, NaClO2 and H2O2 were added to the aldehyde pulp suspension to a final concentration of 1 M, 0.26 M and 0.26 M, respectively. The mixture was stirred at room temperature (24 h, 105 rpm) and the pH was maintained at 5 using NaOH. The nanocrystals were separated from the non-reacted residues by centrifugation (26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 min); the nanocrystals in the supernatant were then precipitated by adding an organic solvent (ethanol), removed by centrifugation (20000g, 10 min) and re-suspended in DI. To prepare CNC fractions with different carboxyl contents, the highly charged CNC fraction was hydrolysed with 3 N HCl at 45 °C for various time periods (30 min to 3 h). The excess HCl was neutralized by NaHCO3 at the end of hydrolysis and the product was purified by dialysis (MWCO: 8000) with 4–5 exchanges of DI. The concentration of each fraction was determined by weighing the samples after drying overnight at 70 °C.
000g, 10 min); the nanocrystals in the supernatant were then precipitated by adding an organic solvent (ethanol), removed by centrifugation (20000g, 10 min) and re-suspended in DI. To prepare CNC fractions with different carboxyl contents, the highly charged CNC fraction was hydrolysed with 3 N HCl at 45 °C for various time periods (30 min to 3 h). The excess HCl was neutralized by NaHCO3 at the end of hydrolysis and the product was purified by dialysis (MWCO: 8000) with 4–5 exchanges of DI. The concentration of each fraction was determined by weighing the samples after drying overnight at 70 °C.
Characterization of CNCs
Fourier transform infrared (FTIR) spectroscopy.
A transmission FTIR spectrometer (Perkin Elmer) with single bounce diamond ATR (Attenuated Total Reflectance) accessory was used to characterize the samples after each step of treatment. Freeze-dried samples were placed directly on the ATR crystal and maximum pressure was applied by lowering the tip of the pressure clamp using a ratchet-type clutch mechanism. All the spectra of measured samples were averaged from 32 scans from 550 to 4000 cm−1 with a resolution of 4 cm−1.
X-ray diffraction (XRD).
XRD measurements were performed on freeze-dried CNCs using a Bruker Discover D8 two dimensional diffractometer with a VANTEC 2D detector and CuKα radiation (λ = 1.54 Å). The X-ray diffractograms were acquired with a 2θ (Bragg angle) range of 10–30° at a scan rate of 0.005 s−1. The crystallinity index (C.I.) of cellulose38 was used to quantify the crystallinity of each fraction:| | | C.I. = 100(I200 − IAM)/I200 | (1) |
Here, I200 is the intensity of the (200) plane reflection, typically located at approximately 2θ = 22.6°; IAM is the intensity at 2θ = 18°, corresponding to the minimum in a diffractogram.
Conductometric titration.
The concentration of carboxyl groups on the nanocrystals was determined for each fraction using conductometric titrations. Conductometric titrations were performed on a Metrohm 836 Titrando instrument. A volume of CNC suspension containing 0.02 g of nanocrystals was diluted to 200 mL with DI. The ionic strength of the suspension was adjusted using NaCl solution (to 0.28 mM) and pH was adjusted to 3.5 using 0.1 M HCl. The mixture was titrated with a 10 mM NaOH solution at a rate of 0.1 mL min−1. The part of the titration curve representing weak acid titration (negligible change in conductivity with time) was used to calculate the carboxyl content.
Dynamic light scattering (DLS).
The hydrodynamic diameter of the nanocrystals was measured using a Malvern Instruments Zetasizer Nano-ZS. The samples were diluted to 0.1 wt% in an appropriate medium (DI, 1× phosphate buffered saline (PBS) or cell culture medium) and filtered using a 0.45 μm syringe filter. Standard quartz cuvettes were used for all measurements and each measurement was repeated five times at room temperature.
Transmission electron microscopy (TEM).
CNC size was further examined by TEM. A glow-discharged carbon coated copper grid was placed on a 5 μL drop of sample (0.1 w/w%) for 5 min and washed 3 times by placing on drops of water for 10 s. Samples were negatively stained using 2% uranyl acetate and examined via a Philips Tecnai 12 120 kV TEM and images were captured with a Gatan 792 Bioscan 1k × 1k Wide Angle Multiscan CCD camera. For measuring particle size using TEM, 30 images were captured from each sample and at least 50 particles were measured in each captured micrograph using Image J.
Preparation of fluoresceinamine-conjugated CNCs
4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM, Sigma) was used as the coupling reagent. The CNC suspension was diluted to a carboxyl concentration of 0.1 g mL−1. Fluoresceinamine (ex/em 496/525, Sigma) was added to the CNC suspension at a ratio of 0.5:1 (carboxyl:fluoresceinamine) for a 0.5 degree of substitution, and dissolved by increasing the pH to 11. The suspension was then mixed with DMTMM at a 1:1 molar ratio with the carboxyl content, and then the reaction mixture was mixed gently for 1 h (in the dark). The resulting fluoresceinamine-conjugated CNC was purified by dialysis (MWCO: 8000) with 4–5 exchanges of DI. To prepare a calibration curve, serial dilutions of CFC-FL in PBS were added to wells of a 96 well plate (Costar, black, clear bottom). The fluorescence intensity was recorded and plotted versus the mass of CFC-FL in each well. The calibration equation was obtained using linear regression (Origin Pro, v.8).
Cell lines and culture conditions
The cell lines used in this study are presented in Table 2. All cell lines, except for Caco-2, were cultured in high glucose DMEM (Invitrogen), supplemented with 5% FBS and 1% penicillin–streptomycin (Life Technologies) and incubated in a humidified incubator (37 °C, 5% CO2). Caco-2 was cultured in α-MEM (Life Technologies) supplemented with 10% FBS, 1% P/S and 2 mM glutamine. All cell lines used have an adherent phenotype. For passaging, J774 cells were scraped from the flask whereas all other cell lines were detached using 0.05% Trypsin-EDTA and passages 1–5 were used for all experiments.
CNC–cell interaction assays
For all the interaction assays, the cells were seeded in flat Costar, tissue culture treated, 96 well plates, clear or black with clear bottom (for fluorescence assays), at a loading of 105 cells per well and incubated overnight in a humidified incubator (37 °C, 5% CO2). The media in the wells were then replaced with α-MEM with 1% P/S to eliminate possible interference of serum with the nanoparticle–cell interaction. Appropriate CNC fractions in DI water were added to each well with a range of concentrations. Negative control wells contained cells incubated with the same volume of DI water without CNCs. After appropriate incubation times (37 °C, 5% CO2), the assays were performed as described below.
MTS assay.
The metabolic activity of cells after incubation with CNC fractions was quantified using the tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS, Sigma). Cells were incubated for 24 h with one of the 6 different fractions of CNC having different carboxyl contents. The wells were washed 3× with PBS (supplemented with MgCl2 and CaCl2) and 100 μL of α-MEM containing 0.5 mg mL−1 MTS was added to each well. The plates were incubated for 1 h (37 °C, 5% CO2) to allow the MTS tetrazolium compound to be bioreduced by cells into a colored formazan product that is soluble in tissue culture medium. Absorbance of the converted dye was measured at a wavelength of 490 nm using a microplate reader (Tecan Infinite M200 Pro).
Membrane integrity assay.
The Live/Dead viability/cytotoxicity kit for mammalian cells (Invitrogen) was used to quantify the number of cells with compromised membranes. The Live/Dead kit consists of calcein AM which stains living cells fluorescent green (ex/em ∼495 nm/∼515 nm) and ethidium homodimer-1 (EthD-1) which binds to nucleic acids of cells with damaged membranes (ex/em ∼495 nm/∼635 nm). The cells were seeded in tissue culture treated black polystyrene 96 well plates (Costar) and incubated for 24 h (37 °C, 5% CO2) with one of the 6 fractions of CNC with varying carboxyl contents. The cells were subsequently washed 3× with PBS+/+ and the fluorescence intensity of the dyes was quantified using a microplate reader operating in fluorescence mode (Tecan Infinite M200 Pro).
LDH assay.
The lactose dehydrogenase (LDH) assay was performed using an LDH kit (Pierce). The assay measured the amount of LDH secreted into the medium by membrane-compromised cells. The cells were seeded in tissue culture treated black polystyrene 96 well plates (Costar) and incubated for 24 h (37 °C, 5% CO2) with one of the 6 fractions of CNCs with varying carboxyl contents. The medium in each well was subsequently mixed with a reaction mixture, as per kit instructions, to allow the formation of a red formazan product (ex/em 490/680) which was quantified using a microplate reader operating in fluorescence mode (Tecan Infinite M200 Pro).
Cellular uptake assay.
For cellular uptake studies, cells were plated into tissue culture treated black polystyrene 96 well plates (Costar) and incubated with CNC-FL for 3 h, 6 h or 24 h. The wells were subsequently washed 3× with PBS+/+, covered with 100 μL of PBS and the fluorescence intensity from each well was determined with the fluorescence microplate reader at excitation and emission wavelengths of 496 nm and 525 nm, respectively. The results were qualitatively confirmed via darkfield-enhanced hyperspectral imaging and confocal laser scanning microscopy.
Microscopy
Hyperspectral imaging.
Optical and hyperspectral images along with hyperspectral data were captured using a research grade optical microscope equipped with an advanced darkfield illumination system integrating hyperspectral analysis (Photon etc., Canada). The cell lines were seeded in Fluorodish cell culture dishes (WPI) at 5 × 104 cells per dish. Cells were incubated with CNCs for 24 h. The hyperspectral imaging camera collected the light directly incident upon the sample. Spatial and spectral data were collected for each pixel in the frame of view. The image created was analyzed to spectrally confirm the presence of CNCs. A reference spectral library was created by collecting spectral data for a suspension containing only CNCs or only cells. The software algorithm compared each spectrum in the reference library with those in the tested samples and provided the final mapped images based on the matching spectral signatures.
Confocal laser scanning microscopy (CLSM).
CLSM was used to image the cell lines before and after incubation with CNCs. The cell lines were seeded in Fluorodish cell culture dishes (WPI) at 5 × 104 cells per dish. The cells were incubated with fluoresceinamine-conjugated CNCs for 24 h, fixed with 3.7% formaldehyde, permeabilized with 0.1% Triton X-100 and stained with EthD-1 (ex/em ∼495 nm/∼635 nm) and Alexa FluorR 635 phalloidin (ex/em ∼633/647). Images of stained cells were captured using a Zeiss CLSM system (Jena, Germany).
Statistical analysis
All results are reported in terms of population mean ± 95% confidence intervals. Significance of difference between treated samples and negative control was analysed using Student's t-test with Bonferroni correction using Statistica 8.0 (Stat Soft. Inc., San Jose, CA) and p-values <0.05 were considered significant.
Conclusions
Cellulose nanocrystals are promising candidates as nanocarriers in nanomedicine and biotechnology. Most studies to date have confirmed that they do not exhibit any significant cyto- or genotoxicity. Our work confirms this notion, but highlights the fact that CNCs are no exception from other nanoparticles in that they can exert stress on cells if they possess a high charge density; we found the threshold to be 3.8 mmol g−1. A high charge density is advantageous, however, for stabilizing the nanoparticle suspension under physiological conditions and once they have been loaded with the desired cargo. Another advantage of the prepared CNCs is their carboxylated poles which appear to enhance their uptake by various cell lines. The exact mechanism for this uptake is the subject of ongoing investigation in our group using model cell membranes. The developed nanocrystals do not aggregate in the presence of high ionic strength and serum, which offers the promise of their applicability to other fields with similar conditions such as the development of green nanocomposites.
Acknowledgements
This work is supported by the Natural Sciences and Engineering Research Council of Canada (NSERC Strategic Research Network on Bioactive Paper – SENTINEL) and the Canada Research Chairs (CRC) program.
Notes and references
- J. P. F. Lagerwall, C. Schutz, M. Salajkova, J. Noh, J. H. Park, G. Scalia and L. Bergstrom, NPG Asia Mater., 2014, 6, e80 CrossRef CAS PubMed.
- M. Jacoby, Chem. Eng. News, 2014, 92, 9–12 Search PubMed.
- H. Wei, K. Rodriguez, S. Renneckar and P. J. Vikesland, Environ. Sci.: Nano, 2014, 1, 302–316 RSC.
- M. Williamson, IWP, 2012, 11–12, 31–36 Search PubMed.
- K. Kümmerer, J. Menz, T. Schubert and W. Thielemans, Chemosphere, 2011, 82, 1387–1392 CrossRef PubMed.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS PubMed.
-
R. Maren, D. Shuping, H. Anjali and L. Yong Woo, in Polysaccharide Materials: Performance by Design, American Chemical Society, 2009, vol. 1017, ch. 4, pp. 81–91 Search PubMed.
- S. Eyley and W. Thielemans, Nanoscale, 2014, 6, 7764–7779 RSC.
- E. Fröhlich, Int. J. Nanomedicine, 2012, 7, 5577–5591 CrossRef PubMed.
- S. Hong, J. A. Hessler, M. M. Banaszak Holl, P. Leroueil, A. Mecke and B. G. Orr, J. Chem. Health Saf., 2006, 13, 16–20 CrossRef PubMed.
- J. S. Teodoro, A. M. Simões, F. V. Duarte, A. P. Rolo, R. C. Murdoch, S. M. Hussain and C. M. Palmeira, Toxicol. In Vitro, 2011, 25, 664–670 CrossRef CAS PubMed.
- A. Isogai, M. Usuda, T. Kato, T. Uryu and R. H. Atalla, Macromolecules, 1989, 22, 3168–3172 CrossRef CAS.
- T. Kovacs, V. Naish, B. O'Connor, C. Blaise, F. Gagné, L. Hall, V. Trudeau and P. Martel, Nanotoxicology, 2010, 4, 255–270 CrossRef CAS PubMed.
- R. H. Marchessault, F. F. Morehead and N. M. Walter, Nature, 1959, 184, 632–633 CrossRef CAS PubMed.
- E. Lam, K. B. Male, J. H. Chong, A. C. W. Leung and J. H. T. Luong, Trends Biotechnol., 2012, 30, 283–290 CrossRef CAS PubMed.
- D. da Silva Perez, S. Montanari and M. R. Vignon, Biomacromolecules, 2003, 4, 1417–1425 CrossRef CAS PubMed.
- A. C. W. Leung, S. Hrapovic, E. Lam, Y. Liu, K. B. Male, K. A. Mahmoud and J. H. T. Luong, Small, 2011, 7, 302–305 CrossRef CAS PubMed.
- S. Dong, A. a. Hirani, K. r. Colacino, Y. w. Lee and M. Roman, Nano LIFE, 2012, 02, 1241006 CrossRef.
- S. Beck-Candanedo, M. Roman and D. G. Gray, Biomacromolecules, 2005, 6, 1048–1054 CrossRef CAS PubMed.
-
T. G. M. van de Ven, A. Tejado, M. N. Alam and M. Antal, U.S. Provisional Patent Application3776923-v3, 2011 Search PubMed.
- S. Safari, A. Sheikhi and T. G. M. van de Ven, J. Colloid Interface Sci., 2014, 432, 151–157 CrossRef CAS PubMed.
- H. Yang, M. N. Alam and T. G. M. van de Ven, Cellulose, 2013, 20, 1865–1875 CrossRef CAS.
- S.-N. Yuen, S.-M. Choi, D. L. Phillips and C.-Y. Ma, Food Chem., 2009, 114, 1091–1098 CrossRef CAS PubMed.
- S. M. A. S. Keshk, Carbohydr. Polym., 2008, 74, 942–945 CrossRef CAS PubMed.
- R. Barbucci, A. Magnani and M. Consumi, Macromolecules, 2000, 33, 7475–7480 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- E. Fröhlich, C. Samberger, T. Kueznik, M. Absenger, E. Roblegg, A. Zimmer and T. R. Pieber, J. Toxicol. Sci., 2009, 34, 363–375 CrossRef.
- M. G. Bexiga, J. A. Varela, F. Wang, F. Fenaroli, A. Salvati, I. Lynch, J. C. Simpson and K. A. Dawson, Nanotoxicology, 2011, 5, 557–567 CrossRef CAS PubMed.
- L. Alexandrescu, K. Syverud, A. Gatti and G. Chinga-Carrasco, Cellulose, 2013, 1–11 Search PubMed.
- M. J. D. Clift, E. J. Foster, D. Vanhecke, D. Studer, P. Wick, P. Gehr, B. Rothen-Rutishauser and C. Weder, Biomacromolecules, 2011, 12, 3666–3673 CrossRef CAS PubMed.
- S. Moreira, N. B. Silva, J. Almeida-Lima, H. A. O. Rocha, S. R. B. Medeiros, C. Alves Jr. and F. M. Gama, Toxicol. Lett., 2009, 189, 235–241 CrossRef CAS PubMed.
- J. M. Dugan, J. E. Gough and S. J. Eichhorn, Biomacromolecules, 2010, 11, 2498–2504 CrossRef CAS PubMed.
- K. B. Male, A. C. W. Leung, J. Montes, A. Kamen and J. H. T. Luong, Nanoscale, 2012, 4, 1373–1379 RSC.
- K. A. Mahmoud, J. A. Mena, K. B. Male, S. Hrapovic, A. Kamen and J. H. T. Luong, ACS Appl. Mater. Interfaces, 2010, 2, 2924–2932 CAS.
- A. Petushkov, J. Intra, J. B. Graham, S. C. Larsen and A. K. Salem, Chem. Res. Toxicol., 2009, 22, 1359–1368 CrossRef CAS PubMed.
- X. Jiang, A. Musyanovych, C. Rocker, K. Landfester, V. Mailander and G. U. Nienhaus, Nanoscale, 2011, 3, 2028–2035 RSC.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional results are presented in the ESI in Fig. S1 through S4. See DOI: 10.1039/c5nr02506k |
| ‡ Current address: Physical Intelligence Department, Max Planck Institute for Intelligent Systems, Stuttgart 70569, Germany. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a